
Submitted:
18 September 2023
Posted:
20 September 2023
You are already at the latest version
Abstract
Characterized by severe and fatal arrhythmias induced by cardiac ischemia/reperfusion (CIR), acute myocardial infarction (AMI) is the main cause of morbidity and mortality worldwide. To investigate the cardioprotective role of cardiac Ca2+/cAMP/adenosine signaling in AMI, the effects of L-type Ca2+ channels (LTCC) blocker Nifedipine (NIF) and Verapamil (VER), in the absence and presence of A1-adenosine receptors (A1R) blocker (DPCPX), on the incidence of ventricular arrhythmias (VA), atrioventricular block (AVB) and lethality (LET) induced by CIR in rats were evaluated. CIR was induced in adult male Wistar rats (290-320 g) by occlusion of the left anterior descendent coronary artery (10 min) followed by reperfusion (75 min). VA, AVB and LET incidences were evaluated by ECG analysis and compared between control (CIR group) and intravenously treated 5 min before CIR with NIF 1, 10, and 30 mg/kg (NIF+CIR groups) or DPCPX 100 µg/kg plus NIF 10 mg/kg (DPCPX+NIF+CIR group) and VER 1 mg/kg (VER+CIR group) or DPCPX 100 µg/kg plus VER 1 mg/kg (DPCPX+VER+CIR group). Serum levels of cardiac injury biomarkers (TCK and CK-MB) were quantified. In CIR group, VA, AVB and LET incidences were 90, 80 and 70%, respectively. In NIF+CIR group, incidence of VA was reduced to 50, 30 and 30%, AVB to 25, 10 and 20%, and LET to 25, 20 and 20%, and respectively. In DPCPX+NIF+CIR group, these incidences were not different from CIR group. TCK and CK-MB levels were similar in all groups. These results indicate that pharmacological modulation of the Ca2+/cAMP/adenosine signaling in cardiac cells by LTCC blockade and A1R stimulation could be a new strategy cardioprotective for human AMI therapy.
Keywords:
cardiac ischemia-reperfusion
; cardiac arrhythmias
; pharmacological cardioprotection
; Ca2+ channels
; adenosine receptors
1. Introduction
After 2023, cardiovascular diseases (CVD) will be responsible for over 26 million annual deaths worldwide, in both industrialized and underdeveloped nations, ischemic heart disorders (IHD) are the leading cause of death, among these diseases, ischemic heart disease (IHD), and, particularly, acute myocardial infarction (AMI) are the main causes of morbidity and mortality in worldwide [1,3]. AMI is one of the life-threatening coronary-related pathologies intimately associated with sudden cardiac death, which has a prevalence of about three million people worldwide [4,5]. AMI results in irreversible damage to the myocardium primarily caused by lack of oxygen in cardiac cells, which may lead to impairment in diastolic and systolic function and make the patient prone to severe and fatal cardiac arrhythmias [4,5,6]. Although AMI can lead to a number of serious complications for cardiac function, there are still few pharmacological resources for the treatment of AMI.
The key to treatment of AMI is rapidly restore coronary blood flow after ischemia (reperfusion) [4,5,6,7]. The earlier the treatment (less than 6 hours from symptom onset), the better the prognosis. Although the main form of AMI treatment is reperfusion of myocardium [4,5,6], this process can cause severe cardiac dysfunctions mainly due to abrupt oxygen entry and severe ionic deregulation in cardiac cells, which in turn may lead to lethal arrhythmias directly related to deregulation of intracellular Ca2+ homeostasis in cardiac cells [6,7,8,9,10,11,12]. This deregulation of Ca2+ homeostasis results from modifications of Ca2+ extrusion or buffering that stimulate the Ca2+ to escape spontaneously from the sarcoplasmic reticulum (SR) and cause delayed after-depolarization activity promoted by cytosolic and mitochondrial Ca2+ overload during ischemia. This deregulation causes suboptimal Ca2+-ATPase performance, resulting in increased cytosolic and mitochondrial Ca2+ concentration that collapses mitochondrial function and ATP synthesis [6,7,8,9,10,11,12]. These Ca2+-related dysfunctions induced by cardiac ischemia followed by reperfusion (CIR) frequently result in death due to significant increase in the incidence of cardiac arrhythmias caused by collapse of myocardial function [6,7,8,9,10,11,12]. Most of these early deaths are caused by complex ventricular arrhythmias (VA) and atrio-ventricular blockade (AVB) resulting from collapse of cardiac function generated by CIR [6,7,8,9,10,11,12].
Additionally, to cellular alterations caused by ischemia, the reperfusion also produces important metabolic and functional alterations in cardiac cells. The reperfusion causes an increase in free radical production and increase Ca2+ entry into the cytosol, exacerbating the Ca2+ influx overload through L-type Ca2+ channels (LTCC). This Ca2+ influx promotes the modulation of ryanodine receptors (RyR) and important enzymes, such as adenylyl cyclase (AC), an enzyme that produces cAMP from ATP, in the T-tubules and intracellular medium [6]. The mitochondria also play a role in maintaining the cellular homeostasis of Ca2+ during brief increases in cytosolic Ca2+ concentration ([Ca2+]c) in cardiac cells, which is crucial in the contraction-relaxation cycle of myocardium [6,7]. The Ca2+ concentration in the mitochondrial matrix ([Ca2+]m) is finely controlled by Ca2+ transporter proteins that are present in the mitochondrial membranes and that control Ca2+ influx and efflux in the mitochondrial matrix [6,7]. The mitochondrial Ca2+ influx in cardiac cells and other excitable cells is primarily controlled by the mitochondrial Ca2+ uniporter (MCU), while its efflux is mostly controlled by the mitochondrial Na+/Ca2+ exchanger (mNCX) [6,7]. As a result, the cardiac cycle and contraction-relaxation process are significantly impacted by mitochondria's role in Ca2+ homeostasis in cardiac cells [6,7]. In addition to this decoupling of the cardiac excitation-contraction (CECC) caused by deregulation of intracellular Ca2+ homeostasis, an important increase in free radical production during reperfusion leads to the oxidation of structural proteins, proteins involved in the respiratory chain, pyridine nucleotides, changes in the permeability of the internal mitochondrial membrane, decoupling of oxidative phosphorylation, and a decrease in mitochondrial ATP production [6,7].
In addition to its role in CECC, Ca2+ modulates 3’5’-cyclic adenosine monophosphate (cAMP) production by isoforms 5 and 6 of adenylyl cyclase (AC), and pharmacological blockade of Ca2+ influx via LTCC produces an increase in production and efflux of intracellular cAMP in cardiac cells [6]. In the extracellular medium, cAMP is transformed into adenosine (ADO) that stimulates A1-type ADO receptors (A1R) located in the plasma membrane of cardiac cells to finely regulate cardiac cell function [6]. It is well known that stimulation of cardiac A1R by ADO is a common and effective strategy used to abolish the cardiac arrhythmias in various clinical situations, and especially in cardiac surgery [4,6]. Thus, we have proposed that the pharmacological modulation of Ca2+/cAMP/ADO signaling in cardiac cells could be a promising strategy in the treatment of AMI and other IHD in humans.
Based on the above proposal, in the present work we investigated the effects of pharmacological modulation Ca2+/cAMP/ADO signaling in cardiac cells on the incidence of severe and fatal arrhythmias related to AMI. Thus, using an animal model of AMI, the effects of the blockade of Ca2+ influx via LTCC produced by nifedipine (NIF) and verapamil (VER), in the presence or absence of blocker of transporter-mediated cAMP efflux probenecid (PROB) or A1R-selective antagonist 8-cyclopentyl-1,3-dipropylxanthine (DPCPX), on the incidence of arrhythmias (VA and AVB) and lethality (LET) induced by CIR were studied. In addition, serum concentration of cardiac injury biomarkers total creatine kinase (CK) and CK-MB were quantified.
