Submitted:
24 August 2023
Posted:
25 August 2023
You are already at the latest version
Abstract
Lupus nephritis (LN) is a severe complication of systemic lupus erythematosus (SLE) and is considered one of the leading causes of mortality. Multiple immunological pathways are involved in the pathogenesis of SLE, which makes it imperative to deepen our knowledge about this disease's immunopathological complexity and explore new therapeutic targets. Since an altered redox state contributes to immune system dysregulation, this document briefly addresses the role of oxidative stress (OS), oxidative DNA damage, antioxidant enzymes, mitochondrial function, and mitophagy in SLE and LN. Although adaptive immunity's participation in the development of autoimmunity is undeniable, increasing data emphasize the importance of innate immunity elements, particularly the Toll-like receptors (TLRs) that recognize nucleic acid ligands, in inflammatory and autoimmune diseases. Here, we discuss the intriguing role of TLR7 and TLR9 in developing SLE and LN. Also included are the essential characteristics of conventional treatments and some other novel and little-explored alternatives that offer options to improve renal function in LN.
Keywords:
systemic lupus erythematosus
; lupus nephritis
; antioxidants
; oxidative stress
; DNA damage
; mitochondrial function
; mitophagy
1. Introduction
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease mainly affecting women of childbearing age. SLE presents a broad spectrum of clinical manifestations involving multiple organs and systems, such as the cardiopulmonary, neuromuscular-psychiatric, endocrine, integumentary, hematological, and genitourinary systems [1]. Figure 1.
SLE characterizes by microvascular inflammation with the development of autoantibodies, particularly against nuclear components [2]. Autoantibody and autoreactive T-cell presentation occur against several ubiquitous nuclear antigens, including chromatin particles and ribonucleoproteins, unleashing systemic autoimmunity and organ damage through additional mechanisms of inflammation and tissue remodeling [3]. In SLE, altering endogenous antigens coupled with the production of autoantibodies promotes the formation of pathogenic immune complexes and organ damage [4].
Oxidative Stress in SLE
Oxidative stress (OS) is the imbalance between the synthesis and neutralization of reactive oxygen species (ROS) [5]. Increasingly, evidence shows that elevated OS correlates with autoimmune disease activity and facilitates tissue damage [6]. The interaction of ROS with carbohydrates, lipids, proteins, and nucleic acids bolsters acute and chronic tissue damage by mediating immunomodulation, which triggers autoimmunity in SLE (Figure 1) [7]. ROS generate by incomplete reduction of oxygen during the redox process, where excessive release and insufficient elimination of ROS promote the development of SLE [8]. The ROS imbalance in SLE is partially explained by abnormal exposure to endogenous antigens and response to characteristic cell death signals [8]. Exogenous ROS that might be involved in the onset of SLE derive mainly from external predisposing factors such as ultraviolet radiation, chemical exposure, and viral or bacterial infection. At the same time, endogenous sources include excessive ROS formation by mitochondria and extra-mitochondrial organelles such as the endoplasmic reticulum (ER) [8]. The transfer of electrons to molecular oxygen in the electron transport chain generates ROS as part of mitochondria's oxidative phosphorylation (OxPhos) process. Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX), nitric oxide synthase (NOS), and xanthine oxidase (XO) are the main causes of extra-mitochondrial OS [9]. The superoxide anion radical (O2•-) and hydrogen peroxide (H2O2) are among the main ROS regularly accompanied by pro-inflammatory signals and capable of interacting with nitric oxide (NO), which explains the appearance of reactive nitrogen species, including the highly reactive nitrosonium cation (NO+), nitroxyl anion (NO-) and peroxynitrites (ONOO-) [10].
Lipids are targets susceptible to oxidation due to the double bonds of unsaturated fatty acids in mitochondria, lysosomes, and cell membranes that serve as the major sites of OS, allowing for increased lipid peroxidation in disease [11]. The lipoperoxidation (LPO) cascade generates many breakdown end-products, including reactive aldehydes such as malondialdehyde (MDA), MDA-modified proteins, 4-hydroxy-nonenal (HNE), HNE-modified proteins, and levels of F2 isoprostanes [12]. LPO generates a wide variety of metabolites, the best known being the saturated mono aldehydes, unsaturated aldehydes, dicarbonyls, MDA, 4-oxo-2-nonenal, hydroxydialdehydes (4-hydroxy-2-nonenal, 4-hydroxy-2-hexenal), and oxidized phospholipids [13]. The defense mechanisms of the human organism can limit the destructive effect of LPO. These include the metabolization of LPO by oxide reductase enzymes (aldo-keto-reductase, aldehyde-dehydrogenase, alcohol-dehydrogenase, glutathione-S-transferase) and cellular antioxidant defense mechanisms that include enzymes such as superoxide dismutase (SOD), catalase (CAT), glutathione peroxidase (GPx), glutathione reductase, thioredoxin reductase, heme oxygenase, and vitamins A, C, E, carotenoids, flavonoids, glutathione (GSH) and other antioxidant minerals [14].
The free radical-mediated peroxidation of arachidonic acid generates isoprostanes independent of cyclooxygenase activity. Measurement of plasma F2-isoprostanes provides a window into the balance between ROS production and antioxidant defenses in vivo [15]. Due to the formation mechanism, their specific structural characteristics distinguish them from other products generated by radicals and their relative chemical stability. The measurement of F2-isoprostanes in plasma is currently accepted as the most reliable index of OS in vivo [16]. Therefore, elevated levels of F2-isoprostanes are associated with oxidative tissue damage in various autoimmune disorders [17]. In SLE, plasmatic levels of F2-isoprostane are related to high disease activity, renal manifestations, and antiphospholipid syndrome [18]. This marker is also helpful in assessing the relationship between OS and mitochondrial function [19]. Evidence suggests a positive correlation between elevated LPO levels and disease activity in patients with SLE [20].
DNA Damage and Repair in SLE
ROS can attack all cellular biomolecules, including DNA and RNA. The formation of 8-hydroxy-2′-deoxyguanosine (8-OHdG) causes oxidative damage to guanine (G). 8-OHdG is the most common form of oxidative DNA damage. Without G repair, adenine (A) can mispair with 8-OHdG instead of cytosine (C) and cause G: C to T: A cross-mutation in human cells [21]. DNA repair and response mechanisms identify genetic faults and ensure proper DNA replacement during the cell cycle. The cell transfers the mutant genome to its descendants in the presence of unrepaired lesions. The determination of the 8-OHdG marker reflects the oxidative status of the whole organism. 8-OHdG accumulates in tissues and induces genomic instability and cellular dysfunction [22]. Moreover, 8-OHdG is a sensitive and specific predictor for determining oxidative DNA damage in SLE flares (Figure 1). 8-OHdG can be repaired primarily by the human enzyme 8-oxoguanine DNA glycosylase 1 (hOGG1) through base excision repair mechanisms. hOGG1 performs preventive actions against inflammatory responses by reconfiguring 8-OHdG levels [23,24].
Antioxidants in SLE
Free radicals (FR) and other ROS can interact with all the biomolecules present in SLE patients to create harmful products that alter immune regulation and trigger autoimmunity. The abundance of ROS correlates with disease activity, and the damaging cascade is controlled by antioxidant repair systems (Figure 1) [25]. The most common enzymatic antioxidants are peroxidase, SOD, CAT, heme oxygenase, GPx, glutathione reductase (GR), and thioredoxin reductase [26,27]. Non-enzymatic antioxidants include vitamins A, C, and E, carotenoids, flavonoids, GSH, uric acid (UA), and various antioxidant minerals [28]. Glutathione (L-γ-glutamyl-L-cysteinylglycine) is required for many biochemical pathways, playing a significant role in maintaining and regulating the thiol-redox state of the cell. In the normal cell, >90% of the total GSH pool is in the reduced form, and <10% is in the oxidized form (GSSG) [29]. The GSH: GSSG ratio could be a valuable tool to define OS because the changes can correlate with disease activity in patients with SLE [30].
The antioxidant enzyme SOD is an active protease containing metallic elements and widely exists in animals and plants [20]. SOD is necessary for its ability to clean oxygen FRs, where its levels are a sensitive index of antioxidant performance [31]. SOD plays a vital role in the genesis of SLE by maintaining the balance of the body's oxidation-antioxidation system [32]. Several SOD isoforms (SOD1, SOD2, and SOD3) have been found in human cells [33]. SOD1 is a metallo detoxifying enzyme and a scavenger of the radical anion O2•- that exists primarily in the cytoplasmic, peroxisomal, and mitochondrial membrane space. SOD1 accounts for approximately 90% of SOD activity in eukaryotic cells [34]. The O2•- anion is converted to H2O2 by the action of copper/zinc SOD (Cu/ZnSOD) in the cytosol and manganese SOD (MnSOD) in the mitochondria. H2O2 is reduced to H2O by CAT- or GSH-supported GPx-1 in the cytosol and GPx-4 in the mitochondria. GSH is the main non-enzymatic antioxidant and redox modulator of human cells. Through GPx, GSH can be oxidized to glutathione disulfide (GSSG) to reduce H2O2 to H2O. GSSG is reduced to GSH to maintain a sufficient antioxidant level [35]. The increase in OS could reflect lower enzymatic activity of CAT in SLE, which could favor the accumulation of harmful H2O2. On the other hand, the oxidant products are considered potential neo-antigens that could be involved in the pathogenesis of SLE.
A decrease in CAT activity could affect the oxidant-antioxidant balance. The chronic alteration of this balance in patients with SLE may favor the premature appearance of atherogenesis with severe vascular effects [36]. There are other organic metabolites with antioxidant capacity, such as UA. UA results from the breakdown of ingested and endogenously synthesized purines and is excreted by the kidneys and the intestinal tract without further metabolism [37]. UA could function as an antioxidant, protecting cells from the adverse effects of ROS [38]. The UA molecule is involved in a complex reaction and has protective functions under OS conditions [39].
Mitochondrial Function in SLE
Mitochondria are the principal organelles responsible for ATP production by integrating the glycolysis process in the cytosol and the Krebs cycle, electron transport, and OxPhos in mitochondria. Although most of the respiratory enzyme complexes (approximately 90 polypeptides) are encoded by nDNA, 13 are encoded by mtDNA. Therefore, mtDNA copy number and expression of mtDNA-encoded polypeptides may be crucial for energy delivery to cells. In general, mtDNA replication and transcription are controlled by the mitochondrial transcription factor A (Tfam) and further regulated by the nuclear respiratory factor (NRF)-1/NRF-2 [42]. In glycolysis, the enzymes hexokinase-II (HK-II), glucose 6-phosphate isomerase (GPI), and phosphofructokinase (PFK) regulate the rate-limiting step and, together with glyceraldehyde 3-phosphate dehydrogenase (GAPDH), participate in the conversion of glucose into pyruvate that is metabolized to acetyl-CoA to enter the Krebs cycle [43]. Under some extreme conditions (inadequate oxygen supply or impaired mitochondrial function), pyruvate is transiently reduced to lactate by lactate dehydrogenase with the help of cytosol-stabilized hypoxia-inducible factor 1 alpha (HIF-1α) [44]. The mitochondrial dysfunction that results in OS could be associated with tissue and cellular damage typical in many T cell-mediated autoimmune diseases. The autoreactive CD4 T cell effector subsets (Th1, Th17) that drive these diseases require increased glycolytic metabolism to regulate the upregulates critical transcription factors (TF) such as T-bet and RORγt that drive differentiation and pro-inflammatory responses. Research in immunometabolism has shown that mitochondrial-derived ROS act as signaling molecules that contribute to T cell fate and function. Eliminating autoreactive T cells is directed to glycolysis or ROS production as a potential strategy to inhibit the activation of autoreactive T cells without compromising systemic immune function. Increasing auto tolerance by promoting functional immunosuppressive CD4 regulatory T cells (Tregs) is a therapeutic alternative for autoimmune disease. Tregs require increased ROS and OxPhos for expression, differentiation, and synthesis of anti-inflammatory cytokines IL-10 from TF Foxp3. Decreasing the glycolytic activity or increasing the antioxidant activity of GSH and the antioxidant enzyme SOD may be beneficial in inhibiting effector responses of cytotoxic CD8 T cells. Current treatment options for T cell-mediated autoimmune diseases such as type 1 diabetes mellitus, multiple sclerosis, rheumatoid arthritis, and SLE include global immunosuppression, antibodies to deplete immune cells, and anti-cytokine therapy. While they effectively decrease autoreactive T cells, they can also compromise other immune responses, increasing susceptibility to other diseases and complications. The impact of mitochondria-derived ROS could be a potential therapeutic target for T cell-mediated autoimmune disorders such as SLE [45].
Mitophagy
Mitochondria form a sophisticated and dynamic network for energy production, primarily through OxPhos in connection with glycolysis and fatty acid oxidation. During OxPhos, electrons are transported in the inner chain of the mitochondria to generate an H+ gradient, which is crucial for the final step of ATP generation. Electron leakage or damaged transport chain can lead to ROS formation. Some ROS are not neutralized and act on cell homeostasis, with low ROS levels contributing to cell proliferation and survival. However, at higher levels, their actions as protein/lipid oxidants and inducers of DNA damage contribute to tumorigenesis and apoptosis. The accumulation of faulty mitochondria directly affects energy production and cellular homeostasis, and cells may attempt to repair them through fusion/fission mechanisms or degrade them through mitophagy [46]. The analysis of autophagy in peripheral B cells of patients with SLE was recently reported, demonstrating maximal activation in naïve B cells. In SLE, naïve B cells encounter a tolerance checkpoint after faulty immunophenotyping of B cells upon exiting the bone marrow [47]. Thus, autophagy could represent a potential therapeutic target in SLE because it may be a clinically relevant mechanism of action of the commonly used immunomodulatory antimalarial hydroxychloroquine as an inhibitor of autophagy by raising lysosomal pH and preventing autophagosome-lysosome fusion (Figure 1) [48].
Toll-like Receptors in SLE
B lymphocytes are central in autoimmune diseases because specific autoantibody patterns often define them and exhibit a loss of tolerance. SLE is a prototypical disease associated with B cell hyperactivity. In SLE patients, loss of B cell tolerance to self-antigens is intrinsically controlled in the cells by TLRs. The TLRs are a family of receptors whose activation is crucial for the induction of innate and adaptive immune responses. Nucleic acid ligands activate a subset of TLRs on endosomes. TLR7 and TLR9 recognize single-stranded RNA fragments and DNA sequences containing unmethylated cytokine-phosphate-guanosine motifs, respectively (Figure 1) [49]. TLR signaling promotes three key activities that on B cells can contribute to autoimmune diseases: a) antibody production, b) antigen presentation to T cells, and c) cytokine production. Genetic association studies implicate TLR signaling in SLE [50]. A TLR7 variant capable of producing greater activation in childhood-onset SLE has even been described [51].
TLR7 drives the extrafollicular B cell response and germinal center reaction in autoantibody production and disease pathogenesis. Surprisingly, TLR9 appears to protect against SLE. TLR9 is required to produce autoantibodies that recognize double-stranded DNA-associated antigens that are abundant in SLE and are the hallmark of the disease. The protective function of TLR9 is mediated, in part, by its ability to limit the stimulatory activity of TLR7. Hence, the role of TLR7 and TLR9 in B cell effector function in patients with SLE and the unique characteristics of TLR signaling in B cells could have beneficial therapeutic effects [52]. Expression of TLR7 is higher in women than in men because of its location on the X chromosome. One X chromosome usually is inactive in women, but some genes on the X chromosome, including TLR7, always seem to escape inactivation. As a result, TLR7 is expressed bi-allelically in plasmacytoid dendritic cells (pDC), monocytes, and B cells. Thus, LR7 is present at higher levels in women's cells than in men's. B cells from women exposed to TLR7 agonists in vitro differentiate more efficiently into CD27hi plasmablasts than B cells from men. This gender difference is not observed with the addition of TLR9 (gene encoding chromosome 3) agonists [53], which is consistent with the higher prevalence of SLE in women than in men [54]. The number of X chromosomes affects susceptibility to SLE. The presence of two X chromosomes in men with Klinefelter syndrome is associated with a greater predisposition to present SLE than in men with only one X chromosome [55]. Similarly, women with only one X chromosome (for example, those with Turner syndrome) are less prone to SLE than women with two X chromosomes [41]. The reduction in the activity of TLR7 could lessen SLE development. TLR7 expression is also modulated by metabolic parameters (for example, a high-fat diet), which exacerbates SLE [56]. It would be possible and desirable to reduce the symptoms of SLE by modulating the function of TLR7 [57]. However, circumstantial evidence supports enhanced TLR7 signaling as a mechanism of human systemic autoimmune disease [58]. TLR7 is a sensor for viral RNA and binds guanosine [59]. SLE patients display phenotypes consistent with increased TLR7 signaling associated with elevated CD27-IgD double-negative B cells and the CXCR5-CD11c+ subset (DN2 B cells or age-associated B cells) [60]. TLR7s are critical receptors for extra-follicular and germinal center responses associated with activating autoreactive B cells implicated in disease. The different endosomal TLRs act as nucleic acid sensors [53].
