
Submitted:
29 August 2023
Posted:
31 August 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Sepsis is a life-threatening condition caused by the body's overwhelming response to an infection, such as pneumonia or urinary tract infection. It occurs when the immune system releases cytokines into the bloodstream, triggering widespread inflammation. If not treated, it can lead to organ failure and death. Unfortunately, sepsis has a high mortality rate, with studies reporting rates ranging from 20% to over 50%, depending on the severity and promptness of treatment. According to the World Health Organization (WHO), the annual death toll in the world is about 11 million. One of the main toxins responsible for inflammation induction are lipopolysaccharides (LPS, endotoxin) from Gram-negative bacteria, which ranks among the most potent immunostimulants found in nature. Antibiotics are consistently prescribed as a part of anti-sepsis-therapy. However, antibiotic therapy (i) is increasingly ineffective due to resistance development and (ii) most antibiotics are unable to bind and neutralize LPS, a prerequisite to inhibit the interaction of endotoxin with its cellular receptor complex, namely Toll-like receptor 4 (TLR4)/MD-2, responsible for the intracellular cascade leading to pro-inflammatory cytokine secretion. The pandemic virus SARS-CoV-2 has infected hundreds of millions of humans worldwide since its emergence in 2019. The COVID-19 (Coronavirus disease-19) caused by this virus is associated with high lethality, particularly for elderly and immunocompromised people. As of August 2023, nearly 7 million deaths were reported worldwide due to this disease. According to some reported studies, upregulation of TLR4 and the subsequent inflammatory signaling detected in COVID-19 patients “mimics bacterial sepsis”. Furthermore, the immune response to SARS-CoV-2 was described by others as “mirror image of sepsis”. Similarly, the cytokine profile in sera from severe COVID-19 patients was very similar to those suffering from the acute respiratory distress syndrome (ARDS) and sepsis. Finally, the severe COVID-19 infection was frequently accompanied by bacterial co-infections, as well as by the presence of significant LPS concentrations. These data indicate similarity and interdependences of both syndromes, but also significant differences which will be discussed in the present review.
Keywords:
sepsis
; lipopolysaccharide
; gram-negative bacteria
; covid pandemic
; hyperinflammation
; TLR4
; cytokines
; ARDS
; Aspidasept
1. Risk factors and complications of COVID-19
The Coronavirus Disease of 2019 (COVID-19) due to the coronavirus SARS-CoV-2, was a pandemic with a high rate of mortality [1,2]. The first cases were reported at the end of 2019 in Wuhan, China, and diagnosed with severe acute respiratory syndrome (SARS) leading to a potentially life-threatening disease. The symptoms of this pathological condition were fever, shortness of breath, cough, and sudden onset of anosmia (“smell blindness”), ageusia (loss of the sense of taste), or dysgeusia (disorder of the sense of taste). In most cases, approximately 80%, COVID-19 was mild or moderate, but it could evolve into severe or critical clinical presentations with a significant risk of mortality in about 14% and 5% of the cases respectively [3].
Numerous studies have analyzed which comorbidities were more commonly associated with Covid-19 severity. Interestingly, all these meta-analyses consistently showed that patients suffering from diabetes type II, hypertension, cancer, and cardiovascular diseases were at higher risk of developing severe COVID-19. Association between other comorbidities and disease burden was also strong, although their relative contribution to disease severity varied among the distinct meta-analyses (Table 1).

Table 1.
Comorbidities of COVID-19 associated with disease severity. Data from non-redundant studies analyzed in references [4,5,6,7,8].
| Risk factor | Number of studies | Total sample size | Association with covid severity |
|---|---|---|---|
| Diabetes | 142 | 59’476 | Yes |
| Hypertension | 140 | 58’808 | Yes |
| Malignancy | 94 | 48’488 | Yes |
| Cerebrovascular disease | 71 | 16’124 | Yes |
| Chronic liver disease | 56 | 27’924 | Yes |
| COPD | 50 | 32’173 | Yes |
| Chronic kidney disease | 43 | 20’103 | Yes |
| Cardiovascular diseases | 37 | 25’016 | Yes |
| Coronary heart disease | 33 | 16’525 | Yes |
| Respiratory disease | 31 | 7’552 | Yes |
| Chronic lung disease | 31 | 3’702 | Yes |
| Chronic heart disease | 9 | 3’583 | Yes |
| Autoimmune disease | 7 | 2’372 | Yes |
| Renal insufficiency | 6 | 2’997 | Yes |
| Stroke | 5 | 1’616 | Yes |
| Cerebral infarction | 4 | 2’647 | Yes |
| Fatty liver | 4 | 992 | Yes |
| Arrhythmia | 4 | 781 | Yes |
| Cardiac insufficiency | 2 | 1’912 | Yes |
| Genital system diseases | 2 | 546 | Yes |
| Kidney failure | 2 | 294 | Yes |
| Coronary atherosclerosis | 1 | 3’044 | Yes |
| Benign prostatic hyperplasia | 1 | 3’044 | Yes |
| Myocardial infarction | 1 | 660 | Yes |
| Aorta sclerosis | 1 | 140 | No |
| Atrial fibrillation | 1 | 112 | No |
| Coronary artery disease | 2 | 1’073 | No |
| Heart failure | 1 | 172 | No |
| Intracerebral hemorrhage | 1 | 1’767 | No |
| Asthma | 3 | 5’359 | No |
| Chronic bronchitis | 2 | 2’525 | No |
| Tuberculosis | 7 | 4’125 | No |
| Nephritis | 1 | 3’044 | No |
| Gallbladder disease | 3 | 779 | No |
| Hepatitis B | 6 | 3’307 | No |
| Gastrointestinal disease | 6 | 4’764 | No |
| Peptic ulcer | 1 | 145 | No |
| Gout | 1 | 134 | No |
| Hyperlipidemia | 7 | 4’131 | No |
| Hyperuricemia | 1 | 172 | No |
| Thyroid disease | 5 | 1’125 | No |
| Cirrhosis | 3 | 5’134 | No |
| Prostatitis | 1 | 3’044 | No |
| Gynecological disease | 1 | 238 | No |
| HIV infection | 7 | 1’099 | No |
| Nervous system disease | 5 | 2’203 | No |
| Rheumatism | 2 | 273 | No |
| Urinary system disease | 2 | 1’075 | No |
| Urolithiasis | 1 | 140 | No |
| Blood system diseases | 3 | 965 | No |
| Bone disease | 1 | 238 | No |
However, to date, the decisive pathophysiologic processes that were responsible for COVID-19 patient morbidity and mortality remain unclear. Acute respiratory distress syndrome (ARDS), respiratory failure, multiple organ dysfunction syndrome (MODS) and septic shock were complications strongly associated with critical cases of coronavirus disease [4]. This meta-analysis was particularly relevant as it examined data from 187 studies describing 77’013 patients. Other studies reached the same conclusions [5], [6], and [7]. Importantly, severe cases of non-Covid sepsis caused by respiratory pathogens lead to complications similar to those described by these authors, thereby suggesting that COVID-19 mortality may be the result of sepsis. To address this hypothesis, Ahlström et al. (2021) compared the impact of comorbidities on mortality in patients with COVID-19 and sepsis [8] [9], [10]). Mortality was significantly reduced in the COVID-19 patients compared with those with sepsis, whereas the use of invasive mechanical ventilation was more common in COVID-19 than in sepsis patients. However, the key conclusion of this study is that almost all the investigated comorbidities were shared between COVID-19 and sepsis patients. Consistent with this finding, sepsis scores have been consistently shown to predict COVID-19 outcomes including death, ICU (intensive care unit) transfer and respiratory failure [11] [12]. For instance, the majority of severely ill COVID-19 patients (78%) met the Sepsis 3.0 criteria for sepsis/septic shock with ARDS (adult respiratory distress Syndrome) being the most frequent organ dysfunction (88%) [13].
