
Submitted:
28 August 2023
Posted:
29 August 2023
You are already at the latest version
Abstract
Polymer hydrogels are 3D networks consisting of hydrophilic crosslinked macromolecular chains, able to swell and retain water. Since their invention in the 1960s, they have become an outstanding pillar in the design, development, and application of engineered polymer systems suitable for biomedical and pharmaceutical applications (such as drug or cell delivery, regeneration of hard and soft tissues, wound healing, and bleeding prevention, among others). Despite several well-established synthetic routes of polymer hydrogels based on batch polymerization techniques, about fifteen years ago academia started to look for alternative methods involving simpler reaction paths, shorter reaction times, and lower energy consumption. In this context, frontal polymerization (FP) undoubtedly has become an alternative and efficient reaction model that allows for converting monomers into polymers via a localized and propagating reaction, by exploiting the formation and propagation of a “hot” polymerization front, able to self-sustain and propagate throughout the monomeric mixture. Therefore, the present work aims to summarize the main research outcomes, achieved during the last years, concerning the design, preparation, and application of FP-derived polymeric hydrogels, demonstrating the feasibility of this technique for the obtainment of functional 3D networks, and providing the reader with some perspectives for the forthcoming years.
Keywords:
Hydrogels
; Frontal polymerization
; Structure-property relationships
; Applications
1. Introduction
In the last 30 years, the biomedical and pharmaceutical sectors have greatly benefited from the design and application of polymeric hydrogels, i.e., three-dimensional hydrophilic networks, made of crosslinked macromolecular chains, exhibiting high swelling in water and water retention, external stimuli responsiveness, and tunable mechanical features as well [1,2,3,4]. Because of these peculiarities, polymeric hydrogels have found several applications in different sectors, ranging from the “traditional” biomedical field to their quite recent use for flexible aqueous energy storage devices (as hydrogel electrolytes) [5,6].
Figure 1 elucidates how polymeric hydrogels can be classified, according to different parameters, namely: the source of hydrogels (i.e., natural or/and synthetic materials), the preparation method (i.e., through free radical polymerization reactions, gamma or UV irradiation, interpenetrating network formation, solution casting, or simple mixing), the type of crosslinking (i.e., chemical, physical, or hybrid), the possible ionic charge (anionic, cationic or neutral systems), and even the type of stimuli responsiveness (i.e., biochemical-, chemical-, or physical-responsive hydrogels).
As reported in the scientific literature, all the aforementioned synthetic strategies for polymer hydrogels are well-established and optimized, although they are usually time consuming, they encompass quite complex reaction routes and the energy required is quite high. In this context, the Frontal Polymerization (FP) technique [7,8,9] may represent an alternative strategy to limit the drawbacks associated with the synthesis of polymeric hydrogels, giving rise, at the same time, to the formation of tailored 3D networks fulfilling the requirements needed for the envisaged applications. FP takes advantage of the formation of a hot polymerization front that, if the exothermicity of the polymerization reaction is sufficient, is capable of self-sustaining and propagating throughout the monomeric medium, hence rapidly converting monomers into polymers. Experiments are most often carried out in test tubes containing monomer mixtures 5 to 10 cm high. Notwithstanding some special monomeric mixtures that front polymerize in really a few seconds [10], the FP systems usually polymerize from tenths of seconds to a few minutes, i.e., much more rapidly than the batch polymerized counterparts; further, the thermal and mechanical properties of the resulting FP polymers are often better than those of the systems obtained through classical polymerization [11,12,13,14].
The interest in the use of frontal polymerization for preparing advanced hydrogels is witnessed by the significant number of peer-reviewed articles published in the scientific literature (Figure 2).
Therefore, in the present review we will give some basics about the general characteristics of frontal polymerization; then, some specific features of the FP technique applied to the synthesis of hydrogels will be provided. Eventually, the most recent outcomes on this topic will be presented and discussed also in view of their possible applications. When possible, a comparison with the hydrogels prepared by the traditional synthetic routes will be given.
2. Fundamentals of the frontal polymerization technique
2.1. Definitions
In the state of the art, there are two main types of frontal polymerization: isothermal and thermal FP (Figure 3).
In isothermal frontal polymerization, [16] the gel effect is exploited. This is a phenomenon observed mainly in the polymerization of some acrylic monomers and occurs when the viscosity of the monomer mixture becomes high enough. Under these conditions, diffusion becomes disfavored. More specifically, a slowing down of the termination rate is observed in radical polymerizations due to the decreased diffusion efficiency of the growing macromolecules. In addition to the increase in the average length of macromolecular chains, an increase in the concentration of radicals and, consequently, in the polymerization rate is also observed.
In practical terms, comparing the polymerization rate of the same monomer mixture in two different media, one with low viscosity and the other with high viscosity, the polymerization rate is higher under the latter conditions. On this basis, the insertion of a preformed polymer seed capable of swelling when placed inside a low-viscosity monomer mixture results in the polymerization rate of the monomer within the polymer being greater than that outside, with the result that the size of the seed is seen to increase. At this point, it is easy to imagine how the conversion of monomer to polymer occurs mainly at the interface between the high-viscosity and low-viscosity phases. Therefore, polymerization is observed to take place due to the formation of a propagating front (Figure 3-B). From a practical point of view, to prevent the amount of heat that develops from being excessive, there is a tendency for this type of reaction to occur at a low and constant temperature (even below 10 °C), hence the name isothermal frontal polymerization.
However, when we talk about frontal polymerization, we tend to refer to its variant that exploits the exothermicity of the reaction, and which in its most complete definition is called thermal frontal polymerization. However, in this review, although the adjective thermal is omitted, in speaking of frontal polymerization or FP we will refer only to this second type of macromolecular synthesis.
In this case, for a FP to occur, it is necessary for the polymerization reaction to be sufficiently exothermic. More specifically, let us initially consider a polymerization reaction promoted by increasing temperature and conducted according to the classical technique and, more specifically, consider the simplest system, i.e., the bulk polymerization of a liquid monomer. The most common approach, or at least the most efficient, is to thermostat the reactor to a certain temperature, the choice of which is determined by kinetic, thermodynamic, energy, safety and cost considerations. Ideally, the goal is to get a reaction going in the shortest possible time while operating safely, and ensuring a polymer product that meets the required characteristics.
This implies that while thermostatting the reactor at a relatively high temperature leads to decreased process times, it also causes higher energy costs, more pollution and greater likelihood of reaction runaway with decreased safety and loss of control over the final material properties. Conversely, the use of low process temperatures solves safety and energy-saving issues, but involves long times with consequent negative effects on efficiency.
Frontal polymerization is an interesting alternative that aims to solve at least part of the problems listed above. In its most common protocol, it operates in an elongated, generally cylindrical reactor (most often a test tube), which is not insulated (but can be if required by process parameters) and not thermostated. Through an external energy source (usually heat or light), polymerization is induced in a localized area of the reaction mixture. Since the reaction is exothermic (this requirement is essential), locally there is sufficient temperature rise for the reaction to proceed spontaneously without further input of external energy.
Given that the hot zone that is polymerizing is in contact with the monomer mixture that has not yet reacted, it can be guessed that enough heat exchange occurs at the interface to cause local initiation of the reaction. The result is the growth of the hot polymerized phase at the expense of the cold monomeric phase. The growth is nothing else than a hot front of polymerization that ideally propagates until the whole monomer is converted into polymer (Figure 3-A).
Some aspects must be taken into account, which we will briefly summarize below. For further discussion, we refer to other publications. [9,16]
Some types and applications of frontal polymerization are reported in Figure 4.
2.2. Reactor characteristics
As mentioned above, in the majority of cases reported in the literature, front polymerization reactions are conducted in cylindrical reactors (test tubes). Actually, this condition is not necessary but is convenient on a laboratory scale. Instead, what matters most is the surface area. In fact, as it increases, the amount of heat dissipated outward by the curing system also increases up to reach a critical limit. At this point, the remaining heat becomes not sufficient to allow the front to self-sustain.
Thus, even tubular reactors that are too thin may not be suitable for FP to take place. Similar considerations apply to obtaining films or otherwise materials in the form of thin layers. If the surface area exposed to the outside is large relative to the total volume of the curing mass, FP may not occur. However, films prepared by FP are reported [9].
2.3. Direction of front propagation and stirring
In general, it is preferred to make a FP occur by triggering it from above so that the front propagates downward. Given the upward direction of convective motions, this might seem counterintuitive, and upward propagation might seem to be the favored one. In fact, to avoid the quenching of the polymerization front, one generally operates under conditions such that any motion of the polymerizing mixture is avoided, at least in the direction perpendicular to the propagation of the front. In the case of parallel motions, and particularly those having the same direction as the front, on the other hand, the risk is that the front will lose its conditions of stability, and assume elongated shapes such that it will result in an increase in the surface area of contact with the outside or with the cold monomer, and will eventually destroy itself (Figure 5) [17].
2.4. Fingering
This is a phenomenon related to the previous aspects, and is another possible reason for front quenching that can occur in the case of descending fronts. The hot polymer that is forming at the expense of the underlying monomer could be easily soluble in it, or it could have such a density that it drips and infiltrates into it. [19] If the refraction index of the monomer is sufficiently different from that of the polymer, such infiltration manifests visually as “fingers” creeping into the underlying phase, hence the name fingering (Figure 6). Since this is a phenomenon that occurs in the same direction as the front, this leads to its quenching, often associated with the initiation of polymerization in the areas of the monomer that have come in contact with the hot fingers, thus causing bulk polymerization.
2.5. Monomers
In the most common cases, the monomers used are liquids. However, examples are known where front polymerization also occurs on solids (e.g., acrylamide, dicyclopentadiene). [9,16] Obviously, in principle, solid monomers are unfavorable since their polymerization passes through their melting, which, being an endothermic process, subtracts heat from the hot front, thus making its propagation more critical. On the other hand, solid monomers have the advantage of not giving fingering phenomena. Therefore, the choice of monomers suitable for descending frontal polymerization is generally limited to those giving rise to thermosets or polymers that have little or no solubility in the polymerization mixture. Generally, this condition is achieved by the addition of multifunctional comonomers, which give rise to crosslinking.
Another diriment characteristic is the boiling temperature of the monomer. Indeed, given the temperatures reached by the polymerization fronts (typically between 120 and 180 °C, [9] although FP reactions occurring outside this range have been widely reported), [20] there is a possibility that the monomer will boil when heated by the approaching front. In this case, bubbles will form and remain entrapped in the final polymeric material. However, since boiling is an endothermic phenomenon, its occurrence implies the removal of heat from the system and the possible quenching of the front. Of course, it is possible to operate under pressure; however, in laboratory experiments, mostly aimed at demonstrating the feasibility of FP under the simplest possible conditions, this possibility is generally ignored.
2.6. Mechanisms, reaction kinetics, initiators and catalysts
Among the requirements for a FP to be successfully carried out, pot-life plays a key role. In fact, the monomer mixture must be stable at relatively low temperature (generally, at room temperature), but must react quickly at the front temperature. While on the one hand this ensures that the monomer does not spontaneously react in areas far from the front, which would result in undesirable bulk polymerization, on the other hand it guarantees that the heat of polymerization is released as quickly as possible, thus avoiding its excessive dissipation that would prevent the front from self-sustaining and propagating.
Most of the monomers successfully used in FP are acrylic or methacrylic, which are polymerized by radical chain polymerization [9]. However, step-growth kinetics or different mechanisms have also been reported (e.g., polyurethanes, [21,22,23] epoxy resins) [24,25,26].
In particular, some systems have been obtained through catalytic processes [9] (e.g., polydicyclopentadiene) [27,28] or through cationic polymerization including radical induced cationic frontal polymerization [26], among others.
Another important aspect is the concentration of initiator or catalyst used. In fact, as already pointed out, in order to exploit as much heat of polymerization as possible, it is important that it be developed quickly so that the system does not have time to dissipate it outward. This is reflected in the concentrations of initiator or catalyst, which are often found to be higher than those typically employed in similar reactions conducted according to traditional protocols. However, as they increase, two phenomena can be observed: a decrease in pot-life, resulting in a tendency for the system to polymerize spontaneously in bulk, and the generation of an excessive amount of heat, such that the achieved temperature of the front turns out to be too high to guarantee a final material having the desired characteristics and to operate safely. On the other hand, too low concentrations of initiator or catalyst imply a slowing down of reaction kinetics, which results in heat amounts that, at a given time, are too low to ignite monomer polymerization, thus generating fronts that are unable to self-sustain or do not form at all.
Therefore, there is a general tendency to define and operate within appropriate initiator or catalyst concentration ranges.
Another aspect of great importance lies in the decomposition products of the initiators. For example, azo-bis-isobutyrronitrile is known to decompose by releasing nitrogen, and benzoylperoxide can release CO2. Therefore, to avoid the formation of bubbles that remain within the final material, efforts are made to use gas-free initiators, which are often synthesized on purpose [9,20,29].
Nevertheless, it is still sometimes necessary to add appropriate activators or inhibitors [27,30] to the system. They respectively aim to promote the reaction under particularly unfavorable conditions or to increase the pot-life of the system.
It is also worth pointing out that, especially in the case of acrylic or methacrylic monomers, these can often be used as is, without purifying them from the inhibitors added by the supplier. In fact, the temperatures reached by the polymerization fronts are generally high enough to make their presence almost kinetically irrelevant.
2.7. Solvents
In many cases, FP is conducted in the presence of solvents, which further complicates its management. In fact, in addition to the well-known considerations about the advantages and disadvantages of polymerization in solution compared to that operated in the absence of solvents, performing FP in the presence of solvents further complicates the polymerization process, which limits the potential use of this technique. Specifically, a first requirement is their boiling temperature, which - wanting to avoid bubbles and heat absorption due to evaporation - must be higher than that of the polymerization front. This limits the use of the most common solvents, including water itself; however, several frontal polymerizations are known to have been successfully performed in this medium, [9] including the obtainment of hydrogels such as the ones that are the subject of this review. By the way, among others, FPs conducted in ether, chloroform, acetone, benzene, etc., are in fact impracticable unless one operates under pressure (see above).
Heat capacity is another issue related to the presence of the solvent (actually, it is so referred to all components of the mixture). In systems in which the exothermicity of the reaction is relatively low, the presence of a solvent - which absorbs some of the heat released - makes FP even more critical, with consequent effects on other process parameters, including the concentration of initiator or catalyst, the geometry of the reactor, and its degree of insulation. On the other hand, the presence of a solvent can be useful in increasing the pot-life of the system and decreasing the front temperature, if excessively high.
Thus, the influence of solvent power is related to the previous considerations. In general, the tendency is to limit the use of solvents as much as possible. Thus, a good solvent is preferred to avoid excessive dilution and consequent heat dissipation.
2.8. Viscosity
In order to stabilize the fronts, especially the descending ones, but also the horizontal ones, one can choose to increase the viscosity of the monomer mixture. This way, such phenomena as fingering turn out to be in some extent prevented as the easy diffusion of the polymeric phase into the monomeric one is disfavored. For this purpose, it is possible to opt for particularly viscous solvents (e.g., glycerol) or for the addition of appropriate viscosifiers (e.g., polymers, fumed silica, etc.) [9].
For particular applications (e.g., in putties) [26], the high viscosity resulting from the addition of various inert components to the monomer is of paramount importance to ensure the propagation of fronts in any direction.
2.9. Composites
One of the most interesting applications of FP refers to the obtainment of composite and nanocomposite materials. Regarding the former, in which the amount of filler can be even greater than that of the monomer, some of the foreseeable consequences include a decrease in temperature and front velocity as the additive content increases. For these systems, in which the monomer is in fact diluted in a heat-absorbing medium, a limiting composition is identified, above which the amount of filler is too high to ensure front self-sustaining.
Obviously, given that the filler concentration is typically no more than a few percent units, in nanocomposites this phenomenon is less likely. For another, nanocomposites obtained by frontal polymerization can benefit from an additional advantage over analogs prepared by traditional techniques as the result of the much greater rate at which monomer is converted into polymer. For these systems this is not insignificant, as examples are reported in which nanoparticles that would otherwise tend to re-aggregate in the monomer-to-polymer conversion, actually remain dispersed as their diffusion turns out to be particularly slow compared to the rate of polymerization. As an example, we cite graphene during the frontal polymerization of N-isopropylacrylamide. [31,32]
3. Hydrogels obtained by frontal polymerization
Obtaining hydrogels by frontal polymerization represents one of the possible applications of FP and, provided the general requirements outlined above and the more specific ones illustrated below are met, is a particularly convenient method because of its simplicity and short time of execution. The latter aspect is particularly useful not only in the study of frontal polymerization per se but also in providing rapid indications about the characteristics of new materials whose obtainment is then scaled up to the industrial production, with considerable savings in time and cost.
3.1. Monomers
The main characteristic of a hydrogel is, of course, its compatibility with water, which - in the case of noncrosslinked systems - can be so high to dissolve the material [4].
In general, the hydrophilicity of a polymeric hydrogel depends on that of the starting monomers, which must therefore be highly polar and, if possible, ionizable. In the next paragraph, a series of polymeric hydrogels will be presented and their characteristics will be discussed in view of their current or potential applications.
It should be specified at once that through frontal polymerization it is not possible to obtain the whole class of hydrogels that can be synthesized by the classical route.
This depends first of all on the possible polymerization reactions involved in the process but also on the intrinsic features of the various individual monomers.
In fact, many hydrogels are obtainable through reactions that are not compatible with the characteristics required by FP, first and foremost the exothermicity of the reaction. In particular, polycondensation reactions are very unlikely to be carried out frontally because the condensation coproduct formation is an endothermic process that subtracts some of the heat made available by the macromolecular chain growth reaction.
Moreover, focusing, for example, on the radical polymerization of acrylic or methacrylic monomers, which are probably the most widely used in FP, there immediately appears a problem related to the molar mass of the monomer to be polymerized. As a general rule, in fact, a small monomer consisting of a C-C double bond and a few other atoms is more likely to be frontally polymerized than a larger one. The reason lies in the number of atoms not involved in the polymerization process, which, if present in a high number, tend to absorb an excessive amount of the heat released due to the exothermicity of the reaction, thus preventing the front from self-sustaining [33]. The obvious consequence related to the low molecular mass of the monomer is thus the limited number of possible compounds and isomers.
As will be seen later by discussing the reviewed publications, the easiest way to increase the number of available systems lies in copolymerization and functionalization [34,35], the latter being generally employed in order to impart particular properties to the final material. Relative to copolymerization, as will be seen, the tendency is to use a reasonably small number of monomers (e.g., acrylamide, acrylic, itaconic acid, N-isopropylacrylamide, 2-hydroxyethylacrylate, etc.), having good hydrophilicity and capable of frontally polymerizing, and copolymerize them with other compounds. However, precisely because of the considerations about front self-sustaining, the volume fraction of the latter cannot be very high.
3.2. Solvents
In the most favorable cases, it is possible to operate in water. Of course, this implies that all the components of the reaction mixture are soluble in this medium and that the amount of heat absorbed by this solvent (known to have high heat capacity and heat of evaporation) does not prevent self-sustaining of the polymerization front.
However, as hydrophilic as monomers may be, these might not be water-soluble, a problem that becomes even more dramatic in the case of copolymers or functionalized systems. Thus, it is often necessary to use other solvent media, usually polar. Examples of these include dimethylsulfoxide, N-methylpyrrolidone, glycerol, ionic liquids, deep eutectic solvents [9].
3.3. Initiators
In the case of operating in water, the preferred initiators are generally belonging to the persulfate class [8,9,20,29]. Whereas, if the solvent is different or not present, more compounds can be used. For another, the presence of any bubbles within the hydrogel may not be a problem or may even be a desired feature. If this is the case, the number of available initiators is greater than in the front polymerization of other materials.
3.4. Front temperature and material characteristics
A large proportion of commonly used polymeric hydrogels find application at near room temperature, including the physiological one. In many cases, these are materials that undergo physical, if not chemical, changes at relatively low temperatures.
For example, poly(N-isopropylacrylamide) exhibits a lower critical solution temperature (LCST) in water at about 30-32 °C. [31] This characteristic, which determines a drastic decrease in hydrophilicity, could be a problem e.g., in the presence of third components. Indeed, the fact that the material is swollen at room temperature (thus below the LCST) while being shrunk at the face temperature could affect the desired characteristics of the final copolymer or composite material.
4. Applications of frontally polymerized hydrogels
Frontal polymerized hydrogels have been finding interesting applications in various advanced sectors. This paragraph will summarize the main outcomes achieved in the last eight years, which clearly demonstrate the importance and feasibility of exploiting the frontal polymerization method for the rapid and low energy-consuming synthesis of hydrogels for advanced applications.
4.1. Biomedical applications
Nuvoli et al. [36] succeeded in synthesizing double-responsive (i.e., pH and temperature) hydrogels based on poly(N-vinylcaprolactam-co-itaconic acid). The itaconic acid content in the comonomer mixture was responsible for the possibility of performing the frontal copolymerization reaction in quantitative yield and for the stimuli-responsive characteristics of the obtained hydrogels. In particular, as this comonomer bears allylic hydrogens inhibiting radical polymerization, frontal copolymerization occurred only when the itaconic acid content was kept below 10 mol %. Further, the hydrogels containing 1 mol % of the comonomer exhibited a lower critical solution temperature at around 30°C, like that of poly(N-isopropylacrylamide), the most well-known thermoresponsive hydrogel. In addition, all the synthesized hydrogels were pH-responsive and this behavior was strictly correlated with the presence of itaconic acid: more specifically, below pH=7, the comonomer units accounted for increased hydrophobicity, hence lowered swelling. Conversely, beyond pH=7, an increase in both hydrophilicity and swelling ratio was observed, due to the itaconic acid units bearing carboxylate groups.