2. Materials and Methods
2.1. Animals
Male Wistar rats (14- to 16-week-old) weighting between 290 and 320 g, were kept at 21 ± 2°C with 12:12-h light/dark cycle and were given food and water ad libitum. All experimental protocols used in this study were approved by the Ethics Committee of the Escola Paulista de Medicina – Universidade Federal de São Paulo (UNIFESP #1130/11 and #0065/12).
2.2. Cardiac Ischemia and Reperfusion (CIR) Induction
In order to produce an animal model of AMI, rats were submitted surgical procedures in accordance with the approach previously published by our laboratory [7,8,9,10,11,12]. Initially, the rats were anesthetized with ketamine (75 mg/kg, intraperitoneally) and xylazine (8 mg/kg, intraperitoneally). After anesthesia, rats were intubated using a Jelco 14G (USA), and mechanically ventilated using an Insight model EFF 312 mechanical ventilator (Insight Equipamentos Cientificos, Brazil) [7,8,9,10,11,12]. A thoracotomy was done to insert a vascular tourniquet (4/0 braided silk suture linked to a 10-mm micropoint reverse cutting needle, Ethicon K-890H, USA) around the left anterior descending coronary artery to induce ischaemia after the patient had been stabilized for 15 minutes. The tourniquet was removed after 10 min of myocardial ischemia to allow 75 min of coronary recirculation (cardiac reperfusion) [7,8,9,10,11,12].
2.3. Assessment of Cardiac Activity during CIR
All animals underwent ECG analysis to evaluate cardiac activity during CIR, in accordance to previously described methodology [7,8,9,10,11,12]. This ECG analysis was performed to evaluate the effects of NIF and VER, in the presence or absence of blocker ABC transporter-mediated cAMP efflux PROB or A1R-selective antagonist DPCPX, on the incidence of arrhythmias (VA and AVB) and lethality (LET) induced by CIR. Initially, ECG was recorded for 15 minutes before ischemia protocol (stabilization period) and during ischemia (10 minutes) and reperfusion (75 minutes) protocol [7,8,9,10,11,12]. A biopotential amplifier was used to record the ECG using needle electrodes inserted subcutaneously on the limbs. ECG changes (increase in R wave and ST segment) brought on by CIR were utilized to confirm that the coronary artery had successfully been blocked by surgery [7,8,9,10,11,12,13,14,15]. A heated operating table and the proper heating lamps were used to keep body temperature at 37.5°C, and a rectal thermometer was regularly used to check the temperature [7,8,9,10,11,12]. ECG data were captured using the AqDados 7.02 collection equipment from Lynx Tecnologia Ltda. in Brazil and examined using AqDAnalysis 7 software. We assessed heart rates and the incidence of VA, AVB, and LET brought on by CIR using this program. All three conditions were regarded as VA: ventricular fibrillation, torsades de pointes, and ventricular tachycardia [6,7,8,9,10,11,12].
2.4. Biochemical Assessment of Heart Lesions' Biomarkers
Blood samples (3–4 mL) were taken from the abdominal aorta and placed in siliconized tubes to determine the serum levels of biomarkers of cardiac lesion, total creatine kinase (CK) and creatine kinase - MB fraction (CK-MB). These samples were taken in rats that survived the entire 75–minute CIR protocol. Centrifugation of blood samples (2,500 rpm for 40 minutes at 5 °C) was performed. The supernatant was removed and kept at -20°C for enzymatic analysis. A commercial kit from Vida Biotecnologia, Belo Horizonte, Brazil, was used to perform a kinetic UV test, measuring at 340 nm the enzymatic activity of CK and CK-MB in serum [12].
2.5. Pharmacological Treatments
To evaluate the effects of with nifedipine (NIF) (1mg/kg, 10 mg/kg, and 30 mg/kg, IV; Sigma-Aldrich, St. Louis, MO, USA) and verapamil (VER) (1mg/kg, IV; Sigma-Aldrich, St. Louis, MO, USA) on the incidence of VA, AVB and LET caused by CIR, rats were treated with this NIF or VER alone or combined with drugs that block ABC transporter-mediated cAMP efflux from cardiac cells (probenecid, PROB; Sigma-Aldrich, St. Louis, MO, USA) or A1R antagonist (8-cyclopentyl-1,3-dipropylxanthine, DPCPX; Sigma-Aldrich, St. Louis, MO, USA). All drugs were intravenously (IV) administered before CIR. We previously showed that LET in control animals treated with 0.9% saline solution (SS) varied from 70-80 % [6,10]. In the present work, the animals used were divided into the following experimental groups:
- (1)
- CIR group (n = 40): animals treated with 0.9% SS vehicle (IV) one minute before were subjected to a surgical procedure to induce cardiac ischemia (10 min), followed by coronary reperfusion (75 min), and subsequent electrocardiogram (ECG) monitoring (100 min) for determination of VA, AVB and LET incidence;
- (2)
- PROB+CIR group (n = 20): animals treated with blocker ABC transporter-mediated cAMP efflux from cardiac cells probenecid (PROB, 100 mg/kg, IV) five minutes before were subjected to a surgical procedure to induce cardiac ischemia (10 min), followed by coronary reperfusion (75 min), and subsequent electrocardiogram (ECG) monitoring (100 min) for determination of VA, AVB and LET incidence;
- (3)
- NIF1+CIR group (n = 20): animals treated with NIF (1mg/kg, IV) one minute before were subjected to a surgical procedure to induce cardiac ischemia (10 min), followed by coronary reperfusion (75 min), and subsequent electrocardiogram (ECG) monitoring (100 min) for determination of VA, AVB and LET incidence;
- (4)
- NIF10+CIR group (n = 20): animals treated with NIF (10 mg/kg, IV) one minute before were subjected to a surgical procedure to induce cardiac ischemia (10 min), followed by coronary reperfusion (75 min), and subsequent electrocardiogram (ECG) monitoring (100 min) for determination of VA, AVB and LET incidence;
- (5)
- NIF30+CIR group (n = 20): animals treated with NIF (30 mg/kg, IV) one minute before were subjected to a surgical procedure to induce cardiac ischemia (10 min), followed by coronary reperfusion (75 min), and subsequent electrocardiogram (ECG) monitoring (100 min) for determination of VA, AVB and LET incidence;
- (6)
- PROB+NIF+CIR group (n = 20): animals treated with block ABC transporter-mediated cAMP efflux from cardiac cells probenecid (PROB; 100 mg/kg, IV), 5 min before administration of NIF (10 mg/kg, IV) for 1 min before induction of cardiac ischemia (10 min), followed by cardiac reperfusion, and subsequent ECG monitoring for 100 min for determination of the VA, AVB and LET incidence;
- (7)
- DPCPX+NIF+CIR group (n = 20): animals treated with A1R antagonist 8-Cyclopentyl-1,3-dipropylxanthine (DPCPX; 100 µg/kg, IV), 5 min before administration of NIF (10 mg/kg, IV) for 1 min before induction of cardiac ischemia (10 min), followed by cardiac reperfusion, and subsequent ECG monitoring for 100 min for determination of the VA, AVB and LET incidence;
- (8)
- VER1+CIR group (n = 20): animals treated with verapamil (VER, 1 mg/kg, IV) for 1 min before induction of cardiac ischemia (10 min), followed by coronary reperfusion (75 min), and subsequent ECG monitoring (100 min) for determination of the VA, AVB and LET incidence.
