
Submitted:
23 August 2023
Posted:
24 August 2023
You are already at the latest version
Abstract
Burkholderia andropogonis is a severe plant pathogen that can infect more than 50 plant species. Although its genome has been sequenced and annotated, various mechanisms underlying its physiology and pathogenicity remain unknown. Some of these mechanisms can be uncovered through understanding its proteome. In this study, sodium dodecyl polyacrylamide gel electrophoresis (SDS-PAGE) and mass spectrometry analysis were used to profile the proteome of B. andropogonis. Exactly 538 proteins, which belong to 190 protein families performing 88 different functions were identified. The resulting STRING network analysis identified 28 proteins having highly connected interactions with adenosine triphosphate (ATP)-binding-cassette and (ABC) transport system. To the knowledge of the authors, this is the first comprehensive proteomic study on B. andropogonis.
Keywords:
Burkholderia andropogonis
; mass spectrometry
; sodium dodecyl polyacrylamide gel electrophoresis
; proteomic profile
1. Introduction
Over the past three decades, research on Burkholderia bacterial species has been steadily expanding. This is because the members of the genus Burkholderia occupy surprisingly diverse ecological niches, including soil and hospital environments [1,2]. Furthermore, these aerobic gram-negative bacteria can be used as plant pest antagonists, plant growth promoters and degradative agents of toxic substances, while others have either no known ecological roles or are nuisances, such as being plant pathogens [1,3,4,5]. Burkholderia species that are pathogenic to plants include among others B. cepacia, found to be an onion pathogen [5], B. gladioli, which is a rice pathogen [4] and B. andropogonis, which is pathogenic to a wide range of crops [6,7,8,9,10]. Burkholderia andropogonis is a devastating pathogen of various crops such as amaranth (Amaranthus cruentus L.), common bean (Phaseolus vulgaris L.), broad bean (Vicia faba L.), hyacinth bean (Dolichos lablab L.), sorghum (Sorghum bicolor L.), sugarcane (Saccharum officinarum L.), rice (Oryza sativa L.) and maize (Zea mays L.) [6,7,8,9,10]. Its infection is associated with symptoms that include, among others, stem lesions, stem distortion, leaf spots, streaks and stripes [8,10,11,12,13]. The devastation caused by B. andropogonis is partly due to its production chlorotic compounds, for an example, amino enol ether rhizobitoxine which is responsible for chlrosis in soyaben [14].
Planting of resistant crops is thought to be the most effective strategy for plant disease control [15]. However, this strategy also requires an understanding of the plant pathogen at a molecular level. Efforts have been undertaken to understand the various molecular characteristics of B. andropogonis. Recently, the 6.29 Mbp genome of B. andropogonis ICMP2807 was sequenced [6]. The availability of this genome data and annotation showed that amongst others, that B. andropogonis has 6,047 protein-coding genes, which has inspired its further high-throughput functional molecular characterization such as transcriptomics, proteomics and metabolomics [6]. The efficient mining of these data will require the development of functional genomic tools, coupled with the determination of the full proteome. Large-scale proteomics have received attention because of its ability to uncover proteome-wide facts which can be directly linked to cell, tissue and organ function. Some of the milestone studies include studies which search for drug targets or discover potential drug targets for plant, animals and humans and as disease markers for patient diagnosis and disease progress monitoring. Through proteomics, Carvalho et al. [16] discovered that extracellular vesicles can be explored as candidates for patient diagnosis, follow-up and disease monitoring. Similarly, through proteomics Bellei et al. [17] disclosed the potential of gingival crevicular fluid as a source of biomarkers for severe periodontitis. Proteomic profiling of pathogenic Burkholderia species, such as B. cepacia and B. cenocepacia has been conducted [18,19]. Studies to elucidate the proteomes of B. cepacia and B. cenocepacia have made it possible to describe at a proteome-level, a significant number of cellular pathways, such as nucleotide synthesis, translation and protein-folding, cell envelope biogenesis, and iron homeostasis [18,19]. Furthermore, these profiles provided insight for understanding the physiology of these species in response to certain stresses, especially those induced by exposure to hazardous compounds [18]. This large scale proteomic can be used, among others, to discover targets for antibacterial drugs to control these plant pathogens similar to the potential applications discovered by Carvallo et al. [16] and Bellei et al. [17] on human health applications. Unfortunately, large scale proteomic studies have not yet been conducted for B. andropogonis. The lack of such studies is a critical gap in existing knowledge, which this study attempted to close. The aim of the present work was, therefore, to profile the proteome of B. andropogonis cultivated in vitro as well as determine protein interaction networks. The results generated in this research can be useful in elucidating the various physiological processes of B. andropogonis.
2. Materials and Methods
Protein profiling of B. andropogonis was determined following the schematic flowchart shown in Figure 1. In general, proteins were extracted from an overnight Luria-Bertani culture of B. andropogonis, the extracted proteins were separated by SDS-PAGE, and individual protein mass analysis and identification was done using AB SCIEX TripleTOF 6600 spectrometer coupled with Dionex Ultimate 3000 RSLCnano System (Thermo Fisher Scientific, United States of America), and UniProt database. Protein interaction networks were determined using STRING.
2.1. Extraction of Burkholderia andropogonis proteins
2.1.1. Growth of bacterial samples
The gram-negative bacterium, B. andropogonis (BD 00074) isolated from Ruscus (Ruscus aculeatus), was obtained from the Agricultural Research Council – Plant Protection Research (ARC-PPR). This bacterial strain was grown in lysogeny broth at 37 ˚C overnight, and the B. andropogonis cells were harvested by centrifugation at 3500 rpm for 6 min at room temperature [20]. The harvested bacterial cells were then lyophilized and incubated at -80 ̊ C for further analysis.
2.1.2. Preparation of protein extract
Lyophilized bacterial cells were transferred into sterile 1.5 ml sterile Eppendorf tubes and suspended in lysis buffer (1% SDS, 50 mM Tris-HCl pH 8.0). Re-suspended bacterial cells were then sonicated on ice (9 pulses, 10 sec per pulse with 10 sec intermission between pulses, 50% power setting) [19]. For the removal of nucleic acid contaminants, two microliters of viscolase (Benzonase®, Germany) was added to the cell lysate and incubated at 37 ̊ C for 30 min. Following incubation, cell lysate was then centrifuged (10 000 rpm, 5 min) to clear the cell debris [21]. The supernatant of the cell lysate was then stored at -20 ̊ C for further analysis.