In a further research effort, Nuvoli and co-workers [37] exploited frontal polymerization for preparing two types of poly(2-hydroxyethylacrylate)-based hydrogels containing β-cyclodextrins, either directly dispersed into the acrylic polymer, or grafted (as acryloyl-β-cyclodextrin) onto poly(2-hydroxyethylacrylate) chains. The presence of 0.5 mol % of cyclodextrins, irrespective of their type, accounted for the maximum swelling ratio (about +300% with respect to neat poly(2-hydroxyethylacrylate)). Concerning the frontal polymerization reaction, both front temperatures (Tmax) and front velocities (Vf) were found to decrease with increasing the cyclodextrin loading (Figure 7): this finding was ascribed to the fact that cyclodextrins behaved as an inert material, favoring heat dissipation. It is worth noticing that heat dissipation was lesser when acryloyl-β-cyclodextrins were utilized; these latter also promoted a significant increase in the glass transition temperature of the dry hydrogels with respect to neat poly(2-hydroxyethylacrylate) (43 °C vs. 20 °C, respectively), because of an increased crosslinking effect exerted by the functionalized β-cyclodextrins.
The same group [8/28/2023 7:06:00 PM] further demonstrated the successful synthesis of poly(N-isopropylacrylamide)-grafted-acryloyl-β-cyclodextrin hydrogels via FP. In particular, it was possible to obtain crosslinked hydrogel structures by exploiting supramolecular interactions only, i.e., without employing any chemical crosslinker, as schematized in Figure 8. The overall behavior of supramolecularly crosslinked hydrogels was compared with that of covalently-crosslinked counterparts, using 0.5 mol% of N,N’-methylene-bis-acrylamide as crosslinker. The presence of AβCD in both the types of hydrogels did not affect the front temperatures, despite a slight increase in the front velocities. Further, as far as their swelling behavior is considered, all the synthesized hydrogels showed thermoresponsive features, with lower critical solution temperatures within 28 and 30°C. Nevertheless, the swelling ratio of the covalently-crosslinked systems was much lower than that of the supramolecular-crosslinked counterpart, these latter exhibiting swelling degrees characteristic of superabsorbent hydrogel networks.
Feng and co-workers [39] succeeded in enhancing the mechanical properties of temperature-sensitive poly(N-isopropylacrylamide) hydrogels through the incorporation of an organo-modified montmorillonite at different loadings (namely, 2, 5, 10, 15, and 20 wt.%). The fast front propagation efficiently inhibited phase separation phenomena, hence providing a homogeneous distribution of the nanoclay within the polymer matrix, resulting in a partially exfoliated structure. Further, the compressive strength of the hydrogel filled with 5 wt.% of clay increased by about 70% with respect to the unfilled counterpart; besides, this composite hydrogel showed the fastest temperature response among all the prepared composite hydrogels. Conversely, the nanoclay, irrespective of its loading, did not modify the lower critical solution temperature, which was around 33 °C. Finally, at constant nanoclay loading (2 wt.%), both front temperature and velocity were found to increase with increasing the reactor tube diameter because of decreased heath losses due to the exchange with the environment; however, at the same time, the compressive strength slightly lowered.
Rassu and co-workers [40] demonstrated the suitability of frontal polymerization for preparing hydrogels made of methyl cellulose and poly(acrylamide), using N-N’-methylene-bis-acrylamide as crosslinker at different concentrations (ranging from 0.1 to 2.0 mol%). To this aim, the reaction was carried out in water and glycerol: this latter accounted for higher front temperatures and front velocities with respect to the counterparts synthesized in H2O, due to the solvent evaporation and the subsequent heat loss. Further, being constant the methyl cellulose content, progressively increasing the crosslinker amount determined a noticeable decrease in swelling ratio and a remarkable increase in the mechanical behavior, irrespective of the employed solvent.
Another interesting approach that can be successfully exploited for preparing hydrogels for biomedical applications refers to the frontal polymerization of polymerizable deep eutectic monomers, i.e., a recent type of deep eutectic solvents that can be obtained by mixing quaternary ammonium and hydrogen bond donor monomers, characterized by high thermal stability, low vapor pressure, low cost, high viscosity, and low toxicity as well [41,42]. In this context, Jiang and co-workers [43] synthesized macroporous polyacrylamide hydrogels via frontal polymerization of deep eutectic monomers obtained by mixing several molar ratios of acrylamide and choline chloride, as schematized in Figure 9.
Increasing the molar ratio of acrylamide to choline chloride accounted for an increase in both front temperatures and front velocities. Further, the presence of choline chloride was beneficial for enhancing the conversion of acrylamide. As assessed by SEM analyses, washing off the water-soluble choline chloride allowed for obtaining a macroporous structure of the hydrogels, which exhibited superfast responsive features in alternate acetone and water media.
4.2. Drug delivery
The first hydrogel designed for drug delivery prepared by frontal polymerization was synthesized by the Mariani’s group in 2009 [44]. Lately, Wang and co-workers [45] frontally polymerized a mixture containing N,N′-methylenebisacrylamide, acrylic acid (used as functional monomer), N-isopropylacrylamide, and gatifloxacin (template molecule) for preparing pH/temperature-sensitive hydrogel-based molecularly imprinted systems, suitable for drug delivery purposes. In particular, molecular imprinted systems show stimuli-sensitive recognition features toward target (template) molecules and can reversibly swell and shrink according to specific environmental changes (like temperature and pH). The imprinting efficiency was strictly dependent on both acrylic acid/gatifloxacin ratio and N-isopropylacrylamide content; besides, compared to the counterparts prepared via classical batch polymerization, the frontally polymerized hydrogels showed higher imprinting effects in terms of imprinting factor.
Feng et al. [46] exploited the frontal polymerization technique for preparing semi-interpenetrating polymer networks made of poly(N-isopropylacrylamide) and polyvinylpyrrolidone. The presence of increasing polyvinylpyrrolidone loadings accounted for shortened response times, without altering the lower critical solution temperature of poly(N-isopropylacrylamide). Besides, both front temperatures and front velocities decreased with increasing the polyvinylpyrrolidone content, because of the increase in the viscosity of the reaction system, which lowered the buoyancy-driven convection, hence decreasing the heat transfer rate. Conversely, the mechanical behavior, and, in particular the compressive strength of the obtained hydrogels increased with increasing the polyvinylpyrrolidone loading, from 26.4 (for poly(N-isopropylacrylamide) hydrogel), to 53.6 kPa (for the semi-interpenetrating hydrogel containing 20 wt.% of polyvinylpyrrolidone). Further, using aspirin as a model drug it was possible to assess the suitability of the designed systems for drug delivery. In particular, it was found not only that their drug loading capacity was raised from 245 ((poly(N-isopropylacrylamide) hydrogel) to 422 mg/g (hydrogel containing 20 wt.% of polyvinylpyrrolidone) but also that the drug release was better controllable in the presence of the semi-interpenetrating networks.
Then, the possibility of synthesizing semi-interpenetrating poly(N-isopropylacrylamide) hydrogels containing methylcellulose via frontal polymerization was demonstrated by Mariani and co-workers [47]. In particular, the effect of increasing amounts of N,N’-methylene-bis-acrylamide (employed as crosslinker within 0.1 and 2.0 mol%) on the mechanical and swelling behavior (in two solvents, namely water and dimethyl sulfoxide, having different polarity) was thoroughly evaluated, elucidating the role of the methylcellulose at a constant loading (15 wt.%). Despite a negligible impact of the crosslinker on front temperatures and velocities, it remarkably affected the Young’s moduli of the hydrogels, regardless of the employed swelling solvent. Further, the lower critical solution temperature of the synthesized hydrogels (which exhibited the highest swelling ratio values at around 15°C) was below 30°C, due to the incorporation of methylcellulose. Finally, the significant decrease (by two orders of magnitude) in the swelling ratios measured at room temperature (about 37200%) and body temperature (around 240%) clearly confirmed the potential of these hydrogels for drug delivery purposes.
An interesting alternative to standard frontal polymerization for the obtainment of thermoresponsive poly(N-isopropylacrylamide) hydrogels suitable for drug delivery was quite recently proposed by Su and co-workers [48]. Indeed, the use of UV radiation allows for operating at low pressure, reducing the temperature of the propagation front to a restrained level, hence avoiding possible risks of explosion and morphological defects in the obtained hydrogels because of the partial monomer evaporation [49]. More specifically, the authors discussed the effects of the adopted experimental conditions (i.e., types and concentrations of photoinitiators - 2,4,6-trimethylbenzoyldiphenyl phosphine oxide or phenyl bis(2,4,6-trimethylbenzoyl)-phosphine oxide – and of crosslinkers - N,N′-methylenebisacrylamide, tri(propylene glycol) diacrylate), and an acrylopropyl polyhedral oligomeric silsesquioxane cage mixture -, as well as radiation intensity) on the frontal polymerization reaction and on the overall properties of the obtained hydrogels. A general scheme of the device employed for the frontal photopolymerization is displayed in Figure 10: in a typical preparation, the monomer (0.05 mol), the photoinitiator (0.5–2.5 mol% relative to NIPAm), and the crosslinking agent (0.5–2.5 mol% of N,N′-methylenebisacrylamide or tri(propylene glycol) diacrylate), or 0.25–1.0 mol% of acrylopropyl polyhedral oligomeric silsesquioxane cage mixture, relative to NIPAm) were ultrasonicated in 10 mL of dimethylsulfoxide for 30 min. Then, the resulting solution was poured into a glass tube encapsulated with a rubber bulb at the bottom to compensate for the polymerization shrinkage. The filled tube was irradiated for 15 min with a medium-pressure mercury lamp and the obtained hydrogels were immersed in deionized water for 24 h. This procedure was repeated three times to eliminate the unreacted monomer and soluble materials. Finally, the hydrogels were dried for 24 h in a vacuum at 60 °C. Among the selected experimental parameters, only the radiation intensity turned out to increase both front temperatures and front velocities. Further, using aspirin as a model drug allowed for assessing the drug delivery behavior of the synthesized hydrogels as a function of the type of crosslinker. In particular, a significant increase in the drug release rates was first observed, regardless of the type of crosslinker employed. Then, after 10 h, an asymptotic plateau was achieved when the temperature was set below the lower critical solution temperature of the poly(N-isopropylacrylamide) (Figure 11). Finally, because of its highly microporous structure, the hydrogel crosslinked with N,N′-methylenebisacrylamide showed a higher drug release rate with respect to the counterparts crosslinked with the other crosslinkers.
Irfan and co-workers [50] exploited frontal polymerization for synthesizing a series of poly(itaconic acid-co-acrylic acid-co-acrylamide) hydrogels, suitable for drug delivery purposes. More specifically, the effects of the itaconic acid/acrylic acid weight ratio (changing from 1:39 to 5:35 wt/wt) on the front temperatures and velocities were thoroughly investigated. As shown in Figure 12, the increase in this weight ratio accounted for a significant decrease in both frontal parameters. This finding was attributed to the limited reactivity of the allylic hydrogens of itaconic acid, which restricts its propensity to homo- or co-polymerize.
Besides, standard gravimetric analyses were exploited for assessing the effect of the presence of itaconic acid on the swelling behavior of the synthesized hydrogels in H2O, keeping constant at 10 wt.% the acrylamide content. In particular, the swelling ratio values at equilibrium were found to increase from about 2500 to 4400% with increasing the itaconic acid/acrylic acid weight ratio respectively from 1:39 to 3:37 wt/wt. Beyond this latter ratio, the swelling capability of the hydrogels significantly lowered, because of the more difficult ionization of the polyelectrolyte, which in turn determined a more tightened and denser hydrogel structure, less prone to swell in water.
Quite recently, Martinez-Serrano and co-workers [51] succeeded in obtaining fluorescent hydrogels based on poly(ethylene glycol) methyl ether acrylate or di(ethylene glycol) methyl ether methacrylate and containing 5-(2-(2-(2-(2-phenoxyethoxy)ethoxy) ethoxy)-10,15,20-triphenylporphyrin methacrylate, synthesized on purpose. First of all, the incorporation of the porphyrin units was found to increase the glass transition temperature of the di(ethylene glycol) methyl ether methacrylate-based hydrogels only, from -61°C (for hydrogel not containing the porphyrin methacrylate monomer) to about -15°C. This finding was ascribed to the large volume of the porphyrin units with respect to the di(ethylene glycol) methyl ether methacrylate, which made the final hydrogels stiffer. Further, front velocities showed an increasing trend with increasing the 5-(2-(2-(2-(2-phenoxyethoxy)ethoxy) ethoxy)-10,15,20-triphenylporphyrin methacrylate content (Figure 13). However, beyond 1 and 0.4 wt.% concentration of the porphyrin methacrylate monomer (respectively for the hydrogels based on poly(ethylene glycol) methyl ether acrylate and di(ethylene glycol) methyl ether methacrylate), the steric effects exerted by the fluorescent monomer did not allow for the self-sustainment of the propagating front. Besides, the presence of increasing content of porphyrin units did not substantially affect the front temperatures.
Finally, as displayed in Figure 14 (for poly(ethylene glycol) methyl ether acrylate series) and Figure 15 (for di(ethylene glycol) methyl ether methacrylate series), the prepared hydrogels were both pH- and temperature-dependent. Indeed, both types of hydrogels showed reduced equilibrium swelling ratios with increasing the testing temperatures, as well as with increasing the pH of the water medium. Besides, di(ethylene glycol) methyl ether methacrylate exhibited swelling reversibility during immersion cycles in alternated low and high pH values (Figure 15c), unlike the systems derived from poly(ethylene glycol) methyl ether acrylate (Figure 14c), which showed a reversibility loss.
Very recently, Li and co-workers [52] exploited the frontal polymerization method for preparing pH-responsive and β-cyclodextrins-containing poly(acrylic acid-co-acrylamide) hydrogels, starting from a deep eutectic solvent mixture made of betaine (as hydrogen bond acceptor), acrylic acid, and acrylamide (both as hydrogen bond donors). The acrylamide/acrylic acid/betaine molar ratio was set at 2:2:1; N,N-methylene bisacrylamide (at 1 wt.%), and potassium persulfate (at 0.5 wt.%) were utilized as crosslinker and initiator, respectively. Different β-cyclodextrins loadings were employed, namely 0.25, 0.50, and 1.0 wt.%. In the adopted experimental conditions, less than 6 min was enough to perform the frontal polymerization reactions, though the presence of β-cyclodextrins accounted for a decrease in the polymerization rates, front velocities, and front temperatures. Conversely, both the crosslinking density and the number of H-bonds in the hydrogels increased with increasing the amount of β-cyclodextrins, hence leading to an enhanced overall mechanical behavior. Further, as assessed by drug loading and release tests carried with tetracycline hydrochloride (employed as a drug model), the incorporation of β-cyclodextrins accounted for a gradual increase in the drug loading and a gradual lowering in the drug release, due to the formation of host–guest inclusion complexes.
4.3. Self-healing
The possibility of designing materials able to repair themselves after damage and recover at least their functionality utilizing the resources intrinsically available to them is very intriguing and has significantly motivated both the academia and industrial world during the last 20-25 years [53,54]. In this context, the past decade witnessed the design, preparation, and application of self-healing injectable hydrogels, for which self-healing relies on reversible chemical approaches: these advanced materials are capable of provisionally fluidizing when subjected to sufficient shear stresses and successively regain their original mechanical features [55,56]. In further research efforts, the possibility of preparing frontally polymerized self-healing hydrogels has been thoroughly investigated: the next paragraphs summarize the most recent successful examples.
Yang and co-workers [57] succeeded in obtaining frontally polymerized (in horizontal configuration) bi-layered anisotropic hydrogels made of poly(acrylamide-co-2-acrylamide-2-methylpropanesulfonic acid) and poly(acrylamide-co-hydroxypropyl acrylate). For the former layer, the weight ratio between acrylamide and 2-acrylamide-2-methylpropanesulfonic acid was set at 4:1, whereas a 1:1 acrylamide/hydroxypropyl acrylate weight ratio was chosen for the latter; N,N,N’,N’-tetramethylethylenediamine was employed as crosslinker. As presented in Figure 16, the bi-layered structure was obtained by pouring first the solution of poly(acrylamide-co-hydroxypropyl acrylate) into a horizontal glass tube, and then the solution of poly(acrylamide-co-2-acrylamide-2-methylpropanesulfonic acid) as the second layer. Then, the left side of the glass tube was heated by means of a soldering iron for 20-30 s, to trigger the reaction and start the propagation fronts.
Both front temperatures and velocities were found to decrease with decreasing the weight ratio between the solutions of poly(acrylamide-co-2-acrylamide-2-methylpropanesulfonic acid) and poly(acrylamide-co-hydroxypropyl acrylate): this finding was ascribed to the higher heat loss in the solution poly(acrylamide-co-2-acrylamide-2-methylpropanesulfonic acid) due to its direct exposure to the atmosphere.
All the bi-layered hydrogels showed high hydrophilicity and excellent water absorption capability: among the different investigated weight ratios between the two polymer solutions, the 5:5 one exhibited the highest equilibrium swelling ratio in water (around 3370%) at neutral pH. Besides, as assessed through tensile tests, the cut surfaces of the hydrogels showed self-healing capabilities, without the use of any external stimulus; the self-healed systems were able to withstand robust stretching (Figure 17).
Finally, the bi-layered hydrogels possessed good cytocompatibility and low cytotoxicity, with 78.13% of cell survival, as demonstrated by means of in vitro cell culture tests of L929 fibroblasts.
Liu and co-workers [58] took advantage of an interfacial ignited gelation process for obtaining Ag+/Fe3+-poly(acrylic acid), Ag+/Fe3+-poly(acrylic acid-co-N,N-dimethylacrylamide), and Ag+/Fe3+-poly(acrylic acid-co-acrylamide) self-healing hydrogels. The reaction occurred in water by means of a frontal polymerization process. As presented in Figure 18, it was possible to exploit the interfacial ignited gelation process for repairing the two types of synthesized hydrogel, by simply utilizing the acrylic acid or acrylic acid/acrylamide aqueous solutions as the repairing medium, working at room temperature in the air atmosphere and achieving healing efficiencies beyond 90% even after 5 repeated healing cycles. In particular, high healing efficiencies and as low as 1 min healing times were observed for the hydrogels at high polymer content (i.e., 92 wt.%).
Recently, Li et al. [59] demonstrated the suitability of frontally polymerized deep eutectic monomers for obtaining poly(urea-co-acrylamide-co-choline chloride) hydrogels containing sodium alginate as a filler (from 0.5 to 8.0 wt.% loading). The effect of sodium alginate on the front velocities and temperatures, as well as on the mechanical, swelling, and self-healing features of the resulting hydrogels was thoroughly investigated. More specifically, because of the decrease in the number of acrylamide units per unit volume and therefore in the heat generation per unit time, both front velocities (1.23 vs. 0.66 cm/min for the unfilled hydrogel and the counterpart containing 8 wt.% of filler, respectively) and temperatures (from about 136 to 100°C, respectively) lowered with increasing the sodium alginate content. Conversely, the presence of increasing filler loadings accounted for an increase in the maximum tensile strengths of the hydrogels (from 56.4 to 163.3 kPa for the unfilled hydrogel and the counterpart containing 8 wt.% of filler, respectively). A similar trend was also observed as far as the swelling ratios and self-healing efficiencies are considered: for the latter, a value as high as 94.4% was achieved after 48 h for the hydrogels containing the highest loading of sodium alginate. Replacing sodium alginate with ZnO nanoparticles (in the range 0.4-1.2 wt.%) allowed for providing the obtained hydrogels with interesting antibacterial features toward both E.coli and S.aureus [60]. More specifically, the inhibition rate progressively increased with increasing the nanofiller content, reaching values beyond 80% for the highest loading.
Pursuing this research, the same group [61] exploited a deep eutectic monomer mixture made of different molar ratios of acrylamide, choline chloride, and glycerol, for obtaining multifunctional frontally polymerized hydrogels, using N,N-methylenebisacrylamide and potassium persulfate (both at 0.5 wt.%) as crosslinker and initiator, respectively. Increasing the acrylamide content in the hydrogel formulation accounted for increased front temperatures and velocities. Further, because of the formation of hydrogen bonding interactions taking place between acrylamide and choline chloride/glycerol, the resulting hydrogels showed very good mechanical features, including high stretchability (with around 350% maximum elongation) and twistability (Figure 19), and good self-healing capability. Finally, the presence of increasing amounts of glycerol in the deep eutectic monomer mixture was responsible for an augmented ionic conductivity of the hydrogels (Figure 20), making them potentially suitable as compression sensors.
In a further research effort [62], a deep eutectic monomer mixture consisting of choline chloride, acrylamide, and acrylic acid (at 1:1:1 molar ratio) was employed for synthesizing frontally polymerized composite hydrogels containing β-cyclodextrins (from 0.25 to 2.0 wt.%). N,N-methylene bisacrylamide and potassium persulfate were employed as crosslinker (at 1 wt.% loading) and initiator (0.5 wt.%), respectively. Increasing the content of β-cyclodextrins favored the increase in the H-bond formation, hence improving the overall mechanical behavior of the hydrogels: in particular, compressive and tensile strengths were raised by about 2.26 and 4.12 times with respect to the unfilled counterparts. Besides, the presence of β-cyclodextrins provided the hydrogels with interesting self-healing properties, with an efficiency approaching 92% for the systems containing the highest filler loading after 48 h (Figure 21).