2. COVID-19-induced sepsis, immunotherapies, and antiviral treatments
During COVID-19 disease, both the innate and the adaptive immune responses experience dysregulation. The first clinical reports from early 2020 highlighted high plasma levels of interleukin-6 (IL-6), IL-1, tumour necrosis factor α (TNF-α), ferritin and increased amounts of other inflammatory biomarkers. This underlined the assumption that COVID-19 was comparable to sepsis and lead to the idea that these biomarker levels were the cause for organ failure and thus, needed to be supressed [14], [15], [16], [17], [18], [19]. Therefore, several clinical trials started using anti-inflammatory therapies to try to reduce the cytokine plasma levels [20], [21], [22], (Table 2). These clinical trials have not been successful so far and, in some cases, have even worsened patient outcomes [23], [24].
Table 2.
Immunotherapies against COVID-19.
| Mechanism | Drug family | Drugs | Status |
|---|---|---|---|
| Anti-inflammatory drugs | Systemic glucocorticoids | Dexamethasone, Prednisone, Hydrocortisone, Methylprednisolone | Recommended for certain hospitalized patients |
| Anti-IL-6 receptor antibodies | Tocilizumab, Sarulimab | Recommended for certain hospitalized patients | |
| Anti-IL-6 antibody | Siltuximab | Not recommended. Under investigation in clinical trials | |
| IL-1 receptor antagonists | Anakinra, Canakinumab | Anakinra received an FDA EUA for certain hospitalized patients.Canakinumab is not recommended | |
| JAK/STAT inhibitors | Baricitinib, Tofacitinib, Ruxolitinib | Baricitinib and Tofacitinib recommended for certain hospitalized patients.Ruxolitinib under investigation in clinical trials | |
| GM-CSF inhibitors | Lenzilumab, Mavrilimumab, Namilumab, Otilimab, Gimsilumab | Not recommended. Under investigation in clinical trials | |
| TNF-alpha inhibitor | XPro1595, CERC-002, Infliximab, Adalimumab | Not recommended. Under investigation in clinical trials | |
| Immune stimulants | Programmed death ligand pathway inhibitors | Nivolumab and Pembrolizumab | Not recommended. Under investigation in clinical trials |
| IL-7 | Not recommended. Under investigation in clinical trials | ||
| IFN-γ | Not recommended. Under investigation in clinical trials | ||
| NKG2D-ACE2 CAR-NK cells | Not recommended. Under investigation in clinical trials |
Currently it is understood, that, for instance, early conclusions based on IL-6 concentration were not robust as predictors of COVID-19 prognosis. Although initial data showed abnormally elevated IL-6 concentrations in COVID-19 patients of a few hundred pg/µl, these levels were modest compared with those measured in septic shock patients undergoing a cytokine storm. Specifically, the levels measured in the plasma of the latter patients exceeded those of COVID-19 patients by a factor of 10-20, leading to IL-6 plasma concentrations of thousands of pg/µl. In addition, it was soon observed that elevation of IL-6 levels associated with COVID-19 was a transient phenomenon. Thus, Gu et al. (2020) showed that wild-type and ACE2-expressing (adenovirus-5/human angiotensin-converting enzyme 2) BALB/c mice challenged with a combination of polyinosinic-polycytidylic acid (an immunostimulant used in the form of its sodium salt to simulate viral infections) [25] and a recombinant SARS-CoV-2 spike-extracellular domain protein expressed high levels of TNF-α and underwent 100-fold increases in IL-6 at 6 h post challenge. However, levels of TNF-α and IL-6 later returned to normal ranges from the bronchoalveolar lavage fluid (BALF) after 24 h of the exposure [26]. As a result of these studies, our current knowledge about the disease evolution considers not only the plasma concentrations of inflammatory markers, but also the phase of the disease (Table 3).
| EarlySepsis | EarlyCOVID-19 | LateSepsis | LateCOVID-19 | |
|---|---|---|---|---|
| IL-6 increase | +++ | + | +++ | |
| Lymphopenia | + | ++ | ++ | +++ |
| Nosocomial infections | +++ | +++ |
According to several studies, the inflammatory phase for patients with severe COVID-19 is limited to the initial period of the disease. The subsequent chronic basal inflammation, which lasts several days, leads the immune system towards a refractory state, which is also observed in protracted sepsis. A comparative study of patients with severe and mild COVID-19 concluded that all cytokines, except IL-6 and IL-10, reached their peak level in serum 3-6 days after beginning of the disease. IL-6 levels on the other hand, began to drop approximately 16 days later, and IL-10 levels were at their lowest 13 days after disease onset. Interestingly, the cytokine levels reached similar points for all patients with severe and mild disease 16 days after disease onset. This observation corresponds to the most advanced phases of sepsis, in which the macrophages develop a refractory state characterized by a strong inhibition of the NF-κB and interferon regulatory factor (IRF) pathways in response to pathogens. In contrast to the systemic response, the lung compartment in the severely ill COVID-19 patients typically undergoes a robust, protracted inflammation.
At the COVID-19 management level, there is no dominant break-through strategy, which would dramatically differ (apart from the antimicrobials/antivirals) from the established sepsis treatment bundle by the US National Institutes of Health guidelines. One important exception is the dissimilar efficacy of glucocorticoids (GC). In the current sepsis guidelines, there is a weak recommendation for glucocorticoids, whereas their use in severe SARS-CoV-2 pneumonia is unequivocally beneficial [13]. The biological mechanism behind this difference is still unclear and must be elucidated since the underlying reasons could lead to a renaissance of GC in bacterial sepsis and critical care in general. Similarly, some immune-therapies appear to confer amelioration for some COVID-19 patients [29], [30], while this fact has not been proven for sepsis cases. As a result of these observations, the National Institutes of Health (NIH), EMA and other international institutions issued a daily updated guideline that summarizes the recommended immunotherapies against COVID-19 and ongoing clinical trials (Table 2) [31].
Besides IL-6, our knowledge about the concentrations of other proinflammatory or anti-inflammatory mediators in patients with COVID-19 is still modest. We have no clear picture regarding the landscape of the cytokine storm, and especially the chemokines that regulate the distribution and activity of effector cell populations. Interpreting changes in cytokine plasma concentrations that seem to be elevated without additional immune cellular parameters does not provide clarity about the molecular basis of COVID-19 [32]. As a consequence, choosing an appropriate treatment strategy becomes a challenge.
IL-10, a pleiotropic cytokine known for its potent anti-inflammatory and immunosuppressive effects, has also been found to be elevated in COVID-19 patients [33]. This could lead to a different conclusion for therapeutic approaches and in understanding the disease pathophysiology [32]. However, the role of IL-10 is currently under re-evaluation. Besides the classical IL-10 role as an anti-inflammatory cytokine, non-classical pro-inflammatory effects of IL-10 provide a plausible explanation for elevated IL-10 levels in the face of systemic inflammation [34].
Profound lymphopenia, an abnormally low level of lymphocytes in the blood similar to levels often seen in septic shock, is a near uniform finding in severely ill patients with COVID-19 and correlates with increased secondary infections and mortality [35]. The reduction and loss of immune effector cells is observed in all lymphocyte types, including CD8+ and natural killer cells, which have important antiviral roles, as well as B cells, which are essential for making antibodies that inactivate the virus [36], [32], [37]. As a consequence, beside the “cytokine storm” hypothesis, another hypothesis has been suggested, namely that severe immunosuppression and not a cytokine storm characterises COVID-19 infections [36]. The authors continue to suggest that treatments that support host protective immunity must be considered. The most rational approach to support the patient’s protective immunity is to evaluate immune stimulants targeting the adaptive immunity and T-cell function [38], [39], [40], [27], [36]. Monoclonal antibody checkpoint inhibitors, nivolumab (Opdivom) and pembrolizumab (Keytruda) targeting PD-1 as well as IL-7 have been studied in clinical trials (Table 2) [41]. The inhibition of IL-7 has shown a beneficial effect in septic patients with an increase in the lymphocyte counts [42], [43]. An aspect about the controversial two hypotheses is the current inability to address them due to a lack of appropriate diagnostic tools to evaluate cell immune function in COVID-19 infections [21].