2.2. Protein quantification
2.2.1. Bicinchoninic acid (BCA) assay
Proteins were quantified following modified bicinchoninic acid (BCA) assay [22]. In a 96 well plate assay, eight parts of the BCA working reagent (4 ml copper sulphide (CuS) and 80 µl BCA (Sigma-Aldrich, United States)) were mixed with one part of a protein sample. The samples were then incubated at 60 ̊ C for 15 min before reading the absorbance at 562 nm using a UV-2600 spectrophotometer (Shimadzu, Kyoto, Japan). The protein concentration was then determined by comparison to a standard curve; Bio-Rad standards (Bovine Gamma Globulin) with known concentrations were used to draw the standard curve.
2.2.2. Sodium Dodecyl Sulfate – Polyacrylamide Gel Electrophoresis (SDS-PAGE)
The protein patterns were separated according to size on a one-dimensional SDS-PAGE according to Laemmli [23]. BoltTM Mini gels (Life Technologies, United States) were used to separate the protein bands. Ten micrograms of each sample were mixed with BoltTM lithium dodecyl sulfate (LDS) sample buffer and BoltTM reducing agent before heating samples at 70 ̊ C for 10 min. After cooling, the samples were then loaded in BoltTM Mini gel and bands were separated at 200 V for 35 min. This was followed by gel staining with Coomassie® Brilliant blue R250 (Merck, Germany) overnight. The gel was then de-stained with successive distilled water (dH2O) washes before generating the gel image using G:Box syngene genesnap (Syngene, United Kingdom).
2.3. Extraction of peptides
2.3.1. Reduction of disulfide bonds and alkylating free cysteines
The protocol for the reduction of disulfide bonds and alkylating free cysteines was derived from Gundry et al. [24]. Selected protein bands from SDS-PAGE were excised manually, by cutting into small pieces (approximately 1 mm x 1 mm) before being transferred into 1.5 ml Eppendorf tubes respectively. Two hundred microliters of de-staining solution (50 mM ammonium hydrogen carbonate (NH4HCO3) and 50% methanol (MeOH)) was added to each sample before centrifugation at 10 000 rpm for 10 min. The supernatant was discarded before adding 100 µl of 75% acetonitrile (ACN) and centrifuging again at 10 000 rpm for 10 min. The supernatant was then discarded before drying the samples using a CentriVap concentrator (Labconco, USA). The dried samples were stored overnight at -20 ̊ C for further analysis.
Following overnight incubation, 200 µl of the working solution (10 mM dithiothreitol (DTT) in 25 mM NH4HCO3) was added to dried gels before homogenizing briefly. The reaction was allowed to proceed for one hour at 60 ̊ C. After an hour, the samples were chilled at room temperature before adding 500 µl of 100% ACN and incubating at 23 ̊ C for 10 min. Following incubation, the samples were centrifuged (10 000 rpm, 5 min) before discarding the supernatant. Two hundred microliters of the working solution (55 mM iodoacetamide in 25 mM NH4HCO3) was then added to the samples, before homogenizing briefly and incubating samples in the dark, at 23 ̊ C. After 20 min of incubation, the samples were centrifuged (10 000 rpm, 5 min) and the supernatant was discarded. To wash the samples, 100 µl of 25 mM NH4HCO3 was added to gel pieces before homogenization for 10 min. After homogenization, the samples were centrifuged (10 000 rpm, 5 min) and the supernatant was discarded before adding 200 µl of dehydration solution (25 mM NH4HCO3 in 50% ACN). The samples were then homogenized for 10 min before centrifugation (10 000 rpm, 5 min). The supernatant was then discarded before drying the gel pieces using a CentriVap concentrator (Labconco, USA) and storing the samples at -20 ̊ C for further analysis.
2.3.2. In-gel digestion
Peptides were extracted following a procedure derived from Gundry et al. [24]. According to this procedure, 25 ml of trypsin solution, enough to cover the gel pieces, was added to each respective gel piece and incubated on ice for one hour to allow diffusion and rehydration of the dried gel pieces. After an hour, 25 ml of 25 mM NH4HCO3 was added into each tube respectively, and the samples were incubated at 37 ̊ C overnight to allow enzymatic cleavage of proteins.
Following overnight incubation, the digested products were transferred into clean protein Lo-Bind tubes (Eppendorf North America, USA). To the respective gel pieces, 30 µl gel extraction solution (50% mass spectrometric (MS) grade acetonitrile and 5% mass spectrometric (MS) grade formic acid) was added before homogenizing the samples at room temperature for one hour. The samples were then sonicated on ice for 10 min before removing solutions and combining them with respective digest solutions in protein Lo-Bind tubes (Eppendorf North America, USA) [21]. The mixture of digest and aqueous extraction solutions were respectively dried using a CentriVap concentrator (Labconco, USA) before storing at -80 ̊ C for further analysis.
2.3.3. Peptide sample clean-up and protein identification
The basic protocol for peptide sample clean-up before MS analysis was derived from Gundry et al. [24]. The column was washed three times with 200 µl 100% ACN before being rinsing three times with 200 µl 0.1% trifluoroacetic acid (TFA) and discarding the flow-through. Ten percent TFA was added to bring the final concentration to 0.1% TFA. The samples were then applied to the column and pushed through slowly. The column was then rinsed five times with 200 µl 0.1% TFA before eluting the peptides from the column using 30 µl reversed-phase liquid chromatography electrospray ionization – tandem mass spectrometry (RP LC-ESI-MS/MS) elution solution.
The clean-up step preceded the analysis with AB Sciex TripleTOF 6600 mass spectrometer conducted at the Council for Scientific and Industrial Research, Pretoria, South Africa.
