
Submitted:
02 September 2023
Posted:
05 September 2023
You are already at the latest version
Abstract
BRAF is one of the most frequently mutated oncogenes, with an overall frequency of about 50%. Targeting BRAF and its effector mitogen-activated protein kinase kinase 1/2 (MEK1/2) is now a key therapeutic strategy for BRAF-mutant tumors, and therapies based on dual BRAF/MEK inhibition showed significant efficacy in a broad spectrum of BRAF tumors. Nonetheless, BRAF/MEK inhibition therapy is not always effective for BRAF tumor suppression, and significant challenges remain to improve its clinical outcomes. First, certain BRAF tumors have an intrinsic ability to rapidly adapt to the presence of BRAF and MEK1/2 inhibitors by bypassing drug effects via rewired signaling, metabolic, and regulatory networks. Second, almost all tumors initially responsive to BRAF and MEK1/2 inhibitors eventually acquire therapy resistance via an additional genetic or epigenetic alteration(s). Overcoming these challenges requires identifying the molecular mechanism underlying tumor cell resistance to BRAF and MEK inhibitors and analyzing their specificity in different BRAF tumors. This review aims to update this information.
Keywords:
BRAF
; MEK
; tumor
; drug resistance
1. Introduction
BRAF is one of the most frequently mutated oncogenes, with an overall frequency of about 50%. Commonly occurring BRAF mutations switch the codon usage in the activation segment of the kinase domain, such as Val600 to Glu, Lys, or Asp, and render the kinase constitutively active independently of its upstream activator RAS, thereby causing the hyperactivation of its downstream effector, the MEK-extracellular signal-regulated kinase (ERK) cascade. Among the BRAF mutations identified thus far, BRAFV600E is most common with frequencies ~50% in melanomas, ~40% of papillary thyroid carcinomas, ~10% of colorectal cancers, ~5% of lung adenocarcinomas while also being detected in a subset of brain and hematological malignancies [1,2,3,4,5]. As such, there has been much effort to develop small molecule inhibitors that selectively target BRAF and its effector cascade MEK/ERK, and many inhibitors have been successfully developed [6,7]. Indeed, BRAF inhibitor (BRAFi) treatment resulted in high response rates in patients. However, the rates were short-lived due to the development of therapy resistance, which involves mainly the reactivation of the MEK/ERK cascade [8,9,10,11]. Subsequently, a MEK inhibitor (MEKi) was combined with BRAFi, significantly extending the median duration of response [9,10]. Since then, increasing evidence supports that dual BRAF/MEK inhibition improves clinical outcomes compared with BRAF inhibition alone in different BRAFV600E tumors [12].
Combining the BRAFi dabrafenib (TAFINLAR®) and the MEKi trametinib (MEKINIST®) is an effective dual BRAF/MEK inhibition for cancer therapy, and the U.S. Food and Drug Administration (FDA) has approved these drugs for mono- and combination-therapy (Figure 1). A dabrafenib and trametinib combination (hereafter D/T combination) significantly improved response rates (76% vs 54%), prolonged progression-free survival (PFS; 9.4 versus 5.8 months), and reduced skin toxicities compared with dabrafenib monotherapy in BRAF melanoma patients [9]. FDA recently granted approvals to the D/T combination therapy for patients with melanoma with BRAFV600E or BRAFV600K mutations [13] and those with metastatic anaplastic thyroid cancer [14] and non-small cell lung cancer (NSCLC) with BRAFV600E mutation [15]. The D/T combination therapy also modestly improved response rates compared to BRAFi monotherapy (12% vs. 5%) in BRAFV600E colon cancer patients [16,17]. However, it is not indicated for patients with colorectal cancer because of the relatively high intrinsic resistance of the tumor type. More recently, the FDA approved D/T combination for the treatment of adult and pediatric patients (≥ six years of age) with unresectable or metastatic solid BRAFV600E tumors who have progressed following prior treatment and have no satisfactory alternative treatment options [18] and for pediatric patients (≥ one year of age) with low-grade BRAFV600E glioma who require systemic therapy [19]. Nevertheless, the D/T combination is not always effective for BRAF tumors because certain BRAF tumors have an intrinsic ability to rapidly adapt to the presence of the drugs by bypassing drug effects via rewired signaling/metabolic/regulatory networks. Moreover, almost all tumors initially responsive to D/T combination therapy eventually acquired therapy resistance via an additional genetic/epigenetic alteration(s). Understanding the similarities and differences in therapy resistance in different tumor types is crucial. The goal of this review is to update this information.
2. MEK/ERK-dependent resistance mechanisms
Many therapy resistance mechanisms have been reported from BRAFi monotherapy cases [20,21,22,23]. ERK1/2 reactivation has been identified as a primary mechanism of BRAFi resistance in BRAFV600E melanoma, colon, and thyroid cancers [24,25]. Accordingly, combining BRAFi with an inhibitor of MEK1/2 or ERK1/2 was evaluated to prevent the MEK/ERK reactivation. Indeed, concomitant inhibition of ERK1/2 or MEK1/2 has been shown to attenuate BRAFi resistance in different cell lines and preclinical models, providing a rationale for dual BRAF/MEK inhibition for therapy [26,27,28]. Although this strategy is successful, ERK1/2 reactivation remains a significant resistance mechanism in the combination therapy. In addition, MEK/ERK-independent resistance mechanisms are also activated, albeit at a lower frequency. These mechanisms are described below and summarized in Table 1 and Table 2.
2.1. MEK/ERK-dependent adaptive resistance
Specific tumor cells can rapidly adapt to the presence of BRAF/MEK inhibitors by turning on a feedback mechanism that can reestablish MEK/ERK signaling, which is often mediated through a receptor tyrosine kinase (RTK) signaling pathway (illustrated in Figure 2). The rates of adaptive resistance development are varied in cancers, and relatively low in melanoma compared to colon and thyroid cancers. For example, the relatively low efficacy of vemurafenib/PLX4032 in BRAFV600E colon cancer is mainly attributed to the ability of the tumor cells to rapidly feedback-upregulate epidermal growth factor receptor (EGFR) signaling in response to the BRAFi, which does not occur as effectively in melanoma cells due to their intrinsically low EGFR expression [29]. Similarly, BRAFV600E thyroid cancer cells can rapidly relieve the negative feedback-regulation of human epidermal growth factor receptor 3 (HER3/ErbB3) transcription and increase autocrine secretion of the HER2 and HER3 ligand neuregulin 1 in response to vemurafenib, which also does not occur as effectively in melanoma cells [30]. This is accompanied by the rebound of ERK1/2 activity, which the HER kinase inhibitor lapatinib prevented [30]. Lapatinib also sensitized these tumor cells to vemurafenib [30]. Although HER3 is also activated through transcriptionally increased neuregulin in BRAFV600E melanoma cell lines following exposure to BRAFi and/or MEKi, HER3 activation mainly leads to protein kinase B (AKT) hyperphosphorylation. Antibodies directed against different HER3 surface epitopes prevented the establishment of resistance to BRAF/MEK inhibitors [31]. Melanoma cells mainly acquired adaptive resistance to vemurafenib via platelet-derived growth factor receptors-β (PDGFR-β) upregulation [32]. Therefore, cellular context-dependent heterogeneity can determine the efficacy of a therapy.
2.2. MEK/ERK-dependent acquired resistance
Alterations of the molecular switches in the Ras/Raf/MEK/ERK pathway have been the primary mechanism of acquired resistance involving pathway reactivation (Figure 2). Various alterations of these switches have been detected in a tumor-specific manner, mainly in skin, colon, and thyroid cancers, as summarized below. While these alterations may develop by the selection pressure of inhibitors, some of them may preexist in tumor cells and become dominant upon the selection pressure.
Alterations at RAS and upstream regulator level BRAFi-resistance of BRAFV600E tumor cells is mainly associated with ERK1/2 reactivation. Intriguingly, earlier versions of BRAFi can drive BRAFV600E binding to wild-type BRAF or CRAF, RAS-dependent wild-type RAF activation, and subsequently MEK/ERK activation [33,34,35]. The emergence of RAS mutation or amplification often facilitated MEK/ERK reactivation via these mechanisms. For example, NRAS mutations such as NRASQ61K and NRASA146T were detected in dabrafenib-resistant melanoma patient tumors and cell lines [27,32]. Similarly, activating mutations on different RAS isoforms, such as KRASG12V, NRASQ61K, and NRASG13D, were also detected in dabrafenib-resistant thyroid cancers of patients [36]. In addition, increased NRAS expression was also found in BRAFV600E vemurafenib-resistant melanoma cell lines [37]. Of note, KRAS amplification and emergence of KRASG12C in cell free DNA have been detected in D/T combination-resistant colon cancer patients, albeit at a much lower frequency than BRAFi monotherapy [28,38], which suggests that trametinib cannot completely suppress the emergency of MEK/ERK-dependent therapy resistance in cancers.