Very recently [63] frontal polymerization was exploited for obtaining highly swellable, stretchable, and self-healing hydrogels through the copolymerization of acrylamide, 3-[Dimethyl-[2-(2-methylprop-2-enoyloxy)ethyl]azaniumyl]propane-1-sulfonate, and acrylic acid. The obtained hydrogels exhibited superabsorbent features and were sensitive to pH: their swelling ratio was as high as 11,802% in water and 13,588% in an alkaline environment. Finally, these hydrogels showed high self-healing capabilities, with a healing efficiency of up to 95% (Figure 22).
4.4. Other applications: electrically conductive and photothermic hydrogels
The versatility and tunability of hydrogels prepared via frontal polymerization are so high that they recently started to be employed for purposes other than those in the biomedical and pharmaceutical sectors. This paragraph will summarize the most recent outcomes.
As far as the electrical conductivity (already mentioned in the previous paragraph as one of the possible features of frontally polymerized multifunctional hydrogels) is considered, Chen and co-workers [64] demonstrated the suitability of starch (containing 25 wt.% of amylose) for enhancing the ionic conductivity of frontally polymerized hydrogels obtained from a deep eutectic monomer mixture made of acrylic acid, acrylamide, and choline chloride (1:1:1 molar ratio), using potassium persulfate and N,N-methylenebisacrylamide as initiator and crosslinker, respectively. A general scheme of the adopted synthetic procedure is shown in Figure 23.
First of all, the presence of increasing starch loadings accounted for a decrease of the front velocities, while its effect on front temperatures was very limited. Further, thanks to the strong H-bond interactions with the deep eutectic monomer system, the biomacromolecule promoted an increase in the tensile and compressive strength of the hydrogels, as well as in the water absorption and the electrical conductivity, as presented in Figure 24.
A similar approach for obtaining electrically conductive hydrogels via frontal polymerization was employed by Li and co-workers [65], who incorporated N-doped carbon nanotubes (from 0.2 to 1.0 wt.% loading) into a deep eutectic monomer mixture consisting of choline chloride, acrylic acid, and acrylamide (at constant 1:1:1 molar ratio). Again, potassium persulfate (at 0.3 wt.% loading) and N,N-methylenebisacrylamide (at 1.0 wt.%) were employed as initiator and crosslinker, respectively. The presence of increasing amounts of the nanofiller accounted for a gradual increase of both front velocities and temperatures, due to the viscosity increase of the reactive medium. Furthermore, because of the strong interfacial connection between the N-doped carbon nanotubes and the hydrogel networks, both tensile and compressive strengths increased, respectively achieving 5.42 and 4.29 MPa, which were about 4.7 and 2.1 times those obtained for the unfilled hydrogel. Finally, the conductivity of the hydrogels embedding 1.0 wt.% of nanofiller was as high as 0.42 mS cm-1, i.e., about 4.2 times higher than that of the unfilled counterpart: a very bright light emission (Figure 25) was observed for this nanocomposite hydrogel when connected to a LED bulb after water absorption.
Liang and co-workers [66] demonstrated the possibility of using solar-initiated frontal polymerization for obtaining poly(acrylamide-co-acrylic acid)/carbon black hydrogels, suitable for photothermic seawater desalination. The filler loading was changed from 0.4 to 2.2 wt.%; the acrylic acid/acrylamide weight ratio was kept constant at 3.7. The performances of the hydrogels obtained via frontal polymerization were compared with the same systems prepared by means of batch polymerization: a general scheme of the adopted strategy is presented in Figure 26. The combination of the peculiar honeycomb-like macroporous structure of the frontally polymerized hydrophilic hydrogels with the hydrophobicity of the carbon black clusters accounted for broadband light absorption and enhanced water transportation. In particular, the evaporation rate was as high as 2.42 kg m-2 h-1, which resulted in 92.8% light-to-vapor efficiency under one sun irradiation. In addition, the solar evaporation flux was significantly boosted thanks to the important swelling behavior of the FP-derived hydrogel evaporator, able to expand to 900 and 2700% of its original surface area and volume, respectively.
5. Conclusions and perspectives
Frontal polymerization was invented in the 1970s [67] and for a couple of decades was considered an interesting example of phenomena related to nonlinear dynamics only [16]. Since the early years of this century, studies related to its feasibility using new monomeric systems, kinetics and mechanisms have been added to, almost to the point of replacing them entirely. By now, FP is a mature technique and, as such, is attracting the interest of an increasing number of research groups around the world. In particular, attention is now focused primarily on its practical applications, of which those involving hydrogels are among the most widely investigated.
As we have pointed out in previous paragraphs, FP allows for obtaining smart hydrogels, among which we like to mention those exhibiting self-healing and stimulus responsive (thermal, pH) characteristics.
However, there are still aspects that limit the possibilities of their use.
In terms of the Technology Readiness Level, the maturity achieved by FP now requires a further significant effort to move from the level of technology validated in relevant environment to subsequent levels that, from prototype demonstration in operational environment, reach its practical use. From the point of view of research developments, it is still necessary to solve the heat dissipation issue. To this aim, catalytic or activating systems need to be developed, which allow the propagation of fronts at relatively low temperatures and are chemically compatible with aqueous media. This will make it possible not only to operate in water, but also to expand the number of exploitable monomers, and consequently polymers. In addition, it will be possible to make polymeric materials having geometries not currently accessible by all the FP systems (e.g., thin films, fibers)
This will also make it possible to focus more on the use of biobased chemicals even if they have relatively high molecular mass.
In addition, more interaction with biological expertises will be required in order to design and prepare biocompatible systems of biomedical interest. It may even be speculated that these materials could be obtainable in situ within living organisms, upon triggering with an appropriate external stimulus.
Last but not least, frontal polymerization appears to be a natural candidate to be implemented in 3D printing techniques used in biomedical applications, in which the generation of the artifact occurs as a result of layer-by-layer deposition of monomers: FP could be exploited for continuously polymerizing the deposited layers.
Author Contributions
For research articles with several authors, a short paragraph specifying their individual contributions must be provided. The following statements should be used “Conceptualization, A.M.; data curation, A.M and G.M.; writing—original draft preparation, A.M. and G.M.; writing—review and editing, A.M. and G.M.; supervision, A.M. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by FAR 2020, 2021, 2022 funds provided by the University of Sassari.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wichterle, O.; Lím, D. Hydrophilic Gels for Biological Use. Nature 1960, 185, 117–118. [Google Scholar] [CrossRef]
- Correa, S.; Grosskopf, A. K.; Lopez Hernandez, H.; Chan, D.; Yu, A. C.; Stapleton, L. M.; Appel, E. A. Translational Applications of Hydrogels. Chem. Rev. 2021, 121, 11385–11457. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Inda, M. E.; Lai, Y.; Lu, T. K.; Zhao, X. Engineered Living Hydrogels. Adv. Mater. 2022, 34. [Google Scholar] [CrossRef] [PubMed]
- Bashir, S.; Hina, M.; Iqbal, J.; Rajpar, A. H.; Mujtaba, M. A.; Alghamdi, N. A.; Wageh, S.; Ramesh, K.; Ramesh, S. Fundamental Concepts of Hydrogels: Synthesis, Properties, and Their Applications. Polymers 2020, 12. [Google Scholar] [CrossRef]
- Wang, Z.; Li, H.; Tang, Z.; Liu, Z.; Ruan, Z.; Ma, L.; Yang, Q.; Wang, D.; Zhi, C. Hydrogel Electrolytes for Flexible Aqueous Energy Storage Devices. Adv. Funct. Mater. 2018, 28. [Google Scholar] [CrossRef]
- Chan, C. Y.; Wang, Z.; Jia, H.; Ng, P. F.; Chow, L.; Fei, B. Recent Advances of Hydrogel Electrolytes in Flexible Energy Storage Devices. J. Mater. Chem. A 2021, 9, 2043–2069. [Google Scholar] [CrossRef]
- Davtyan, S. P.; Tonoyan, A. O. The Frontal Polymerization Method in High Technology Applications. Rev. J. Chem. 2019, 9, 71–94. [Google Scholar] [CrossRef]
- Li, Q.; Shen, H.-X.; Liu, C.; Wang, C.-F.; Zhu, L.; Chen, S. Advances in Frontal Polymerization Strategy: From Fundamentals to Applications. Prog. Polym. Sci. 2022, 127, 101514. [Google Scholar] [CrossRef]
- Suslick, B. A.; Hemmer, J.; Groce, B. R.; Stawiasz, K. J.; Geubelle, P. H.; Malucelli, G.; Mariani, A.; Moore, J. S.; Pojman, J. A.; Sottos, N. R. Frontal Polymerizations: From Chemical Perspectives to Macroscopic Properties and Applications. Chem. Rev. 2023, 123, 3237–3298. [Google Scholar] [CrossRef]
- Mariani, A.; Fiori, S.; Bidali, S.; Alzari, V.; Malucelli, G. Frontal Polymerization of Diurethane Diacrylates. J. Polym. Sci. Part Polym. Chem. 2008, 46, 3344–3352. [Google Scholar] [CrossRef]
- Illescas, J.; Ortíz-Palacios, J.; Esquivel-Guzmán, J.; Ramirez-Fuentes, Y. S.; Rivera, E.; Morales-Saavedra, O. G.; Rodríguez-Rosales, A. A.; Alzari, V.; Nuvoli, D.; Scognamillo, S.; Mariani, A. Preparation and Optical Characterization of Two Photoactive Poly(Bisphenol a Ethoxylate Diacrylate) Copolymers Containing Designed Amino-Nitro-Substituted Azobenzene Units, Obtained via Classical and Frontal Polymerization, Using Novel Ionic Liquids as In. J. Polym. Sci. Part Polym. Chem. 2012, 50, 1906–1916. [Google Scholar] [CrossRef]
- Zhong, D.-D.; Liu, X.; Pang, Q.-Q.; Huang, Y.-P.; Liu, Z.-S. Rapid Preparation of Molecularly Imprinted Polymer by Frontal Polymerization. Anal. Bioanal. Chem. 2013, 405, 3205–3214. [Google Scholar] [CrossRef] [PubMed]
- Holt, T.; Fazende, K.; Jee, E.; Wu, Q.; Pojman, J. A. Cure-on-demand Wood Adhesive Based on the Frontal Polymerization of Acrylates. J. Appl. Polym. Sci. 2016, 133, app.44064. [Google Scholar] [CrossRef]
- Robertson, I. D.; Yourdkhani, M.; Centellas, P. J.; Aw, J. E.; Ivanoff, D. G.; Goli, E.; Lloyd, E. M.; Dean, L. M.; Sottos, N. R.; Geubelle, P. H.; Moore, J. S.; White, S. R. Rapid Energy-Efficient Manufacturing of Polymers and Composites via Frontal Polymerization. Nature 2018, 557, 223–227. [Google Scholar] [CrossRef]
- Lewis, L. L.; Massey, K. N.; Meyer, E. R.; McPherson, J. R.; Hanna, J. S. New Insight into Isothermal Frontal Polymerization Models: Wiener’s Method to Determine the Diffusion Coefficients for High Molecular-Weight Poly(Methyl Methacrylate) with Neat Methyl Methacrylate. Opt. Lasers Eng. 2008, 46, 900–910. [Google Scholar] [CrossRef]
- Pojman, J. A. Frontal Polymerization. In Polymer Science: A Comprehensive Reference; Elsevier, 2012; pp. 957–980. [Google Scholar] [CrossRef]
- Pojman, J. A.; Ilyashenko, V. M.; Khan, A. M. Free-Radical Frontal Polymerization: Self-Propagating Thermal Reaction Waves. J. Chem. Soc. Faraday Trans. 1996, 92. [Google Scholar] [CrossRef]
- Bowden, G.; Garbey, M.; Ilyashenko, V. M.; Pojman, J. A.; Solovyov, S. E.; Taik, A.; Volpert, V. A. Effect of Convection on a Propagating Front with a Solid Product: Comparison of Theory and Experiments. J. Phys. Chem. B 1997, 101, 678–686. [Google Scholar] [CrossRef]
- Pojman, J. A.; Craven, R.; Khan, A.; West, W. Convective Instabilities in Traveling Fronts of Addition Polymerization. J. Phys. Chem. 1992, 96, 7466–7472. [Google Scholar] [CrossRef]
- Mariani, A.; Nuvoli, D.; Alzari, V.; Pini, M. Phosphonium-Based Ionic Liquids as a New Class of Radical Initiators and Their Use in Gas-Free Frontal Polymerization. Macromolecules 2008, 41, 5191–5196. [Google Scholar] [CrossRef]
- Fiori, S.; Mariani, A.; Ricco, L.; Russo, S. First Synthesis of a Polyurethane by Frontal Polymerization. Macromolecules 2003, 36, 2674–2679. [Google Scholar] [CrossRef]
- Mariani, A.; Bidali, S.; Fiori, S.; Malucelli, G.; Sanna, E. Synthesis and Characterization of a Polyurethane Prepared by Frontal Polymerization. E-Polym. 2003, 3. [Google Scholar] [CrossRef]