Regardless of the differences with respect to immunotherapy, the importance of antimicrobial treatments and supportive therapies (e.g. oxygen for hypoxaemia and ventilatory support) are lessons learned from sepsis that can be transferred to COVID-19 patients. As in other infections leading to sepsis, successful treatment against COVID-19 involves eradication of the causative organism, namely SARS-CoV-2. Since the WHO declared the COVID-19 pandemic on March 2019, scientists and clinicians around the world have worked around the clock to develop therapies, diagnostic kits, and vaccines against SARS-CoV-2. Many of those discoveries were first approved globally as temporary emergency use authorizations (EUA) by the Food and Drug Administration (FDA) in the USA and its international counterparts worldwide. As such, several EUAs were issued to treat COVID-19 that allowed the use of unapproved drugs or unapproved uses of approved drugs in the absence of alternatives. The European Medicines Agency (EMA) took a similar approach by granting conditional marketing authorization (CMA) to those types of drugs including both antivirals and antibodies. Some EUAs or CMA were later revised after some of the antibodies became ineffective against the Omicron variant of the virus (Table 4).
Table 4.
Antivirals and antibodies granted an emergency use authorization (EUA) by the FDA or a conditional marketing authorization (CMA) by the European Medicines Agency (EMA) during COVID-19 pandemic.
Table 4.
Antivirals and antibodies granted an emergency use authorization (EUA) by the FDA or a conditional marketing authorization (CMA) by the European Medicines Agency (EMA) during COVID-19 pandemic.
| Drug | Brand name | FDAEUA | EMA CMA | Rescinded-revised by FDA/EMA | |
|---|---|---|---|---|---|
| Antivirals | Hydroxychloroquine sulfateChloroquine phosphate | Several | Ma 2020 | Jun 2020 | |
| Remdesivir | Veklury | May 2020 | Jun 2020 | ||
| Nirmatrelvir/ Ritonavir | Paxlovid | Dec 2021 | Jan 2022 | ||
| Molnupiravir | Lagevrio | Dec 2021 | |||
| Anti-SARS-CoV-2-antibodies | Convalescent plasma | Aug 2020 | |||
| Bamlanivimab | Nov 2020 | Mar 2021 | Jan 2022/Nov 2021 | ||
| Casirivimab/Imdevimab | Regn-cov2 | Nov 2020 | Feb 2021 | Jan 2022 | |
| Etesevimab | Dec 2021 | Mar 2021 | Jan 2022/Nov 2021 | ||
| Tixagevimab/Cilgavimab | Evusheld | Dec 2021 | Mar 2022 | ||
| Sotrovimab | Xevudy | Jan 2022 | May 2021 | ||
| Regdanvimab | Regkirona | Nov 2021 |
Additionally, the use of combination therapies has been proposed [44]. In this context, it was found that the antiviral activity of lactoferrin makes it a potential immunity enhancer which could be administered along with conventional antivirals [45]. Interestingly, this compound shows anti-SARS-CoV2 activity by itself [46], which seems to be mechanistically independent from its antibacterial and LPS-binding activities [47]. On the other hand, Sohn et al. (2020) [45] discovered that drugs that have been described as inhibitors of the LPS-induced cytokine storm such as the polypeptide Aspidasept (Pep19-2.5) [48,49,50,51,52] may also be useful against SARS-Cov2 induced hyperinflammation [45] [53]. This may open the door to a new therapeutic approach against SARS-CoV-2.
3. Bacterial coinfections and the relationship between LPS and SARS-CoV-2
Bacterial coinfections with SARS-CoV-2 seem to be as prevalent as they once were with influenza virus from serotype H1N1, the etiological agent that caused the 1918 influenza pandemic, and they are believed to have played a significant role in the lethality of both diseases.
Bacterial coinfections or secondary bacterial infections are indeed critical risk factors for the severity and mortality rates of COVID-19 [54]. In addition, there is evidence supporting that most of the deaths during the 1918 influenza pandemic were due to the bacterial coinfections rather than the flu virus. In accordance with this hypothesis, serotype H1N1 influenza virus continues to have widespread prevalence worldwide without the devastating consequences of the 1918 pandemic. Nevertheless, there are many important differences between COVID-19 and the 1918 influenza pandemic. For instance, whereas the latter mainly affected young and fully immune-competent people, morbi-mortality due to COVID-19 was strongly associated with aging [55], comorbidities (see above) and immune deficiencies [56].
On the other hand, the cell mediators induced in the case of Gram-negative (lipopolysaccharide, LPS) [57], Gram-positive (lipoproteins or peptides) [58] and SARS-CoV-2 infections [59], [57] (see above) are remarkably similar. In this regard, it is worth noting that the most potent pathogen associated molecular patterns (PAMPs) from Gram-negative bacteria and SARS-CoV-2 induce inflammation through the same cell receptor, namely Toll-like receptor 4 (TLR4)/MD-2. Importantly, TLR4 is responsible for the intracellular cascade leading to pro-inflammatory cytokine secretion and its canonical agonist is LPS (endotoxin). Bacterial endotoxin ranks among the most potent immunostimulants found in nature and is the main triggering factor of Gram-negative sepsis, which affects millions of people worldwide [60].
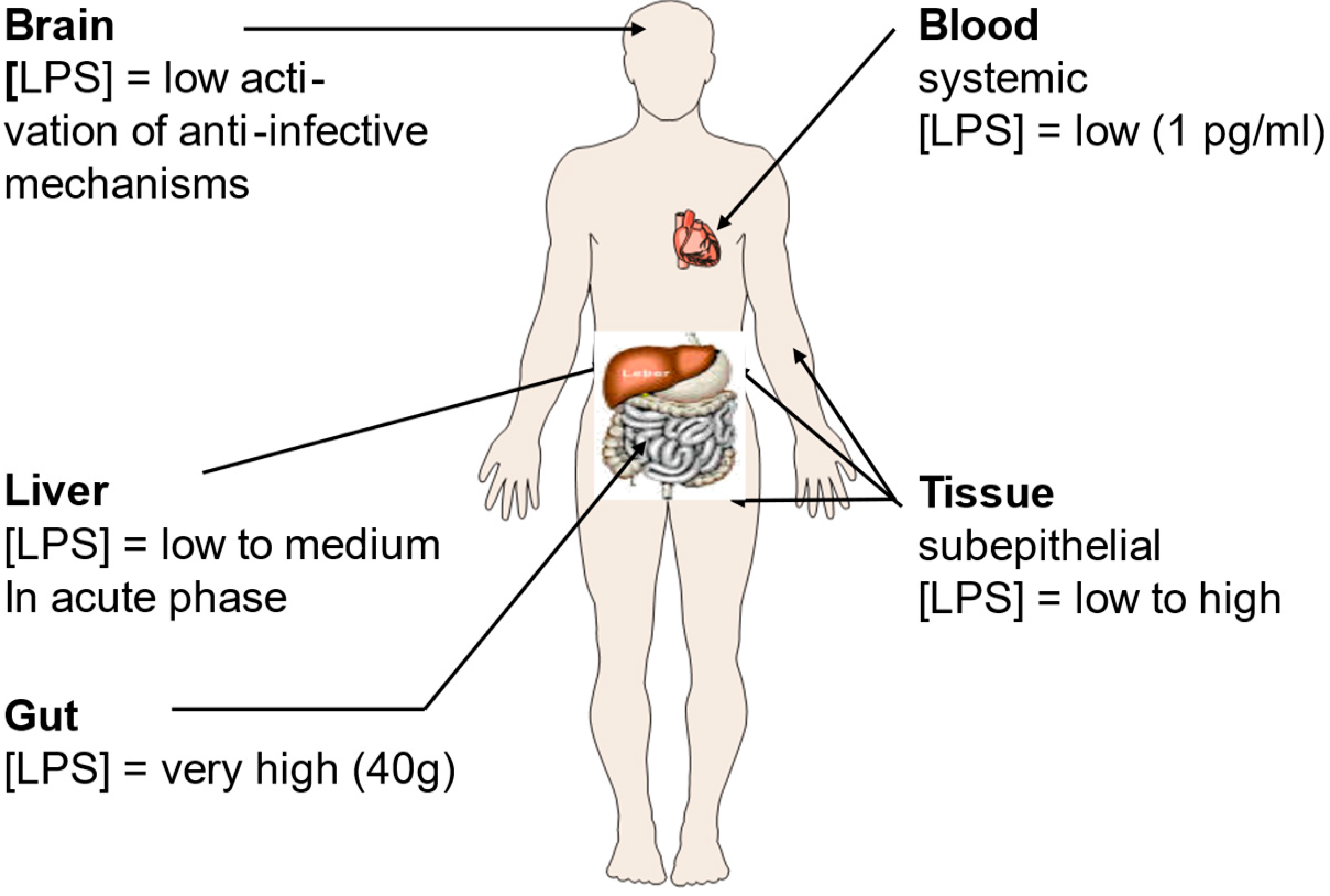
Besides well-known or presumed disorders triggered by bacteria such as colitis and Crohn’s disease, a variety of additional pathologies are due to the interaction of LPS with the immune system [61]. Such interaction can be the consequence of infections with Gram-negative bacteria, and/or be due to contact with commensal bacteria (Figure 1). The main concentration of this molecule is found in the gut that can contain up to 1.0 to 1.5 kg of bacteria [62]. But there might also be significant concentrations in subepithelial tissues and in the liver [63].