Alterations at BRAF level BRAF splicing variation and amplification have been detected in D/T combination therapy-resistant melanoma in patients [39,40]. BRAF amplification has also been associated with D/T combination resistance in colon cancer patients [28,38]. A novel BRAF splicing isoform lacking exons 2-10 was detected in one out of five patients with D/T combination resistant melanoma tumor that was undetectable in the pre-treatment tumor [40]. Similarly, in-frame deletion mutations involving exons 2-8, which includes the Ras-binding domain, were also detected in D/T combination-resistant BRAF melanomas in patients, albeit at a low frequency of 0.4% [41]. BRAF-activating deletion mutations were also detected at low frequencies (0.6–1%) in pancreatic, lung, ovarian, and thyroid tumors [42,43]. These deletions shorten the β3/αC-helix loop of BRAF and hinder its flexibility by locking the helix in the active αC-helix-in conformation that favors dimer formation [42,43]. The influence of the β3-αC deletion mutation on the binding profiles of three BRAF inhibitors (AZ628, dabrafenib, and vemurafenib) indicated that the β3-αC deletion mutation enhances the flexibility of the αC helix and alters structural conformation, which weakens the interactions between BRAF and these inhibitors [44].
Alterations at MEK1/2 level MEK2 mutations such as MEK2C125S and MEK2Q60P have been detected at higher frequencies in D/T combination therapy-resistant melanoma of patients than in BRAFi- or MEKi-monotherapy-resistant tumors [39,40]. Interestingly, MEK1 mutations were detected at a less frequency in these studies, and only MEK2C125S, but not the synonymous MEK1C121S, conferred resistance to the D/T combination [39,40]. Nonetheless, MEK1C121S exhibited increased kinase activity and conferred resistance to RAF and MEK inhibitors in melanoma cell cultures [45]. Of note, an in vitro screening revealed that a mutation on the allosteric drug binding pocket or αC-helix of MEK confers resistance to allosteric MEK inhibition, and, consistent with this, MEKi-resistant MEK1P124L mutation was detected in selumetinib/AZD6244-resistant BRAFV600E melanoma of patients [46]. In colon cancer, MEK1F53L mutation was detected in D/T combination-resistant tumor biopsies, albeit at lower frequencies [28]. However, MEK1 or MEK2 alterations were not detected in human colon cancer cell lines that have developed MEKi resistance in vitro [47]. These observations suggest that trametinib is the main selection pressure driving MEK1 and MEK2 mutations in tumor cells treated with D/T-combination and that these kinases may have a functional difference in therapy-resistance.
Alterations at ERK1/2 level MEK1/2 are considered the only ERK1/2 activators, and, in that context, a constitutively active ERK mutation would be an effective strategy for tumor cells to bypass the effects of MEK1/2 inhibition. Nevertheless, D/T combination therapy-resistant ERK1/2 mutations have rarely been reported. Of note, unlike MEK1/2 or most other kinases, the threonine–glutamic acid–tyrosine residue (TEY) site in the activation loop of ERK1/2 cannot be replaced by phosphomimetic amino acids to generate a constitutively active mutant [48]. Autophosphorylation is the only way for ERK to increase its activity autonomously, and its rate can increase upon several synergistic mutations that facilitate hydrogen bonding between the phosphoryl acceptor and catalytic nucleophile and different mutations that affect the gatekeeper residue [49,50,51]. Nevertheless, these ERK mutants display substantially lower activity than MEK1/2-activated ERK and produce limited effects in cells that are not cell-proliferative [52,53]. Considering this, constitutively active ERK mutation, but not other ERK mutations that affect ERK interaction with MEK1/2, phosphatases, or scaffolds, is probably not feasible for tumor cells to resist BRAF and MEK1/2 inhibition. Of note, different ERK mutations arise in ERK inhibitor-resistant tumor cells in culture, and it is important to understand by what mechanism these mutations facilitate restoring ERK activity in the tumor cells [54].
Interestingly, the combination of trametinib with the BRAFi PLX4720 induced ERK1/2 translocation to endoplasmic reticulum in BRAF mutant melanoma cells, and the protein kinase R-like endoplasmic reticulum kinase (PERK) phosphorylated ERK1/2 upon exiting endoplasmic reticulum. Activated ERK1/2 by this mechanism phosphorylated activating transcription factor 4 to activate cytoprotective autophagy, eventually driving resistance to dual BRAF and MEK1/2 inhibition [55]. A separate study also reported the involvement of PERK-mediated ERK1/2 activation in BRAFi resistance [56]. Consistent with this, upregulation of glucose-regulated protein 78 and phosphorylation of activating transcription factor 4 were detected in tumors of patients resistant to PLX4720 and trametinib combination [55]. This suggests that certain tumor cells can activate ERK1/2 via a non-canonical mechanism. It is important to address whether similar non-canonical mechanisms may exist and decrease the efficacy of D/T combination and whether these mechanisms may vary in tumors and underlie tumor-specific heterogeneous outcomes of the therapy. Since dual BRAF and ERK1/2 inhibition effectively abrogates clonal outgrowth of BRAFV600E colorectal cancer cells, which have relatively high intrinsic resistance to BRAFi/MEKi combination [57], addition of ERK inhibition to the combination strategy is promising. As such, predicting possible bypass mechanisms at ERK1/2 level is critical. Advanced ERK inhibitors have been recently reviewed elsewhere [58].
3. MEK/ERK-independent resistance mechanisms
In addition to MEK/ERK reactivation, other mechanisms also drive therapy resistance (Figure 2). For example, about 30% of patients develop MEK/ERK-independent resistance to BRAF inhibition in melanoma [59,60]. Numerous MEK/ERK-independent resistance mechanisms to BRAFi have been identified through preclinical studies, although many of these remain to be determined for clinical relevance. Meanwhile, much less MEK/ERK-independent resistance mechanisms to BRAFi/MEKi combination are known. Many of these resistance mechanisms are likely to overlap substantially between BRAFi resistance and BRAFi/MEKi resistance, given the common convergent evolutionary context, i.e., overcoming MEK/ERK inhibition, between them. These mechanisms are summarized below and listed in Table 1 and Table 2.
Loss of phosphatase and tensin homolog (PTEN): The status of PTEN, an important regulator of phosphoinositide 3-kinase (PI3K), is important for determining the propensity of BRAFV600E tumor cells to acquire BRAFi resistance through ERK1/2 reactivation. For example, wild-type PTEN-carrying tumors required hyperactivation of ERK1/2 and AKT to resist BRAFi [61], whereas PTEN-inactivated cells required only ERK1/2 activity for the resistance [56]. The PTEN status also affects mobilization of the mammalian target of rapamycin (mTOR) pathway for drug resistance. For example, dual BRAF/MEK inhibition initially suppressed the mTOR complex I signaling pathway in melanoma cells in culture and patient-derived tumor xenografts in mice (PDX), but the pathway activity rebounded upon the acquisition of drug resistance in an AKT-dependent manner in PTEN-deficient melanoma cells [62].
Activation of PI3K/AKT pathway: BRAFi monotherapy or D/T combination therapy frequently led to rebound of AKT phosphorylation at an early stage of treatment in melanomas, suggesting that adaptive resistance involving upregulation of the PI3K/AKT pathway is developed and may affect clinical outcomes of BRAFi therapy [63]. While it is unclear how AKT mediates drug resistance in response to D/T combination therapy, studies of BRAFi resistance demonstrated that it can upregulate embryonic stem cell expressed Ras to elicit a prosurvival signal though the Bcl-2-associated death promoter (BAD) pathway [64]. The insulin-like growth factor 1 receptor (IGF1R)/PI3K pathway was activated as an acquired resistance mechanism to the BRAFi SB-590885 in BRAFV600E melanoma cells [65]. Similarly, IGF1R/Insulin Receptor (IR) expression increased in D/T combination-resistant melanoma cells in correlation with poor patient survival. Moreover, a treatment with the IGF1R/IR inhibitor BMS-754807 reduced phosphorylation of AKT but not ERK1/2 [66]. This suggests an involvement of the IGF1R pathway in tumor cell resistance to BRAFi monotherapy and BRAFi/MEKi combination therapy. Of note, these pathways may be monitored to predict patient response to D/T combination. For example, unsupervised clustering of a large cohort of BRAFV600E colorectal cancer patients identified molecular subgroups not associated with known clinical characteristics. One subgroup exhibited elevated PI3K/mTOR/AKT/eukaryotic initiation factor 4E-binding protein 1 signaling, whereas the other subgroup dysregulated cell cycle and checkpoint pathways [67]. Interestingly, in response to D/T-combination, the PI3K-upregulated subtype showed higher confirmed response rates, median progression-free survival, and median overall survival, as well as greater immune reactivity than the other group [68].