TLR9 has protective functions against SLE, though TLR9 must recognize the abundant double-stranded DNA-associated antigens in SLE, which are the disease's trademark. Therefore, the protective process of TLR9 is mediated, at least in part, by its ability to limit the stimulatory activity of TLR7. The roles of TLR7 and TLR9 in B cell effector function in SLE and the key features of TLR signaling in B cells suggest that both TLRs could be therapeutically beneficial targets [52]. Antagonism between TLR7 and TLR9 can occur within a single B cell if it expresses a B cell antigen receptor (BCR) that recognizes self-antigens comprising TLR7 and TLR9 agonists. However, it was recently reported that TLR9 restricts the differentiation of B cells instructed by TLR7 in vitro [61].
Intracellular nucleic acids are detected by TLR8, which also detects single-stranded RNA [62]. TLR8 and TLR9 restrict TLR7 activity in dendritic and B cells, respectively [63]. Although the different endosomal TLRs act as sensors for nucleic acid, TLR7, TLR8, and TLR9 have distinct functions in patients with SLE. TLR8 and TLR9 appear to have beneficial functions for SLE patients, making it relevant to discover the biochemistry of these molecular processes, which could lead to identifying new targets for drug development [52]. The antagonistic interaction between TLR7 and TLR9 within a B cell is highlighted by the competition of TLRs for the intracellular protein UNC93B1 that promotes their transit into endosomal compartments [64].
Lupus Nephritis
Lupus nephritis (LN) is a form of glomerulonephritis that constitutes one of SLE's most severe and disturbing organic manifestations. Most patients with the disease develop LN within five years of diagnosis, and in many cases, LN is the initial diagnostic manifestation of the disease [65]. LN is one of the leading causes of morbidity and mortality in most patients at some point in the course of the disease, characterized primarily by long-lasting relentless proteinuria. Therefore, proteinuria is considered a serious and frequent SLE complication and a poor prognostic sign [66]. LN patients have varying degrees of kidney injury and proteinuria, and proteinuria levels are also unstable in LN patients, which is associated with a robust glomerular filtration membrane self-renewal and repair system (Figure 2) [67]. In early LN, it is possible to repair pathologic damage to the filtration membrane caused by various complement proteins and cytokines through autoregulation. However, the injury cannot be fully repaired as the disease progresses, leading to severe proteinuria. Thus, to prevent proteinuria and alleviate kidney damage in patients with LN, it is urgent to explore the self-repair mechanism of the glomerular filtration membrane in early-stage LN and its failure mechanism in late-stage LN [68].
There are various mechanisms of proteinuria, podocyte dysfunction or injury being the most crucial cause. Podocyte-induced proteinuria is associated with pore membrane protein expression [69]. Podocytes are highly specific innate glomerular cells and play a role in the glomerular basement membrane filtration barrier, pore size, and energy charge [70]. The autophagy process is activated in LN, especially in podocytes. One of the principal causes of proteinuria in LN is podocyte damage, so searching for therapeutic targets to prevent podocyte damage is essential from a clinical perspective in treating LN [71].
The deposition of immune complexes in the glomerulus and the activation of the complement system are involved in the pathogenesis of LN. Additionally, evidence suggests that OS significantly contributes to renal failure over time in SLE [72]. Patients with LN in the active phase have an unbalanced redox state that causes lipid peroxidation of the glomerular basement membrane, altering its integrity and affecting tubular function in patients with SLE. OS can inflict renal damage by promoting tumor necrosis factor-alpha (TNF-α) production by infiltrating macrophages, excessive Th17 cell differentiation, secretion of the inflammatory cytokine IL-17, and activation of neutrophils. By blocking TNF-α, it has been possible to alleviate OS and renal damage in NZB/NZW mice with interferon-alpha (IFNα)-induced LN [5]. Because kidney injury acts as a risk factor for cardiovascular disease (CVD), OS is the basis of the pathogenesis of CVD in SLE. It is possible to observe an increased risk of atherosclerosis and CVD in patients with SLE compared to healthy individuals, and the disruption of lipid and lipoprotein metabolism induced by OS is of critical importance [73]. MDA-modified oxidized low-density lipoprotein (OxLDL), autoantibodies against endothelial cells, and phospholipids may also contribute to endothelial damage in early lupus directly or through activation of the type I IFN pathway [74]. Monocytes phagocytosed OxLDL to differentiate into foam cells and release inflammatory substances, predisposing to atherosclerosis. Neutrophil extracellular traps (NETs) are central components of neutrophils involved in OS with the ability to damage endothelial cells, activate macrophages and release myeloperoxidase (NADPH oxidase capable of oxidizing HDL), and decrease levels of the antioxidant HDL [75]. However, OS is essential for forming anti-β2GPI antibodies and thrombotic events in patients with SLE. Moreover, the OxLDL/β2GPI/anti-2GPI complex induces the differentiation of macrophages in foam cells, which promotes the development of atherosclerosis, possibly through the mechanism by which 4-hydroxy-alkenes oxidize the β2GPI antigen, thereby that it enhances immunogenicity in response to increased anti-β2GPI antibodies and that antiphospholipid (APL) antibody cross-react with OxLDL, facilitating its entry into macrophages and promoting the progression of atherosclerosis [76]. Most atherosclerosis risk factors increase OS and accelerate disease progression. Arterial hypertension is associated with OS, and its presence in SLE implies a dysregulated ratio of Th1/Th2 cytokines, increased production of IL-17, and insulin resistance. By augmenting the susceptibility of the tiny renal inlet arteries to angiotensin II and the expression of sodium chloride co-transporters, OS may enhance the reabsorption of water and sodium in the renal tubules, precipitating the development of hypertension [77,78]. Nuclear factor E2-related factor 2 (Nrf2) is a vital regulator of the antioxidant response in LN. Nrf2 has an antioxidant role that promotes cell protection and prevention of tissue damage [79]. It was recently revealed that the expression of Nrf2 and downstream molecules, heme oxygenase-1 (HO-1) and GPx, are downregulated in mouse models of diabetic nephropathy and IgA nephropathy [80].
Oxidative Stress in LN
Oxidation is key in developing the organic damage characteristic of SLE, especially in LN (Figure 3) [81]. LPOs generate a variety of metabolites, the best known being saturated aldehydes such as MDA, unsaturated aldehydes, dicarbonyls, MDA, 4-oxo-2-nonenal, hydroxydialdehydes (4-hydroxy-2-nonenal, 4-hydroxy-2-hexenal), oxidized phospholipids, and others [82]. In LN, high levels of OS were previously detected in patients with active SLE, suggesting the link between LPO and disease activity. Increased activity of MDA, HNE, MDA protein adduct, HNE protein adduct, SOD, inducible nitric oxide synthase (iNOS), anti-MDA, and anti-HNE antibodies correlate with the SLE Disease Activity Index (SLEDAI) in patients during follow-up [86]. The destructive effect of LPO could be limited by the defense mechanisms of the human organism, such as the metabolization of LPO by oxide reductase (aldo-keto-reductase, aldehyde-dehydrogenase, alcohol-dehydrogenase, glutathione-S-transferase) and mechanisms of cellular antioxidant defense including enzymes (SOD, chloramphenicol acetyltransferase—CAT, GPx—GPx, reductase—GR, S-transferases—GST, thioredoxin-reductase, heme oxygenase), non-enzymatic vitamins A, C, E, and carotenoids, flavonoids, glutathione, and other antioxidant minerals [83]. F2 isoprostanes (8-iso-PGF2) levels are a result of LPO and are correlated with disease activity in subjects with SLE [84].
The increase in protein carbonyls may result from the presence of multiple oxidants. Carbonyl formation indicates that total protein oxidation levels are high in patients with SLE and LN, thus suggesting there is an altered balance between oxidation and repair in these patients [85]. However, increased levels of protein-bound carbonyls have been detected in aging and various diseases, including rheumatoid arthritis, pulmonary fibrosis, diabetes, Parkinson's disease, and Alzheimer's disease [86].
Nitric oxide (NO) is a ubiquitous intra and extracellular messenger molecule involved in various signaling pathways for different physiological processes, including inflammation, immunity, ion channel modulation, gene transcription, and vascular tone [87]. NO is synthesized from L-arginine by a group of enzymes called NO synthases (NOS) [88]. There are three isoforms of NOS, often described based on their tissue expression: neuronal NOS (nNOS or NOS1), inducible NOS (iNOS or NOS2), and endothelial NOS (eNOS or NOS3) [89]. The kidney expresses all three isoforms, which are known to affect natriuresis and diuresis and contribute to blood pressure control [90]. Various renal cells can release NO, including endothelial and mesangial cells, the macula densa, and podocytes [91]. NO is a potent signaling molecule involved in many physiological and pathophysiological processes in the kidney. NO plays a complex role in ultrafiltration, vasodilation, and glomerular inflammation. Changes in NO bioavailability in pathophysiological conditions such as hypertension or diabetes can lead to podocyte damage, proteinuria, and the rapid development of chronic kidney disease (CKD). Despite the wealth of data emphasizing the essential roles of NO in health and pathology, related signaling in glomerular cells, particularly podocytes, needs to be better studied. Various reports indicate that the bioavailability of NO in glomerular cells decreases during the development of renal pathology. Restoring NO levels may be beneficial for glomerular function. At the same time, the compromised activity of the NOS enzyme can lead to the formation of peroxynitrite (ONOO-). ONOO- is related to autoimmune diseases such as SLE. Changes in the distribution of NO sources due to altered expression of NOS subunits or shifts in the activity of NADPH oxidases can link or promote the development of pathologies. However, the mechanisms describing NO's production and release in glomerular cells, the interaction of NO and other ROS in podocytes, and how crosstalk between NO and calcium regulates glomerular cell function are still not fully described [92]. iNOS is upregulated in mouse models of LN and is overexpressed in the glomerulus in proliferative human LN [93]. Administration of iNOS competitive inhibitors reduced reactive OS markers in two mouse models of LN, strongly suggesting that most systemic OS in these models results from iNOS activity. The relative swiftness of ROS reduction with iNOS inhibitors points to a direct effect on the iNOS enzyme rather than the known impacts of more extended treatment with iNOS inhibitors on cell proliferation and glomerular infiltration in murine models [94]. It was previously reported that brief therapy with iNOS inhibitors does not significantly affect renal pathology or that deletion of the iNOS gene in the absence of infection reduces systemic OS levels [95].
Oxidative Damage to DNA in LN
In normal cells, 2-deoxyguanosine (dG) associates with 2′-deoxycytosine (dC) during DNA replication. In tissues with high OS levels, dG could mispair 2′-deoxyadenine (dA) and induce G → C to T → A transversion. If 8-OHdG is not cleared efficiently, it accumulates in the tissues causing genomic instability and cellular dysfunction [96]. Usually, OGG1 is responsible for the clearance of 8-OH-dG and OGG1 deficiency induces high levels of 8-OH-dG in DNA. OGG1 overexpression improves mitochondrial function and cell survival while reducing the number of DNA lesions by 8-OH-dG repair under OS conditions in vitro. OGG1 could play a protective role in inflammatory diseases. Furthermore, the OGG1 polymorphism could offer susceptibility to LN and modulate the serum level of 8-OH-dG in patients with SLE [97].
Antioxidants in LN
Antioxidants may protect against the development of SLE by opposing OS [98]. Surface thiols and GSH participate in the redox quenching of cells, protecting against OS. Chronic LN inflammation is associated with OS and decreased levels of cellular antioxidant sulfhydryls [99]. The imbalance of the oxidative state, represented by an increase in plasma MDA and a reduction in GSH, is a possible cause of LN activity in SLE (Figure 3). However, treating LN with antioxidants is rarely documented [100,101]. N-acetylcysteine (NAC) is a reliable antioxidant often applied for clinical treatment. In two recent cases of LN treated with antioxidants, the authors reported their beneficial effect in modulating oxidative status, though the underlying mechanisms require further investigation [102]. Studies have indicated that antioxidants can reduce oxygen consumption in mitochondria and block mTOR (regulator of mitochondria oxygen consumption and OS). In LN, antioxidants increase GSH and lower F2-8-isoprostane levels [103].
Mitochondrial Function in LN
The mitochondrion is an intracellular organelle that regulates numerous cellular functions, among which the best known are ATP production and programmed cell death [104]. Mitochondria are derived from the endosymbiosis of an α-proteobacterium and a precursor of the eucaryotic cell, giving the organelles many bacterial characteristics [105]. Unlike other organelles, mitochondria form by binary fission and cannot be produced directly by the cell. Mitochondria consist of an intermembrane space and a mitochondrial matrix separated by the outer mitochondrial membrane and the inner mitochondrial membrane. Mitochondria are assembled through the interaction between the nuclear and mitochondrial genomes. Mammalian mitochondrial DNA (mtDNA) encodes 37 genes, 13 of which encode polypeptide components of the oxidative phosphorylation machinery, and the 22 tRNAs and two rRNAs required for gene transcription and translation within the organelle [106]. Approximately 99% of mitochondrial proteins are encoded by nuclear genes, translated on cytoplasmic ribosomes, and imported into mitochondria by the translocase outer membrane (TOM) and translocase inner membrane (TIM) complexes [107]. Mitochondria are involved in the synthesis of fatty acids, the production of amino acids, the synthesis of heme, and the biogenesis of iron and sulfur groups [108]. Mitochondria communicate with the ER through the mitochondria-associated ER membrane (MAM) to regulate Ca2+ homeostasis, lipids, and apoptosis [109]. Mitochondria are an important site of ROS (mtROS) production. Under normal conditions, mtROS are rapidly cleared by the enzyme SOD2 in the mitochondrial matrix and SOD1 in the mitochondrial intermembrane space, as well as by other antioxidant enzymes, GPX, GSH, and glutathione disulfide [110]. Physiological levels of mtROS are essential for various signaling functions by maintaining the functional state of mitochondria, while excess mtROS causes oxidative damage to proteins, lipids, and DNA, leading to ATP depletion and a further increase in the production of mtROS and activation of the inflammasome, which exacerbates cell damage and initiates programmed cell death [111]. Mitochondria contain numerous copies of a compact circular genome that encodes RNA molecules and proteins involved in mitochondrial OxPhos. The mtDNA activates the immune system present in the cytosol or the extracellular environment. Because mitochondria retain several features of their ancestral prokaryotic origin, releasing mitochondrial components into the extracellular milieu can activate the innate immune system [112]. Cardiolipin, N-formylated peptides, mtDNA, ATP, and ROS, are known damage molecular patterns (DAMPs) associated with mitochondria that activate cells through nuclear oligomerization domain-like receptors, TLR-like receptors (e.g., TLR9 for mtDNA) or formyl peptide receptors [113]. The mitochondria are also the target of circulating autoantibodies in SLE. However, whether mtRNA is also recognized by autoantibodies in SLE is unknown [114]. Anti-mitochondrial autoantibodies recognize proteins, such as those involved in OxPhos, phospholipids, or unidentified epitopes present on the mitochondrial membrane. Despite the extensive literature on antibodies directed to cardiolipin (mitochondrial M1 antigen) in SLE, the repertoire of anti-mitochondrial autoantibodies and their antigenic targets must still be characterized [115].
Metabolic abnormalities influence the nature and state of activation of the kidney's immune cell infiltrate, highlighting the importance of mitochondrial function in developing diseases such as LN [116]. An analysis of gene expression profiles in lupus-affected human and mouse kidneys revealed increases in gene sets characteristic of myeloid cells, accompanied by decreases in genes that control glucose and lipid metabolism [117]. Even metabolism-linked transcriptional alterations were found in LN patients with less severe glomerular damage, indicating that metabolic dysfunction is an early and common change in lupus-affected tissues that results from immunological processes and contributes to tissue damage. At the same time, the expression of tubular damage markers was negatively correlated with the tricarboxylic acid (TCA) cycle in murine models of LN. Likewise, transcriptional studies show that defects in regulating fatty acid oxidation in renal tubular epithelial cells facilitate important intracellular damage mechanisms in LN such as lipid deposition, ATP depletion, cell death, and fibrosis [118].
