
Submitted:
08 February 2024
Posted:
09 February 2024
You are already at the latest version
Abstract
Reconstructing the Tree of Life remains a central goal in biology. Early methods, relying on small numbers of morphological or genetic characters, often yielded conflicting evolutionary histories, undermining confidence in the results. Investigations based on phylogenomics, which use hundreds to thousands of loci for phylogenetic inquiry, have provided a clearer picture of life's history, but certain branches remain problematic. To resolve difficult nodes on the Tree of Life, two recent studies tested the utility of synteny, the conserved collinearity of orthologous genetic loci in two or more organisms, for phylogenetics. Synteny exhibits compelling phylogenomic potential while also raising new challenges. We identify and discuss specific opportunities and challenges that bear on the value of synteny data and other rare genomic changes for phylogenomic studies. Synteny-based analyses of highly contiguous genome assemblies mark a new chapter in the phylogenomic era and the quest to reconstruct the Tree of Life.
Keywords:
phylogenomics
; synteny
; chromosomes
; long-read sequencing
; tree of life
; orthology
; phylogenetic trees
; incongruence
“A comparison of the different gene arrangements in the same chromosome may, in certain cases, throw light on the historical relationships of these structures, and consequently on the history of the species as a whole.” Dobzhansky & Sturtevant, 1938, Genetics [1].
Arguably, the most ambitious goal in phylogenetics is to reconstruct the entire Tree of Life. To build phylogenetic trees, diverse data types have been used and our understanding of the Tree of Life has undergone significant transformations with each methodological advance.
Early studies relied on aligning single or few loci to reconstruct evolutionary histories [2], but analyses of different loci often yielded phylogenies with conflicting or poorly-supported topologies [3,4] (Figure 1A–C). This may happen due to, for example, incomplete lineage sorting, the random sorting of ancestral alleles, a phenomenon pronounced among introns and loci under balancing selection (Prüfer et al. 2012). The advent of cost-effective whole genome sequencing paved the way for the phylogenomics era, in which hundreds to thousands of orthologous loci are analyzed in a total evidence approach [5,6].
Phylogenomics has successfully delineated previously problematic branches within the Tree of Life, including the monophyletic grouping of nematodes and arthropods within Ecdysozoa [7,8], the placement of turtles as sister to archosaurs (crocodiles and birds) [9], and the placement of eukaryotes within Archaea [10]. These successes have positioned phylogenomics as the current standard for reconstructing most evolutionary histories. However, certain deep branches remain unresolved.
Therefore, phylogeneticists have sought to identify new genomic characters that accurately reflect evolutionary history, in part because they are unlikely to evolve independently in unrelated groups of organisms [11,12,13]. To this end, two recent studies [14,15] have tested the utility of gene synteny as a character for phylogenetics. Here we review the challenges that inspired these studies, evaluate the current utility of gene synteny as a new character for phylogenetics, and offer a roadmap for future use of gene synteny to reconstruct the Tree of Life.
Tangled branches in the Tree of Life
Although there are many unresolved branches in the Tree of Life, two major challenges concern how to root the tree of animals and how major clades of teleost fish, a group encompassing nearly half of all vertebrates, are related. These evolutionary questions exemplify how analyses of genome-scale data can yield incongruent phylogenies and undermine our ability to fully reconstruct the Tree of Life.
The controversy surrounding the root of the animal tree was somewhat unexpected, as morphological comparisons consistently favored the placement of sponges (Figure 1D), not ctenophores (Figure 1E), as the earliest-branching lineage [16,17]; this hypothesis garnered nearly universal support during the single-locus era of phylogenetics [16,18,19,20] (Figure 1F). The dawn of phylogenomics, however, changed the situation. A 2008 study based on 150 genes from 77 taxa, including two sponges and two ctenophores, provided the first support for placing ctenophores at the root of the animal tree (Figure 1G) [21]. Then, in 2009, the sponge-first hypothesis was supported by a study using 128 genes and 55 taxa, including nine sponges and three ctenophores [22]. Since then, investigations powered by ever larger datasets (including dozens of ctenophores and sponges) analyzed using the latest methods in phylogenomics have provided compelling and contradictory evidence for the two competing hypotheses [22,23,24,25,26,27,28].
Early branching patterns in the teleost fish phylogeny are also intensely debated. Teleosts encompass three major clades: Elopomorpha (mostly slim-headed fish like bonefish, eels, and skipjacks; Figure 1H), Osteoglossomorpha (mostly bony-tongued fish like elephantnose fish, doublesash butterflyfish, and mormyrids; Figure 1I), and Clupeocephala (the remaining extant teleosts like pufferfish and sticklebacks; Figure 1J). Phylogenetics of some single-locus data suggested a sister relationship between Elopomorpha and Osteoglossomorpha—the Eloposteoglossocephala (EO-sister) hypothesis—in which the slim-headed and bony-tongued fish are thought to form a sister clade relative to all other teleosts [29]. However, all possible topologies (Figure 1K–M) have received support in the phylogenomic era. Challenged by a history of conflict, some have suggested that the base of the teleost fish phylogeny is one of the most important unresolved questions in ray-finned fish evolution [30].
Rare genomic changes as phylogenomic markers
Amidst these and other ongoing debates, the value of alternative phylogenetic markers, such as rare genomic changes, has been explored [31]. The phylogenetic distributions of some rare genomic changes, including insertions and deletions, gene duplications and losses, and alternative genetic codes, often mirror the inferred evolutionary relationships among major vertebrate, insect, fungal, and related lineages [32,33,34,35]. The earliest studies underscoring the promise of rare genomic changes for phylogenetics were conducted before widely available whole-genome sequences. For example, Sturtevant and Dobzhansky successfully reconstructed phylogenetic relationships among populations of Drosophila pseudoobscura by analyzing chromosomal inversions detected in the polytene chromosomes of salivary glands [1,36].
Since then, several other cases of rare genomic changes recapitulating phylogeny have been identified. For example, copy number variants (duplicated or deleted loci) can mirror population structure in humans, zebrafish, and fungal pathogens among other species [37,38,39,40]. Gene presence-absence polymorphisms can also reflect phylogeny. For example, lineage-specific gene duplication and loss events have been detected in humans [41] and in lineages of the bipolar budding yeast Hanseniaspora [35]. Another rare genomic change is the genetic recoding of CUG to alanine and serine, rather than leucine, in a monophyletic lineage of yeast [42]. Among more ancient divergences, the root of the angiosperm phylogeny has been successfully examined using duplication patterns of phytochrome genes [43], an observation that has received continued support in the phylogenomic era [44].
Transposable element insertions and deletions represent another class of rare genomic change extensively used in phylogenetic studies. For example, retrotransposon insertions have proven useful in elucidating the evolutionary relationships among several vertebrate lineages [45,46,47,48,49]. Importantly, sources of locus-tree-species-tree discordance, such as incomplete lineage sorting and homoplasy (similar insertion events that do not share ancestry), have been examined [45,46,50]. A better understanding of these errors has allowed phylogeneticists to account for and ameliorate their impact. Taken with the previous examples, these studies demonstrate the utility of structural variants to illuminate phylogeny across scales ranging from populations to deep time.
Nonetheless, rare genomic changes can evolve convergently. One example includes repeated convergent losses of gene duplicates in flatworms [51]. Similarly, genetic recoding of the CUG codon from leucine to serine in Saccharomycotina fungi occurred twice independently [52]. Convergence has also been observed among structural genomic features. For example, distributions of mitochondrial genome size, structure, and content have converged among Placozoa, chytrid fungi, and choanoflagellates [53] leading briefly to the inference that Placozoa diverged from all other animals first—a hypothesis largely refuted by phylogenomic analyses of nuclear genes [26,54]. Even in closely related species of walnuts, phylogenies inferred from large amounts of local gene order data, DNA-sequence alignments, and gene family content yield differing tree topologies [55].
Thus, the utility of rare genomic changes has been mixed. Several examples demonstrate that rare genomic changes can recapitulate evolutionary history, while others contradict generally accepted evolutionary relationships established using other data types. A barrier to the widespread usage of rare genomic changes is the sparsity of methods for detecting rare genomic changes and algorithms for analyzing their informativeness.
Synteny emerges in the phylogenomic era
As abundant genome assemblies have become available and algorithm development has followed suit, the field of phylogenomics is primed to revisit the value of rare genomic changes—specifically synteny—for phylogenetic inference. User-friendly software has enabled the detection of collinear DNA sequences in genomes from related organisms [56,57,58,59,60,61], thereby streamlining robust orthology inference [5] and analyses of changes in microsynteny and macrosynteny (Figure 2A,B). Although there is no widely accepted consensus for what differentiates micro- from macrosynteny, microsynteny typically concerns only a handful of genes, whereas macrosynteny typically refers to hundreds to thousands of collinear genes, at times spanning whole chromosomes [62]. Shared rearrangements in gene order would be predicted to indicate a common evolutionary history, so long as convergence is not at play.
A major molecular mechanism driving syntenic variation is unequal homologous recombination [63]. Genomes with multiple copies of similar sequences, like transposable elements in plant genomes, can be particularly prone to unequal homologous recombination [64]. Similarly, recombination between highly similar but nonallelic sequences (nonhomologous recombination) can result in major mutational events, such as recurrent deletions or duplications [65]. Whether a recombination event results in a micro- or macrosyntenic change depends on the spacing between recombinant regions. Other error-prone DNA repair mechanisms—including nonhomologous end joining—can also result in syntenic changes [66].
Comparison of the relationships among shared syntenic blocks in Saccharomycotina yeast with an evolutionary history previously inferred using concatenated multiple sequence alignments, provides insight into the potential value of synteny for phylogenetics. In a data set of 120 yeast, nearly 99% of microsyntenic blocks were more likely to be shared among closely related species than expected by random chance [67], reinforcing the notion that synteny can reflect phylogeny [68]. Subsequent developments in software and bioinformatic pipelines, vetted through simulations and examinations of empirical data, have facilitated the inference of organismal histories based on syntenic blocks [69,70,71]. Although promising, these studies primarily focused on establishing the utility of synteny through proof-of-principle approaches—that is, reevaluating well-established relationships or using simulated scenarios. Applying these methodologies to address challenging Tree of Life debates has been a more recent development.
Synteny brings fresh perspectives to the Tree of Life
Synteny and the Root of the Animal Tree. Reconstruction of ancient gene linkages by Schultz et al. has brought new data to bear on the sponge- versus ctenophore-first debate at the base of the animal Tree of Life [15](Figure 1F,G). This study relied on a new ensemble of genome assemblies from select sponges, ctenophores, bilaterians, cnidarians, and three outgroup taxa—a choanoflagellate (Salpingoeca rosetta), a filasterean (Capsaspora owczarzaki), and an ichthyosporean (Creolimax fragrantissima). Although detecting synteny is complicated by the accumulation of chromosomal rearrangements across deep time, comparative analyses identified syntenic blocks conserved between outgroup and animal taxa using three- or four-way reciprocal-best-BLAST hits; 29 and 20 different syntenic blocks were shared between animals and the filasterean or choanoflagellate, respectively. Notably, all 20 syntenic regions identified in the choanoflagellate were also present in the filasterean.