2.4. Extraction of peptides
Identified proteins were submitted to the STRING (version 11.0) for network analysis [26]. This database is one of the largest databases of known and predicted protein interactions [27,28]. Burkholderia phytofirmans was used as a related bacterium during the STRING search. Proteins were represented with nodes, and direct interactions (physical) were represented by continuous lines, while interrupted lines represented indirect interactions (functional). Cluster networks were then created using the Markov Cluster algorithm (MCL), which is included in the STRING website, and a value of 2 (confidence level) was selected for all the analyses [27].
3. Results and discussion
3.1. Protein profile of Burkholderia andropogonis
Five hundred and thirty-eight proteins were reproducibly present in all three technical replicates of B. andropogonis (Table 1). Functional classification of these 538 proteins revealed that 24% undertake translational functions, 13% protein biosynthesis processes, 5% is responsible for response to stress and the remaining constitute <5% in their respective functions (Figure 2). Furthermore, the protein-protein network of these proteins as analyzed by STRING revealed a multitude of interconnected pathways (Figure 3). As it was expected based on its genome sequence, B. andropogonis multitude of proteins which could be classified under type II topoisomerase GyrA and type II topoisomerase GyrB family are expressed (Table 1). These bacterial protein families modulate DNA topology by working against DNA topological problems generated from transcription, replication and repair events, thus are essential for cell survival [29,30,31]. As a result of being essential for cell survival and conferring antibacterial resistance, DNA gyrase, which is an enzyme needed for the production of these essential protein families, has been exploited as an important drug targets for anticancer and antibacterial agents [32,33].
Heat shock protein 70 and 90 protein families are also expressed protein families, which their respective genes dnaK and htpG, appear to be potential drug targets [34,35,36,37]. Their expression is required for survival of bacteria under stressful conditions, such as thermal stress and challenge with heavy metals, antibiotics or anticancer agents [38,39,40,41]. Other protein families that are expressed by B. andropogonis for stress response include CIpA/CIpB, GrpE, Hfq and peptidase S16 protein families (Table 1). The expression of these protein families contributes to resistance to stress, protein folding and various housekeeping functions [42,43,44]. This is supported by the genome sequence analysis of B. andropogonis that revealed nine genes for the peptide antibiotic colicin V and 94 genes associated with resistance to antibiotics and toxic compounds [6]. Furthermore, there is also iron/manganese superoxide dismutase and LptD family of proteins which are expressed during the removal of superoxide radicals and in response to organic substances. This was also expected as genes related to the degradation of aromatic compounds were also identified during postgenomic analysis of the B. andropogonis genome sequence [6].
In addition to these expressed protein families, other essential protein families that are expressed for DNA repair which include amongst others RecA and DNA mismatch repair MutS protein families. All these protein families contribute to the flexibility, plasticity and versatility of B. andropogonis, hence this bacterium can withstand cellular stress and show resistance to nearly all classes of antimicrobial agents.
Our results have highlighted the expressed proteins that confer antibiotic resistance and stress. Targeting these enzymes and their homologues might be an effective way to control this devastating plant pathogen. This is supported by the work reported by Wolska et al. [38], that Eschericia coli dnaK and dnaJ mutants showed antibiotic susceptibility. DnaK is Hsp70, which is a molecular chaperone expressed in response to stress [45,46]. Hence, targeting molecular chaperones of such nature expressed by B. andropogonis, suppressing their expression and their resultant proteins thereof, might consequently result in finding an effective control method for this devastating plant pathogen.
3.2. Functional information: Interactome analysis
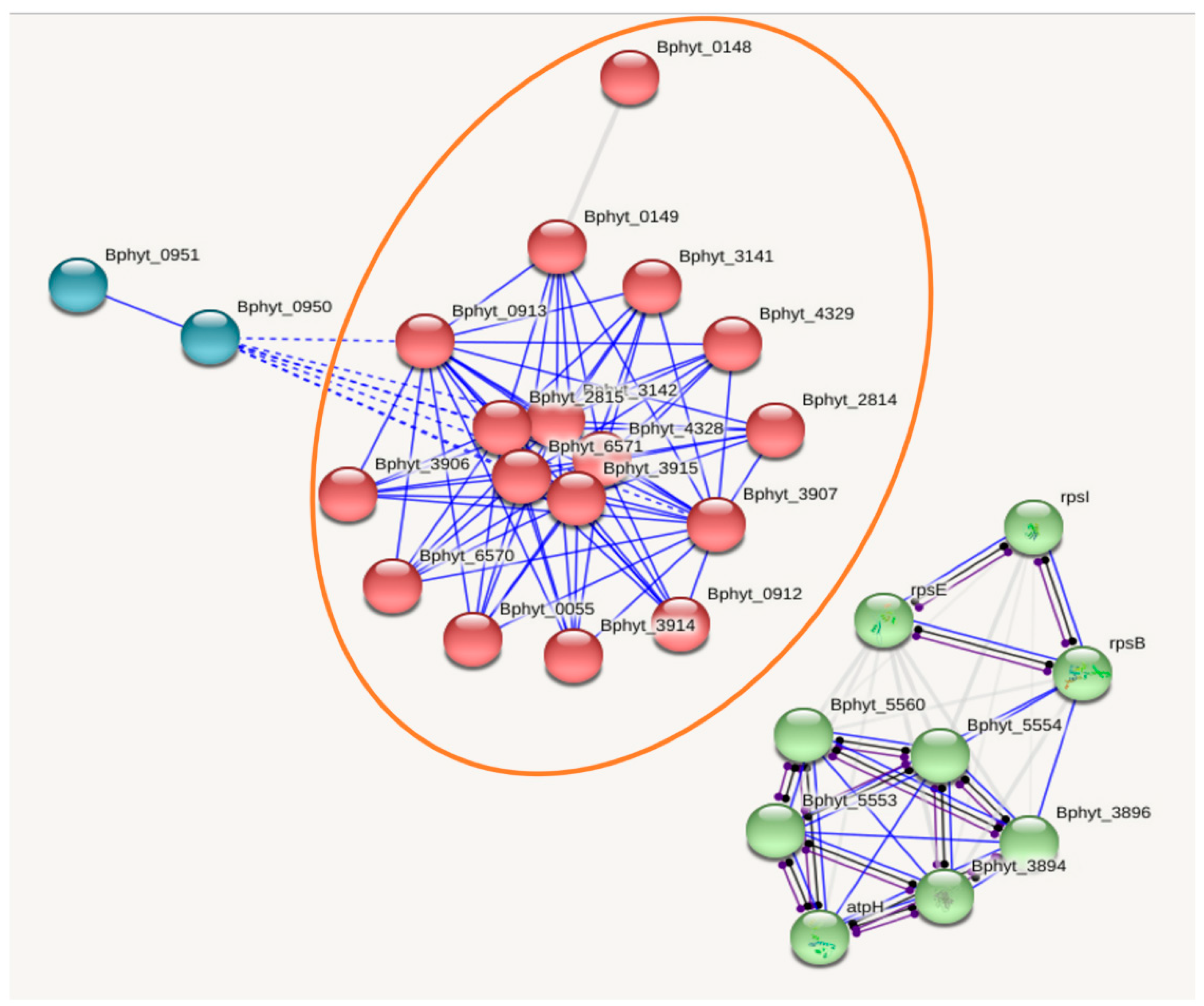
The resulting network of selected proteins identified during profiling composed 28 nodes (proteins) and 116 edges (interactions) (Figure 3). This network represents the first comprehensive interactomics map for the selected part of B. andropogonis proteome and provides an excellent starting point for navigating through the proteome. The topological analysis of this network demonstrated a central interplay with highly connected interactions (physical) and indirectly connected sub-networks. The main central subset is composed of 17 nodes and is related to the adenosine triphosphate (ATP)-binding-cassette (ABC) transport system (Figure 3). ABC transporters are widespread among all living organisms and accomplish not only the uptake of nutrients in bacteria, but also operate in diverse processes, which include among others signal transduction, protein secretion, bacterial pathogenesis, drug and antibiotic resistance [47,48].