- (9)
- PROB+VER+CIR group (n = 20): animals treated with blocker ABC transporter-mediated cAMP efflux from cardiac cells probenecid (PROB; 100 mg/kg, IV), 5 min before administration of VER (1 mg/kg, IV) for 1 min before induction of cardiac ischemia (10 min), followed by cardiac reperfusion, and subsequent ECG monitoring for 100 min for determination of the VA, AVB and LET incidence;
- (10)
- DPCPX+VER+CIR group (n = 20): animals treated with A1R antagonist 8-Cyclopentyl-1,3-dipropylxanthine (DPCPX; 100 µg/kg, IV), 5 min before administration of VER (1 mg/kg, IV) for 1 min before induction of cardiac ischemia (10 min), followed by cardiac reperfusion, and subsequent ECG monitoring for 100 min for determination of the VA, AVB and LET incidence;
Data analysis
Data corresponding to VA, AVB, and LET incidences were expressed as percentages and statistically analyzed using Fisher’s exact test with the Prism 5.0 software (GraphPad, USA) [7,8,9,10,11,12]. Data corresponding to serum concentration (mg/dL) of biomarkers of cardiac lesion (total CK and CK-MB) were expressed as mean ± standard error of mean (SEM) and statistically analyzed by an analysis of variance test using Prism [12]. Results were considered statistically significant when p < 0.05.
3. Results
Incidence of VA, AVB and LET induced by CIR
No arrhythmias were detected during the stabilization periods of any animal (15 min). During CIR, VA and AVB were detected and measured in different experimental groups. After CIR, the incidence of VA, AVB and LET were 90%, 80% and 70%, respectively (Figure 1).
Effects of the NIF and VER on the incidence of VA, AVB and LET induced by CIR
Figure 1 shows that incidences of AVB and LET induced by CIR was significantly reduced by treatment with NIF (1, 10 and 30 mg/kg, IV) and VER (1 mg/kg, IV). VA incidence was reduced from 90% to 30% in NIF10+CIR and 30% in NIF30+CIR groups, compared to CIR group. AVB incidence was reduced from 80% to 30% in NIF1+CIR, 20% in NIF10+CIR, and 20% in NIF30+CIR groups, compared to CIR group. LET incidence was reduced from 70% to 30% in NIF1+CIR, 10% in NIF10+CIR, and 20% in NIF30+CIR, compared to CIR group. In addition, treatment with VER was also able to reduce the incidences of VA (90% to 20%), AVB (90% to 20%), and LET (90% to 20%) induced by CIR. These results confirm previous studies [6,7,11] that demonstrated that the blockade of Ca2+ influx via LTCC in cardiac cells before CIR attenuates cardiac collapse and reduces the incidence of severe and fatal arrhythmias induced by CIR.
Effects of the pre-treatment with PROB or DPCPX before administration of NIF or VER on the incidence of VA, AVB and LET induced by CIR
To investigate whether the Ca2+/cAMP/ADO signaling in cardiac cells is involved in the cardioprotective effect of NIF and VER (Figure 1), we pretreated rats submitted to CIR with DPCPX (100 µg/kg, IV) or PROB (100 mg/kg, IV), as well as NIF (10 mg/kg, IV) and VER (1 mg/kg, IV). Figure 2 shows that the reduction of VA, AVB and LET incidence in the PROB+NIF+CIR, PROB+VER+CIR, DPCPX+NIF+CIR, and DPCPX+VER+CIR groups was not statistically different from the CIR group, indicating that pretreatment with DPCPX and PROB completely abolished the cardioprotective effects of NIF and VER. These results indicate that an increment in extracellular levels of ADO due to cAMP transport to extracellular environment combined with an increase of activation by ADO of A1R receptors in cardiac cells directly participate in the cardioprotective response stimulated by NIF and VER in rats submitted to CIR.
Effects of the pre-treatment with DPCPX before administration of NIF and VER on biochemical markers of cardiac injury
Figure 3A shows that serum concentration of biomarkers of cardiac lesion, total CK and CK-MB, were not statistically different in CIR (5,487 ± 449 mg/dL, n = 3), NIF1+CIR (5,395 ± 876 mg/dL, n = 5), NIF10+CIR (5,344 ± 193 mg/dL, n = 5), NIF30+CIR (5,018 ± 508 mg/dL, n = 5), DPCPX+NIF+CIR (4,864 ± 445 mg/dL, n = 5), VER+CIR (4,437 ± 771 mg/dL, n = 5), and DPCPX+VER+CIR (4,802 ± 254 mg/dL, n = 5) groups. Similarly, Figure 3B shows that serum CK-MB concentrations were also not statistically different in CIR (2,225 ± 290 mg/dL, n = 3), NIF1+CIR (2,087 ± 61 mg/dL, n = 5), NIF10+CIR (2,054 ± 106.3 mg/dL, n = 5), NIF30+CIR (2,112 ± 102 mg/dL, n = 5), and DPCPX+NIF+CIR (1,954 ± 161 mg/dL, n = 5), VER+CIR (2,905 ± 656 mg/dL, n = 5), and DPCPX+VER+CIR (2,701 ± 350 mg/dL, n = 5) groups. Thus, NIF, DPCPX, and VER seem to modulate cardiac electric activity to attenuate arrhythmias and LET post-CIR, with no impact in the extent or severity of ischemic cell lesions.
4. Discussion
In the present work, we showed that the pharmacological modulation of Ca2+/cAMP/ADO signaling in cardiac cells by means the attenuation of Ca2+ influx via LTCC combined with an increase of activation of A1R by ADO generated by increment of extracellular transport of cAMP reduced the incidence of severe and fatal arrhythmias induced by CIR (see Figure 1 and Figure 2). This cardioprotective effect stimulated by pharmacological modulation of Ca2+/cAMP/ADO signaling in cardiac cells has been supported by several experimental studies that showed in cellular and animal models of CIR [6,7,10]. It is well known that the attenuation of cytosolic Ca2+ overload produced by L-type Ca2+ channel blockers and stimulation of cardiac A1R produced by ADO and others A1R agonists and constitutes a common and effective pharmacological strategy used to abolish the cardiac arrhythmias in various clinical situations, and especially in cardiac surgery [4,6]. Thus, pharmacological modulation of Ca2+/cAMP/ADO signaling in cardiac cells could be a promising therapeutic strategy to reduce the incidence of severe and fatal arrhythmias caused by AMI in humans.
The dynamic equilibrium between the concentration of Ca2+ into cytosol, sarcoplasmic reticulum and mitochondria is crucial to finely control cardiac excitation-contraction coupling (CECC) [6,16,17,18]. Thus, deregulation of cellular Ca2+ homeostasis causes decoupling of CECC, increasing the incidence of cardiac arrhythmias [6]. ATP deficit during ischemia inhibits ATP-dependent ionic transporters, like Na+/K+-ATPase, Ca2+-ATPase plasmalemmal (PMCA) and sarco-endoplasmic reticulum Ca2+-ATPase (SERCA), leading to accumulation of Na+ and Ca2+ in the cytosol [6,17] and cytosolic Ca2+overload [6,19]. This process induces an increase in mitochondrial Ca2+ influx, which further reduces ATP production and, consequently, collapses the cardiac function [6]. Cytosolic and mitochondrial Ca2+ overload severely compromises CECC, favoring the development of severe and fatal arrhythmias [6,17,20]. Thus, drugs that reduce Ca2+ influx through L-type Ca2+ channels (LTCC) in cardiac cells significantly reduced the incidence of severe and fatal arrhythmias induced by CIR, strengthening the idea that the attenuation of cytosolic and mitochondrial Ca2+ overload reduces cardiac collapse caused by CIR [6].
In addition to its role in CECC, LTCC-mediated Ca2+ influx in cardiac cells modulates cAMP production by AC isoforms 5 (AC5) and 6 (AC6) [6,21,22], and pharmacological block of Ca2+ via LTCC increases production and efflux of intracellular cAMP [6,21,22]. In the extracellular medium, cAMP is transformed into ADO that can stimulate A1R located in the plasma membrane of cardiac cells to finely regulate cardiac function [6,22]. A1R stimulation with ADO is a common and effective strategy used to abolish the cardiac arrhythmias in various clinical situations, and especially in cardiac surgery [6].