Figure 1.
Lipopolysaccharide (LPS) concentrations in the human body. LPS is the main constituent of the outer leaflet of the outer membrane of Gram-negative bacteria, and it may induce inflammation even in the nanomolar range [64]. Its presence in the body is tightly associated with locations where bacteria are particularly abundant such as the gut, and the subepithelial tissue. Figure kindly provided by Robert Munford, Oxon, USA.
Figure 1.
Lipopolysaccharide (LPS) concentrations in the human body. LPS is the main constituent of the outer leaflet of the outer membrane of Gram-negative bacteria, and it may induce inflammation even in the nanomolar range [64]. Its presence in the body is tightly associated with locations where bacteria are particularly abundant such as the gut, and the subepithelial tissue. Figure kindly provided by Robert Munford, Oxon, USA.
Teixeira et al. (2021) found that severe COVID-19 patients have increased LPS levels and systemic inflammation that result in monocyte activation [65]. Since mucosal barrier alterations may play a role in the pathogenesis of several diseases, COVID-19 included, the authors evaluated the association between bacterial translocation markers and systemic inflammation at the earliest time-point after hospitalization and during the last 72 h of hospitalization in surviving and non-surviving COVID-19 patients. Sixty-six SARS-CoV-2 RT-PCR positive patients and nine non-COVID-19 pneumonia controls were admitted in this study. Blood samples were collected at hospital admission (T1) and 0–72 h before hospital discharge (T2, alive or dead) to analyze systemic cytokines and chemokines, LPS concentrations and soluble CD14 (sCD14) levels. The THP-1 human monocytic cell line was incubated with plasma from survivors and non-survivors, and their phenotype, activation status, TLR4, and chemokine receptors were analyzed by flow cytometry. It was found that COVID-19 patients exhibited higher IL-6, IFN-γ, TNF-α, TGF-β1, CCL2/MCP-1, CCL4/MIP-1β, and CCL5/RANTES levels than controls. Moreover, LPS and sCD14 were higher at hospital admission in SARS-CoV-2-infected patients. Non-surviving COVID-19 patients had increased LPS levels concomitant with higher IL-6, TNF-α, CCL2/MCP-1, and CCL5/ RANTES levels at T2. Increased expression of CD16 and CCR5 were identified in THP-1 cells incubated with the plasma of surviving patients obtained at T2. The incubation of THP-1 with T2 plasma of non-surviving COVID-19 patients led to higher TLR4, CCR2, CCR5, CCR7, and CD69 expression. This confirmed that microbial translocation and hyperinflammation were directly correlated in patients with severe COVID-19.
Animal models based on mice, hamsters, ferrets, and nonhuman primates were developed to study COVID-19 infection and potential therapies. Since mouse ACE2 does not effectively bind the viral S protein, transgenic mouse models expressing human ACE2 were used [66]. These mice were susceptible to infection by SARS-CoV-2, however, they differed in disease severity. More recently, new animal models were developed to recapitulate all aspects of COVID-19 in humans and, in particular, pulmonary vascular disease and ARDS [67].
A mouse inflammation model based on the coadministration of aerosolized SARS-CoV-2 spike (S) protein together with LPS to the lungs has been developed [68]. Using nuclear factor-kappa B (NF-κB) luciferase reporter and C57BL/6 mice followed by combinations of bioimaging, cytokine, chemokine, FACS, and histochemistry analyses, the model showed severe pulmonary inflammation and a cytokine profile similar to that observed in COVID-19. Using this animal model, a previously unknown high-affinity interaction between the SARS-CoV-2 S protein and LPS from E. coli and P. aeruginosa has been demonstrated, leading to a hyperinflammation in vitro as well as in vivo [68]. Very importantly, the molecular mechanism underlying this effect was dependent on specific and distinct interactions between the S protein and LPS, enabling LPS’s transfer to CD14 and subsequent downstream NF-κB activation. The resulting synergism between the S protein and LPS has clinical relevance, providing new insights into comorbidities that may increase the risk for ARDS during COVID-19. In addition, microscale thermophoresis assays have yielded a KD of 47 nM for the interaction between LPS and SARS-CoV-2 spike (S) protein. Computational modeling and all-atom molecular dynamics simulations further substantiated the experimental results, identifying a main LPS-binding site in SARS-CoV-2 S protein. S protein, when combined with low levels of LPS, boosted (NF-kB) activation in monocytic THP-1 cells and cytokine responses in human blood and peripheral blood mononuclear cells, respectively [63].
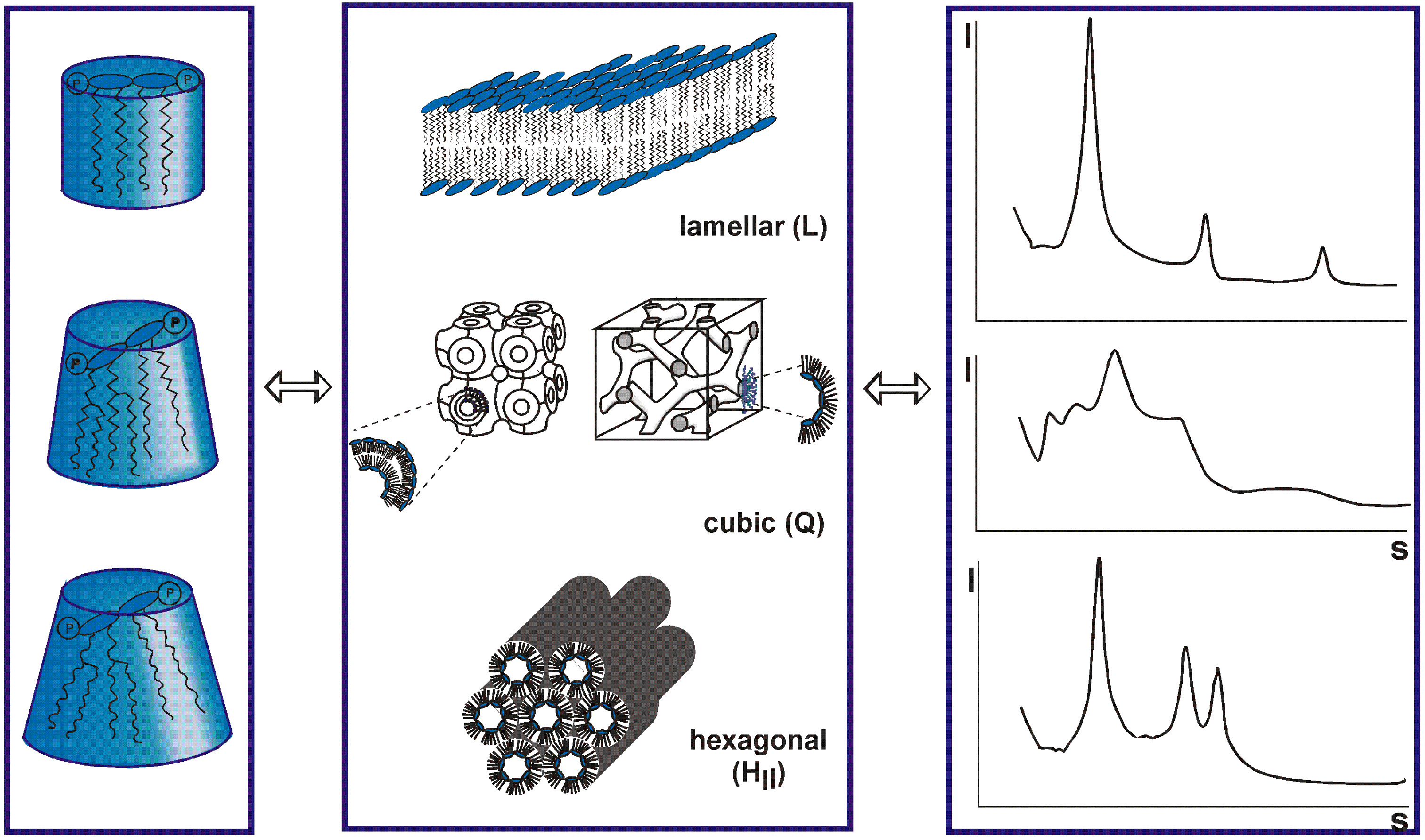
The data of the interaction of the S protein with LPS should be discussed in the light of immune stimulation induced by LPS. There are various scenarios possible, one hypothesis is that LPS is transferred to CD14 which then induces cell activation via the interaction of LPS with the complex of TLR4 and myeloid differentiation factor 2 (MD-2) [69]. A role of the LPS-binding protein LBP is also envisioned, although cell activation may also take place in the absence of LBP [70]. In any case, today it is assumed that for cell stimulation the aggregate structure of LPS is decisive [71]. It has been shown that LPS monomers are biologically inactive [72]. LPS molecules naturally form aggregates that can lead to high activity when they are in a non-lamellar geometry, and display no activity in a lamellar form [73,74] [75]. The different possible aggregate structures for LPS depend on the chemical structures of the monomers (Figure 2). In standard LPS, the lipid A part, the endotoxin principle, has a hexa-acylated diglucosamine backbone which is highly active. Other LPS that are under-acylated, for example with a tetra- or a penta-acylated lipid A, lack bioactivity [76], [77] [77], [78,79]. In an analogy to this behavior, biologically active LPS converts, when it is inactivated by the addition of for example antimicrobial peptides such as compound Pep19-2.5 or polymyxin B, into a (multi)lamellar and thus, inactive aggregate [80], [81],[82].
