Submitted:
03 August 2023
Posted:
04 August 2023
You are already at the latest version
Abstract
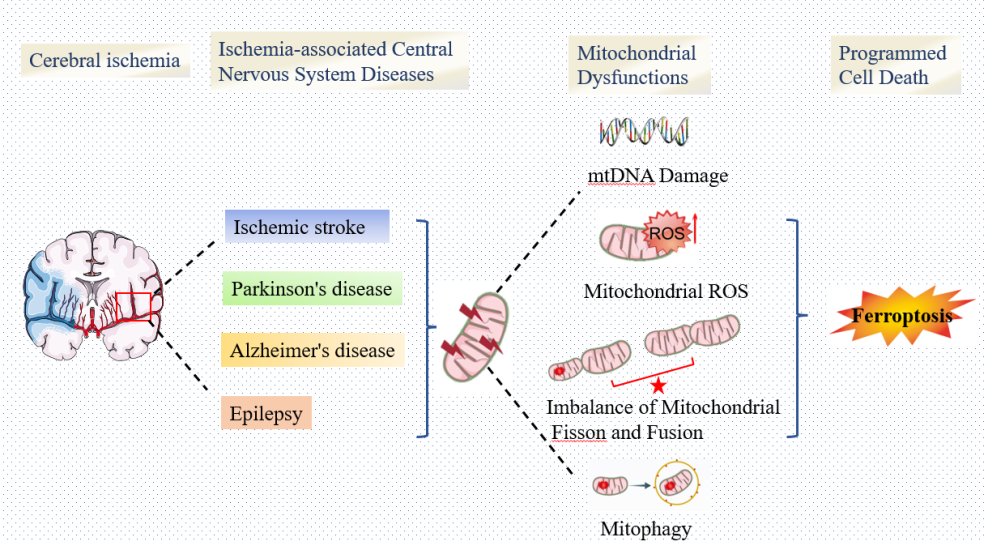
Cerebral ischemia, which is a leading cause of disability and mortality worldwide, sets off a chain of molecular and cellular pathologies that are associated with some central nervous system (CNS) disorders, mainly including ischemic stroke, Alzheimer's disease (AD), Parkinson's disease (PD), epilepsy and other CNS diseases. In recent times, despite significant advancements in the treatment of the pathological processes underlying various neurological illnesses, effective therapeutic approaches specifically targeted at minimizing the damage of such diseases remain absent. Remarkably, ischemia causes severe damage to cells in Ischemia-associated CNS Diseases. Cerebral ischemia initiates oxygen and glucose deprivation, which subsequently promotes mitochondrial dysfunction, including MPTP opening, mitophagy dysfunction, and excessive mitochondrial fission, triggering various forms of cell death, such as autophagy, apoptosis, as well as ferroptosis. Ferroptosis, a novel type of regulated cell death (RCD), is characterized by iron-dependent accumulation of lethal reactive oxygen species and lipid peroxidation. Mitochondrial dysfunction and ferroptosis both play critical roles in the pathogenic progression of Ischemia-associated CNS Diseases. In recent years, growing evidence has indicated that mitochondrial dysfunction interplays with ferroptosis to aggravate cerebral ischemia injury. However, the potential connections between mitochondrial dysfunction and ferroptosis in Cerebral Ischemia have not yet been clarified. Thus, we analyze the underlying mechanism between mitochondrial dysfunction and ferroptosis in Ischemia-associated CNS Diseases. We also discovered that GSH depletion and GPX4 inactivation cause lipoxygenase activation and calcium influx following cerebral ischemia injury, resulting in MPTP opening and mitochondrial dysfunction. Additionally, dysfunction in mitochondrial electron transport and an imbalanced fusion-to-fission ratio can lead to the accumulation of reactive oxygen species and iron overload, which further contribute to the occurrence of ferroptosis. This creates a vicious cycle that continuously worsens cerebral ischemia injury. In this study, our focus is on exploring the interplay between mitochondrial dysfunction and ferroptosis, which may offer new insights into potential therapeutic approaches for the treatment of Ischemia-associated CNS Diseases.