The inferred evolutionary changes to otherwise conserved syntenic blocks were placed in one of three categories based on outgroup taxa—no fusion, fusion-without-mixing, and fusion-with-mixing (Figure 2C,D)—which were then encoded and utilized in a phylogenetic framework. “No fusion” referred to syntenic blocks that remain on separate chromosomes. For example, imagine that an ancestral organism contains genes A1, B1, and C1 in one chromosome and genes A2, B2, and C2 in another (Figure 2E). If these blocks are on separate chromosomes (chromosomes 1 and 2) in two descendent organisms, there was “no fusion.” In the case of “fusion-without-mixing,” syntenic blocks A and B now co-exist on the same chromosome in a descendent genome compared to the ancestor. This phenomenon is relatively well documented among acrocentric human chromosomes, which have a centromere near the end of one chromosome arm and is termed a Robertsonian Translocation [72]. Finally, “fusion-with-mixing” refers to a rearrangement pattern involving multiple steps between the ancestral genome and the descendent genome—first, chromosomal fusion followed by one or more rearrangements that cause the syntenic blocks to interweave. For example, a single chromosome might contain a contiguous stretch of DNA encoding genes A1, C2, A2, B1, B2, and C1 in that order.
For reconstructing the animal Tree of Life, the codified matrix of fusion events was then used for phylogenetic inference. Compared to fusion and fission events, the transition probability from fusion-with-mixing to other states was inferred to be unlikely (Figure 2E). Bayesian analysis of this data matrix supported the ctenophore-first hypothesis, as did direct examination of fusions analyzed using parsimony. Specifically, the ctenophore-first hypothesis was supported by seven fusion events shared by bilaterians, cnidarians, and sponges, but missing from extant ctenophores and outgroup taxa. Four of these events occurred with mixing; under the sponge-first hypothesis, convergent fusions with mixing or precise reversions are required to explain these data. Thus, the absence of these fusions from ctenophores and outgroup taxa (with the exception of variation in region 7) was interpreted as evidence that ctenophores diverged from all other animals before the fusion and mixing events (Figure 3A). Region 7 may have independently undergone fusion and mixing events in the Filasterean lineage. An alternative but less likely scenario is region 7 was already in a “mixed” state in the ancestor of all sampled taxa and subsequently underwent de-mixing and de-fusion events followed by a complex pattern of fusion and mixing events.
Nonetheless, other findings from the synteny analysis contradict well-established evolutionary relationships. For example, despite phylogenomic analyses robustly supporting choanoflagellates as the closest living relatives of animals [73,74,75,76,77], animals shared more syntenic blocks with the filasterean than with the choanoflagellate (29 syntenic blocks compared to 20). There are also more unique syntenic blocks shared between the filasterean and animals than the choanoflagellate (nine syntenic blocks compared to two). The incongruence between the pattern of synteny conservation and prior findings from phylogenomics either suggests a previously undetected close evolutionary relationship between filastereans and animals or, more likely, a lineage-specific loss of synteny in choanoflagellates.
Indeed, some choanoflagellates have undergone unique, accelerated genome evolution. Specifically, the choanoflagellate S. rosetta used in the Schultz et al. study has experienced rapid gene family evolution compared with other choanoflagellates, resulting in a reduced gene repertoire relative to that of the last common ancestor of animals and choanoflagellates [78]. Accordingly, S. rosetta may not be the best representative of choanoflagellates for phylogenetics, highlighting the importance of expanded taxon sampling.
Similarly, unbiased phylogenetic analysis of fusion states did not recover the monophyly of Porifera, which contradicts more recent phylogenomic studies supporting the monophyly of the lineage [21,22,79]. Although some analyses support paraphyly among Porifera [80,81], the exemplar sponges in the Schultz et al. study [15] belong to the class Demospongiae, which most analyses support as a monophyletic clade [82]. These observations call for caution in using syntenic blocks, especially when synteny has been lost.
Synteny and the Evolutionary Relationships Among Major Groups of Teleost Fish. Early branching patterns in the teleost fish phylogeny were recently reexamined by Parey et al. [14] using a combination of expanded taxon sampling and analysis of syntenic blocks. Synteny was detected using the position of orthologous genes along chromosomes for every pairwise comparison of species. Phylogenetic analyses of the resulting macro- and microsynteny data (Figure 2A,B)—wherein lack of syntenic conservation was used to measure distance—supported the EO-sister hypothesis. Using macrosyntenies, nearly 20% of breakpoints supported the EO-sister hypothesis. Using microsynteny data, the sister relationship between these lineages received full bootstrap support. Evidence of a single chromosome fusion event unique to slim-headed and bony-tongued fish and another unique to other teleosts corroborated the EO-sister hypothesis; specifically, after a whole genome duplication event along the stem lineage of teleosts, one chromosome pair fused among slim-headed and bony-tongued fish, whereas the other chromosome pair fused and mixed among other teleosts (Figure 3C,D).
In addition to synteny-based analyses, standard phylogenomic approaches were employed. Phylogenomic analyses and distributions of support frequencies based on analyses of single genes supported the EO-sister hypothesis (Figure 1K). Interestingly, this finding was not supported by previous studies examining single-gene support frequencies and ultraconserved elements under a maximum likelihood framework [83,84]. Thus, with this expanded set of taxa, the EO-sister hypothesis is supported by synteny analysis as well as gene sequence concatenation and coalescence, pointing to the influence of expanded taxon sampling.
Analyzing data from more taxa generally improves phylogenetic inference, particularly among close relatives of phylogenetically unstable taxa [4,85,86]. For example, when represented by a single taxon, the placement of the Saccharomycotina family Ascoideaceae conflicted between two phylogenomic studies that likely did not suffer from insufficient locus sampling [87,88]. However, expanded sampling of genomes from three Ascoideaceae and close relatives robustly supported one hypothesis [89]. Additional analyses suggested that increased taxon sampling resulted in improved model fit and greater phylogenetic stability of focal lineages. These studies demonstrate how additional taxon sampling can improve phylogenetic inference. Moreover, the benefits of high-quality, chromosome-scale genome assemblies are multifold. For example, standard phylogenomic analyses have benefitted from synteny data to improve orthology predictions, and multiple data types, such as patterns of macro- and microsynteny, provide additional lines of evidence for phylogenomic inquiry [90].
Toward high-quality synteny-based Tree of Life reconstructions
As highly contiguous genome assemblies become more commonplace, our understanding of synteny as a phylogenomic marker will mature. Here, we provide a roadmap of research opportunities and identify challenges that will shape the use of synteny as a phylogenomic character (Figure 4A).
A roadmap to infer synteny-based phylogenies.
Taxon sampling/selection. Taxon sampling influences numerous downstream steps, such as orthology inference. Generally, the more taxon sampling, the better [4,85]. Fortunately, there are a growing number of chromosome-level or highly-contiguous genome assemblies that are publicly available for downloading and analysis. However, representatives from undersampled lineages may require genome sequencing. Thus, taxon sampling should be guided by the phylogenetic question at hand. For example, determining evolutionary relationships among vertebrates does not require taxon sampling among fungi; in fact, poor taxon sampling of distantly related taxa may introduce long branches and contribute to long-branch attraction artifacts [91,92].
Long-read sequencing and chromosomal conformation analyses. Much like traditional phylogenomics using collections of multiple sequence alignments, synteny-based phylogenomics starts with data acquisition. However, unlike multiple sequence alignment-based phylogenomics, high-quality genomes (ideally assembled accurately from telomere-to-telomere on all chromosomes) are necessary. The state of the art for genome assembly requires long-read sequencing (e.g. using Oxford Nanopore or PacBio) [93,94], which in turn requires acquisition of high molecular weight DNA from each organism to be sequenced. For more complex genomes, chromosomal interactions detected from Hi-C analyses will help provide additional lines of evidence for subsequent steps, namely genome assembly [95].
Genome assembly. With long-read sequences and chromosomal conformation data in hand, the next step for synteny-based phylogenomics is to generate an accurate and precise genome for each species to be analyzed. Poor genome assembly quality can be a source of error when detecting synteny [96] and, in turn, introduce errors in synteny-based phylogenomics. While there is no broadly accepted definition of a “high quality” assembly, researchers should consider three important metrics—completeness, contiguity, and accuracy. Completeness can be assessed by comparing inferred gene content with expectations from transcriptome sequences and the presence/absence of nearly-universal single-copy orthologs [97]. Incomplete genomes may be difficult to incorporate into synteny-based phylogenomics and may necessitate further efforts to improve the original genome assembly. When highly contiguous genomes are difficult to achieve, macrosyntenic blocks that are broken up across several scaffolds should be removed from the data matrix. Alternatively, microsyntenies may be more appropriate to use because they are more likely to be preserved, even in a discontiguous genome assembly. Examining assembly accuracy is difficult without physical mapping data from, for example, fluorescence in situ hybridization or optical maps [98]. However, these data can be useful not only to validate but also to improve genome assembly quality, even helping achieve near-complete genomes [98]. Of note, other measures of assembly quality, such as degree of contamination, should be taken into account, particularly when loss of synteny is inferred.
Genome annotation. To detect syntenic blocks across the resulting set of genomes, the relative positions of orthologous genes are often used [67,71]. Thus, phylogeneticists must predict gene boundaries accurately to prevent, for example, erroneously combining two genes into a single gene model or missing genes entirely (Figure 4). Many phylogenomic studies rely on the outputs of genomes annotated using different methods, but recent studies have shown that the outputs of different gene annotation methods can substantially vary [99]. A troubling result of comparing genomes annotated using different annotation methods is the artifactual inflation of the number of unique or lineage-specific genes [99]. Therefore, a single high-quality annotation method trained on the individual organism or methods that combine the results from multiple gene annotation algorithms, like EVidenceModeler [100], may prove helpful. Moreover, incorporating transcriptomic reads will help refine and provide evidence for gene boundary predictions [101]
Orthology inference. The resulting gene predictions are subsequently used to infer orthologous relationships among genes (Figure 4). Orthology relationships are inferred using all-vs-all sequence similarity information [102]. Researchers face several challenges during orthology inference, stemming from both analytical and biological sources of error [4,103]. Analytical errors may stem from genes that are absent from annotation predictions but that are genuinely encoded in the organism's genome. Other sources of error may stem from complex evolutionary histories, such as gene duplication and loss, convergence, or saturation [4,104].
Alternatively, whole-genome alignment methods, like Progressive Cactus and SibekliaZ [105,106], may overcome potential errors stemming from gene annotation errors. One major innovation offered by Progressive Cactus is that it allows reference-free multiple genome alignment (ameliorating reference-based bias) and detecting multicopy orthology relationships, rather than only single-copy orthology [106]. Furthermore, Progressive Cactus can also handle large datasets, such as 600 or more animal genomes.
Establishing best practices in synteny detection. Typically, the distributions of gene orthologs along chromosomes in different species are used to detect potential syntenic blocks. Therefore, differences in the quality of ortholog prediction and in the density of syntenic orthologs detected should profoundly shape the accuracy of syntenic block detection. Both factors – accuracy of ortholog detection and density of syntenic orthologs – will likely drop off when comparing genomes separated by long evolutionary time scales.
Care must be taken, therefore, in the selection of software and analysis parameters [96]. Two key parameters are the minimum number and density of genes necessary to define orthologous syntenic blocks. Higher thresholds are expected to result in more conservative estimates of syntenic blocks (i.e., fewer false positives), but at the cost of potentially having a smaller number of syntenic blocks to analyze. Several software packages facilitate synteny detection, including MCScanX, SynChro, and syntenet [58,59,60]. Notably, each employs different methodology; for example, Synchro identifies pairwise syntenies using reciprocal best BLAST hits of protein sequence similarity, whereas MCScanX detects synteny blocks across two or more genomes [58,59]. MCScanX also provides additional utilities to further classify syntenic blocks based on putative evolutionary origins, such as those originating from whole genome duplication events or tandem duplication. Although these algorithms vary in efficacy, genome contiguity appears to be a major driver of error, underscoring the importance of obtaining highly contiguous genome assemblies [96].