Figure 2.
Varying conformations of Lipopolysaccharide (LPS) monomers (left column) aggregate in different structures (middle panel) according to the theory of Israelachvili [76], [77]. These different structures produce distinct small-angle X-ray patterns (right panel [77], [78,79]) and result from different degree of acylation of the lipid A molecule (left panel).
Figure 2.
Varying conformations of Lipopolysaccharide (LPS) monomers (left column) aggregate in different structures (middle panel) according to the theory of Israelachvili [76], [77]. These different structures produce distinct small-angle X-ray patterns (right panel [77], [78,79]) and result from different degree of acylation of the lipid A molecule (left panel).
From the foregoing, it is apparent that the binding of the S protein to LPS changes the conformation of the latter in a way that increases the stimulation potency of LPS. Therefore, an analysis of the S protein:LPS complex would give more insight for an understanding of the changes in bioactivity. Recently, biophysical investigations with the S protein have been performed [63] [83]. Incubation of 100 μg/ml of LPS with the SARS-CoV-2 S protein, yielded a significant reduction of the hydrodynamic radii of the particles in solution. The aggregate size was similar to the one observed in the sample with SARS-CoV-2 S protein alone, suggesting a complete dispersion of LPS aggregates by the S protein. Using transmission electron microscopy (TEM) to further characterize the LPS, a marked disaggregating effect on the LPS aggregates was detected using 100 μg/ml LPS.
In the samples with 250 and 500 μg/ml LPS, the authors observed larger aggregates, suggestive of the LPS–SARS-CoV-2 S protein complexes identified by blue native (BN)-PAGE. In a further analysis, a gradual increase in fluorescence was observed by adding sub-nanomolar amounts of S protein, indicating a reduction in fluorescein self-quenching due to S protein-induced disaggregation of LPS. With increasing S protein concentrations, the fluorescence level was found to increase up to a maximum level, indicating a complete dispersion of LPS aggregates. Using higher levels of S protein, however, a decrease in fluorescence intensity of LPS-FITC was noticed, indicating subsequent aggregation. Plotting the fluorescence intensity at 515 nm as function of different concentrations of S protein demonstrated a dose-dependence of the disaggregation and aggregation processes. In summary, these data show complicated, dynamic, and dose-dependent interactions within SARS-CoV-2 S protein–LPS complexes. Notably, SARS-CoV-2 S protein induced a marked disaggregation of LPS at sub-nanomolar to nanomolar levels.
These findings indicate that the interaction of S protein with LPS complexes is concentration-dependent leading first to disaggregation and then again to an increase with corresponding differences in biological activity. For a biophysical understanding of these processes, analyses based on the methodology of the publications quoted in the legends of Figure 2 (e.g. small-angle X-ray scattering) would be necessary.
According to the various papers cited above, it seems that LPS has a fundamental role in the expression of infectivity: In each case an enhancing action of LPS can be found. Interestingly, higher amounts of LPS and soluble CD14, a transport protein of LPS, was found in COVID-19 patients. Therefore, the question arises whether the infection caused by the SARS-Cov2 virus influences the metabolism of LPS in a way that leads to the observed detrimental effects of the infection.
4. Influence of SARS-CoV-2 on the coagulation system
Coagulopathy, with an incidence as high as 50% in patients with severe COVID-19, is frequent during both conventional sepsis and COVID-19. Coagulopathy in COVID-19 can be triggered by an increase in the vasoconstrictor angiotensin II, a decrease in the vasodilator angiotensin, and the sepsis-induced release of cytokines [85]. However, the effects of COVID-19 on the coagulation system are far from the typical disseminated intravascular coagulation (DIC) seen during bacterial sepsis [86]. While bacterial coagulopathy is associated with coagulation factor X, COVID-19-associated coagulopathy is characterized by elevated circulating fibrinogen, high levels of D-dimer, thrombocytopenia and mildly affected clotting times [87]. In addition, pulmonary microvascular thrombosis has been reported and may play a role in progressive lung failure.
Unlike during conventional sepsis, anticoagulation seems to play a key role in the treatment of COVID-19. However, there is a lack of practice guidelines tailored to these patients. A scoring system for COVID-19-coagulopathy and stratification of patients for the purpose of anticoagulation therapy based on risk categories has been proposed [34]. In patients with shock, it was observed that antithrombin (AT) alone, but not the combined action of heparin and AT showed therapeutically favorable effects. Their proposed scoring system and therapeutic guidelines are likely to undergo revisions in the future as new data becomes available in this evolving field.
5. Long COVID-19 syndrome
There is a striking parallelism between bacterial sepsis and COVID-19 phenotypes: the long-term sequelae. In both patient groups, the hospital discharge does not equal full recovery, and it is frequently followed by protracted, incapacitating consequences. While in bacterial sepsis, the post-discharge complications are referred to as post-sepsis syndrome and/or Persistent inflammation, Immunosuppression, and Catabolism Syndrome (PICS), in SARS-CoV-2 infected patients, they are known as “long COVID” [88] ([89]. The most common persistent symptoms for both, long COVID and post-sepsis syndrome, include fatigue, muscle pain, poor sleep, and cardiac or cognitive disturbances (e.g. arrhythmias, short-term memory loss). Remarkably, a troubling difference exists between the two conditions. Unlike in post-sepsis syndrome, long-COVID is frequently diagnosed in mildly SARS-CoV-2 infected patients (i.e. those with no hospital stay). The presence of the “long-phenotype” in both illnesses strongly indicates a severe and prolonged deregulation of the immune-inflammatory system (with clear immunosuppression features) and organ homeostasis. In the context of the slowly subsiding severe COVID-19 manifestations, one should re-focus on the long-term sequalae to evaluate a potential risk of increase in chronic debilitation.