- Scognamillo, S.; Gioffredi, E.; Piccinini, M.; Lazzari, M.; Alzari, V.; Nuvoli, D.; Sanna, R.; Piga, D.; Malucelli, G.; Mariani, A. Synthesis and Characterization of Nanocomposites of Thermoplastic Polyurethane with Both Graphene and Graphene Nanoribbon Fillers. Polymer 2012, 53, 4019–4024. [Google Scholar] [CrossRef]
- Mariani, A.; Bidali, S.; Fiori, S.; Sangermano, M.; Malucelli, G.; Bongiovanni, R.; Priola, A. UV-Ignited Frontal Polymerization of an Epoxy Resin. J. Polym. Sci. Part Polym. Chem. 2004, 42, 2066–2072. [Google Scholar] [CrossRef]
- Mariani, A.; Bidali, S.; Caria, G.; Monticelli, O.; Russo, S.; Kenny, J. M. Synthesis and Characterization of Epoxy Resin-Montmorillonite Nanocomposites Obtained by Frontal Polymerization. J. Polym. Sci. Part Polym. Chem. 2007, 45, 2204–2211. [Google Scholar] [CrossRef]
- Scognamillo, S.; Bounds, C.; Thakuri, S.; Mariani, A.; Wu, Q.; Pojman, J. A. Frontal Cationic Curing of Epoxy Resins in the Presence of Defoaming or Expanding Compounds. J. Appl. Polym. Sci. 2014, 131, n/a–n/a. [Google Scholar] [CrossRef]
- Mariani, A.; Fiori, S.; Chekanov, Y.; Pojman, J. A. Frontal Ring-Opening Metathesis Polymerization of Dicyclopentadiene. Macromolecules 2001, 34, 6539–6541. [Google Scholar] [CrossRef]
- Ruiu, A.; Sanna, D.; Alzari, V.; Nuvoli, D.; Mariani, A. Advances in the Frontal Ring Opening Metathesis Polymerization of Dicyclopentadiene. J. Polym. Sci. Part Polym. Chem. 2014, 52, 2776–2780. [Google Scholar] [CrossRef]
- Masere, J.; Chekanov, Y.; Warren, J. R.; Stewart, F. D.; Al-Kaysi, R.; Rasmussen, J. K.; Pojman, J. A. Gas-Free Initiators for High-Temperature Free-Radical Polymerization. J. Polym. Sci. Part Polym. Chem. 2000, 38, 3984–3990. [Google Scholar] [CrossRef]
- Alzari, V.; Nuvoli, D.; Sanna, D.; Ruiu, A.; Mariani, A. Effect of Limonene on the Frontal Ring Opening Metathesis Polymerization of Dicyclopentadiene. J. Polym. Sci. Part Polym. Chem. 2016, 54, 63–68. [Google Scholar] [CrossRef]
- Alzari, V.; Nuvoli, D.; Scognamillo, S.; Piccinini, M.; Gioffredi, E.; Malucelli, G.; Marceddu, S.; Sechi, M.; Sanna, V.; Mariani, A. Graphene-Containing Thermoresponsive Nanocomposite Hydrogels of Poly(N-Isopropylacrylamide) Prepared by Frontal Polymerization. J. Mater. Chem. 2011, 21. [Google Scholar] [CrossRef]
- Alzari, V.; Nuvoli, D.; Sanna, R.; Scognamillo, S.; Piccinini, M.; Kenny, J. M.; Malucelli, G.; Mariani, A. In Situ Production of High Filler Content Graphene-Based Polymer Nanocomposites by Reactive Processing. J. Mater. Chem. 2011, 21. [Google Scholar] [CrossRef]
- Bynum, S.; Tullier, M.; Morejon-Garcia, C.; Guidry, J.; Runnoe, E.; Pojman, J. A. The Effect of Acrylate Functionality on Frontal Polymerization Velocity and Temperature. J. Polym. Sci. Part Polym. Chem. 2019, 57, 982–988. [Google Scholar] [CrossRef]
- Damonte, G.; Cozzani, M.; Di Lisa, D.; Pastorino, L.; Mariani, A.; Monticelli, O. Mechanically-Reinforced Biocompatible Hydrogels Based on Poly(N-Isopropylacrylamide) and Star-Shaped Polycaprolactones. Eur. Polym. J. 2023, 195, 112239. [Google Scholar] [CrossRef]
- Damonte, G.; Maddalena, L.; Fina, A.; Cavallo, D.; Müller, A. J.; Caputo, M. R.; Mariani, A.; Monticelli, O. On Novel Hydrogels Based on Poly(2-Hydroxyethyl Acrylate) and Polycaprolactone with Improved Mechanical Properties Prepared by Frontal Polymerization. Eur. Polym. J. 2022, 171, 111226. [Google Scholar] [CrossRef]
- Nuvoli, L.; Sanna, D.; Alzari, V.; Nuvoli, D.; Sanna, V.; Malfatti, L.; Mariani, A. Double Responsive Copolymer Hydrogels Prepared by Frontal Polymerization. J. Polym. Sci. Part Polym. Chem. 2016, 54, 2166–2170. [Google Scholar] [CrossRef]
- Nuvoli, D.; Alzari, V.; Nuvoli, L.; Rassu, M.; Sanna, D.; Mariani, A. Synthesis and Characterization of Poly(2-Hydroxyethylacrylate)/β-Cyclodextrin Hydrogels Obtained by Frontal Polymerization. Carbohydr. Polym. 2016, 150, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Sanna, D.; Alzari, V.; Nuvoli, D.; Nuvoli, L.; Rassu, M.; Sanna, V.; Mariani, A. β-Cyclodextrin-Based Supramolecular Poly( N -Isopropylacrylamide) Hydrogels Prepared by Frontal Polymerization. Carbohydr. Polym. 2017, 166, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Chen, X.; Zhao, Y.; Hu, S.; Xia, Z.; Yan, Q.-Z. Preparation of Poly(N-Isopropylacrylamide)/Montmorillonite Composite Hydrogel by Frontal Polymerization. Colloid Polym. Sci. 2017, 295, 883–890. [Google Scholar] [CrossRef]
- Rassu, M.; Alzari, V.; Nuvoli, D.; Nuvoli, L.; Sanna, D.; Sanna, V.; Malucelli, G.; Mariani, A. Semi-Interpenetrating Polymer Networks of Methyl Cellulose and Polyacrylamide Prepared by Frontal Polymerization. J. Polym. Sci. Part Polym. Chem. 2017, 55, 1268–1274. [Google Scholar] [CrossRef]
- Smith, E. L.; Abbott, A. P.; Ryder, K. S. Deep Eutectic Solvents (DESs) and Their Applications. Chem. Rev. 2014, 114, 11060–11082. [Google Scholar] [CrossRef]
- Clarke, C. J.; Tu, W.-C.; Levers, O.; Bröhl, A.; Hallett, J. P. Green and Sustainable Solvents in Chemical Processes. Chem. Rev. 2018, 118, 747–800. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Li, S.; Chen, Y.; Yan, S.; Tao, M.; Wen, P. Facile and Green Preparation of Superfast Responsive Macroporous Polyacrylamide Hydrogels by Frontal Polymerization of Polymerizable Deep Eutectic Monomers. Ind. Eng. Chem. Res. 2020, 59, 1526–1533. [Google Scholar] [CrossRef]
- Gavini, E.; Mariani, A.; Rassu, G.; Bidali, S.; Spada, G.; Bonferoni, M. C.; Giunchedi, P. Frontal Polymerization as a New Method for Developing Drug Controlled Release Systems (DCRS) Based on Polyacrylamide. Eur. Polym. J. 2009, 45, 690–699. [Google Scholar] [CrossRef]
- Wang, X.-L.; Yao, H.-F.; Li, X.-Y.; Wang, X.; Huang, Y.-P.; Liu, Z.-S. PH/Temperature-Sensitive Hydrogel-Based Molecularly Imprinted Polymers (HydroMIPs) for Drug Delivery by Frontal Polymerization. RSC Adv. 2016, 6, 94038–94047. [Google Scholar] [CrossRef]
- Feng, Q.; Zhao, Y.; Li, H.; Zhang, Y.; Xia, X.; Yan, Q. Frontal Polymerization and Characterization of Interpenetrating Polymer Networks Composed of Poly(N-Isopropylacrylamide) and Polyvinylpyrrolidone. Colloid Polym. Sci. 2018, 296, 165–172. [Google Scholar] [CrossRef]
- Mariani, A.; Nuvoli, L.; Sanna, D.; Alzari, V.; Nuvoli, D.; Rassu, M.; Malucelli, G. Semi-Interpenetrating Polymer Networks Based on Crosslinked Poly( N -Isopropyl Acrylamide) and Methylcellulose Prepared by Frontal Polymerization. J. Polym. Sci. Part Polym. Chem. 2018, 56, 437–443. [Google Scholar] [CrossRef]
- Su, J.; Yang, Y.; Chen, Z.; Zhou, J.; Liu, X.; Fang, Y.; Cui, Y. Preparation and Performance of Thermosensitive Poly( N -isopropylacrylamide) Hydrogels by Frontal Photopolymerization. Polym. Int. 2019, 68, 1673–1680. [Google Scholar] [CrossRef]
- Fortenberry, D. I.; Pojman, J. A. Solvent-Free Synthesis of Polyacrylamide by Frontal Polymerization. J. Polym. Sci. Part Polym. Chem. 2000, 38, 1129–1135. [Google Scholar] [CrossRef]
- Irfan, M.; Du, X.; Xu, X. R.; Shen, R. Q.; Chen, S.; Xiao, J. J. Synthesis and Characterization of PH-sensitive Poly(IA- Co -AAc- Co -AAm) Hydrogels via Frontal Polymerization. J. Polym. Sci. Part Polym. Chem. 2019, 57, 2214–2221. [Google Scholar] [CrossRef]
- Martínez-Serrano, R. D.; Ugone, V.; Porcu, P.; Vonlanthen, M.; Sorroza-Martínez, K.; Cuétara-Guadarrama, F.; Illescas, J.; Zhu, X.-X.; Rivera, E. Novel Porphyrin-Containing Hydrogels Obtained by Frontal Polymerization: Synthesis, Characterization and Optical Properties. Polymer 2022, 247, 124785. [Google Scholar] [CrossRef]
- Li, B.; Qin, H.; Ma, M.; Xu, X.; Zhou, M.; Hao, W.; Hu, Z. Preparation of Novel β-CD/P(AA- Co -AM) Hydrogels by Frontal Polymerization. RSC Adv. 2023, 13, 5667–5673. [Google Scholar] [CrossRef] [PubMed]
- Blaiszik, B. J.; Kramer, S. L. B.; Olugebefola, S. C.; Moore, J. S.; Sottos, N. R.; White, S. R. Self-Healing Polymers and Composites. Annu. Rev. Mater. Res. 2010, 40, 179–211. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, L.; Han, Z.; Li, Q.; He, J.; Wang, Q. Self-Healing Polymers for Electronics and Energy Devices. Chem. Rev. 2023, 123, 558–612. [Google Scholar] [CrossRef] [PubMed]
- Bertsch, P.; Diba, M.; Mooney, D. J.; Leeuwenburgh, S. C. G. Self-Healing Injectable Hydrogels for Tissue Regeneration. Chem. Rev. 2023, 123, 834–873. [Google Scholar] [CrossRef] [PubMed]
- Talebian, S.; Mehrali, M.; Taebnia, N.; Pennisi, C. P.; Kadumudi, F. B.; Foroughi, J.; Hasany, M.; Nikkhah, M.; Akbari, M.; Orive, G.; Dolatshahi-Pirouz, A. Self-Healing Hydrogels: The Next Paradigm Shift in Tissue Engineering? Adv. Sci. 2019, 6. [Google Scholar] [CrossRef]
- Yang, Y.; Hu, J.; Liu, J.; Qin, Y.; Mao, J.; Liang, Y.; Wang, G.; Shen, H.; Wang, C.; Chen, S. Rapid Synthesis of Biocompatible Bilayer Hydrogels via Frontal Polymerization. J. Polym. Sci. 2022, 60, 2784–2793. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, L.; Jiang, J.; Wang, X.; Dai, C.; Weng, G. Fast Healing of Covalently Cross-Linked Polymeric Hydrogels by Interfacially Ignited Fast Gelation. Macromolecules 2023, 56, 49–58. [Google Scholar] [CrossRef]
- Li, B.; Wu, A.; Hao, W.; Liu, J.; Hu, Z.; Wang, Y. Preparation of SA/P(U-AM-ChCl) Composite Hydrogels by Frontal Polymerization and Its Performance Study. RSC Adv. 2023, 13, 11530–11536. [Google Scholar] [CrossRef]
- Li, B.; Zhou, M.; Cheng, M.; Liu, J.; Xu, X.; Xie, X. Rapid Preparation of ZnO Nanocomposite Hydrogels by Frontal Polymerization of a Ternary DES and Performance Study. RSC Adv. 2022, 12, 12871–12877. [Google Scholar] [CrossRef]
- Liu, J.; Li, B.; Wu, A.; Qin, H.; Xu, X.; Yan, S. Rapid Preparation of Liquid-free, Antifreeze, Stretchable, and Ion-conductive Eutectic Gels with Good Compression Resistance and Self-healing Properties by Frontal Polymerization. J. Appl. Polym. Sci. 2023, 140, e54231. [Google Scholar] [CrossRef]
- Li, B.; Zhou, M.; Xu, X.; Liu, J.; Hao, W.; Wu, A. Preparation and Properties of β-CD /P( AM- co -AA ) Composite Hydrogel by Frontal Polymerization of Ternary Deep Eutectic Solvent. Polym. Int. 2023, 72, 664–670. [Google Scholar] [CrossRef]
- Qin, Y.; Li, H.; Shen, H.-X.; Wang, C.-F.; Chen, S. Rapid Preparation of Superabsorbent Self-Healing Hydrogels by Frontal Polymerization. Gels 2023, 9. [Google Scholar] [CrossRef]
- Chen, Y.; Li, S.; Yan, S. Starch as a Reinforcement Agent for Poly(Ionic Liquid) Hydrogels from Deep Eutectic Solvent via Frontal Polymerization. Carbohydr. Polym. 2021, 263, 117996. [Google Scholar] [CrossRef]
- Li, B.; Xu, X.; Hu, Z.; Li, Y.; Zhou, M.; Liu, J.; Jiang, Y.; Wang, P. Rapid Preparation of N-CNTs/P(AA- Co -AM) Composite Hydrogel via Frontal Polymerization and Its Mechanical and Conductive Properties. RSC Adv. 2022, 12, 19022–19028. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Bai, Y.; Xie, A.-Q.; Mao, J.; Zhu, L.; Chen, S. Solar-Initiated Frontal Polymerization of Photothermic Hydrogels with High Swelling Properties for Efficient Water Evaporation. Sol. RRL 2022, 6. [Google Scholar] [CrossRef]
- Chechilo, N. M.; Khvilivitskii, R. J.; Enikolopyan, N. S. On the Phenomenon of Polymerization Reaction Spreading. Dokl. Akad. Nauk 1972, 204, 1180–1181. [Google Scholar]
Figure 1.
Schematic classification of polymeric hydrogels. Reprinted from ref. [4] under Creative Commons CC-BY 4.0 License.
Figure 1.
Schematic classification of polymeric hydrogels. Reprinted from ref. [4] under Creative Commons CC-BY 4.0 License.
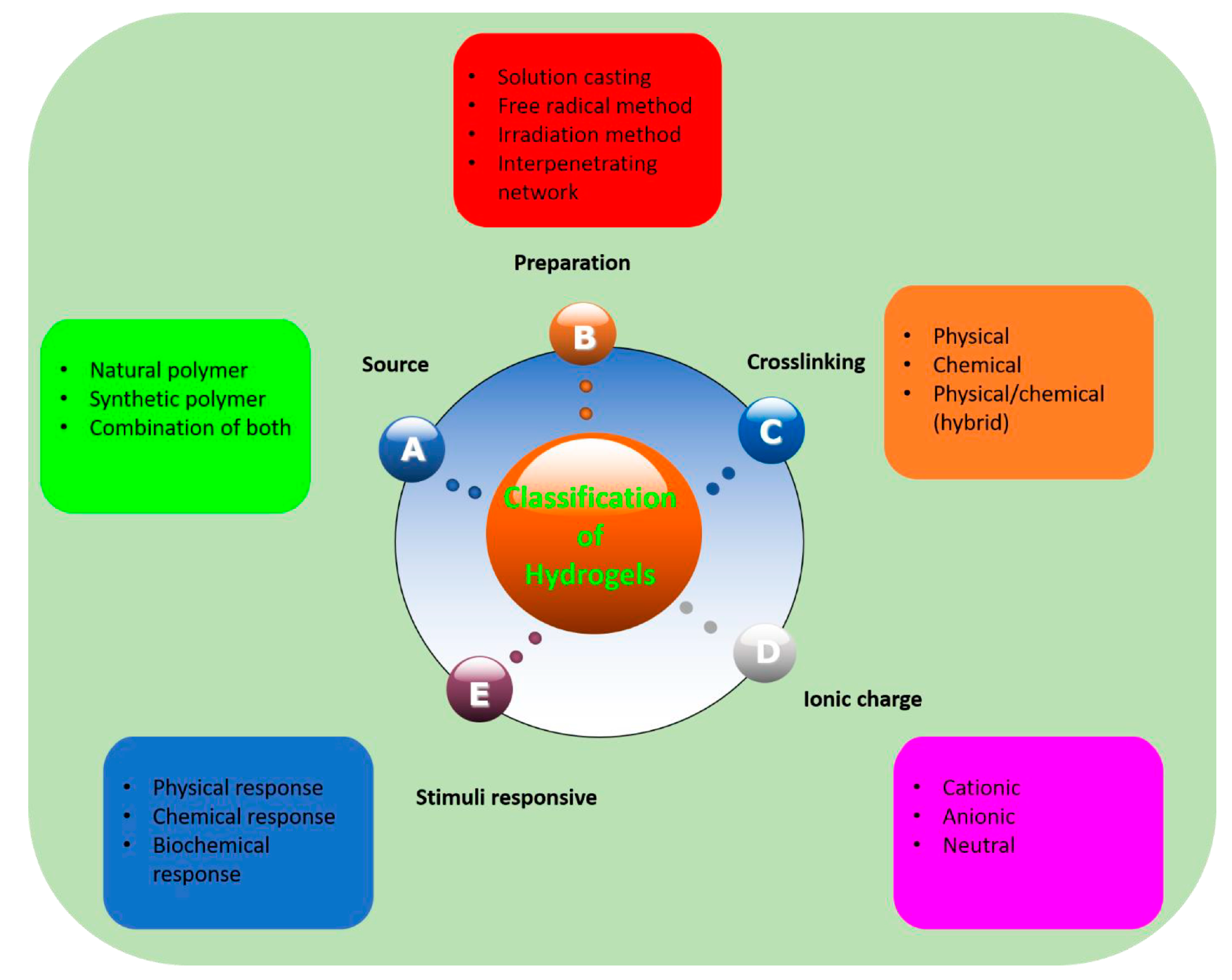
Figure 2.
Number of publications (from 2014 to 2023) in peer-reviewed journals, dealing with the hydrogels obtained through frontal polymerization (data collected from the Web of ScienceTM database, accessed on June 25, 2023).
Figure 2.
Number of publications (from 2014 to 2023) in peer-reviewed journals, dealing with the hydrogels obtained through frontal polymerization (data collected from the Web of ScienceTM database, accessed on June 25, 2023).
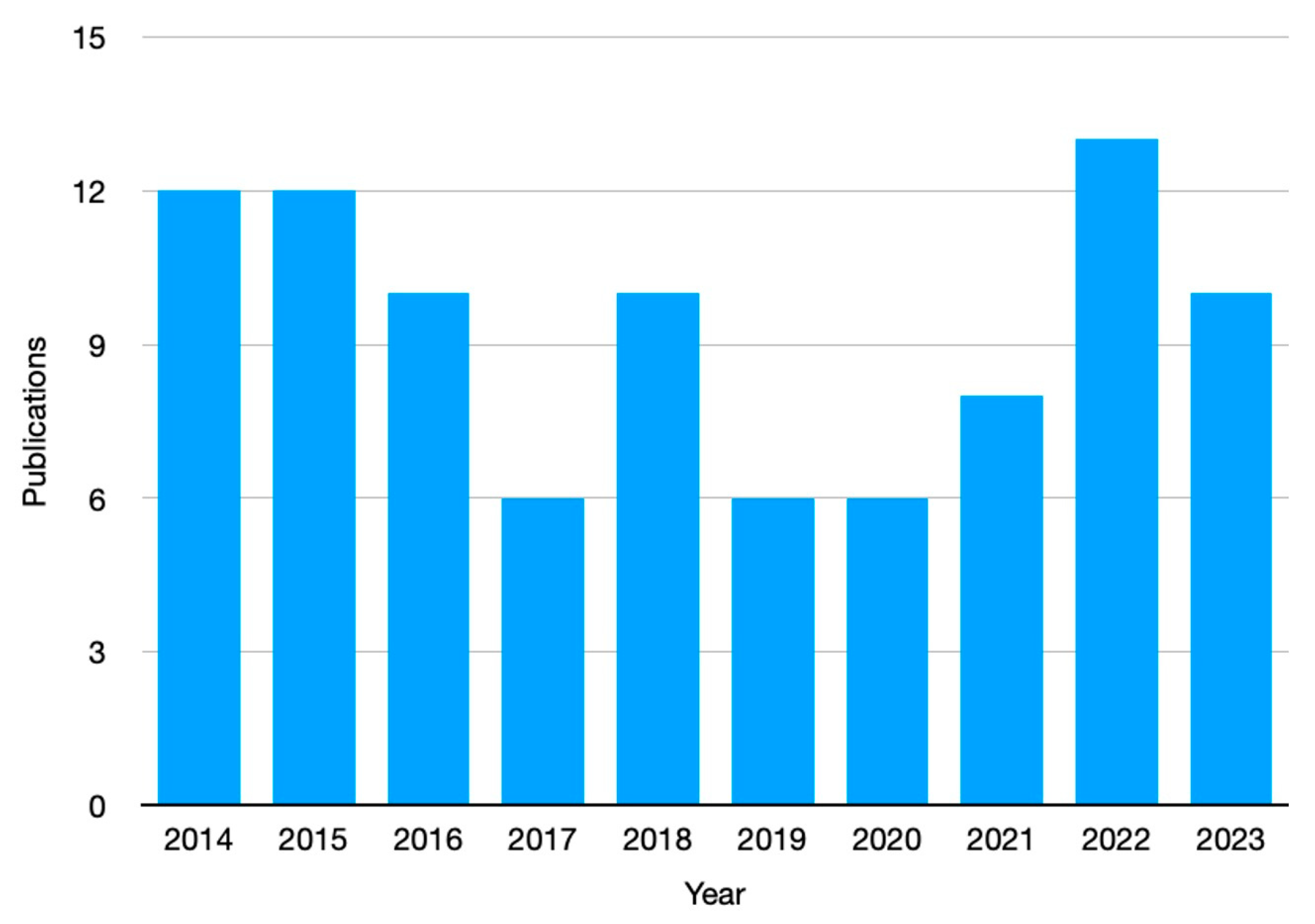
Figure 3.
(A) Thermal frontal polymerization and (B) isothermal frontal polymerization. (Reprinted with permission from Ref. [15]. Copyright Elsevier, 2008).
Figure 3.
(A) Thermal frontal polymerization and (B) isothermal frontal polymerization. (Reprinted with permission from Ref. [15]. Copyright Elsevier, 2008).
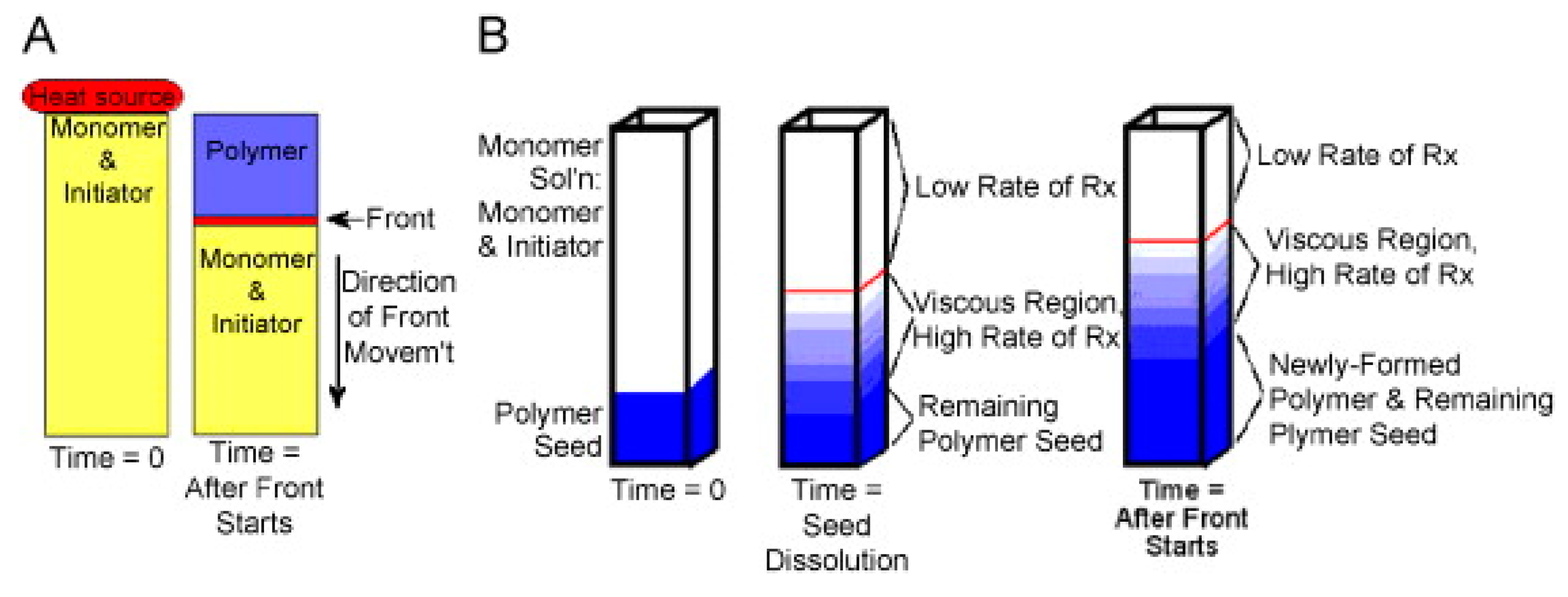
Figure 4.
Some types and applications of frontal polymerization. Reprinted with permission from Ref. [8]. Copyright Elsevier, 2022.
Figure 4.
Some types and applications of frontal polymerization. Reprinted with permission from Ref. [8]. Copyright Elsevier, 2022.
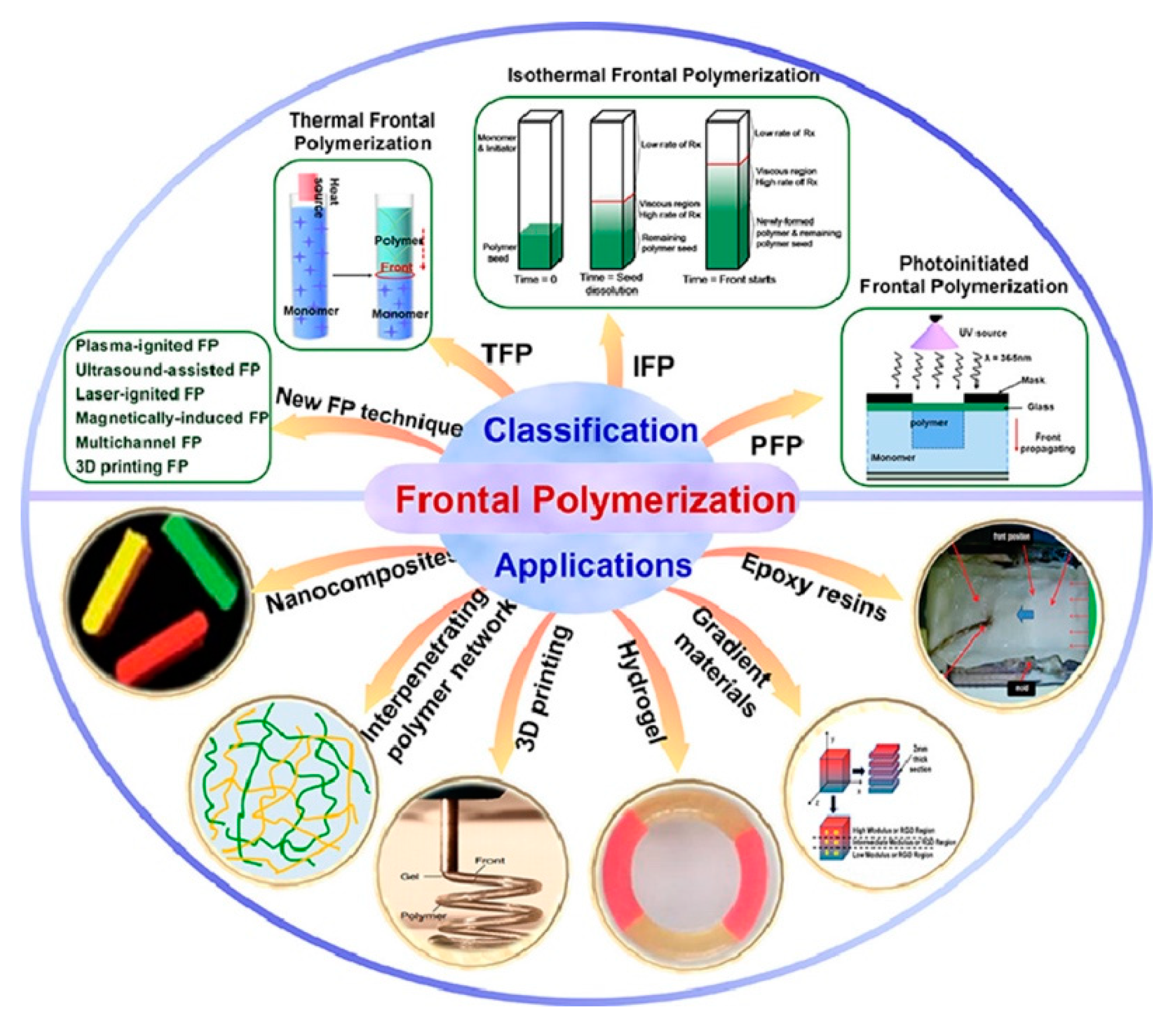
Figure 5.