In bacteria, the ABC transport system transports diverse substrates, which include virulence factors, [47,48,49] substrates that confer bactericidal resistance [50] or cellular defence factors on pathogenic bacteria [51]. Transportation of these substrates results in antibacterial resistant bacterial strains that pose a threat to food security and contribute to a growing threat to public health [52]. This is supported by the reports of antibacterial resistant bacterial strains such as B. andropogonis that has shown emergence of resistance to nearly all classes of antimicrobial agents [42,43,44]. Genome annotation of this bacterium showed that B. andropogonis has nine genes for the peptide antibiotic colicin V and 94 genes associated with resistance to antibiotic and toxic compounds [53]. Our STRING analysis results suggest that some of the antibacterial resistance factors are transported via the ABC transport system. Therefore, this system confers to the antibacterial resistance of this bacterium; hence B. andropogonis have shown the emergence of resistance to nearly all classes of antimicrobial agents [42,43,44].
3.3. Cellular pathways expressed in B. andropogonis
As it was expected based on protein families expressed by B. andropogonis, both catabolic and anabolic pathways are expressed respectively. The catabolic pathways expressed included amongst others glycolysis, which involves a series of metabolic processes by which one molecule of glucose is catabolized to two molecules of pyruvate with a net gain of two ATP molecules [54]. Also known as the Embden-Meyerhof pathway, this process was reported to be increased in cancer cells [55,56,57,58]. This suggested that a high level of glycolytic activity is essential for cancer cells to survive and grow. Moreover, this metabolic feature has led to the hypothesis that inhibition of glycolysis may severely eliminate ATP generation in cancer cells and thus may preferentially kill the malignant cells [59,60,61]. Since the increased expression of glycolytic enzymes provides a survival advantage for the cancer cells [54], it can be hypothesized that in the presence of the antibacterial agents, B. andropogonis also increases its glycolytic activity resulting in survival advantage and consequently displays the antibacterial resistance. This is supported by the fact that B. andropogonis has shown an emergence of resistance to nearly all classes of antimicrobial agents [49,50,51].
Conversely, the anabolic pathways expressed included, amongst others, gluconeogenesis, which involves synthesizing glucose from non-glucose precursors such as lactate, amino acids and glycerol [62]. This pathway has been reported to be providing growth advantage to bacteria in intestinal fluids by utilizing aspartate, serine, glycerol and lactate as the main gluconeogenic substrates [62]. Thus, this suggests that B. andropogonis can also utilize this pathway for growth advantage and survival. In addition, tricarboxylic acid cycle (TCA), also known as Krebs cycle, is an amphibolic pathway that is shown to be expressed by B. andropogonis. This pathway serves in both catabolic and anabolic processes, feeding substrates to various pathways including glycolysis and gluconeogenesis [62]. This means that the higher the activity of TCA, the higher the activity of glycolysis and gluconeogenesis. And consequently, the higher the growth and survival advantage of B. andropogonis even in the presence of the antibacterial agents. This study generated large-scale data because of the choice of applications used. Our choice of SDS-PAGE and mass spectrometry as analytical tools was suitable to uncover the multitude of knowledge we have generated. SDS-PAGE coupled with mass spectrometry has been useful and validated as a useful proteomics technique. The coupling of these techniques proved useful in the study of Darie-Ion et al., [63] who identified zein proteins upon eco-friendly ultrasound-based extraction, similarly, the study of Sousa et al., [64], Isani et al., [65] and a plethora of other investigations as reviewed by Egragán et al., [66] and Cordwell et al., [67] and Saleh et al., [68] who focussed specifically on bcterial pathogens.
4. Conclusion and future perspectives
The identification of the 538 proteins produced by B. andropogonis provides new and important insight into the pathogen’s in vitro growth and development (excluding proteins associated with its pathogenesis). More importantly, the functional characterization of molecular chaperones proteins reveals that they are essential for the growth, adaptation and survival of B. andropogonis even in the presence of antibacterial agents. This is in line with the previous reports that the increase of glycolysis enhances drug resistance [61]. Hence, targeting these proteins (stress responses and antibiotic resistance) may consequently result in determining an effective control method for this devastating plant pathogen.