Keywords:
cerebral ischemia
; central nervous system diseases
; mitochondrial dysfunction
; ferroptosis
; therapeutics
1. Introduction
Cerebral ischemia, characterized by insufficient blood flow and oxygen delivery to the brain, is a major cause of morbidity and mortality worldwide (Levchenkova et al., 2021). It can trigger various pathological processes, including inflammation, oxidative stress, excitotoxicity, increased blood-brain barrier (BBB) permeability, and mitochondrial dysfunction. These processes are also implicated in neurodegenerative diseases of the central nervous system (CNS) such as Alzheimer's disease (AD), Parkinson's disease (PD), and others. There is a wealth of evidence suggesting that cerebral ischemia is associated with the development and progression of CNS diseases, including ischemic stroke (Sveinsson et al., 2014), AD (Pluta et al., 2021), PD (Lohmann et al., 2022), epilepsy (L. Jiang et al., 2022)and others (Goulay et al., 2020) related conditions collectively referred to as ischemia-associated CNS diseases (X. Liu et al., 2019).
Mitochondria, as regulators of energy generation, play a crucial role in maintaining cellular homeostasis and function. Additionally, they serve as the primary energy source for the brain and their dysfunction is closely associated with cerebral ischemic injury, which is essential for the pathophysiology of ischemia-associated CNS diseases (Koch et al., 2017), During ischemia, there are significant reductions in ATP levels for brief periods of time. Mitochondria play a key role in various cellular processes, including ATP production, Ca2+ homeostasis, reactive oxygen species (ROS) generation, and apoptosis. Studies have reported that resveratrol can significantly increase SOD activity and improve mitochondrial integrity by preventing the loss of mitochondrial membrane potential, thereby reducing neuronal apoptosis after cerebral ischemia (R. Wang et al., 2014). Furthermore, selenium has been shown to protect against cell death induced by glutamate and hypoxia by reducing glutamate-induced ROS production, preserving mitochondrial membrane potential, and increasing mitochondrial biogenesis (Mehta et al., 2012).
Due to mitochondrial dysfunction and other pathophysiological responses induced by cerebral ischemia, neurons are unable to maintain the normal ionic gradient across the neuronal membrane. This pathological process can lead to various types of cell death, including apoptosis, necrosis, autophagy, and ferroptosis (Figure 1) (Khoshnam, Winlow, & Farzaneh, 2017; Khoshnam, Winlow, Farzaneh, et al., 2017; P. Y. Li et al., 2016; Luo et al., 2013; van der Worp et al., 2010). Ferroptosis is a recently discovered form of regulated cell death characterized by the abnormal accumulation of iron-dependent lipid peroxidation and lipophilic ROS in cellular membranes (Dixon et al., 2012; Stockwell et al., 2020). Previous research has shown that the mechanisms of ferroptosis are involved in glutathione (GSH) depletion (Cao et al., 2016; Stockwell, et al., 2020; Sun et al., 2018), glutathione peroxidase 4 (GPX4) inactivation (Cao, et al., 2016; Forcina et al., 2019; Stockwell, et al., 2020; Vuckovic et al., 2020), and metabolic imbalances of iron and lipids (Cao, et al., 2016; Dixon, et al., 2012; Forcina, et al., 2019). Previous studies have shown that ferroptosis has pathological roles in a wide variety of diseases, including stroke, PD, AD (Toyokuni et al., 2017). Additionally, activation of ferroptosis has been found to trigger mitochondrial swelling and mitochondrial membrane potential collapse via MPTP opening, ultimately leading to cell death (Kwong et al., 2015). Increasing evidence has prompted various interplay between ferroptosis and mitochondrial dysfunction following cerebral ischemia. In this case, mitochondrial iron metabolism is impaired, which results in excessive free iron accumulation in mitochondria and lipid peroxidation of their membranes. Another pathway of lipid peroxidation accumulation in the mitochondria is cysteine deprivation, which promotes glutaminolysis, thereby effectively enhancing mitochondrial respiration. Stimulating the activity of the tricarboxylic acid cycle gives rise to mitochondrial hyperpolarization and increased ROS production, which promotes the induction of lipid peroxidation and ferroptosis (Gao et al., 2019). It is crucial to understand the interplay between mitochondrial dysfunction and ferroptosis in the context of ischemia-associated CNS diseases. In this review, we aim to summarize and discuss the mechanisms underlying this interplay and its relevance to ischemia-associated CNS diseases. By clarifying this mechanism, we hope to provide a new perspective that can contribute to the development of treatments for ischemia-associated CNS diseases.
