
Submitted:
23 August 2023
Posted:
23 August 2023
You are already at the latest version
Abstract
Hepatocellular Carcinoma (HCC) continues to pose a substantial global health challenge due to its high incidence and limited therapeutic options. In recent years, the Janus Kinase (JAK) and Signal Transducer and Activator of Transcription (STAT) pathway has emerged as a critical signaling cascade in HCC pathogenesis. The review commences with an overview of the JAK/STAT pathway, delving into the dynamic interplay between the JAK/STAT pathway and its numerous upstream activators, such as cytokines and growth factors enriched in pathogenic livers afflicted with chronic inflammation and cirrhosis. This paper also elucidates how the persistent activation of JAK/STAT signaling leads to diverse oncogenic processes during hepatocarcinogenesis, including uncontrolled cell proliferation, evasion of apoptosis, and immune escape. In the context of therapeutic implications, this review summarizes recent advancements in targeting the JAK/STAT pathway for HCC treatment. Preclinical and clinical studies investigating inhibitors and modulators of JAK/STAT signaling are discussed, highlighting their potential in suppressing the deadly disease. The insights presented herein underscore the necessity for continued research into targeting the JAK/STAT signaling pathway as a promising avenue for HCC therapy.
Keywords:
Hepatocellular Carcinoma
; JAK/STAT Signaling
; Cytokine
; Targeted Therapy
1. Introduction
Hepatocellular Carcinoma (HCC) is the most prevalent primary liver malignancy and continues to be a global health concern due to its rising incidence and limited treatment options [1,2,3]. Despite recent advancements in understanding the molecular pathogenesis of HCC, the intricate molecular mechanisms underlying its initiation, progression, and therapeutic resistance remain unresolved. One critical pathway that has emerged as a pivotal player in HCC development is the Janus Kinase (JAK) and Signal Transducer and Activator of Transcription (STAT) pathway, commonly referred to as the JAK/STAT pathway.
The JAK/STAT pathway is a conserved and tightly regulated signaling cascade that translates extracellular signals from a plethora of cytokines and growth factors into intracellular responses governing various cellular processes, such as proliferation, differentiation, apoptosis, and immune response [4,5]. Dysregulation of this pathway has been implicated in numerous diseases, including cancers, autoimmune disorders, and inflammatory conditions [6]. In the context of HCC, the aberrant activation of the JAK/STAT pathway has garnered significant attention due to its profound impact on tumorigenesis, metastasis, angiogenesis, and immune evasion.
This review paper aims to provide a comprehensive overview of the current state of knowledge regarding the role of the JAK/STAT pathway in HCC. We will explore the implications of JAK/STAT pathway dysregulation in HCC, which is triggered by a variety of cytokines and growth factors present in livers with chronic inflammation and cirrhosis. Additionally, we will discuss the results from recent preclinical and clinical studies that target the JAK/STAT signaling cascade as a potential therapeutic approach for treating HCC.
2. JAK/STAT signaling pathway
2.1. Overview of JAK/STAT signaling
The JAK/STAT pathway is a vital cellular cascade that controls a wide range of processes, including cell proliferation, differentiation, and apoptosis [7,8]. Its significance is particularly pronounced in modulating immune and inflammatory responses [9,10]. Additionally, it plays a role in hematopoiesis and the functioning of hematopoietic stem cells (HSCs) [11,12,13]. Notably, the JAK/STAT pathway contributes to diverse liver functions, such as hepatic proliferation, regeneration, hepatoprotection, and metabolic processes like gluconeogenesis [14,15]. This signaling pathway also plays a crucial role in various stages of tumorigenesis, including epithelial-mesenchymal transition (EMT) and metastasis. Moreover, it is highly associated with the generation and maintenance of cancer stem cells (CSCs), which play pivotal roles in therapy resistance and metastasis [16,17].
The activation of this pathway begins when specific cytokines, such as interleukins, interferons, or growth factors, bind to their corresponding cell surface receptors [18]. These receptors are often physically associated with Janus kinases (JAKs), which possess tyrosine kinase activity. Upon cytokine binding, a conformational change induces the dimerization of the receptor molecules, leading to the transphosphorylation and activation of JAKs that are non-covalently attached to the receptors (Figure 1). Activated JAKs phosphorylate tyrosine residues at the cytoplasmic tail of the receptor, and this event, in turn, recruits the Signal Transducers and Activators of Transcription (STAT) protein family to the receptor by creating docking sites for STATs, which have Src homology 2 (SH2) domains. Once recruited to the receptor, STAT proteins are phosphorylated by JAKs at specific tyrosine residues, leading to their dimerization. These phosphorylated and dimerized STATs can now translocate into the nucleus, where they regulate the expression of their target genes (Figure 1).
2.2. JAKs and STATs
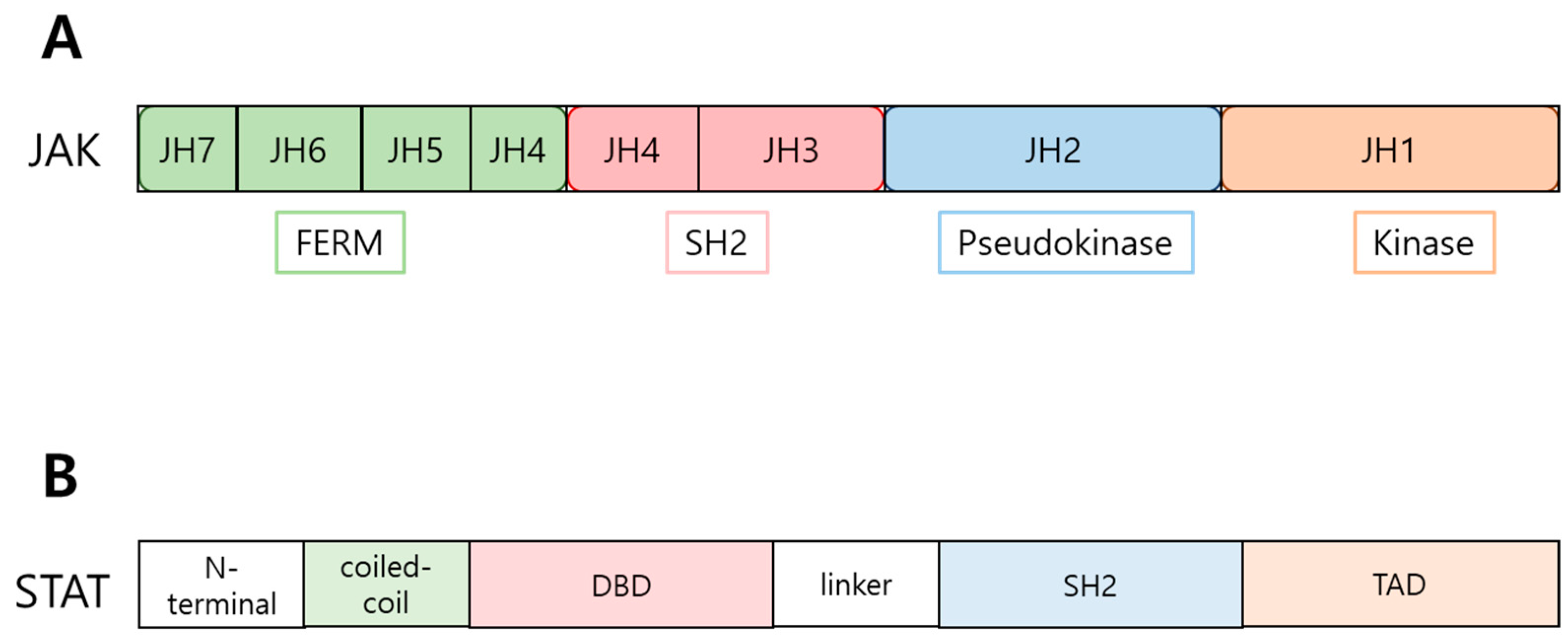
There are four types of JAK proteins: JAK 1, 2, 3, and tyrosine kinase 2 (TYK2), as well as seven types of STAT proteins: STAT 1, 2, 3, 4, 5A, 5B, and 6. Although their roles differ slightly, they share common structures. Each JAK protein consists of seven homology domains known as Janus homology (JH) domains [19]. These seven JH domains are divided into four functional domains (Figure 2A). The kinase domain, corresponding to JH domain 1, is responsible for the kinase activity, providing the ATP binding site [20]. The pseudokinase domain, containing JH domain 2, shares structural similarity with the kinase domain but does not participate in kinase activity. Instead, this domain facilitates interactions between JAK and STAT molecules. The Src homology 2 (SH2) domain and the four-point one, ezrin, radixin, moesin (FERM) domain regulate protein-protein interactions, including the non-covalent attachment of JAK to cytokine receptors [5,21]. While the SH2 domain typically recognizes and binds to phosphotyrosine residues, there is a perspective that the SH2 domain in JAK differs from the standard SH2 domain because JAK can bind to cytokine receptors without relevant phosphotyrosine residues. Instead, it is speculated that the SH2 domain in JAK acts as a scaffold [22]. The FERM domain mediates protein interactions with membrane-associated proteins [22,23].
STAT proteins consist of several domains: N-terminal domain, coiled-coil domain, DNA-binding domain (DBD), linker domain, Src homology 2 (SH2) domain, and transcription activation domain (TAD) [5,20] (Figure 2B). The N-terminal domain plays a crucial role in the dimerization and phosphorylation of STAT [22,24]. The coiled-coil domain is involved in nuclear import and export. The DNA-binding domain (DBD) facilitates binding to the target DNA sequence. The linker domain connects the DBD and the SH2 domain, providing structural support [25]. The SH2 domain of STATs recognizes the phosphotyrosine residue in the cytoplasmic tail of cytokine receptors, which is phosphorylated by JAK following ligand-mediated receptor dimerization (Figure 1). The binding of STATs to cytokine receptors through the interaction between the SH2 domain and phosphotyrosine residues renders STATs physically proximal to JAK. This proximity enables JAK to efficiently phosphorylate STAT, leading to dimer formation and nuclear import of the transcriptional factors [19,20,25]. Inside the nucleus, the activated STAT dimers interact with specific DNA sequences called STAT response elements (SREs) in the promoters or enhancers of target genes. This interaction triggers transcriptional activation, leading to the expression of genes that modulate essential cellular functions, including growth, differentiation, and homeostasis (Figure 1).
2.3. Cytokines activating the JAK/STAT signaling pathway
Various ligands trigger the initiation of the JAK/STAT signaling pathway, with cytokines being one of the most representative examples. Cytokines are small molecules secreted by a variety of cells that facilitate interactions and communication between cells [26]. They play a pivotal role in mediating a wide range of cellular functions, including cell growth, adhesion, differentiation, proliferation, apoptosis, as well as the regulation of immunity and inflammatory responses. Cytokines have been intensively studied in the context of cancer as well [27]. Cytokines known to ignite the JAK/STAT signaling pathway encompass interleukins (IL), colony-stimulating factors, growth factors, and interferons (IFN) [28,29,30]. (See Table 1)
The IL-2 family cytokines, including IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21, engage their respective receptors, which consist of heterodimeric or heterotrimeric complexes composed of various cytokine-specific receptor chains. Upon ligand binding, these receptors undergo conformational changes that facilitate the recruitment and activation of JAK family members, primarily JAK1 and JAK3 [31,32]. These receptor-associated JAKs phosphorylate each other and the cytoplasmic tails of cytokine receptors, creating docking sites for STATs. Notably, the IL-2 family cytokines activate specific STAT proteins, such as STAT5 for IL-2, IL-15, and IL-21, and STAT6 for IL-4 [32,33,34]. This culminates in the transcriptional regulation of genes critical for cell survival, proliferation, differentiation, and immune responses [19,35].