To determine how much of the genome is captured during detection, synteny coverage can be calculated, which is the sum length of blocks divided by genome size [96]. Synteny coverage may differ between genomes due to biological phenomena such as genome size, content variation, or analytical factors that can relax the definition of a syntenic block; thus, it will be important to report syntenic coverage for individual genomes as well as summary statistics across them. Ideally, syntenic coverage will be high and cover nearly the entire genome for closely related organisms. However, syntenic coverage may be reduced based on the threshold applied for detecting synteny, the rate of evolution among chromosomes, the rate of evolution of local gene order, and the evolutionary distance between species analyzed.
Accounting for sources of phylogenomic error. Diverse factors can lead to erroneous species tree inference. Although these are well studied in analyses of multiple sequence alignments [4,103,107], they are underexplored for synteny-based phylogenomics. Here we discuss potential sources of error for synteny analysis and methods for taking them into account.
Saturation. In nucleotide and amino acid sequence evolution, stepwise evolution of a sequence away from, and then back to, an ancestral state is described as “saturation” and this phenomenon can mask evolutionary history. Saturation may also occur during synteny evolution, whereby multiple sequential rearrangements may interfere with tracing the step-wise evolution of syntenic blocks. To overcome saturation, one solution may be to purge data matrices of rapidly evolving syntenic blocks, wherein the evolutionary history may be harder to trace.
Incomplete lineage sorting. The random sorting of ancestral polymorphisms can lead to genealogies that differ from the species tree, especially during rapid radiation events [108,109]. Incomplete lineage sorting among structural variants may also be a source of synteny-based phylogenomic error. Incomplete lineage sorting among gene trees is particularly prevalent during radiation events and in large populations [108,110]. Given that genome rearrangement can occur rapidly in a population [111,112], it raises the possibility that some structural variants may coalesce before a speciation event; that is, be subject to incomplete lineage sorting. Determining the prevalence (if any) of incomplete lineage sorting among structural variants will elucidate if incomplete lineage sorting is a source of incongruence.
Reticulate evolution. Reticulate evolution refers to non-vertical inheritance of loci, resulting in loci with an evolutionary history that deviates from a strictly bifurcating tree model, such as horizontal gene transfer and introgression/hybridization [113,114,115]. This issue will have varying influences across different lineages; for example, horizontal gene transfer occurs more frequently among bacteria and archaea than many eukaryotic lineages [116,117]. Similarly, hybridization is common among plant lineages [118,119,120], and has also been observed in other lineages like animals and fungi [114,121,122,123].
The non-vertical acquisition of loci may interfere with the detection of otherwise conserved syntenic regions [124]. In the case of horizontal gene transfer, synteny analysis would suggest an erroneous phylogenetic placement of a lineage; for example, synteny analysis of the horizontally acquired bacterial siderophore gene cluster in yeast [125] would suggest a close affinity between yeast and bacteria, a hypothesis that is incontrovertibly refuted. Loci with signatures of horizontal gene transfer can be pruned from a data matrix. However, in some cases, horizontally acquired loci that undergo vertical inheritance may be helpful markers for synteny-based phylogenomics [126].
Modeling syntenic changes. In standard molecular phylogenetics, substitution models approximate the evolutionary process of transitions between character states. These models vary in complexity and ability to capture biological reality [127,128,129]. Analogous substitution models for syntenic data have yet, to our knowledge, to be developed. However, structural variants can segregate among human populations [40] and recent developments of reference-free pangenomes may help facilitate their detection and illuminate their evolutionary dynamics [130], paving the way for creating models that capture exchange rates between syntenic states. The empirical determination of best practices for model selection will be important for future studies. Assuming overfitting is not an issue, highly parameterized models may be appropriate for synteny-based tree inference.
Other potential sources of error. Several other sources of error may come into play. For example, although few examples of convergent evolution in genome structure are known [131,132,133], they nonetheless demonstrate how independent rearrangements that result in the same structure could contribute to error in synteny-based phylogenomics. Specifically, the currently accepted evolutionary relationships among the major rodent clades of Hystricomorpha (e.g., capybaras and naked-mole rats), Sciuromorpha (e.g., squirrels and marmots), and Myomorpha (e.g., rats and mice) indicate that Hystricomorpha diverged first and that Sciuromorpha and Myomorpha are sister lineages [132]. However, independent splitting events in the ortholog of human 3p21.31 in the Hystricomorpha (e.g., Capybaras) and Sciuromorpha (e.g., squirrels) lineages would incorrectly suggest a sister relationship between each lineage [132]. Other sources of error may include an underpowered number of syntenic blocks and intraspecies heterogeneity in karyotype and chromosome structure due to, for example, Robertsonian chromosome fusions and copy number variants [72,111].
For phylogenomic analyses based on collections of multiple sequence alignments, researchers have demonstrated that not all loci have equal phylogenetic information. For example, genes displaying a clock-like pattern of evolution have often been favored for divergence time analysis [134,135,136]. Measures have been developed to quantify the information encoded in multiple sequence alignments and phylogenetic trees inferred from them. Fortunately, some methods may be easily adapted to synteny data. For example, treeness—a measure of signal-to-noise based on the proportion of tree distance observed among internal branches [137]—may help identify syntenic blocks with robust phylogenetic signal. Similarly, rogue taxa, organisms with unstable phylogenetic placement among syntenic blocks, can be pruned from a data matrix [86]. Developing methods to measure the phylogenetic informativeness of different syntenic blocks will help increase signal-to-noise ratios among datasets and aid in refining their usage and interpretation within phylogenomic analyses.
Research opportunities using synteny data and species trees.
Developing best practices for accurate synteny-based phylogenomics will help address current knowledge gaps in our understanding of genome evolution. For example, not only will synteny-based phylogenomics offer a new perspective for Tree of Life reconstructions, but the underlying synteny data may help provide functional insights into gene clusters (Figure 4B). Synteny-based phylogenomics will also help trace the evolution of chromosomes and gene clusters along phylogenies. Such reconstructions will help identify whole genome duplication events, which have been of longstanding interest to biologists because they provide fodder for molecular innovation, such as functional divergence of the resultant ohnologs [138,139].
Synteny-based phylogenomics may also facilitate ancestral genome reconstruction, potentially enabling (near) reference-level assemblies given sufficiently sequenced and assembled genomes from extant species (Figure 4B). Accurate reconstructions of ancestral genomes, coupled with ancient DNA sequencing, may help resurrect the genomes of extinct lineages. More broadly, a complete understanding of synteny evolution across time and species will contribute to a unified theory of genome architecture evolution.
While these opportunities present only a few exciting research prospects, phylogeneticists must first prioritize evaluating the efficacy of synteny-based phylogenomics for reconstructing ancient and recent divergences, spanning species and populations.
Conclusion
Improvements in genome sequencing, assembly, and annotation have revolutionized the quest to reconstruct the Tree of Life. With cutting-edge technologies and algorithms that enable the inference of highly contiguous genomes, synteny has reemerged as a powerful character for Tree of Life inquiries. Two studies tackling longstanding debates in animal phylogeny serve as key early studies for demonstrating the potential utility and caveats of using synteny to reconstruct life’s history [14,15]. These studies mark a new chapter wherein synteny-based phylogenomics promises to bring fresh insights, albeit after a series of technical challenges have been overcome. Tackling these challenges head-on will help shape best practices and deepen our understanding of synteny-based phylogenomics.
It is unlikely that Sturtevant and Dobzhansky, pioneers of their time in the 1930s, could have foreseen the far-reaching implications of their work on synteny as a phylogenetic marker. Nonetheless, their efforts have laid the groundwork for discoveries that continue to unfold today, nearly a century later, as technological advancements enable the realization of their ambition. Uniting phylogenomics with comparisons of genome architecture in a whole-evidence approach promises to illuminate the detailed topology of the Tree of Life.
Funding
JLS is a Howard Hughes Medical Institute Awardee of the Life Sciences Research Foundation.
Acknowledgments
JLS thanks Drs. Antonis Rokas, Xing-Xing Shen, and Yuanning Li for fruitful discussions about phylogenomics over the years. In particular, JLS thanks Dr. Rokas for teaching him much of what he knows about phylogenomics. JLS and NK thank Drs. Thibaut Brunet, Maxwell C Coyle, and Xing-Xing Shen for reading the manuscript and providing helpful comments and suggestions prior to submission.
Conflicts of interest
JLS is an advisor for ForensisGroup Inc.
References
- Dobzhansky T, Sturtevant AH. INVERSIONS IN THE CHROMOSOMES OF DROSOPHILA PSEUDOOBSCURA. Genetics. 1938, 23, 28–64. [CrossRef]
- Fitch WM, Margoliash E. Construction of Phylogenetic Trees: A method based on mutation distances as estimated from cytochrome c sequences is of general applicability. Science. 1967, 155, 279–284. [CrossRef]
- Haggerty LS, Martin FJ, Fitzpatrick DA, McInerney JO. Gene and genome trees conflict at many levels. Phil Trans R Soc B. 2009, 364, 2209–2219. [CrossRef]
- Steenwyk JL, Li Y, Zhou X. Incongruence in the phylogenomics era. Nature Reviews Genetics. 2023. [Google Scholar] [CrossRef] [PubMed]
- Rokas A, Williams BL, King N, Carroll SB. Genome-scale approaches to resolving incongruence in molecular phylogenies. Nature. 2003, 425, 798–804. [CrossRef]
- Kapli P, Yang Z, Telford MJ. Phylogenetic tree building in the genomic age. Nat Rev Genet. 2020, 21, 428–444. [CrossRef]
- Philippe H, Lartillot N, Brinkmann H. Multigene Analyses of Bilaterian Animals Corroborate the Monophyly of Ecdysozoa, Lophotrochozoa, and Protostomia. Molecular Biology and Evolution. 2005, 22, 1246–1253. [CrossRef]
- Giribet G, Edgecombe GD. Current Understanding of Ecdysozoa and its Internal Phylogenetic Relationships. Integrative and Comparative Biology. 2017, 57, 455–466. [CrossRef]
- Crotty SM, Minh BQ, Bean NG; et al. GHOST: Recovering Historical Signal from Heterotachously Evolved Sequence Alignments. Systematic Biology 2019, (syz051). [Google Scholar] [CrossRef]
- Williams TA, Cox CJ, Foster PG, Szöllősi GJ, Embley TM. Phylogenomics provides robust support for a two-domains tree of life. Nat Ecol Evol. 2019, 4, 138–147. [CrossRef]
- Jarvis ED, Mirarab S, Aberer AJ, Li B, Houde P, Li C, et al. Whole-genome analyses resolve early branches in the tree of life of modern birds. Science. 2014, 346, 1320–1331. [CrossRef]
- Choi B, Crisp MD, Cook LG, Meusemann K, Edwards RD, Toon A, et al. Identifying genetic markers for a range of phylogenetic utility–From species to family level. Brewer MS, editor. PLoS ONE. 2019, 14, e0218995. [CrossRef]
- Debray K, Marie-Magdelaine J, Ruttink T, Clotault J, Foucher F, Malécot V. Identification and assessment of variable single-copy orthologous (SCO) nuclear loci for low-level phylogenomics: a case study in the genus Rosa (Rosaceae). BMC Evol Biol. 2019, 19, 152. [CrossRef]
- Parey E, Louis A, Montfort J, Bouchez O, Roques C, Iampietro C, et al. Genome structures resolve the early diversification of teleost fishes. Science. 2023, 379, 572–575. [CrossRef]
- Schultz DT, Haddock SHD, Bredeson JV, Green RE, Simakov O, Rokhsar DS. Ancient gene linkages support ctenophores as sister to other animals. Nature. 2023 [cited ]. 21 May. [CrossRef]
- Wainright PO, Hinkle G, Sogin ML, Stickel SK. Monophyletic Origins of the Metazoa: an Evolutionary Link with Fungi. Science. 1993, 260, 340–342. [CrossRef]
- Brusca RC, Brusca GJ. Invertebrates. Sinauer Associates Incorporated; 2002.