Future work must focus on the molecular responses of this bacterium to exposure of various control agents. Various combinations of antibiotics, using antibiotic adjuvants or even botanical extracts can be investigated on how they alter the level of protein expressions that confers stress responses and antibiotic resistance. Lastly, various B. andropogonis mutants could be created and subjected to antibacterial agents. This will highlight the essential genes that should be targeted for its control and in order of their importance. This study, therefore, has added to the protein database of B. andropogonis, which will benefit functional characterization and future proteome level investigations of other phytopathogenic fungi.
Author Contributions
L.L.K., M.L.B.T. and N.K. writing – original draft preparation; N.K. and O.S. writing – review and editing; L.L.K., O.S. and M.S. data curation. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Agricultural Research Council-Vegetable, Indigenous and Medicinal Plants of South Africa and the University of South Africa. Funding was also provided partially by the National Research Foundation (grant number TTK170413227119).
Data Availability Statement
Data generated in this work was submitted into the MassIVE Data submission platform under ID MassIVE MSV000090102.
Conflicts of Interest
The authors declare that they have no known competing financial interest or personal relationships that could have appeared to influence the work reported in this paper.
References
- Estrada-De Los Santos, P.; Bustillos-Cristales, R.; Caballero-Mellado, J. Burkholderia, a genus rich in plant-associated nitrogen fixers with wide environmental and geographic distribution. Appl. Environ. Microbiol. 2001, 67, 2790–2798. [Google Scholar] [CrossRef] [PubMed]
- Coenye, T.; Vandamme, P. Diversity and significance of Burkholderia species occupying diverse ecological niches. Environ. Microbiol. 2003, 5, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Chiarini, L.; Bevivino, A.; Dalmastri, C.; Tabacchioni, S.; Visca, P. Burkholderia cepacia complex species: health hazards and biotechnological potential. Trends Microbiol. 2006, 14, 277–286. [Google Scholar] [CrossRef]
- Ura, H.; Furuya, N.; Iiyama, K.; Hidaka, M.; Tsuchiya, K.; Matsuyama, N. Burkholderia gladioli associated with symptoms of bacterial grain rot and leaf-sheath browning of rice. J. Gen. Plant Pathol. 2006, 72, 98–103. [Google Scholar] [CrossRef]
- Jacobs, J.L.; Fasi, A.C.; Ramette, A.; Smith, J.J.; Hammerschmidt, R.; Sundin, G.W. Identification and onion pathogenicity of Burkholderia cepacia complex isolates from the onion rhizosphere and onion field soil. Appl. Environ. Microbiol. 2008, 74, 3121–3129. [Google Scholar] [CrossRef]
- Lopes-Santos, L.; Castro, D.B.A.; Ottoboni, L.M.M.; Park, D.; Weir, B.S.; Destéfano, S.A.L. Draft genome sequence of Burkholderia andropogonis type strain ICMP2807, isolated from Sorghum bicolor. Genome Announc. 2015, 3, e00455–15. [Google Scholar] [CrossRef] [PubMed]
- Cother, E.J.; Noble, D.; Peters, B.J.; Albiston, A.; Ash, G.J. A new bacterial disease of jojoba (Simmondsia chinensis) caused by Burkholderia andropogonis. Plant Pathol. 2004, 53, 129–135. [Google Scholar] [CrossRef]
- Li, X.; De Boer, S.H. First report of Burkholderia andropogonis causing leaf spots of Bougainvillea sp. in Hong Kong and Clover in Canada. Plant Dis. 2005, 89, 1132. [Google Scholar] [CrossRef]
- Compant, S.; Nowak, J.; Coenye, T.; Clément, C.; Barka, E.A. Diversity and occurrence of Burkholderia spp. in the natural environment. FEMS Microbiol. Rev. 2008, 32, 607–626. [Google Scholar] [CrossRef]
- Duan, Y.P.; Sun, X.; Zhou, L.J.; Gabriel, D.W.; Benyon, L.S.; Gottwald, T. Bacterial brown leaf spot of citrus, a new disease caused by Burkholderia andropogonis. Plant Dis. 2009, 93, 607–614. [Google Scholar] [CrossRef]
- Ullstrup, A.J. Bacterial stripe of corn. Phytopathology. 1960, 50, 906–910. [Google Scholar]
- Bradbury, J.F. Pseudomonas andropogonis; In IMI Descriptions of Fungi and Bacteria; CAB International: Wallingford, UK, 1973; Volume 38, p. 372. [Google Scholar]
- Gitaitis, R.D.; Miller, J.; Wells, H.D. Bacterial leaf spot of white clover in Georgia. Plant Dis. 1983, 67, 913–914. [Google Scholar] [CrossRef]
- Coenye, T.; Laevens, S.; Gillis, M.; Vandamme, P. Genotypic and chemotaxonomic evidence for the reclassification of Pseudomonas woodsii (Smith 1911) Stevens 1925 as Burkholderia andropogonis (Smith 1911) Gillis et al. 1995. Int. J. Syst. Evol. Microbiol. 2001, 51, 183–185. [Google Scholar] [CrossRef]
- Ham, J.H.; Melanson, R.A.; Rush, M.C. Burkholderia glumae: next major pathogen of rice? Mol. Plant Pathol. 2011, 12, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.S.; Baeta, H.; Henriques, A.F.A.; Ejtehadifar, M.; Tranfield, E.M.; Sousa, A.L.; Farinho, A.; Silva, B.C.; Cabeçadas, J.; Gameiro, P.; Silva, M.G.; Beck, H.C.; Matthiesen, R. Proteomic Landscape of Extracellular Vesicles for Diffuse Large B-Cell Lymphoma Subtyping. Int. J. Mol. Sci. 2021, 22, 11004. [Google Scholar] [CrossRef]
- Bellei, E.; Bertoldi, C.; Monari, E.; Bergamini, S. Proteomics Disclose the Potential of Gingival Crevicular Fluid (GCF) as a Source of Biomarkers for Severe Periodontitis. Materials 2022, 15, 2161. [Google Scholar] [CrossRef]
- Kwok, S.Y.; Siu, A.F.M.; Ngai, S.M.; Che, C.M.; Tsang, J.S.H. Proteomic analysis of Burkholderia cepacia MBA4 in the degradation of monochloroacetate. Proteomics. 2007, 7, 1107–1116. [Google Scholar] [CrossRef]
- Madeira, A.; dos Santos, S.C.; Santos, P.M.; Coutinho, C.P.; Tyrell, J.; McClean, S.; Callaghan, M.; Sá-Correia, L. Proteomic profiling of Burkholderia cenocepacia clonal isolates with different virulence potential retrieved from a cystic fibrosis patient during chronic lung infection. PLoS ONE. 2013, 8, e83065. [Google Scholar] [CrossRef]
- Rojas-Rojas, F.U.; López-Sánchez, D.; Meza-Radilla, G.; Méndez-Canarios, A.; Ibarra, J.A.; Estrada-de Los Santos, P. The controversial Burkholderia cepacia complex, a group of plant growth promoting species and plant, animals and human pathogens. Rev. Argent. Microbiol. 2019, 51, 84–92. [Google Scholar]
- Stoychev, S.; Naicker, P.; Mamputha, S.; Khumalo, F.; Gerber, I.; van der Westhuyzen, C.; Jordan, J. Universal unbiased pre-MS clean-up using magnetic HILIC micro-particles for SPE. 2017. Available online: https://www.bioinfor.com/wp-content/uploads/2017/06/Universal-unbiased-pre-ms-clean-up-using-magnetic-hilic.pdf. (accessed on 14 August 2017).