The IL-6 family of cytokines, including IL-6, IL-11, IL-27, IL-31, and leukemia inhibitory factor (LIF), engages their cognate receptors, which generally consist of gp130 as a common receptor subunit, along with specific ligand-binding receptor chains [36,37]. IL-6 family proteins induce the activation of JAK1 and JAK2 primarily, as well as tyrosine kinase 2 (TYK2) for some members like IL-6 and IL-11. Notably, the IL-6 family cytokines predominantly lead to the activation of STAT3 [14,38,39,40,41]. It's worth noting that certain members, like IL-27, can also activate STAT1, influencing specific immune responses [42,43].

Table 1.
Ligands triggering the activation of JAK/STAT signaling in HCC.
| Ligand | JAK | STAT | Reference |
|---|---|---|---|
|
IL-2 family (IL-2, 4, 7, 9, 15, 21) |
JAK1, JAK3 | STAT1, STAT3, STAT5, STAT6 | [18,44,45,46,47] |
|
IL-6 family (IL-6, 11, 27, 31, LIF) |
JAK1, JAK2, TYK2 | STAT1, STAT3 | [18,44,45,46,48] |
|
IL-10 family (IL-10, 19, 20, 22, 24, 26) |
JAK1, TYK2 | STAT1, STAT3, STAT5 | [18,45,49,50,51] |
|
IL-12 family (IL-12, 23) |
JAK2 | STAT3, STAT4 | [44,45,46] |
|
IL-17 family (IL-17A-F) |
JAK2 | STAT3 | [52,53] |
|
β common cytokine family (IL-3, 5, GM-CSF) |
JAK2 | STAT3, STAT5 | [18,44,45,46] |
|
Hematopoietic growth factors (EPO, G-CSF, TPO) |
JAK2 | STAT3, STAT5 | [44,45,46,47] |
|
Type1 IFN (α, β) |
JAK1, TYK2 | STAT1, STAT2 | [29,44,45,46,48] |
|
Type2 IFN (γ) |
JAK1, JAK2 | STAT1 | [18,29,44,45,46,47,48] |
The table lists cytokines, growth factors, and interferons that are predominantly enriched in HCC. It also provides information about the major JAKs and STATs affected by each type of ligand.
The IL-10 family includes IL-10 itself, as well as IL-19, IL-20, IL-22, IL-24, and IL-26. These cytokines engage specific receptor complexes, typically composed of receptor subunits such as IL-10R1, IL-10R2, and IL-22R1 [54]. Upon ligand binding, the receptor complex activates JAK1 and TYK2, in particular. The IL-10 family cytokines predominantly activate STAT3, in addition to STAT1 and STAT5 [55,56,57,58]. Similarly, the IL-12 family, consisting of IL-12 and IL-23, leads to the activation of JAK2. IL-12 predominantly signals through STAT4, while IL-23 induces STAT3 phosphorylation [59,60]. The IL-17 family cytokines, which include IL-17A, IL-17B, IL-17C, IL-17D, IL-17E (also known as IL-25), and IL-17F, trigger JAK/STAT signaling cascade through the activation of JAK2 and STAT3 [52,53].
The β common cytokine family includes cytokines such as IL-3, IL-5, and granulocyte-macrophage colony-stimulating factor (GM-CSF), which share a common β subunit (CD131) in their receptor complexes [61]. The ligand-receptor complex primarily activates JAK2. The β common cytokines mainly activate STAT5, which becomes phosphorylated, dimerizes, and translocates into the nucleus to drive gene expression [62]. Additionally, these cytokines can also induce activation of STAT3 to varying degrees [63,64].
Hematopoietic growth factors, including erythropoietin (EPO), granulocyte colony-stimulating factor (G-CSF), and thrombopoietin (TPO), engage their respective receptors to initiate signal transduction cascades. These receptors typically consist of single or multiple subunits, with some sharing the common β subunit (CD131), and predominantly activate JAK2, leading to the activation of specific members of STATs [11,13,65,66,67,68,69]. For instance, EPO activates JAK2 and STAT5, while G-CSF triggers JAK2 activation and subsequent STAT3 phosphorylation, stimulating the proliferation and differentiation of granulocyte lineage cells. TPO, engaging JAK2, primarily activates STAT3, promoting the development and maturation of platelets.
Type 1 IFNs, including IFN-α and IFN-β, are produced in response to viral infections and engage a heterodimeric receptor complex composed of IFNAR1 and IFNAR2 subunits. Upon ligand binding, this receptor complex triggers the activation of JAK1 and TYK2. These JAKs primarily phosphorylate STAT1 and, to a lesser extent, STAT2 [70]. On the other hand, Type 2 IFN, mainly IFN-γ, engages a receptor complex consisting of IFNGR1 and IFNGR2 subunits. Ligand binding leads to the activation of JAK1 and JAK2, which phosphorylate STAT1, leading to the formation of homodimers or heterodimers with STAT3 [71].
2.4. Receptors and negative regulators of the JAK/STAT signaling pathway
Initiation of JAK/STAT signaling occurs when the aforementioned ligands, primarily cytokines, bind to transmembrane receptors. The types of transmembrane receptors involved in the JAK/STAT pathway include cytokine receptors, G-protein coupled receptors, and growth factor receptors [72]. Given the diverse range of ligands that induce JAK/STAT signaling transduction, there exists a multitude of receptors. These receptors can be categorized into Class I and Class II receptors [22,23,54,73].
Class I receptors feature conserved pairs of cysteines connected by disulfide bonds. This class of receptors is further divided into the glycoprotein 130 (gp130) family, the γ Chain family, the β Chain family, the single-chain family, receptor tyrosine kinases, and other interleukin receptors. Within the glycoprotein 130 (gp130) family, members include the IL-6R family (IL-6R, IL-11R, IL-12R, IL-23R), the G-CSF receptor, and the leukemia inhibitory factor receptor chain (LIFR). The interaction of gp130 with JAK1, JAK2, and TYK2 leads to the activation of STAT1, STAT3, and STAT5. Additionally, the γ Chain family encompasses receptors for the IL-2 family (IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21). Another subset of Class I receptors is the β Chain family, which involves IL-3R, IL-5R, and the granulocyte-macrophage colony-stimulating factor (GM-CSF) receptor [74]. The single-chain family, characterized by homomeric receptors, includes receptors such as the thrombopoietin receptor (TPOR), erythropoietin receptor (EPOR), prolactin receptor (PRLR), and growth hormone receptor (GHR), linked with JAK2 and STAT5. On the other hand, Class II receptors are divided into antiviral and non-antiviral categories. The antiviral subset primarily engages with interferons (IFNs), while the non-antiviral subset includes receptors for IL-10 family cytokines such as IL-10, IL-20, IL-22, and IL-24. This comprehensive classification sheds light on the intricate landscape of JAK-STAT signaling receptors and their connections.
The JAK/STAT signaling pathway is crucial for transmitting signals from cell surface receptors, primarily those of cytokines, to the nucleus, where it regulates the expression of numerous genes involved in various cellular processes. To maintain appropriate cellular responses, the pathway requires tight regulation. In addition to the numerous activators of this signaling pathway, including various growth factors and cytokines, there are negative regulators that play a vital role in inhibiting the JAK/STAT pathway. There are three categories of negative regulators: suppressors of cytokine signaling (SOCS), protein inhibitors of activated STAT (PIAS), and protein tyrosine phosphatases (PTPs) (Figure 1).
The SOCS family encompasses eight distinct protein members: cytokine-inducible SH2 domain protein (CISH), SOCS1, SOCS2, SOCS3, SOCS4, SOCS5, SOCS6, and SOCS7. The SOCS proteins bind to the activation loop within the kinase domain of JAK or the cytoplasmic tail of the receptor. For instance, SOCS1 binds to activated JAKs, while SOCS3 binds to tyrosine residues on receptors. This binding efficiently blocks the phosphorylation of the receptor by JAKs [5]. Additionally, SOCS proteins function as E3 ubiquitin ligases and mediate ubiquitination-driven degradation of signaling components of the JAK/STAT pathway [75,76]. The PIAS family consists of four protein members: PIAS1, PIAS2 (PIASx), PIAS3, and PIAS4 (PIASy). These molecules have been shown to inhibit the JAK/STAT signaling pathway by preventing STAT transcription factors from binding to their target genes [20,24]. Each PIAS protein interacts with a specific type of STAT protein. For example, PIAS1 inhibits the binding of STAT1 to DNA [77]. The final group consists of protein tyrosine phosphatases (PTPs). Given the significance of phosphorylation within the JAK/STAT signaling pathway, removing phosphate groups from tyrosine residues significantly affects the transmission of the signal. The prominent proteins in this category are SH2 domain-containing phosphatase-1 (SHP-1), SH2 domain-containing phosphatase-2 (SHP-2), and CD45 [78]. SHP proteins are involved in dephosphorylating both JAKs and STATs, with SHP-2 specifically interacting with STAT1, STAT3, and STAT5 [78]. CD45 is responsible for removing phosphates that are attached to JAKs [20].
3. Roles of JAK/STAT signaling in HCC
3.1. Dysregulation of the JAK/STAT signaling pathway in human HCC
The JAK/STAT signaling pathway, activated by numerous cytokines and growth factors, serves as a central communication hub in cellular activities [5,79]. Components of the JAK/STAT signaling can also affect the expression of a plethora of genes via epigenetic modifications of chromatin. For example, JAK2 directly phosphorylates Tyr 41 on histone H3, which prevents the binding of heterochromatin protein 1alpha (HP1alpha) [80]. Aberrant activation of the JAK/STAT signaling pathway confers malignant properties on cancer cells by dysregulating cellular proliferation and survival, as well as enhancing stem cell properties and inflammatory responses. The JAK/STAT signaling also promotes invasion and metastasis of cancer cells, partly achieved through the activation of the epithelial-to-mesenchymal transition (EMT) [81].
In HCC, the abnormal activation of the JAK/STAT pathway is frequently found and known to play multifaceted roles in tumorigenesis, including tumor growth, angiogenesis, invasion, and metastasis. This pathway is also involved in maintaining cancer stem cells with tumor-propagating capabilities and creating an immunosuppressive microenvironment [14,82]. Analysis of protein expression data from the Human Protein Atlas indicates that STAT3 exhibits the highest levels of expression within the JAK/STAT pathway proteins in HCC. Approximately 60% of HCC samples demonstrated activated STAT3 [82,83]. STAT3 significantly influences cell survival, proliferation, angiogenesis, immunity, inflammation, cancer stem cells, invasion, metastasis, drug resistance, and apoptosis evasion in HCC [4,14].