It has been described that glycolysis regulates macrophage polarization and the association between the intrarenal presence of macrophage markers with increased pentose phosphate pathway activity linked to renal dysfunction and increased cytokines in patients affected by SLE [119]. Similarly, T cells can increase glycolysis in response to their activation, and this increase, while essential to carry out their effector functions, can lead to autoimmunity [120,121,122]. Some studies suggest that the highest percentage of kidney-infiltrating cells correspond to T cells with an activated phenotype [123,124]. However, recent evidence demonstrates that in LN, CD4 and CD8 T cells from renal tissue are not functional effector cells but have reduced ability to proliferate and produce cytokines [125]. This hypofunctional phenotype observed in preclinical models of LN has been linked to the presence of mitochondrial dysfunction and an "exhausted" transcriptional signature.
Interferon-gamma (IFNγ) produced by CD4 T cells and nicotinamide phosphoribosyl transferase (NAMPT), a rate-limiting enzyme in the NAD+ biosynthetic pathway, are crucial elements in the pathogenesis of LN [126]. In CD4 T cells from LN patients or MRL/lpr NAMPT mice, aerobic glycolysis and mitochondrial respiration are promoted through the production of NAD+. NAMPT inhibition suppresses IFNγ production in CD4 T cells, thus decreasing inflammatory cell infiltration and renal damage. NAMPT can potentially normalize metabolic competence and pathogenicity of CD4 T cells in LN. It has also been observed that normalization of glycolysis and oxidative metabolism in CD4 T cells by treatment with metformin and 2-deoxy-D-glucose leads to disease improvement in murine models of lupus [126,127,128]. This evidence supports the development of targeted therapies to control mitochondrial metabolism in T cell subsets to treat systemic autoimmune diseases such as LN.
Mitophagy in LN
Mitophagy is the process by which dysfunctional or superfluous mitochondria are selectively removed by autophagy to control their quality and quantity [129]. Recent mitophagy studies reveal that mitochondrial priming is mediated by the phosphatase and tensin homolog (PTEN) induced kinase 1 (PINK1)/Parkin signaling pathway and mitophagy receptors [130]. Mitophagy is potently induced during OS and facilitates mitochondrial quality control to mediate metabolic adjustments to external challenges. At the same time, impaired mitophagy is responsible for mitochondrial dysfunction and the progressive accumulation of defective organelles, leading to cell death and tissue damage. The mild and transient OS induced by H2O2 at low concentrations for a short time leads to low levels of ROS capable of inducing mitophagy, suggesting that mitophagy functions as an early cytoprotective response that favors OS adaptation by eliminating damaged mitochondria [131]. Once cellular mitochondria are damaged by increased OS and apoptotic proteases exceed the range that mitophagy can eliminate, the programmed cell death pathway is activated [132]. The balance between mitophagy and apoptosis plays a critical role in determining cell fate under conditions of OS, hypoxia, DNA damage, and loss of growth factors [93]. The kidney is an energetically demanding organ rich in mitochondria; even renal function depends to a large extent on mitophagy [133].
Mitochondrial antigens can be generated by degrading old or damaged mitochondria through mitophagy. Mitochondria-containing auto phagosomes travel through the endo-lysosomal system, leading to the degradation of their cargo and allowing the production of mitochondrial peptides that can be processed and expressed by the major histocompatibility complex (MHC). Both MHC-I and MHC-II have been implicated. However, a recent study revealed that mitochondrial antigen processing can also occur independent of mitophagy. In this case, mitochondrial antigens are transported to endosomes by mitochondria-derived vesicles formed by a mechanism regulated by PINK1 and Parkin proteins [134]. There is an intense effort to discover new biomarkers that make it possible to discriminate patients with SLE specifically. Anti-mtDNA antibodies (AmtDNA) are positively associated with nephritis, whereas anti-mtRNA antibodies (AmtRNA) show a negative association. The presence of AmtDNA and AmtRNA can help predict the risk of kidney damage in patients with SLE (Figure 3) [135].
Emerging evidence suggests that aberrant or defective mitophagy is central to many renal diseases, including LN pathology [136]. Multiple signaling pathways are involved in the deterioration of mitophagy in SLE and LN, so multi-targeted therapies are required to induce remission and prevent flare-ups. Rapamycin prevents LN development in lupus-prone mice [137] and patients with SLE [138] by inhibiting mTOR and enhancing autophagosome formation and autolysosomal degradation [139]. Recent studies have demonstrated the protective effects of rapamycin on mitochondrial function in the context of SLE. Rapamycin increases Drp1 through mTOR inhibition in lupus-prone mice and decreases mitochondrial dysfunction by activating mitophagy. Competent mitophagy may have therapeutic effects in LN, and drugs that induce mitophagy, such as rapamycin and 3-PEHPC, deserve further exploration as therapeutic strategies to enhance clearance of fragmented mitochondria that promote injury and speed recovery from mitochondria. SLE and LN outbreaks [140].
Innate Immunity in LN
The pathogenesis of LN involves a variety of mechanisms. The extra-renal etiology of SLE is based on multiple combinations of genetic variants that compromise the mechanisms that generally ensure immune tolerance to nuclear autoantigens. The loss of immune tolerance becomes clinically detectable by the presence of antinuclear antibodies. The oxidant/antioxidant imbalance in SLE due to exposure to endogenous or exogenous toxic factors produced by alteration of the response/repair mechanisms of tissue damage induces aberrant activity of the innate and adaptive immune response with high production of autoantibodies and multiple lesions of the target tissues and organs [141]. The nucleic acid component of the immune complexes activates intrarenal inflammation by TLRs in intrarenal macrophages and dendritic cells. Immunostimulatory nucleic acids activate glomerular endothelium, mesangial cells, and macrophages to produce large amounts of pro-inflammatory cytokines, IFN-α, and interferon-beta (IFN-β) [142]. Nucleic acids released from the network or apoptotic neutrophils activate innate and adaptive immunity via viral nucleic acid-specific TLRs. Therefore, many clinical manifestations of SLE resemble a viral infection [126]. In SLE, endogenous nuclear particles activate IFN-α signaling in the same way as viral particles during viral infection. Dendritic, helper T, B, and plasma cells contribute to aberrant polyclonal autoimmunity. The intrarenal etiology of LN involves binding antibodies to multiple intrarenal autoantigens rather than the deposition of circulating immune complexes. Tertiary lymphoid tissue formation and local antibody production add to intrarenal complement activation as renal immunopathology progresses [143]. The delay in eliminating dead cells leads to the degeneration of their components, compromising the elements that distinguish self-nucleic acids from viral nucleic acids [144]. However, nature evolved DNA and RNA methylation to inhibit RNA and DNA recognition by TLRs 3, 7, and 9 (endosomal viral nucleic acid recognition receptor set) that trigger antiviral immunity during viral infection [145]. Dendritic and B cells can process antigens, present antigens to T cells, and substitute for each other for that purpose [146]. Dendritic cells have a limited lifespan, but their persistent activation by SLE autoantigens through TLR7 and TLR9 improves their survival and renders them resistant to glucocorticoid-induced killing [147]. The nucleic acid component of immune complexes also activates intrarenal inflammation via TLR in intrarenal macrophages and dendritic cells (Figure 4) [148]. Immunostimulatory nucleic acids activate glomerular endothelium, mesangial cells, and macrophages to produce large amounts of pro-inflammatory cytokines IFN-α and IFN-β [149]. The functional importance of intraglomerular IFN signaling needs to be better understood. Still, it contributes to renal damage in LN and would trigger the formation of tubuloreticular structures or inclusions as an ultrastructural feature of IFN signaling [150]. The binding of TLRs and complement receptors activate renal cells to release pro-inflammatory cytokines and chemokines and induce luminal expression of selectins and adhesion molecules within the microvasculature [151]. The concept of pseudo-antiviral immunity is based on molecular mimicry of endogenous nucleic acids in the viral nucleic acid recognition receptors TLR7 and TLR9 [152]. TLR blockade and subsequent IFN signaling add to the established therapeutic targets in SLE [153].
Toll-like Receptors in LN
Current therapeutic approaches may fail due to the inability to direct distal innate immune responses to immune complex deposits adequately. Understanding the mechanisms that drive innate downstream responses is essential for developing new therapies for SLE and LN. A key mediator of innate immune responses is increased ROS production in response to inflammatory stimuli [154].
The local release of pro-inflammatory cytokines blocks TLRs with the capacity to improve the efficacy of treatment in autoimmunity without increasing systemic immunosuppression [155]. TLR9 is in the endosomal compartment where it can detect unmethylated CpG motifs in endogenous DNA sequences or exogenous DNA from viruses or bacteria [156]. There are endogenous TLR9 ligands like hypomethylated self-DNA within immune complexes, NETs, oxidized mitochondrial nucleoids, and other chromatin formats [157]. Defects in lysosomal maturation support TLR9 activation [148]. Additionally, Type I IFNs promote receptor crosslinking responsiveness in B cells and make dendritic cells responsive to endogenous nucleic acids after upregulating TLR7 and TLR9 [158]. Blockade of endogenous TLR9 ligands with a TLR9 antagonist in MRL/lpr mice ameliorates systemic autoimmunity and immune complex glomerulonephritis [159]. Consistently, CpG and LPS application to anti-dsDNA transgenic mice exacerbate the disease [160]. Extrinsic and intrinsic ligands can bind TLRs on infiltrating monocytes, dendritic cells, and B cells to enhance cytokine secretion [161].
TLR7 in LN
TLRs are mediators of innate antimicrobial and antiviral immunity in self-defense. The role of TLR in non-infectious disease states, such as autoimmunity and autoimmune tissue injury, remains to be fully elucidated. TLR7s were first described as innate pathogen recognition receptors that trigger appropriate antimicrobial immune responses upon exposure to pathogen-associated molecules. In parallel with ongoing studies on TLR biology, growing experimental evidence suggests that endogenous RNA-related self-antigens may also activate dendritic and B cells via TLR7. TLR7-mediated dendritic cell activation, autoantibody secretion, lymph proliferation, and autoimmune tissue injury are frequently observed in several murine models of SLE and LN (Figure 4) [162]. It was recently published that polymorphisms in TLR7 are related to the development of lupus [168]. Murine models of lupus have provided genetic and experimental evidence to support a role for TLR7 activation in LN pathogenesis [163]. Male BXSB mice, which contain an extra copy of TLR7 on the Y chromosome, develop LN, whereas females are protected [164]. Transgenic mice that overexpress TLR7 also develop lupus [165]. Murine lupus models, such as the pristane-induced lupus model, depend on TLR7 signaling for lupus pathogenesis [166]. Repeated epicutaneous application of the TLR7 agonist to wild-type mice leads to lupus-like features, including mild LN. Despite these supporting data, the mechanisms by which TLR7 signaling leads to LN remain unclear [167]. TLR7 activation can result in the production of type I IFNs and activation of nuclear factor κB (NFκB) in various cell populations, including dendritic cells, monocytes, macrophages, and B cells. Type I IFNs, including IFNα, can promote the development of lupus and are sufficient to accelerate nephritis in lupus-prone mice [168]. Several genetic and inducible models are protected from developing lupus when type I IFN signaling is knocked down [169].
Previously published research has shown that a moderate increase in TLR7 is sufficient for developing nephritis. Normalization of B cell TLR7 expression or temporary pDC depletion slows the progression of LN. Conventional dendritic cell expression of TLR7 is essential for severe autoimmunity in SLE. A new expanding CD11b(+) conventional CD subpopulation dominates the renal infiltrating inflammatory milieu with localization in the glomeruli. Exposure of human myeloid dendritic cells to IFN-α or Flu increases TLR7 expression, suggesting that they may have a role in self-RNA recognition pathways in clinical disease [170]. TLR7 recognizes single-stranded RNA (ssRNA), which induces downstream activation of signaling molecules, including Jnk and NFκB, through a myeloid differentiation primary response gene 88 (MyD88)-dependent cascade. This process is essential for the host's defense against invading viruses. However, TLR7 hyperactivity may also drive the initiation and progression of autoimmunity [171]. Multiple investigations have shown that TLR7 and the MyD88 signaling pathway are critical for initiating autoimmunity and developing auto reactivity because genetic ablation of TLR7 or MyD88 prevents the development of anti-nuclear antibodies and immune pathology. This signaling pathway is required explicitly within B cells [172]. TLR7 has emerged as a significant regulator of autoantibody production in murine LN. Tonic interactions between TLRs and environmental agonists derived from commensal microbes and endogenous sources may also influence autoimmune diseases and inflammatory disorders affecting the kidney [173]. The contribution of TLRs and other innate immune receptors in regulating inflammation, immune responses, and kidney injury remains to be elucidated. That knowledge will pave the way for novel therapeutic interventions.
TLR9 in LN
TLR9 regulates SLE pathogenesis and seems to be able to repress the disease. In recent studies of selective TLR9 deletion or over-expression, TLR9 deficiency in B cells was sufficient to exacerbate LN while quenching anti-nucleosome antibodies, whereas TLR9 deficiency in dendritic cells, pDCs, and neutrophils had no discernible effect on disease. B-cell-specific TLR9 deficiency appears to decouple the condition from autoantibody production. The authors highlight the non-redundant role of TLR9 expressed in B cells in regulating lupus and suggest therapeutic potential to modulate or enhance TLR9 signaling in B cells [174]. TLR9 and TLR7 have paradoxical effects on the pathogenesis of SLE, especially since both receptors involved in downstream signaling pathways are believed to be nearly identical (Figure 4). Deciphering why TLR7 and TLR9 play such different roles in SLE provides fundamental insights into the biology of critical TLRs and TLR signaling.
One hypothesis to explain the dichotomous effects of TLR7 and TLR9 is that there are cell type-specific functions for each TLR. TLR9 may be protective due to its effects on one cell type, and TLR7 may accelerate disease due to its impact on another. Alternatively, and non-exclusively, TLR9 can regulate TLR7 in a cis manner within the same cell type by competing for shared rate-limiting downstream signaling components [175]. The authors recently reported that, although TLR9 overexpression had a significant protective effect in two disease models, the scope of this protection may be limited by some technical aspects of the model used. TLR9 overexpression by both alleles did not occur in an average of 15.6% of B cells, so some B cells lacked the suppressive effect and might have dominantly promoted the disease, for example, by serving as antigen-presenting cells for autoreactive T cells. There is a precedent for this effect from escaped B cells when CD19-Cre was used to knock down MHCII in MRL/lpr mice conditionally [176]. Systemically administered TLR9 agonists have been used in clinical trials in the treatment of cancer and were generally well tolerated [177]. TLR9 agonists could be designed only to activate B cells or even DNA-specific B cells. Therefore, understanding how cell populations regulate SLE could allow for a more targeted therapeutic design [178].
Management of SLE and LN
Early and accurate diagnosis of LN and early initiation of therapy is of vital importance to improve outcomes in patients with SLE [179]. The primary goal of treatment for this immunologic disease includes long-term patient survival, prevention of target organ damage recurrences, and optimization of health-related quality of life. Treatment generally consists of an initial period of high-intensity immunosuppressive therapy to control disease activity, followed by a more extended period of less-intensive therapy to consolidate the response and prevent relapses. Managing disease and treatment-related comorbidities, especially infections and atherosclerosis, is paramount. Conventional agents and new disease-modifying biologic agents, alone, in combination, or sequential, have improved rates of short- and long-term treatment goal achievement, including minimization of glucocorticoid use [180].
The goal of LN therapy was established with a reduction in proteinuria by ≥25% with a stable glomerular filtration rate (GFR, ± 10% of baseline) in the first three months after the start of treatment, reduction in ≥50% proteinuria at six months and proteinuria < 0.5-0.7 g/24 h at 12-24 months (all with stable GFR) (Figure 5) [181]. All SLE patients with LN should receive hydroxychloroquine at a dose not exceeding 5 mg/kg of actual body weight [182]. Antimalarial drugs such as hydroxychloroquine inhibit lysosomal acidification, thereby blocking the adjuvant effect of endogenous nucleic acids by TLR7 and TLR9 during lysosomal processing of nuclear particles in the endolysosomal compartments of antigen-presenting cells [183].