2. Mitochondrial Dysfunction and Ischemia-Associated Central Nervous System Diseases
Mitochondria play a critical role not only in other cells but also in neurons, and their structural and functional integrity is particularly important. Cerebral ischemia leads to damage to the structure and function of mitochondria, which is characterized by the opening of MPTP, oxidative stress, and disturbances in mitochondrial quality control. In this context, we aim to investigate these characteristics and their relationships with ischemia-associated CNS diseases.
2.1. Mitochondrial Membrane Permeability Transition Pore
The mitochondrial permeability transition pore (MPTP) complex, which is a non-specific and voltage-dependent channel, consists of the voltage-dependent anion channel (VDAC), adenine nucleotide translocator (ANT), and cyclophilin D (CypD) (Kalani et al., 2018). The opening of MPTP results in the uncoupling of oxidative phosphorylation, mitochondrial depolarization, swelling and mechanistic disruption of the outer mitochondrial membrane (Gustafsson et al., 2008; Sesso et al., 2012; Weiss et al., 2003). The swollen organelle matrix preceding uncoupling of oxidative phosphorylation releases cytochrome c, which in turn activates apoptosis-inducing factor (AIF) and endonuclease G (Endo G), subsequently translocating to the nucleus and causing chromatin condensation and DNA breakage (Bonora et al., 2014).
Previous studies have demonstrated that MPTP opening occurs in response to mitochondrial Ca2+ overload (Varanyuwatana et al., 2012). In addition, it also can sense oxidative stress, inflammation, pH imbalance, and iron disturbance generated by ischemic tissue and participate in this process to promote mitochondrial damage in neuronal cells (Bonora et al., 2019; Matsumoto et al., 2018). MPTP induction induces the activation of an inflammatory response by the release of succinate and mitochondrial damage-associated molecular patterns (DAMPs) that will induce tissue damage after cerebral ischemia (Dhalla et al., 2000). BCL2 family proteins (Bax, Bid, Puma, and BNIP3) can increase the permeability of the outer mitochondrial membrane,which results in the creation of permeability pores and upregulates other apoptotic factors (Gustafsson, et al., 2008; Orrenius et al., 2007). Moreover, the formation of MPTP was aided by the presence of oxidizing agents, inorganic phosphate (Pi), and alkaline pH (Boczek et al., 2015; Varanyuwatana, et al., 2012). Therefore, blocking MPTP to maintain mitochondrial integrity and function may be beneficial in cerebral ischemia.
2.2. ROS Production and Mitochondrial Dysfunction
Mitochondria represent a crucial source of ROS, as some of the electrons are instead leaked out of the mitochondrial electron transport chain (Niizuma et al., 2010; Sanderson et al., 2013). Under physiological conditions, ROS, as the second messenger involved in a variety of cellular pathways, allows cells to undergo normal physiological activities, and sufficient antioxidant systems limit the damage to cellular components (Finkel, 2011). However, following cerebral ischemia, mitochondrial dysfunction disrupts the balance between ROS production and scavenging, leading to oxidative stress and cell injury (Radak et al., 2014).