Biochemical analyses of membrane preparations in overexpression systems have been used to establish the paradigm for Ca2+-mediated inhibition of AC5 and AC6 in the submicromolar range [23]. In fact, the crystal structure of an AC5-catalytic domain-containing high affinity Ca2+-pyrophosphate (PPi) complex was just recently published [23]. Although there are many papers reporting Ca2+-mediated modulation of AC6 activity in an endogenous setting, whole cell overexpression experiments provide the majority of the evidence for Ca2+ inhibition of AC6 [6,21,22]. Although less thorough (six research in overexpression systems and three in endogenous systems), the evidence for Ca2++ inhibition of the extremely comparable AC5 is consistent in its assertion. As a result, there is strong support for the idea that Ca2+ inhibits both AC5 and AC6 and that this inhibition occurs both in vitro and in vivo [6,21,22,23].
Physiological effects of knocking down AC5 and AC6 have been seen in several investigations, although none of these can be clearly linked to the enzymes' Ca2+- inhibitability. Mice lacking the AC5 gene have impaired pain perception, diminished motor activity, and altered heart function [23]. The preponderance of AC5 and AC6 in cardiac tissue has been hypothesized to have a substantial role in the rhythmicity of sympathetic regulation of inotropy [23]. The AC5 mutant animals exhibit a lower left ventricular ejection fraction, attenuated baroreflexes, and a lack of acetylcholine-mediated Gi inhibition of AC activity [23]. They also have a reduced Ca2+-mediated inhibition of cAMP. Reduced left ventricular function is shown in AC6 knockout mice, as well as a diminished Ca2+-mediated suppression of cAMP [22,23].
Additionally, cytosolic Ca2+ also regulates several intracellular second messengers and various cellular responses [6,17,18]. For instance, increased Ca2+ influx through LTCC modulates the β1-adrenoceptors (β1AR)-mediated excitatory response of cardiac cells, due to Ca2+-induced inhibition of AC activity [6,22]. Thus, cytosolic cAMP increases due to cardiac β1AR stimulation are even higher when Ca2+ influx is decreased, such as by the action of LTCC blockers [22]. In addition, cAMP efflux mediated by multidrug resistance proteins transporters (MRPT) in cardiac cells leads cAMP to the extracellular medium, where it is degraded into ADO, which in turn regulates cellular functions [6,7,24].
In stress conditions, such as hypoxia or ischemia, increased extracellular ADO levels are responsible for cardioprotective effects, which involve, at least in part, activation of Gi-coupled A1R [25,26,27]. Activation of A1R and A3R has been shown to decrease cardiac infarction lesion size, as well as to consistently improve functional recovery in isolated hearts [28,29,30,31]. A1R mediates the direct negative chronotropic and dromotropic actions of ADO, as well as indirect anti-β1AR actions [32,33,34,35]. It is significant to note that pharmacological stimulation of cardiac A1R lowers cardiac cell excitability [36,39], perhaps reducing the likelihood of fatal AVB. There is substantial evidence that activation of all four AR (A1, A2A, A2B and A3) are importantly involved cardioprotective response in different pathological conditions, including the CIR [33,36].
In adult male rats submitted to in vivo regional myocardial ischemia (25 min) and reperfusion (120 min), treatment with the A1R-selective agonist 2-chloro-N6-cyclopentyladenosine (CCPA) (10 µg/kg) or the nonselective AR agonist 5’-N-Ethylcarboxamidoadenosine (NECA) (10 µg/kg) reduced myocardial infarction size by 50% and 35%, respectively [31]. These cardioprotective effects were blocked by pretreatment with selective antagonists of A1R (DPCPX, 100 µg/kg) or A2aR (ZM241385, 1.5 mg/kg) [31]. In a cardiac H9c2(2-1) cell ischemia model, AR agonists, such as N6-cyclopentyladenosine (CPA) and (N(6)-(3-iodobenzyl)-adenosine-5’-N- methylcarboxamide (IB-MECA), reduced the proportion of nonviable cells [37]. However, these cardioprotective effects of CPA were decreased in the presence of ADO deaminase, which reduces the endogenous levels of ADO [37]. In addition, these cardioprotective effects mediated by AR were also attenuated by DPCPX, ZM241385 or A2bR-selective antagonist MRS1191 [37]. Despite the likely cardioprotective role of exogenous or endogenous ADO in prevention of cardiomyocyte necrosis, in this work we observed a significant antiarrhythmic effect produced by endogenous ADO and LTCC blockers NIF and VER (see Figure 1 and Figure 2), independent of any effect on biochemical markers of cardiomyocyte lesion (see Figure 3).
The results obtained in this study (see Figure 1 and Figure 2) also demonstrate that there is a positive correlation between Ca2+ influx via LTCC and activity of the purinergic pathway through A1R activation in cardiac cells. This cross-communication between Ca2+ influx and the purinergic signaling mediated by A1R is importantly involved in the regulation of the electrophysiology and contractile activity of cardiac cells, attenuating the severe and fatal arrhythmias induced by CIR. It was shown that the positive chronotropic response induced by activation of cardiac β1AR is attenuated by an increase in extracellular levels of ADO produced by enzymatic degradation of ATP released from intracardiac sympathetic neurons combined with transport of cAMP to the extracellular medium from cardiac cells during stimulation [37]. According to a number of lines of evidence, the adrenergic-purinergic communication that is critical for controlling cardiac chronotropism also plays a significant role in cardioprotective responses under various pathological circumstances [3,20,31,37]. However, like other xanthine derivatives, DPCPX also functions as a phosphodiesterase (PDE) inhibitor and is virtually as powerful as rolipram at inhibiting PDE [6,38,39]. DPCPX exhibits a high selectivity for A1R over other AR subtypes [37,38,39,40]. Figure 2 showed that DPCPX inhibited cardioprotective effects of NIF and VER, indicating that the A1R is involved in these effects (see Figure 2).
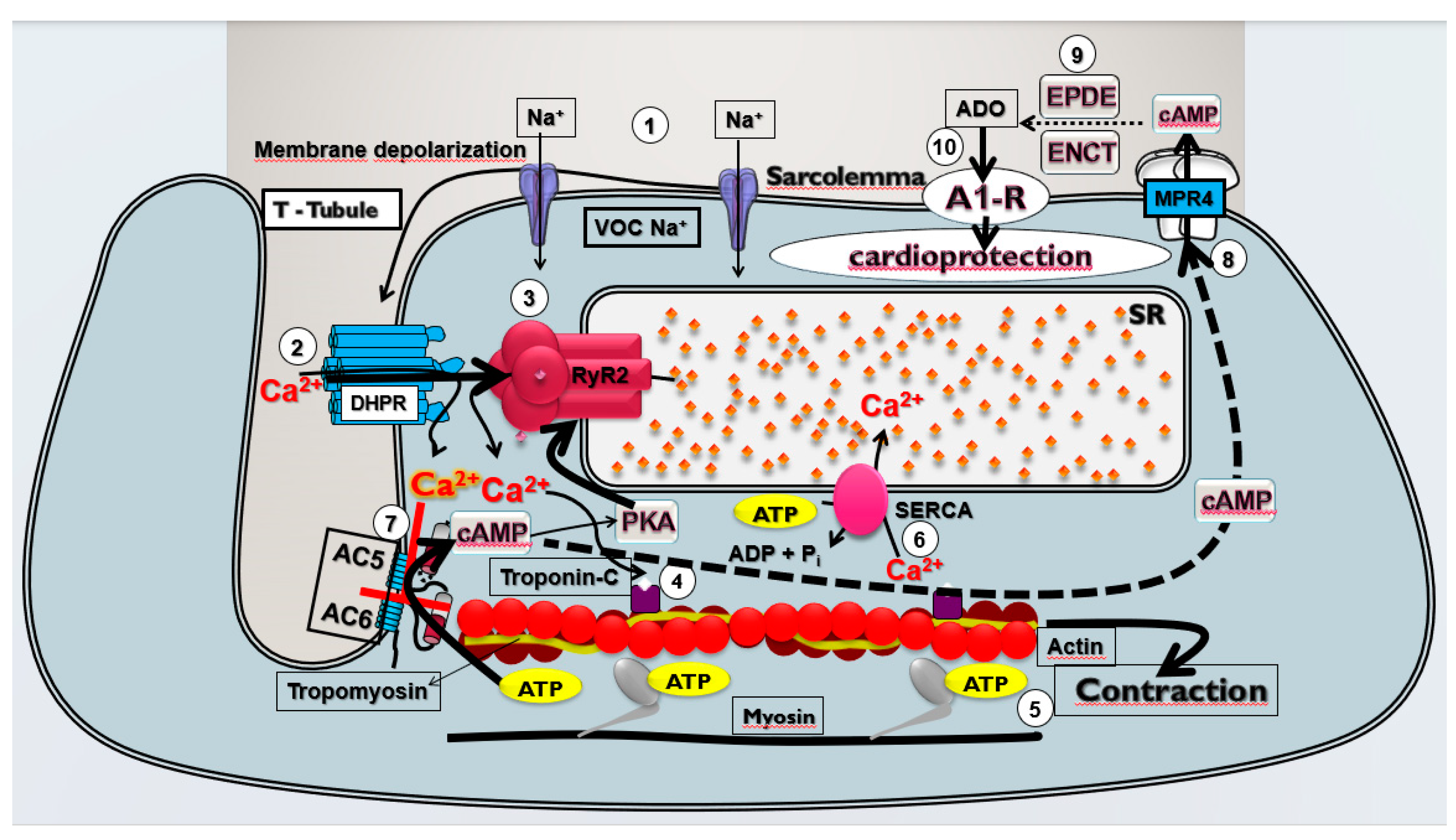
We have proposed that this pharmacological modulation of Ca2+/cAMP/ADO signaling in cardiac cells by means the attenuation of Ca2+ influx via LTCC combined with an increase of activation of A1R by ADO generated by increment of extracellular transport of cAMP may be effective to prevent sudden mortality in individuals with AMI due to severe arrhythmias brought on by cardiac collapse. Bringing together all the results obtained in the present study and the existing data in the literature, we built a theoretical model of cardioprotective response stimulated by pharmacological modulation of the Ca2+/cAMP/ADO signaling in cardiac cells (see Figure 4).