Suppressor of cytokine signaling (SOCS) proteins function as inhibitors of the JAK/STAT signaling (Figure 1). SOCS hinder the ability of STAT proteins to bind with JAK and subsequently modulates the signaling pathways triggered by cytokines, thus suppressing the phosphorylation of STAT by JAK. SOCS can also suppress the JAK/STAT signaling by acting as ubiquitin ligases, leading to proteasomal degradation of JAK [81]. Hypermethylation and gene silencing of SOCS have been frequently observed in subsets of HCC, notably in hepatitis B virus-related HCC, with a frequency ranging from 40.9% to 67% [84,85]. Similarly, downregulation of other negative regulators such as PIA and SHP via promoter hypermethylation has been observed in HCC [86,87].
3.2. Overproduction of inflammatory cytokines activating JAK/STAT signaling in HCC
The livers of HCC patients are often infiltrated with different types of inflammatory cells that secrete various types of cytokines, growth factors, chemokines, and other molecules [39,88]. Dysregulated production of inflammatory cytokines is considered a major trigger for HCC carcinogenesis and tumor progression [89]. Among the numerous inflammatory cytokines associated with the activation of the JAK/STAT signaling in HCC, this review focuses on IL-6 family cytokines, IL-10 family cytokines, IL-23 as a member of IL-12 family cytokines, and IL-17, which play critical roles in the pathogenesis of HCC.
3.2.1. IL-6 family (IL-6, IL-11, IL-27, LIF)
IL-6 family cytokines, which activate the JAK/STAT signaling pathway, have been linked to the initiation and progression of HCC [39]. Both T cells and macrophages secrete IL-6 [90], and its expression plays a role in the growth, invasion, and progression of various cancer types, including HCC [91,92,93]. Elevated levels of interleukin-6 (IL-6) have been documented in the bloodstream across various hepatic disorders susceptible to the development of hepatocellular carcinoma (HCC) [94]. Elevated serum IL-6 levels have shown a positive correlation with an increased propensity for HCC development [95]. Moreover, individuals with HCC display elevated serum levels of both IL-6 and the IL-6 receptor [96]. Besides its oncogenic properties conferred on tumor cells through the JAK/STAT signaling pathway, IL-6 promotes PD-L1 expression in monocytes and macrophages in human HCC, contributing to immunosuppression in patients [90]. Similarly, IL-27 is significantly increased in HCC patients [39]. IL-27 plays a role in regulating inflammation within the tumor microenvironment and influences the proliferation, survival, and invasiveness of tumor cells. These effects are mediated through downstream signaling pathways, notably the JAK/STAT3 pathway [97]. Another IL-6 family cytokine, LIF, has also been observed to be significantly upregulated in HCC [98].
3.2.2. IL-10 family (IL-10, IL-19, IL-20, IL-22, IL-24)
Interleukin-10 (IL-10) family cytokines play a pivotal role in the progression of liver diseases. Beyond their diverse immunoregulatory functions, IL-10 influences susceptibility to HCV infection and hepatic fibrogenesis [99,100]. A study revealed higher IL-10 secretion in liver cancer cells compared to normal hepatocytes [101]. In terms of anti-inflammatory response, IL-10 binds to its receptor complex (IL10R1, IL10R2), which associates with JAK1 and TYK2, subsequently engaging STAT3 as a transcription factor [102]. IL-10 levels were found to be positively correlated with p-STAT3 levels. IL-10 serves as a critical negative regulator of NK cell activity. The release of IL-10 by HCC cells activates the STAT3 signaling pathway in NK cells, inhibiting their killing activity. This results in promotion of HCC recurrence and metastasis [103]. Inhibiting STAT3 in HCC cells led to decreased production of immunosuppressive IL-10 cytokines and reduced the number of regulatory T (Treg) cells, thereby alleviating their combined inhibitory effect on NK cells [104].
Despite sharing structural similarities and utilizing similar or partially identical receptors, different members of the IL-10 family exhibit distinct biological functions [105,106]. Interleukin-19 (IL-19) triggers its own expression (auto-induction) and the expression of other pro-inflammatory cytokines, leading to the activation of cell apoptosis. It specifically targets hepatocytes and stimulates the production of TNF-α and IL-10 [107]. IL-19 is also involved in liver disease-associated wound healing, serving as an initial step in processes such as fibrogenesis and cirrhosis, which subsequently promote compensatory proliferation of hepatocytes [108]. In primary murine hepatocytes, IL-19 binds to the IL-20R1/IL-20R2 heteroreceptor complex, activating STAT3 and triggering cell proliferation [109,110]. STAT3 plays a dominant role in negatively regulating the immune response, as well as governing cell growth, differentiation, apoptosis, and tumor development and metastasis [5].
IL-20 signals through its binding to either IL-22R/IL-20R2 or IL-20R1/IL-20R2. Both of these heterodimer receptor complexes activate the JAK/STAT pathway. Interestingly, the presence of constitutive IL-20R2 expression on hepatocytes alone is enough to activate STAT3 [111,112]. IL-20 plays a crucial role in the development of liver injury. In liver biopsies from patients with fibrosis, cirrhosis, and hepatocellular carcinoma, IL-20 levels were significantly elevated in hepatocytes and hepatic stellate cells compared to normal liver tissue from healthy individuals [113]. IL-20 shows a positive correlation with cyclin D1 expression in both HCC patients and human HCC cell lines [114]. Moreover, the treatment with an anti-IL-20 monoclonal antibody effectively hindered HCC cell proliferation, migration, and invasion in vitro and demonstrated the suppression of liver tumor growth in vivo [115].
3.2.3. IL-23 and IL-17
IL-23 belongs to the IL-12 family of cytokines and acts as a crucial molecular link between tumor-promoting pro-inflammatory processes and the impairment of adaptive immune surveillance [116]. Upon binding to its receptor IL-23R, JAK2 and TYK2 are activated, leading to subsequent STAT3 phosphorylation [117]. In the context of HCC, IL-23 plays a pivotal role in promoting cancer growth, progression, and metastasis. It also diminishes CD8+ cell infiltration in tumors while enhancing the immunosuppressive function of Treg cells. Moreover, elevated IL-23 expression levels in HCC are correlated with advanced TNM stage and metastasis [118].
Interleukin-17 (IL-17), which exhibits increased expression in HCC, can facilitate the invasion and migration of HCC cells [52,119]. The tumor-promoting effects of IL-17 and its receptor IL-17R are achieved through direct influence on tumor cells via the IL-6/JAK2/STAT3-related pathway. Aberrantly activated STAT3 in HCC cells, in turn, leads to the upregulation of IL-17 [120]. IL-17 plays a vital role in attracting neutrophils to the peritumoral stroma of HCC tissues and promoting pro-angiogenic activity in HCC cells [121]. The interaction between IL-17 and IFN-γ has significant effects on HCC cell apoptosis and growth in both in vitro and in vivo settings. IL-17 counteracts the anti-tumor effects of IFN-γ in HCC cells, thereby promoting HCC development [53].
4. In vitro and in vivo studies targeting the JAK/STAT signaling in HCC
As discussed above, the upregulation of JAK/STAT signaling significantly contributes to the increased proliferation, migration, and invasion of HCC cells. Furthermore, this up-regulation leads to enhanced cell survival and metastasis. Given that the JAK/STAT signaling pathway exerts strong tumor-promoting effects on HCC, it is reasonable to consider inhibiting the pathway for the treatment of HCC. In vitro investigations have consistently indicated that inhibiting JAK/STAT activity results in increased apoptosis in HCC cells, thereby reducing cell survival. Similarly, in vivo preclinical studies have also convincingly demonstrated that blocking JAK/STAT activity can effectively impede tumor growth or even delay the progression of tumors.
4.1. In vitro studies targeting the JAK/STAT signaling pathway in HCC cells
The SOCS proteins act as endogenous inhibitors of the JAK/STAT signaling pathway. Frequent observations of hypermethylation or histone modification in the promoter regions of SOCS have been made in HCC [122,123]. These events result in the loss of normal SOCS function, contributing to the activation of the JAK/STAT signaling pathway and the subsequent development of HCC. The epigenetic suppression of SOCS in HCC can be countered by inhibiting DNA methyltransferase (DNMT) using Decitabine (5-aza-2'-deoxycytidine or 5-Aza-CdR) and inhibiting histone deacetylase (HDAC) using Vorinostat (suberoylanilide hydroxamic acid or SAHA). Treatment with these agents elevates the expression levels of SOCS, leading to JAK/STAT pathway inhibition. Upon treatment with these agents, human HCC cells (HLE, LCL-PI 11) exhibited increased expression of SOCS1 and SOCS3, resulting in reduced activation of JAK2 and STAT3. Consequently, cell growth was hindered, and apoptosis was enhanced [124].
JAK1 was identified as a target of MicroRNA 30e (miR-30e). Transfection of HCC cells (HepG2 and Huh-7) with miR-30e led to decreases in phosphorylated levels of JAK1 and STAT3. Additionally, overexpression of miR-30e resulted in reduced cell proliferation, migration, and invasion, along with increased apoptosis in the HCC cells [125]. Pharmacological inhibition of JAK1 was also attempted using baricitinib, a small-molecule and selective inhibitor of JAK1 [126]. Treating HepG2 and Huh-7 cells with baricitinib downregulated phosphorylation of JAK1 and STAT3, resulting in reduced nuclear translocation of STAT3. Baricitinib administration led to decreased cell growth and increased apoptosis in the HCC cells [127].
Etomidate inhibits phosphorylation and activation of JAK2 by competing with ATP for binding to the ATP-binding pocket of JAK2. Application of etomidate to HCC cells (HepG2 and Huh-7) suppressed phosphorylation of JAK2 and STAT3, resulting in the inhibition of cell proliferation, migration, and invasion, coupled with enhanced apoptosis [128]. Similarly, momelotinib, another small-molecule JAK2 inhibitor, suppressed phosphorylated JAK2 and STAT3 levels upon treatment of HCC cells (SNU-378 and SNU-475), subsequently leading to reduced cell viability, migration, and invasion [129].
Targeting STAT proteins also exerts similar anti-cancer effects in HCC cells. Pravastatin, a non-peptide small molecule, effectively impedes STAT1 phosphorylation upon the activation by cytokine-induced pathways. Notably, pravastatin effectively dampened the phosphorylation of STAT1 induced by IFN-γ [130]. The proliferation of HepG2 and Huh-7 cells was significantly decreased following the treatment with pravastatin [131].
Napabucasin (BBI608) is a novel small-molecule inhibitor of STAT3. It has been shown to impair cancer stemness in various types of cancers, such as colorectal, pancreatic, and prostate cancers [132,133]. In a variety of HCC cells, including Huh-7, Hepa1-6, and HepG2, treatment with napabucasin led to decreases in cell viability and cell growth. Importantly, the treatment resulted in the downregulation of pSTAT3 and stemness markers such as Nanog, CD133, Klf4, Oct4, and Sox2 [134]. In another study, tumors induced by activated beta-catenin and mTOR signaling were highly susceptible to napabucasin, which strongly inhibited the proliferation of the tumor cells [135].
4.2. Preclinical animal studies targeting the JAK/STAT signaling pathway in HCC
Studies have demonstrated that miR-515-5p directly binds to the 3'-untranslated region (3'-UTR) of interleukin 6 (IL-6). Treatment of HCC cells (HepG2) with miR-515-5p suppressed the JAK/STAT signaling pathway along with reduced IL-6 expression. Overexpression of miR-515-5p inhibited migration and invasion of HepG2 cells that were transplanted into nude mice [136]. Similarly, in xenograft mice transplanted with HepG2 cells, treatment with etomidate, an inhibitor of JAK2, led to the inhibition of tumor growth and subsequent increases in animal survival [128].