Examples of descending (left) and ascending (right) fronts. (Reprinted with permission from Ref. [18]. Copyright American Chemical Society, 1997.
Figure 5.
Examples of descending (left) and ascending (right) fronts. (Reprinted with permission from Ref. [18]. Copyright American Chemical Society, 1997.
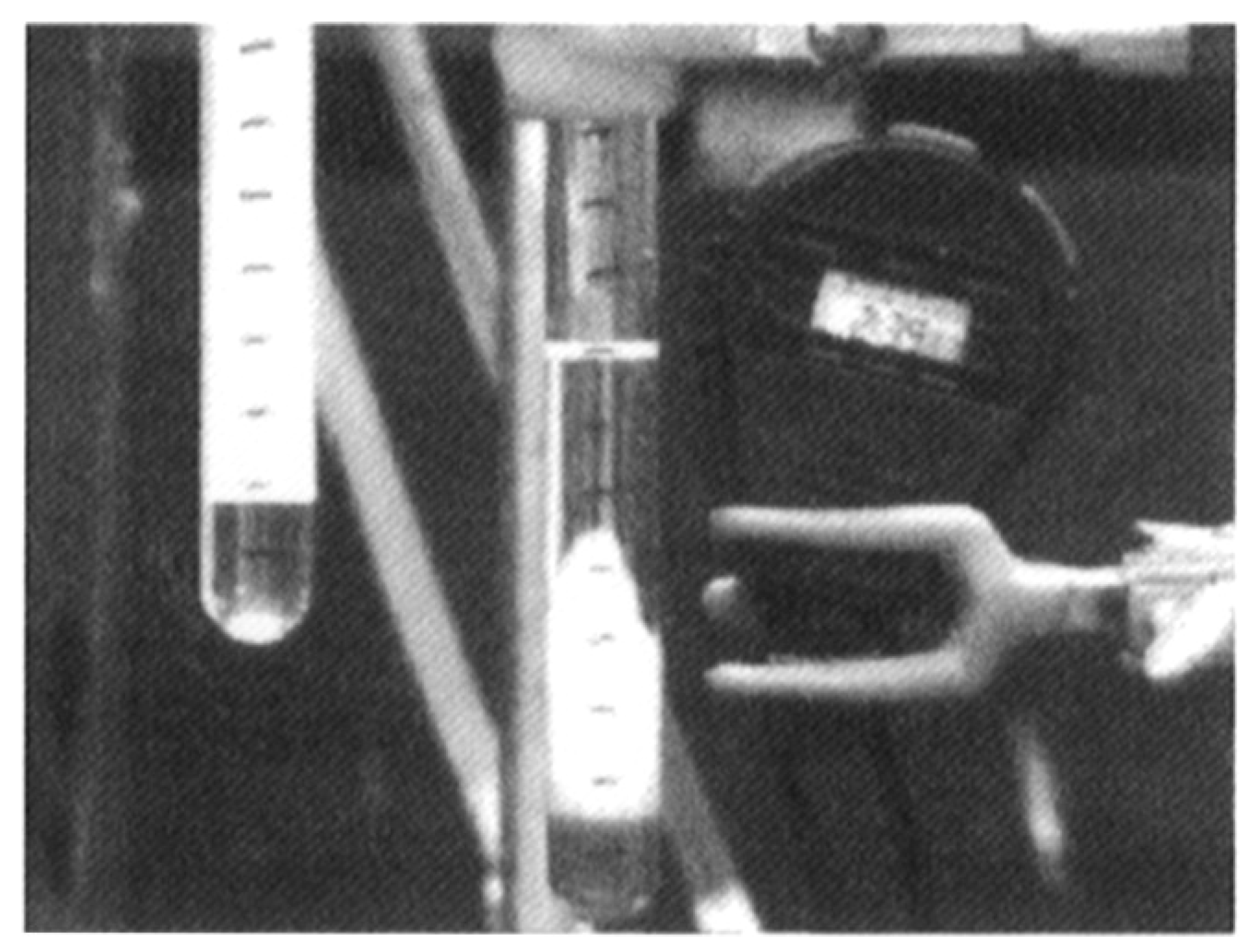
Figure 6.
Fingering observed during the frontal polymerization of methacrylic acid. Reprinted with permission from Ref. [19]. Copyright American Chemical Society, 1992.
Figure 6.
Fingering observed during the frontal polymerization of methacrylic acid. Reprinted with permission from Ref. [19]. Copyright American Chemical Society, 1992.
Figure 7.
Front temperatures Tmax (a) and front velocities Vf (b) as a function of the molar amount of β-cyclodextrin (βCD, white symbols) and acryloyl-β-cyclodextrin (AβCD, black symbols). Reprinted with permission from [37]. Copyright Elsevier, 2016.
Figure 7.
Front temperatures Tmax (a) and front velocities Vf (b) as a function of the molar amount of β-cyclodextrin (βCD, white symbols) and acryloyl-β-cyclodextrin (AβCD, black symbols). Reprinted with permission from [37]. Copyright Elsevier, 2016.
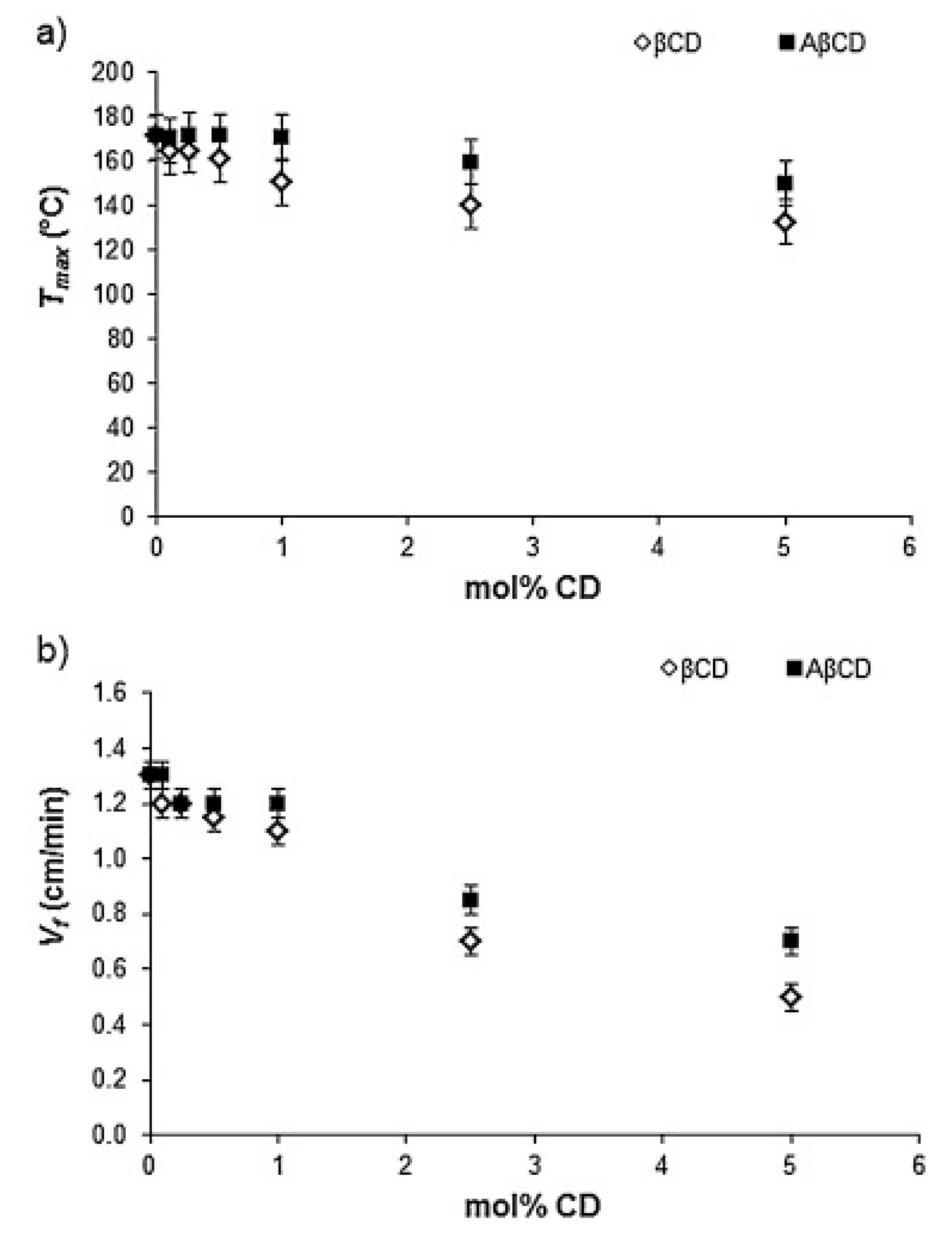
Figure 8.
Supramolecular crosslinking in poly(N-isopropylacrylamide) hydrogels due to both mutual β-cyclodextrin interaction (A) and host-guest (i.e., inclusion complex) formation (B) with isopropylamide groups. Legend: PNIPAAM= poly(N-isopropylacrylamide); AβCD= acryloyl-β-cyclodextrin. Reprinted with permission from [38]. Copyright Elsevier, 2017.
Figure 8.
Supramolecular crosslinking in poly(N-isopropylacrylamide) hydrogels due to both mutual β-cyclodextrin interaction (A) and host-guest (i.e., inclusion complex) formation (B) with isopropylamide groups. Legend: PNIPAAM= poly(N-isopropylacrylamide); AβCD= acryloyl-β-cyclodextrin. Reprinted with permission from [38]. Copyright Elsevier, 2017.
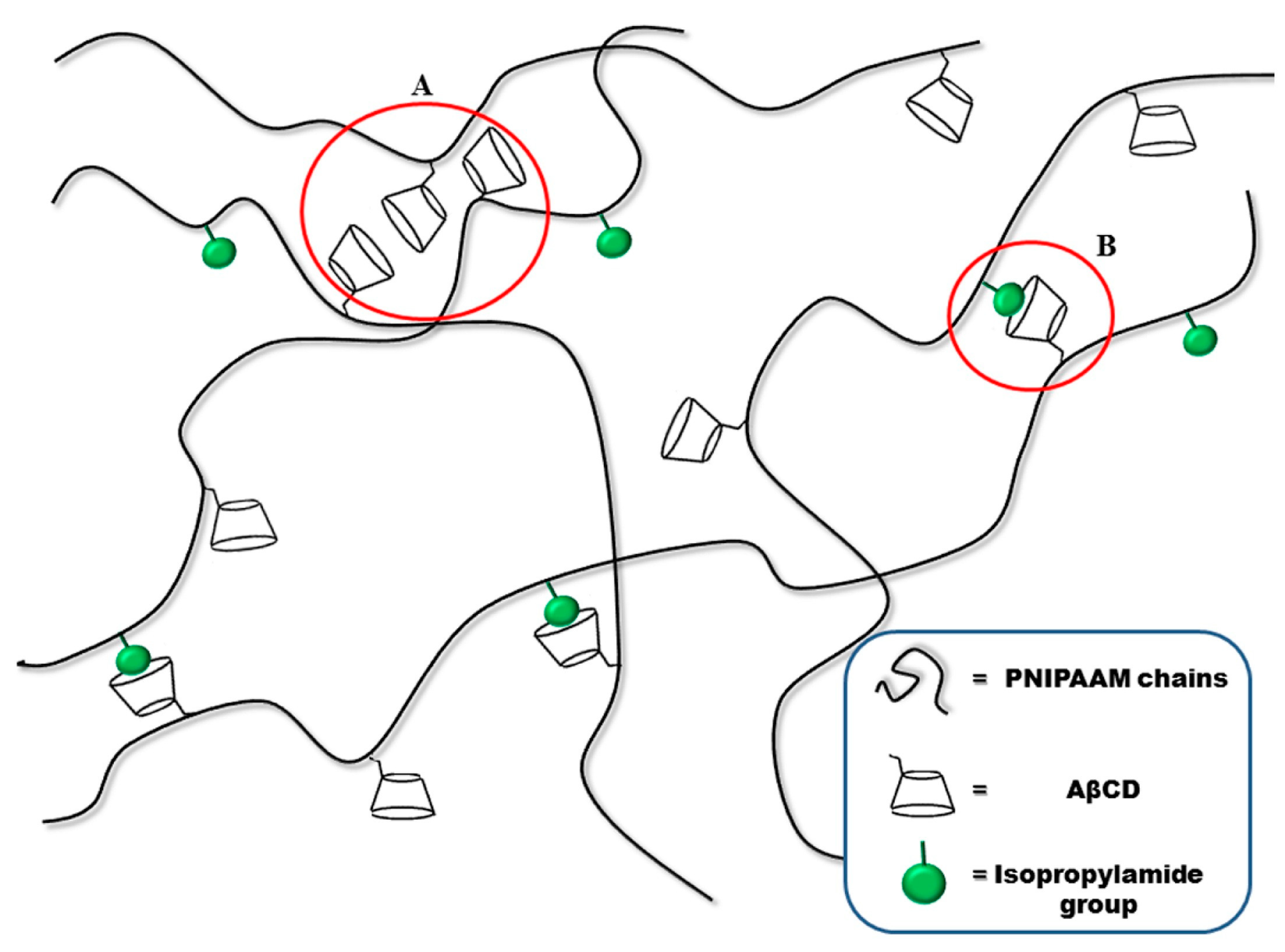
Figure 9.
Scheme of the synthesis of macroporous polyacrylamide hydrogels by FP of deep eutectic monomers. Legend: AM= acrylamide; ChCl= choline chloride; DEM= deep eutectic monomer; FP= frontal polymerization; KPS= potassium persulfate; RT= room temperature. Reprinted with permission from [43]. Copyright American Chemical Society, 2020.
Figure 9.
Scheme of the synthesis of macroporous polyacrylamide hydrogels by FP of deep eutectic monomers. Legend: AM= acrylamide; ChCl= choline chloride; DEM= deep eutectic monomer; FP= frontal polymerization; KPS= potassium persulfate; RT= room temperature. Reprinted with permission from [43]. Copyright American Chemical Society, 2020.
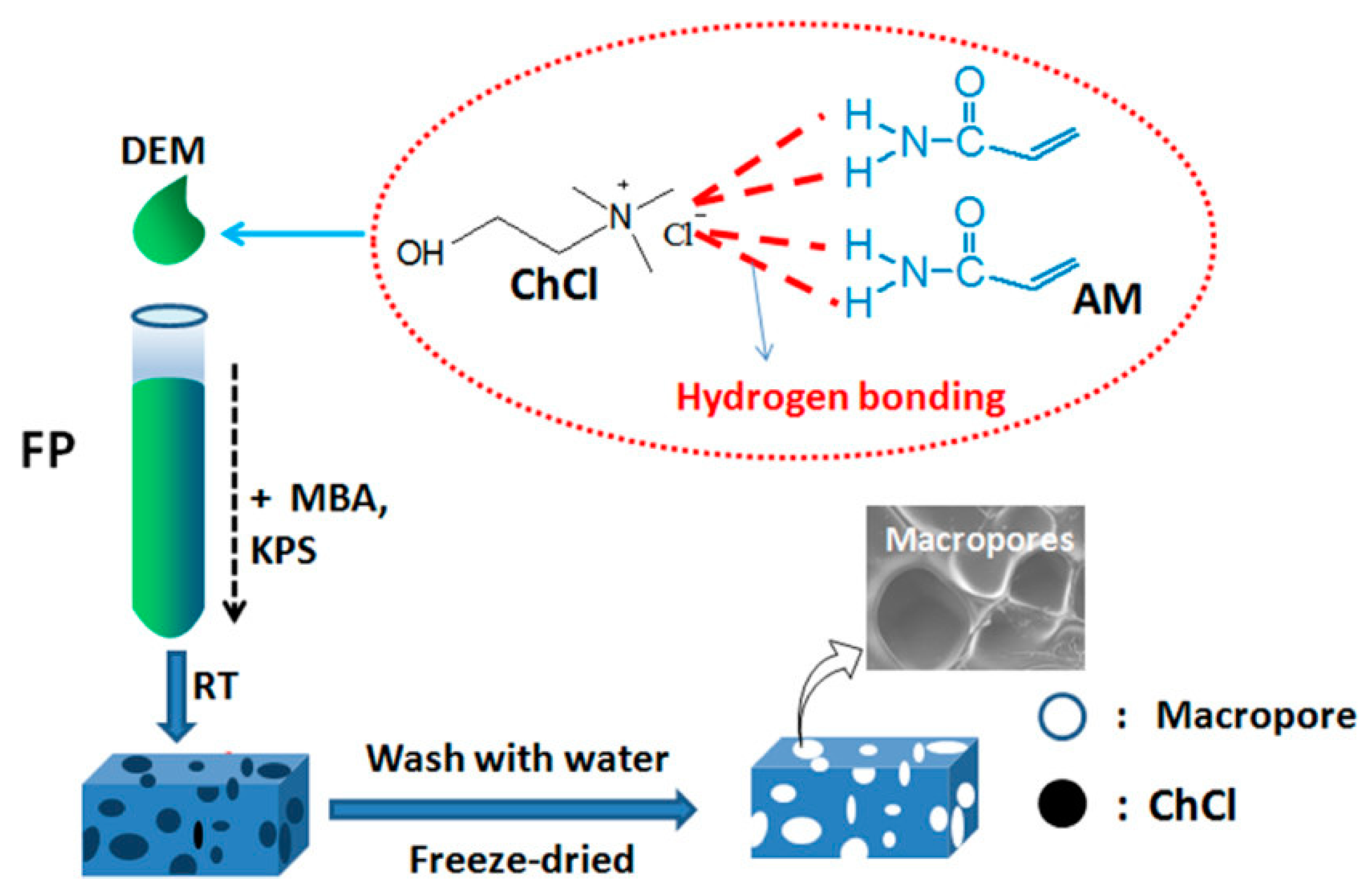
Figure 10.
The device employed for the frontal photopolymerization. Reprinted with permission from [48]. Copyright Wiley, 2019.
Figure 10.
The device employed for the frontal photopolymerization. Reprinted with permission from [48]. Copyright Wiley, 2019.
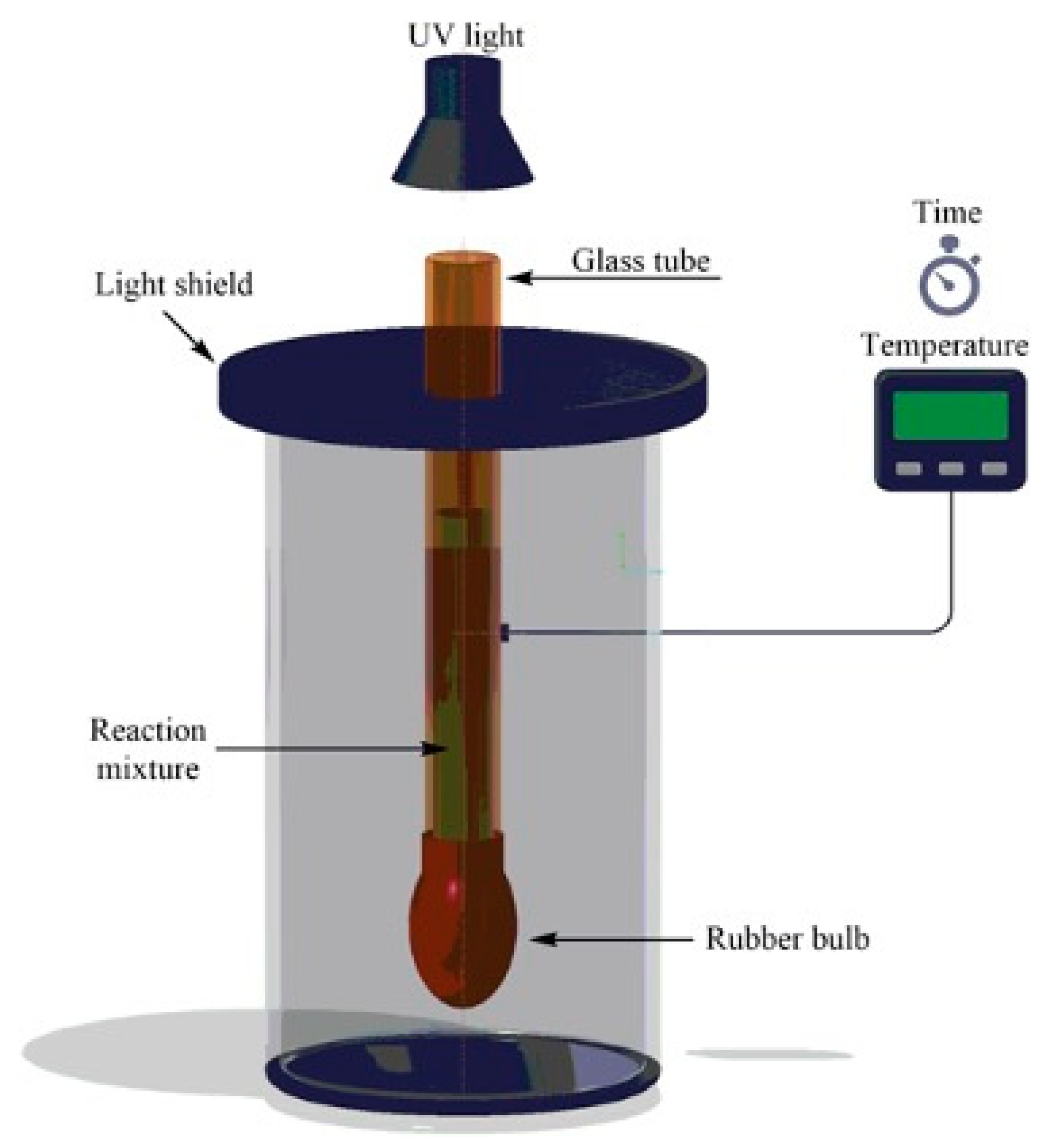
Figure 11.