- Collins, AG. Evaluating multiple alternative hypotheses for the origin of Bilateria: An analysis of 18S rRNA molecular evidence. Proc Natl Acad Sci USA. 1998, 95, 15458–15463. [Google Scholar] [CrossRef]
- Medina M, Collins AG, Silberman JD, Sogin ML. Evaluating hypotheses of basal animal phylogeny using complete sequences of large and small subunit rRNA. Proc Natl Acad Sci USA. 2001, 98, 9707–9712. [CrossRef]
- Podar M, Haddock SHD, Sogin ML, Harbison GR. A Molecular Phylogenetic Framework for the Phylum Ctenophora Using 18S rRNA Genes. Molecular Phylogenetics and Evolution. 2001, 21, 218–230. [CrossRef]
- Dunn CW, Hejnol A, Matus DQ, Pang K, Browne WE, Smith SA, et al. Broad phylogenomic sampling improves resolution of the animal tree of life. Nature. 2008, 452, 745–749. [CrossRef]
- Philippe H, Derelle R, Lopez P, Pick K, Borchiellini C, Boury-Esnault N, et al. Phylogenomics Revives Traditional Views on Deep Animal Relationships. Current Biology. 2009, 19, 706–712. [CrossRef]
- Simion P, Philippe H, Baurain D, Jager M, Richter DJ, Di Franco A, et al. A Large and Consistent Phylogenomic Dataset Supports Sponges as the Sister Group to All Other Animals. Current Biology. 2017, 27, 958–967. [CrossRef]
- Shen X-X, Hittinger CT, Rokas A. Contentious relationships in phylogenomic studies can be driven by a handful of genes. Nat Ecol Evol. 2017, 1, 0126. [CrossRef]
- King N, Rokas A. Embracing Uncertainty in Reconstructing Early Animal Evolution. Current Biology. 2017, 27, R1081–R1088. [CrossRef]
- Whelan NV, Kocot KM, Moroz TP, Mukherjee K, Williams P, Paulay G, et al. Ctenophore relationships and their placement as the sister group to all other animals. Nat Ecol Evol. 2017, 1, 1737–1746. [CrossRef]
- Li Y, Shen X-X, Evans B, Dunn CW, Rokas A. Rooting the Animal Tree of Life. Tamura K, editor. Molecular Biology and Evolution. 2021, 38, 4322–4333. [CrossRef]
- Whelan NV, Halanych KM. Available data do not rule out Ctenophora as the sister group to all other Metazoa. Nat Commun. 2023, 14, 711. [CrossRef]
- Le HLV, Lecointre G, Perasso R. A 28S rRNA-Based Phylogeny of the Gnathostomes: First Steps in the Analysis of Conflict and Congruence with Morphologically Based Cladograms. Molecular Phylogenetics and Evolution. 1993, 2, 31–51. [CrossRef]
- Dornburg A, Near TJ. The emerging phylogenetic perspective on the evolution of actinopterygian fishes. Annual Review of Ecology, Evolution, and Systematics. 2021, 52, 427–452.
- Rokas A, Holland PWH. Rare genomic changes as a tool for phylogenetics. Trends in Ecology & Evolution. 2000, 15, 454–459. [CrossRef]
- Castresana J, Feldmaier-Fuchs G, Yokobori S, Satoh N, Pääbo S. The Mitochondrial Genome of the Hemichordate Balanoglossus carnosus and the Evolution of Deuterostome Mitochondria. Genetics. 1998, 150, 1115–1123. [CrossRef]
- Venkatesh B, Ning Y, Brenner S. Late changes in spliceosomal introns define clades in vertebrate evolution. Proc Natl Acad Sci USA. 1999, 96, 10267–10271. [CrossRef]
- Rokas A, Kathirithamby J, Holland PWH. Intron insertion as a phylogenetic character: the engrailed homeobox of Strepsiptera does not indicate affinity with Diptera. Insect Mol Biol. 1999, 8, 527–530. [CrossRef]
- Steenwyk JL, Opulente DA, Kominek J, Shen X-X, Zhou X, Labella AL, et al. Extensive loss of cell-cycle and DNA repair genes in an ancient lineage of bipolar budding yeasts. Kamoun S, editor. PLoS Biol. 2019, 17, e3000255. [CrossRef]
- Sturtevant AH, Dobzhansky Th. Inversions in the Third Chromosome of Wild Races of Drosophila Pseudoobscura, and Their Use in the Study of the History of the Species. Proc Natl Acad Sci USA. 1936, 22, 448–450. [CrossRef]
- Steenwyk JL, Soghigian JS, Perfect JR, Gibbons JG. Copy number variation contributes to cryptic genetic variation in outbreak lineages of Cryptococcus gattii from the North American Pacific Northwest. BMC Genomics. 2016, 17, 700. [CrossRef]
- Lee Y-L, Bosse M, Mullaart E, Groenen MAM, Veerkamp RF, Bouwman AC. Functional and population genetic features of copy number variations in two dairy cattle populations. BMC Genomics. 2020, 21, 89. [CrossRef]
- Brown KH, Dobrinski KP, Lee AS, Gokcumen O, Mills RE, Shi X, et al. Extensive genetic diversity and substructuring among zebrafish strains revealed through copy number variant analysis. Proc Natl Acad Sci USA. 2012, 109, 529–534. [CrossRef]
- Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J, et al. An integrated map of structural variation in 2,504 human genomes. Nature. 2015, 526, 75–81. [CrossRef]
- Fortna A, Kim Y, MacLaren E, Marshall K, Hahn G, Meltesen L, et al. Lineage-Specific Gene Duplication and Loss in Human and Great Ape Evolution. Chris Tyler-Smith, editor. PLoS Biol. 2004, 2, e207. [CrossRef]
- Mühlhausen S, Schmitt HD, Pan K-T, Plessmann U, Urlaub H, Hurst LD, et al. Endogenous Stochastic Decoding of the CUG Codon by Competing Ser- and Leu-tRNAs in Ascoidea asiatica. Current Biology. 2018, 28, 2046–2057e5. [CrossRef]
- Mathews S, Donoghue MJ. The Root of Angiosperm Phylogeny Inferred from Duplicate Phytochrome Genes. Science. 1999, 286, 947–950. [CrossRef]
- One Thousand Plant Transcriptomes Initiative. One thousand plant transcriptomes and the phylogenomics of green plants. Nature. 2019, 574, 679–685. [CrossRef]
- Doronina L, Reising O, Clawson H, Ray DA, Schmitz J. True Homoplasy of Retrotransposon Insertions in Primates. Susko E, editor. Systematic Biology. 2019, 68, 482–493. [CrossRef]
- Cloutier A, Sackton TB, Grayson P, Clamp M, Baker AJ, Edwards SV. Whole-Genome Analyses Resolve the Phylogeny of Flightless Birds (Palaeognathae) in the Presence of an Empirical Anomaly Zone. Faircloth B, editor. Systematic Biology. 2019, 68, 937–955. [CrossRef]
- Murphy WJ, Foley NM, Bredemeyer KR, Gatesy J, Springer MS. Phylogenomics and the Genetic Architecture of the Placental Mammal Radiation. Annu Rev Anim Biosci. 2021, 9, 29–53. [CrossRef]
- Takahashi K, Terai Y, Nishida M, Okada N. A novel family of short interspersed repetitive elements (SINEs) from cichlids: the patterns of insertion of SINEs at orthologous loci support the proposed monophyly of four major groups of cichlid fishes in Lake Tanganyika. Molecular Biology and Evolution. 1998, 15, 391–407. [CrossRef]
- Takahashi K, Nishida M, Yuma M, Okada N. Retroposition of the AFC Family of SINEs (Short Interspersed Repetitive Elements) Before and During the Adaptive Radiation of Cichlid Fishes in Lake Malawi and Related Inferences About Phylogeny. Journal of Molecular Evolution. 2001, 53, 496–507. [CrossRef]
- Springer MS, Molloy EK, Sloan DB, Simmons MP, Gatesy J. ILS-Aware Analysis of Low-Homoplasy Retroelement Insertions: Inference of Species Trees and Introgression Using Quartets. Murphy W, editor. Journal of Heredity. 2020, 111, 147–168. [CrossRef]
- Martín-Durán JM, Ryan JF, Vellutini BC, Pang K, Hejnol A. Increased taxon sampling reveals thousands of hidden orthologs in flatworms. Genome Res. 2017, 27, 1263–1272. [CrossRef]
- Krassowski T, Coughlan AY, Shen X-X, Zhou X, Kominek J, Opulente DA, et al. Evolutionary instability of CUG-Leu in the genetic code of budding yeasts. Nat Commun. 2018, 9, 1887. [CrossRef]
- Dellaporta SL, Xu A, Sagasser S, Jakob W, Moreno MA, Buss LW, et al. Mitochondrial genome of Trichoplax adhaerens supports Placozoa as the basal lower metazoan phylum. Proc Natl Acad Sci USA. 2006, 103, 8751–8756. [CrossRef]
- Laumer CE, Gruber-Vodicka H, Hadfield MG, Pearse VB, Riesgo A, Marioni JC, et al. Support for a clade of Placozoa and Cnidaria in genes with minimal compositional bias. eLife. 2018, 7, e36278. [CrossRef]
- Ding Y-M, Pang X-X, Cao Y, Zhang W-P, Renner SS, Zhang D-Y, et al. Genome structure-based Juglandaceae phylogenies contradict alignment-based phylogenies and substitution rates vary with DNA repair genes. Nat Commun. 2023, 14, 617. [CrossRef]
- Haas BJ, Delcher AL, Wortman JR, Salzberg SL. DAGchainer: a tool for mining segmental genome duplications and synteny. Bioinformatics. 2004, 20, 3643–3646.
- Proost S, Fostier J, De Witte D, Dhoedt B, Demeester P, Van de Peer Y, et al. i-ADHoRe 3.0—fast and sensitive detection of genomic homology in extremely large data sets. Nucleic acids research. 2012, 40, e11–e11.
- Wang Y, Tang H, Debarry JD, Tan X, Li J, Wang X, et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic acids research. 2012, 40, e49–e49.
- Drillon G, Carbone A, Fischer G. SynChro: a fast and easy tool to reconstruct and visualize synteny blocks along eukaryotic chromosomes. PloS one. 2014, 9, e92621.