Activation of survival pathway and altered translation via persistent formation of eukaryotic translation initiation factor 4F (eIF4F) complex: Myeloid leukemia 1 (Mcl-1) overexpression was detected in D/T combination-resistant progressive melanoma biopsies [69]. Indeed, Mcl-1 overexpression conferred resistance to vemurafenib or D/T combination in melanoma cells [69]. Consistent with this, silencing of BH3-only protein conferred resistance to PLX4720 in human melanoma cell lines [70]. As stated below, the apoptotic activator, Bcl-2 modifying factor (BMF), is upregulated upon vemurafenib treatment and may contribute to drug resistance by facilitating eIF4F -mediated translation [71]. Persistent formation of eIF4F complex has been suggested to be a nexus of resistance to anti-BRAF and anti-MEK cancer therapies regardless of whether the resistance mechanisms rely on reactivation of the Raf/MEK/ERK pathway, activation of the PI3K/AKT/mTOR pathway, or modulation of the caspase-dependent apoptotic cascade [71]. This study demonstrated that all these pathways converge on regulating the formation of the eIF4F eukaryotic translation initiation complex, thereby modulating the translation of specific mRNAs. Further, the persistent formation of the eIF4F complex, comprising the eIF4E cap-binding protein, the eIF4G scaffolding protein, and the eIF4A RNA helicase, was associated with resistance to BRAFi, MEKi, and BRAFi/MEKi combination in BRAFV600E melanoma, colon, and thyroid cancer cells. The apoptotic activator, BMF, regulated this complex formation by acting on eIF4G cleavage. While vemurafenib induced BMF overexpression, BMF silencing conferred BRAFi resistance and was detected in drug-resistant melanoma cells [71]. Therefore, BMF may be a good surrogate marker indicating the status of eIF4 complex formation and translational activity in tumor cells and, subsequently, drug resistance potential.
Activation of a G-protein-coupled receptors (GPCR)/cyclic AMP-dependent signaling network: At low frequencies, mutation or overexpression of the transcription factors E26 transformation-specific (ETS) and sterile alpha motif domain containing 4B (SAMD4B) were detected in melanoma relapse after D/T combination therapy [40]. A study confirmed the ability of these transcription factors to confer drug resistance by conducting a “gain-of-function” study in human melanoma cell lines [72]. Alongside, this study demonstrated that a cyclic AMP-dependent melanocytic signaling pathway that consists of GPCR, adenyl cyclase, protein kinase A and cyclic AMP response element binding protein (CREB) regulates these and several other transcription factors, including c-FOS, NR4A1, NR4A2, and MITF, which were also segregated to BRAFi-resistance. Indeed, preliminary analysis of BRAFV600E melanoma biopsies revealed that CREB phosphorylation decreases upon BRAF inhibition but is restored in relapsing tumors [72]. Given that MEK/ERK also regulates these transcription factors, it is conceivable that tumor cells mobilize the cyclic AMP pathway to overcome MEK/ERK deficiency in the context of convergent evolution.
Development of c-JUN-mediated mesenchymal-like phenotype: Vemurafenib resistance in BRAFV600E melanoma cell lines is associated with a high abundance of c-JUN and characteristics of a mesenchymal-like phenotype [73]. Early adaptation of tumor cells to the drug was correlated with upregulation of JUN and downregulation of lymphoid enhancer binding factor 1 (LEF1) and sprouty RTK signaling antagonist 4 (SPRY4), and changes in the markers for epithelial-mesenchymal transition (EMT), as determined in cell cultures, xenografts in mice, and patient tumors [73]. Importantly, disrupting the signaling between ERK2 and JUNB and Fos related antigen-1 transcription factors enabled vemurafenib-addicted tumor cells to survive on treatment discontinuation [74], suggesting the involvement of these transcription factors in developing tumor cell addiction to vemurafenib. EMT is an indication of feedback activation of RTK signaling in response to MEK1/2 inhibition in KRAS-mutant lung cancers [75], and it has been proposed as a marker for MEKi resistance [76].
Activation of signal transducer and activator of transcription 3 (STAT3) signaling pathway: The EGFR-SRC family kinase (SFK)-STAT3 pathway is involved in vemurafenib resistance of melanoma. For example, increased EGFR and SFK activity was detected in association with increased tumor cell proliferation, invasion, and metastasis in tumor biopsies from patients with intrinsic or acquired vemurafenib resistance, and EGFR inhibitors cooperated with BRAFi to block the growth of the resistant cells in vitro and in vivo [77]. In line with this, interleukin 6 (IL6) secreted by cancer-associated fibroblasts can induce EMT and drug resistance of esophageal adenocarcinoma [78]. Given that IL6 activates STAT3 via its canonical effector janus kinase (JAK), activation of STAT3 may also underlie the EMT-mediated drug resistance [79].
Upregulation of Hippo and yes-associated protein 1 (YAP1) signaling: YAP was identified as a vemurafenib resistance gene by shRNA-mediated loss of function screening in the BRAFV600E NSCLC line HCC364 [80]. In this study, combined YAP inhibition with RAF or MEK inhibition induced synthetic lethality not only in BRAF tumor cells but also in RAS tumor cells [80]. This study also proposed YAP1 upregulation as a biomarker of poor initial response to BRAF and MEK inhibition in BRAFV600E tumor patients [80]. The significance of YAP1 is supported by other studies that also identified YAP1 as a biomarker and a drug resistance mediator [81,82,83,84].
Reprogramed metabolic processes: Oncogenic BRAF regulates oxidative metabolism via peroxisome proliferator-activated receptor γ coactivator 1-α (PGC1α), whose transcription is directly regulated by microphthalmia-associated transcription factor (MITF), a target of BRAF for negative regulation [85]. This metabolic alteration is a lineage program in melanoma cells resistant to BRAF/MEK inhibition [64,72,74,85,86,87,88,89]. Indeed, MITF alteration is a part of the genetic landscape of clinical resistance to BRAF inhibition in metastatic melanoma [88]. Another example of cell lineage-specific drug resistance is found in thyroid cancer. RAS or RAF mutations leading to malignant thyroid epithelium transformation are accompanied by dedifferentiation and a decrease in the sodium-iodide symporter (SLC5A5) expression, which results in resistance to radioactive iodine therapy. Indeed, D/T combination, but not dabrafenib alone, upregulated sodium-iodide symporter expression in patient-derived thyroid tumor cells in culture, suggesting the possibility that D/T combination may increase tumor cell uptake of radioactive 131I [90]. Intriguingly, this effect was more significant in tumor cells from younger patients, implicating the involvement of a developmental biological aspect. This concept has been recently proven in a clinical trial [91]. More in depth review on the use of MAPK pathway inhibitors in thyroid cancer is available elsewhere [92].
4. Co-evolution of intra-tumoral immunity
Increasing evidence suggests that BRAF and MEK inhibitors have immune-modulating effects and can enhance antitumor immunity (illustrated in Figure 3). For example, advanced melanoma patients treated with BRAFi or BRAFi/MEKi combination exhibited increased expression of programmed cell death 1 (PD-1) and its ligand, PD-L1 [93]. BRAFi/MEKi combination also expanded memory and activated/exhausted CD8+ T cells, which was required for durable tumor regression elicited by the inhibitor combination [94]. This suggests that these inhibitors and the immune-therapeutic modality can synergize for tumor suppression. Indeed, multiple clinical trials have shown that D/T combination and the PD-1 antibody pembrolizumab can be combined to increase long-lasting antitumor responses and prolonged progression-free survival of BRAF-mutant melanoma patients [95,96]. Similarly, a combination of D/T and spartalizumab, a monoclonal PD-1 antibody, also showed long-term benefit potential for BRAF-mutant melanoma and colorectal cancer patients [97,98]. Of note, single-cell RNA sequencing analysis revealed that the latter drug combination induced greater tumor cell-intrinsic immune programs and more complete MEK/ERK inhibition in BRAF-mutant colon cancer patients with better clinical outcome [98]. Nevertheless, BRAFi/MEKi-resistant melanoma failed to induce cleavage of pyroptosis marker gasdermin E (GSDME), did not undergo pyroptosis, and showed decreased intra-tumoral T cell infiltration [99]. In line with acquired resistance to BRAFi or BRAFi/MEKi resistance, melanoma tumor biopsies showed CD8+ T cell deficiency and exhaustion, and downregulated PD-1 expression [81]. Similarly, combinatorial treatment of melanoma cells with vemurafenib or vemurafenib/trametinib combination impaired T cell activation [100]. Moreover, the spartalizumab and D/T combination did not show overall survival differences for BRAF-mutant melanoma patients in a phase III trial [101]. Therefore, post-hoc analyses of these trials have been suggested to identify tumor type-specific biomarkers for precise selection of patients for the triple combination of immune checkpoint inhibitors and BRAF/MEK inhibitors [102]. More extensive review in this area is available elsewhere [103].