Drug release behaviors of poly(N-isopropylacrylamide) hydrogels containing different crosslinkers at 37 °C (solid line) and 20 °C (dotted line), respectively. Legend: MBA= N,N′-methylenebisacrylamide, used at 2 mol% loading; TPGDA= tri(propylene glycol) diacrylate at 2 mol% loading; MA-POSS= acrylopropyl polyhedral oligomeric silsesquioxane cage mixture at 0.5 mol% loading. Reprinted with permission from [48]. Copyright Wiley, 2019.
Figure 11.
Drug release behaviors of poly(N-isopropylacrylamide) hydrogels containing different crosslinkers at 37 °C (solid line) and 20 °C (dotted line), respectively. Legend: MBA= N,N′-methylenebisacrylamide, used at 2 mol% loading; TPGDA= tri(propylene glycol) diacrylate at 2 mol% loading; MA-POSS= acrylopropyl polyhedral oligomeric silsesquioxane cage mixture at 0.5 mol% loading. Reprinted with permission from [48]. Copyright Wiley, 2019.
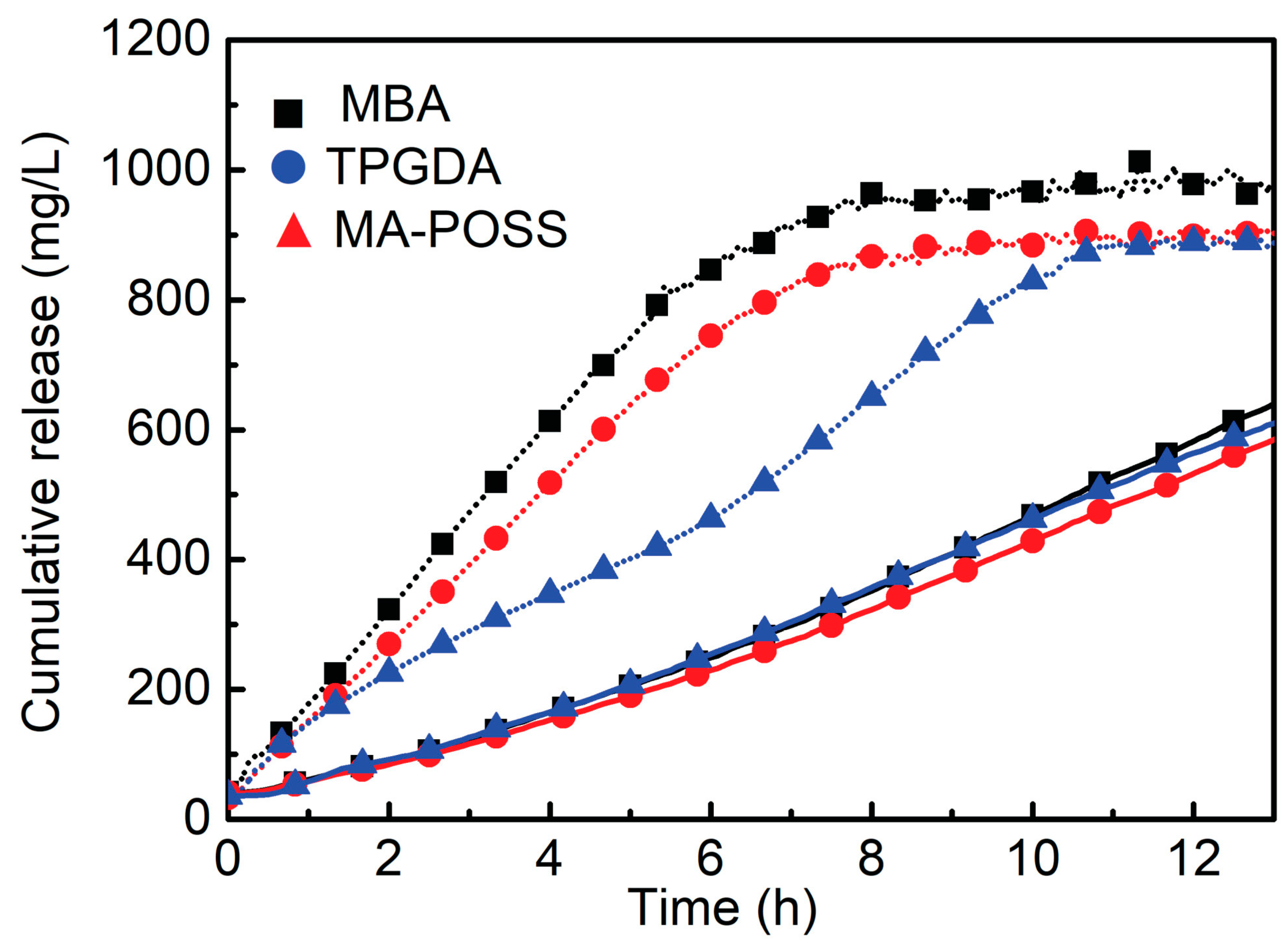
Figure 12.
(a) Front position versus time for poly(itaconic acid-co-acrylic acid-co-acrylamide) hydrogels by the FP at different IA/AAc weight ratios of (1) 1:39, (2) 2:38, (3) 3:37, (4) 4:36, and (5) 5:35 wt./wt. (b) Frontal velocity (FV) and frontal temperature (Tmax) as a function of the itaconic acid/acrylic acid (IA/AAc) weight ratios at 10 wt.% acrylamide, 50 wt. %, glycerol, 0.24 wt.% N,N-methylenebisacrylamide, 0.1 wt.% ammonium persulfate, and 10 μL N,N,N′,N′-tetramethylethylenediamine. Reprinted with permission from [50]. Copyright Wiley, 2019.
Figure 12.
(a) Front position versus time for poly(itaconic acid-co-acrylic acid-co-acrylamide) hydrogels by the FP at different IA/AAc weight ratios of (1) 1:39, (2) 2:38, (3) 3:37, (4) 4:36, and (5) 5:35 wt./wt. (b) Frontal velocity (FV) and frontal temperature (Tmax) as a function of the itaconic acid/acrylic acid (IA/AAc) weight ratios at 10 wt.% acrylamide, 50 wt. %, glycerol, 0.24 wt.% N,N-methylenebisacrylamide, 0.1 wt.% ammonium persulfate, and 10 μL N,N,N′,N′-tetramethylethylenediamine. Reprinted with permission from [50]. Copyright Wiley, 2019.
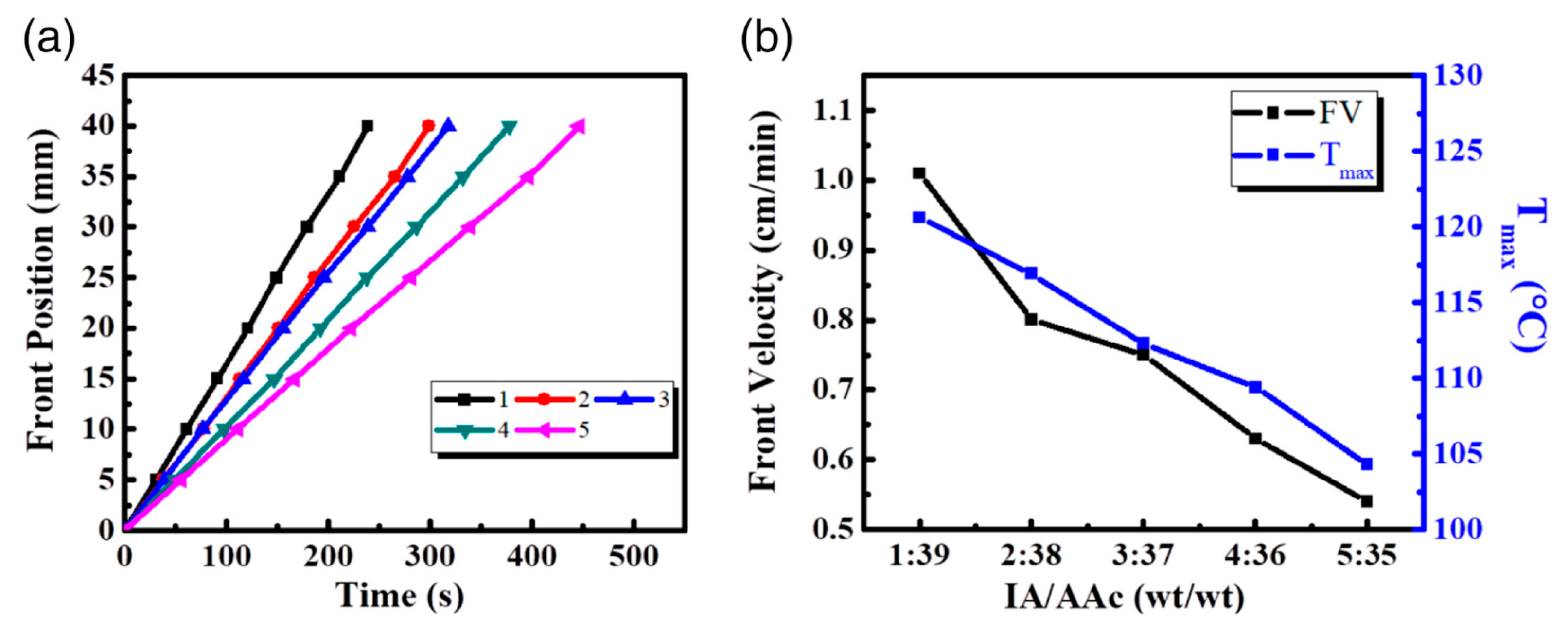
Figure 13.
Front velocity vs. 5-(2-(2-(2-(2-phenoxyethoxy)ethoxy) ethoxy)-10,15,20-triphenylporphyrin methacrylate (TPP-TEG-MA) comonomer concentration of the frontal polymerization of poly(ethylene glycol) methyl ether acrylate (a) and di(ethylene glycol) methyl ether methacrylate (b) series. Reprinted with permission from [51]. Copyright Elsevier, 2022.
Figure 13.
Front velocity vs. 5-(2-(2-(2-(2-phenoxyethoxy)ethoxy) ethoxy)-10,15,20-triphenylporphyrin methacrylate (TPP-TEG-MA) comonomer concentration of the frontal polymerization of poly(ethylene glycol) methyl ether acrylate (a) and di(ethylene glycol) methyl ether methacrylate (b) series. Reprinted with permission from [51]. Copyright Elsevier, 2022.
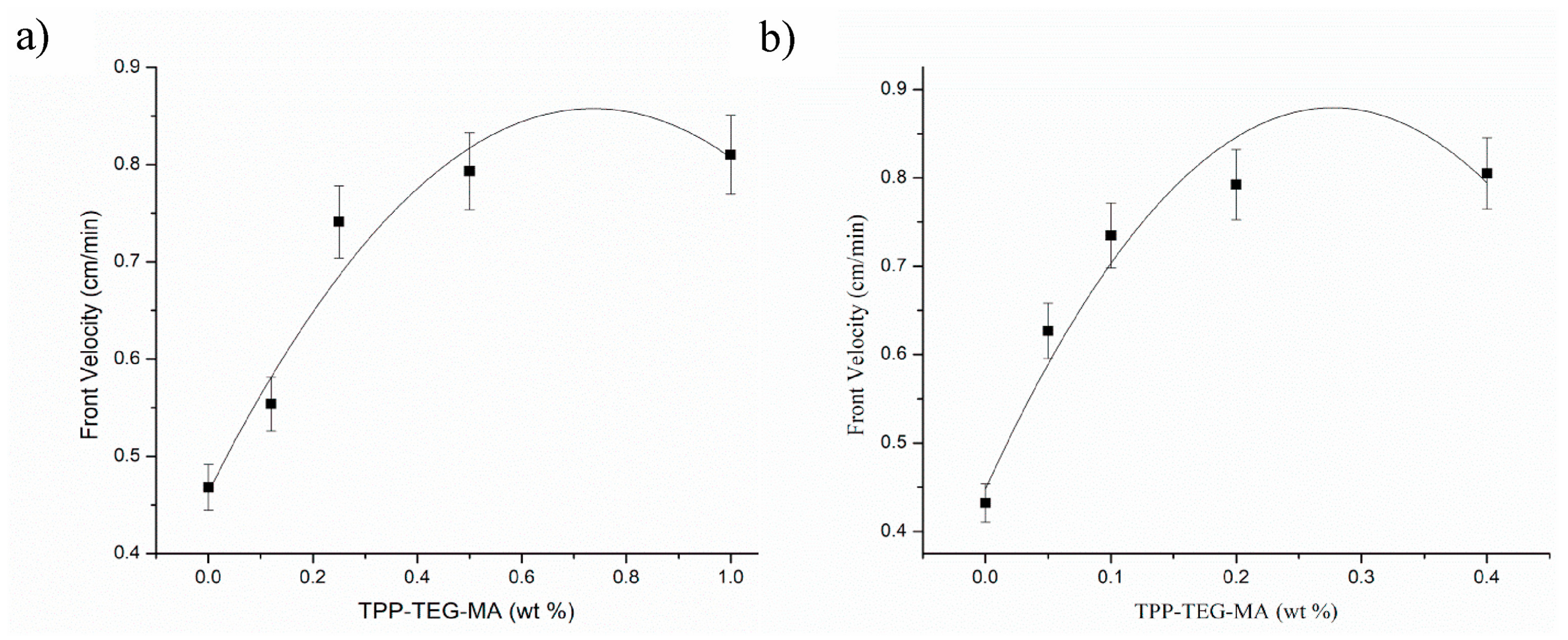
Figure 14.
Swelling behavior of poly(ethylene glycol) methyl ether acrylate (PEGTPP-X) series, where X represents the mol.% loading of the porphyrin methacrylate monomer: a) at constant temperature, 25 °C and pH = 7; b) pH response; c) pH reversibility response for PEGTPP-1.0, measurements were taken after equilibrium was reached; d) temperature response. Reprinted with permission from [51]. Copyright Elsevier, 2022.
Figure 14.
Swelling behavior of poly(ethylene glycol) methyl ether acrylate (PEGTPP-X) series, where X represents the mol.% loading of the porphyrin methacrylate monomer: a) at constant temperature, 25 °C and pH = 7; b) pH response; c) pH reversibility response for PEGTPP-1.0, measurements were taken after equilibrium was reached; d) temperature response. Reprinted with permission from [51]. Copyright Elsevier, 2022.
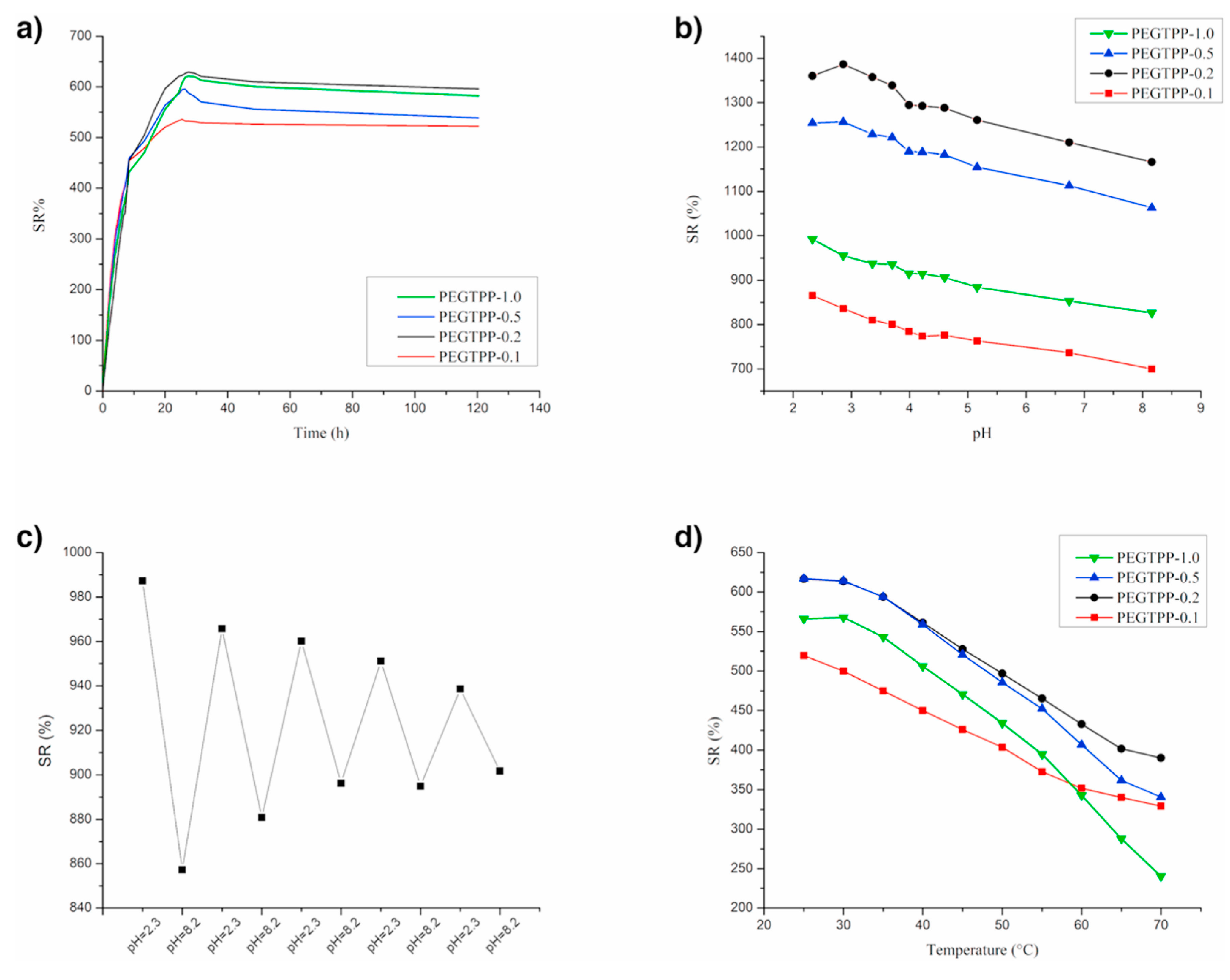
Figure 15.
Swelling behavior of di(ethylene glycol) methyl ether methacrylate (DEGTPP-X) series, where X represents the mol.% loading of the porphyrin methacrylate monomer: a) at constant temperature, 25 °C and pH = 7; b) pH response; c) pH reversibility response for DEGTPP-0.4, measurements were taken after equilibrium was reached; d) temperature response. Reprinted with permission from [51]. Copyright Elsevier, 2022.
Figure 15.
Swelling behavior of di(ethylene glycol) methyl ether methacrylate (DEGTPP-X) series, where X represents the mol.% loading of the porphyrin methacrylate monomer: a) at constant temperature, 25 °C and pH = 7; b) pH response; c) pH reversibility response for DEGTPP-0.4, measurements were taken after equilibrium was reached; d) temperature response. Reprinted with permission from [51]. Copyright Elsevier, 2022.