- Almeida-Silva F, Zhao T, Ullrich KK, Schranz ME, Van De Peer Y. syntenet: an R/Bioconductor package for the inference and analysis of synteny networks. Martelli PL, editor. Bioinformatics. 2023, 39, btac806. [CrossRef]
- Mackintosh A, De La Rosa PMG, Martin SH, Lohse K, Laetsch DR. Inferring inter-chromosomal rearrangements and ancestral linkage groups from synteny. Evolutionary Biology; 2023 Sep. [CrossRef]
- Hane JK, Rouxel T, Howlett BJ, Kema GH, Goodwin SB, Oliver RP. A novel mode of chromosomal evolution peculiar to filamentous Ascomycete fungi. Genome Biol. 2011, 12, R45. [CrossRef]
- Robberecht C, Voet T, Esteki MZ, Nowakowska BA, Vermeesch JR. Nonallelic homologous recombination between retrotransposable elements is a driver of de novo unbalanced translocations. Genome Res. 2013, 23, 411–418. [CrossRef]
- Ma J, Bennetzen JL. Recombination, rearrangement, reshuffling, and divergence in a centromeric region of rice. Proc Natl Acad Sci USA. 2006, 103, 383–388. [CrossRef]
- Liu P, Lacaria M, Zhang F, Withers M, Hastings PJ, Lupski JR. Frequency of Nonallelic Homologous Recombination Is Correlated with Length of Homology: Evidence that Ectopic Synapsis Precedes Ectopic Crossing-Over. The American Journal of Human Genetics. 2011, 89, 580–588. [CrossRef]
- Ferguson S, Jones A, Murray K, Schwessinger B, Borevitz JO. Interspecies genome divergence is predominantly due to frequent small scale rearrangements in Eucalyptus. Molecular Ecology. 2023, 32, 1271–1287. [CrossRef]
- Li Y, Liu H, Steenwyk JL, LaBella AL, Harrison M-C, Groenewald M, et al. Contrasting modes of macro and microsynteny evolution in a eukaryotic subphylum. Current Biology. 2022, S0960982222016700. [CrossRef]
- Delsuc F, Brinkmann H, Philippe H. Phylogenomics and the reconstruction of the tree of life. Nat Rev Genet. 2005, 6, 361–375. [CrossRef]
- Zheng C, Sankoff D. Gene order in rosid phylogeny, inferred from pairwise syntenies among extant genomes. BMC Bioinformatics. 2012, 13, S9. [CrossRef]
- Drillon G, Champeimont R, Oteri F, Fischer G, Carbone A. Phylogenetic Reconstruction Based on Synteny Block and Gene Adjacencies. Battistuzzi FU, editor. Molecular Biology and Evolution. 2020, 37, 2747–2762. [CrossRef]
- Zhao T, Zwaenepoel A, Xue J-Y, Kao S-M, Li Z, Schranz ME, et al. Whole-genome microsynteny-based phylogeny of angiosperms. Nat Commun. 2021, 12, 3498. [CrossRef]
- Therman E, Susman B, Denniston C. The nonrandom participation of human acrocentric chromosomes in Robertsonian translocations. Annals of Human Genetics. 1989, 53, 49–65. [CrossRef]
- Fairclough SR, Chen Z, Kramer E, Zeng Q, Young S, Robertson HM, et al. Premetazoan genome evolution and the regulation of cell differentiation in the choanoflagellate Salpingoeca rosetta. Genome Biol. 2013, 14, R15. [CrossRef]
- King N, Westbrook MJ, Young SL, Kuo A, Abedin M, Chapman J, et al. The genome of the choanoflagellate Monosiga brevicollis and the origin of metazoans. Nature. 2008, 451, 783–788. [CrossRef]
- Ocaña-Pallarès E, Williams TA, López-Escardó D, Arroyo AS, Pathmanathan JS, Bapteste E, et al. Divergent genomic trajectories predate the origin of animals and fungi. Nature. 2022, 609, 747–753. [CrossRef]
- Torruella G, Derelle R, Paps J, Lang BF, Roger AJ, Shalchian-Tabrizi K, et al. Phylogenetic Relationships within the Opisthokonta Based on Phylogenomic Analyses of Conserved Single-Copy Protein Domains. Molecular Biology and Evolution. 2012, 29, 531–544. [CrossRef]
- Ruiz-Trillo I, Roger AJ, Burger G, Gray MW, Lang BF. A Phylogenomic Investigation into the Origin of Metazoa. Molecular Biology and Evolution. 2008, 25, 664–672. [CrossRef]
- Richter DJ, Fozouni P, Eisen MB, King N. Gene family innovation, conservation and loss on the animal stem lineage. eLife. 2018, 7, e34226. [CrossRef]
- Whelan NV, Kocot KM, Moroz LL, Halanych KM. Error, signal, and the placement of Ctenophora sister to all other animals. Proc Natl Acad Sci USA. 2015, 112, 5773–5778. [CrossRef]
- Sperling EA, Pisani D, Peterson KJ. Poriferan paraphyly and its implications for Precambrian palaeobiology. SP. 2007, 286, 355–368. [CrossRef]
- Borchiellini C, Manuel M, Alivon E, Boury-Esnault N, Vacelet J, Le Parco Y. Sponge paraphyly and the origin of Metazoa: Sponge paraphyly. Journal of Evolutionary Biology. 2001, 14, 171–179. [CrossRef]
- Kenny NJ, Francis WR, Rivera-Vicéns RE, Juravel K, De Mendoza A, Díez-Vives C, et al. Tracing animal genomic evolution with the chromosomal-level assembly of the freshwater sponge Ephydatia muelleri. Nat Commun. 2020, 11, 3676. [CrossRef]
- Hughes LC, Ortí G, Huang Y, Sun Y, Baldwin CC, Thompson AW, et al. Comprehensive phylogeny of ray-finned fishes (Actinopterygii) based on transcriptomic and genomic data. Proc Natl Acad Sci USA. 2018, 115, 6249–6254. [CrossRef]
- Faircloth BC, Sorenson L, Santini F, Alfaro ME. A Phylogenomic Perspective on the Radiation of Ray-Finned Fishes Based upon Targeted Sequencing of Ultraconserved Elements (UCEs). Moreau CS, editor. PLoS ONE. 2013, 8, e65923. [CrossRef]
- Pollock DD, Zwickl DJ, McGuire JA, Hillis DM. Increased Taxon Sampling Is Advantageous for Phylogenetic Inference. Crandall K, editor. Systematic Biology. 2002, 51, 664–671. [CrossRef]
- Aberer AJ, Krompass D, Stamatakis A. Pruning Rogue Taxa Improves Phylogenetic Accuracy: An Efficient Algorithm and Webservice. Systematic Biology. 2013, 62, 162–166. [CrossRef]
- Shen X-X, Zhou X, Kominek J, Kurtzman CP, Hittinger CT, Rokas A. Reconstructing the Backbone of the Saccharomycotina Yeast Phylogeny Using Genome-Scale Data. G3 Genes|Genomes|Genetics. 2016, 6, 3927–3939. [CrossRef]
- Riley R, Haridas S, Wolfe KH, Lopes MR, Hittinger CT, Göker M, et al. Comparative genomics of biotechnologically important yeasts. Proc Natl Acad Sci USA. 2016, 113, 9882–9887. [CrossRef]
- Shen X-X, Opulente DA, Kominek J, Zhou X, Steenwyk JL, Buh KV, et al. Tempo and Mode of Genome Evolution in the Budding Yeast Subphylum. Cell. 2018, 175, 1533–1545e20. [CrossRef]
- Scannell DR, Byrne KP, Gordon JL, Wong S, Wolfe KH. Multiple rounds of speciation associated with reciprocal gene loss in polyploid yeasts. Nature. 2006, 440, 341–345. [CrossRef]
- Pisani D, Pett W, Dohrmann M, Feuda R, Rota-Stabelli O, Philippe H, et al. Genomic data do not support comb jellies as the sister group to all other animals. Proc Natl Acad Sci USA. 2015, 112, 15402–15407. [CrossRef]
- Brinkmann H, Van Der Giezen M, Zhou Y, De Raucourt GP, Philippe H. An Empirical Assessment of Long-Branch Attraction Artefacts in Deep Eukaryotic Phylogenomics. Hedin M, editor. Systematic Biology. 2005, 54, 743–757. [CrossRef]
- Marx, V. Method of the year: long-read sequencing. Nat Methods. 2023, 20, 6–11. [Google Scholar] [CrossRef]
- Giani AM, Gallo GR, Gianfranceschi L, Formenti G. Long walk to genomics: History and current approaches to genome sequencing and assembly. Computational and Structural Biotechnology Journal. 2020, 18, 9–19. [CrossRef]
- Belton J-M, McCord RP, Gibcus JH, Naumova N, Zhan Y, Dekker J. Hi–C: A comprehensive technique to capture the conformation of genomes. Methods. 2012, 58, 268–276. [CrossRef]
- Liu D, Hunt M, Tsai IJ. Inferring synteny between genome assemblies: a systematic evaluation. BMC Bioinformatics. 2018, 19, 26. [CrossRef]
- Waterhouse RM, Seppey M, Simão FA, Manni M, Ioannidis P, Klioutchnikov G, et al. BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics. Molecular Biology and Evolution. 2018, 35, 543–548. [CrossRef]
- Rhie A, McCarthy SA, Fedrigo O, Damas J, Formenti G, Koren S, et al. Towards complete and error-free genome assemblies of all vertebrate species. Nature. 2021, 592, 737–746. [CrossRef]
- Weisman CM, Murray AW, Eddy SR. Mixing genome annotation methods in a comparative analysis inflates the apparent number of lineage-specific genes. Current Biology. 2022, 32, 2632–2639e2. [CrossRef]
- Haas BJ, Salzberg SL, Zhu W, Pertea M, Allen JE, Orvis J, et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, R7. [CrossRef]
- Rosato M, Hoelscher B, Lin Z, Agwu C, Xu F. Transcriptome analysis provides genome annotation and expression profiles in the central nervous system of Lymnaea stagnalis at different ages. BMC Genomics. 2021, 22, 637. [CrossRef]
- Fernández R, Gabaldón T, Dessimoz C. Orthology: definitions, inference, and impact on species phylogeny inference. 2019 [cited ]. 25 May. [CrossRef]
- Philippe H, Vienne DMD, Ranwez V, Roure B, Baurain D, Delsuc F. Pitfalls in supermatrix phylogenomics. EJT. 2017 [cited 5 Dec 2023]. [CrossRef]
- Stolzer M, Lai H, Xu M, Sathaye D, Vernot B, Durand D. Inferring duplications, losses, transfers and incomplete lineage sorting with nonbinary species trees. Bioinformatics. 2012, 28, i409–i415. [CrossRef]
- Minkin I, Medvedev P. Scalable multiple whole-genome alignment and locally collinear block construction with SibeliaZ. Nat Commun. 2020, 11, 6327. [CrossRef]
- Armstrong J, Hickey G, Diekhans M, Fiddes IT, Novak AM, Deran A, et al. Progressive Cactus is a multiple-genome aligner for the thousand-genome era. Nature. 2020, 587, 246–251. [CrossRef]
- Philippe H, Brinkmann H, Lavrov DV, Littlewood DTJ, Manuel M, Wörheide G, et al. Resolving Difficult Phylogenetic Questions: Why More Sequences Are Not Enough. Penny D, editor. PLoS Biol. 2011, 9, e1000602. [CrossRef]
- Maddison WP, Knowles LL. Inferring Phylogeny Despite Incomplete Lineage Sorting. Collins T, editor. Systematic Biology. 2006, 55, 21–30. [CrossRef]
- Feng S, Bai M, Rivas-González I, Li C, Liu S, Tong Y, et al. Incomplete lineage sorting and phenotypic evolution in marsupials. Cell. 2022, 185, 1646–1660e18. [CrossRef]
- Avise JC, Robinson TJ. Hemiplasy: A New Term in the Lexicon of Phylogenetics. Kubatko L, editor. Systematic Biology. 2008, 57, 503–507. [CrossRef]
- Jeffares DC, Jolly C, Hoti M, Speed D, Shaw L, Rallis C, et al. Transient structural variations have strong effects on quantitative traits and reproductive isolation in fission yeast. Nat Commun. 2017, 8, 14061. [CrossRef]