5. Future perspectives and conclusion
Precision medicine cancer treatment has greatly advanced by accumulating data on the genotype–phenotype relationship of various oncogenic mutations. Targeting BRAF and MEK1/2 in combination is now a key therapeutic strategy for BRAF tumors, as D/T combination therapy showed efficacy in a broad spectrum of tumors. Nonetheless, significant challenges remain for D/T combination therapy. First, certain BRAF tumors have an intrinsic ability to rapidly adapt to the presence of these drugs by bypassing drug effects via rewired signaling, metabolic, and regulatory networks. Second, almost all tumors initially responsive to D/T combination eventually acquire therapy resistance via an additional genetic/epigenetic alteration(s). Overcoming these challenges requires identifying the molecular background of a tumor type, other than BRAF mutations, that also determines clinical outcomes. Indeed, many potential gene signatures of MEK/ERK functional outputs have been identified from therapy-resistant tumor cells. For example, a 13-RAS effector gene signature has been identified to predict the existence of compensatory signaling in selumetinib-resistant tumor cells [104]. Several of the genes in this signature have been functionally validated [47]. A 147-gene expression signature was also identified to predict RAS-mutant tumor responsiveness to PI3K and RAS pathway inhibition [105]. Multiple somatic mutations in patients have also been detected in association with therapy resistance [39,40]. In vitro ‘gain- or loss-of-function’ studies have been conducted to identify many candidate resistance genes [72,80]. The status of these genes might need to be analyzed comparatively in patient exome and RNA sequencing data from clinical trials. Data analysis should also consider the off-target effect of a drug. For example, dabrafenib but not vemurafenib can inhibit NIMA (Never In Mitosis Gene A)-related kinase and cyclin-dependent kinase-16 in addition to BRAF [106].
Identifying reliable prognosis markers through active correlative and functional analysis of molecular alterations associated with clinical outcomes will enable the establishment of a reliable guideline for companion diagnostics. Whether similar across tumor types or tumor type-specific, knowledge of these alterations is expected to refine patient selection and improve clinical outcomes, eventually providing the maximal benefit of BRAF/MEK/ERK targeted therapies.

Table 1.
Resistance mechanisms to combination therapy of BRAF and MEK1/2 inhibitors.
| Drugs | Tumor types | Source of study | Alterations for resistance | Resistance types | Consequence | Reference |
|---|---|---|---|---|---|---|
| Dabra/Tram* | Melanoma | Patient biopsy | BRAF amplification, NRAS mutations, MEK2C125S | Acquired | ERK1/2 reactivation | [39] |
| Dabra/Tram | Melanoma | Patient biopsy |
BRAF splicing isoform lacking exons 2-10, MEK2Q60P, Somatic mutations of ETS, SAMD4B |
Acquired | ERK1/2 reactivation | [40] |
| Dabra/Tram | Melanoma | Patient biopsy | Activating BRAF in-frame deletion | Acquired | ERK1/2 reactivation | [41] |
| Dabra/Tram | Melanoma | Patient biopsy, cell lines | AKT1Q79K that activates PI3K-AKT signaling, PDGFR- β upregulation | Adaptive | MEK/ERK-independent resistance | [63] |
| Dabra/Tram | Melanoma | Patient biopsy | MCL-1 overexpression, activation of survival pathway | Adaptive | MEK/ERK-independent resistance | [69] |
| Dabra/Tram | Colorectal cancer | Patient biopsy | KRAS amplification, BRAF amplification, MEK1F53L | Acquired | ERK1/2 reactivation | [28] |
| Dabra/Tram | Colorectal cancer | Patient biopsy | KRASG12C, BRAFV600E allele frequency increase | Acquired | ERK1/2 reactivation | [38] |
| Dabra/Tram | Melanoma | Cell lines, PDX model, biopsy | Increase of IGF1R/IR expression | Acquired | MEK/ERK-independent resistance | [66] |
| PLX4720/PD0325901 | Melanoma | Cell lines, PDX model | Rebound of mTOC1 pathway | Acquired | AKT or ERK contributes to the activation of mTORC1 depending on PTEN status | [62] |
| PLX4720/Tram Dabra/Tram |
Melanoma | Cell lines, PDX model | Upregulation of ATF4 | Acquired | ERK1/2 reactivation | [55] |
| PLX4720/PD0325901 | Melanoma | Synergetic mouse model, cell lines, | Failed to induce GSDME, decreased intra-tumoral T cell infiltration |
Acquired | MEK/ERK-independent resistance | [99] |
| BRAFi/EGFRi (dabrafenib + panitumumab), BRAFi/EGFRi/MEKi (dabrafenib + panitumumab + trametinib) | Colorectal cancer | Patient biopsy, cell lines | One or more RAS mutations (KRAS or NRAS) | Acquired | ERK1/2 reactivation | [57] |
| PLX4720+ AZD6244 | Melanoma | Gain of function screen, Patient biopsy | GPCR-PKA-cAMP, CREB phosphorylation | Adaptive | MEK/ERK-independent resistance | [72] |
| PLX4720+ AZD6244 | Melanoma | Gain of function screen, patient biopsy | c-Fos, NR4A1, NR4A2, MITF, activation of MEK/ERK downstream effectors | Intrinsic, adaptive, acquired | MEK/ERK-independent resistance | [72] |
| Vemurafenib only or Vemurafenib/Tram |
Melanoma | Cell lines, Patient biopsy | Decreased ability to induce IFNγ release by CD8+ TILs | Acquired | Decreases T cell activation | [100] |
| Vemurafenib only or Vemurafenib/Tram |
Melanoma | Cell lines, Patient biopsy | Decreased TOP1 expression | Acquired | unclear | [107] |
*Dabrafenib/trametinib combination.
Table 2.
Resistance mechanisms to BRAF or MEK1/2 inhibitors.
| Drug | Tumor types | Source of study | Alterations for resistance | Resistance types | Consequence | Reference |
|---|---|---|---|---|---|---|
| Vemurafenib | Melanoma | Patient biopsy, cell lines | PDGFR-β upregulation, NRASQ61K | Acquired | ERK1/2 reactivation | [32] |
| Dabrafenib | Melanoma | Cell lines | MEK1K59del, NRASQ61K and/or NRASA146T with and without MEK1P387S | Acquired | ERK1/2 reactivation | [27] |
| SB590885 | Melanoma | Patient biopsy, cell lines | IGF1R-PI3K-AKT activation | Acquired | MEK/ERK-independent resistance | [65] |
| Dabrafenib or vemurafenib | Melanoma | Patient biopsy | RAS mutations, mutant BRAF amplification, and alternative splicing | Acquired | ERK1/2 reactivation | [59] |
| Dabrafenib or vemurafenib | Melanoma | Patient biopsy | AKT1E17K and AKT1Q79K | Acquired | MEK/ERK-independent resistance | [59] |
| Vemurafenib | Melanoma | Cell lines | FGFR3-Ras activation | Acquired | ERK1/2 reactivation | [108] |
| Vemurafenib | Melanoma | Cell lines | SHOC-2/Sur-8 expression for N-Ras/C-Raf interaction | Acquired | ERK1/2 reactivation | [109] |
| Vemurafenib | Melanoma | Cell lines | Bcl-2 modifying factor (BMF) downregulation, increased eIF4F complex formation, reprogrammed translation | Acquired, adaptive | MEK/ERK-independent resistance | [71] |
| Vemurafenib | Melanoma | Cell lines | Relief of feedback inhibition of mitogenic signaling | Adaptive | ERK1/2 reactivation | [110] |
| Vemurafenib | Melanoma | Patient biopsy, Cell lines | c-JUN upregulation, LEF1 and SPRY4 downregulation, activation of downstream effector | Acquired, adaptive | MEK/ERK-independent resistance | [73] |
| Vemurafenib | NSCLC, Melanoma | Cell lines, Patient biopsy | YAP upregulation, activation of downstream effectors | Intrinsic, adaptive | MEK/ERK-independent resistance | [80] |
| PLX4720 | Melanoma | Gain of function screen | MAP3K8/COT/TPL-2 | Secondary tumor development | ERK1/2 reactivation | [111] |
| PLX4720 | Melanoma | Cell lines | BH-3 only protein silencing, activation of survival pathway | Acquired | MEK/ERK-independent resistance | [70] |
| Vemurafenib | Melanoma | Cell lines, Patient biopsy | EGFR-SFK-STAT3, activation of downstream effector | Acquired, adaptive | ERK1/2 reactivation | [77] |
| Vemurafenib | Melanoma | Cell lines | Activation of MAPKs and the PI3K pathways, enhanced NRAS expression | Acquired | Activation of all the three MAPKs, ERK, JNK, and p38 | [37] |
| Vemurafenib | Melanoma | Cell lines | Upregulated AXL in PTEN wild-type cells | Acquired | Hyperactivation of AXL/AKT and ERK pathways | [61] |
| Vemurafenib | Melanoma | Cell lines | Upregulated PERK in PTEN-inactivated | Acquired | Hyperactivation of ERK pathway | [56] |
| Vemurafenib | Thyroid cancer | Cell lines | ERBB/HER3 transcription, autocrine secretion of neuregulin 1 | Adaptive | ERK1/2 reactivation | [30] |
| Vemurafenib | Colorectal cancer | Cell lines | EGFR activation | Adaptive | ERK1/2 reactivation | [29] |
| Selumetinib | Colorectal cancer | Cell lines | KRAS or BRAF amplification | Acquired | ERK1/2 reactivation | [47] |
| Selumetinib | Melanoma | Patient biopsy | MEK1P124L | Acquired | ERK1/2 reactivation | [46] |
| Selumetinib | Melanoma | Patient biopsy, cell lines | c-MET up-expression, LEF1 down-expression, YAP1 signature enrichment | Acquired | ERK1/2 reactivation | [81] |
| Selumetinib | Colorectal cancer | Cell lines | BRAF amplification | Acquired | ERK1/2 reactivation | [112] |
Funding
This work was supported by the National Cancer Institute (R01CA138441 and R01CA269452) to J.I.P.