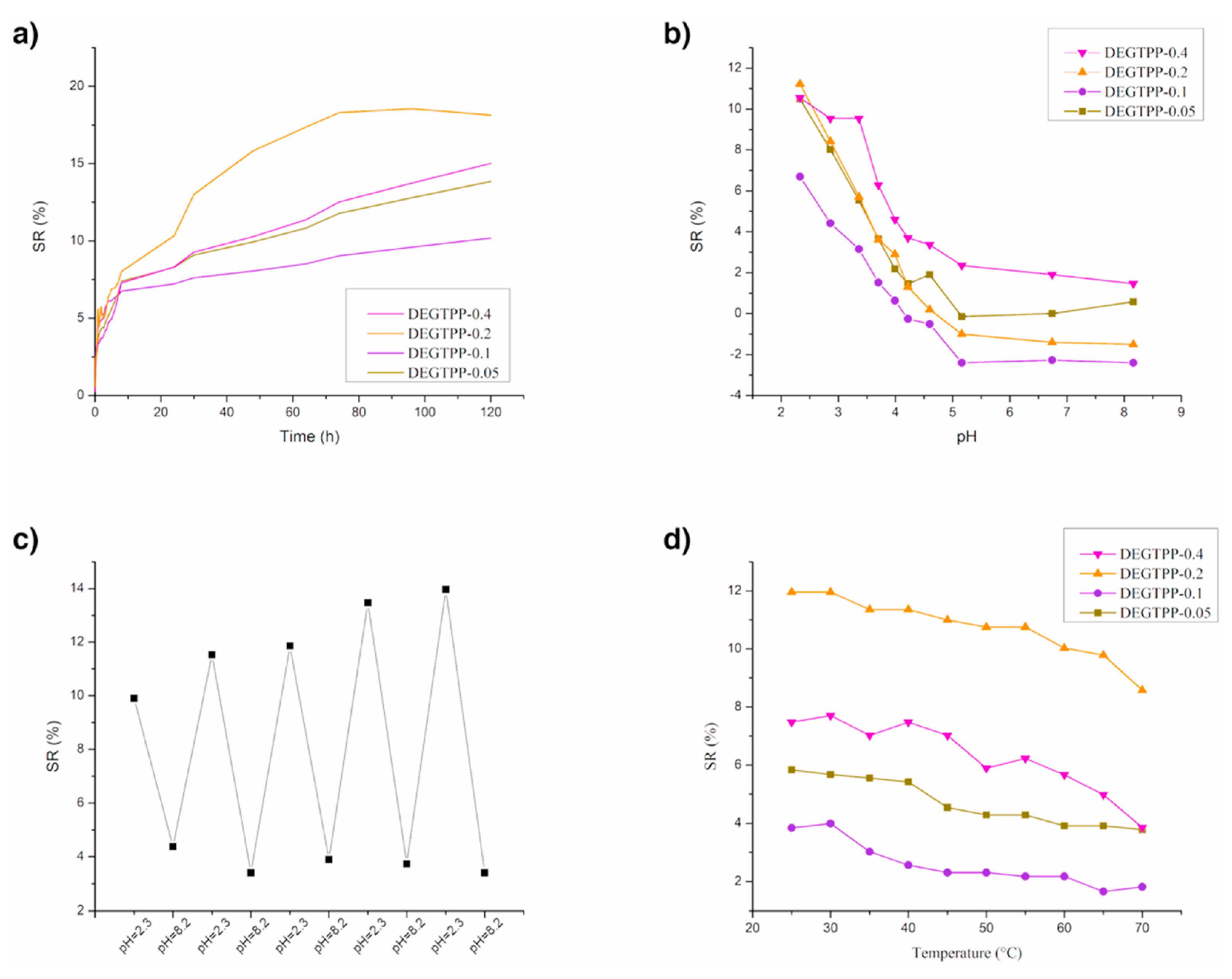
Figure 16.
(A) Schematic illustrations for the synthesis of bi-layered hydrogels via horizontal biphase frontal polymerization. Rapid synchronized preparation of (B) poly(acrylamide-co-2-acrylamide-2-methylpropanesulfonic acid) (poly(AM-co-AMPS)) (G-1) and (C) poly(acrylamide-co-hydroxypropyl acrylate) (poly(AM-co-HPA)) (G-2) to form bilayer hydrogels. Reprinted with permission from [57]. Copyright Wiley, 2022.
Figure 16.
(A) Schematic illustrations for the synthesis of bi-layered hydrogels via horizontal biphase frontal polymerization. Rapid synchronized preparation of (B) poly(acrylamide-co-2-acrylamide-2-methylpropanesulfonic acid) (poly(AM-co-AMPS)) (G-1) and (C) poly(acrylamide-co-hydroxypropyl acrylate) (poly(AM-co-HPA)) (G-2) to form bilayer hydrogels. Reprinted with permission from [57]. Copyright Wiley, 2022.
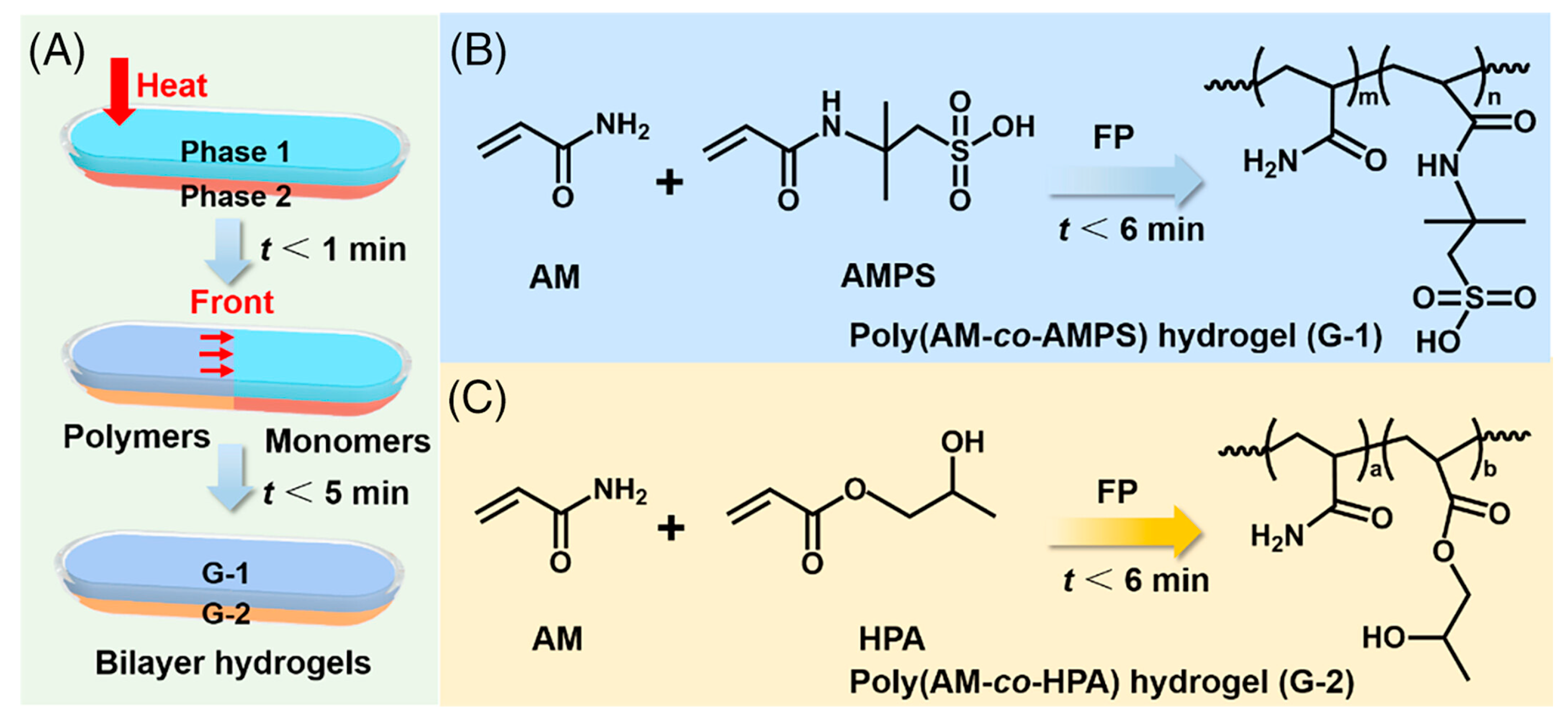
Figure 17.
Digital photographs of self-healed samples of (A) poly(acrylamide-co-2-acrylamide-2-methylpropanesulfonic acid) and (B) poly(acrylamide-co-hydroxypropyl acrylate) hydrogels obtained by slicing the bi-layered system (prepared with 5:5 weight ratio between the two polymer solutions). Each sample was cut into two separate halves, one half of which was dyed, then put together for 24 h at room temperature, and finally merged into a complete sample sustaining robust stretching. Reprinted with permission from [57]. Copyright Wiley, 2022.
Figure 17.
Digital photographs of self-healed samples of (A) poly(acrylamide-co-2-acrylamide-2-methylpropanesulfonic acid) and (B) poly(acrylamide-co-hydroxypropyl acrylate) hydrogels obtained by slicing the bi-layered system (prepared with 5:5 weight ratio between the two polymer solutions). Each sample was cut into two separate halves, one half of which was dyed, then put together for 24 h at room temperature, and finally merged into a complete sample sustaining robust stretching. Reprinted with permission from [57]. Copyright Wiley, 2022.
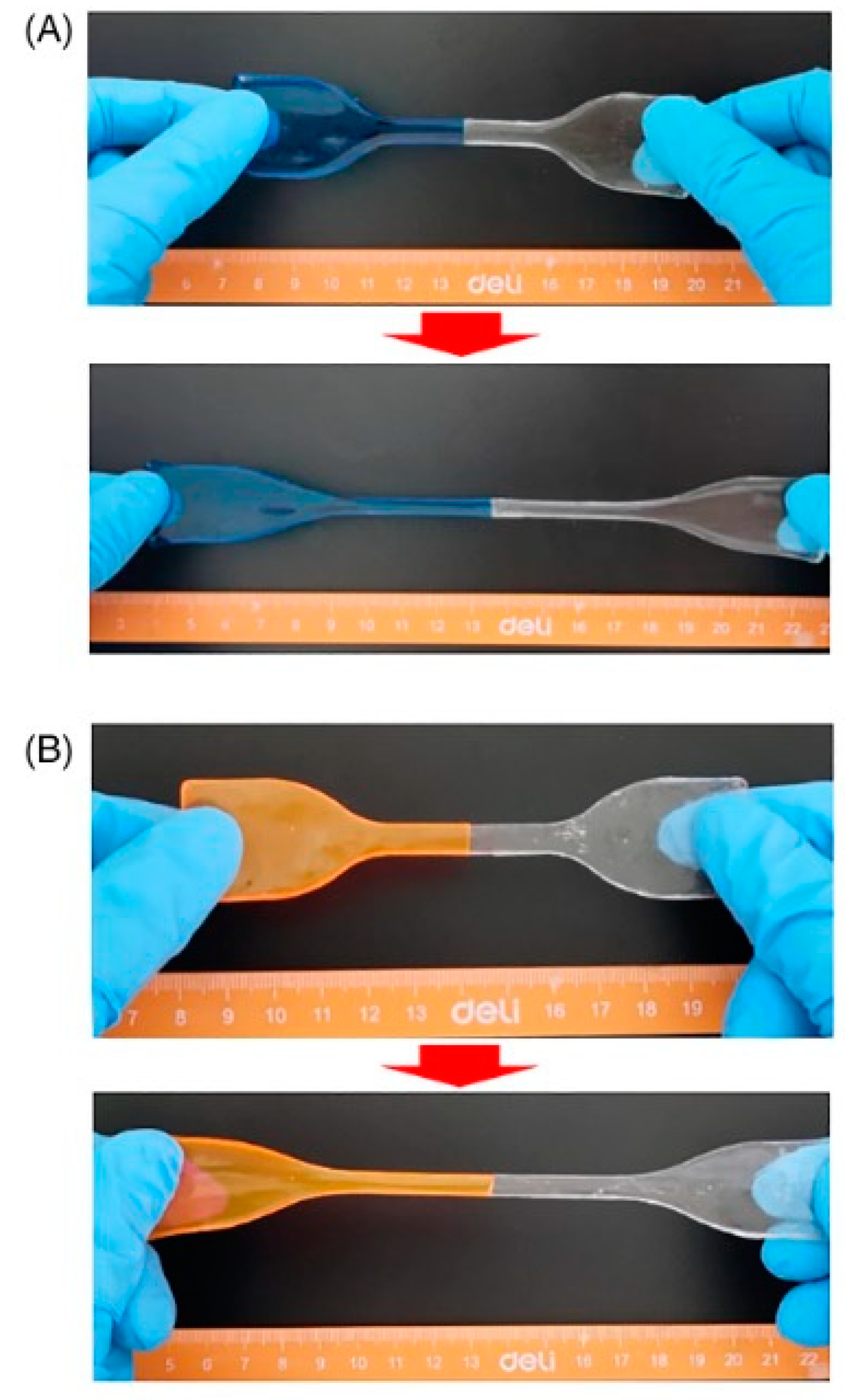
Figure 18.
(a) Schematic illustration of the interfacial ignited gelation-promoted healing of a covalently cross-linked polymer hydrogel. (b–d) Pictures showing the interfacial ignited gelation-healing of the covalently cross-linked Ag+/Fe3+-poly(acrylic acid) (AgFe-PAA; polymer concentration: 20 wt.%), Ag+/Fe3+-poly(acrylic acid-co-acrylamide) (AgFe-P(AA-co-AM); polymer concentration: 20 wt.%; 2:1 acrylic acid/acrylamide mole ratio), and Ag+/Fe3+-poly(acrylic acid-co-N,N-dimethylacrylamide) (AgFe-P(AA-co-DMA; polymer concentration: 20 wt.%; 2:1 acrylic acid/ N,N-dimethylacrylamide mole ratio) hydrogels. (e–g) Stress–strain curves illustrating the interfacial ignited gelation-healing of different hydrogels in panels (b–d). (h and j) Stress–strain curves showing the repeated interfacial ignited gelation-healing of the AgFe-PAA hydrogels using “repairing liquids” without and with Fe3+. (i) Healing efficiencies of the hydrogels in (h and j) at different healing cycles (up to 5 cycles). Reprinted with permission from [58]. Copyright American Chemical Society, 2023.
Figure 18.
(a) Schematic illustration of the interfacial ignited gelation-promoted healing of a covalently cross-linked polymer hydrogel. (b–d) Pictures showing the interfacial ignited gelation-healing of the covalently cross-linked Ag+/Fe3+-poly(acrylic acid) (AgFe-PAA; polymer concentration: 20 wt.%), Ag+/Fe3+-poly(acrylic acid-co-acrylamide) (AgFe-P(AA-co-AM); polymer concentration: 20 wt.%; 2:1 acrylic acid/acrylamide mole ratio), and Ag+/Fe3+-poly(acrylic acid-co-N,N-dimethylacrylamide) (AgFe-P(AA-co-DMA; polymer concentration: 20 wt.%; 2:1 acrylic acid/ N,N-dimethylacrylamide mole ratio) hydrogels. (e–g) Stress–strain curves illustrating the interfacial ignited gelation-healing of different hydrogels in panels (b–d). (h and j) Stress–strain curves showing the repeated interfacial ignited gelation-healing of the AgFe-PAA hydrogels using “repairing liquids” without and with Fe3+. (i) Healing efficiencies of the hydrogels in (h and j) at different healing cycles (up to 5 cycles). Reprinted with permission from [58]. Copyright American Chemical Society, 2023.
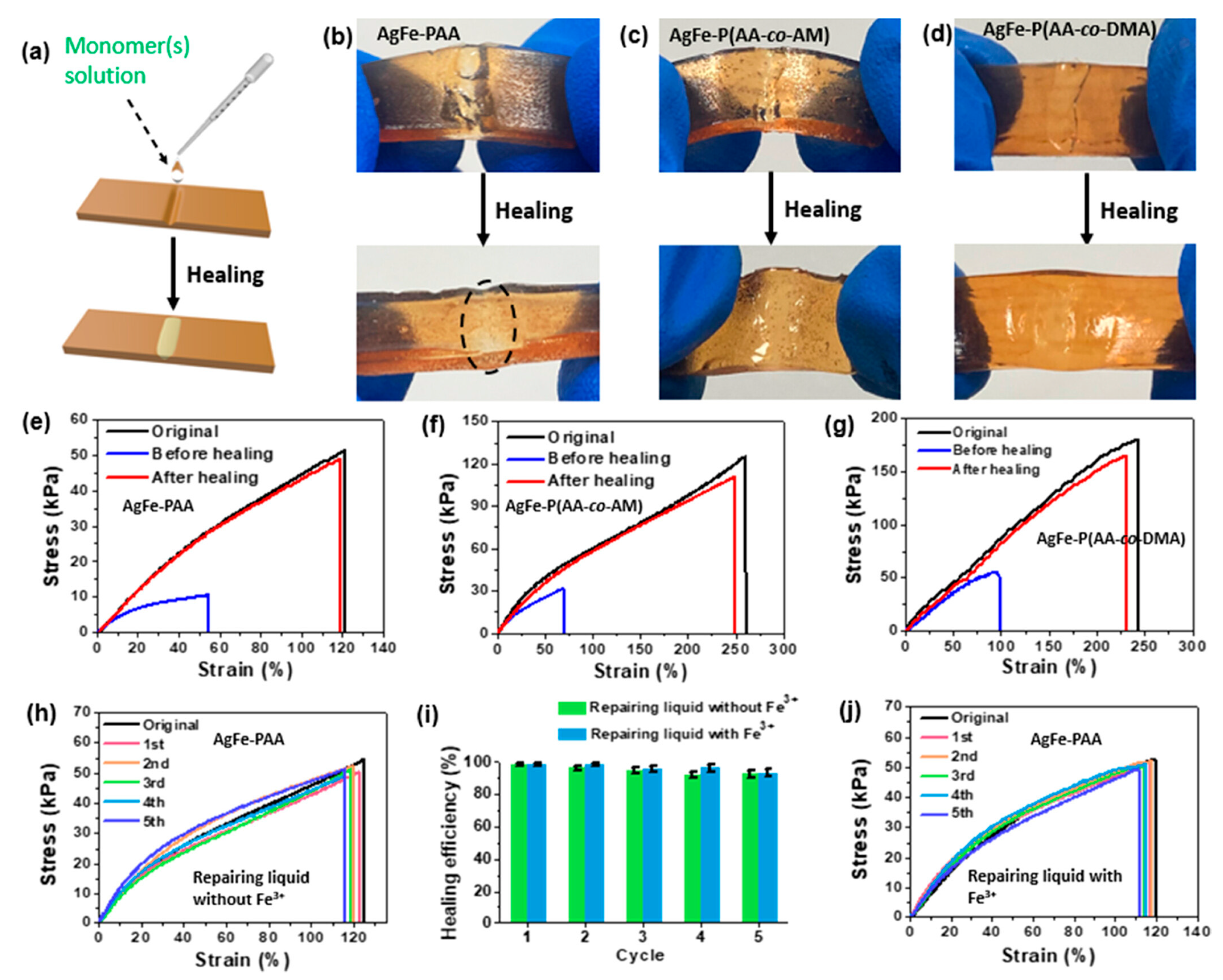
Figure 19.
Hydrogel (2:1:1 acrylamide/choline chloride/glycerol molar ratio): (a) Original length, (b) Easily stretches to more than twice its original length, (c) Fold at will, (d) Twistable after folding without damage. Reprinted with permission from [61]. Copyright Wiley, 2023.
Figure 19.
Hydrogel (2:1:1 acrylamide/choline chloride/glycerol molar ratio): (a) Original length, (b) Easily stretches to more than twice its original length, (c) Fold at will, (d) Twistable after folding without damage. Reprinted with permission from [61]. Copyright Wiley, 2023.

Figure 20.
Photographs of LED light brightness after connecting different hydrogel samples: (a, b) hydrogel (1:1:1 acrylamide/choline chloride/glycerol molar ratio) before and after water absorption, (c, d) the same hydrogel before and after compression. Reprinted with permission from [61]. Copyright Wiley, 2023.
Figure 20.
Photographs of LED light brightness after connecting different hydrogel samples: (a, b) hydrogel (1:1:1 acrylamide/choline chloride/glycerol molar ratio) before and after water absorption, (c, d) the same hydrogel before and after compression. Reprinted with permission from [61]. Copyright Wiley, 2023.

Figure 21.
(a) Self-healing of the hydrogel without (P(AM-co-AA)) and with 2.0 wt.% of β-cyclodextrins (β-CD/P(AM-co-AA)): concerning the former, an obvious dividing line at the cross-section appears, while for the latter the line is almost invisible at the cross-section. (b) scheme of the self-healing process of β-CD/P(AM-co-AA) hydrogels through hydrogen bonding; (c) β-CD/P(AM-co-AA) hydrogen bonds formed between the molecular chains of the hydrogel. Reprinted with permission from [62]. Copyright Wiley, 2023.
Figure 21.
(a) Self-healing of the hydrogel without (P(AM-co-AA)) and with 2.0 wt.% of β-cyclodextrins (β-CD/P(AM-co-AA)): concerning the former, an obvious dividing line at the cross-section appears, while for the latter the line is almost invisible at the cross-section. (b) scheme of the self-healing process of β-CD/P(AM-co-AA) hydrogels through hydrogen bonding; (c) β-CD/P(AM-co-AA) hydrogen bonds formed between the molecular chains of the hydrogel. Reprinted with permission from [62]. Copyright Wiley, 2023.