Author Contributions
Conceptualization, K.B., G.M.T., G. W., P. G.; R.F, and C.N. ; methodology, S. F., K.M., and R.F. ; software, K. B.,, G. W. C.N. ; validation, C.N. S.F., K.M. ; formal analysis, S.F., C.N., G.W. ; data curation, K.B., G.M.T., and P,G. ; writing—original draft preparation, K.B., G.M.T., G.W, P.G. ; writing—review and editing,P.G. , S.F., K.M., R.F. ; visualization, K.B. ; supervision, P.G., ; project administration, P.G., ; All authors have read and agreed to the published version of the manuscript.
Funding
C.N. Phospholipid Research Center, grant number CNE-2022-104/1-1
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data on X-ray scattering of bacterial toxins and on the biological activities of the antimicrobial peptide Aspidasept (Pep19-2.5) can be obtained from “Brandenburg Antiinfectiva GmbH”.(K.B. and K.M.)
Conflicts of Interest
The authors declare no conflict of interest.
References
- Crespi BJ: Evolutionary and genetic insights for clinical psychology. Clin Psychol Rev 2020, 78:101857. [CrossRef]
- Ziegler-Heitbrock HW, Appl B, Kafferlein E, Loffler T, Jahn-Henninger H, Gutensohn W, Nores JR, McCullough K, Passlick B, Labeta MO et al: The antibody MY4 recognizes CD14 on porcine monocytes and macrophages. Scand J Immunol 1994, 40(5):509-514. [CrossRef]
- Gallo CG, Fiorino S, Posabella G, Antonacci D, Tropeano A, Pausini E, Pausini C, Guarniero T, Hong W, Giampieri E et al: COVID-19, what could sepsis, severe acute pancreatitis, gender differences, and aging teach us? Cytokine 2021, 148:155628. [CrossRef]
- Chen Z, Peng Y, Wu X, Pang B, Yang F, Zheng W, Liu C, Zhang J: Comorbidities and complications of COVID-19 associated with disease severity, progression, and mortality in China with centralized isolation and hospitalization: A systematic review and meta-analysis. Front Public Health 2022, 10:923485. [CrossRef]
- Ahlstrom B, Frithiof R, Larsson IM, Strandberg G, Lipcsey M, Hultstrom M: A comparison of impact of comorbidities and demographics on 60-day mortality in ICU patients with COVID-19, sepsis and acute respiratory distress syndrome. Sci Rep 2022, 12(1):15703. [CrossRef]
- Nalbandian A, Sehgal K, Gupta A, Madhavan MV, McGroder C, Stevens JS, Cook JR, Nordvig AS, Shalev D, Sehrawat TS et al: Post-acute COVID-19 syndrome. Nature medicine 2021, 27(4):601-615. [CrossRef]
- Terpos E, Ntanasis-Stathopoulos I, Elalamy I, Kastritis E, Sergentanis TN, Politou M, Psaltopoulou T, Gerotziafas G, Dimopoulos MA: Hematological findings and complications of COVID-19. Am J Hematol 2020, 95(7):834-847. [CrossRef]
- Drake TM, Riad AM, Fairfield CJ, Egan C, Knight SR, Pius R, Hardwick HE, Norman L, Shaw CA, McLean KA et al: Characterisation of in-hospital complications associated with COVID-19 using the ISARIC WHO Clinical Characterisation Protocol UK: a prospective, multicentre cohort study. Lancet 2021, 398(10296):223-237. [CrossRef]
- Shappell C, Rhee C, Klompas M: Update on Sepsis Epidemiology in the Era of COVID-19. Semin Respir Crit Care Med 2023, 44(1):173-184. [CrossRef]
- Cavaillon JM: During Sepsis and COVID-19, the Pro-Inflammatory and Anti-Inflammatory Responses Are Concomitant. Clin Rev Allergy Immunol 2023. [CrossRef]
- Kostakis I, Smith GB, Prytherch D, Meredith P, Price C, Chauhan A, Portsmouth Academic ConsortIum For Investigating C: The performance of the National Early Warning Score and National Early Warning Score 2 in hospitalised patients infected by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Resuscitation 2021, 159:150-157. [CrossRef]
- Lalueza A, Lora-Tamayo J, Maestro-de la Calle G, Folgueira D, Arrieta E, de Miguel-Campo B, Diaz-Simon R, Lora D, de la Calle C, Mancheno-Losa M et al: A predictive score at admission for respiratory failure among hospitalized patients with confirmed 2019 Coronavirus Disease: a simple tool for a complex problem. Intern Emerg Med 2022, 17(2):515-524. [CrossRef]
- Herminghaus A, Osuchowski MF: How sepsis parallels and differs from COVID-19. EBioMedicine 2022, 86:104355.
- Stasi A, Franzin R, Fiorentino M, Squiccimarro E, Castellano G, Gesualdo L: Multifaced Roles of HDL in Sepsis and SARS-CoV-2 Infection: Renal Implications. Int J Mol Sci 2021, 22(11). [CrossRef]
- Coudereau R, Waeckel L, Cour M, Rimmele T, Pescarmona R, Fabri A, Jallades L, Yonis H, Gossez M, Lukaszewicz AC et al: Emergence of immunosuppressive LOX-1+ PMN-MDSC in septic shock and severe COVID-19 patients with acute respiratory distress syndrome. J Leukoc Biol 2022, 111(2):489-496. [CrossRef]
- Stasi A, Castellano G, Ranieri E, Infante B, Stallone G, Gesualdo L, Netti GS: SARS-CoV-2 and Viral Sepsis: Immune Dysfunction and Implications in Kidney Failure. J Clin Med 2020, 9(12). [CrossRef]
- Dong X, Wang C, Liu X, Gao W, Bai X, Li Z: Lessons Learned Comparing Immune System Alterations of Bacterial Sepsis and SARS-CoV-2 Sepsis. Front Immunol 2020, 11:598404. [CrossRef]
- Gorski A, Borysowski J, Miedzybrodzki R: Sepsis, Phages, and COVID-19. Pathogens 2020, 9(10). [CrossRef]
- Limmer A, Engler A, Kattner S, Gregorius J, Pattberg KT, Schulz R, Schwab J, Roth J, Vogl T, Krawczyk A et al: Patients with SARS-CoV-2-Induced Viral Sepsis Simultaneously Show Immune Activation, Impaired Immune Function and a Procoagulatory Disease State. Vaccines (Basel) 2023, 11(2). [CrossRef]
- Plocque A, Mitri C, Lefevre C, Tabary O, Touqui L, Philippart F: Should We Interfere with the Interleukin-6 Receptor During COVID-19: What Do We Know So Far? Drugs 2023, 83(1):1-36. [CrossRef]
- Remy KE, Mazer M, Striker DA, Ellebedy AH, Walton AH, Unsinger J, Blood TM, Mudd PA, Yi DJ, Mannion DA et al: Severe immunosuppression and not a cytokine storm characterizes COVID-19 infections. JCI Insight 2020, 5(17).