Acknowledgments
We thank the members of the Park lab for critical reading of the manuscript. The authors wish to apologize to those whose work is not cited owing to space limitations.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cancer Genome Atlas, N., Genomic Classification of Cutaneous Melanoma. Cell, 2015. 161(7): p. 1681-96.
- Cheng, L., et al., Molecular testing for BRAF mutations to inform melanoma treatment decisions: a move toward precision medicine. Mod Pathol, 2018. 31(1): p. 24-38. [CrossRef]
- Cohen, Y., et al., BRAF mutation in papillary thyroid carcinoma. J Natl Cancer Inst, 2003. 95(8): p. 625-7. [CrossRef]
- Gonsalves, W.I., et al., Patient and tumor characteristics and BRAF and KRAS mutations in colon cancer, NCCTG/Alliance N0147. J Natl Cancer Inst, 2014. 106(7). [CrossRef]
- Xu, X., et al., High prevalence of BRAF gene mutation in papillary thyroid carcinomas and thyroid tumor cell lines. Cancer Res, 2003. 63(15): p. 4561-7.
- Karoulia, Z., E. Gavathiotis, and P.I. Poulikakos, New perspectives for targeting RAF kinase in human cancer. Nat Rev Cancer, 2017. 17(11): p. 676-691. [CrossRef]
- Wu, P.K. and J.I. Park, MEK1/2 Inhibitors: Molecular Activity and Resistance Mechanisms. Semin Oncol, 2015. 42(6): p. 849-62.
- Flaherty, K.T., et al., Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med, 2010. 363(9): p. 809-19.
- Flaherty, K.T., et al., Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med, 2012. 367(18): p. 1694-703. [CrossRef]
- Long, G.V., et al., Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. Lancet, 2015. 386(9992): p. 444-51. [CrossRef]
- Hauschild, A., et al., Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet, 2012. 380(9839): p. 358-65. [CrossRef]
- Salama, A.K.S., et al., Dabrafenib and Trametinib in Patients With Tumors With BRAF(V600E) Mutations: Results of the NCI-MATCH Trial Subprotocol H. J Clin Oncol, 2020. 38(33): p. 3895-3904.
- FDA. FDA approves dabrafenib plus trametinib for adjuvant treatment of melanoma with BRAF V600E or V600K mutations. 2018; Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-dabrafenib-plus-trametinib-adjuvant-treatment-melanoma-braf-v600e-or-v600k-mutations.
- FDA. FDA approves new uses for two drugs administered together for the treatment of BRAF-positive anaplastic thyroid cancer. 2018; Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-new-uses-two-drugs-administered-together-treatment-braf-positive-anaplastic-thyroid.
- FDA. FDA grants regular approval to dabrafenib and trametinib combination for metastatic NSCLC with BRAF V600E mutation. 2017; Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-regular-approval-dabrafenib-and-trametinib-combination-metastatic-nsclc-braf-v600e.
- Corcoran, R.B., et al., Combined BRAF and MEK Inhibition With Dabrafenib and Trametinib in BRAF V600-Mutant Colorectal Cancer. J Clin Oncol, 2015. 33(34): p. 4023-31. [CrossRef]
- Kopetz, S., et al., Phase II Pilot Study of Vemurafenib in Patients With Metastatic BRAF-Mutated Colorectal Cancer. J Clin Oncol, 2015. 33(34): p. 4032-8. [CrossRef]
- FDA. FDA grants accelerated approval to dabrafenib in combination with trametinib for unresectable or metastatic solid tumors with BRAF V600E mutation. 2022; Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-dabrafenib-combination-trametinib-unresectable-or-metastatic-solid.
- FDA. FDA approves dabrafenib with trametinib for pediatric patients with low-grade glioma with a BRAF V600E mutation. 2023; Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-dabrafenib-trametinib-pediatric-patients-low-grade-glioma-braf-v600e-mutation.
- Puzanov, I., P. Burnett, and K.T. Flaherty, Biological challenges of BRAF inhibitor therapy. Mol Oncol, 2011. 5(2): p. 116-23. [CrossRef]
- Solit, D.B. and N. Rosen, Resistance to BRAF inhibition in melanomas. N Engl J Med, 2011. 364(8): p. 772-4. [CrossRef]
- Villanueva, J., A. Vultur, and M. Herlyn, Resistance to BRAF inhibitors: unraveling mechanisms and future treatment options. Cancer Res, 2011. 71(23): p. 7137-40. [CrossRef]
- Alcala, A.M. and K.T. Flaherty, BRAF inhibitors for the treatment of metastatic melanoma: clinical trials and mechanisms of resistance. Clin Cancer Res, 2012. 18(1): p. 33-9. [CrossRef]
- Paraiso, K.H., et al., Recovery of phospho-ERK activity allows melanoma cells to escape from BRAF inhibitor therapy. Br J Cancer, 2010. 102(12): p. 1724-30. [CrossRef]
- Sala, E., et al., BRAF silencing by short hairpin RNA or chemical blockade by PLX4032 leads to different responses in melanoma and thyroid carcinoma cells. Mol Cancer Res, 2008. 6(5): p. 751-9. [CrossRef]
- Hatzivassiliou, G., et al., ERK inhibition overcomes acquired resistance to MEK inhibitors. Mol Cancer Ther, 2012. 11(5): p. 1143-54.
- Greger, J.G., et al., Combinations of BRAF, MEK, and PI3K/mTOR inhibitors overcome acquired resistance to the BRAF inhibitor GSK2118436 dabrafenib, mediated by NRAS or MEK mutations. Mol Cancer Ther, 2012. 11(4): p. 909-20.
- Ahronian, L.G., et al., Clinical Acquired Resistance to RAF Inhibitor Combinations in BRAF-Mutant Colorectal Cancer through MAPK Pathway Alterations. Cancer Discov, 2015. 5(4): p. 358-67.
- Prahallad, A., et al., Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature, 2012. 483(7387): p. 100-3. [CrossRef]
- Montero-Conde, C., et al., Relief of feedback inhibition of HER3 transcription by RAF and MEK inhibitors attenuates their antitumor effects in BRAF-mutant thyroid carcinomas. Cancer Discov, 2013. 3(5): p. 520-33.
- Fattore, L., et al., Combination of antibodies directed against different ErbB3 surface epitopes prevents the establishment of resistance to BRAF/MEK inhibitors in melanoma. Oncotarget, 2015. 6(28): p. 24823-41. [CrossRef]
- Nazarian, R., et al., Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature, 2010. 468(7326): p. 973-7. [CrossRef]
- Heidorn, S.J., et al., Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell, 2010. 140(2): p. 209-21. [CrossRef]
- Hatzivassiliou, G., et al., RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature, 2010. 464(7287): p. 431-5. [CrossRef]
- Poulikakos, P.I., et al., RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature, 2010. 464(7287): p. 427-30.
- Cabanillas, M.E., et al., Acquired Secondary RAS Mutation in BRAF(V600E)-Mutated Thyroid Cancer Patients Treated with BRAF Inhibitors. Thyroid, 2020. 30(9): p. 1288-1296.
- Lidsky, M., et al., Mitogen-activated protein kinase (MAPK) hyperactivation and enhanced NRAS expression drive acquired vemurafenib resistance in V600E BRAF melanoma cells. J Biol Chem, 2014. 289(40): p. 27714-26. [CrossRef]
- Oddo, D., et al., Molecular Landscape of Acquired Resistance to Targeted Therapy Combinations in BRAF-Mutant Colorectal Cancer. Cancer Res, 2016. 76(15): p. 4504-15.
- Long, G.V., et al., Increased MAPK reactivation in early resistance to dabrafenib/trametinib combination therapy of BRAF-mutant metastatic melanoma. Nat Commun, 2014. 5: p. 5694. [CrossRef]
- Wagle, N., et al., MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov, 2014. 4(1): p. 61-8.
- Johnson, D.B., et al., BRAF internal deletions and resistance to BRAF/MEK inhibitor therapy. Pigment Cell Melanoma Res, 2018. 31(3): p. 432-436. [CrossRef]
- Chen, S.H., et al., Oncogenic BRAF Deletions That Function as Homodimers and Are Sensitive to Inhibition by RAF Dimer Inhibitor LY3009120. Cancer Discov, 2016. 6(3): p. 300-15.
- Foster, S.A., et al., Activation Mechanism of Oncogenic Deletion Mutations in BRAF, EGFR, and HER2. Cancer Cell, 2016. 29(4): p. 477-493. [CrossRef]
- Niu, Y., Y. Zhang, and X. Yao, Resistance mechanism of the oncogenic beta3-alphaC deletion mutation in BRAF kinase to dabrafenib and vemurafenib revealed by molecular dynamics simulations and binding free energy calculations. Chem Biol Drug Des, 2019. 93(2): p. 177-187.
- Wagle, N., et al., Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol, 2011. 29(22): p. 3085-96.
- Emery, C.M., et al., MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci U S A, 2009. 106(48): p. 20411-6.