- Steenwyk J, Rokas A. Extensive Copy Number Variation in Fermentation-Related Genes Among Saccharomyces cerevisiae Wine Strains. G3 Genes|Genomes|Genetics. 2017, 7, 1475–1485. [CrossRef]
- Abbott R, Albach D, Ansell S, Arntzen JW, Baird SJE, Bierne N, et al. Hybridization and speciation. J Evol Biol. 2013, 26, 229–246. [CrossRef]
- Irisarri I, Singh P, Koblmüller S, Torres-Dowdall J, Henning F, Franchini P, et al. Phylogenomics uncovers early hybridization and adaptive loci shaping the radiation of Lake Tanganyika cichlid fishes. Nat Commun. 2018, 9, 3159. [CrossRef]
- Bjornson S, Upham N, Verbruggen H, Steenwyk J. Phylogenomic Inference, Divergence-Time Calibration, and Methods for Characterizing Reticulate Evolution. Biology and Life Sciences; 2023 Sep. [CrossRef]
- Arnold BJ, Huang I-T, Hanage WP. Horizontal gene transfer and adaptive evolution in bacteria. Nat Rev Microbiol. 2022, 20, 206–218. [CrossRef]
- Gophna U, Altman-Price N. Horizontal Gene Transfer in Archaea—From Mechanisms to Genome Evolution. Annu Rev Microbiol. 2022, 76, 481–502. [CrossRef]
- Buck R, Ortega-Del Vecchyo D, Gehring C, Michelson R, Flores-Rentería D, Klein B, et al. Sequential hybridization may have facilitated ecological transitions in the Southwestern pinyon pine syngameon. New Phytologist. 2023, 237, 2435–2449. [CrossRef]
- Goulet BE, Roda F, Hopkins R. Hybridization in Plants: Old Ideas, New Techniques. Plant Physiol. 2017, 173, 65–78. [CrossRef]
- Rieseberg LH, Kim S-C, Randell RA, Whitney KD, Gross BL, Lexer C, et al. Hybridization and the colonization of novel habitats by annual sunflowers. Genetica. 2007, 129, 149–165. [CrossRef]
- Steenwyk JL, Lind AL, Ries LNA, dos Reis TF, Silva LP, Almeida F, et al. Pathogenic Allodiploid Hybrids of Aspergillus Fungi. Current Biology. 2020, 30, 2495–2507e7. [CrossRef]
- Marcet-Houben M, Gabaldón T. Beyond the Whole-Genome Duplication: Phylogenetic Evidence for an Ancient Interspecies Hybridization in the Baker’s Yeast Lineage. Hurst LD, editor. PLoS Biol. 2015, 13, e1002220. [CrossRef]
- Adavoudi R, Pilot M. Consequences of Hybridization in Mammals: A Systematic Review. Genes. 2021, 13, 50. [CrossRef]
- League GP, Slot JC, Rokas A. The ASP3 locus in Saccharomyces cerevisiae originated by horizontal gene transfer from Wickerhamomyces. FEMS Yeast Res. 2012, 12, 859–863. [CrossRef]
- Kominek J, Doering DT, Opulente DA, Shen X-X, Zhou X, DeVirgilio J, et al. Eukaryotic Acquisition of a Bacterial Operon. Cell. 2019, 176, 1356–1366e10. [CrossRef]
- Davín AA, Tannier E, Williams TA, Boussau B, Daubin V, Szöllősi GJ. Gene transfers can date the tree of life. Nat Ecol Evol. 2018, 2, 904–909. [CrossRef]
- Jukes TH, Cantor CR. Evolution of Protein Molecules. Mammalian Protein Metabolism. Elsevier; 1969. pp. 21–132. [CrossRef]
- Tavaré, S. Some probabilistic and statistical problems in the analysis of DNA sequences. Lect Math Life Sci (Am Math Soc). 1986, 17, 57–86. [Google Scholar]
- Philippe H, Zhou Y, Brinkmann H, Rodrigue N, Delsuc F. Heterotachy and long-branch attraction in phylogenetics. BMC Evol Biol. 2005, 5, 50. [CrossRef]
- Liao W-W, Asri M, Ebler J, Doerr D, Haukness M, Hickey G, et al. A draft human pangenome reference. Nature. 2023, 617, 312–324. [CrossRef]
- Svedberg J, Hosseini S, Chen J, Vogan AA, Mozgova I, Hennig L, et al. Convergent evolution of complex genomic rearrangements in two fungal meiotic drive elements. Nat Commun. 2018, 9, 4242. [CrossRef]
- Jain Y, Chandradoss KR, A. V. A, Bhattacharya J, Lal M, Bagadia M, et al. Convergent evolution of a genomic rearrangement may explain cancer resistance in hystrico- and sciuromorpha rodents. npj Aging Mech Dis. 2021, 7, 20. [CrossRef]
- Mezzasalma M, Streicher JW, Guarino FM, Jones MEH, Loader SP, Odierna G, et al. Microchromosome fusions underpin convergent evolution of chameleon karyotypes. Gaitan-Espitia JD, Chapman T, editors. Evolution. 2023, 77, 1930–1944. [CrossRef]
- Steenwyk JL, Shen X-X, Lind AL, Goldman GH, Rokas A. A Robust Phylogenomic Time Tree for Biotechnologically and Medically Important Fungi in the Genera Aspergillus and Penicillium. Boyle JP, editor. mBio. 2019, 10, e00925–19. [CrossRef]
- Liu L, Zhang J, Rheindt FE, Lei F, Qu Y, Wang Y, et al. Genomic evidence reveals a radiation of placental mammals uninterrupted by the KPg boundary. Proc Natl Acad Sci USA. 2017, 114. [CrossRef]
- Smith SA, Brown JW, Walker JF. So many genes, so little time: A practical approach to divergence-time estimation in the genomic era. Escriva H, editor. PLoS ONE. 2018, 13, e0197433. [CrossRef]
- Phillips MJ, Penny D. The root of the mammalian tree inferred from whole mitochondrial genomes. Molecular Phylogenetics and Evolution. 2003, 28, 171–185. [CrossRef]
- Ortiz-Merino RA, Kuanyshev N, Braun-Galleani S, Byrne KP, Porro D, Branduardi P, et al. Evolutionary restoration of fertility in an interspecies hybrid yeast, by whole-genome duplication after a failed mating-type switch. Hurst L, editor. PLoS Biol. 2017, 15, e2002128. [CrossRef]
- Clark JW, Donoghue PCJ. Whole-Genome Duplication and Plant Macroevolution. Trends in Plant Science. 2018, 23, 933–945. [CrossRef]
Figure 1.
Depictions of incongruence and alternate hypotheses for primates, the base of the animal tree, and teleost fish phylogenies. (A) Example of tree incongruence. The weight of evidence strongly supports a sister relationship between bonobos and chimps, to the exclusion of humans. Phylogenies that are incongruent would suggest a sister relationship between (B) humans and chimps or (C) humans and bonobos. The debate concerning early animal evolution has largely focused on whether (D) sponges or (E) ctenophores diverged first from all other animals: the (F) sponge- and (G) ctenophore-first hypotheses, respectively. Among teleost fish, the debate centers on the relationships among three major lineages—the (H) Elopomorpha (mostly slim-headed fish), (I) Osteoglossomorpha (mostly bony-tongued fish), and (J) Clupeocephala (all other teleost fish). (K) The Eloposteoglossocephala (EO-sister) hypothesis suggests a sister relationship between slim-headed and bony-tongued fish, whereas the (L) Elopomorpha-first and (M) Osteoglossomorpha-first hypotheses suggest that slim-headed fish or bony-tongued fish, respectively, diverged before the other lineages split from one another. Recent studies that employed synteny as a phylogenomic marker supported the (G) ctenophore-first and (K) EO-sister hypotheses [14,15]. Image credits: (D) Bruno C. Vellutini and (E) Nick Hobgood via the Creative Commons Attribution-Share Alike 3.0 Unported license; (H) opencage via the Creative Commons Attribution-Share Alike 2.5 Generic license; (I) Viktor Kravtchenko via the CC BY 3.0 DEED Attribution 3.0 Unported; and (J) Gilles San Martin via the Creative Commons Attribution-Share Alike 2.0 license. (A, B, C, F, G, K, L, and M) Silhouette images were obtained from PhyloPic (https://www.phylopic.org); Bonobo: Kai R. Caspar, CC BY 3.0 DEED Attribution 3.0 Unported; Chimp: T. Michael Keesey and Tony Hisgett, CC BY 3.0 DEED Attribution 3.0 Unported; Human and Sponge: T. Michael Keesey, CC0 1.0 Universal Public Domain Dedication; Ctenophore: Steven Haddock, Public Domain Mark 1.0; Other animals (jellyfish): Walker Pett, Public Domain Mark 1.0; Slim-headed fish (Anguilla rostrata): Steven Traver, CC0 1.0 Universal Public Domain Dedication; Bony-tongued fish (Campylomormyrus numenius): Maija Karala, Attribution-NonCommercial-ShareAlike 3.0 Unported; and Clupeocephala (Siphateles bicolor): xgirouxb, Public Domain Mark 1.0.
Figure 1.
Depictions of incongruence and alternate hypotheses for primates, the base of the animal tree, and teleost fish phylogenies. (A) Example of tree incongruence. The weight of evidence strongly supports a sister relationship between bonobos and chimps, to the exclusion of humans. Phylogenies that are incongruent would suggest a sister relationship between (B) humans and chimps or (C) humans and bonobos. The debate concerning early animal evolution has largely focused on whether (D) sponges or (E) ctenophores diverged first from all other animals: the (F) sponge- and (G) ctenophore-first hypotheses, respectively. Among teleost fish, the debate centers on the relationships among three major lineages—the (H) Elopomorpha (mostly slim-headed fish), (I) Osteoglossomorpha (mostly bony-tongued fish), and (J) Clupeocephala (all other teleost fish). (K) The Eloposteoglossocephala (EO-sister) hypothesis suggests a sister relationship between slim-headed and bony-tongued fish, whereas the (L) Elopomorpha-first and (M) Osteoglossomorpha-first hypotheses suggest that slim-headed fish or bony-tongued fish, respectively, diverged before the other lineages split from one another. Recent studies that employed synteny as a phylogenomic marker supported the (G) ctenophore-first and (K) EO-sister hypotheses [14,15]. Image credits: (D) Bruno C. Vellutini and (E) Nick Hobgood via the Creative Commons Attribution-Share Alike 3.0 Unported license; (H) opencage via the Creative Commons Attribution-Share Alike 2.5 Generic license; (I) Viktor Kravtchenko via the CC BY 3.0 DEED Attribution 3.0 Unported; and (J) Gilles San Martin via the Creative Commons Attribution-Share Alike 2.0 license. (A, B, C, F, G, K, L, and M) Silhouette images were obtained from PhyloPic (https://www.phylopic.org); Bonobo: Kai R. Caspar, CC BY 3.0 DEED Attribution 3.0 Unported; Chimp: T. Michael Keesey and Tony Hisgett, CC BY 3.0 DEED Attribution 3.0 Unported; Human and Sponge: T. Michael Keesey, CC0 1.0 Universal Public Domain Dedication; Ctenophore: Steven Haddock, Public Domain Mark 1.0; Other animals (jellyfish): Walker Pett, Public Domain Mark 1.0; Slim-headed fish (Anguilla rostrata): Steven Traver, CC0 1.0 Universal Public Domain Dedication; Bony-tongued fish (Campylomormyrus numenius): Maija Karala, Attribution-NonCommercial-ShareAlike 3.0 Unported; and Clupeocephala (Siphateles bicolor): xgirouxb, Public Domain Mark 1.0.