In a chemically induced HCC model, rats treated with pravastatin, an inhibitor of STAT1 phosphorylation, showed significantly smaller tumor sizes in the liver. Immunohistochemistry studies revealed that the level of cellular proliferation in HCC from mice treated with pravastatin was significantly diminished compared to that from untreated mice [137].
Additionally, attempts to inhibit STAT3 have been made in HCC murine models. Mice transplanted with Hepa1-6 cells were intraperitoneally injected with napabucasin, a STAT3 inhibitor, at a dose of 20 mg per kilogram of body weight, or vehicle. Napabucasin treatment significantly reduced tumor volumes compared to the control group. Notably, approximately 25% of mice showed tumor regression when examined 21 days after administration [134]. OPB-31121 is another potent inhibitor of STAT3 phosphorylation that has demonstrated significant growth inhibition in various hematopoietic malignant cells [138]. Immunodeficient mice transplanted with Huh-7 or HepG2 cells were orally administered OPB-31121 at a daily dose of 60 mg per kg of body weight for 14 days. Treatment with OPB-31121 significantly inhibited HCC growth in the murine models [139].
5. Clinical studies targeting the JAK/STAT signaling pathway in HCC
The significant role of the JAK/STAT signaling pathway in HCC suggests that targeted inhibition of this pathway could hold therapeutic potential for treating patients with HCC [81]. Various types of JAK/STAT inhibitors are currently under investigation for their clinical relevance in HCC. Itacitinib, an oral selective inhibitor of JAK1, has demonstrated no serious treatment-related adverse events [140,141]. A single-arm Phase Ib study is currently underway to evaluate the effect of itacitinib on 25 patients with advanced HCC (NCT04358185).
Considering the tumor-suppressive effects of pravastatin in preclinical animal models of HCC [137], the STAT1 inhibitor was assessed for its anti-cancer effects in clinical studies (NCT01418729, NCT01903694, and NCT01075555). In a Phase III clinical trial, patients with HCC received either sorafenib alone, a standard therapeutic agent for HCC, or sorafenib plus pravastatin. However, the combination of pravastatin with sorafenib failed to provide additional benefits to patients, as the mean overall survivals were not significantly different between the sorafenib alone and sorafenib plus pravastatin groups (10.7 months and 10.5 months, respectively) [142].
Danvatirsen (also known as AZD9150) is an antisense oligonucleotide that binds to STAT3 mRNA, thereby inhibiting the translation of STAT3 mRNA [143]. In patients with lymphoma and lung cancer, danvatirsen exhibited anti-tumor activity [144]. A Phase I trial assessed its safety and anti-tumor activity in patients with advanced or metastatic HCC (NCT01839604). Although the drug appeared relatively safe, it did not consistently show an antitumor effect [145].
In vitro and in vivo animal studies have demonstrated that napabucasin has a strong anti-tumor effect on HCC, as the STAT3 inhibitor suppresses cell viability and proliferation, as well as downregulates expression of stemness genes [134,135]. Based on promising preclinical results, a clinical trial evaluated the co-administration of sorafenib and napabucasin for potential benefits in advanced HCC patients over sorafenib treatment alone. However, a Phase I/II clinical trial revealed that co-administration of sorafenib and napabucasin achieved no improvement in overall response rates in HCC patients, compared to the treatment with sorafenib alone (NCT02279719).
Similarly, due to its effective suppression of tumor growth in murine HCC models [139], OPB-31121, a potent STAT3 inhibitor, was administered to 23 patients with progressive HCC at varying doses. To evaluate its anti-tumor efficacy, overall responses were assessed (NCT01406574). From the clinical trial, a marginal effect was observed in only 6 out of the 23 patients for a maximum of 8 weeks [139].
It's important to note that while inhibition of the JAK/STAT signaling pathway has shown promise in preclinical studies, its efficacy in patients with advanced HCC remains uncertain. Current clinical trials focused on this strategy are still in their early stages, and more studies are needed to better assess the effectiveness of targeting this signaling pathway. In summary, the ongoing clinical studies targeting the JAK/STAT pathway emphasize the complexity of this approach in HCC treatment and underscore the necessity for further investigation to identify more effective treatment strategies.
Table 2.
Clinical studies targeting the JAK/STAT signaling pathway in HCC.
| Drug | Target | NCT number | Phase | Status | Clinical outcomes |
|---|---|---|---|---|---|
| Itacitinib | JAK1 | 04358185 | Ib | In progress, | Not yet determined |
| Pravastatin | STAT1 | 01075555 | III | Completed (September 2015) |
MOS 10.7m vs 10.5m (sorafenib vs pravastatin + sorafenib) |
| Danvatirsen | STAT3 | 01839604 | I | Completed (February 2015) |
Limited antitumor activity. |
| Napabucasin | STAT3 | 02279719 | I/II | Completed (October 2019) |
No significant difference in overall response rates between sorafenib alone and napabucasin + sorafenib groups |
| OPB-31121 | STAT3 | 01406574 | I/II | Completed (March 2014) |
Limited anti-tumor effects and low overall responses. |
MOS, mean overall survival.
6. Perspectives and Conclusions
In the realm of clinical translation, it is noteworthy that while targeting the JAK/STAT pathway in hepatocellular carcinoma (HCC) holds great promise, ongoing clinical trials focused on this strategy are still in their infancy. Despite the well-established significance of aberrant JAK/STAT signaling in HCC pathogenesis, along with preclinical observations of the anti-cancer effects of targeting the pathway in HCC, the journey toward clinical implementation requires a more systematic approach and refinement. Notably, adverse effects routinely observed in administering anti-cancer agents are particularly problematic in patients with HCC, given their pre-existing liver dysfunction due to sustained ailments from underlying chronic hepatitis and cirrhosis [1,2].
To explain the failure of therapeutic drugs that demonstrated anti-tumor effects in preclinical models of HCC to achieve effectiveness in clinical studies, several complex factors must be considered. In vitro and animal models of HCC do not perfectly mimic the human tumor microenvironment [146]. Factors like immune response, blood supply, and interactions with neighboring tissues significantly influence HCC in humans [147]. Preclinical models might not accurately capture these aspects, leading to differences in drug response [148]. Additionally, human HCCs are genetically and molecularly diverse [149,150]. Genetic mutations, gene expression patterns, and signaling pathways can vary significantly among human patients. It is also noteworthy that animals and humans metabolize drugs differently due to varying pharmacokinetics between species. Thus, drugs that are effectively metabolized in animals to fully function as anti-cancer agents might not exhibit the desired effects on cancer in human due to imperfect metabolism of the drugs in the human body.
While current clinical studies targeting the JAK/STAT signaling pathway might not be yielding optimistic results, it's crucial to point out that these studies are still in the early stages. As researchers navigate the complexities of this pathway and its clinical translation, it's imperative that these efforts be met with the clinical successes they deserve. By shedding light on the underlying mechanisms driving JAK/STAT pathway activation and its contributions to HCC pathophysiology, we aim to pave the way for the development of novel JAK/STAT targeted therapies that hold promise in effectively combating this aggressive malignancy.
Funding
This work was supported by a grant from Kyung Hee University in 2023 (KHU- 20230902 awarded to S.W.R.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat Rev Dis Primers 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Singal, A.G.; Lampertico, P.; Nahon, P. Epidemiology and surveillance for hepatocellular carcinoma: New trends. J Hepatol 2020, 72, 250–261. [Google Scholar] [CrossRef]
- Villanueva, A. Hepatocellular Carcinoma. N Engl J Med 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [PubMed]
- Rah, B.; Rather, R.A.; Bhat, G.R.; Baba, A.B.; Mushtaq, I.; Farooq, M.; Yousuf, T.; Dar, S.B.; Parveen, S.; Hassan, R.; et al. JAK/STAT Signaling: Molecular Targets, Therapeutic Opportunities, and Limitations of Targeted Inhibitions in Solid Malignancies. Front Pharmacol 2022, 13, 821344. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct Target Ther 2021, 6, 402. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, U.; Kasembeli, M.M.; Robinson, P.; Tweardy, D.J. Targeting Janus Kinases and Signal Transducer and Activator of Transcription 3 to Treat Inflammation, Fibrosis, and Cancer: Rationale, Progress, and Caution. Pharmacol Rev 2020, 72, 486–526. [Google Scholar] [CrossRef]