- Kapoor, K.N.; Barry, D.T.; Rees, R.C.; Dodi, L.A.; McArdle, S.E.B.; Creaser, C.S.; Bonner, P.L.R. Estimation of peptide concentration by a modified bicinchoninic acid assay. Anal. Biochem. 2009, 393, 138–140. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Gundry, R.L.; White, M.Y.; Murray, C.I.; Kane, L.A.; Fu, Q.; Stanley, B.A.; Van Eyk, J.E. Preparation of proteins and peptides for mass spectrometry analysis in a bottom-up proteomics workflow. Curr. Protoc. Mol. Biol. 2010, 90, 10.25.1–10.25.23. [Google Scholar] [CrossRef]
- Available online: https://www.uniprot.org (accessed on 1 February 2019).
- Available online: https://string-db.org (accessed on 12 February 2019).
- Szklarczyk, D.; Franceschini, A.; Kuhn, M.; Simonovic, M.; Roth, A.; Minguez, P.; Doerks, T.; Stark, M.; Muller, J.; Bork, P.; Jensen, J.J.; von Mering, C. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011, 39, 561–568. [Google Scholar] [CrossRef]
- Carrera, M.; Cañas, B.; Gallardo, J.M. The sarcoplasmic fish proteome: pathways, metabolic networks and potential bioactive peptide for nutritional inferences. J. Proteomics. 2013, 78, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C. DNA topoisomerases. Annu. Rev. Biochem. 1996, 65, 635–692. [Google Scholar] [CrossRef] [PubMed]
- Champoux, J.J. DNA topoisomerases: structure, function, and mechanism. Ann. Rev. Biochem. 2001, 70, 369–413. [Google Scholar] [CrossRef] [PubMed]
- Corbett, K.D.; Berger, J.M. Structure, molecular mechanisms, and evolutionary relationships in DNA topoisomerases. Ann. Rev. Biophy. Biomol. Struct. 2004, 33, 95–118. [Google Scholar] [CrossRef]
- Maxwell, A. DNA gyrase as a drug target. Trends Microbiol. 1997, 5, 102–109. [Google Scholar] [CrossRef]
- Mdluli,K. ; Ma, Z. Mycobacterium tuberculosis DNA gyrase as a target for drug discovery, Infect. Disord. Drug Targets. 2007, 7, 159–168. [Google Scholar] [CrossRef]
- Hanawa, T.; Fukuda, M.; Kawakamis, H.; Hirano, H.; Kamiya, S.; Yamamoto, T. The Listeria monocytogenes DnaK chaperone is required for stress tolerance and efficient phagocytosis with macrophages. Cell Stress Chaperones. 1999, 4, 118–128. [Google Scholar] [CrossRef]
- Takaya, A.; Tomoyasu, T.; Matsui, H.; Yamamoto, T. The DnaK/DnaJ chaperone machinery of Salmonella enterica serovar typhimurium is essential for invasion of epithelial cells and survival within macrophages, leading to systemic infection. Infect. Immun. 2004, 72, 1364–1373. [Google Scholar] [CrossRef] [PubMed]
- da Rocha Dias, S.; Friedlos, F.; Light, Y.; Springer, C.; Workman, P.; Marais, R. Activated B-RAF is an Hsp90 client protein that is targeted by the anticancer drug 17-Allylamino-17-demethoxygeldanamycin. Cancer Res. 2005, 65, 10686–10691. [Google Scholar] [CrossRef]
- Powers, M.V.; Workman, P. Targeting of multiple signaling pathways by heat shock protein 90 molecular chaperone inhibitors. Endocr. Relat. Cancer. 2006, 13, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Wolska, K.I.; Bugajska, E.; Jurkiewicz, D.; Kuć, M.; Jŏźwik, A. Antibiotic susceptibility of Escherichia coli dnaK and dnaJ mutants. Microb. Drug Resist. 2004, 6, 119–126. [Google Scholar] [CrossRef]
- Whitesell, L.; Bagatell, R.; Falsey, R. The stress response: implications for the clinical development of Hsp90 inhibitors. Curr. Cancer Drug Targets. 2003, 3, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Henderson, B.; Allan, E.; Coates, A.R.M. Stress wars: the direct role of host and bacterial molecular chaperones in bacterial infection. Infect. Immun. 2006, 74, 3693–3706. [Google Scholar] [CrossRef]
- Lemos, J.A.; Luzardo, Y.; Burne, R.A. Physiologic effects of forced down-regulation of dnaK and groEL expression in Streptococcus mutans. J. Bacteriol. 2007, 189, 1582–1588. [Google Scholar] [CrossRef]
- Mah, T.F.; O’Toole, G.A. Mechanisms of biofilm resistance to antimicrobial agents. Trends in Microbiol. 2001, 9, 34–39. [Google Scholar] [CrossRef]
- BurmØlle, M.; Webb, J.S.; Rao, D.; Hansen, L.H.; SØrensen, S.J.; Kjelleberg, S. Enhanced biofilm formation and increased resistance to antimicrobial agents and bacterial invasion are caused by synergistic interactions in multispecies biofilms. Appl. Environ. Microbiol. 2006, 72, 3916–3923. [Google Scholar] [CrossRef]
- Yura, T; Nakahigashi, K. Regulation of the heat-shock response. Curr. Opin. Microbiol. 1999, 2, 153–158. [Google Scholar] [CrossRef]
- Lu, P. , Xue, J., Chen, X; Ji, X. DnaK-Mediated Protein Deamidation: a Potential Mechanism for Virulence and Stress Adaptation in Cronobacter sakazakii. Appl. Environ. Microbiol. 2023, 89, e00505-23. [Google Scholar] [CrossRef] [PubMed]
- Oberto, J. , Bonnefoy, E., Mouray, E., Pellegrini, O., Wikström, P.M., Rouvière-Yaniv, J. The Escherichia coli ribosomal protein S16 is an endonuclease. Mol. Microbiol. 1996, 19, 1319–1330. [Google Scholar] [CrossRef] [PubMed]