5. Conclusions
The results obtained in the present study indicate that the pharmacological modulation of Ca2+/cAMP/ADO signaling in cardiac cells by means the attenuation of Ca2+ influx via LTCC and activation of A1R by endogenous ADO could be a promising therapeutic strategy to reduce the incidence of severe and fatal arrhythmias caused by AMI in humans.
Author Contributions
Conception and design of the study and technical procedures—F.S.T., P.O.S., S.A.G.P and E.A.d.A.; interpretation of electrocardiogram and manuscript— F.S.T. and P.O.S.; analysis and interpretation of data and statistical analysis—R.Y.L., C.E.B. F., A.H.P.B.; analysis and interpretation of data and manuscript—L.A.D.N., J.V.R.M. and M.P.-O.; conception and design of the study and interpretation of electrocardiogram—J.G.P.T., M.O.T. and A.I.D.; conception and design of the study and critical revision— F.S.T., M.P.-O., A.M.C.; conception and design of the study, interpretation of data, and critical revision— L-F.B, A.G.W, A.C.-N. and F.S.M.-R. All authors have read and agreed to the published version of the manuscript.
Funding
This research received external funding from CNPq, CAPES, and FAPESP (#2017/25565-1).
Institutional Review Board Statement
The animal study protocol was approved by the Ethics Committee on the Use of Animals of the Universidade Federal de São Paulo (protocol codes #1130/11 and #0065/12 approved in 2011 and 2012, respectively).
Informed Consent Statement
Not applicable.
Data Availability Statement
The data will be available upon justified request and agreement of the authors. Conflicts of Interest: The authors declare no conflict of interest.
References
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; de Ferranti, S.; Després, J.-P.; Fullerton, H.J.; Howard, V.J.; et al. Heart Disease and Stroke Statistics--2015 Update: A Report from the American Heart Association. Circulation 2015, 131, e29–e322. [Google Scholar] [CrossRef] [PubMed]
- Writing Group Members; Mozaffarian, D. ; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; Das, S.R.; de Ferranti, S.; Després, J.-P.; et al. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation 2016, 133, e38–e360. [Google Scholar] [CrossRef]
- Rutledge, T.; Reis, V.A.; Linke, S.E.; Greenberg, B.H.; Mills, P.J. Depression in Heart Failure a Meta-Analytic Review of Prevalence, Intervention Effects, and Associations with Clinical Outcomes. J. Am. Coll. Cardiol. 2006, 48, 1527–1537. [Google Scholar] [CrossRef]
- Frampton, J.; Ortengren, A.R.; Zeitler, E.P. Arrhythmias After Acute Myocardial Infarction. Yale J. Biol. Med. 2023, 96, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Naryzhnaya, N. V.; Maslov, L.N.; Derkachev, I.A.; Ma, H.; Zhang, Y.; Prasad, N.R.; Singh, N.; Fu, F.; Pei, J.-M.; Sarybaev, A.; et al. The Effect of an Adaptation to Hypoxia on Cardiac Tolerance to Ischemia/Reperfusion. J. Biomed. Res. 2022, 1–25. [Google Scholar] [CrossRef]
- Caricati-Neto, A.; Errante, P.R.; Menezes-Rodrigues, F.S. Recent Advances in Pharmacological and Non-Pharmacological Strategies of Cardioprotection. Int. J. Mol. Sci. 2019, 20, 1–24. [Google Scholar] [CrossRef]
- Menezes-Rodrigues, F.S.; Tavares, J.G.P.; Vasques, E.R.; Errante, P.R.; Araújo, E.A. de; Pires-Oliveira, M.; Scorza, C.A.; Scorza, F.A.; Taha, M.O.; Caricati-Neto, A. Cardioprotective Effects of Pharmacological Blockade of the Mitochondrial Calcium Uniporter on Myocardial Ischemia-Reperfusion Injury. Acta Cir. Bras. 2020, 35, e202000306. [Google Scholar] [CrossRef]
- Menezes-Rodrigues, F.S.; Errante, P.R.; Ferreira, R.M.; Tavares, J.G.P.; Paula, L. de; Araújo, E.A.; Govato, T.C.P.; Tikazawa, E.H.; Reis, M. do C.M.; Luna-Filho, B.; et al. Cardioprotective Effect of Lipstatin Derivative Orlistat on Normotensive Rats Submitted to Cardiac Ischemia and Reperfusion. Acta Cir. Bras. 2018, 33, 524–532. [Google Scholar] [CrossRef]
- Menezes-Rodrigues, F.S.; Errante, P.R.; Tavares, J.G.P.; Ferraz, R.R.N.; Gomes, W.J.; Taha, M.O.; Scorza, C.A.; Scorza, F.A.; Caricati-Neto, A. Pharmacological Modulation of B-Adrenoceptors as a New Cardioprotective Strategy for Therapy of Myocardial Dysfunction Induced by Ischemia and Reperfusion. Acta Cir. Bras. 2019, 34, e201900505. [Google Scholar] [CrossRef]
- Tavares, J.G.P.; Vasques, E.R.; Arida, R.M.; Cavalheiro, E.A.; Cabral, F.R.; Torres, L.B.; Menezes-Rodrigues, F.S.; Jurkiewicz, A.; Caricati-Neto, A.; Godoy, C.M.G.; et al. Epilepsy-Induced Electrocardiographic Alterations Following Cardiac Ischemia and Reperfusion in Rats. Brazilian J. Med. Biol. Res. 2015, 48, 140–145. [Google Scholar] [CrossRef]
- Padrao Tavares, J.G.; Menezes Rodrigues, F.S.; Vasques, E.R.; Maia Reis, M. do C.; Paula, L. de; Luna Filho, B.; Errante, P.R.; Caricati Neto, A.; Bergantin, L.B. A Simple and Efficient Methodology for the Study of Cardioprotective Drugs in Animal Model of Cardiac Ischemia-Reperfusion. J. Mol. Imaging Dyn. 2017, 7. [Google Scholar] [CrossRef]
- Tavares, J.G.P.; Errante, P.R.; Govato, T.C.P.; Vasques, Ê.R.; Ferraz, R.R.N.; Taha, M.O.; Menezes-Rodrigues, F.S.; Caricati-Neto, A. Cardioprotective Effect of Preconditioning Is More Efficient than Postconditioning in Rats Submitted to Cardiac Ischemia and Reperfusion1. Acta Cir. Bras. 2018, 33, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Di Diego, J.M.; Antzelevitch, C. Cellular Basis for ST-Segment Changes Observed during Ischemia. J. Electrocardiol. 2003, 36, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.-X.; Lankipalli, R.S.; Burke, J.F.; Musco, S.; Kowey, P.R. Ventricular Repolarization Components on the Electrocardiogram: Cellular Basis and Clinical Significance. J. Am. Coll. Cardiol. 2003, 42, 401–409. [Google Scholar] [CrossRef]
- Walker, M.J.; Curtis, M.J.; Hearse, D.J.; Campbell, R.W.; Janse, M.J.; Yellon, D.M.; Cobbe, S.M.; Coker, S.J.; Harness, J.B.; Harron, D.W. The Lambeth Conventions: Guidelines for the Study of Arrhythmias in Ischaemia Infarction, and Reperfusion. Cardiovasc. Res. 1988, 22, 447–455. [Google Scholar] [CrossRef]