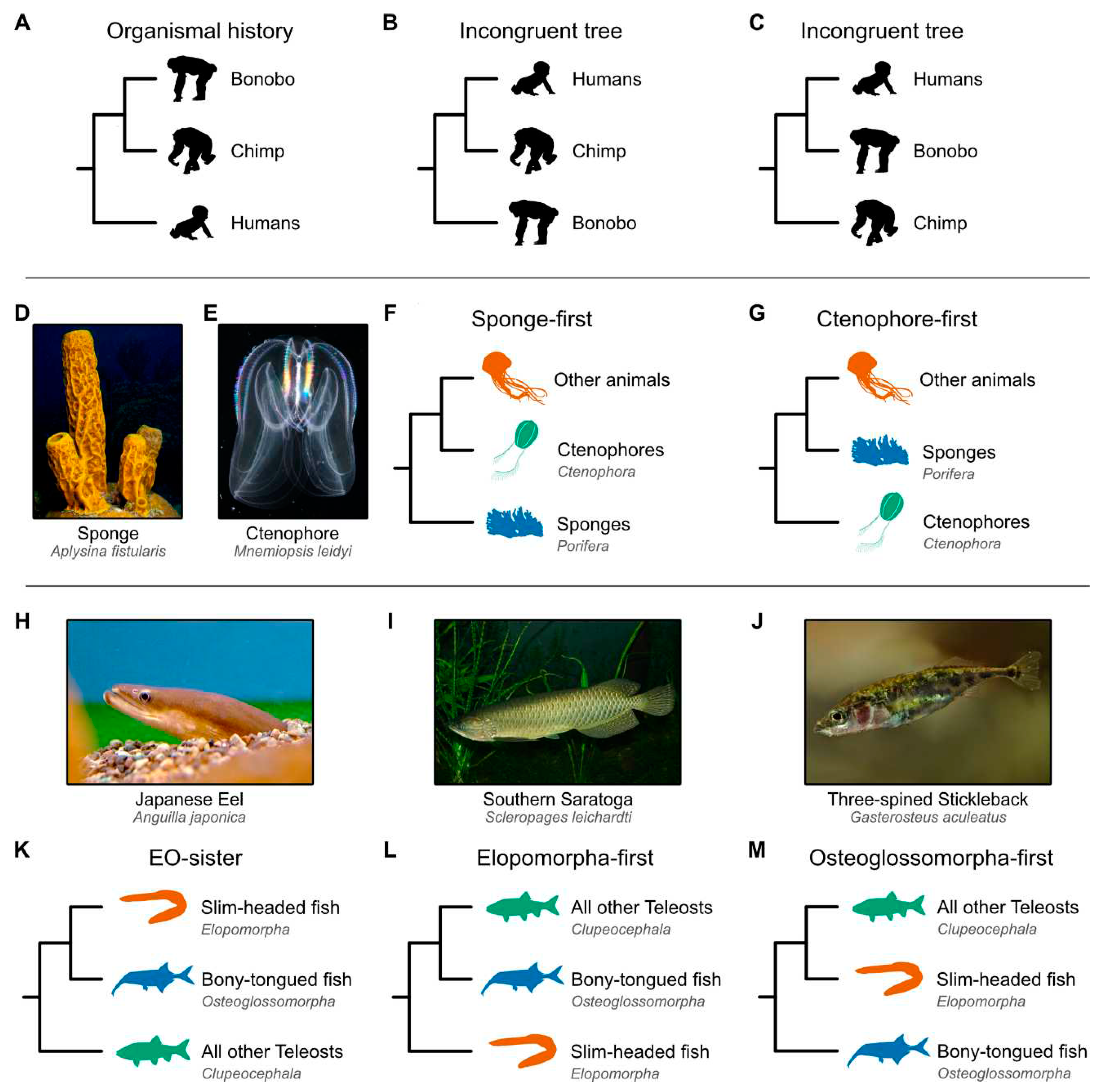
Figure 2.
Data types for sequence-based phylogenetics. Consider the relationships among four taxa (represented as T1, T2, T3, and T4) wherein the pairs T1 and T2 and T3 and T4 are sister to one another. Changes in genome architecture can be examined at the scale of (A) microsynteny (short stretches of orthologous loci) or (B) macrosynteny (long stretches of orthologous loci). Changes in synteny can be described by different processes, such as (C) fusion events without- and (D) with-mixing. (A, top) In the case of microsynteny, evidence of an inversion may occur between the blue and orange loci, (A, bottom) which happened in the ancestor of T1 and T2. (B) The same phenomenon can happen in the case of macrosynteny. (C, top) Fusion-without-mixing events between two chromosomes may also reflect phylogeny. (C, bottom) In this case, a fusion event may have occurred in the ancestor between T1 and T2. (D, top) Fusion-with-mixing can also be used to recapitulate phylogeny. Note, the evolutionary scenarios at the bottom of panels A, B, C, D depict one possible scenario. (E) Fusion-with-mixing events may occur in two steps. First, there is a fusion event, then rearrangements occur, scrambling the order of genes that once were encoded on separate chromosomes. As a result, the probability of going from a “no fusion” to “fusion-without-mixing” state, vice versa, and going from a “fusion-without-mixing” state to a “fusion-with-mixing” state is relatively higher compared to going from a “fusion-with-mixing” to a “fusion-without-mixing” state. Transitioning directly from a “no fusion” to a “fusion-with-mixing” state is highly unlikely and may require an intermediate “fusion-without-mixing” state. Transition probabilities may vary depending on the underlying genome biology of the organism, size of the syntenic region, and other parameters; only one possible set of relative probabilities are depicted here.
Figure 2.
Data types for sequence-based phylogenetics. Consider the relationships among four taxa (represented as T1, T2, T3, and T4) wherein the pairs T1 and T2 and T3 and T4 are sister to one another. Changes in genome architecture can be examined at the scale of (A) microsynteny (short stretches of orthologous loci) or (B) macrosynteny (long stretches of orthologous loci). Changes in synteny can be described by different processes, such as (C) fusion events without- and (D) with-mixing. (A, top) In the case of microsynteny, evidence of an inversion may occur between the blue and orange loci, (A, bottom) which happened in the ancestor of T1 and T2. (B) The same phenomenon can happen in the case of macrosynteny. (C, top) Fusion-without-mixing events between two chromosomes may also reflect phylogeny. (C, bottom) In this case, a fusion event may have occurred in the ancestor between T1 and T2. (D, top) Fusion-with-mixing can also be used to recapitulate phylogeny. Note, the evolutionary scenarios at the bottom of panels A, B, C, D depict one possible scenario. (E) Fusion-with-mixing events may occur in two steps. First, there is a fusion event, then rearrangements occur, scrambling the order of genes that once were encoded on separate chromosomes. As a result, the probability of going from a “no fusion” to “fusion-without-mixing” state, vice versa, and going from a “fusion-without-mixing” state to a “fusion-with-mixing” state is relatively higher compared to going from a “fusion-with-mixing” to a “fusion-without-mixing” state. Transitioning directly from a “no fusion” to a “fusion-with-mixing” state is highly unlikely and may require an intermediate “fusion-without-mixing” state. Transition probabilities may vary depending on the underlying genome biology of the organism, size of the syntenic region, and other parameters; only one possible set of relative probabilities are depicted here.
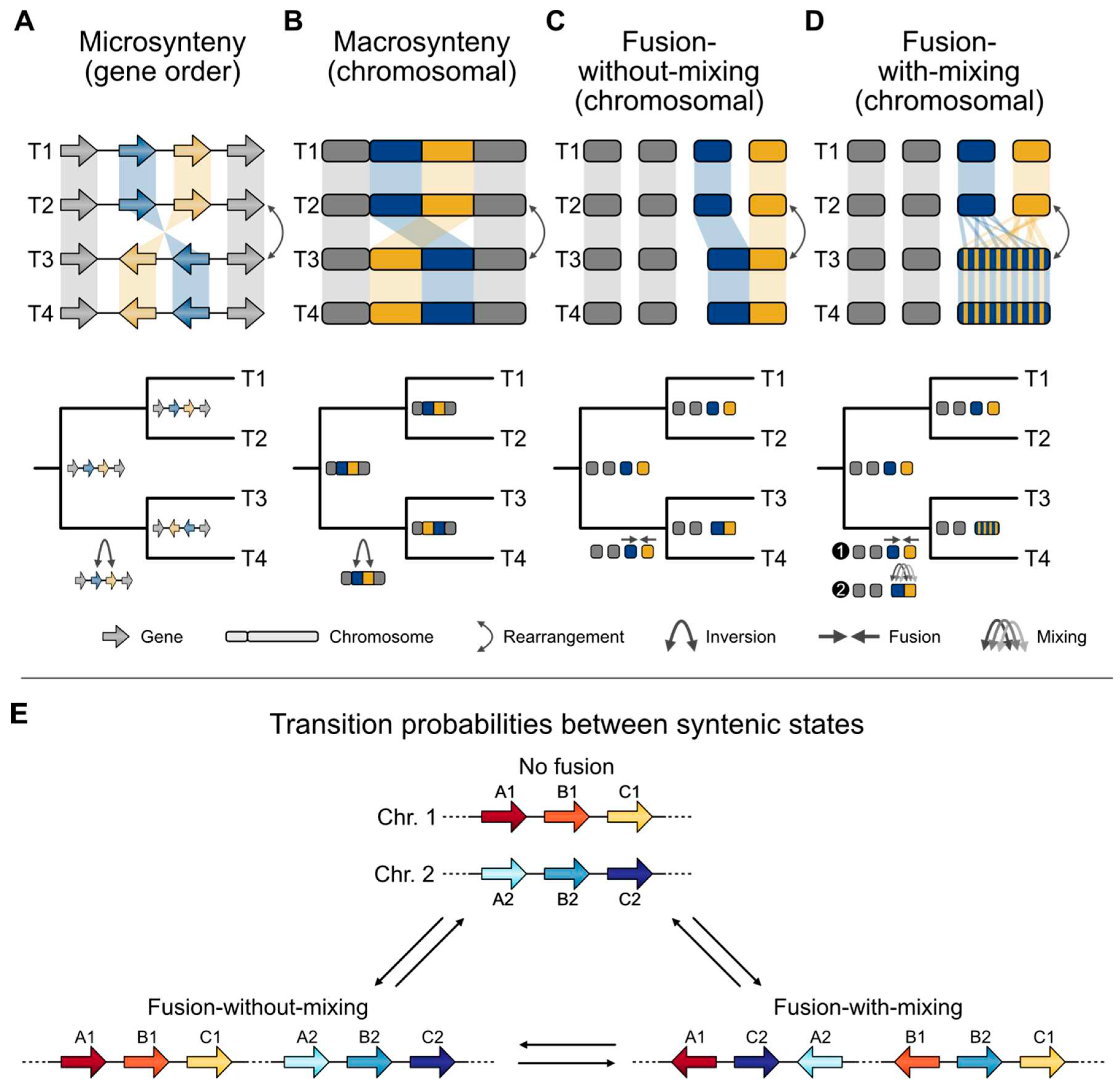
Figure 3.