- Dodington, D.W.; Desai, H.R.; Woo, M. JAK/STAT - Emerging Players in Metabolism. Trends Endocrinol Metab 2018, 29, 55–65. [Google Scholar] [CrossRef]
- Gu, Y.J.; Sun, W.Y.; Zhang, S.; Li, X.R.; Wei, W. Targeted blockade of JAK/STAT3 signaling inhibits proliferation, migration and collagen production as well as inducing the apoptosis of hepatic stellate cells. Int J Mol Med 2016, 38, 903–911. [Google Scholar] [CrossRef]
- O'Shea, J.J.; Plenge, R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity 2012, 36, 542–550. [Google Scholar] [CrossRef]
- O'Shea, J.J.; Holland, S.M.; Staudt, L.M. JAKs and STATs in immunity, immunodeficiency, and cancer. N Engl J Med 2013, 368, 161–170. [Google Scholar] [CrossRef]
- Akada, H.; Akada, S.; Hutchison, R.E.; Sakamoto, K.; Wagner, K.U.; Mohi, G. Critical role of Jak2 in the maintenance and function of adult hematopoietic stem cells. Stem Cells 2014, 32, 1878–1889. [Google Scholar] [CrossRef] [PubMed]
- Fasouli, E.S.; Katsantoni, E. JAK-STAT in Early Hematopoiesis and Leukemia. Front Cell Dev Biol 2021, 9, 669363. [Google Scholar] [CrossRef]
- Staerk, J.; Constantinescu, S.N. The JAK-STAT pathway and hematopoietic stem cells from the JAK2 V617F perspective. Jakstat 2012, 1, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Hin Tang, J.J.; Hao Thng, D.K.; Lim, J.J.; Toh, T.B. JAK/STAT signaling in hepatocellular carcinoma. Hepat Oncol 2020, 7, Hep18. [Google Scholar] [CrossRef] [PubMed]
- Svinka, J.; Mikulits, W.; Eferl, R. STAT3 in hepatocellular carcinoma: New perspectives. Hepat Oncol 2014, 1, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Bian, Q.; Rong, D.; Wang, L.; Song, J.; Huang, H.S.; Zeng, J.; Mei, J.; Wang, P.Y. JAK/STAT pathway: Extracellular signals, diseases, immunity, and therapeutic regimens. Front Bioeng Biotechnol 2023, 11, 1110765. [Google Scholar] [CrossRef]
- Jin, W. Role of JAK/STAT3 Signaling in the Regulation of Metastasis, the Transition of Cancer Stem Cells, and Chemoresistance of Cancer by Epithelial-Mesenchymal Transition. Cells 2020, 9. [Google Scholar] [CrossRef]
- Zhou, Q.; Ren, Q.; Jiao, L.; Huang, J.; Yi, J.; Chen, J.; Lai, J.; Ji, G.; Zheng, T. The potential roles of JAK/STAT signaling in the progression of osteoarthritis. Front Endocrinol (Lausanne) 2022, 13, 1069057. [Google Scholar] [CrossRef]
- Schindler, C.; Levy, D.E.; Decker, T. JAK-STAT signaling: From interferons to cytokines. J Biol Chem 2007, 282, 20059–20063. [Google Scholar] [CrossRef]
- Seif, F.; Khoshmirsafa, M.; Aazami, H.; Mohsenzadegan, M.; Sedighi, G.; Bahar, M. The role of JAK-STAT signaling pathway and its regulators in the fate of T helper cells. Cell Commun Signal 2017, 15, 23. [Google Scholar] [CrossRef]
- Ferrao, R.; Lupardus, P.J. The Janus Kinase (JAK) FERM and SH2 Domains: Bringing Specificity to JAK-Receptor Interactions. Front Endocrinol (Lausanne) 2017, 8, 71. [Google Scholar] [CrossRef]
- Jatiani, S.S.; Baker, S.J.; Silverman, L.R.; Reddy, E.P. Jak/STAT pathways in cytokine signaling and myeloproliferative disorders: Approaches for targeted therapies. Genes Cancer 2010, 1, 979–993. [Google Scholar] [CrossRef]
- Kisseleva, T.; Bhattacharya, S.; Braunstein, J.; Schindler, C.W. Signaling through the JAK/STAT pathway, recent advances and future challenges. Gene 2002, 285, 1–24. [Google Scholar] [CrossRef]
- Xin, P.; Xu, X.; Deng, C.; Liu, S.; Wang, Y.; Zhou, X.; Ma, H.; Wei, D.; Sun, S. The role of JAK/STAT signaling pathway and its inhibitors in diseases. Int Immunopharmacol 2020, 80, 106210. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, N.; Liongue, C.; Ward, A.C. STAT proteins: A kaleidoscope of canonical and non-canonical functions in immunity and cancer. J Hematol Oncol 2021, 14, 198. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.M.; An, J. Cytokines, inflammation, and pain. Int Anesthesiol Clin 2007, 45, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Berraondo, P.; Sanmamed, M.F.; Ochoa, M.C.; Etxeberria, I.; Aznar, M.A.; Pérez-Gracia, J.L.; Rodríguez-Ruiz, M.E.; Ponz-Sarvise, M.; Castañón, E.; Melero, I. Cytokines in clinical cancer immunotherapy. Br J Cancer 2019, 120, 6–15. [Google Scholar] [CrossRef]
- Kupsa, T.; Horacek, J.M.; Jebavy, L. The role of cytokines in acute myeloid leukemia: A systematic review. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub 2012, 156, 291–301. [Google Scholar] [CrossRef]
- Morris, R.; Kershaw, N.J.; Babon, J.J. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci 2018, 27, 1984–2009. [Google Scholar] [CrossRef]
- Luo, Y.; Alexander, M.; Gadina, M.; O'Shea, J.J.; Meylan, F.; Schwartz, D.M. JAK-STAT signaling in human disease: From genetic syndromes to clinical inhibition. J Allergy Clin Immunol 2021, 148, 911–925. [Google Scholar] [CrossRef]
- Mitra, S.; Leonard, W.J. Biology of IL-2 and its therapeutic modulation: Mechanisms and strategies. J Leukoc Biol 2018, 103, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Lin, J.X.; Leonard, W.J. IL-2 family cytokines: New insights into the complex roles of IL-2 as a broad regulator of T helper cell differentiation. Curr Opin Immunol 2011, 23, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.M.; Read, K.A.; Oestreich, K.J. Dynamic Roles for IL-2-STAT5 Signaling in Effector and Regulatory CD4(+) T Cell Populations. J Immunol 2020, 205, 1721–1730. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Jiang, L.; Hao, S.; Liu, Z.; Ding, S.; Zhang, W.; Yang, X.; Li, S. Activation of the IL-4/STAT6 Signaling Pathway Promotes Lung Cancer Progression by Increasing M2 Myeloid Cells. Front Immunol 2019, 10, 2638. [Google Scholar] [CrossRef]
- Verhoeven, Y.; Tilborghs, S.; Jacobs, J.; De Waele, J.; Quatannens, D.; Deben, C.; Prenen, H.; Pauwels, P.; Trinh, X.B.; Wouters, A.; et al. The potential and controversy of targeting STAT family members in cancer. Semin Cancer Biol 2020, 60, 41–56. [Google Scholar] [CrossRef]
- Unver, N.; McAllister, F. IL-6 family cytokines: Key inflammatory mediators as biomarkers and potential therapeutic targets. Cytokine Growth Factor Rev 2018, 41, 10–17. [Google Scholar] [CrossRef]
- Rose-John, S. Interleukin-6 Family Cytokines. Cold Spring Harb Perspect Biol 2018, 10. [Google Scholar] [CrossRef]
- Johnson, D.E.; O'Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol 2018, 15, 234–248. [Google Scholar] [CrossRef]
- Lokau, J.; Schoeder, V.; Haybaeck, J.; Garbers, C. Jak-Stat Signaling Induced by Interleukin-6 Family Cytokines in Hepatocellular Carcinoma. Cancers (Basel) 2019, 11. [Google Scholar] [CrossRef]
- Jones, S.A.; Scheller, J.; Rose-John, S. Therapeutic strategies for the clinical blockade of IL-6/gp130 signaling. J Clin Invest 2011, 121, 3375–3383. [Google Scholar] [CrossRef]
- Xu, J.; Lin, H.; Wu, G.; Zhu, M.; Li, M. IL-6/STAT3 Is a Promising Therapeutic Target for Hepatocellular Carcinoma. Front Oncol 2021, 11, 760971. [Google Scholar] [CrossRef] [PubMed]
- Kourko, O.; Seaver, K.; Odoardi, N.; Basta, S.; Gee, K. IL-27, IL-30, and IL-35: A Cytokine Triumvirate in Cancer. Front Oncol 2019, 9, 969. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.W.; Hill, D.G.; Cardus, A.; Jones, S.A. IL-27: A double agent in the IL-6 family. Clin Exp Immunol 2018, 193, 37–46. [Google Scholar] [CrossRef]
- O'Shea, J.J.; Schwartz, D.M.; Villarino, A.V.; Gadina, M.; McInnes, I.B.; Laurence, A. The JAK-STAT pathway: Impact on human disease and therapeutic intervention. Annu Rev Med 2015, 66, 311–328. [Google Scholar] [CrossRef]
- Kiu, H.; Nicholson, S.E. Biology and significance of the JAK/STAT signalling pathways. Growth Factors 2012, 30, 88–106. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Biehl, A.; Gadina, M.; Hasni, S.; Schwartz, D.M. JAK-STAT Signaling as a Target for Inflammatory and Autoimmune Diseases: Current and Future Prospects. Drugs 2017, 77, 521–546. [Google Scholar] [CrossRef]
- Aittomäki, S.; Pesu, M. Therapeutic targeting of the Jak/STAT pathway. Basic Clin Pharmacol Toxicol 2014, 114, 18–23. [Google Scholar] [CrossRef]
- Aaronson, D.S.; Horvath, C.M. A road map for those who don't know JAK-STAT. Science 2002, 296, 1653–1655. [Google Scholar] [CrossRef]
- Wang, X.; Xin, W.; Zhang, H.; Zhang, F.; Gao, M.; Yuan, L.; Xu, X.; Hu, X.; Zhao, M. Aberrant expression of p-STAT3 in peripheral blood CD4+ and CD8+ T cells related to hepatocellular carcinoma development. Mol Med Rep 2014, 10, 2649–2656. [Google Scholar] [CrossRef]
- Wang, C.J.; Xiao, C.W.; You, T.G.; Zheng, Y.X.; Gao, W.; Zhou, Z.Q.; Chen, J.; Xue, X.B.; Fan, J.; Zhang, H. Interferon-α enhances antitumor activities of oncolytic adenovirus-mediated IL-24 expression in hepatocellular carcinoma. Mol Cancer 2012, 11, 31. [Google Scholar] [CrossRef]