During chronic maintenance treatment, glucocorticoids should be minimized to <7.5 mg/day (prednisone equivalent) and withdrawn when possible. Appropriate initiation of immunomodulatory agents (methotrexate, azathioprine, and mycophenolate mofetil (MMF)) may accelerate the tapering/discontinuation of glucocorticoids. In persistently active or exacerbating disease, belimumab should be considered as an add-on therapy for adult patients with active LN. Calcineurin inhibitors (voclosporin and tacrolimus) also show promising results for SLE with renal manifestations [184]. Even rituximab or cyclophosphamide (CY) may be considered in refractory organ-threatening disease [185]. It is a fact that immunosuppressive therapy commonly used to treat SLE improves the LN condition and patient outcomes. However, kidney damage is not reversible [186]. Despite increasing knowledge regarding the pathogenesis of the disease and the availability of better treatment options, within ten years of the initial diagnosis, 5-20% of patients with LN develop end-stage renal disease (ESRD) with multiple comorbidities associated with the immunosuppressive therapy used to treat the underlying condition, including infections, osteoporosis, cardiovascular effects, and reproductive effects [3]. Understanding the pathophysiological manifestations of renal function in LN has improved substantially in recent decades. Even more specific TLR7 and TLR9 agents that effectively suppress LN in mouse models of SLE have been developed and are now in clinical trials [187]. In active proliferative LN, initial treatment (induction) with low-dose intravenous CY (500 mg × 6 biweekly doses) or MMF, 2–3 g/day, or mycophenolic acid at an equivalent dose, both combined with glucocorticoids (pulses of intravenous methylprednisolone, then oral prednisone 0.3-0.5 mg/kg/day). Combination of MMF with calcineurin inhibitors or high doses of CY are considered alternative regimens for patients with nephrotic range proteinuria and adverse prognostic factors, subsequent long-term maintenance treatment with MMF or azathioprine [188]. Minimizing patients' exposure to glucocorticoids has received more attention after IV methylprednisolone pulses, the recommended starting dose being 0.3–0.5 mg/day prednisone equivalent. Prednisone should be gradually reduced to ≤7.5mg/day at 3-6 months [189]. Due to its immunomodulatory and antifibrotic effects, vitamin D should be supplemented in all SLE patients with insufficiency or deficiency. The immunomodulatory properties of vitamin D are mediated by the vitamin D3 receptor (VDR) on multiple lineages of immune cells, including monocytes, dendritic cells, and activated T cells, as well as in the skin, vasculature, and other tissues. In vitro, vitamin D exerts an anti-inflammatory and antiproliferative effect by promoting Th1 (TNF-α, IL-2, IFN-γ) to Th2 (IL-4, IL-5, IL-10, GATA3) polarization as well as, from Th17 (IL12, IL23, IL-6, 17) to Treg (IL-10, TGF-β, FoxP3, CTLA4) status. Additionally, vitamin D affects the development and function of NKT cells [190].
Taurine administration improved renal function, reversed cell death, suppressed OS, and adjusted the immune response of LN mice to a more balanced state. Taurine could be considered a novel strategy as a therapy in LN, which could overcome the disadvantages of traditional immunosuppression and hormone treatments with greater efficacy and fewer side effects [191]. Therefore, more studies are required on the mechanism of action of taurine as there is an urgent need for therapies involving renal function recovery for patients with severe and terminal LN [188]. Luteolin has also been identified as a possible therapeutic option for preventing and treating LN due to its effect by suppressing the expression of HIF-1α in macrophages [192]. The bidirectional relationship between OS and the immune response could change the paradigm for diseases characterized by perturbation of the immune system and high production of autoantibodies [193].
Though multiple treatment options exist according to the focus of each clinical manifestation and patient complications, there are better treatments for this immunological disease.
Conclusions
SLE is a complex multifactorial autoimmune disease characterized by multiple and diverse cellular and molecular aberrations. The pathogenesis of SLE, like other autoimmune diseases and diabetes mellitus, is still far from being fully understood. The dysregulated immune response in SLE has been extensively studied, including innate and adaptive immunity. The B lymphocyte plays a central role in the production of autoantibodies, presentation of autoantigens, and activation of autoreactive T cells. In addition, T lymphocytes participate by activating signaling pathways mediated by costimulatory and cytokines secreted by T cell subsets. The role of the innate immune response in the pathogenesis of SLE, especially the discovery of TLRs capable of being activated by immune complexes, induces the production of IFN-α. Understanding SLE's immune pathophysiology has led to new biologic agents that specifically target abnormal immune processes that reduce unwanted adverse events associated with conventional broad-spectrum immunosuppressive therapies. Although our understanding of SLE remains incomplete and most new drugs are in clinical trials, they may lead to the development of safer and more effective therapies. The precise characterization of the SLE phenotypes based on the molecular and clinical characteristics is crucial to design a more personalized treatment, which can help redefine how LN is classified and will facilitate the identification of more precise predictors of the response to treatment. We hope that understanding the heterogeneity of autoimmunity in the behavior of SLE will soon lead to more effective and less toxic regimens that favor the clinical response of patients.
References
- Smallwood, M.J.; Nissim, A.; Knight, A.R.; Whiteman, M.; Haigh, R.; Winyard, P.G. Oxidative stress in autoimmune rheumatic diseases. Free. Radic. Biol. Med. 2018, 125, 3–14. [CrossRef]
- Heshmat, T.S.; Khalil, N.M.; Elhamid, H.A.; Labib, S.; Mahfouz, M. Assessment of premature coronary atherosclerosis in patients with systemic lupus erythematosus disease. Egypt. Rheumatol. 2015, 37, S43–S47. [CrossRef]
- Kotzin BL. Systemic lupus erythematosus. Cell 1996;8:843-851.
- Fujii J, Kurahashi T, Konno T, Homma T, Iuchi Y. Oxidative stress as a potential causal factor for autoimmune hemolytic anemia and systemic lupus erythematosus. World J Nephrol. 2015;4(2):213-222.
- Shruthi, S.; Thabah, M.M.; Zachariah, B.; Negi, V.S. Association of Oxidative Stress with Disease Activity and Damage in Systemic Lupus Erythematosus: A Cross Sectional Study from a Tertiary Care Centre in Southern India. Indian J. Clin. Biochem. 2021, 36, 185–193. [CrossRef]
- Wójcik, P.; Gęgotek, A.; Žarković, N.; Skrzydlewska, E. Oxidative Stress and Lipid Mediators Modulate Immune Cell Functions in Autoimmune Diseases. Int. J. Mol. Sci. 2021, 22, 723. [CrossRef]
- Perl, A. Oxidative stress in the pathology and treatment of systemic lupus erythematosus. Nat. Rev. Rheumatol. 2013, 9, 674–686. [CrossRef]
- Ahmad, R.; Ahsan, H. Singlet oxygen species and systemic lupus erythematosus: a brief review. J. Immunoass. Immunochem. 2019, 40, 343–349. [CrossRef]
- Wang, S.; Liu, Y.; Liu, J.; Tian, W.; Zhang, X.; Cai, H.; Fang, S.; Yu, B. Mitochondria-derived methylmalonic acid, a surrogate biomarker of mitochondrial dysfunction and oxidative stress, predicts all-cause and cardiovascular mortality in the general population. Redox Biol. 2020, 37, 101741. [CrossRef]
- Costa JH, Mohanapriya G, Bharadwaj R, Noceda C, Thiers KLL, Aziz S, et al. ROS/RNS balancing, aerobic fermentation regulation and cell cycle control - a complex early trait (‘CoV-MAC-TED’) for combating SARS-CoV-2-induced cell reprogramming. Front Immunol. 2021;12:673692.
- Hu, C.; Zhang, J.; Hong, S.; Li, H.; Lu, L.; Xie, G.; Luo, W.; Du, Y.; Xie, Z.; Han, X.; et al. Oxidative stress-induced aberrant lipid metabolism is an important causal factor for dysfunction of immunocytes from patients with systemic lupus erythematosus. Free. Radic. Biol. Med. 2021, 163, 210–219. [CrossRef]
- Bona, N.; Pezzarini, E.; Balbi, B.; Daniele, S.M.; Rossi, M.F.; Monje, A.L.; Basiglio, C.; Pelusa, H.F.; Arriaga, S.M.M. Oxidative stress, inflammation and disease activity biomarkers in lupus nephropathy. Lupus 2020, 29, 311–323. [CrossRef]
- Mitran, M.I.; Nicolae, I.; Tampa, M.; Mitran, C.I.; Caruntu, C.; Sarbu, M.I.; Ene, C.D.; Matei, C.; Georgescu, S.R.; Popa, M.I. Reactive Carbonyl Species as Potential Pro-Oxidant Factors Involved in Lichen Planus Pathogenesis. Metabolites 2019, 9, 213. [CrossRef]
- Tampa, M.; Nicolae, I.; Ene, C.D.; Sarbu, I.; Matei, C.; Georgescu, S.R. Vitamin C and Thiobarbituric Acid Reactive Substances (TBARS) in Psoriasis Vulgaris Related to Psoriasis Area Severity Index (PASI). Rev. de Chim. 2017, 68, 43–47. [CrossRef]
- Morrow, J.D.; Awad, J.A.; Boss, H.J.; Blair, I.A.; Roberts, L.J., 2nd. Non-cyclooxygenase-derived prostanoids (F2-isoprostanes) are formed in situ on phospholipids.. Proc. Natl. Acad. Sci. USA 1992, 89, 10721–10725. [CrossRef]
- Praticò, D.; Rokach, J.; Lawson, J.; FitzGerald, G.A. F2-isoprostanes as indices of lipid peroxidation in inflammatory diseases. Chem. Phys. Lipids 2004, 128, 165–171. [CrossRef]
- Leitinger N. The role of phospholipid oxidation products in inflammatory and autoimmune disease: evidence from animal models and in humans. Subcell Biochem 2008;49:325-350.
- Ames, P.R.; Alves, J.; Murat, I.; Isenberg, D.A.; Nourooz-Zadeh, J. Oxidative stress in systemic lupus erythematosus and allied conditions with vascular involvement. Rheumatology 1999, 38, 529–534. [CrossRef]
- 3: 2021;70(1), 2021.
- Zavadskiy, S.; Sologova, S.; Moldogazieva, N. Oxidative distress in aging and age-related diseases: Spatiotemporal dysregulation of protein oxidation and degradation. Biochimie 2022, 195, 114–134. [CrossRef]
- Peluso, M.; Russo, V.; Mello, T.; Galli, A. Oxidative Stress and DNA Damage in Chronic Disease and Environmental Studies. Int. J. Mol. Sci. 2020, 21, 6936. [CrossRef]
- Ntouros PA, Vlachogiannis NI, Pappa M, Nezos A, Mavragani CP, Tektonidou MG, et al. Effective DNA damage response after acute but not chronic immune challenge: SARS-CoV-2 vaccine versus systemic lupus erythematosus. Clin Immunol. 2021;229:108765.
- Tumurkhuu, G.; Chen, S.; Montano, E.N.; Laguna, D.E.; Santos, G.D.L.; Yu, J.M.; Lane, M.; Yamashita, M.; Markman, J.L.; Blanco, L.P.; et al. Oxidative DNA Damage Accelerates Skin Inflammation in Pristane-Induced Lupus Model. Front. Immunol. 2020, 11. [CrossRef]
- Banerjee, A.; Yang, W.; Karplus, M.; Verdine, G.L. Structure of a repair enzyme interrogating undamaged DNA elucidates recognition of damaged DNA. Nature 2005, 434, 612–618. [CrossRef]
- Lightfoot, Y.L.; Blanco, L.P.; Kaplan, M.J. Metabolic abnormalities and oxidative stress in lupus. Curr. Opin. Rheumatol. 2017, 29, 442–449. [CrossRef]
- Halliwell, B. Reactive oxygen species in living systems: Source, biochemistry, and role in human disease. Am. J. Med. 1991, 91, S14–S22. [CrossRef]
- Zhang VX, Sze KM, Chan LK, Ho DW, Tsui YM, Chiu YT, et al. Antioxidant supplements promote tumor formation and growth and confer drug resistance in hepatocellular carcinoma by reducing intracellular ROS and induction of TMBIM1. Cell Biosci. 2021;11(1):217.
- Sies, H. Role of reactive oxygen species in biological processes. Klin Wochenschr 1991, 69, 965–968. [CrossRef]
- Schafer, F.Q.; Buettner, G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free. Radic. Biol. Med. 2001, 30, 1191–1212. [CrossRef]
- Shah, D.; Sah, S.; Wanchu, A.; Wu, M.X.; Bhatnagar, A. Altered redox state and apoptosis in the pathogenesis of systemic lupus erythematosus. Immunobiology 2013, 218, 620–627. [CrossRef]
- Yan, L.; Wang, B.; Chen, S.; Zhou, H.; Li, P.; Zhou, L.; Zhao, Q.; Chen, W. The ratio of superoxide dismutase to standard deviation of erythrocyte distribution width as a predictor of systemic lupus erythematosus. J. Clin. Lab. Anal. 2020, 34, e23230. [CrossRef]
- Lv, Y.; He, S.; Zhang, Z.; Li, Y.; Hu, D.; Zhu, K.; Cheng, H.; Zhou, F.; Chen, G.; Zheng, X.; et al. Confirmation of C4 gene copy number variation and the association with systemic lupus erythematosus in Chinese Han population. Rheumatol. Int. 2012, 32, 3047–3053. [CrossRef]
- Fridovich, I. The trail to superoxide dismutase. Protein Sci. 1998, 7, 2688–2690. [CrossRef]
- Sun, Y.; Oberley, L.W.; Li, Y. A simple method for clinical assay of superoxide dismutase.. Clin. Chem. 1988, 34, 497–500. [CrossRef]
- Lee, H.; Lin, C.; Lee, C.; Tsai, C.; Wei, Y. Increased 8-hydroxy-2′-deoxyguanosine in plasma and decreased mRNA expression of human 8-oxoguanine DNA glycosylase 1, anti-oxidant enzymes, mitochondrial biogenesis-related proteins and glycolytic enzymes in leucocytes in patients with systemic lupus erythematosus. Clin. Exp. Immunol. 2014, 176, 66–77. [CrossRef]
- D'Souza, A.; Kurien, B.T.; Rodgers, R.; Shenoi, J.; Kurono, S.; Matsumoto, H.; Hensley, K.; Nath, S.K.; Scofield, R.H. Detection of Catalase as a major protein target of the lipid peroxidation product 4-HNE and the lack of its genetic association as a risk factor in SLE. BMC Med Genet. 2008, 9, 62. [CrossRef]
- Martinon, F.; Pétrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241. [CrossRef]
- Nieto, F.; Iribarren, C.; Gross, M.D.; Comstock, G.W.; Cutler, R.G. Uric acid and serum antioxidant capacity: a reaction to atherosclerosis?. Atherosclerosis 2000, 148, 131–139. [CrossRef]
- Drulović J, Dujmović I, Stojsavljević N, Mesaros S, Andjelković S, Miljković D, et al. Uric acid levels in sera from patients with multiple sclerosis. J Neurol. 2001;248:121-126.
- Collins AR, Horvathova E. Oxidative DNA damage, antioxidants and DNA repair: applications of the comet assay. Biochem Soc Trans. 2001;29:337-341.