Summary depictions of syntenies supporting the ctenophore-sister and EO-sister hypothesis. (A) Phylogeny of animal and outgroup taxa used to examine the root of the animal tree. Under the ctenophore-sister hypothesis, regions 1 through 7 each resulted from fusion events between two distinct chromosomes. The syntenic block depicted in orange for region R3 underwent a fission event in the choanoflagellate lineage, resulting in two chromosomes. Regions 4 through 7 underwent subsequent mixing events. Underneath each higher-order lineage name, the names of representatives used in the study are listed. For example, among Bilateria, species from the genera Pecten and Branchiostoma were included in the study [15]. Note, only fusion and mixing events relevant to rooting the animal tree are depicted. (B) Patterns of synteny in seven different regions most parsimoniously support the ctenophore-sister hypothesis. Examination of these regions indicates that all underwent fusion events and four also underwent mixing events. Each region is abbreviated as “R” along the phylogeny (for example, R1 refers to region 1). The number of genes in each syntenic region is listed at the bottom of the panel. (C) Phylogeny of the three teleost fish groups, including an outgroup taxon, the chicken. Cartoon summary drawings of chromosomes are included for representative species. Common names of these species are provided below the taxonomic names. Highly contiguous genome assemblies facilitated the detection of chromosome fusing and mixing events after a whole genome duplication event. Chr is used as an abbreviation for chromosome. (D) Chromosomes observed in extant species are depicted as cartoon summaries. Complete chromosome sequences are drawn with a solid line; dashed lines indicate scaffolds. Separate chromosomes or regions are drawn with orange and blue colors. Duplicated chromosomes from a whole genome duplication event are darkened. Fused chromosomes have two blocks of colors while fused and mixed chromosomes have intermixed blue and orange blocks of sequences. Silhouette images were obtained from PhyloPic (https://www.phylopic.org) and are dedicated to the public domain, except the Atlantic Tarpon (Erika Schumacher, Attribution-NonCommercial 3.0 Unported license); all credit goes to their respective contributors.
Figure 3.
Summary depictions of syntenies supporting the ctenophore-sister and EO-sister hypothesis. (A) Phylogeny of animal and outgroup taxa used to examine the root of the animal tree. Under the ctenophore-sister hypothesis, regions 1 through 7 each resulted from fusion events between two distinct chromosomes. The syntenic block depicted in orange for region R3 underwent a fission event in the choanoflagellate lineage, resulting in two chromosomes. Regions 4 through 7 underwent subsequent mixing events. Underneath each higher-order lineage name, the names of representatives used in the study are listed. For example, among Bilateria, species from the genera Pecten and Branchiostoma were included in the study [15]. Note, only fusion and mixing events relevant to rooting the animal tree are depicted. (B) Patterns of synteny in seven different regions most parsimoniously support the ctenophore-sister hypothesis. Examination of these regions indicates that all underwent fusion events and four also underwent mixing events. Each region is abbreviated as “R” along the phylogeny (for example, R1 refers to region 1). The number of genes in each syntenic region is listed at the bottom of the panel. (C) Phylogeny of the three teleost fish groups, including an outgroup taxon, the chicken. Cartoon summary drawings of chromosomes are included for representative species. Common names of these species are provided below the taxonomic names. Highly contiguous genome assemblies facilitated the detection of chromosome fusing and mixing events after a whole genome duplication event. Chr is used as an abbreviation for chromosome. (D) Chromosomes observed in extant species are depicted as cartoon summaries. Complete chromosome sequences are drawn with a solid line; dashed lines indicate scaffolds. Separate chromosomes or regions are drawn with orange and blue colors. Duplicated chromosomes from a whole genome duplication event are darkened. Fused chromosomes have two blocks of colors while fused and mixed chromosomes have intermixed blue and orange blocks of sequences. Silhouette images were obtained from PhyloPic (https://www.phylopic.org) and are dedicated to the public domain, except the Atlantic Tarpon (Erika Schumacher, Attribution-NonCommercial 3.0 Unported license); all credit goes to their respective contributors.
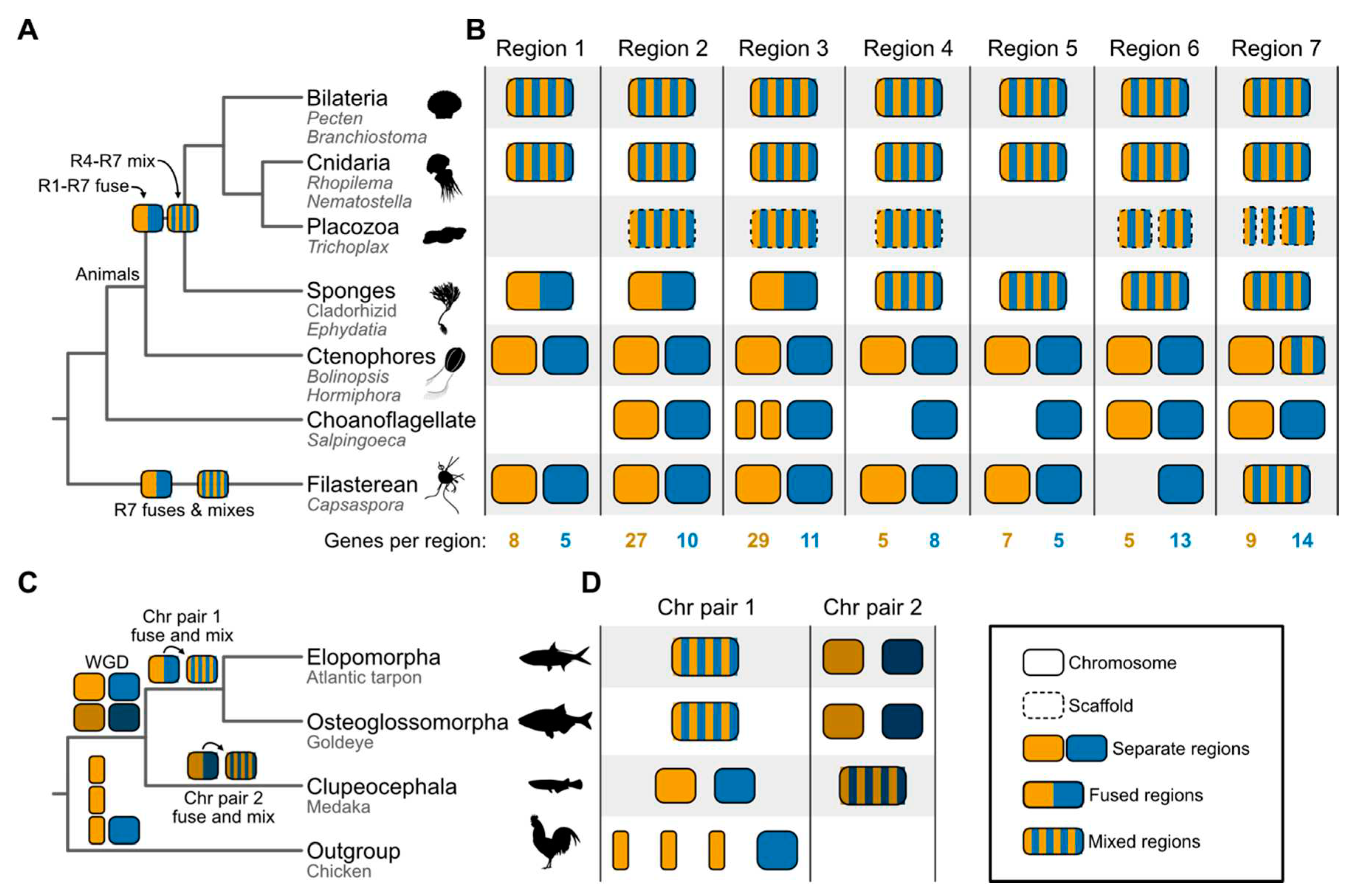
Figure 4.
A roadmap of challenges and opportunities for synteny-based phylogenomics. (A) We provide here a high-level summary of steps toward best practices in synteny-based phylogenomics. Limitations in resource availability (computational power and researcher time) dictate that each project begin with a selection of taxa that are most relevant to the phylogenetic question at hand. For those taxa that lack high-quality genome assemblies, it will be necessary to sequence each genome (using long-read sequencing technology) and assemble the reads. In other cases, previously sequenced and assembled genomes may be publicly available. In either case, the next step is to annotate the genes in all selected genomes using a single high-quality annotation method. Comparisons among the gene complements of each organism should then be used to identify gene orthologs. (Orthologous loci are depicted in green, yellow, and blue.) Orthologs can then be used in whole genome alignment and synteny detection. In addition, alignments of orthologs can be trimmed, assembled into multiple sequence alignments, and used for traditional phylogenenomics. After accounting for various sources of error, synteny blocks and multiple sequence alignments can be used to infer the topology of the Tree of Life. Note that obstacles in one step may be overcome by backtracking in the roadmap; for example, insufficient genome assembly completeness may benefit from additional genome sequencing. (B) Synteny data and organismal histories can be used for numerous research opportunities, including a better understanding of gene cluster function and evolution, reconstructing chromosome evolution, and inferring whole genome duplication events and ancestral genomes. For functional insights into gene clusters, fly embryos are depicted alongside gene clusters indicating how gene cluster organization may influence fly development. Silhouette images were obtained from PhyloPic (https://www.phylopic.org) and are dedicated to the public domain. Additional icons were obtained from bioicons (https://bioicons.com). Credit for silhouette images and icons goes to their respective contributors. The image of the long-read sequencer and chromosome with gene annotations are available via the CC BY 4.0 DEED Attribution 4.0 International.
Figure 4.
A roadmap of challenges and opportunities for synteny-based phylogenomics. (A) We provide here a high-level summary of steps toward best practices in synteny-based phylogenomics. Limitations in resource availability (computational power and researcher time) dictate that each project begin with a selection of taxa that are most relevant to the phylogenetic question at hand. For those taxa that lack high-quality genome assemblies, it will be necessary to sequence each genome (using long-read sequencing technology) and assemble the reads. In other cases, previously sequenced and assembled genomes may be publicly available. In either case, the next step is to annotate the genes in all selected genomes using a single high-quality annotation method. Comparisons among the gene complements of each organism should then be used to identify gene orthologs. (Orthologous loci are depicted in green, yellow, and blue.) Orthologs can then be used in whole genome alignment and synteny detection. In addition, alignments of orthologs can be trimmed, assembled into multiple sequence alignments, and used for traditional phylogenenomics. After accounting for various sources of error, synteny blocks and multiple sequence alignments can be used to infer the topology of the Tree of Life. Note that obstacles in one step may be overcome by backtracking in the roadmap; for example, insufficient genome assembly completeness may benefit from additional genome sequencing. (B) Synteny data and organismal histories can be used for numerous research opportunities, including a better understanding of gene cluster function and evolution, reconstructing chromosome evolution, and inferring whole genome duplication events and ancestral genomes. For functional insights into gene clusters, fly embryos are depicted alongside gene clusters indicating how gene cluster organization may influence fly development. Silhouette images were obtained from PhyloPic (https://www.phylopic.org) and are dedicated to the public domain. Additional icons were obtained from bioicons (https://bioicons.com). Credit for silhouette images and icons goes to their respective contributors. The image of the long-read sequencer and chromosome with gene annotations are available via the CC BY 4.0 DEED Attribution 4.0 International.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