- Higgins, C.F. ABC transporters: from microorganisms to man. Ann. Rev. Cell Biol. 1992, 8, 67–113. [Google Scholar] [CrossRef] [PubMed]
- Schneider, E.; Hunke, S. ATP-binding-cassette (ABC) transport systems: functional and structural aspects of the ATP-hydrolyzing subunits/domains. FEMS Microbiol. Rev. 1998, 22, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Fath, M.J.; Kolter, R. ABC transporters: bacterial exporters. Microbiol. Mol. Biol. Rev. 1993, 57, 995–1017. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.I.; Eady, E.A.; Cove, J.H.; Cunliffe, W.J.; Baumberg, S.; Wootton, J.C. Inducible erythromycin resistance in staphylococci is encoded by a member of the ATP-binding transport super-gene family. Mol. Microbiol. 1990, 4, 1207–1214. [Google Scholar] [CrossRef]
- Parra-Lopez, C.; Baer, M.T.; Groisman, E.A. Molecular genetic analysis of a locus required for resistance to antimicrobial peptides in Salmonella typhimurium. EMBO J. 1993, 12, 4053–4062. [Google Scholar] [CrossRef]
- Travis, J. Reviving the antibiotic miracle? Science. 1994, 264, 360–362. [Google Scholar] [CrossRef]
- Pelicano, H.; Martin, D.S.; Xu, R.H.; Huang, P. Glycolysis inhibition for anticancer treatment. Oncogene. 2006, 25, 4633–4646. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef]
- Warburg, O. Warburg report on the metabolism of tumors. J. Chem. Educ. 1930, 7, 179. [Google Scholar]
- Warburg, O. On the origin of cancer cells. Science. 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Semenza, G.L.; Artemov, D.; Bedi, A.; Bhujwalla, Z.; Chiles, K.; Feldser, D.; Laughner, E.; Ravi, R.; Simons, J.; Taghavi, P.; Zhong, H. ‘The metabolism of tumours’: 70 years later. Novartis Foundation Symp. 2001, 240, 251–264. [Google Scholar]
- Muñoz-Pinedo, C.; Ruiz-Ruiz, C.; de Almodóvar, C.R.; Palacios, C.; López-Rivas, A. Inhibition of glucose metabolism sensitizes tumor cells to death receptor-triggered apoptosis through enhancement of death-inducing signaling complex formation and apical procaspace-8 processing. J. Biol. Chem. 2003, 278, 12759–12768. [Google Scholar] [CrossRef] [PubMed]
- Izyumov, D.S.; Avetisyan, A.V.; Pletjushkina, O.Y.; Sakharov, D.V.; Wirtz, K.W.; Chernyak, B.V.; Skulachev, P.V. “Wages of Fear”: transient threefold decrease in intracellular ATP level imposes apoptosis. Biochim. Biophys. Acta Bioenerg. 2004, 1658, 141–147. [Google Scholar] [CrossRef]
- Xu, R.H.; Pelicano, H.; Zhou, Y.; Carew, J.S.; Feng, L.; Bhalla, K.N.; Keating, J.; Huang, P. Inhibition of glycolysis in cancer cells: a novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia. Cancer Res. 2005, 65, 613–621. [Google Scholar] [CrossRef]
- Mohan, C.; Bessman, S.P. Anabolic regulation of gluconeogenesis by insulin in isolated rat hepatocytes. Arch. Biochem. Biophys. 1985, 242, 563–573. [Google Scholar] [CrossRef]
- Bertin, Y.; Deval, C.; de la Foye, A.; Masson, L.; Gannon, V.; Harel, J.; Martin, C.; Desvaux, M.; Forano, E. The gluconeogenesis pathway is involved in maintenance of enterohaemorrhagic Escherichia coli O157:H7 in bovine intestinal content. PLoS ONE. 2014, 9, e98367. [Google Scholar] [CrossRef]
- Darie-Ion, L.; Jayathirtha, M.; Hitruc, G.E.; Zaharia, M.-M.; Gradinaru, R.V.; Darie, C.C.; Pui, A.; Petre, B.A. A proteomic approach to identify zein proteins upon eco-friendly ultrasound-based extraction. Biomolecules 2021, 11, 1838. [Google Scholar] [CrossRef] [PubMed]
- Sousa, P. , Camacho, I., Câmara, J.S.; Perestrelo, R.. Urinary proteomic/peptidomic biosignature of breast cancer patients using 1D SDS-PAGE combined with matrix-assisted laser desorption/ionization-time of flight mass spectrometry. Separations. 2023, 10, 291. [Google Scholar] [CrossRef]
- Isani, G. , Bellei, E., Rudelli, C., Cabbri, R., Ferlizza, E. Andreani, G. SDS-PAGE-Based Quantitative assay of hemolymph proteins in honeybees: Progress and Prospects for Field Application. Int. J. Mol. Sci. 2023, 24, 10216. [Google Scholar] [CrossRef] [PubMed]
- Agregán, R.; Echegaray, N.; López-Pedrouso, M.; Kharabsheh, R.; Franco, D.; Lorenzo, J.M. Proteomic advances in milk and dairy products. Molecules. 2021, 26, 3832. [Google Scholar] [CrossRef] [PubMed]
- Cordwell, S.J. , Nouwens, A.S.; Walsh, B.J. Comparative proteomics of bacterial pathogens. PROTEOMICS: International Edition, 1(4), 2001; 461-472. [Google Scholar]
- Saleh, S. , Staes, A., Deborggraeve, S.; Gevaert, K. Targeted proteomics for studying pathogenic bacteria. Proteomics. 2019, 19, 1800435. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The schematic flowchart of Materials and Methods used for protein profiling of Burkholderia andropogonis. It represents a work process for a high-throughput proteomic profiling experiment. The workflow comprises seven key steps: sample preparation, protein extraction, protein quantification, peptide extraction, peptide clean-up, mass spectrometry and down-stream analysis.