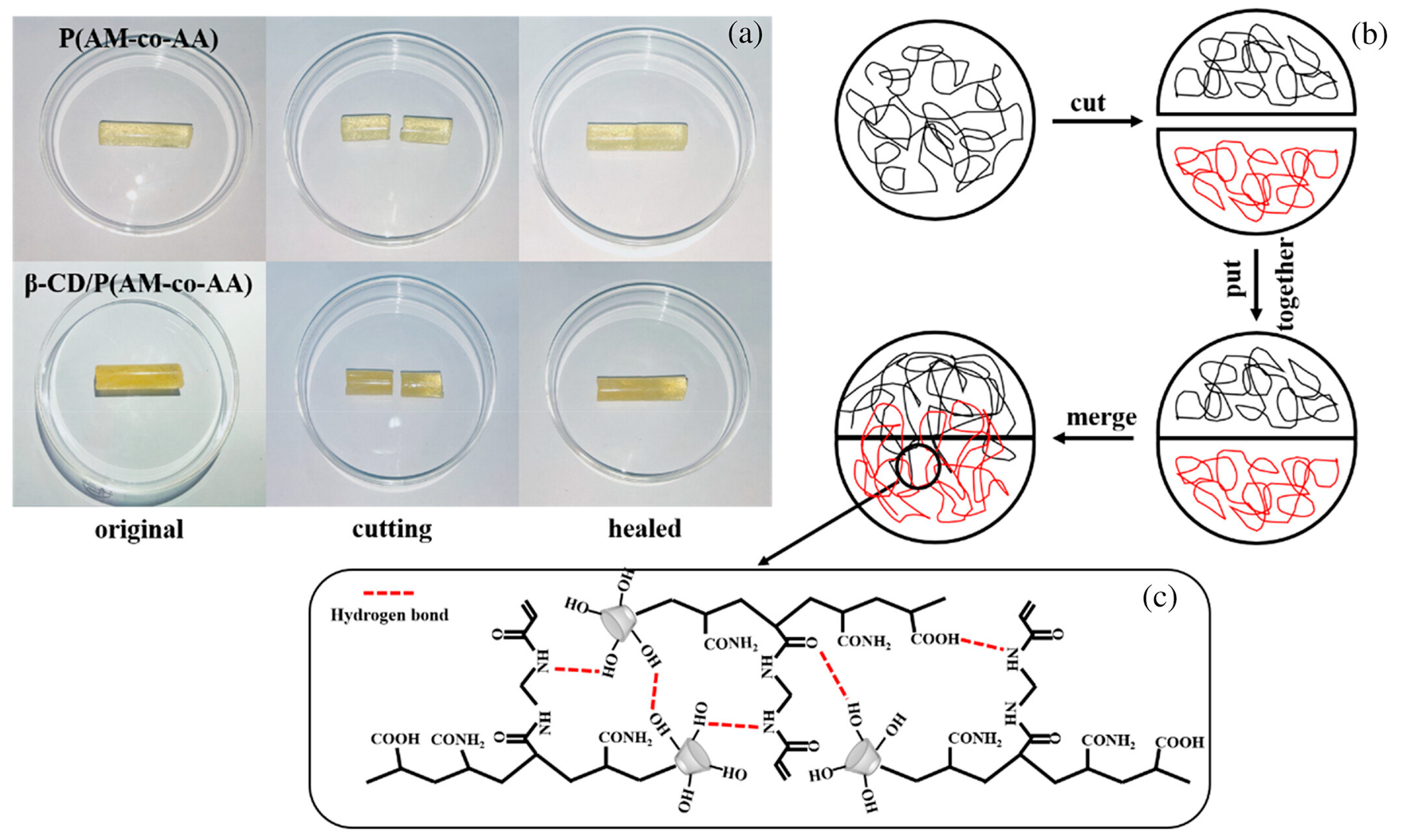
Figure 22.
(a) Photographs of a healed sample of a poly(acrylamide-co-acrylic acid-co-3-[Dimethyl-[2-(2-methylprop-2-enoyloxy)ethyl]azaniumyl]propane-1-sulfonate) hydrogel under stretching. (b,c) Self-healing efficiency of the hydrogels prepared with (b) different acrylamide/acrylic acid (AM/AA) mass ratios (at 10 wt.% of 3-[Dimethyl-[2-(2-methylprop-2-enoyloxy)ethyl]azaniumyl]propane-1-sulfonate (SBMA)) and (c) different SBMA concentrations (AM/AA mass ratio: 8:5, after 24 h at room temperature. (d) Self-healing mechanism via ionic association and hydrogen bonding interactions shown in schematic. (e–h) FT-IR spectroscopy and infrared pictures of these hydrogels before and during self-healing. Reprinted from [63] under CC-BY 4.0 License.
Figure 22.
(a) Photographs of a healed sample of a poly(acrylamide-co-acrylic acid-co-3-[Dimethyl-[2-(2-methylprop-2-enoyloxy)ethyl]azaniumyl]propane-1-sulfonate) hydrogel under stretching. (b,c) Self-healing efficiency of the hydrogels prepared with (b) different acrylamide/acrylic acid (AM/AA) mass ratios (at 10 wt.% of 3-[Dimethyl-[2-(2-methylprop-2-enoyloxy)ethyl]azaniumyl]propane-1-sulfonate (SBMA)) and (c) different SBMA concentrations (AM/AA mass ratio: 8:5, after 24 h at room temperature. (d) Self-healing mechanism via ionic association and hydrogen bonding interactions shown in schematic. (e–h) FT-IR spectroscopy and infrared pictures of these hydrogels before and during self-healing. Reprinted from [63] under CC-BY 4.0 License.
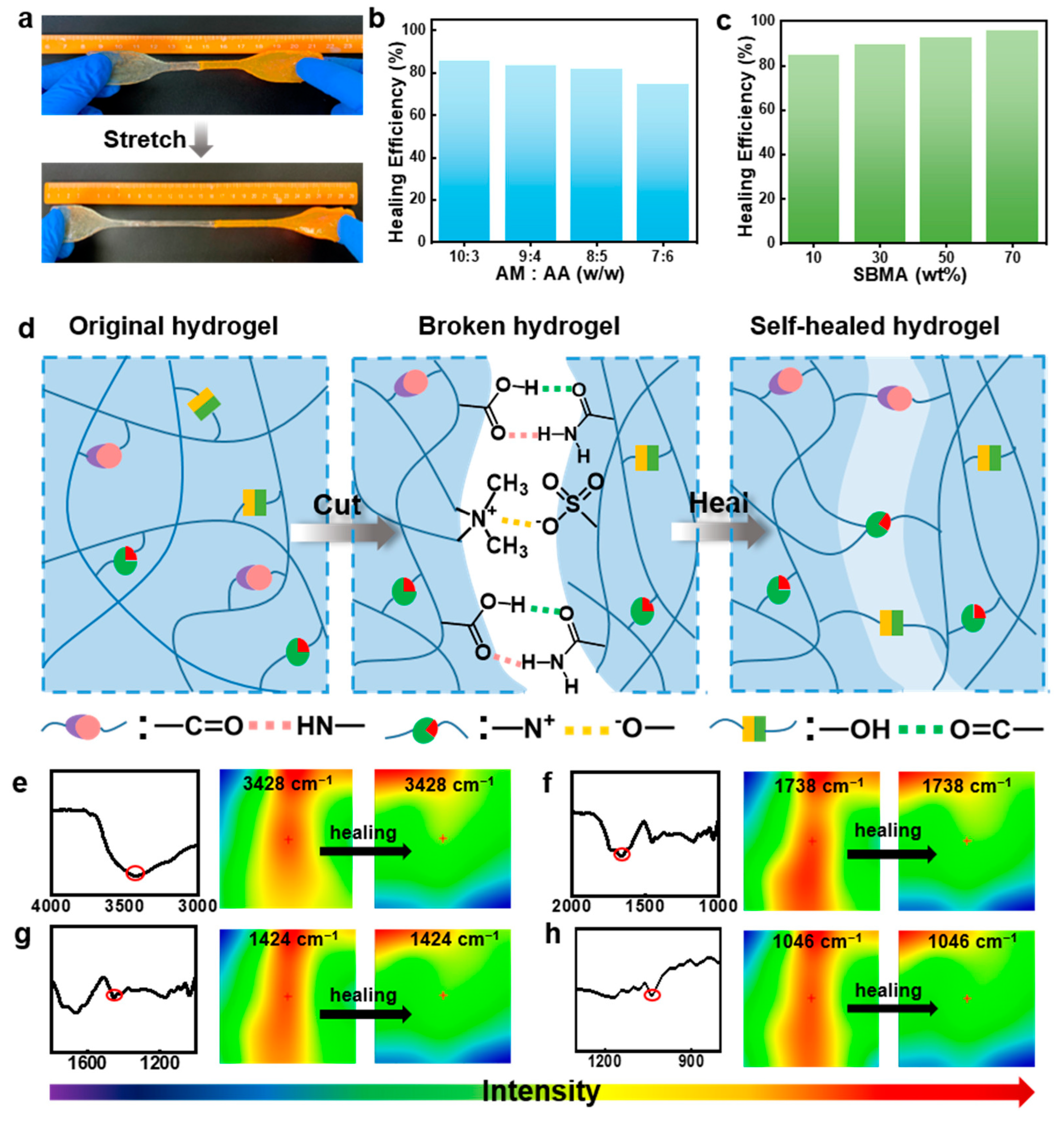
Figure 23.
Scheme of the preparation of poly(ionic liquid)/starch hydrogels. Legend: DES= deep eutectic monomer mixture; FP= frontal polymerization; KPS= potassium persulfate; MBA= N,N-methylenebisacrylamide. Reprinted with permission from [64]. Copyright Elsevier, 2021.
Figure 23.
Scheme of the preparation of poly(ionic liquid)/starch hydrogels. Legend: DES= deep eutectic monomer mixture; FP= frontal polymerization; KPS= potassium persulfate; MBA= N,N-methylenebisacrylamide. Reprinted with permission from [64]. Copyright Elsevier, 2021.
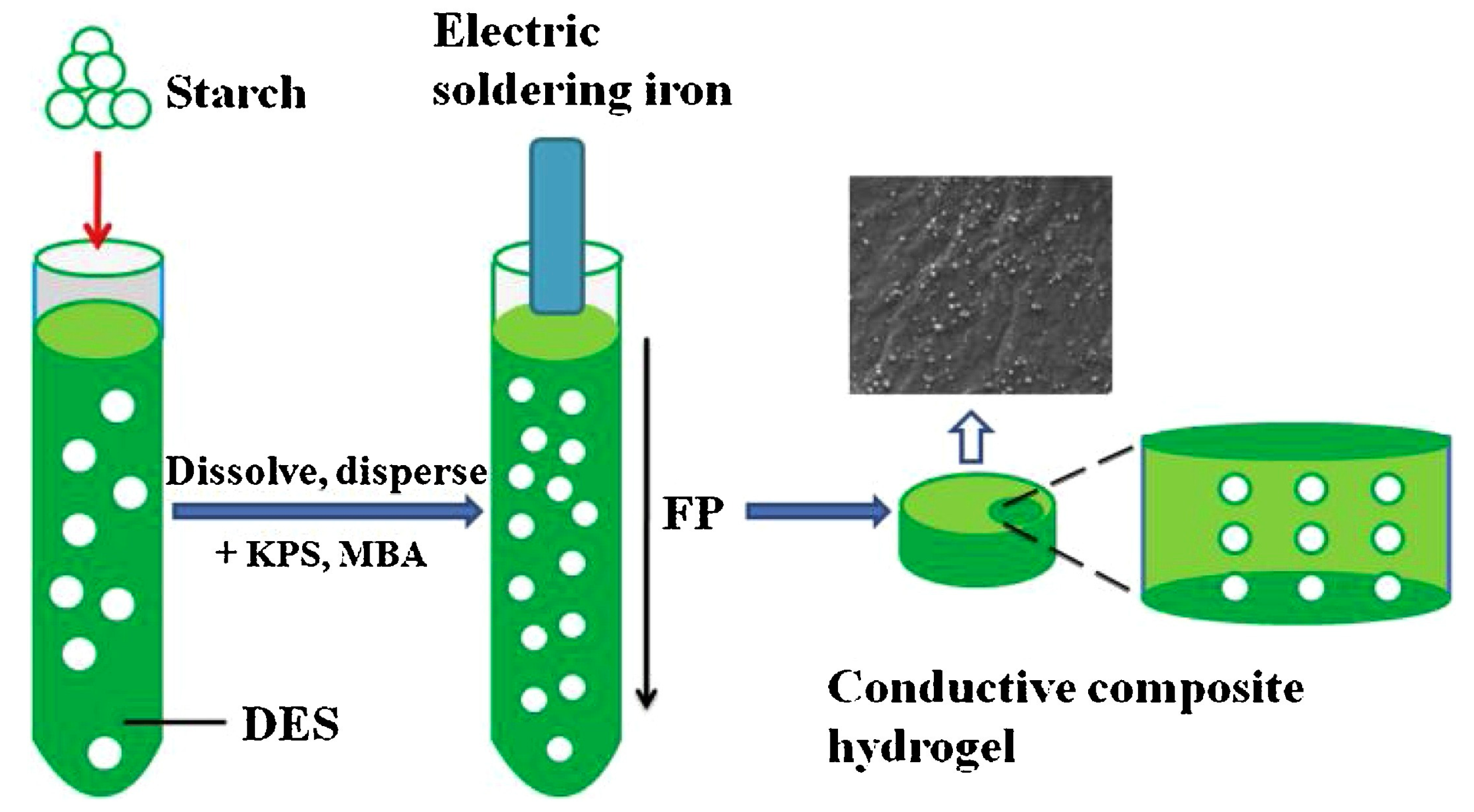
Figure 24.
(a) Conductivity and water absorption (WA) vs. time for the hydrogel containing 10 wt.% of starch; (b) Digital photographs of the brightness of the same hydrogel connected to a LED bulb before and after water absorption. Reprinted with permission from [64]. Copyright Elsevier, 2021.
Figure 24.
(a) Conductivity and water absorption (WA) vs. time for the hydrogel containing 10 wt.% of starch; (b) Digital photographs of the brightness of the same hydrogel connected to a LED bulb before and after water absorption. Reprinted with permission from [64]. Copyright Elsevier, 2021.
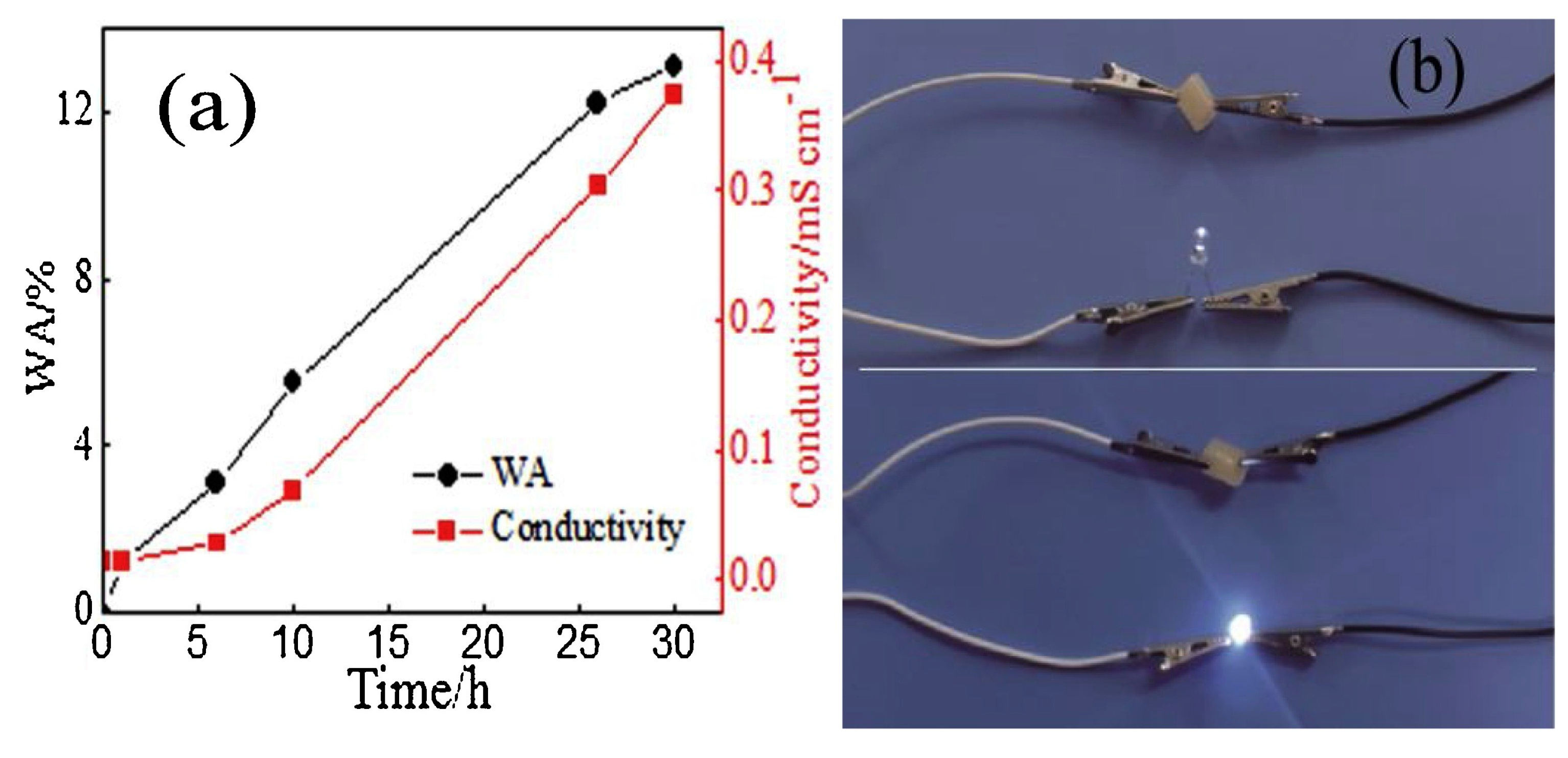
Figure 25.
Brightness change of LED connected to hydrogel circuit before and after absorbing water; (a) unfilled hydrogel before water absorption; (c) hydrogel containing 1.0 wt.% of N-doped carbon nanotubes before water absorption; (d) unfilled hydrogel after water absorption; (f) hydrogel containing 1.0 wt.% of N-doped carbon nanotubes after water absorption. Adapted from [65] under CC-BY 3.0 license.
Figure 25.
Brightness change of LED connected to hydrogel circuit before and after absorbing water; (a) unfilled hydrogel before water absorption; (c) hydrogel containing 1.0 wt.% of N-doped carbon nanotubes before water absorption; (d) unfilled hydrogel after water absorption; (f) hydrogel containing 1.0 wt.% of N-doped carbon nanotubes after water absorption. Adapted from [65] under CC-BY 3.0 license.

Figure 26.
Schematic of solar-initiated frontal polymerization (a) and batch polymerization (b) for preparing hydrogel evaporator and evaporation processes. Reprinted with permission from [66]. Copyright Wiley, 2022.
Figure 26.
Schematic of solar-initiated frontal polymerization (a) and batch polymerization (b) for preparing hydrogel evaporator and evaporation processes. Reprinted with permission from [66]. Copyright Wiley, 2022.
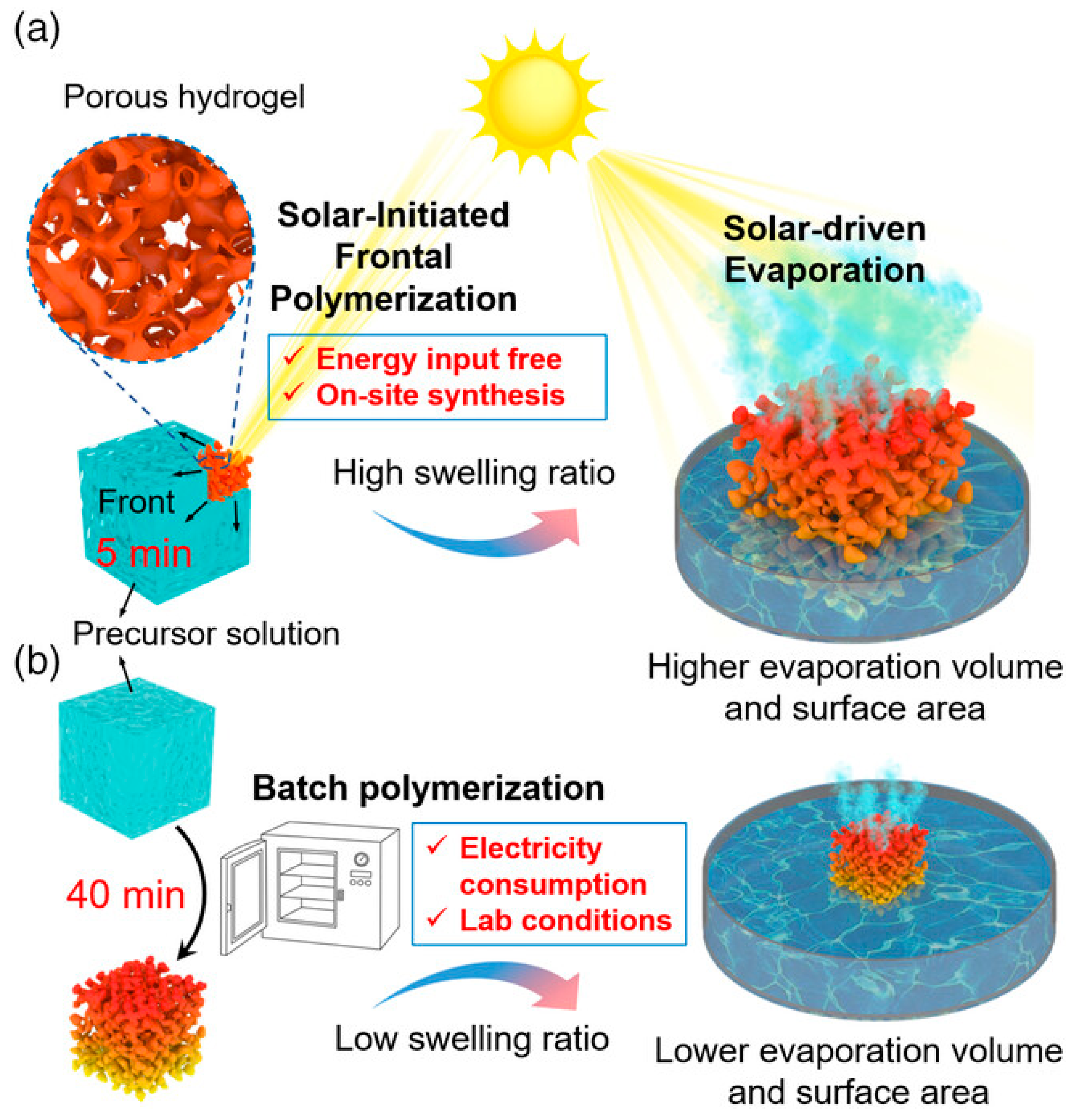
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