- Little, A.S., et al., Amplification of the driving oncogene, KRAS or BRAF, underpins acquired resistance to MEK1/2 inhibitors in colorectal cancer cells. Sci Signal, 2011. 4(166): p. ra17.
- Askari, N., et al., MAP-quest: could we produce constitutively active variants of MAP kinases? Mol Cell Endocrinol, 2006. 252(1-2): p. 231-40.
- Emrick, M.A., et al., Constitutive activation of extracellular signal-regulated kinase 2 by synergistic point mutations. J Biol Chem, 2001. 276(49): p. 46469-79. [CrossRef]
- Emrick, M.A., et al., The gatekeeper residue controls autoactivation of ERK2 via a pathway of intramolecular connectivity. Proc Natl Acad Sci U S A, 2006. 103(48): p. 18101-6. [CrossRef]
- Levin-Salomon, V., et al., Isolation of intrinsically active (MEK-independent) variants of the ERK family of mitogen-activated protein (MAP) kinases. J Biol Chem, 2008. 283(50): p. 34500-10. [CrossRef]
- Wu, P.K., A. Becker, and J.I. Park, Growth Inhibitory Signaling of the Raf/MEK/ERK Pathway. Int J Mol Sci, 2020. 21(15). [CrossRef]
- Wu, P.K., et al., Active ERK2 is sufficient to mediate growth arrest and differentiation signaling. FEBS J, 2015. 282(6): p. 1017-30. [CrossRef]
- Smorodinsky-Atias, K., N. Soudah, and D. Engelberg, Mutations That Confer Drug-Resistance, Oncogenicity and Intrinsic Activity on the ERK MAP Kinases-Current State of the Art. Cells, 2020. 9(1).
- Ojha, R., et al., ER Translocation of the MAPK Pathway Drives Therapy Resistance in BRAF-Mutant Melanoma. Cancer Discov, 2019. 9(3): p. 396-415. [CrossRef]
- Qin, Y., et al., PERK mediates resistance to BRAF inhibition in melanoma with impaired PTEN. NPJ Precis Oncol, 2021. 5(1): p. 68. [CrossRef]
- Hazar-Rethinam, M., et al., Convergent Therapeutic Strategies to Overcome the Heterogeneity of Acquired Resistance in BRAF(V600E) Colorectal Cancer. Cancer Discov, 2018. 8(4): p. 417-427.
- Song, Y., et al., Targeting RAS-RAF-MEK-ERK signaling pathway in human cancer: Current status in clinical trials. Genes Dis, 2023. 10(1): p. 76-88.
- Shi, H., et al., Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov, 2014. 4(1): p. 80-93. [CrossRef]
- Solit, D.B. and N. Rosen, Towards a unified model of RAF inhibitor resistance. Cancer Discov, 2014. 4(1): p. 27-30. [CrossRef]
- Zuo, Q., et al., AXL/AKT axis mediated-resistance to BRAF inhibitor depends on PTEN status in melanoma. Oncogene, 2018. 37(24): p. 3275-3289. [CrossRef]
- Wang, B., et al., Targeting mTOR signaling overcomes acquired resistance to combined BRAF and MEK inhibition in BRAF-mutant melanoma. Oncogene, 2021. 40(37): p. 5590-5599. [CrossRef]
- Shi, H., et al., A novel AKT1 mutant amplifies an adaptive melanoma response to BRAF inhibition. Cancer Discov, 2014. 4(1): p. 69-79. [CrossRef]
- Perna, D., et al., BRAF inhibitor resistance mediated by the AKT pathway in an oncogenic BRAF mouse melanoma model. Proc Natl Acad Sci U S A, 2015. 112(6): p. E536-45. [CrossRef]
- Villanueva, J., et al., Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell, 2010. 18(6): p. 683-95.
- Patel, H., et al., IGF1R/IR Mediates Resistance to BRAF and MEK Inhibitors in BRAF-Mutant Melanoma. Cancers (Basel), 2021. 13(22). [CrossRef]
- Barras, D., et al., BRAF V600E Mutant Colorectal Cancer Subtypes Based on Gene Expression. Clin Cancer Res, 2017. 23(1): p. 104-115. [CrossRef]
- Middleton, G., et al., BRAF-Mutant Transcriptional Subtypes Predict Outcome of Combined BRAF, MEK, and EGFR Blockade with Dabrafenib, Trametinib, and Panitumumab in Patients with Colorectal Cancer. Clin Cancer Res, 2020. 26(11): p. 2466-2476.
- Fofaria, N.M., et al., Overexpression of Mcl-1 confers resistance to BRAFV600E inhibitors alone and in combination with MEK1/2 inhibitors in melanoma. Oncotarget, 2015. 6(38): p. 40535-56.
- Shao, Y. and A.E. Aplin, BH3-only protein silencing contributes to acquired resistance to PLX4720 in human melanoma. Cell Death Differ, 2012. 19(12): p. 2029-39. [CrossRef]
- Boussemart, L., et al., eIF4F is a nexus of resistance to anti-BRAF and anti-MEK cancer therapies. Nature, 2014. 513(7516): p. 105-9. [CrossRef]
- Johannessen, C.M., et al., A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature, 2013. 504(7478): p. 138-42. [CrossRef]
- Ramsdale, R., et al., The transcription cofactor c-JUN mediates phenotype switching and BRAF inhibitor resistance in melanoma. Sci Signal, 2015. 8(390): p. ra82. [CrossRef]
- Kong, X., et al., Cancer drug addiction is relayed by an ERK2-dependent phenotype switch. Nature, 2017. 550(7675): p. 270-274. [CrossRef]
- Kitai, H., et al., Epithelial-to-Mesenchymal Transition Defines Feedback Activation of Receptor Tyrosine Kinase Signaling Induced by MEK Inhibition in KRAS-Mutant Lung Cancer. Cancer Discov, 2016. 6(7): p. 754-69.
- Brighton, H.E., et al., New Mechanisms of Resistance to MEK Inhibitors in Melanoma Revealed by Intravital Imaging. Cancer Res, 2018. 78(2): p. 542-557. [CrossRef]
- Girotti, M.R., et al., Inhibiting EGF receptor or SRC family kinase signaling overcomes BRAF inhibitor resistance in melanoma. Cancer Discov, 2013. 3(2): p. 158-67.
- Ebbing, E.A., et al., Stromal-derived interleukin 6 drives epithelial-to-mesenchymal transition and therapy resistance in esophageal adenocarcinoma. Proc Natl Acad Sci U S A, 2019. [CrossRef]
- Manore, S.G., et al., IL-6/JAK/STAT3 Signaling in Breast Cancer Metastasis: Biology and Treatment. Front Oncol, 2022. 12: p. 866014. [CrossRef]
- Lin, L., et al., The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat Genet, 2015. 47(3): p. 250-6. [CrossRef]
- Hugo, W., et al., Non-genomic and Immune Evolution of Melanoma Acquiring MAPKi Resistance. Cell, 2015. 162(6): p. 1271-85. [CrossRef]
- Kim, M.H., et al., Actin remodeling confers BRAF inhibitor resistance to melanoma cells through YAP/TAZ activation. EMBO J, 2016. 35(5): p. 462-78. [CrossRef]
- Muranen, T., et al., ERK and p38 MAPK Activities Determine Sensitivity to PI3K/mTOR Inhibition via Regulation of MYC and YAP. Cancer Res, 2016. 76(24): p. 7168-7180. [CrossRef]
- Fisher, M.L., et al., Inhibition of YAP function overcomes BRAF inhibitor resistance in melanoma cancer stem cells. Oncotarget, 2017. 8(66): p. 110257-110272. [CrossRef]
- Haq, R., et al., Oncogenic BRAF regulates oxidative metabolism via PGC1alpha and MITF. Cancer Cell, 2013. 23(3): p. 302-15. [CrossRef]
- Haq, R., D.E. Fisher, and H.R. Widlund, Molecular pathways: BRAF induces bioenergetic adaptation by attenuating oxidative phosphorylation. Clin Cancer Res, 2014. 20(9): p. 2257-63. [CrossRef]
- Smith, M.P., et al., The immune microenvironment confers resistance to MAPK pathway inhibitors through macrophage-derived TNFalpha. Cancer Discov, 2014. 4(10): p. 1214-29.
- Van Allen, E.M., et al., The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov, 2014. 4(1): p. 94-109. [CrossRef]
- Smith, M.P., et al., Inhibiting Drivers of Non-mutational Drug Tolerance Is a Salvage Strategy for Targeted Melanoma Therapy. Cancer Cell, 2016. 29(3): p. 270-284. [CrossRef]
- Ullmann, T.M., et al., Dual inhibition of BRAF and MEK increases expression of sodium iodide symporter in patient-derived papillary thyroid cancer cells in vitro. Surgery, 2020. 167(1): p. 56-63. [CrossRef]
- Leboulleux, S., et al., A Phase II Redifferentiation Trial with Dabrafenib-Trametinib and 131I in Metastatic Radioactive Iodine Refractory BRAF p.V600E-Mutated Differentiated Thyroid Cancer. Clin Cancer Res, 2023. 29(13): p. 2401-2409.