Figure 1.
The schematic flowchart of Materials and Methods used for protein profiling of Burkholderia andropogonis. It represents a work process for a high-throughput proteomic profiling experiment. The workflow comprises seven key steps: sample preparation, protein extraction, protein quantification, peptide extraction, peptide clean-up, mass spectrometry and down-stream analysis.
Figure 2.
Functional characterization of all (538) proteins extracted from Burkholderia andropogonis. The listed percentage corresponds only to the proportion of detected proteins profiled from B. andropogonis that are significantly involved in both molecular functions and biological processes.
Figure 2.
Functional characterization of all (538) proteins extracted from Burkholderia andropogonis. The listed percentage corresponds only to the proportion of detected proteins profiled from B. andropogonis that are significantly involved in both molecular functions and biological processes.
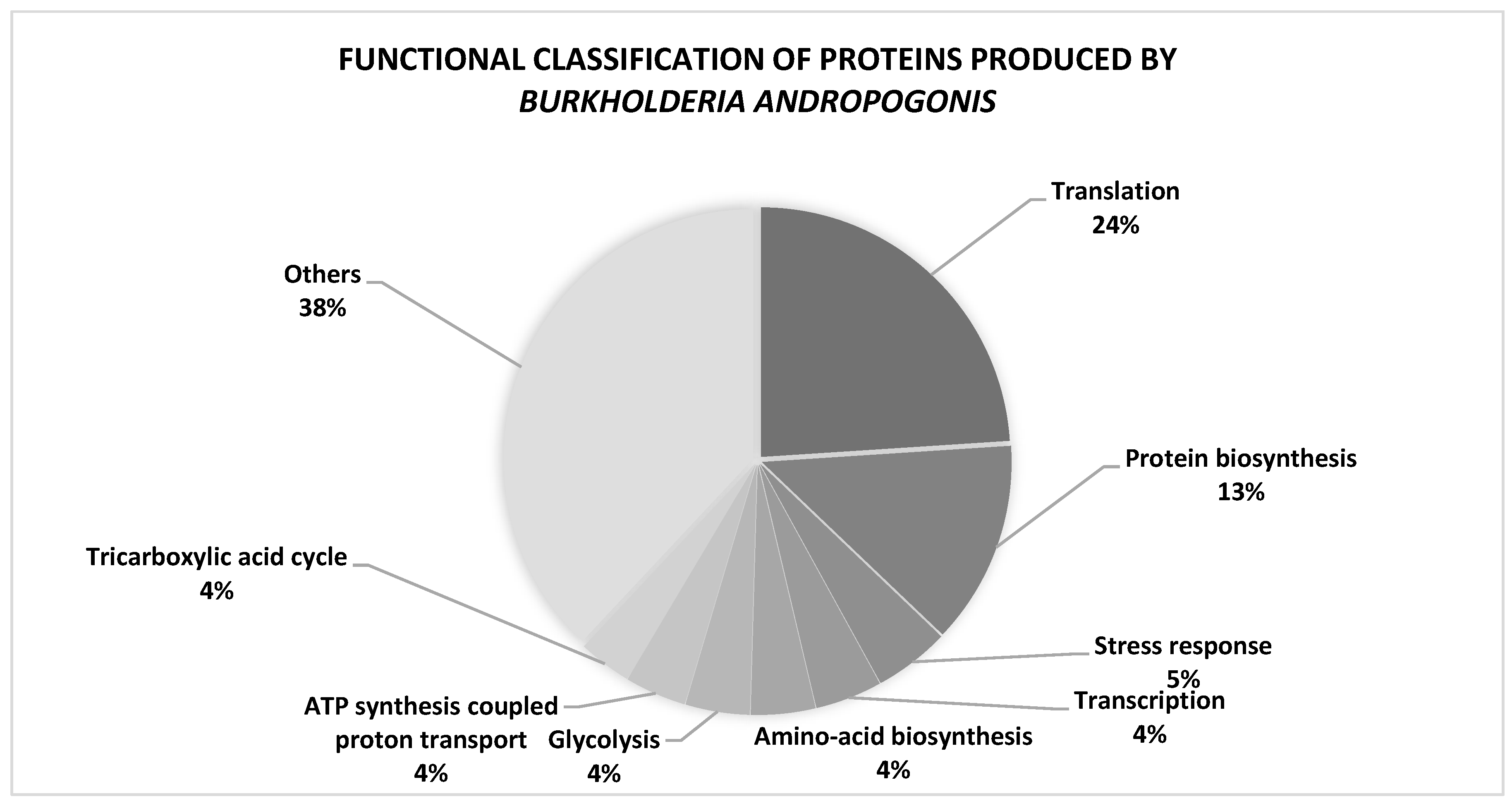
Figure 3.
Functional characterization of all (538) proteins extracted from Burkholderia andropogonis. The listed percentage corresponds only to the proportion of detected proteins profiled from B. andropogonis that are significantly involved in both molecular functions and biological processes.
Figure 3.
Functional characterization of all (538) proteins extracted from Burkholderia andropogonis. The listed percentage corresponds only to the proportion of detected proteins profiled from B. andropogonis that are significantly involved in both molecular functions and biological processes.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