- Chousterman BG, Swirski FK, Weber GF: Cytokine storm and sepsis disease pathogenesis. Semin Immunopathol 2017, 39(5):517-528. [CrossRef]
- Tang H, Qin S, Li Z, Gao W, Tang M, Dong X: Early immune system alterations in patients with septic shock. Front Immunol 2023, 14:1126874. [CrossRef]
- Giamarellos-Bourboulis EJ: Failure of treatments based on the cytokine storm theory of sepsis: time for a novel approach. Immunotherapy 2013, 5(3):207-209. [CrossRef]
- Fortier ME, Kent S, Ashdown H, Poole S, Boksa P, Luheshi GN: The viral mimic, polyinosinic:polycytidylic acid, induces fever in rats via an interleukin-1-dependent mechanism. Am J Physiol Regul Integr Comp Physiol 2004, 287(4):R759-766. [CrossRef]
- Gu T, Zhao S, Jin G, Song M, Zhi Y, Zhao R, Ma F, Zheng Y, Wang K, Liu H et al: Cytokine Signature Induced by SARS-CoV-2 Spike Protein in a Mouse Model. Front Immunol 2020, 11:621441. [CrossRef]
- Allaouchiche B: Immunotherapies for COVID-19: Restoring the immunity could be the priority. Anaesth Crit Care Pain Med 2020, 39(3):385. [CrossRef]
- Gallo RL, Huttner KM: Antimicrobial peptides: an emerging concept in cutaneous biology. The Journal of Investigative Dermatology 1998, 111(5):739-743. [CrossRef]
- Perlin DS, Neil GA, Anderson C, Zafir-Lavie I, Raines S, Ware CF, Wilkins HJ: Randomized, double-blind, controlled trial of human anti-LIGHT monoclonal antibody in COVID-19 acute respiratory distress syndrome. The Journal of clinical investigation 2022, 132(3). [CrossRef]
- Guo Y, Hu K, Li Y, Lu C, Ling K, Cai C, Wang W, Ye D: Targeting TNF-alpha for COVID-19: Recent Advanced and Controversies. Front Public Health 2022, 10:833967. [CrossRef]
- Pau AK, Aberg J, Baker J, Belperio PS, Coopersmith C, Crew P, Grund B, Gulick RM, Harrison C, Kim A et al: Convalescent Plasma for the Treatment of COVID-19: Perspectives of the National Institutes of Health COVID-19 Treatment Guidelines Panel. Ann Intern Med 2021, 174(1):93-95. [CrossRef]
- Remy KE, Brakenridge SC, Francois B, Daix T, Deutschman CS, Monneret G, Jeannet R, Laterre PF, Hotchkiss RS, Moldawer LL: Immunotherapies for COVID-19: lessons learned from sepsis. Lancet Respir Med 2020, 8(10):946-949. [CrossRef]
- Islam H, Chamberlain TC, Mui AL, Little JP: Elevated Interleukin-10 Levels in COVID-19: Potentiation of Pro-Inflammatory Responses or Impaired Anti-Inflammatory Action? Front Immunol 2021, 12:677008. [CrossRef]
- Dalinghaus M, Willem J, Gratama C, Koers JH, Gerding AM, Zijlstra WG, Kuipers JR: Left ventricular oxygen and substrate uptake in chronically hypoxemic lambs. Pediatric research 1993, 34(4):471-477. [CrossRef]
- Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, Xiang J, Wang Y, Song B, Gu X et al: Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 2020, 395(10229):1054-1062. [CrossRef]
- Davitt E, Davitt C, Mazer MB, Areti SS, Hotchkiss RS, Remy KE: COVID-19 disease and immune dysregulation. Best Pract Res Clin Haematol 2022, 35(3):101401. [CrossRef]
- Riva G, Nasillo V, Tagliafico E, Trenti T, Comoli P, Luppi M: COVID-19: more than a cytokine storm. Crit Care 2020, 24(1):549.
- Gartenhaus RB, Wang P, Hoffmann P: Induction of the WAF1/CIP1 protein and apoptosis in human T-cell leukemia virus type I-transformed lymphocytes after treatment with adriamycin by using a p53-independent pathway. Proceedings of the National Academy of Sciences of the United States of America 1996, 93(1):265-268. [CrossRef]
- Dueck R, Schroeder JP, Parker HR, Rathbun M, Smolen K: Carotid artery exteriorization for percutaneous catheterization in sheep and dogs. Am J Vet Res 1982, 43(5):898-901.
- Mayor S: Intensive immunosuppression reduces deaths in covid-19-associated cytokine storm syndrome, study finds. Bmj 2020, 370:m2935. [CrossRef]
- Zheng HY, Zhang M, Yang CX, Zhang N, Wang XC, Yang XP, Dong XQ, Zheng YT: Elevated exhaustion levels and reduced functional diversity of T cells in peripheral blood may predict severe progression in COVID-19 patients. Cell Mol Immunol 2020, 17(5):541-543. [CrossRef]
- Daix T, Mathonnet A, Brakenridge S, Dequin PF, Mira JP, Berbille F, Morre M, Jeannet R, Blood T, Unsinger J et al: Intravenously administered interleukin-7 to reverse lymphopenia in patients with septic shock: a double-blind, randomized, placebo-controlled trial. Ann Intensive Care 2023, 13(1):17. [CrossRef]
- Francois B, Jeannet R, Daix T, Walton AH, Shotwell MS, Unsinger J, Monneret G, Rimmele T, Blood T, Morre M et al: Interleukin-7 restores lymphocytes in septic shock: the IRIS-7 randomized clinical trial. JCI Insight 2018, 3(5).
- Lai CC, Wang CY, Hsueh PR: Co-infections among patients with COVID-19: The need for combination therapy with non-anti-SARS-CoV-2 agents? J Microbiol Immunol Infect 2020, 53(4):505-512. [CrossRef]
- Elnagdy S, AlKhazindar M: The Potential of Antimicrobial Peptides as an Antiviral Therapy against COVID-19. ACS Pharmacol Transl Sci 2020, 3(4):780-782. [CrossRef]
- Hu Y, Meng X, Zhang F, Xiang Y, Wang J: The in vitro antiviral activity of lactoferrin against common human coronaviruses and SARS-CoV-2 is mediated by targeting the heparan sulfate co-receptor. Emerg Microbes Infect 2021, 10(1):317-330. [CrossRef]
- Brandenburg K, Jrgens G, Mller M, Fukuoka S, Koch MHJ: Biophysical characterization of lipopolysaccharide and lipid A inactivation by lactoferrin. BiolChem 2001, 382:1215-1225. [CrossRef]
- Barcena-Varela S, Martinez-de-Tejada G, Martin L, Schuerholz T, Gil-Royo AG, Fukuoka S, Goldmann T, Droemann D, Correa W, Gutsmann T et al: Coupling killing to neutralization: combined therapy with ceftriaxone/Pep19-2.5 counteracts sepsis in rabbits. Exp Mol Med 2017, 49(6):e345. [CrossRef]
- Brandenburg K, Andra J, Garidel P, Gutsmann T: Peptide-based treatment of sepsis. Appl Microbiol Biotechnol 2011, 90(3):799-808. [CrossRef]
- Brandenburg K, Schromm AB, Weindl G, Heinbockel L, Correa W, Mauss K, Martinez de Tejada G, Garidel P: An update on endotoxin neutralization strategies in Gram-negative bacterial infections. Expert Rev Anti Infect Ther 2021, 19(4):495-517. [CrossRef]
- utsmann T, Razquin-Olazaran I, Kowalski I, Kaconis Y, Howe J, Bartels R, Hornef M, Schurholz T, Rossle M, Sanchez-Gomez S et al: New antiseptic peptides to protect against endotoxin-mediated shock. Antimicrob Agents Chemother 2010, 54(9):3817-3824. [CrossRef]
- Kaconis Y, Kowalski I, Howe J, Brauser A, Richter W, Razquin-Olazaran I, Inigo-Pestana M, Garidel P, Rossle M, Martinez de Tejada G et al: Biophysical mechanisms of endotoxin neutralization by cationic amphiphilic peptides. Biophys J 2011, 100(11):2652-2661.
- Sohn KM, Lee SG, Kim HJ, Cheon S, Jeong H, Lee J, Kim IS, Silwal P, Kim YJ, Paik S et al: COVID-19 Patients Upregulate Toll-like Receptor 4-mediated Inflammatory Signaling That Mimics Bacterial Sepsis. J Korean Med Sci 2020, 35(38):e343. [CrossRef]
- Mirzaei R, Goodarzi P, Asadi M, Soltani A, Aljanabi HAA, Jeda AS, Dashtbin S, Jalalifar S, Mohammadzadeh R, Teimoori A et al: Bacterial co-infections with SARS-CoV-2. IUBMB Life 2020, 72(10):2097-2111. [CrossRef]
- Conti G, Amadori F, Bordanzi A, Majorana A, Bardellini E: The Impact of the COVID-19 Pandemic on Pediatric Dentistry: Insights from an Italian Cross-Sectional Survey. Dent J (Basel) 2023, 11(6). [CrossRef]
- Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X et al: Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395(10223):497-506. [CrossRef]
- Rietschel ET, Brade H, Holst O, Brade L, Muller-Loennies S, Mamat U, Zahringer U, Beckmann F, Seydel U, Brandenburg K et al: Bacterial endotoxin: Chemical constitution, biological recognition, host response, and immunological detoxification. Curr Top Microbiol Immunol 1996, 216:39-81. [CrossRef]
- Martinez de Tejada G, Heinbockel L, Ferrer-Espada R, Heine H, Alexander C, Barcena-Varela S, Goldmann T, Correa W, Wiesmuller KH, Gisch N et al: Lipoproteins/peptides are sepsis-inducing toxins from bacteria that can be neutralized by synthetic anti-endotoxin peptides. Sci Rep 2015, 5:14292. [CrossRef]
- Wilson JG, Simpson LJ, Ferreira AM, Rustagi A, Roque J, Asuni A, Ranganath T, Grant PM, Subramanian A, Rosenberg-Hasson Y et al: Cytokine profile in plasma of severe COVID-19 does not differ from ARDS and sepsis. JCI Insight 2020, 5(17).