- Huggins, C.E.; Bell, J.R.; Pepe, S.; Delbridge, L.M.D. Benchmarking Ventricular Arrhythmias in the Mouse--Revisiting the “Lambeth Conventions” 20 Years On. Heart. Lung Circ. 2008, 17, 445–450. [Google Scholar] [CrossRef]
- Bers, D.M. Calcium Cycling and Signaling in Cardiac Myocytes. Annu. Rev. Physiol. 2008, 70, 23–49. [Google Scholar] [CrossRef]
- Wagner, S.; Maier, L.S.; Bers, D.M. Role of Sodium and Calcium Dysregulation in Tachyarrhythmias in Sudden Cardiac Death. Circ. Res. 2015, 116, 1956–1970. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Clarke, S.J.; Khaliulin, I. The Role of Mitochondria in Protection of the Heart by Preconditioning. Biochim. Biophys. Acta 2007, 1767, 1007–1031. [Google Scholar] [CrossRef]
- Xie, L.-H.H.; Weiss, J.N. Arrhythmogenic Consequences of Intracellular Calcium Waves. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H997–H1002. [Google Scholar] [CrossRef]
- Menezes-Rodrigues, F.S.; Pires-Oliveira, M.; Duarte, T.; Paredes-Gamero, E.J.; Chiavegatti, T.; Godinho, R.O. Calcium Influx through L-Type Channels Attenuates Skeletal Muscle Contraction via Inhibition of Adenylyl Cyclases. Eur. J. Pharmacol. 2013, 720, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.J.; Ma, H.; Green, R.D.; Yu, H.; Ma, H.; Green, R.D.; Yu, H.J.; Ma, H.; Green, R.D. Calcium Entry via L-Type Calcium Channels Acts as a Negative Regulator of Adenylyl Cyclase Activity and Cyclic AMP Levels in Cardiac Myocytes. Mol. Pharmacol. 1993, 44, 689–693. [Google Scholar]
- Halls, M.L.; Cooper, D.M.F. Regulation by Ca2+-Signaling Pathways of Adenylyl Cyclases. Cold Spring Harb. Perspect. Biol. 2011, 3, a004143. [Google Scholar] [CrossRef] [PubMed]
- Sellers, Z.M.; Naren, A.P.; Xiang, Y.; Best, P.M. MRP4 and CFTR in the Regulation of cAMP and β-Adrenergic Contraction in Cardiac Myocytes. Eur. J. Pharmacol. 2012, 681, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Safran, N.; Shneyvays, V.; Balas, N.; Jacobson, K.A.; Nawrath, H.; Shainberg, A. Cardioprotective Effects of Adenosine A1 and A3 Receptor Activation during Hypoxia in Isolated Rat Cardiac Myocytes. Mol. Cell. Biochem. 2001, 217, 143–152. [Google Scholar] [CrossRef]
- Du, L.; Gao, Z.-G.; Nithipatikom, K.; Ijzerman, A.P.; Veldhoven, J.P.D. van; Jacobson, K.A.; Gross, G.J.; Auchampach, J.A. Protection from Myocardial Ischemia/Reperfusion Injury by a Positive Allosteric Modulator of the A₃ Adenosine Receptor. J. Pharmacol. Exp. Ther. 2012, 340, 210–217. [Google Scholar] [CrossRef]
- Filho, C.E.B.; Barbosa, A.H.P.; Nicolau, L.A.D.; Medeiros, J.V.R.; Pires-Oliveira, M.; Póvoa, R.M.; Govato, T.C.P.; Júnior, H.J.F.; de Carvalho, R.G.; Luna-Filho, B.; et al. Pharmacological Modulation by Low Molecular Weight Heparin of Purinergic Signaling in Cardiac Cells Prevents Arrhythmia and Lethality Induced by Myocardial Infarction. J. Cardiovasc. Dev. Dis. 2023, 10, 103. [Google Scholar] [CrossRef]
- Peart, J.; Flood, A.; Linden, J.; Matherne, G.P.; Headrick, J.P. Adenosine-Mediated Cardioprotection in Ischemic-Reperfused Mouse Heart. J. Cardiovasc. Pharmacol. 2002, 39, 117–129. [Google Scholar] [CrossRef]
- Lasley, R.D.; Rhee, J.W.; Van Wylen, D.G.; Mentzer, R.M. Adenosine A1 Receptor Mediated Protection of the Globally Ischemic Isolated Rat Heart. J. Mol. Cell. Cardiol. 1990, 22, 39–47. [Google Scholar] [CrossRef]
- Lasley, R.D.; Mentzer, R.M. Dose-Dependent Effects of Adenosine on Interstitial Fluid Adenosine and Postischemic Function in the Isolated Rat Heart. J. Pharmacol. Exp. Ther. 1998, 286, 806–811. [Google Scholar]
- Lasley, R.D.; Kristo, G.; Keith, B.J.; Mentzer, R.M. The A2a/A2b Receptor Antagonist ZM-241385 Blocks the Cardioprotective Effect of Adenosine Agonist Pretreatment in in Vivo Rat Myocardium. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H426–H431. [Google Scholar] [CrossRef] [PubMed]
- Dhalla, A.K.; Shryock, J.C.; Shreeniwas, R.; Belardinelli, L. Pharmacology and Therapeutic Applications of A1 Adenosine Receptor Ligands. Curr. Top. Med. Chem. 2003, 3, 369–385. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G.; Pelleg, A. Cardiac Purinergic Signalling in Health and Disease. Purinergic Signal. 2015, 11, 1–46. [Google Scholar] [CrossRef]
- Belardinelli, L.; Lerman, B.B. Adenosine: Cardiac Electrophysiology. Pacing Clin. Electrophysiol. 1991, 14, 1672–1680. [Google Scholar] [CrossRef] [PubMed]
- Shryock, J.C.; Belardinelli, L. Adenosine and Adenosine Receptors in the Cardiovascular System: Biochemistry, Physiology, and Pharmacology. Am. J. Cardiol. 1997, 79, 2–10. [Google Scholar] [CrossRef]
- Zhan, E.; McIntosh, V.J.; Lasley, R.D. Adenosine A₂A and A₂B Receptors Are Both Required for Adenosine A₁ Receptor-Mediated Cardioprotection. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1183–9. [Google Scholar] [CrossRef]
- Urmaliya, V.B.; Church, J.E.; Coupar, I.M.; Rose’Meyer, R.B.; Pouton, C.W.; White, P.J. Cardioprotection Induced by Adenosine A1 Receptor Agonists in a Cardiac Cell Ischemia Model Involves Cooperative Activation of Adenosine A2A and A2B Receptors by Endogenous Adenosine. J. Cardiovasc. Pharmacol. 2009, 53, 424–433. [Google Scholar] [CrossRef]
- Menezes-Rodrigues, F.S.; de Oliveira, M.P.; Araújo, EA.; Ferraz, HB.; Finsterer, J.; Olszewer, E.; Taha, M.O.; Scorza, C.A.; Caricati-Neto, A.; Scorza, F.A. Role of cardiac β1-adrenergic and A1-adenosine receptors in severe arrhythmias related to Parkinson's disease. Clinics. 2023, 78, 100243. [Google Scholar] [CrossRef]
- Menezes-Rodrigues, F.S.; Tavares, J.G.P.; Errante, P.R.; Vasques, E.V.; Reis, M.C.M.; Luna-Filho, B.; Scorza, F.A.; Caricati-Neto, A.; Bergantin, L.B. Role of the Extracellular Ca2+/cyclic AMP-Adenosine Signaling Pathways in Cardioprotection. J. Thrombo. Cir. 2017, 3, 1. [Google Scholar] [CrossRef]
- Muthal, A.P.; Kulkarni, R.; Kumar, D.; Bagul, C.; Mukherjee-Kandhare, A.A.; Kandhare, A.D.; Ambavade, S.D.; Wagh, V.; Bodhankar, S.L. Cyclic adenosine monophosphate: Recent and future perspectives on various diseases. J. Appl. Pharm. Sci. 2022, 12, 1–15. [Google Scholar] [CrossRef]
Figure 1.