ROS can lead to protein modification or degradation via interacting with amino acids in protein molecules. It can also react with the side chains and the backbone of the protein, causing protein oxidation, peptide bond breaking, and protein degradation (W. Li et al., 2016). Oxidation of proteins by ROS can result in crosslink and aggregation between proteins, which induces functional alterations of the proteins, including enzyme inactivation or modification of ion channel activity (Aizenman et al., 1990; Fucci et al., 1983; Gray et al., 2015; Grune et al., 1995). Cerebral ischemia leads to extensive protein oxidation in human and animal models of stroke (Dominguez et al., 2010; S. Li et al., 2005). Glutamine synthetase can catalyze the conversion of glutamate to glutamine in astrocytes to protect neurons from excitotoxicity, and oxidative inactivation of glutamine synthetase has been found as a significant factor in the neurotoxicity induced by cerebral ischemia in gerbils (Oliver et al., 1990; Suarez et al., 2002). ROS surges also disrupt DNA double strands, and lead to interchain crosslinks, protein-DNA crosslinks, DNA mutations, and DNA structural alterations, which cause irreversible damage to cells (Jena, 2012). 8 hydroxy 2V deoxyguanosine (8OHdG) is a common product of DNA oxidative damage, and elevated levels of 8OHdG during cerebral ischemia highlighted extensive DNA oxidation in cerebral ischemia precedes DNA fragmentation (Cooke et al., 2003; Cui et al., 2000; S. Li, et al., 2005; MacManus et al., 1999). In addition, ROS was found to directly damage the mitochondrial proteins and mtDNA, resulting in membrane integrity destruction, which in turn depolarizes and induces cell death (Droge, 2002). Oxidative degradation of lipids, known as lipid peroxidation, closely involves ferroptosis (Stockwell et al., 2017). ROS can attack lipids containing carbon-carbon double bonds, including polyunsaturated fatty acids, and produce unstable lipid radicals, reacting with oxygen to generate lipid peroxyl radicals. Lipid peroxyl radicals can react with other lipid acids to form another lipid radical and lipid peroxide (W. Li, et al., 2016). The end products of lipid peroxidation-reactive aldehydes are generated in this process, including malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE), which have been employed as markers for lipid peroxidation (Gueraud et al., 2006; Picaud et al., 2004; Serteser et al., 2002). MDA can react with amino acids in proteins and other molecules to form adducts, which can cause protein modification and DNA damage or mutation by promoting intramolecular or intermolecular protein or DNA cross-linking, resulting in secondary injury (J. Cheng et al., 2011; Z. Feng et al., 2006; Niedernhofer et al., 2003; Pizzimenti et al., 2013; Slatter et al., 2004). 4-HNE, a second messenger, can regulate a variety of transcription factors and major cell signaling pathways. Low levels of 4HNE promote cell proliferation, and differentiation as well as protect them against free radicals and inflammation. On the other hand, high concentrations of 4HNE cause cell apoptosis. Elevated lipid peroxidation has been found in rat cerebral ischemia models (Bromont et al., 1989; Yamamoto et al., 1983), and is identified as playing a material role in cell death in cerebral ischemia.
2.3. Disturbed Mitochondrial Quality Control
Mitochondrial quality control, including mitochondrial fission and fusion, mitochondrial biogenesis, and mitophagy, is the cornerstone of preserving the integrity and stability of morphology, quantity and function, and plays an indispensable role in alleviating the pathological process of ischemia-associated CNS diseases (Qiu et al., 2019). However, recent reports found that prolonged ischemic times, directly related to mitochondrial quality control, lead to the disruption of dynamic balance, which ultimately intensifies mitochondrial damage and causes cell death (Simmons et al., 2020).
In order to pre-existing mitochondria to proliferate and divide, a process known as mitochondrial biogenesis coordinates the transcription of mitochondrial and nuclear genes (A. Cheng et al., 2012). The peroxisome proliferator-activated receptor-coactivator-1α(PGC-1α) protein family plays an indispensable role in a multistep process that permits coordination (Scarpulla, 2011). PGC-1α, the chief regulator of mitochondrial biogenesis, is activated by global cerebral ischemia and stimulates the expression and activation of a number of nuclear proteins, including UCP2 and SOD2, which induces mitochondrial biogenesis and has neuroprotective effects (S. D. Chen et al., 2010; Su et al., 2017).