- Ahsan, H.; Ali, A.; Ali, R. Oxygen free radicals and systemic autoimmunity. Clin. Exp. Immunol. 2003, 131, 398–404. [CrossRef]
- Lee, H.-C.; Wei, Y.-H. Mitochondrial biogenesis and mitochondrial DNA maintenance of mammalian cells under oxidative stress. Int. J. Biochem. Cell Biol. 2005, 37, 822–834. [CrossRef]
- Kim, J.-W.; Dang, C.V. Multifaceted roles of glycolytic enzymes. Trends Biochem. Sci. 2005, 30, 142–150. [CrossRef]
- Kim, J.-W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [CrossRef]
- Chávez, M.D.; Tse, H.M. Targeting Mitochondrial-Derived Reactive Oxygen Species in T Cell-Mediated Autoimmune Diseases. Front. Immunol. 2021, 12. [CrossRef]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [CrossRef]
- Yurasov, S.; Wardemann, H.; Hammersen, J.; Tsuiji, M.; Meffre, E.; Pascual, V.; Nussenzweig, M.C. Defective B cell tolerance checkpoints in systemic lupus erythematosus. J. Exp. Med. 2005, 201, 703–711. [CrossRef]
- van Loosdregt, J.; Spreafico, R.; Rossetti, M.; Prakken, B.J.; Lotz, M.; Albani, S. Hydroxychloroquine preferentially induces apoptosis of CD45RO+ effector T cells by inhibiting autophagy: A possible mechanism for therapeutic modulation of T cells. J. Allergy Clin. Immunol. 2013, 131, 1443–1446.e1. [CrossRef]
- Lind, N.A.; Rael, V.E.; Pestal, K.; Liu, B.; Barton, G.M. Regulation of the nucleic acid-sensing Toll-like receptors. Nat. Rev. Immunol. 2022, 22, 224–235. [CrossRef]
- Lampropoulou, V.; Hoehlig, K.; Roch, T.; Neves, P.; Gómez, E.C.; Sweenie, C.H.; Hao, Y.; Freitas, A.A.; Steinhoff, U.; Anderton, S.M.; et al. TLR-Activated B Cells Suppress T Cell-Mediated Autoimmunity. J Immunol. 2008, 180, 4763–4773. [CrossRef]
- Brown, G.J.; Cañete, P.F.; Wang, H.; Medhavy, A.; Bones, J.; Roco, J.A.; He, Y.; Qin, Y.; Cappello, J.; Ellyard, J.I.; et al. TLR7 gain-of-function genetic variation causes human lupus. Nature 2022, 605, 349–356. [CrossRef]
- Fillatreau, S.; Manfroi, B.; Dörner, T. Toll-like receptor signalling in B cells during systemic lupus erythematosus. Nat. Rev. Rheumatol. 2021, 17, 98–108. [CrossRef]
- Souyris, M.; Cenac, C.; Azar, P.; Daviaud, D.; Canivet, A.; Grunenwald, S.; Pienkowski, C.; Chaumeil, J.; Mejía, J.E.; Guéry, J.-C. TLR7 escapes X chromosome inactivation in immune cells. Sci. Immunol. 2018, 3. [CrossRef]
- Margery-Muir, A.A.; Bundell, C.; Nelson, D.; Groth, D.M.; Wetherall, J.D. Gender balance in patients with systemic lupus erythematosus. Autoimmun. Rev. 2017, 16, 258–268. [CrossRef]
- Scofield, R.H.; Bruner, G.R.; Namjou, B.; Kimberly, R.P.; Ramsey-Goldman, R.; Petri, M.; Reveille, J.D.; Alarcón, G.S.; Vilá, L.M.; Reid, J.; et al. Klinefelter's syndrome (47,XXY) in male systemic lupus erythematosus patients: Support for the notion of a gene-dose effect from the X chromosome. Arthritis Rheum. 2008, 58, 2511–2517. [CrossRef]
- Hanna Kazazian, N.; Wang, Y.; Roussel-Queval, A.; Marcadet, L.; Chasson, L.; Laprie, C.; Desnues, B.; Charaix, J.; Irla, M.; Alexopoulou, L. Lupus Autoimmunity and Metabolic Parameters Are Exacerbated Upon High Fat Diet-Induced Obesity Due to TLR7 Signaling. Front. Immunol. 2019, 10, 2015. [CrossRef]
- Bekeredjian-Ding IB, Wagner M, Hornung V, Giese T, Schnurr M, Endres S, et al. Plasmacytoid dendritic cells control TLR7 sensitivity of naive B cells via type I IFN. J Immunol. 2005;174:40434050.
- Berland R, Fernandez L, Kari E, Han JH, Lomakin I, Akira S, et al. Toll-like receptor 7-dependent loss of B cell tolerance in pathogenic autoantibody knock-in mice. Immunity. 2006;25:429-440.
- Diebold, S.S.; Kaisho, T.; Hemmi, H.; Akira, S.; Reis E Sousa, C. Innate Antiviral Responses by Means of TLR7-Mediated Recognition of Single-Stranded RNA. Science 2004, 303, 1529–1531. [CrossRef]
- Caielli, S.; Veiga, D.T.; Balasubramanian, P.; Athale, S.; Domic, B.; Murat, E.; Banchereau, R.; Xu, Z.; Chandra, M.; Chung, C.-H.; et al. A CD4+ T cell population expanded in lupus blood provides B cell help through interleukin-10 and succinate. Nat. Med. 2019, 25, 75–81. [CrossRef]
- Nündel, K.; Green, N.M.; Shaffer, A.L.; Moody, K.L.; Busto, P.; Eilat, D.; Miyake, K.; Oropallo, M.A.; Cancro, M.P.; Marshak-Rothstein, A. Cell-Intrinsic Expression of TLR9 in Autoreactive B Cells Constrains BCR/TLR7-Dependent Responses. J. Immunol. 2015, 194, 2504–2512. [CrossRef]
- Miyake, K. Nucleic acid-sensing Toll-like receptors: Beyond ligand search. Adv. Drug Deliv. Rev. 2008, 60, 782–785. [CrossRef]
- Tilstra, J.S.; John, S.; Gordon, R.A.; Leibler, C.; Kashgarian, M.; Bastacky, S.; Nickerson, K.M.; Shlomchik, M.J. B cell–intrinsic TLR9 expression is protective in murine lupus. J. Clin. Investig. 2020, 130, 3172–3187. [CrossRef]
- Fukui, R.; Saitoh, S.-I.; Kanno, A.; Onji, M.; Shibata, T.; Ito, A.; Onji, M.; Matsumoto, M.; Akira, S.; Yoshida, N.; et al. Unc93B1 Restricts Systemic Lethal Inflammation by Orchestrating Toll-like Receptor 7 and 9 Trafficking. Immunity 2011, 35, 69–81. [CrossRef]
- 7: 2020;6(1), 2020.
- Doria, A.; Gatto, M.; Zen, M.; Iaccarino, L.; Punzi, L. Optimizing outcome in SLE: treating-to-target and definition of treatment goals. Autoimmun. Rev. 2014, 13, 770–777. [CrossRef]
- Hajji, M.; Harzallah, A.; Kaaroud, H.; Barbouch, S.; Ben Hamida, F.; Ben Abdallah, T. Factors associated with relapse of lupus nephritis: A single center study of 249 cases. Saudi J. Kidney Dis. Transplant. 2017, 28, 1349–1355. [CrossRef]
- Ayoub, I.; Birmingham, D.; Rovin, B.; Hebert, L. Commentary on the Current Guidelines for the Diagnosis of Lupus Nephritis Flare. Curr. Rheumatol. Rep. 2019, 21, 12. [CrossRef]
- Matsui, I.; Hamano, T.; Tomida, K.; Inoue, K.; Takabatake, Y.; Nagasawa, Y.; Kawada, N.; Ito, T.; Kawachi, H.; Rakugi, H.; et al. Active vitamin D and its analogue, 22-oxacalcitriol, ameliorate puromycin aminonucleoside-induced nephrosis in rats. Nephrol. Dial. Transplant. 2009, 24, 2354–2361. [CrossRef]
- Shankland, S. The podocyte's response to injury: Role in proteinuria and glomerulosclerosis. Kidney Int. 2006, 69, 2131–2147. [CrossRef]
- Qi, Y.-Y.; Zhou, X.-J.; Cheng, F.-J.; Hou, P.; Ren, Y.-L.; Wang, S.-X.; Zhao, M.-H.; Yang, L.; Martinez, J.; Zhang, H. Increased autophagy is cytoprotective against podocyte injury induced by antibody and interferon-α in lupus nephritis. Rheumatol. 2018, 77, 1799–1809. [CrossRef]
- Ene, C.D.; Georgescu, S.R.; Tampa, M.; Matei, C.; Mitran, C.I.; Mitran, M.I.; Penescu, M.N.; Nicolae, I. Cellular Response against Oxidative Stress, a Novel Insight into Lupus Nephritis Pathogenesis. J. Pers. Med. 2021, 11, 693. [CrossRef]
- Akiyama, M.; Kaneko, Y.; Takeuchi, T. Lupus aortitis: A fatal, inflammatory cardiovascular complication in systemic lupus erythematosus. Lupus 2020, 29, 1652–1654. [CrossRef]
- Martin N, ¿Tu X, Egan AJ, Stover C. Complement activation on endothelial cell-derived microparticles-A key determinant for cardiovascular risk in patients with systemic lupus erythematosus? Medicina. 2020;56(10):533.
- Giannelou, M.; Mavragani, C.P. Cardiovascular disease in systemic lupus erythematosus: A comprehensive update. J. Autoimmun. 2017, 82, 1–12. [CrossRef]
- López-Pedrera, C.; Barbarroja, N.; Jimenez-Gomez, Y.; Collantes-Estevez, E.; Aguirre, M.A.; Cuadrado, M.J. Oxidative stress in the pathogenesis of atherothrombosis associated with anti-phospholipid syndrome and systemic lupus erythematosus: new therapeutic approaches. Rheumatology 2016, 55, 2096–2108. [CrossRef]
- Lozovoy MA, Simão AN, Morimoto HK, Iryioda TM, Panis C, Reiche EM, et al. Hypertension is associated with serologically active disease in patients with systemic lupus erythematosus: role of increased Th1/Th2 ratio and oxidative stress. Scand J Rheumatol. 2014;43(1):59-62.
- Yan, Z.; Chen, Q.; Xia, Y. Oxidative Stress Contributes to Inflammatory and Cellular Damage in Systemic Lupus Erythematosus: Cellular Markers and Molecular Mechanism. J. Inflamm. Res. 2023, 16, 453–465. [CrossRef]
- Li D, Shi G, Wang J, Zhang D, Pan Y, Dou H, et al. Baicalein ameliorates pristane-induced lupus nephritis via activating Nrf2/HO-1 in myeloid-derived suppressor cells. Arthritis Res Ther. 2019;21(1):105.
- Yang SM, Chan YL, Hua KF, Chang JM, Chen HL, Tsai YJ, et al. Osthole improves an accelerated focal segmental glomerulosclerosis model in the early stage by activating the Nrf2 antioxidant pathway and subsequently inhibiting NF-kappaB-mediated COX-2 expression and apoptosis. Free Radic Biol Med. 2014;73:260-269.
- Li Y, Li W, Liu C, Yan M, Raman I, Du Y, et al. Delivering Oxidation Resistance-1 (OXR1) to Mouse Kidney by Genetic Modified Mesenchymal Stem Cells Exhibited Enhanced Protection against Nephrotoxic Serum Induced Renal Injury and Lupus Nephritis. J Stem Cell Res Ther. 2014;4(9):231.
- Mitran, M.I.; Nicolae, I.; Tampa, M.; Mitran, C.I.; Caruntu, C.; Sarbu, M.I.; Ene, C.D.; Matei, C.; Georgescu, S.R.; Popa, M.I. Reactive Carbonyl Species as Potential Pro-Oxidant Factors Involved in Lichen Planus Pathogenesis. Metabolites 2019, 9, 213. [CrossRef]
- Shah D, Mahajan N Sah S., Nath SK, Paudial B. Oxidative stress and its biomarkers in systemic lupus erythematosus. J Biomed Sci. 2014;21:23.
- Ene CD, Ceausu E, Nicolae I, Tampa M, Matei C, Georgescu SR. Ultraviolet radiation, Vitamin D and autoimmune disorders. Rev Chim Buc. 2015;66:1068-1073.
- Dalle-Donne, I.; Giustarini, D.; Colombo, R.; Rossi, R.; Milzani, A. Protein carbonylation in human diseases. Trends Mol. Med. 2003, 9, 169–176. [CrossRef]
- Beal, M. Oxidatively modified proteins in aging and disease. Free. Radic. Biol. Med. 2002, 32, 797–803. [CrossRef]
- Tuteja, N.; Chandra, M.; Tuteja, R.; Misra, M.K. Nitric Oxide as a Unique Bioactive Signaling Messenger in Physiology and Pathophysiology. J. Biomed. Biotechnol. 2004, 2004, 227–237. [CrossRef]
- Lee, J. Nitric Oxide in the Kidney : Its Physiological Role and Pathophysiological Implications. Electrolytes Blood Press. 2008, 6, 27–34. [CrossRef]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: regulation and function. Eur. Heart J. 2012, 33, 829–837. [CrossRef]
- Herrera, M.; Garvin, J.L. Recent Advances in the Regulation of Nitric Oxide in the Kidney. Hypertension 2005, 45, 1062–1067. [CrossRef]
- Ratliff, B.B.; Abdulmahdi, W.; Pawar, R.; Wolin, M.S. Oxidant Mechanisms in Renal Injury and Disease. Antioxidants Redox Signal. 2016, 25, 119–146. [CrossRef]
- Semenikhina, M.; Stefanenko, M.; Spires, D.R.; Ilatovskaya, D.V.; Palygin, O. Nitric-Oxide-Mediated Signaling in Podocyte Pathophysiology. Biomolecules 2022, 12, 745. [CrossRef]
- Wanchu, A.; Khullar, M.; Deodhar, S.D.; Bambery, P.; Sud, A. Nitric oxide synthesis is increased in patients with systemic lupus erythematosus.. Rheumatol. Int. 1998, 18, 41–43. [CrossRef]
- Oates, J.C.; Ruizb, P.; Alexanderc, A.; Pippen, A.M.; Gilkeson, G.S. Effect of Late Modulation of Nitric Oxide Production on Murine Lupus. Clin. Immunol. Immunopathol. 1997, 83, 86–92. [CrossRef]
- Ramsey, K.H.; Sigar, I.M.; Rana, S.V.; Gupta, J.; Holland, S.M.; Byrne, G.I.; Morrow, J.D. Inducible Nitric Oxide Synthase Regulates Production of Isoprostanes In Vivo during Chlamydial Genital Infection in Mice. Infect. Immun. 2003, 71, 7183–7187. [CrossRef]
- Dinu L, Ene CD, Nicolae I, Tampa M, Matei C, Georgescu SR. The serum levels of 8-hydroxy-deoxyguanosine under the chemicals influence. Rev Chim. 2014;65:1319-1326.
- Wang, K.; Maayah, M.; Sweasy, J.B.; Alnajjar, K.S. The role of cysteines in the structure and function of OGG1. J. Biol. Chem. 2020, 296, 100093. [CrossRef]
- Costenbader, K.H.; Kang, J.H.; Karlson, E.W. Antioxidant Intake and Risks of Rheumatoid Arthritis and Systemic Lupus Erythematosus in Women. Am. J. Epidemiology 2010, 172, 205–216. [CrossRef]
- Pedersen-Lane, J.H.; Zurier, R.B.; A Lawrence, D.; Lawrence, D.A. Analysis of the thiol status of peripheral blood leukocytes in rheumatoid arthritis patients. J. Leukoc. Biol. 2007, 81, 934–941. [CrossRef]
- Tewthanom K, Janwityanuchit S, Totemchockchyakarn K, Panomvana D. Correlation of lipid peroxidation and glutathione levels with severity of systemic lupus erythematosus: a pilot study from single center. J Pharm Pharm Sci. 2008;11:30-4.
- Morgan, P.E.; Sturgess, A.D.; Hennessy, A.; Davies, M.J. Serum protein oxidation and apolipoprotein CIII levels in people with systemic lupus erythematosus with and without nephritis. Free. Radic. Res. 2007, 41, 1301–1312. [CrossRef]
- Li, M.; Gao, W.; Ma, J.; Zhu, Y.; Li, X. Early-stage lupus nephritis treated with N-acetylcysteine: A report of two cases. Exp. Ther. Med. 2015, 10, 689–692. [CrossRef]
- Schieke, S.M.; Phillips, D.; McCoy, J.P.; Aponte, A.M.; Shen, R.-F.; Balaban, R.S.; Finkel, T. The Mammalian Target of Rapamycin (mTOR) Pathway Regulates Mitochondrial Oxygen Consumption and Oxidative Capacity. J Biol Chem. 2006, 281, 27643–27652. [CrossRef]
- Andersen, J.L.; Kornbluth, S. The Tangled Circuitry of Metabolism and Apoptosis. Mol. Cell 2013, 49, 399–410. [CrossRef]
- Friedman JR, Nunnari J. Mitochondrial form and function. Nature. 2014;505:335-43.
- Barshad, G.; Marom, S.; Cohen, T.; Mishmar, D. Mitochondrial DNA Transcription and Its Regulation: An Evolutionary Perspective. Trends Genet. 2018, 34, 682–692. [CrossRef]
- Pfanner, N.; Warscheid, B.; Wiedemann, N. Mitochondrial proteins: from biogenesis to functional networks. Nat. Rev. Mol. Cell Biol. 2019, 20, 267–284. [CrossRef]
- Murphy MP, Hartley RC. Mitochondria as a therapeutic target for common pathologies. Nat Rev Drug Discov. 2018;17:865-886.