Histograms representing the (A) Incidence of ventricular arrhythmias (AV), (B) Atrioventricular block (AVB) and (C) Lethality (LET) in the CIR, NIF1+CIR, NIF10+CIR, NIF30+CIR, VER1+CIR, PROB+CIR and DPCPX+CIR groups. Results were expressed as mean and statistical analysis was performed using Fisher's exact test (*p < 0.05). The incidence of VA, AVB and LET were significantly reduced in all groups of animals treated with NIF (1, 10 and 30 mg/kg or VER 1 mg/kg) and submitted to the CIR protocol when compared to the CIR.
Figure 1.
Histograms representing the (A) Incidence of ventricular arrhythmias (AV), (B) Atrioventricular block (AVB) and (C) Lethality (LET) in the CIR, NIF1+CIR, NIF10+CIR, NIF30+CIR, VER1+CIR, PROB+CIR and DPCPX+CIR groups. Results were expressed as mean and statistical analysis was performed using Fisher's exact test (*p < 0.05). The incidence of VA, AVB and LET were significantly reduced in all groups of animals treated with NIF (1, 10 and 30 mg/kg or VER 1 mg/kg) and submitted to the CIR protocol when compared to the CIR.
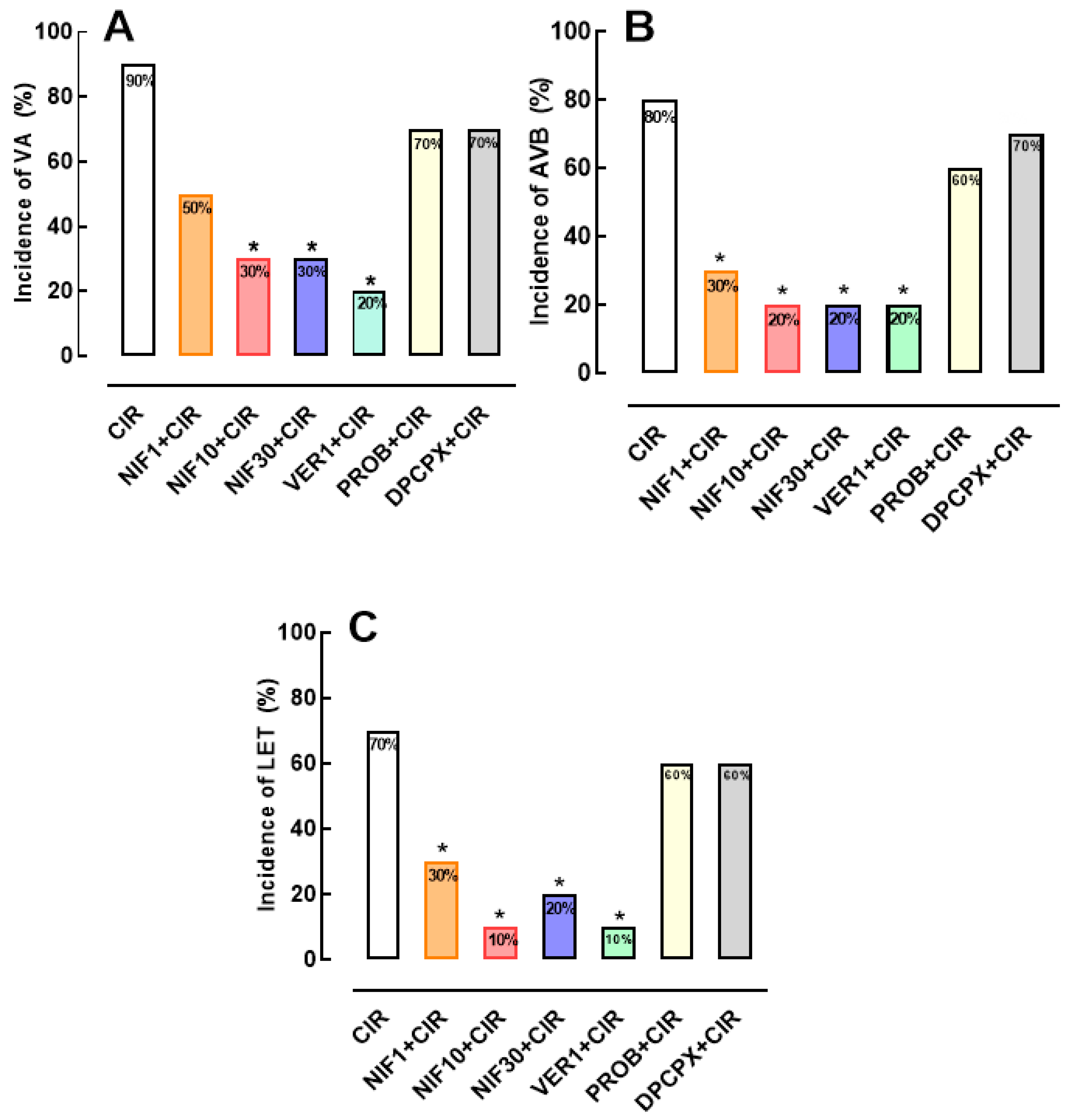
Figure 2.
Histograms representing the (A) incidence of ventricular arrhythmias (AV), (B) Atrioventricular block (AVB) and (C) Lethality (LET) in the CIR, NIF10+CIR, PROB+NIF10+CIR, DPCPX+NIF10+CIR; VER1+CIR, PROB+VER1+CIR, and DPCPX+VER1+CIR groups. Results were expressed as mean and statistical analysis was performed using Fisher's exact test (*p < 0.05). The pre-treatment with DPCPX 4 minutes before administration of NIF or VER was able to abolish the effects cardioprotective of NIF and VER in rats submitted to CIR.
Figure 2.
Histograms representing the (A) incidence of ventricular arrhythmias (AV), (B) Atrioventricular block (AVB) and (C) Lethality (LET) in the CIR, NIF10+CIR, PROB+NIF10+CIR, DPCPX+NIF10+CIR; VER1+CIR, PROB+VER1+CIR, and DPCPX+VER1+CIR groups. Results were expressed as mean and statistical analysis was performed using Fisher's exact test (*p < 0.05). The pre-treatment with DPCPX 4 minutes before administration of NIF or VER was able to abolish the effects cardioprotective of NIF and VER in rats submitted to CIR.