- Schubert, L., et al., MAPK Pathway Inhibitors in Thyroid Cancer: Preclinical and Clinical Data. Cancers (Basel), 2023. 15(3). [CrossRef]
- Frederick, D.T., et al., BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res, 2013. 19(5): p. 1225-31.
- Hong, A., et al., Durable Suppression of Acquired MEK Inhibitor Resistance in Cancer by Sequestering MEK from ERK and Promoting Antitumor T-cell Immunity. Cancer Discov, 2021. 11(3): p. 714-735. [CrossRef]
- Ribas, A., et al., Combined BRAF and MEK inhibition with PD-1 blockade immunotherapy in BRAF-mutant melanoma. Nat Med, 2019. 25(6): p. 936-940. [CrossRef]
- Ascierto, P.A., et al., Dabrafenib, trametinib and pembrolizumab or placebo in BRAF-mutant melanoma. Nat Med, 2019. 25(6): p. 941-946. [CrossRef]
- Dummer, R., et al., Combined PD-1, BRAF and MEK inhibition in advanced BRAF-mutant melanoma: safety run-in and biomarker cohorts of COMBI-i. Nat Med, 2020. 26(10): p. 1557-1563. [CrossRef]
- Tian, J., et al., Combined PD-1, BRAF and MEK inhibition in BRAF(V600E) colorectal cancer: a phase 2 trial. Nat Med, 2023. 29(2): p. 458-466.
- Erkes, D.A., et al., Mutant BRAF and MEK Inhibitors Regulate the Tumor Immune Microenvironment via Pyroptosis. Cancer Discov, 2020. 10(2): p. 254-269. [CrossRef]
- Pieper, N., et al., Evolution of melanoma cross-resistance to CD8(+) T cells and MAPK inhibition in the course of BRAFi treatment. Oncoimmunology, 2018. 7(8): p. e1450127. [CrossRef]
- Dummer, R., et al., Randomized Phase III Trial Evaluating Spartalizumab Plus Dabrafenib and Trametinib for BRAF V600-Mutant Unresectable or Metastatic Melanoma. J Clin Oncol, 2022. 40(13): p. 1428-1438.
- Maeda, T., T. Yanagi, and H. Ujiie, Lessons from clinical trials on triple combination of immune checkpoint inhibitors and BRAF/MEK inhibitors in BRAF-mutant melanoma. Ann Transl Med, 2023. 11(9): p. 326. [CrossRef]
- Ascierto, P.A. and R. Dummer, Immunological effects of BRAF+MEK inhibition. Oncoimmunology, 2018. 7(9): p. e1468955. [CrossRef]
- Dry, J.R., et al., Transcriptional pathway signatures predict MEK addiction and response to selumetinib (AZD6244). Cancer Res, 2010. 70(6): p. 2264-73.
- Loboda, A., et al., A gene expression signature of RAS pathway dependence predicts response to PI3K and RAS pathway inhibitors and expands the population of RAS pathway activated tumors. BMC Med Genomics, 2010. 3: p. 26. [CrossRef]
- Phadke, M., et al., Dabrafenib inhibits the growth of BRAF-WT cancers through CDK16 and NEK9 inhibition. Mol Oncol, 2018. 12(1): p. 74-88. [CrossRef]
- Oliveira, E.A., et al., TOP1 modulation during melanoma progression and in adaptative resistance to BRAF and MEK inhibitors. Pharmacol Res, 2021. 173: p. 105911.
- Yadav, V., et al., Reactivation of mitogen-activated protein kinase (MAPK) pathway by FGF receptor 3 (FGFR3)/Ras mediates resistance to vemurafenib in human B-RAF V600E mutant melanoma. J Biol Chem, 2012. 287(33): p. 28087-98.
- Kaplan, F.M., et al., SHOC2 and CRAF mediate ERK1/2 reactivation in mutant NRAS-mediated resistance to RAF inhibitor. J Biol Chem, 2012. 287(50): p. 41797-807.
- Lito, P., et al., Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell, 2012. 22(5): p. 668-82. [CrossRef]
- Johannessen, C.M., et al., COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature, 2010. 468(7326): p. 968-72.
- Corcoran, R.B., et al., BRAF gene amplification can promote acquired resistance to MEK inhibitors in cancer cells harboring the BRAF V600E mutation. Sci Signal, 2010. 3(149): p. ra84. [CrossRef]
Figure 1.
The history of FDA approval of dabrafenib and trametinib. Dabrafenib and trametinib have been approved by the FDA for monotherapy and combination therapy for BRAFV600 mutant solid tumors.
Figure 1.
The history of FDA approval of dabrafenib and trametinib. Dabrafenib and trametinib have been approved by the FDA for monotherapy and combination therapy for BRAFV600 mutant solid tumors.
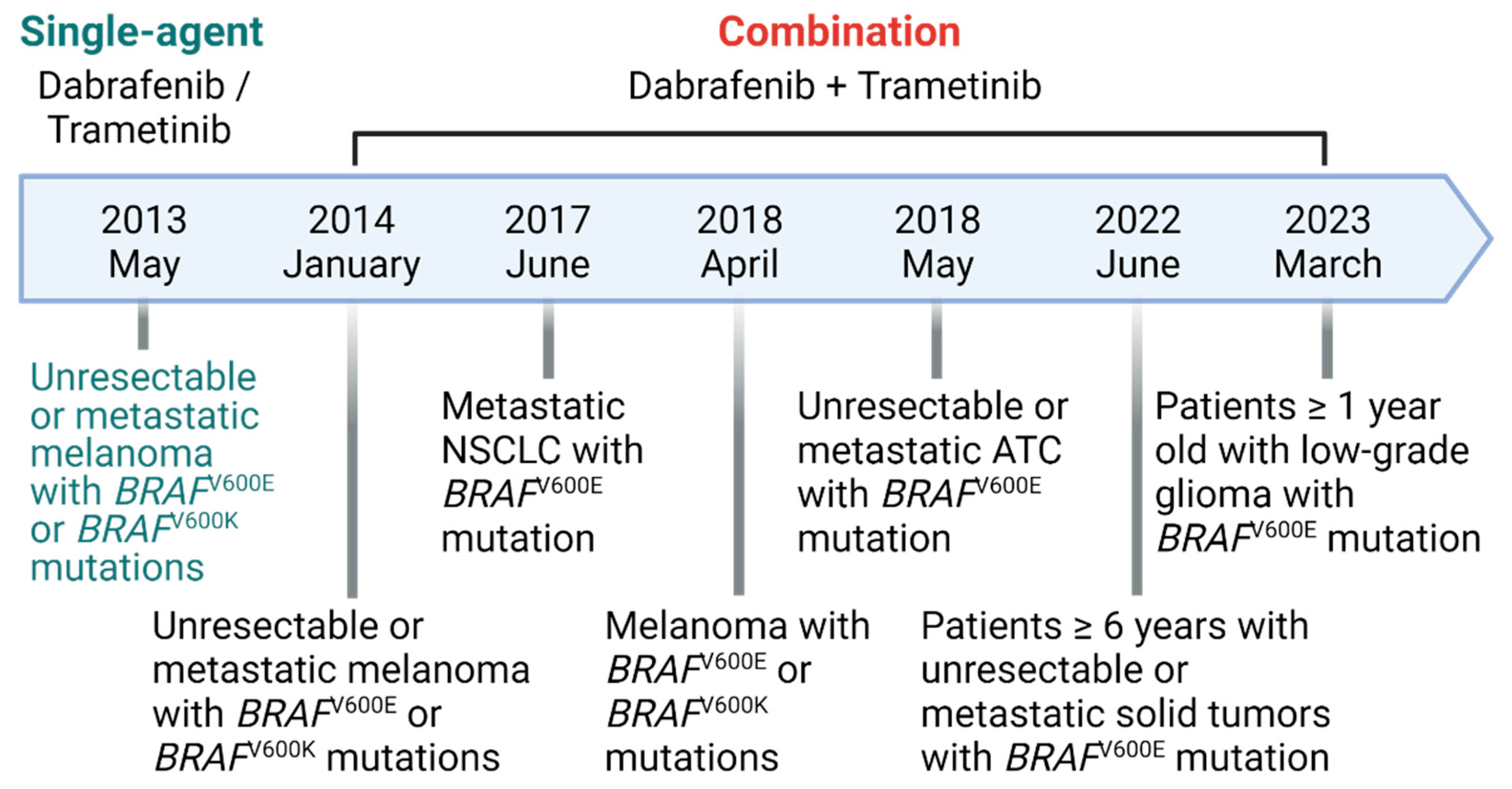
Figure 2.
Intracellular mechanisms of tumor cell resistance to BRAFi and MEKi. Tumor cells can develop resistance to BRAFi and MEKi mainly by reactivating the MEK/ERK pathway by altering the regulators and molecular switches in the RAS/RAF/MEK pathway. Tumor cells can also develop drug resistance in an MEK/ERK-independent manner through various pathways illustrated. This figure is created by biorender.com.
Figure 2.