- Lee, S.; Min, K.-T. The Interface Between ER and Mitochondria: Molecular Compositions and Functions. Mol. Cells 2018, 41, 1000–1007. [CrossRef]
- Li, X.; Fang, P.; Mai, J.; Choi, E.T.; Wang, H.; Yang, X.-F. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J. Hematol. Oncol. 2013, 6, 19–19. [CrossRef]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [CrossRef]
- Krysko, D.V.; Agostinis, P.; Krysko, O.; Garg, A.D.; Bachert, C.; Lambrecht, B.N.; Vandenabeele, P. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. 2011, 32, 157–164. [CrossRef]
- Weinberg, S.E.; Sena, L.A.; Chandel, N.S. Mitochondria in the Regulation of Innate and Adaptive Immunity. Immunity 2015, 42, 406–417. [CrossRef]
- Becker, Y.; Marcoux, G.; Allaeys, I.; Julien, A.-S.; Loignon, R.-C.; Benk-Fortin, H.; Rollet-Labelle, E.; Rauch, J.; Fortin, P.R.; Boilard, E. Autoantibodies in Systemic Lupus Erythematosus Target Mitochondrial RNA. Front. Immunol. 2019, 10, 1026. [CrossRef]
- Wang, H.; Li, T.; Chen, S.; Gu, Y.; Ye, S. Neutrophil Extracellular Trap Mitochondrial DNA and Its Autoantibody in Systemic Lupus Erythematosus and a Proof-of-Concept Trial of Metformin. Arthritis Rheumatol. 2015, 67, 3190–3200. [CrossRef]
- Basso, P.J.; Andrade-Oliveira, V.; Câmara, N.O.S. Targeting immune cell metabolism in kidney diseases. Nat. Rev. Nephrol. 2021, 17, 465–480. [CrossRef]
- Kingsmore, K.M.; Bachali, P.; Catalina, M.D.; Daamen, A.R.; Heuer, S.E.; Robl, R.D.; Grammer, A.C.; Lipsky, P.E. Altered expression of genes controlling metabolism characterizes the tissue response to immune injury in lupus. Sci. Rep. 2021, 11, 1–18. [CrossRef]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.-A.; Han, S.H.; Chinga, F.; Park, A.S.D.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [CrossRef]
- Grayson PC, Eddy S, Taroni JN, Lightfoot YL, Mariani L, Parikh H, et al. Vasculitis Clinical Research Consortium, the European Renal cDNA Bank cohort, and the Nephrotic Syndrome Study Network. Metabolic pathways and immunometabolism in rare kidney diseases. Ann Rheum Dis. 2018, 77, 1226–1233. [CrossRef]
- MacIver, N.J.; Michalek, R.D.; Rathmell, J.C. Metabolic Regulation of T Lymphocytes. Annu. Rev. Immunol. 2013, 31, 259–283. [CrossRef]
- Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. 2011;208(7):1367-76.
- Jacobs SR, Herman CE, Maciver NJ, Wofford JA, Wieman HL, Hammen JJ, et al. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J Immunol. 2008;180(7):4476-86.
- Alexopoulos, E.; Seron, D.; Hartley, R.B.; Cameron, J.S. Lupus nephritis: Correlation of interstitial cells with glomerular function. Kidney Int. 1990, 37, 100–109. [CrossRef]
- Couzi, L.; Merville, P.; Deminière, C.; Moreau, J.-F.; Combe, C.; Pellegrin, J.-L.; Viallard, J.-F.; Blanco, P. Predominance of CD8+ T lymphocytes among periglomerular infiltrating cells and link to the prognosis of class III and class IV lupus nephritis. Arthritis Rheum. 2007, 56, 2362–2370. [CrossRef]
- Tilstra, J.S.; Avery, L.; Menk, A.V.; Gordon, R.A.; Smita, S.; Kane, L.P.; Chikina, M.; Delgoffe, G.M.; Shlomchik, M.J. Kidney-infiltrating T cells in murine lupus nephritis are metabolically and functionally exhausted. J. Clin. Investig. 2018, 128, 4884–4897. [CrossRef]
- Li, M.; Lai, Y.; Chen, B.; Guo, C.; Zhou, M.; Zhao, S.; Wang, S.; Li, J.; Yang, N.; Zhang, H. NAMPT is a metabolic checkpoint of IFNγ-producing CD4+ T cells in lupus nephritis. Mol. Ther. 2023, 31, 193–210. [CrossRef]
- Yin, Y.; Choi, S.-C.; Xu, Z.; Perry, D.J.; Seay, H.; Croker, B.P.; Sobel, E.S.; Brusko, T.M.; Morel, L. Normalization of CD4 + T cell metabolism reverses lupus. Sci. Transl. Med. 2015, 7, 274ra18–274ra18. [CrossRef]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022. [CrossRef]
- Frank, M.; Duvezin-Caubet, S.; Koob, S.; Occhipinti, A.; Jagasia, R.; Petcherski, A.; Ruonala, M.O.; Priault, M.; Salin, B.; Reichert, A.S. Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochim. et Biophys. Acta (BBA) - Mol. Cell Res. 2012, 1823, 2297–2310. [CrossRef]
- Kubli DA, Gustafsson AB. Mitochondria and mitophagy: The yin and yang of cell death control. Circ Res. 2012;111:1208-1221.
- Sun, N.; Yun, J.; Liu, J.; Malide, D.; Liu, C.; Rovira, I.I.; Holmström, K.M.; Fergusson, M.M.; Yoo, Y.H.; Combs, C.A.; et al. Measuring In Vivo Mitophagy. Mol. Cell 2015, 60, 685–696. [CrossRef]
- Matheoud, D.; Sugiura, A.; Bellemare-Pelletier, A.; Laplante, A.; Rondeau, C.; Chemali, M.; Fazel, A.; Bergeron, J.J.; Trudeau, L.-E.; Burelle, Y.; et al. Parkinson’s Disease-Related Proteins PINK1 and Parkin Repress Mitochondrial Antigen Presentation. Cell 2016, 166, 314–327. [CrossRef]
- Nicolaou, O.; Kousios, A.; Hadjisavvas, A.; Lauwerys, B.; Sokratous, K.; Kyriacou, K. Biomarkers of systemic lupus erythematosus identified using mass spectrometry-based proteomics: a systematic review. J. Cell. Mol. Med. 2017, 21, 993–1012. [CrossRef]
- Wang, Y.; Cai, J.; Tang, C.; Dong, Z. Mitophagy in Acute Kidney Injury and Kidney Repair. Cells 2020, 9, 338. [CrossRef]
- Lui, S.L.; Tsang, R.; Chan, K.W.; Zhang, F.; Tam, S.; Yung, S.; Chan, T.M. Rapamycin attenuates the severity of established nephritis in lupus-prone NZB/W F1 mice. Nephrol. Dial. Transplant. 2008, 23, 2768–2776. [CrossRef]
- Fernandez, D.R.; Crow, M.K. CD8 T cells and mTOR: new concepts and targets for systemic lupus erythematosus. Lancet 2018, 391, 1126–1127. [CrossRef]
- Chen, J.; Fang, Y. A novel pathway regulating the mammalian target of rapamycin (mTOR) signaling. Biochem. Pharmacol. 2002, 64, 1071–1077. [CrossRef]
- Oaks, Z.; Winans, T.; Caza, T.; Fernandez, D.; Liu, Y.; Landas, S.K.; Banki, K.; Perl, A. Mitochondrial Dysfunction in the Liver and Antiphospholipid Antibody Production Precede Disease Onset and Respond to Rapamycin in Lupus-Prone Mice. Arthritis Rheumatol. 2016, 68, 2728–2739. [CrossRef]
- Wang, L.; Law, H.K.W. Immune Complexes Impaired Glomerular Endothelial Cell Functions in Lupus Nephritis. Int. J. Mol. Sci. 2019, 20, 5281. [CrossRef]
- Flür, K.; Allam, R.; Zecher, D.; Kulkarni, O.P.; Lichtnekert, J.; Schwarz, M.; Beutler, B.; Vielhauer, V.; Anders, H.-J. Viral RNA Induces Type I Interferon-Dependent Cytokine Release and Cell Death in Mesangial Cells via Melanoma-Differentiation-Associated Gene-5: Implications for Viral Infection-Associated Glomerulonephritis. Am. J. Pathol. 2009, 175, 2014–2022. [CrossRef]
- Lech, M.; Anders, H.-J. The Pathogenesis of Lupus Nephritis. J. Am. Soc. Nephrol. 2013, 24, 1357–1366. [CrossRef]
- Hof, D.; Raats, J.M.; Pruijn, G.J. Apoptotic modifications affect the autoreactivity of the U1 snRNP autoantigen. Autoimmun. Rev. 2005, 4, 380–388. [CrossRef]
- Karikó, K.; Ni, H.; Capodici, J.; Lamphier, M.; Weissman, D. mRNA Is an Endogenous Ligand for Toll-like Receptor 3. J Biol Chem. 2004, 279, 12542–12550. [CrossRef]
- Chan, O.T.; Hannum, L.G.; Haberman, A.M.; Madaio, M.P.; Shlomchik, M.J. A Novel Mouse with B Cells but Lacking Serum Antibody Reveals an Antibody-independent Role for B Cells in Murine Lupus. J. Exp. Med. 1999, 189, 1639–1648. [CrossRef]
- Guiducci, C.; Gong, M.; Xu, Z.; Gill, M.; Chaussabel, D.; Meeker, T.; Chan, J.H.; Wright, T.; Punaro, M.; Bolland, S.; et al. TLR recognition of self nucleic acids hampers glucocorticoid activity in lupus. Nature 2010, 465, 937–941. [CrossRef]
- Allam, R.; Lichtnekert, J.; Moll, A.G.; Taubitz, A.; Vielhauer, V.; Anders, H.-J. Viral RNA and DNA Trigger Common Antiviral Responses in Mesangial Cells. J. Am. Soc. Nephrol. 2009, 20, 1986–1996. [CrossRef]
- 5: 2004;18, 2004.
- Rich, S.A. Human Lupus Inclusions and Interferon. Science 1981, 213, 772–775. [CrossRef]
- Wang, Y.; Ito, S.; Chino, Y.; Goto, D.; Matsumoto, I.; Murata, H.; Tsutsumi, A.; Hayashi, T.; Uchida, K.; Usui, J.; et al. Laser microdissection-based analysis of cytokine balance in the kidneys of patients with lupus nephritis. Clin. Exp. Immunol. 2010, 159, 1–10. [CrossRef]
- Anders, H.-J. Pseudoviral immunity – a novel concept for lupus. Trends Mol. Med. 2009, 15, 553–561. [CrossRef]
- Barrat, F.J.; Coffman, R.L. Development of TLR inhibitors for the treatment of autoimmune diseases. Immunol. Rev. 2008, 223, 271–283. [CrossRef]
- Oates, J.; Mashmoushi, A.; Shaftman, S.; Gilkeson, G. NADPH oxidase and nitric oxide synthase-dependent superoxide production is increased in proliferative lupus nephritis. Lupus 2013, 22, 1361–1370. [CrossRef]
- Allam, R.; Anders, H.-J. The role of innate immunity in autoimmune tissue injury. Curr. Opin. Rheumatol. 2008, 20, 538–544. [CrossRef]
- Lorenz G, Anders HJ. Neutrophils, dendritic cells, toll-like receptors, and interferon-alpha in lupus nephritis. Semin Nephrol. 2015;35:410-426.
- Rönnblom, L.; Pascual, V. The innate immune system in SLE: type I interferons and dendritic cells. Lupus 2008, 17, 394–399. [CrossRef]
- Rodriguez-Pla, A.; Patel, P.; Maecker, H.T.; Rossello-Urgell, J.; Baldwin, N.; Bennett, L.; Cantrell, V.; Baisch, J.; Punaro, M.; Gotte, A.; et al. IFN Priming Is Necessary but Not Sufficient To Turn on a Migratory Dendritic Cell Program in Lupus Monocytes. J Immunol. 2014, 192, 5586–5598. [CrossRef]
- Patole PS, Zecher D, Pawar RD, Grone HJ, Schlondorff D, Anders HJ. G-rich DNA suppresses systemic lupus. J Am Soc Nephrol. 2005, 16, 3273–3280. [CrossRef]
- Lee TP, Huang JC, Liu CJ, Chen HJ, Chen YH, Tsai YT, et al. Interactions of surface-expressed TLR-4 and endosomal TLR-9 accelerate lupus progression in anti-dsDNA antibody transgenic mice. Exp Biol Med (Maywood) 2014;239:715-723.
- Pawar, R.D.; Patole, P.S.; Wörnle, M.; Anders, H.-J. Microbial nucleic acids pay a Toll in kidney disease. Am. J. Physiol. Physiol. 2006, 291, F509–F516. [CrossRef]
- Anders, H.; Krug, A.; Pawar, R.D. Molecular mimicry in innate immunity? The viral RNA recognition receptor TLR7 accelerates murine lupus. Eur. J. Immunol. 2008, 38, 1795–1799. [CrossRef]
- Ye, D.-Q.; Tian, J.; Ma, Y.; Li, J.; Cen, H.; Wang, D.-G.; Feng, C.-C.; Li, R.-J.; Leng, R.-X.; Pan, H.-F. The TLR7 7926A>G polymorphism is associated with susceptibility to systemic lupus erythematosus. Mol. Med. Rep. 2012, 6, 105–110. [CrossRef]
- Horton, C.G.; Farris, A.D. Toll-like Receptors in Systemic Lupus Erythematosus: Potential Targets for Therapeutic Intervention. Curr. Allergy Asthma Rep. 2012, 12, 1–7. [CrossRef]
- Murphy, E.D.; Roths, J.B. A y chromosome associated factor in strain bxsb producing accelerated autoimmunity and lymphoproliferation. Arthritis Rheum. 1979, 22, 1188–1194. [CrossRef]
- Deane, J.A.; Pisitkun, P.; Barrett, R.S.; Feigenbaum, L.; Town, T.; Ward, J.M.; Flavell, R.A.; Bolland, S. Control of Toll-like Receptor 7 Expression Is Essential to Restrict Autoimmunity and Dendritic Cell Proliferation. Immunity 2007, 27, 801–810. [CrossRef]
- Lee PY, Kumagai Y, Li Y, Takeuchi O, Yoshida H, Weinstein J, et al. TLR7-dependent and FcgammaR-independent production of type I interferon in experimental mouse lupus. The Journal of experimental medicine. 2008;205:2995-300.
- Yokogawa, M.; Takaishi, M.; Nakajima, K.; Kamijima, R.; Fujimoto, C.; Kataoka, S.; Terada, Y.; Sano, S. Epicutaneous Application of Toll-like Receptor 7 Agonists Leads to Systemic Autoimmunity in Wild-Type Mice: A New Model of Systemic Lupus Erythematosus. Arthritis Rheumatol. 2014, 66, 694–706. [CrossRef]
- Liu, Z.; Bethunaickan, R.; Huang, W.; Lodhi, U.; Solano, I.; Madaio, M.P.; Davidson, A. Interferon-α accelerates murine systemic lupus erythematosus in a T cell-dependent manner. Arthritis Rheum. 2011, 63, 219–229. [CrossRef]
- Agrawal, H.; Jacob, N.; Carreras, E.; Bajana, S.; Putterman, C.; Turner, S.; Neas, B.; Mathian, A.; Koss, M.N.; Stohl, W.; et al. Deficiency of Type I IFN Receptor in Lupus-Prone New Zealand Mixed 2328 Mice Decreases Dendritic Cell Numbers and Activation and Protects from Disease. J. Immunol. 2009, 183, 6021–6029. [CrossRef]
- Celhar, T.; Hopkins, R.; Thornhill, S.I.; De Magalhaes, R.; Hwang, S.-H.; Lee, H.-Y.; Yasuga, H.; Jones, L.A.; Casco, J.; Lee, B.; et al. RNA sensing by conventional dendritic cells is central to the development of lupus nephritis. Proc. Natl. Acad. Sci. 2015, 112, E6195–E6204. [CrossRef]
- Barton GM, Medzhitov R. Toll-like receptor signaling pathways. Science. 2003;300(5625):1524-1525.
- Soni, C.; Wong, E.B.; Domeier, P.P.; Khan, T.N.; Satoh, T.; Akira, S.; Rahman, Z.S.M. B Cell–Intrinsic TLR7 Signaling Is Essential for the Development of Spontaneous Germinal Centers. J Immunol. 2014, 193, 4400–4414. [CrossRef]
- Smith, K.D. Toll-like receptors in kidney disease. Curr. Opin. Nephrol. Hypertens. 2009, 18, 189–196. [CrossRef]
- Tilstra, J.S.; John, S.; Gordon, R.A.; Leibler, C.; Kashgarian, M.; Bastacky, S.; Nickerson, K.M.; Shlomchik, M.J. B cell–intrinsic TLR9 expression is protective in murine lupus. J. Clin. Investig. 2020, 130, 3172–3187. [CrossRef]
- Hosseini, A.M.; Majidi, J.; Baradaran, B.; Yousefi, M. Toll-Like Receptors in the Pathogenesis of Autoimmune Diseases. Adv. Pharm. Bull. 2015, 5, 605–614. [CrossRef]
- Giles JR, Kashgarian M, Koni PA, Shlomchik MJ. B cell-specific MHC class II deletion reveals multiple nonredundant roles for B cell antigen presentation in murine lupus. J Immunol. 2015;195(6):2571-2579.