- Andoh, A.; Shioya, M.; Nishida, A.; Bamba, S.; Tsujikawa, T.; Kim-Mitsuyama, S.; Fujiyama, Y. Expression of IL-24, an activator of the JAK1/STAT3/SOCS3 cascade, is enhanced in inflammatory bowel disease. J Immunol 2009, 183, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Gu, F.M.; Li, Q.L.; Gao, Q.; Jiang, J.H.; Zhu, K.; Huang, X.Y.; Pan, J.F.; Yan, J.; Hu, J.H.; Wang, Z.; et al. IL-17 induces AKT-dependent IL-6/JAK2/STAT3 activation and tumor progression in hepatocellular carcinoma. Mol Cancer 2011, 10, 150. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zeng, M.; Yan, K.; Yang, Y.; Li, H.; Xu, X. IL-17 promotes hepatocellular carcinoma through inhibiting apoptosis induced by IFN-γ. Biochem Biophys Res Commun 2020, 522, 525–531. [Google Scholar] [CrossRef] [PubMed]
- O'Sullivan, L.A.; Liongue, C.; Lewis, R.S.; Stephenson, S.E.; Ward, A.C. Cytokine receptor signaling through the Jak-Stat-Socs pathway in disease. Mol Immunol 2007, 44, 2497–2506. [Google Scholar] [CrossRef]
- Bedke, T.; Muscate, F.; Soukou, S.; Gagliani, N.; Huber, S. Title: IL-10-producing T cells and their dual functions. Semin Immunol 2019, 44, 101335. [Google Scholar] [CrossRef]
- Wilbers, R.H.P.; van Raaij, D.R.; Westerhof, L.B.; Bakker, J.; Smant, G.; Schots, A. Re-evaluation of IL-10 signaling reveals novel insights on the contribution of the intracellular domain of the IL-10R2 chain. PLoS ONE 2017, 12, e0186317. [Google Scholar] [CrossRef]
- Hutchins, A.P.; Diez, D.; Miranda-Saavedra, D. The IL-10/STAT3-mediated anti-inflammatory response: Recent developments and future challenges. Brief Funct Genomics 2013, 12, 489–498. [Google Scholar] [CrossRef]
- Li, L.; Zhang, J.; Chen, J.; Xu-Monette, Z.Y.; Miao, Y.; Xiao, M.; Young, K.H.; Wang, S.; Medeiros, L.J.; Wang, M.; et al. B-cell receptor-mediated NFATc1 activation induces IL-10/STAT3/PD-L1 signaling in diffuse large B-cell lymphoma. Blood 2018, 132, 1805–1817. [Google Scholar] [CrossRef]
- von Essen, M.R.; Søndergaard, H.B.; Petersen, E.R.S.; Sellebjerg, F. IL-6, IL-12, and IL-23 STAT-Pathway Genetic Risk and Responsiveness of Lymphocytes in Patients with Multiple Sclerosis. Cells 2019, 8. [Google Scholar] [CrossRef]
- Floss, D.M.; Moll, J.M.; Scheller, J. IL-12 and IL-23-Close Relatives with Structural Homologies but Distinct Immunological Functions. Cells 2020, 9. [Google Scholar] [CrossRef]
- Dougan, M.; Dranoff, G.; Dougan, S.K. GM-CSF, IL-3, and IL-5 Family of Cytokines: Regulators of Inflammation. Immunity 2019, 50, 796–811. [Google Scholar] [CrossRef] [PubMed]
- Hercus, T.R.; Kan, W.L.T.; Broughton, S.E.; Tvorogov, D.; Ramshaw, H.S.; Sandow, J.J.; Nero, T.L.; Dhagat, U.; Thompson, E.J.; Shing, K.; et al. Role of the β Common (βc) Family of Cytokines in Health and Disease. Cold Spring Harb Perspect Biol 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Thorn, M.; Guha, P.; Cunetta, M.; Espat, N.J.; Miller, G.; Junghans, R.P.; Katz, S.C. Tumor-associated GM-CSF overexpression induces immunoinhibitory molecules via STAT3 in myeloid-suppressor cells infiltrating liver metastases. Cancer Gene Ther 2016, 23, 188–198. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Yuan, C.; Ji, Q.; Chen, D.; Zhao, H.; Jiang, W.; Ma, K.; Liu, L. JAK2/STAT3 is associated with the inflammatory process in periapical granuloma. Int J Clin Exp Pathol 2019, 12, 190–197. [Google Scholar] [PubMed]
- Reusswig, F.; Fazel Modares, N.; Brechtenkamp, M.; Wienands, L.; Krüger, I.; Behnke, K.; Lee-Sundlov, M.M.; Herebian, D.; Scheller, J.; Hoffmeister, K.M.; et al. Efficiently Restored Thrombopoietin Production by Ashwell-Morell Receptor and IL-6R Induced Janus Kinase 2/Signal Transducer and Activator of Transcription Signaling Early After Partial Hepatectomy. Hepatology 2021, 74, 411–427. [Google Scholar] [CrossRef] [PubMed]
- Mu, W.; Ao, J.; Li, Y.; Zhang, J.; Duan, C. Exploring the protective mechanisms of total tannins from Geum japonicum var. chinense F.Bolle in mice with hematopoietic dysfunction via the JAK2/STAT3/5 signaling pathway. J Ethnopharmacol 2022, 296, 115507. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Liu, X.; Hoffman, R.D.; Shi, R.Z.; Lv, G.Y.; Gao, J.L. G-CSF/GM-CSF-induced hematopoietic dysregulation in the progression of solid tumors. FEBS Open Bio 2022, 12, 1268–1285. [Google Scholar] [CrossRef]
- Karagiannidis, I.; Salataj, E.; Said Abu Egal, E.; Beswick, E.J. G-CSF in tumors: Aggressiveness, tumor microenvironment and immune cell regulation. Cytokine 2021, 142, 155479. [Google Scholar] [CrossRef] [PubMed]
- Tóthová, Z.; Tomc, J.; Debeljak, N.; Solár, P. STAT5 as a Key Protein of Erythropoietin Signalization. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Wang, H.; Lafdil, F.; Feng, D. STAT proteins - key regulators of anti-viral responses, inflammation, and tumorigenesis in the liver. J Hepatol 2012, 57, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Jorgovanovic, D.; Song, M.; Wang, L.; Zhang, Y. Roles of IFN-γ in tumor progression and regression: A review. Biomark Res 2020, 8, 49. [Google Scholar] [CrossRef] [PubMed]
- Bousoik, E.; Montazeri Aliabadi, H. "Do We Know Jack" About JAK? A Closer Look at JAK/STAT Signaling Pathway. Front Oncol 2018, 8, 287. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, F.; Luo, F. The Role of JAK/STAT Pathway in Fibrotic Diseases: Molecular and Cellular Mechanisms. Biomolecules 2023, 13. [Google Scholar] [CrossRef] [PubMed]
- Broughton, S.E.; Dhagat, U.; Hercus, T.R.; Nero, T.L.; Grimbaldeston, M.A.; Bonder, C.S.; Lopez, A.F.; Parker, M.W. The GM-CSF/IL-3/IL-5 cytokine receptor family: From ligand recognition to initiation of signaling. Immunol Rev 2012, 250, 277–302. [Google Scholar] [CrossRef]
- Durham, G.A.; Williams, J.J.L.; Nasim, M.T.; Palmer, T.M. Targeting SOCS Proteins to Control JAK-STAT Signalling in Disease. Trends Pharmacol Sci 2019, 40, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Tamiya, T.; Kashiwagi, I.; Takahashi, R.; Yasukawa, H.; Yoshimura, A. Suppressors of cytokine signaling (SOCS) proteins and JAK/STAT pathways: Regulation of T-cell inflammation by SOCS1 and SOCS3. Arterioscler Thromb Vasc Biol 2011, 31, 980–985. [Google Scholar] [CrossRef]
- Ungureanu, D.; Vanhatupa, S.; Grönholm, J.; Palvimo, J.J.; Silvennoinen, O. SUMO-1 conjugation selectively modulates STAT1-mediated gene responses. Blood 2005, 106, 224–226. [Google Scholar] [CrossRef]
- Xu, D.; Qu, C.K. Protein tyrosine phosphatases in the JAK/STAT pathway. Front Biosci 2008, 13, 4925–4932. [Google Scholar] [CrossRef]
- Calvisi, D.F.; Ladu, S.; Gorden, A.; Farina, M.; Conner, E.A.; Lee, J.S.; Factor, V.M.; Thorgeirsson, S.S. Ubiquitous activation of Ras and Jak/Stat pathways in human HCC. Gastroenterology 2006, 130, 1117–1128. [Google Scholar] [CrossRef]
- Dawson, M.A.; Bannister, A.J.; Göttgens, B.; Foster, S.D.; Bartke, T.; Green, A.R.; Kouzarides, T. JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature 2009, 461, 819–822. [Google Scholar] [CrossRef]
- Thomas, S.J.; Snowden, J.A.; Zeidler, M.P.; Danson, S.J. The role of JAK/STAT signalling in the pathogenesis, prognosis and treatment of solid tumours. Br J Cancer 2015, 113, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liao, X.; Yu, T.; Gong, Y.; Zhang, L.; Huang, J.; Yang, C.; Han, C.; Yu, L.; Zhu, G.; et al. Analysis of clinical significance and prospective molecular mechanism of main elements of the JAK/STAT pathway in hepatocellular carcinoma. Int J Oncol 2019, 55, 805–822. [Google Scholar] [CrossRef]
- He, G.; Karin, M. NF-κB and STAT3 - key players in liver inflammation and cancer. Cell Res 2011, 21, 159–168. [Google Scholar] [CrossRef]
- Yoshikawa, H.; Matsubara, K.; Qian, G.S.; Jackson, P.; Groopman, J.D.; Manning, J.E.; Harris, C.C.; Herman, J.G. SOCS-1, a negative regulator of the JAK/STAT pathway, is silenced by methylation in human hepatocellular carcinoma and shows growth-suppression activity. Nat Genet 2001, 28, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, J.; Cheng, J.; Ding, S.; Li, M.; Sun, S.; Zhang, L.; Liu, S.; Chen, X.; Zhuang, H.; et al. An integrated analysis of SOCS1 down-regulation in HBV infection-related hepatocellular carcinoma. J Viral Hepat 2014, 21, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Calvisi, D.F.; Ladu, S.; Gorden, A.; Farina, M.; Lee, J.S.; Conner, E.A.; Schroeder, I.; Factor, V.M.; Thorgeirsson, S.S. Mechanistic and prognostic significance of aberrant methylation in the molecular pathogenesis of human hepatocellular carcinoma. J Clin Invest 2007, 117, 2713–2722. [Google Scholar] [CrossRef]
- Calvisi, D.F. Dr. Jekyll and Mr. Hyde: A paradoxical oncogenic and tumor suppressive role of signal transducer and activator of transcription 3 in liver cancer. Hepatology 2011, 54, 9–12. [Google Scholar] [CrossRef]
- Chung, S.I.; Moon, H.; Ju, H.L.; Cho, K.J.; Kim, D.Y.; Han, K.H.; Eun, J.W.; Nam, S.W.; Ribback, S.; Dombrowski, F.; et al. Hepatic expression of Sonic Hedgehog induces liver fibrosis and promotes hepatocarcinogenesis in a transgenic mouse model. J Hepatol 2016, 64, 618–627. [Google Scholar] [CrossRef]