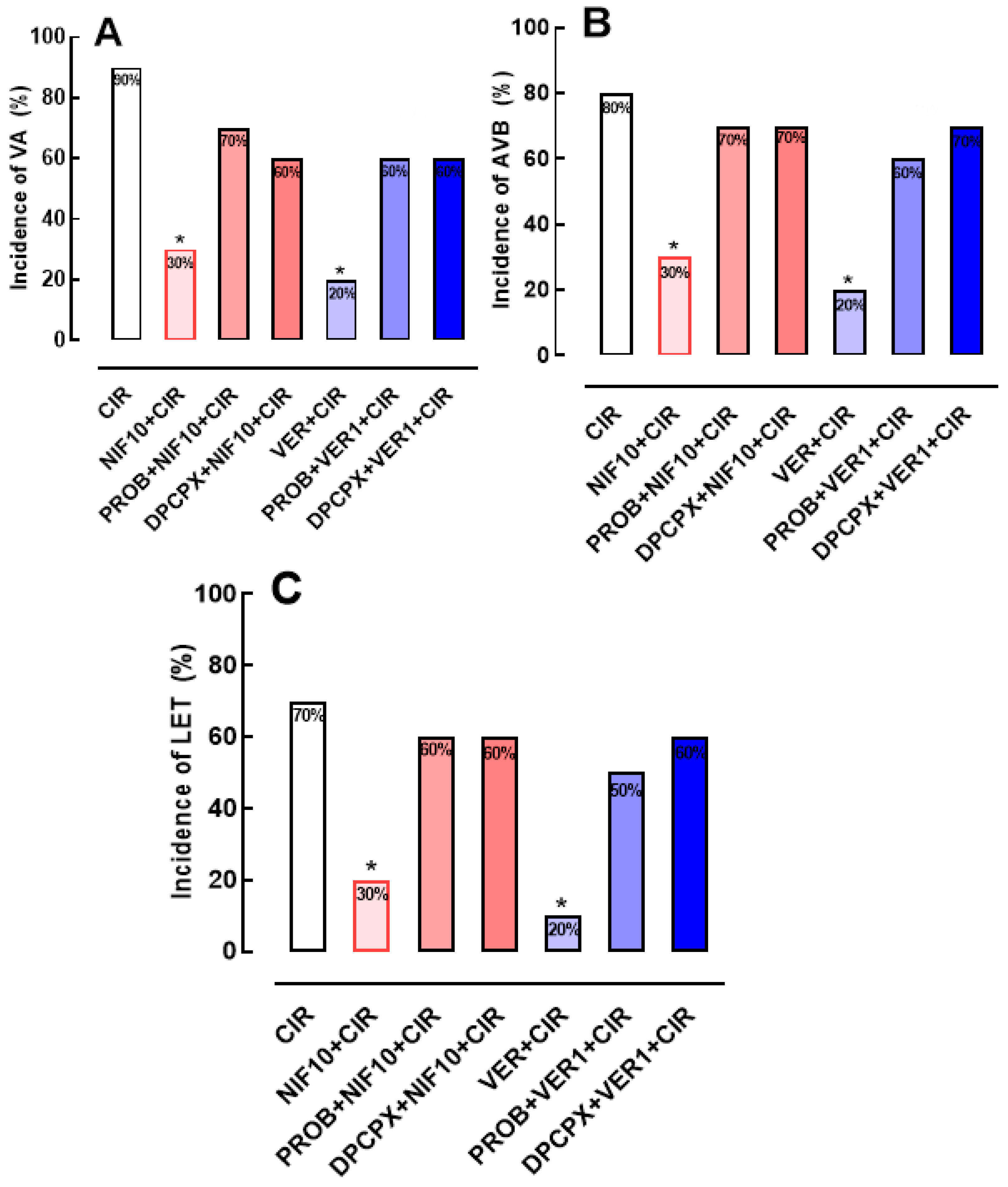
Figure 3.
Histograms representing the serum concentrations of total CK (A) and CK-MB (B) in the CIR, NIF+CIR, NIF10+CIR, NIF30+CIR, DPCPX+NIF10+CIR, VER 1+CIR, and DPCPX+VER1+CIR groups. Results were expressed as mean ± standard error of mean, and one-way analysis of variance (ANOVA) was applied, followed by Tukey's post-test. There was no statistical difference between the different groups.
Figure 3.
Histograms representing the serum concentrations of total CK (A) and CK-MB (B) in the CIR, NIF+CIR, NIF10+CIR, NIF30+CIR, DPCPX+NIF10+CIR, VER 1+CIR, and DPCPX+VER1+CIR groups. Results were expressed as mean ± standard error of mean, and one-way analysis of variance (ANOVA) was applied, followed by Tukey's post-test. There was no statistical difference between the different groups.
Figure 4.
Theoretical model of cardioprotective response stimulated by pharmacological modulation of the Ca2+/cAMP/ADO signaling in cardiac cells. The membrane depolarization of cardiac cells generates the (1) Na+ influx via voltage-operated Na+ channels (VOC Na+) that induces (2) opening of the L-type Ca2+ channels (LTCC) containing dihydropyridine receptors (DHPR) located in the T-tubules and depolarization of the sarcolemma that lead to Ca2+ influx that (3) stimulates the release of Ca2+ from sarcoplasmic reticulum (SR) mediated by activation by Ca2+ of the R2-type ryanodine receptors (RYR2); this Ca2+ released from SR (4) binds to troponin C and (5) activates the contractile machinery generating contraction of cardiac cells. At the same time, (6) the Ca2+ concentration in the cytosol is restored by Ca2+ sequestration by SR via SERCA; Ca2+ influx via LTCC also (7) inhibits the isoforms 5 and 6 of adenylyl cyclase (AC) producing reduction in the intracellular production of cAMP and phosphorylation of RYR2 by cAMP-dependent kinases (PKA), and then reducing cardiac contraction frequency and strength; in addition (8) efflux of cAMP through membrane transporters MRP4 and (9) extracellular degradation of cAMP to adenosine (ADO) by ectonucleotides (ENCT) and ectophosphodiesterases (EPDE) cause increment of extracellular ADO levels that lead to (10) activation of A1R, culminating in the cardioprotection resulting from prevention of CECC collapse and consequent reduction of incidence of severe and fatal cardiac arrhythmias induced by CIR.
Figure 4.
Theoretical model of cardioprotective response stimulated by pharmacological modulation of the Ca2+/cAMP/ADO signaling in cardiac cells. The membrane depolarization of cardiac cells generates the (1) Na+ influx via voltage-operated Na+ channels (VOC Na+) that induces (2) opening of the L-type Ca2+ channels (LTCC) containing dihydropyridine receptors (DHPR) located in the T-tubules and depolarization of the sarcolemma that lead to Ca2+ influx that (3) stimulates the release of Ca2+ from sarcoplasmic reticulum (SR) mediated by activation by Ca2+ of the R2-type ryanodine receptors (RYR2); this Ca2+ released from SR (4) binds to troponin C and (5) activates the contractile machinery generating contraction of cardiac cells. At the same time, (6) the Ca2+ concentration in the cytosol is restored by Ca2+ sequestration by SR via SERCA; Ca2+ influx via LTCC also (7) inhibits the isoforms 5 and 6 of adenylyl cyclase (AC) producing reduction in the intracellular production of cAMP and phosphorylation of RYR2 by cAMP-dependent kinases (PKA), and then reducing cardiac contraction frequency and strength; in addition (8) efflux of cAMP through membrane transporters MRP4 and (9) extracellular degradation of cAMP to adenosine (ADO) by ectonucleotides (ENCT) and ectophosphodiesterases (EPDE) cause increment of extracellular ADO levels that lead to (10) activation of A1R, culminating in the cardioprotection resulting from prevention of CECC collapse and consequent reduction of incidence of severe and fatal cardiac arrhythmias induced by CIR.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