- Iribarren, K.; Bloy, N.; Buqué, A.; Cremer, I.; Eggermont, A.; Fridman, W.H.; Fucikova, J.; Galon, J.; Špíšek, R.; Zitvogel, L.; et al. Trial Watch: Immunostimulation with Toll-like receptor agonists in cancer therapy. OncoImmunology 2016, 5, e108863. [CrossRef]
- Tilstra, J.S.; John, S.; Gordon, R.A.; Leibler, C.; Kashgarian, M.; Bastacky, S.; Nickerson, K.M.; Shlomchik, M.J. B cell–intrinsic TLR9 expression is protective in murine lupus. J. Clin. Investig. 2020, 130, 3172–3187. [CrossRef]
- Fanouriakis, A.; Kostopoulou, M.; Alunno, A.; Aringer, M.; Bajema, I.; Boletis, J.; Cervera, R.; Doria, A.; Gordon, C.; Govoni, M.; et al. 2019 update of the EULAR recommendations for the management of systemic lupus erythematosus. Ann Rheum Dis 2019;78:736-45. [CrossRef]
- Fanouriakis, A.; Tziolos, N.; Bertsias, G.; Boumpas, D.T. Update οn the diagnosis and management of systemic lupus erythematosus. Ann Rheum Dis. 2021, 80, 14–25. [CrossRef]
- Moroni, G.; Gatto, M.; Tamborini, F.; Quaglini, S.; Radice, F.; Saccon, F.; Frontini, G.; Alberici, F.; Sacchi, L.; Binda, V.; et al. Lack of EULAR/ERA-EDTA response at 1 year predicts poor long-term renal outcome in patients with lupus nephritis. Ann Rheum Dis 2020, 79, 1077–1083. [CrossRef]
- Pons-Estel, G.J.; Alarcón, G.S.; McGwin, G.; Danila, M.I.; Zhang, J.; Bastian, H.M.; Reveille, J.D.; Vilá, L.M.; Lumina Study Group Protective effect of hydroxychloroquine on renal damage in patients with lupus nephritis: LXV, data from a multiethnic US cohort. Arthritis Rheum. 2009, 61, 830–839. [CrossRef]
- Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. 2002;416:603-607.
- Takeuchi, T.; Wakasugi, N.; Uno, S.; Makino, H. Long-term Safety and Effectiveness of Tacrolimus in Patients With Lupus Nephritis: 5-year Interim Postmarketing Surveillance Study in Japan (TRUST). J. Rheumatol. 2021, 48, 74–81. [CrossRef]
- Dall'Era, M.; Bruce, I.N.; Gordon, C.; Manzi, S.; McCaffrey, J.; E Lipsky, P. Current challenges in the development of new treatments for lupus. Ann. Rheum. Dis. 2019, 78, 729–735. [CrossRef]
- Navarra SV, Guzmán RM, Gallacher AE, Hall S, Levy RA, Jimenez RE, et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: A randomised, placebo-controlled, phase 3 trial. Lancet 2011;377:721-731. [CrossRef]
- Barrat, F.J.; Meeker, T.; Chan, J.H.; Guiducci, C.; Coffman, R.L. Treatment of lupus-prone mice with a dual inhibitor of TLR7 and TLR9 leads to reduction of autoantibody production and amelioration of disease symptoms. Eur. J. Immunol. 2007, 37, 3582–3586. [CrossRef]
- Zen, M.; Iaccarino, L.; Gatto, M.; Saccon, F.; Larosa, M.; Ghirardello, A.; Punzi, L.; Doria, A. Lupus low disease activity state is associated with a decrease in damage progression in Caucasian patients with SLE, but overlaps with remission. Ann. Rheum. Dis. 2018, 77, 104–110. [CrossRef]
- Yu, S.; Cantorna, M.T. The vitamin D receptor is required for iNKT cell development. Proc. Natl. Acad. Sci. USA 2008, 105, 5207–5212. [CrossRef]
- Luo H, Geng CJ, Miao SM, Wang LH, Li Q. Taurine attenuates the damage of lupus nephritis mouse via inactivation of the NF-κB pathway. Ann Palliat Med. 2021;10(1):137-147.
- Zhang, L.; Zhang, M.; Chen, X.; He, Y.; Chen, R.; Zhang, J.; Huang, J.; Ouyang, C.; Shi, G. Identification of the tubulointerstitial infiltrating immune cell landscape and immune marker related molecular patterns in lupus nephritis using bioinformatics analysis. Ann. Transl. Med. 2020, 8, 1596. [CrossRef]
- Ding, T.; Yi, T.; Li, Y.; Zhang, W.; Wang, X.; Liu, J.; Fan, Y.; Ji, J.; Xu, L. Luteolin attenuates lupus nephritis by regulating macrophage oxidative stress via HIF-1α pathway. Eur. J. Pharmacol. 2023, 953, 175823. [CrossRef]
- Popp, H.D.; Kohl, V.; Naumann, N.; Flach, J.; Brendel, S.; Kleiner, H.; Weiss, C.; Seifarth, W.; Saussele, S.; Hofmann, W.-K.; et al. DNA Damage and DNA Damage Response in Chronic Myeloid Leukemia. Int. J. Mol. Sci. 2020, 21, 1177. [CrossRef]
Figure 1.
CLINICAL MANIFESTATIONS OF SLE AND FACTORS THAT INFLUENCE ITS DEVELOPMENT. SLE presents a broad spectrum of clinical manifestations in multiple organs and systems, including lupus nephritis (LN). Microvascular inflammation with the development of autoantibodies, the creation of pathogenic immune complexes, and the activation of autoreactive T cells against ubiquitous nuclear antigens are characteristic of SLE. Oxidative stress (OS), antioxidant capacity, DNA damage and repair, mitochondrial function, mitophagy, and innate immunity mechanisms mediated by TLR7 and TLR9 influence the induction of systemic autoimmunity and tissue damage in SLE.
Figure 1.
CLINICAL MANIFESTATIONS OF SLE AND FACTORS THAT INFLUENCE ITS DEVELOPMENT. SLE presents a broad spectrum of clinical manifestations in multiple organs and systems, including lupus nephritis (LN). Microvascular inflammation with the development of autoantibodies, the creation of pathogenic immune complexes, and the activation of autoreactive T cells against ubiquitous nuclear antigens are characteristic of SLE. Oxidative stress (OS), antioxidant capacity, DNA damage and repair, mitochondrial function, mitophagy, and innate immunity mechanisms mediated by TLR7 and TLR9 influence the induction of systemic autoimmunity and tissue damage in SLE.
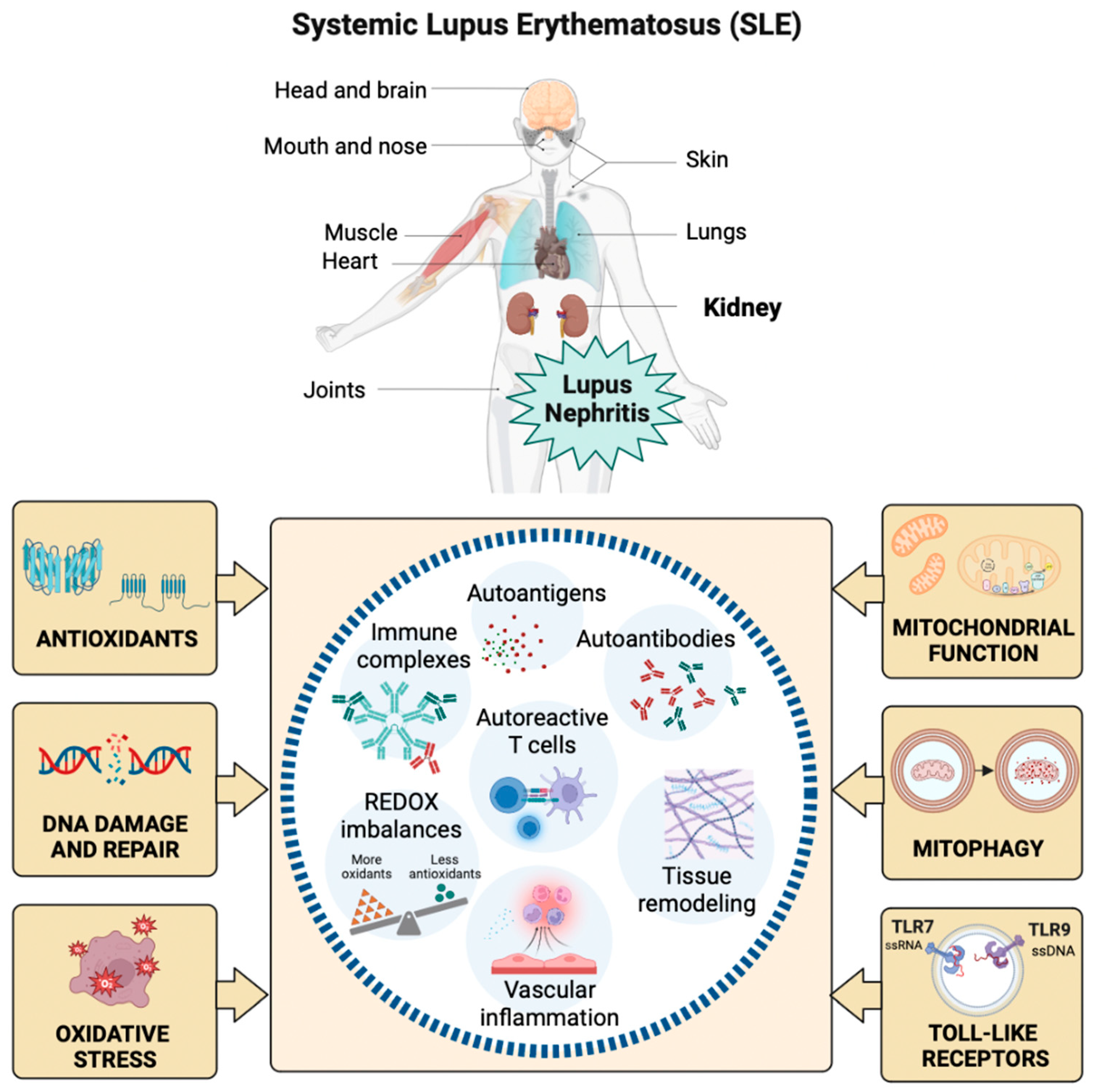
Figure 2.
LN AND ITS DAMAGE MECHANISMS. Glomerular damage in LN features endothelial injury, glomerular membrane damage, and podocyte dysfunction mediated by autoantibodies, immune complexes, complement system activation, inflammation, autophagy, neutrophil extracellular traps (NETs), and redox imbalances. In patients with LN, a robust self-renewal and repair system of the glomerular filtration membrane results in instability regarding the degrees of renal injury and proteinuria. In early LN, it is possible to repair pathological damage to the filtration membrane caused by complement proteins and cytokines through autoregulation. However, tissue damage cannot be repaired as the disease progresses, leading to severe proteinuria, increased risk of cardiovascular disease, and high mortality.
Figure 2.
LN AND ITS DAMAGE MECHANISMS. Glomerular damage in LN features endothelial injury, glomerular membrane damage, and podocyte dysfunction mediated by autoantibodies, immune complexes, complement system activation, inflammation, autophagy, neutrophil extracellular traps (NETs), and redox imbalances. In patients with LN, a robust self-renewal and repair system of the glomerular filtration membrane results in instability regarding the degrees of renal injury and proteinuria. In early LN, it is possible to repair pathological damage to the filtration membrane caused by complement proteins and cytokines through autoregulation. However, tissue damage cannot be repaired as the disease progresses, leading to severe proteinuria, increased risk of cardiovascular disease, and high mortality.
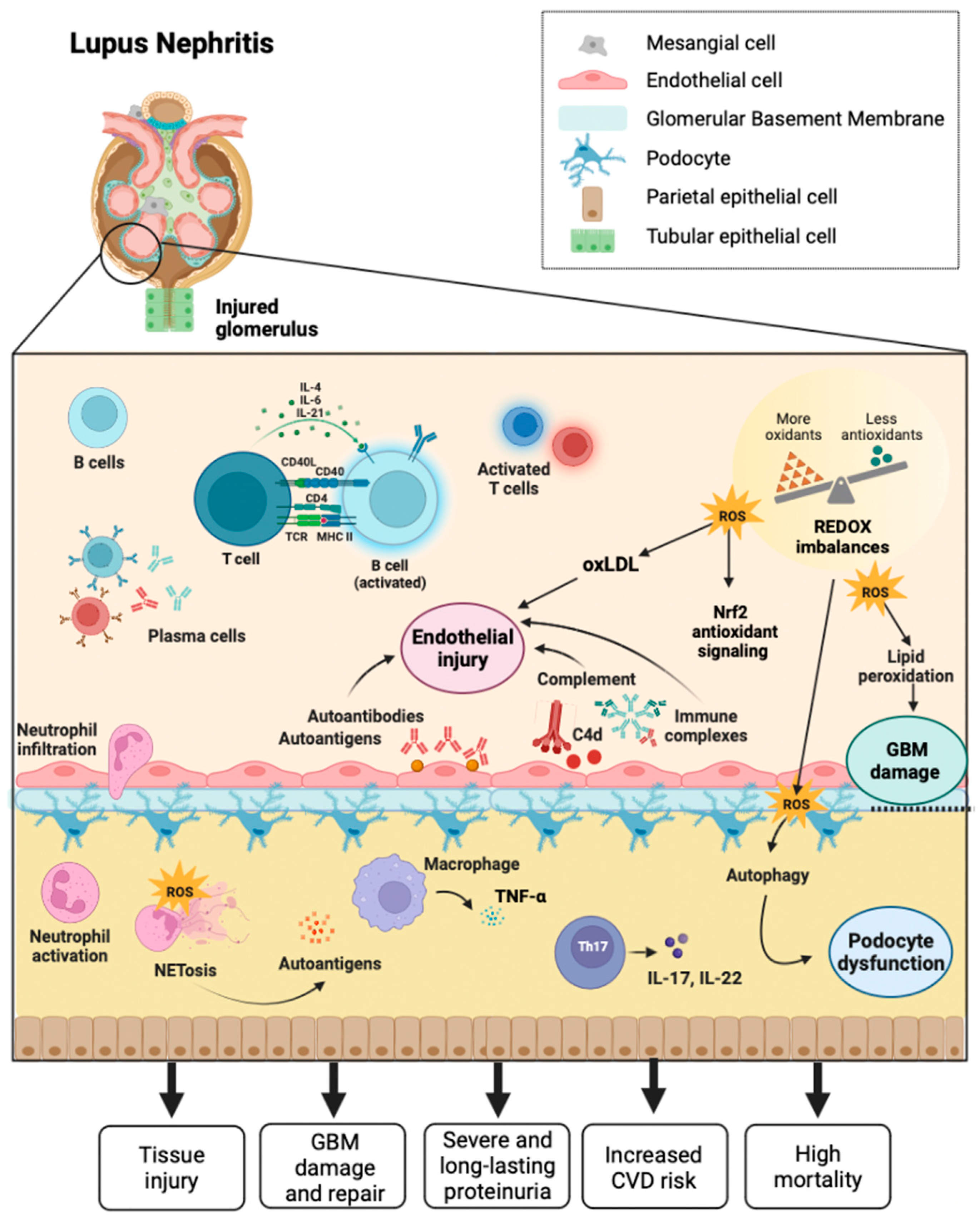
Figure 3.
INFLUENCE OF OS, DNA DAMAGE, ANTIOXIDANTS, MITOCHONDRIAL FUNCTION AND MITOPHAGY ON THE DEVELOPMENT OF LN. Oxidation leads to organic damage characteristic of SLE, especially in LN. Increases in lipid peroxidation due to OS, limited antioxidant defense, and low NO availability contribute to the injury observed in LN. The balance between oxidation and DNA repair is altered in patients with LN, accumulating the marker of oxidative DNA damage (8-OHdG), genomic instability, and cellular dysfunction. Additionally, metabolic abnormalities influence the nature and state of activation of the kidney's immune cell infiltrate, highlighting the importance of mitochondrial function in developing diseases such as LN through Danger Associated Molecular Pattern (DAMPs), TLR activation, and the production of autoantibodies. Finally, aberrant, or defective mitophagy is central to the pathology of LN as it facilitates the release of mitochondrial antigens, promotes their processing by major histocompatibility complex (MHC) molecules, and contributes to mitochondrial dysfunction.