- Luderitz O, Galanos C, Rietschel ET: Endotoxins of Gram-negative bacteria. Pharmacol Ther 1981, 15(3):383-402.
- Mohammad S, Al Zoubi S, Collotta D, Krieg N, Wissuwa B, Ferreira Alves G, Purvis GSD, Norata GD, Baragetti A, Catapano AL et al: A Synthetic Peptide Designed to Neutralize Lipopolysaccharides Attenuates Metaflammation and Diet-Induced Metabolic Derangements in Mice. Front Immunol 2021, 12:701275. [CrossRef]
- Munford RS: Endotoxin(s) and the liver. Gastroenterology 1978, 75(3):532-535.
- Petruk G, Puthia M, Petrlova J, Samsudin F, Stromdahl AC, Cerps S, Uller L, Kjellstrom S, Bond PJ, Schmidtchen AA: SARS-CoV-2 spike protein binds to bacterial lipopolysaccharide and boosts proinflammatory activity. J Mol Cell Biol 2020, 12(12):916-932. [CrossRef]
- Andra J, Gutsmann T, Garidel P, Brandenburg K: Mechanisms of endotoxin neutralization by synthetic cationic compounds. J Endotoxin Res 2006, 12(5):261-277.
- Teixeira PC, Dorneles GP, Santana Filho PC, da Silva IM, Schipper LL, Postiga IAL, Neves CAM, Rodrigues Junior LC, Peres A, Souto JT et al: Increased LPS levels coexist with systemic inflammation and result in monocyte activation in severe COVID-19 patients. Int Immunopharmacol 2021, 100:108125. [CrossRef]
- Fan C, Wu Y, Rui X, Yang Y, Ling C, Liu S, Liu S, Wang Y: Animal models for COVID-19: advances, gaps and perspectives. Signal Transduct Target Ther 2022, 7(1):220. [CrossRef]
- Hong W, Yang J, Bi Z, He C, Lei H, Yu W, Yang Y, Fan C, Lu S, Peng X et al: A mouse model for SARS-CoV-2-induced acute respiratory distress syndrome. Signal Transduct Target Ther 2021, 6(1):1. [CrossRef]
- Puthia M, Tanner L, Petruk G, Schmidtchen A: Experimental Model of Pulmonary Inflammation Induced by SARS-CoV-2 Spike Protein and Endotoxin. ACS Pharmacol Transl Sci 2022, 5(3):141-148. [CrossRef]
- Tobias PS, Soldau K, Gegner JA, Mintz D, Ulevitch RJ: Lipopolysaccharide binding protein-mediated complexation of lipopolysaccharide with soluble CD14. J Biol Chem 1995, 270(18):10482-10488. [CrossRef]
- Schumann RR, Latz E: Lipopolysaccharide-binding protein. ChemImmunol 2000, 74:42-60.
- Richter W, Vogel V, Howe J, Steiniger F, Brauser A, Koch MH, Roessle M, Gutsmann T, Garidel P, Mantele W et al: Morphology, size distribution, and aggregate structure of lipopolysaccharide and lipid A dispersions from enterobacterial origin. Innate Immun 2011, 17(5):427-438. [CrossRef]
- Mueller M, Lindner B, Kusumoto S, Fukase K, Schromm AB, Seydel U: Aggregates are the biologically active units of endotoxin. J Biol Chem 2004, 279(25):26307-26313. [CrossRef]
- Brandenburg K, Schromm AB, Gutsmann T: Endotoxins: relationship between structure, function, and activity. Subcell Biochem 2010, 53:53-67. [CrossRef]
- Schromm AB, Brandenburg K, Loppnow H, Moran AP, Koch MH, Rietschel ET, Seydel U: Biological activities of lipopolysaccharides are determined by the shape of their lipid A portion. European journal of biochemistry / FEBS 2000, 267(7):2008-2013. [CrossRef]
- Gutsmann T, Howe J, Zahringer U, Garidel P, Schromm AB, Koch MH, Fujimoto Y, Fukase K, Moriyon I, Martinez-de-Tejada G et al: Structural prerequisites for endotoxic activity in the Limulus test as compared to cytokine production in mononuclear cells. Innate Immun 2010, 16(1):39-47. [CrossRef]
- Israelachvili JN: Thermodynamic principles of self-assembly. In: Intermolecular & Surface Forces. vol. 2. London, San Diego, New York, Boston, Sydney, Tokyo, Toronto: Academic Press Ltd.; 1991: 341-365.
- Brandenburg K, Wiese A: Endotoxins: relationships between structure, function, and activity. CurrTopMedChem 2004, 4(11):1127-1146. [CrossRef]
- Andra J, Garidel P, Majerle A, Jerala R, Ridge R, Paus E, Novitsky T, Koch MH, Brandenburg K: Biophysical characterization of the interaction of Limulus polyphemus endotoxin neutralizing protein with lipopolysaccharide. European journal of biochemistry / FEBS 2004, 271(10):2037-2046. [CrossRef]
- Brandenburg K: Fourier transform infrared spectroscopy characterization of the lamellar and nonlamellar structures of free lipid A and Re lipopolysaccharides from Salmonella minnesota and Escherichia coli Biophys J 1993, 64:1215-1231.
- Howe J, Andra J, Conde R, Iriarte M, Garidel P, Koch MH, Gutsmann T, Moriyon I, Brandenburg K: Thermodynamic analysis of the lipopolysaccharide-dependent resistance of gram-negative bacteria against polymyxin B. Biophys J 2007, 92(8):2796-2805.
- Brandenburg K, David A, Howe J, Koch MH, Andra J, Garidel P: Temperature dependence of the binding of endotoxins to the polycationic peptides polymyxin B and its nonapeptide. Biophys J 2005, 88(3):1845-1858. [CrossRef]
- Garidel P, Brandenburg, K.: understanding of polymyxin B applications in bacteraemia/sepsis therapy prevention: Clinical, pharmaceutical, structural and mechanistic aspects. Anti-Infective Agents in Medicinal Chemistry 2009, 8(4):18. [CrossRef]
- Petrlova J, Samsudin F, Bond PJ, Schmidtchen A: SARS-CoV-2 spike protein aggregation is triggered by bacterial lipopolysaccharide. FEBS Lett 2022, 596(19):2566-2575. [CrossRef]
- Chen X, Liao B, Cheng L, Peng X, Xu X, Li Y, Hu T, Li J, Zhou X, Ren B: The microbial coinfection in COVID-19. Applied microbiology and biotechnology 2020, 104(18):7777-7785. [CrossRef]
- Miesbach W, Makris M: COVID-19: Coagulopathy, Risk of Thrombosis, and the Rationale for Anticoagulation. Clin Appl Thromb Hemost 2020, 26:1076029620938149. [CrossRef]
- Hadid T, Kafri Z, Al-Katib A: Coagulation and anticoagulation in COVID-19. Blood Rev 2021, 47:100761. [CrossRef]
- Jose RJ, Williams A, Manuel A, Brown JS, Chambers RC: Targeting coagulation activation in severe COVID-19 pneumonia: lessons from bacterial pneumonia and sepsis. Eur Respir Rev 2020, 29(157). [CrossRef]
- Szabo S, Zayachkivska O, Hussain A, Muller V: What is really ’Long COVID’? Inflammopharmacology 2023, 31(2):551-557.
- Natarajan A, Shetty A, Delanerolle G, Zeng Y, Zhang Y, Raymont V, Rathod S, Halabi S, Elliot K, Shi JQ et al: A systematic review and meta-analysis of long COVID symptoms. Syst Rev 2023, 12(1):88. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