- Refolo, M.G.; Messa, C.; Guerra, V.; Carr, B.I.; D'Alessandro, R. Inflammatory Mechanisms of HCC Development. Cancers (Basel) 2020, 12. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, Y.; Yan, Z.; Yang, H.; Sun, W.; Yao, Y.; Chen, Y.; Jiang, R. IL-6 promotes PD-L1 expression in monocytes and macrophages by decreasing protein tyrosine phosphatase receptor type O expression in human hepatocellular carcinoma. J Immunother Cancer 2020, 8. [Google Scholar] [CrossRef]
- Lin, Y.; He, Z.; Ye, J.; Liu, Z.; She, X.; Gao, X.; Liang, R. Progress in Understanding the IL-6/STAT3 Pathway in Colorectal Cancer. Onco Targets Ther 2020, 13, 13023–13032. [Google Scholar] [CrossRef] [PubMed]
- Manore, S.G.; Doheny, D.L.; Wong, G.L.; Lo, H.W. IL-6/JAK/STAT3 Signaling in Breast Cancer Metastasis: Biology and Treatment. Front Oncol 2022, 12, 866014. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.; Wang, R.; Chen, Q.; Luo, J.; Wang, J.; Zhao, Z.; Li, Y.; Wang, Y.; Wang, X.; Cheng, B. Cancer-associated fibroblasts promote stem cell-like properties of hepatocellular carcinoma cells through IL-6/STAT3/Notch signaling. Am J Cancer Res 2018, 8, 302–316. [Google Scholar] [PubMed]
- Schmidt-Arras, D.; Rose-John, S. IL-6 pathway in the liver: From physiopathology to therapy. J Hepatol 2016, 64, 1403–1415. [Google Scholar] [CrossRef]
- Aleksandrova, K.; Boeing, H.; Nöthlings, U.; Jenab, M.; Fedirko, V.; Kaaks, R.; Lukanova, A.; Trichopoulou, A.; Trichopoulos, D.; Boffetta, P.; et al. Inflammatory and metabolic biomarkers and risk of liver and biliary tract cancer. Hepatology 2014, 60, 858–871. [Google Scholar] [CrossRef]
- Soresi, M.; Giannitrapani, L.; D'Antona, F.; Florena, A.M.; La Spada, E.; Terranova, A.; Cervello, M.; D'Alessandro, N.; Montalto, G. Interleukin-6 and its soluble receptor in patients with liver cirrhosis and hepatocellular carcinoma. World J Gastroenterol 2006, 12, 2563–2568. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Chen, J.; Quan, M.; Li, L.; Li, Y.; Gao, Y. CD63 negatively regulates hepatocellular carcinoma development through suppression of inflammatory cytokine-induced STAT3 activation. J Cell Mol Med 2021, 25, 1024–1034. [Google Scholar] [CrossRef]
- Zhang, J.F.; He, M.L.; Fu, W.M.; Wang, H.; Chen, L.Z.; Zhu, X.; Chen, Y.; Xie, D.; Lai, P.; Chen, G.; et al. Primate-specific microRNA-637 inhibits tumorigenesis in hepatocellular carcinoma by disrupting signal transducer and activator of transcription 3 signaling. Hepatology 2011, 54, 2137–2148. [Google Scholar] [CrossRef]
- Aroucha, D.C.; do Carmo, R.F.; Moura, P.; Silva, J.L.; Vasconcelos, L.R.; Cavalcanti, M.S.; Muniz, M.T.; Aroucha, M.L.; Siqueira, E.R.; Cahú, G.G.; et al. High tumor necrosis factor-α/interleukin-10 ratio is associated with hepatocellular carcinoma in patients with chronic hepatitis C. Cytokine 2013, 62, 421–425. [Google Scholar] [CrossRef]
- Swiątek, B.J. Is interleukin-10 gene polymorphism a predictive marker in HCV infection? Cytokine Growth Factor Rev 2012, 23, 47–59. [Google Scholar] [CrossRef]
- Zhang, S.; Gan, X.; Qiu, J.; Ju, Z.; Gao, J.; Zhou, J.; Shi, C.; Zhu, Y.; Li, Z. IL-10 derived from Hepatocarcinoma cells improves human induced regulatory T cells function via JAK1/STAT5 pathway in tumor microenvironment. Mol Immunol 2021, 133, 163–172. [Google Scholar] [CrossRef]
- Buchert, M.; Burns, C.J.; Ernst, M. Targeting JAK kinase in solid tumors: Emerging opportunities and challenges. Oncogene 2016, 35, 939–951. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Fu, K.; Yang, L.; Wu, S.; Cen, Z.; Meng, X.; Huang, Q.; Xie, Z. Hypoxia-inducible gene 2 promotes the immune escape of hepatocellular carcinoma from nature killer cells through the interleukin-10-STAT3 signaling pathway. J Exp Clin Cancer Res 2019, 38, 229. [Google Scholar] [CrossRef]
- Sui, Q.; Zhang, J.; Sun, X.; Zhang, C.; Han, Q.; Tian, Z. NK cells are the crucial antitumor mediators when STAT3-mediated immunosuppression is blocked in hepatocellular carcinoma. J Immunol 2014, 193, 2016–2023. [Google Scholar] [CrossRef] [PubMed]
- Langer, J.A.; Cutrone, E.C.; Kotenko, S. The Class II cytokine receptor (CRF2) family: Overview and patterns of receptor-ligand interactions. Cytokine Growth Factor Rev 2004, 15, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Sabat, R. IL-10 family of cytokines. Cytokine Growth Factor Rev 2010, 21, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.H.; Li, H.H.; Sung, J.M.; Chen, W.T.; Hou, Y.C.; Chang, M.S. Interleukin-19 mediates tissue damage in murine ischemic acute kidney injury. PLoS ONE 2013, 8, e56028. [Google Scholar] [CrossRef]
- Caparrós, E.; Francés, R. The Interleukin-20 Cytokine Family in Liver Disease. Front Immunol 2018, 9, 1155. [Google Scholar] [CrossRef]
- Hsing, C.H.; Cheng, H.C.; Hsu, Y.H.; Chan, C.H.; Yeh, C.H.; Li, C.F.; Chang, M.S. Upregulated IL-19 in breast cancer promotes tumor progression and affects clinical outcome. Clin Cancer Res 2012, 18, 713–725. [Google Scholar] [CrossRef]
- Fujimoto, Y.; Kuramoto, N.; Yoneyama, M.; Azuma, Y.T. Interleukin-19 as an Immunoregulatory Cytokine. Curr Mol Pharmacol 2021, 14, 191–199. [Google Scholar] [CrossRef]
- Wang, H.H.; Hsu, Y.H.; Chang, M.S. IL-20 bone diseases involvement and therapeutic target potential. J Biomed Sci 2018, 25, 38. [Google Scholar] [CrossRef]
- Wegenka, U.M.; Dikopoulos, N.; Reimann, J.; Adler, G.; Wahl, C. The murine liver is a potential target organ for IL-19, IL-20 and IL-24: Type I Interferons and LPS regulate the expression of IL-20R2. J Hepatol 2007, 46, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.S.; Wei, C.C.; Lin, Y.J.; Hsu, Y.H.; Chang, M.S. IL-20 and IL-20R1 antibodies protect against liver fibrosis. Hepatology 2014, 60, 1003–1014. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Xuefeng, Y.; Jianhua, X. Systematic review of the roles of interleukins in hepatocellular carcinoma. Clin Chim Acta 2020, 506, 33–43. [Google Scholar] [CrossRef]
- Ding, W.Z.; Han, G.Y.; Jin, H.H.; Zhan, C.F.; Ji, Y.; Huang, X.L. Anti-IL-20 monoclonal antibody suppresses hepatocellular carcinoma progression. Oncol Lett 2018, 16, 6156–6162. [Google Scholar] [CrossRef] [PubMed]
- Langowski, J.L.; Zhang, X.; Wu, L.; Mattson, J.D.; Chen, T.; Smith, K.; Basham, B.; McClanahan, T.; Kastelein, R.A.; Oft, M. IL-23 promotes tumour incidence and growth. Nature 2006, 442, 461–465. [Google Scholar] [CrossRef]
- Silvagni, E.; Missiroli, S.; Perrone, M.; Patergnani, S.; Boncompagni, C.; Bortoluzzi, A.; Govoni, M.; Giorgi, C.; Alivernini, S.; Pinton, P.; et al. From Bed to Bench and Back: TNF-α, IL-23/IL-17A, and JAK-Dependent Inflammation in the Pathogenesis of Psoriatic Synovitis. Front Pharmacol 2021, 12, 672515. [Google Scholar] [CrossRef]
- Kang, Y.; Su, G.; Sun, J.; Zhang, Y. Activation of the TLR4/MyD88 signaling pathway contributes to the development of human hepatocellular carcinoma via upregulation of IL-23 and IL-17A. Oncol Lett 2018, 15, 9647–9654. [Google Scholar] [CrossRef]
- Wang, J.; Lu, L.; Luo, Z.; Li, W.; Lu, Y.; Tang, Q.; Pu, J. miR-383 inhibits cell growth and promotes cell apoptosis in hepatocellular carcinoma by targeting IL-17 via STAT3 signaling pathway. Biomed Pharmacother 2019, 120, 109551. [Google Scholar] [CrossRef]
- Wu, J.; Guo, J.; Cao, Q.; Wang, Y.; Chen, J.; Wang, Z.; Yuan, Z. Autophagy impacts on oxaliplatin-induced hepatocarcinoma apoptosis via the IL-17/IL-17R-JAK2/STAT3 signaling pathway. Oncol Lett 2017, 13, 770–776. [Google Scholar] [CrossRef]
- Kuang, D.M.; Zhao, Q.; Wu, Y.; Peng, C.; Wang, J.; Xu, Z.; Yin, X.Y.; Zheng, L. Peritumoral neutrophils link inflammatory response to disease progression by fostering angiogenesis in hepatocellular carcinoma. J Hepatol 2011, 54, 948–955. [Google Scholar] [CrossRef] [PubMed]
- He, B.; You, L.; Uematsu, K.; Zang, K.; Xu, Z.; Lee, A.Y.; Costello, J.F.; McCormick, F.; Jablons, D.M. SOCS-3 is frequently silenced by hypermethylation and suppresses cell growth in human lung cancer. Proc Natl Acad Sci U S A 2003, 100, 14133–14138. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.S.; Ali, I.; Afridi, U.K.; Ishtiaq, M.; Mehmood, R. Epigenetic mechanisms regulating the development of hepatocellular carcinoma and their promise for therapeutics. Hepatol Int 2017, 11, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Sanaei, M.; Kavoosi, F.; Pourahmadi, M. Effect of Decitabine (5-aza-2'-deoxycytidine, 5-aza-CdR) in Comparison with Vorinostat (Suberoylanilide Hydroxamic Acid, SAHA) on DNMT1, DNMT3a and DNMT3b, HDAC 1-3, SOCS 1, SOCS 3, JAK2, and STAT3 Gene Expression in Hepatocellular Carcinoma HLE and LCL-PI 11 Cell Lines. Asian Pac J Cancer Prev 2021, 22, 2089–2098. [Google Scholar] [CrossRef]
- Mao, J.; Hu, X.; Pang, P.; Zhou, B.; Li, D.; Shan, H. miR-30e acts as a tumor suppressor in hepatocellular carcinoma partly via JAK1/STAT3 pathway. Oncol Rep 2017, 38, 393–401. [Google Scholar] [CrossRef]