Figure 3.
INFLUENCE OF OS, DNA DAMAGE, ANTIOXIDANTS, MITOCHONDRIAL FUNCTION AND MITOPHAGY ON THE DEVELOPMENT OF LN. Oxidation leads to organic damage characteristic of SLE, especially in LN. Increases in lipid peroxidation due to OS, limited antioxidant defense, and low NO availability contribute to the injury observed in LN. The balance between oxidation and DNA repair is altered in patients with LN, accumulating the marker of oxidative DNA damage (8-OHdG), genomic instability, and cellular dysfunction. Additionally, metabolic abnormalities influence the nature and state of activation of the kidney's immune cell infiltrate, highlighting the importance of mitochondrial function in developing diseases such as LN through Danger Associated Molecular Pattern (DAMPs), TLR activation, and the production of autoantibodies. Finally, aberrant, or defective mitophagy is central to the pathology of LN as it facilitates the release of mitochondrial antigens, promotes their processing by major histocompatibility complex (MHC) molecules, and contributes to mitochondrial dysfunction.
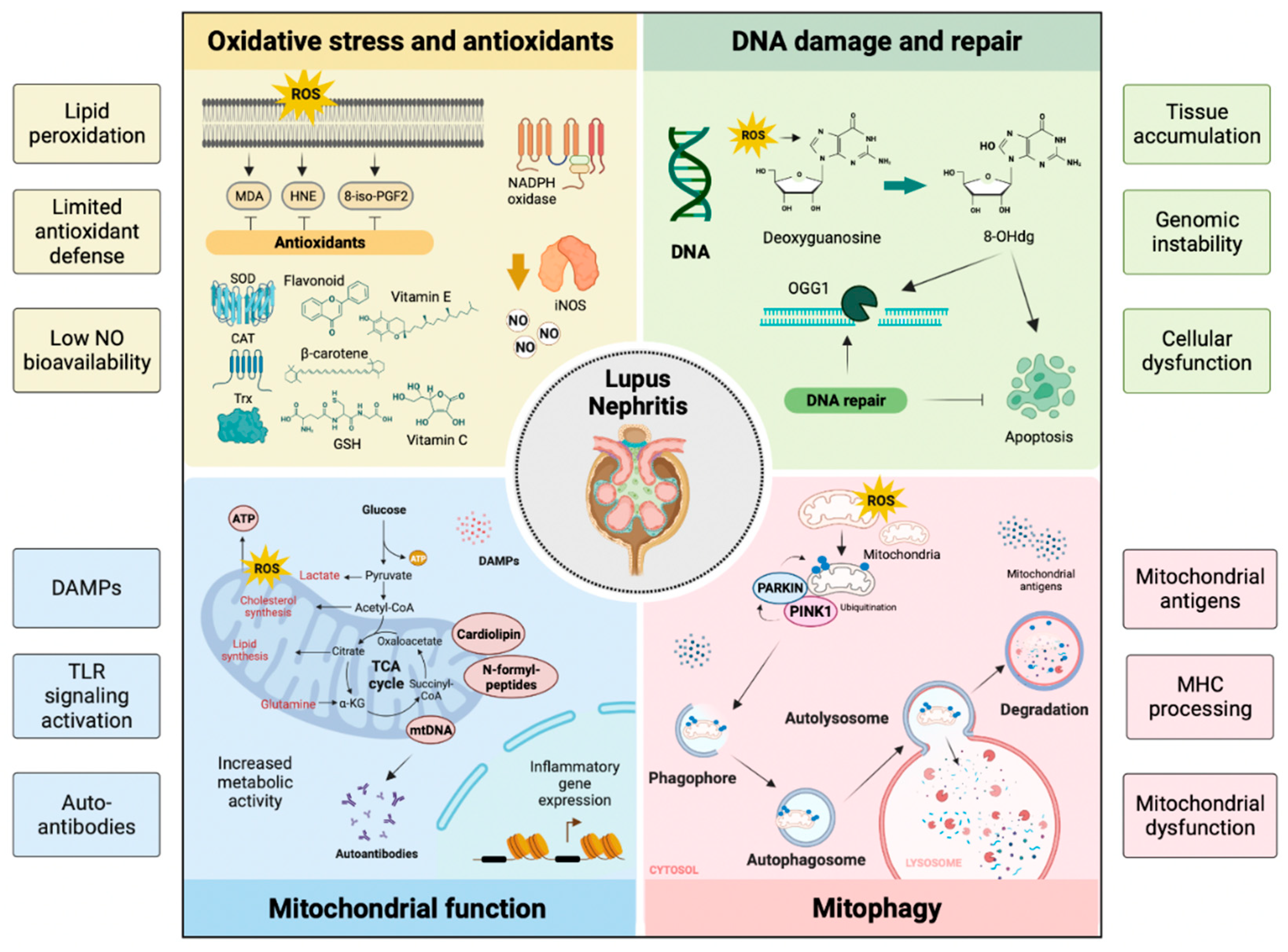
Figure 4.
PARTICIPATION OF INNATE IMMUNITY AND TLRs IN THE DEVELOPMENT OF LN. Some TLRs, such as TLR3, TLR7, and TLR9, are in the endosomal compartment where they detect endogenous ligands such as RNA, DNA, hypomethylated self-DNA within immune complexes, chromatin released from neutrophil extracellular traps (NETs), oxidized mitochondrial nucleoids, and other chromatin formats. Dendritic and B cells can process antigens and present antigens to T cells. In LN, TLR7 and TLR9 allow persistent activation of dendritic and B cells by autoantigens, thereby promoting autoantibody production, systemic autoimmunity, and glomerulonephritis mediated by immune complexes. In addition, the nucleic acid component of the immune complexes also activates intrarenal inflammation via TLR7 and TLR9 in intrarenal macrophages, resulting in the activation of glomerular endothelium and mesangial cells that are characteristic of LN.
Figure 4.
PARTICIPATION OF INNATE IMMUNITY AND TLRs IN THE DEVELOPMENT OF LN. Some TLRs, such as TLR3, TLR7, and TLR9, are in the endosomal compartment where they detect endogenous ligands such as RNA, DNA, hypomethylated self-DNA within immune complexes, chromatin released from neutrophil extracellular traps (NETs), oxidized mitochondrial nucleoids, and other chromatin formats. Dendritic and B cells can process antigens and present antigens to T cells. In LN, TLR7 and TLR9 allow persistent activation of dendritic and B cells by autoantigens, thereby promoting autoantibody production, systemic autoimmunity, and glomerulonephritis mediated by immune complexes. In addition, the nucleic acid component of the immune complexes also activates intrarenal inflammation via TLR7 and TLR9 in intrarenal macrophages, resulting in the activation of glomerular endothelium and mesangial cells that are characteristic of LN.
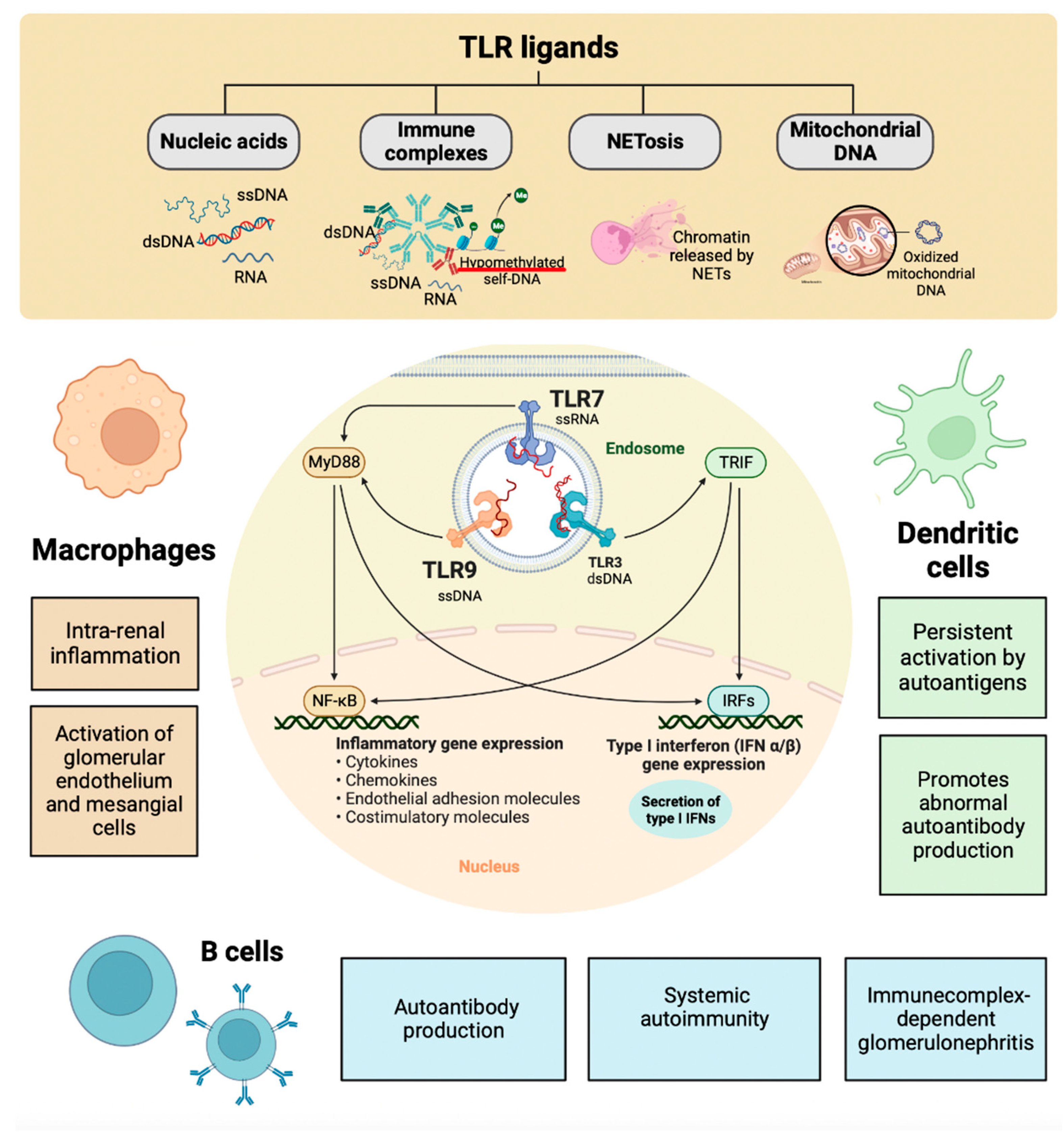
Figure 5.
THERAPEUTIC OBJECTIVES AND MANAGEMENT OF LN IN SLE. The goal of therapy for LN was established with a reduction in proteinuria by ≥25% with a stable glomerular filtration rate (GFR, ± 10% of baseline) in the first three months after the start of treatment, reduction in ≥50% proteinuria at six months and proteinuria <0.5-0.7 g/24 h at 12-24 months (all with stable GFR). Immunosuppressive therapies, immunomodulatory drugs, and vitamin D are currently used to control disease activity, prevent relapses, reduce the use of glucocorticoids, and lessen inflammation and fibrosis. Voclosporin, tacrolimus, rituximab, belimumab, and cyclophosphamide limit damage to target organs, including the kidney, in patients with SLE. However, new therapies focused on reducing the effect of endogenous nucleic acids mediated by TLR7 and TLR9 present promising results in preclinical studies, suggesting that their translation to the clinic could significantly benefit the quality of life, survival, and management of comorbidities in patients with SLE and LN.
Figure 5.
THERAPEUTIC OBJECTIVES AND MANAGEMENT OF LN IN SLE. The goal of therapy for LN was established with a reduction in proteinuria by ≥25% with a stable glomerular filtration rate (GFR, ± 10% of baseline) in the first three months after the start of treatment, reduction in ≥50% proteinuria at six months and proteinuria <0.5-0.7 g/24 h at 12-24 months (all with stable GFR). Immunosuppressive therapies, immunomodulatory drugs, and vitamin D are currently used to control disease activity, prevent relapses, reduce the use of glucocorticoids, and lessen inflammation and fibrosis. Voclosporin, tacrolimus, rituximab, belimumab, and cyclophosphamide limit damage to target organs, including the kidney, in patients with SLE. However, new therapies focused on reducing the effect of endogenous nucleic acids mediated by TLR7 and TLR9 present promising results in preclinical studies, suggesting that their translation to the clinic could significantly benefit the quality of life, survival, and management of comorbidities in patients with SLE and LN.
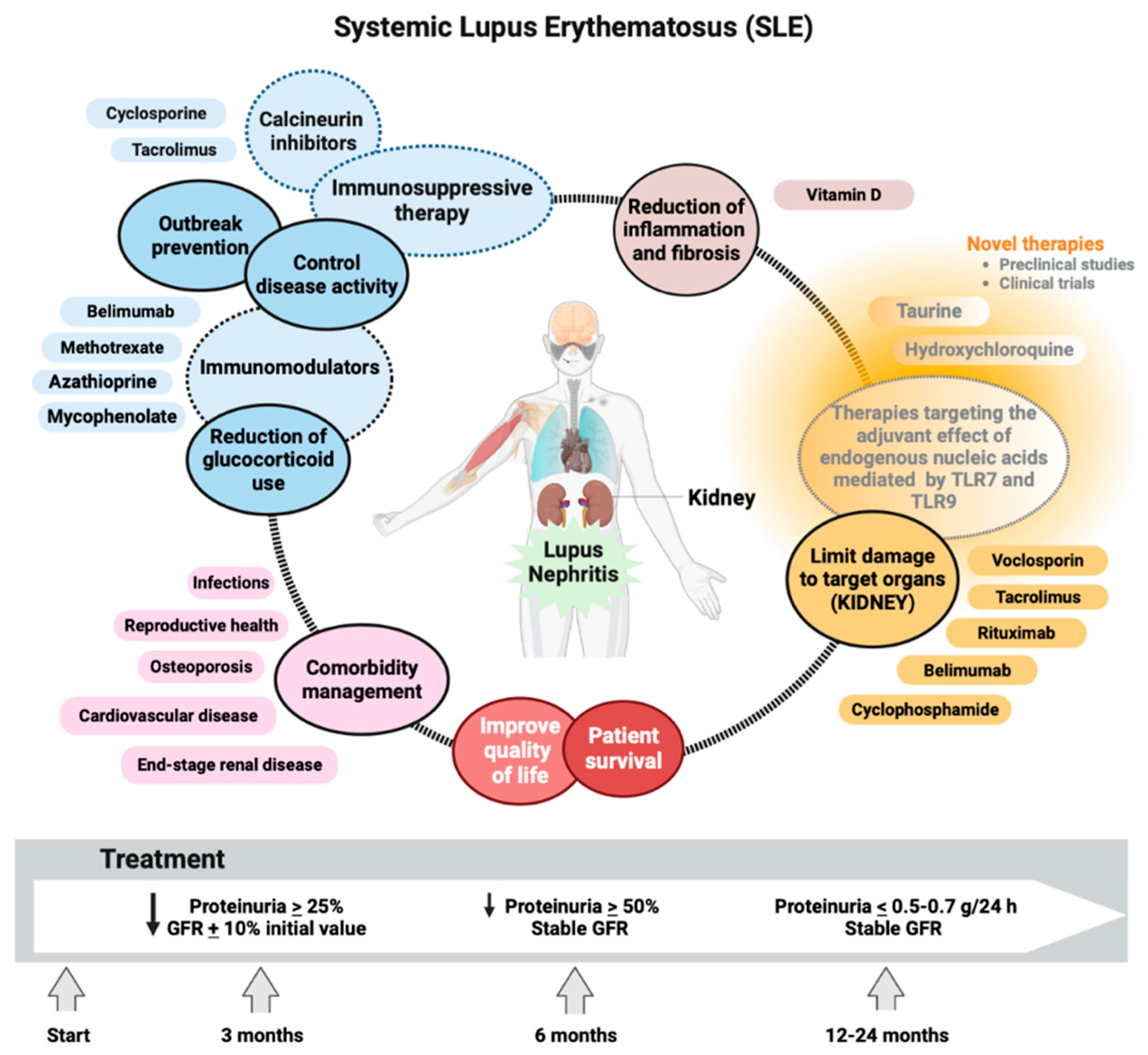
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