Intracellular mechanisms of tumor cell resistance to BRAFi and MEKi. Tumor cells can develop resistance to BRAFi and MEKi mainly by reactivating the MEK/ERK pathway by altering the regulators and molecular switches in the RAS/RAF/MEK pathway. Tumor cells can also develop drug resistance in an MEK/ERK-independent manner through various pathways illustrated. This figure is created by biorender.com.
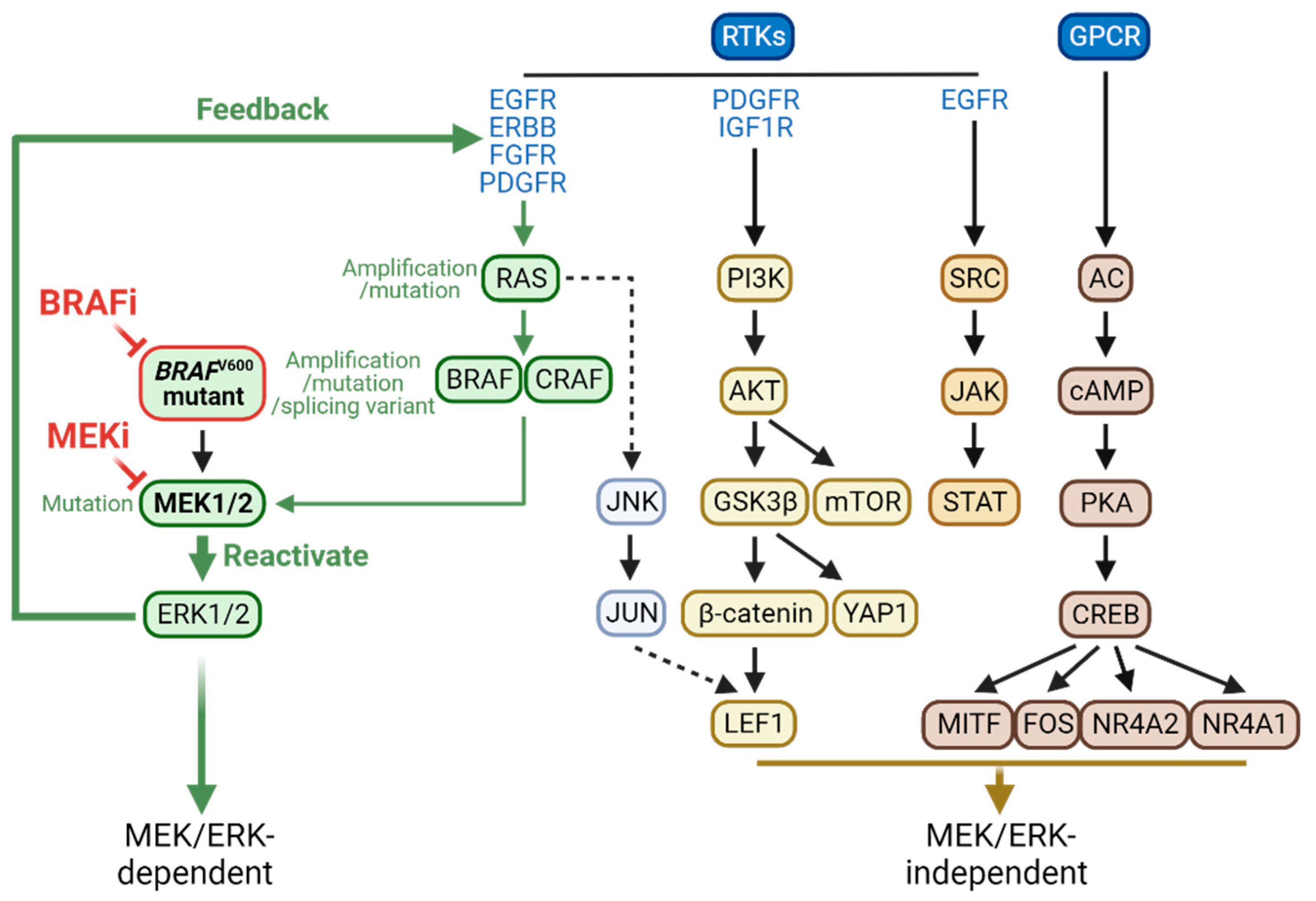
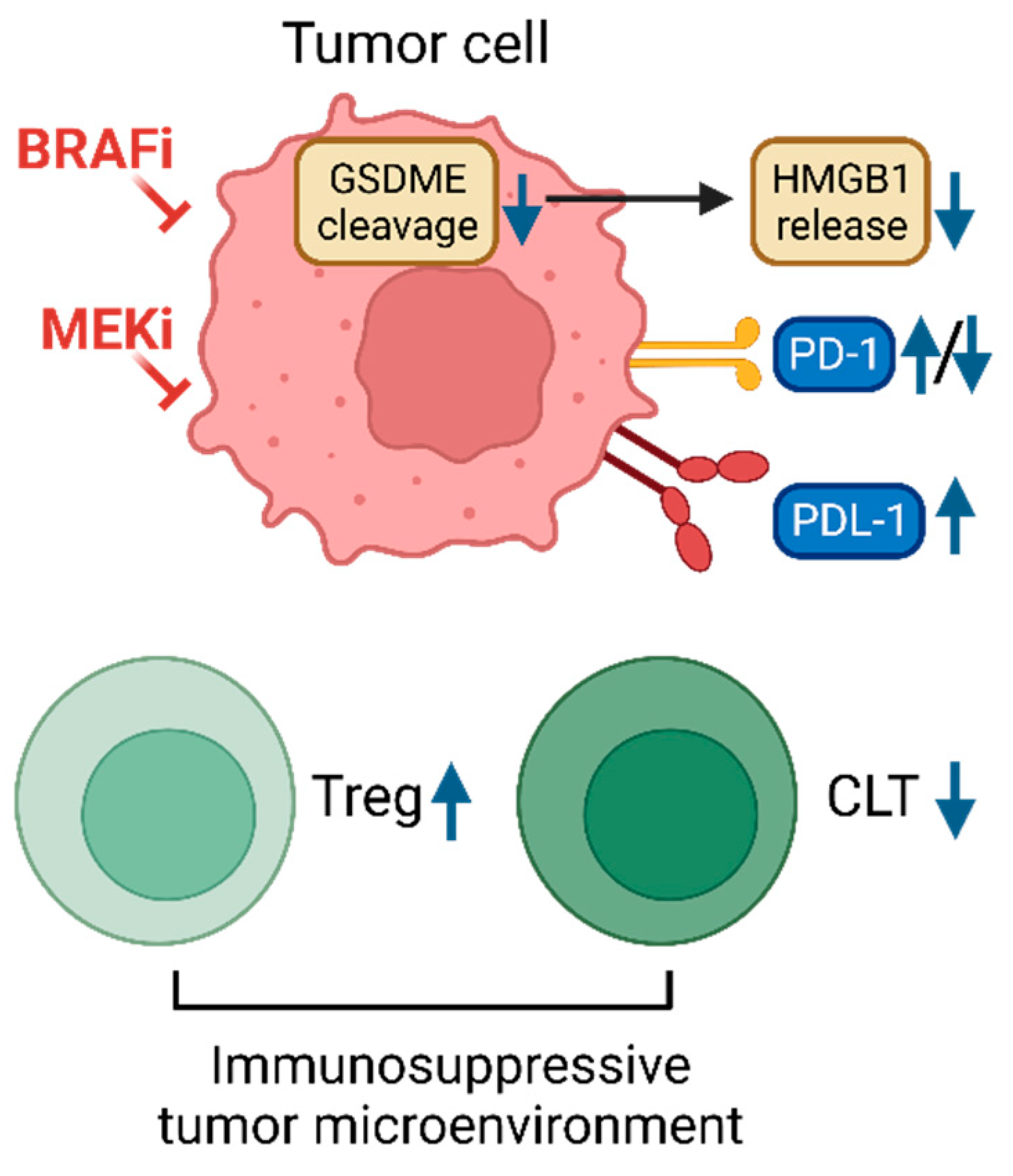
Figure 3.
Immune-modulating effects of BRAFi and MEKi. Tumor cells can develop resistance to BRAFi and MEKi by creating an immunosuppressive tumor microenvironment. Drug-resistant tumor cells fail to undergo pyroptosis induced by BRAFi and MEKi, exhibiting decreased GSDME cleavage and high mobility group box 1 (HMGB1) release. They also display dysregulated PD-1/PDL-1 expressions which affects antitumor immune responses, including increased regulatory T cells (Treg), and decreased cytotoxic T cells (CLT). This figure is created by biorender.com.
Figure 3.
Immune-modulating effects of BRAFi and MEKi. Tumor cells can develop resistance to BRAFi and MEKi by creating an immunosuppressive tumor microenvironment. Drug-resistant tumor cells fail to undergo pyroptosis induced by BRAFi and MEKi, exhibiting decreased GSDME cleavage and high mobility group box 1 (HMGB1) release. They also display dysregulated PD-1/PDL-1 expressions which affects antitumor immune responses, including increased regulatory T cells (Treg), and decreased cytotoxic T cells (CLT). This figure is created by biorender.com.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