- Norman, P. Selective JAK inhibitors in development for rheumatoid arthritis. Expert Opin Investig Drugs 2014, 23, 1067–1077. [Google Scholar] [CrossRef]
- Yang, L.; Xue, H.; Sun, Y.; Zhang, L.; Xue, F.; Ge, R. CircularRNA-9119 protects hepatocellular carcinoma cells from apoptosis by intercepting miR-26a/JAK1/STAT3 signaling. Cell Death Dis 2020, 11, 605. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, L.; Li, N.; Dai, J.; Zhang, R.; Yao, F.; Zhou, S.; Wu, Z.; Zhou, H.; Zhou, L.; et al. Etomidate elicits anti-tumor capacity by disrupting the JAK2/STAT3 signaling pathway in hepatocellular carcinoma. Cancer Lett 2023, 552, 215970. [Google Scholar] [CrossRef]
- Cherng, Y.G.; Chu, Y.C.; Yadav, V.K.; Huang, T.Y.; Hsieh, M.S.; Lee, K.F.; Lee, W.H.; Yeh, C.T.; Ong, J.R. Induced Mitochondrial Alteration and DNA Damage via IFNGR-JAK2-STAT1-PARP1 Pathway Facilitates Viral Hepatitis Associated Hepatocellular Carcinoma Aggressiveness and Stemness. Cancers (Basel) 2021, 13. [Google Scholar] [CrossRef]
- Zhou, X.X.; Gao, P.J.; Sun, B.G. Pravastatin attenuates interferon-gamma action via modulation of STAT1 to prevent aortic atherosclerosis in apolipoprotein E-knockout mice. Clin Exp Pharmacol Physiol 2009, 36, 373–379. [Google Scholar] [CrossRef]
- Sutter, A.P.; Maaser, K.; Höpfner, M.; Huether, A.; Schuppan, D.; Scherübl, H. Cell cycle arrest and apoptosis induction in hepatocellular carcinoma cells by HMG-CoA reductase inhibitors. Synergistic antiproliferative action with ligands of the peripheral benzodiazepine receptor. J Hepatol 2005, 43, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Yu, T.; Dong, N.; Wang, B.; Sun, F.; Jiang, D. Napabucasin, a novel STAT3 inhibitor suppresses proliferation, invasion and stemness of glioblastoma cells. J Exp Clin Cancer Res 2019, 38, 289. [Google Scholar] [CrossRef]
- Li, Y.; Rogoff, H.A.; Keates, S.; Gao, Y.; Murikipudi, S.; Mikule, K.; Leggett, D.; Li, W.; Pardee, A.B.; Li, C.J. Suppression of cancer relapse and metastasis by inhibiting cancer stemness. Proc Natl Acad Sci U S A 2015, 112, 1839–1844. [Google Scholar] [CrossRef]
- Li, Y.; Han, Q.; Zhao, H.; Guo, Q.; Zhang, J. Napabucasin Reduces Cancer Stem Cell Characteristics in Hepatocellular Carcinoma. Front Pharmacol 2020, 11, 597520. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Zheng, X.; Qian, Y.; Liu, M.; Nian, Z.; Cui, Q.; Zhou, Y.; Fu, B.; Sun, R.; Tian, Z.; et al. Interactions between driver genes shape the signaling pathway landscape and direct hepatocellular carcinoma therapy. Cancer Sci 2023, 114, 2386–2399. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.S.; Zheng, H.; Ou, Y.L.; Tao, Y.P.; Wang, Z.G.; Song, L.H.; Yan, H.L.; Zhou, W.P. miR-515-5p suppresses HCC migration and invasion via targeting IL6/JAK/STAT3 pathway. Surg Oncol 2020, 34, 113–120. [Google Scholar] [CrossRef]
- Hijona, E.; Banales, J.M.; Hijona, L.; Medina, J.F.; Arenas, J.; Herreros-Villanueva, M.; Aldazabal, P.; Bujanda, L. Pravastatin inhibits cell proliferation and increased MAT1A expression in hepatocarcinoma cells and in vivo models. Cancer Cell Int 2012, 12, 5. [Google Scholar] [CrossRef]
- Hayakawa, F.; Sugimoto, K.; Harada, Y.; Hashimoto, N.; Ohi, N.; Kurahashi, S.; Naoe, T. A novel STAT inhibitor, OPB-31121, has a significant antitumor effect on leukemia with STAT-addictive oncokinases. Blood Cancer J 2013, 3, e166. [Google Scholar] [CrossRef]
- Okusaka, T.; Ueno, H.; Ikeda, M.; Mitsunaga, S.; Ozaka, M.; Ishii, H.; Yokosuka, O.; Ooka, Y.; Yoshimoto, R.; Yanagihara, Y.; et al. Phase 1 and pharmacological trial of OPB-31121, a signal transducer and activator of transcription-3 inhibitor, in patients with advanced hepatocellular carcinoma. Hepatol Res 2015, 45, 1283–1291. [Google Scholar] [CrossRef]
- Covington, M.; He, X.; Scuron, M.; Li, J.; Collins, R.; Juvekar, A.; Shin, N.; Favata, M.; Gallagher, K.; Sarah, S.; et al. Preclinical characterization of itacitinib (INCB039110), a novel selective inhibitor of JAK1, for the treatment of inflammatory diseases. Eur J Pharmacol 2020, 885, 173505. [Google Scholar] [CrossRef]
- Barbour, A.M.; Rockich, K.; Cimino, E.; Zhou, G.; Leonetti-Whalen, C.; Chen, X.; Yeleswaram, S.; Epstein, N.; Punwani, N. Effect of Hepatic Impairment on the Pharmacokinetics of Itacitinib. J Clin Pharmacol 2021, 61, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Jouve, J.L.; Lecomte, T.; Bouché, O.; Barbier, E.; Khemissa Akouz, F.; Riachi, G.; Nguyen Khac, E.; Ollivier-Hourmand, I.; Debette-Gratien, M.; Faroux, R.; et al. Pravastatin combination with sorafenib does not improve survival in advanced hepatocellular carcinoma. J Hepatol 2019, 71, 516–522. [Google Scholar] [CrossRef] [PubMed]
- Burel, S.A.; Han, S.R.; Lee, H.S.; Norris, D.A.; Lee, B.S.; Machemer, T.; Park, S.Y.; Zhou, T.; He, G.; Kim, Y.; et al. Preclinical evaluation of the toxicological effects of a novel constrained ethyl modified antisense compound targeting signal transducer and activator of transcription 3 in mice and cynomolgus monkeys. Nucleic Acid Ther 2013, 23, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; Zhou, T.; Schmidt, J.; Jo, M.; et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci Transl Med 2015, 7, 314ra185. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Cheung, S.T. STAT3: An Emerging Therapeutic Target for Hepatocellular Carcinoma. Cancers (Basel) 2019, 11. [Google Scholar] [CrossRef]
- Yim, S.Y.; Lee, J.S. Genomic Perspective on Mouse Liver Cancer Models. Cancers (Basel) 2019, 11. [Google Scholar] [CrossRef]
- Brown, Z.J.; Heinrich, B.; Greten, T.F. Mouse models of hepatocellular carcinoma: An overview and highlights for immunotherapy research. Nat Rev Gastroenterol Hepatol 2018, 15, 536–554. [Google Scholar] [CrossRef]
- Macek Jilkova, Z.; Kurma, K.; Decaens, T. Animal Models of Hepatocellular Carcinoma: The Role of Immune System and Tumor Microenvironment. Cancers (Basel) 2019, 11. [Google Scholar] [CrossRef]
- Llovet, J.M.; Bruix, J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology 2008, 48, 1312–1327. [Google Scholar] [CrossRef]
- Dimri, M.; Satyanarayana, A. Molecular Signaling Pathways and Therapeutic Targets in Hepatocellular Carcinoma. Cancers (Basel) 2020, 12. [Google Scholar] [CrossRef]
Figure 1.
Schematic illustration of the JAK/STAT signaling pathway. Ligand binding to cytokine receptor leads to the formation of receptor dimerization (1). The dimerization induces the transphosphorylation of JAK as well as phosphorylation of the cytoplasmic tails of the receptors (2). STAT binds to the phosphorylated site of the receptor (3). Subsequently, JAK phosphorylates STAT recruited to the receptors (4). Phosphorylated STATs dissociate from the receptors and form a dimer (5). STAT dimers translocate into the nucleus and induce the transcription of target genes (6). There are three types of negative regulators of the JAK/STAT signaling: suppressors of cytokine signaling (SOCS), inhibiting the phosphorylation of the receptor by JAK; protein inhibitors of activated STAT (PIAS), preventing the activity of STAT transcription factors; and protein tyrosine phosphatases (PTPs), removing phosphate groups from JAKs and STATs.
Figure 1.
Schematic illustration of the JAK/STAT signaling pathway. Ligand binding to cytokine receptor leads to the formation of receptor dimerization (1). The dimerization induces the transphosphorylation of JAK as well as phosphorylation of the cytoplasmic tails of the receptors (2). STAT binds to the phosphorylated site of the receptor (3). Subsequently, JAK phosphorylates STAT recruited to the receptors (4). Phosphorylated STATs dissociate from the receptors and form a dimer (5). STAT dimers translocate into the nucleus and induce the transcription of target genes (6). There are three types of negative regulators of the JAK/STAT signaling: suppressors of cytokine signaling (SOCS), inhibiting the phosphorylation of the receptor by JAK; protein inhibitors of activated STAT (PIAS), preventing the activity of STAT transcription factors; and protein tyrosine phosphatases (PTPs), removing phosphate groups from JAKs and STATs.
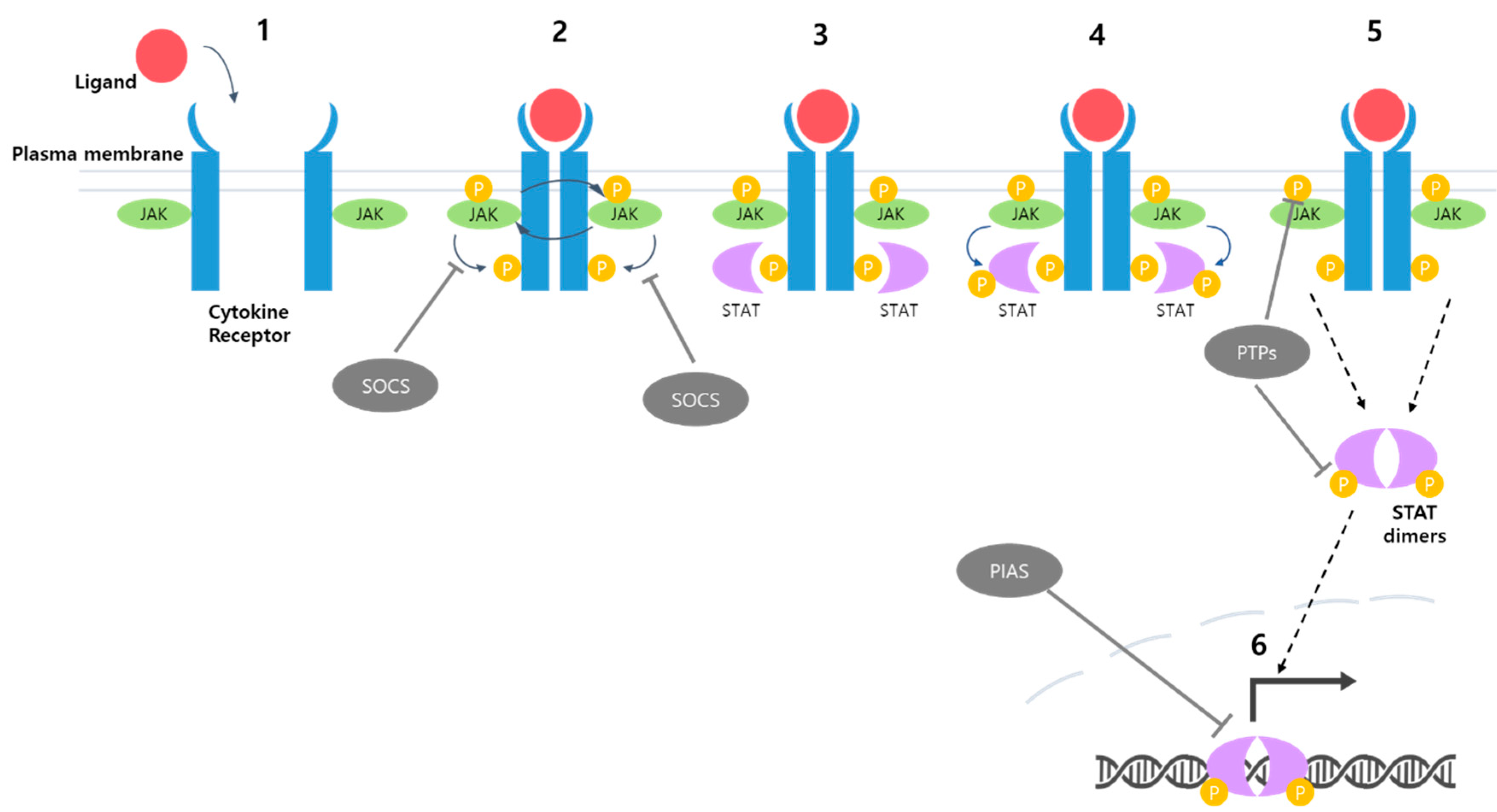
Figure 2.
Domain structures of JAK and STAT proteins. A. JAK proteins consist of seven homology domains known as Janus homology (JH) domains 1 to 7 (JH1 to JH7). B. STAT proteins consist of the N-terminal domain, coiled-coil domain, DNA-binding domain (DBD), linker domain, Src homology 2 (SH2) domain, and transcription activation domain (TAD).
Figure 2.
Domain structures of JAK and STAT proteins. A. JAK proteins consist of seven homology domains known as Janus homology (JH) domains 1 to 7 (JH1 to JH7). B. STAT proteins consist of the N-terminal domain, coiled-coil domain, DNA-binding domain (DBD), linker domain, Src homology 2 (SH2) domain, and transcription activation domain (TAD).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