Aside from mitochondrial biogenesis, mitochondrial dynamics, which includes morphological changes associated with fission and fusion, is also a precisely regulated mechanism (Chung et al., 2021). ATP supply deficiency following cerebral ischemia leads to neuronal plasma membrane depolarization, which leads to glutamate release and subsequent excitotoxicity (Doyle et al., 2008). Glutamate excitotoxicity influences mitochondrial fission and fusion, as well as the imbalance in mitochondrial fission and fusion, resulting in NMDA receptor upregulation and oxidative stress. Additionally, due to increased glutamate release from neurons and astrocytes induced by cerebral ischemia, NMDA and amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid (AMPA) receptors, which are closely associated with ischemia-associated CNS diseases were overactivated (Babaei, 2021; Guo et al., 2021; Hanada, 2020; Lai et al., 2019; Nguyen et al., 2011). Furthermore, mitochondrial fission and ROS interact with one another. On the one hand, increased mitochondrial fission can enhance ROS; correspondingly, inhibition of mitochondrial fission will restore ROS levels to normal (Y. Zhang et al., 2020). On the other hand, the increase of ROS can encourage the activation of Drp1, causing an increase in mitochondrial fission, which further increases ROS production, while knocking out Drp1 can reduce mitochondrial fragmentation caused by oxidative stress (Youle et al., 2012).
Selective mitochondrial autophagy, known as mitophagy, promotes the control of oxidative stress and recovery of damaged mitochondria composition. Excessive mitophagy may provoke uncontrolled degradation of mitochondria, and defective mitophagy may result in the accumulation of mitochondria and excessive ROS production, which have been proposed to contribute to cell degeneration and activation of cell death pathways (Bhatia et al., 2019; Khalifa et al., 2017). Moderate autophagy is protective, while excessive autophagy may contribute to cell deaths during ischemia(Mo et al., 2020). It has been reported that BNIP3L rescues mitophagy deficiency and protects against cerebral ischemia(X. Wu et al., 2021). And inhibiting Peroxynitrite-mediated mitochondrial activation could reduce neuronal damage in ischemic stroke(J. Feng et al., 2018). Additionally, previous studies have demonstrated defective mitophagy in AD and PD, resulting in the aggregation of autophagosomes, abnormal endosomes and abnormal lysosomes, which contributes to the pathology of AD and PD (Franco-Iborra et al., 2018; Ni et al., 2015).
3. Ferroptosis and Ischemia-Associated Central Nervous System Diseases
After cerebral ischemia, ferroptosis has been shown to have detrimental effects on the brain by promoting rapid neuronal death and dysfunction (Tuo et al., 2017). Moreover, inhibiting ferroptosis has been found to significantly reduce disease severity and promote functional recovery (Zhou et al., 2020). Numerous lines of evidence have demonstrated that the mechanisms of ferroptosis involve lipid peroxidation triggered by GSH depletion, inactivation of GPX4, and iron overload, all of which contribute to the development of ischemia-associated CNS diseases (Dixon, et al., 2012; Kagan et al., 2017; Lei et al., 2019). In this context, we aim to elucidate the mechanisms of ferroptosis and their underlying roles in ischemia-associated CNS diseases.
3.1. Lipid Peroxidation
Lipid peroxidation, the prototype of a free radical chain reaction that inserts oxygen into a C-H bond in oxidizable free polyunsaturated fatty acids (PUFAs), accumulates and causes ferroptosis (D'Herde et al., 2017). It is noteworthy that among the PUFAs-related phospholipids, phosphatidylethanolamine (PEs) containing arachidonoyl (AA) and its derivative adrenaline adrenoyl (AdA) moieties have been indicated to be the indispensable substrates of oxidation in ferroptosis (Kagan, et al., 2017).A increasing body of work implicates lipid peroxides as important mediators of many pathological states including stroke, PD and AD (Gaschler et al., 2017).
Ferroptosis induced by lipid peroxidation involves the following steps: acyl-CoA synthetase long-chain family member 4 (ACSL4) acts as a crucial regulator, and catalyzes the esterification of AA or AdA into PEs to form AA-CoA or AdA-CoA. Then AA-CoA or AdA-CoA can be incorporated into membrane phospholipids with the catalysis of lysophosphatidylcholine acyltransferase 3 (LPCAT3) to generate AA-PE or AdA-PE. Finally, AA-PE and AdA-PE were the preferred substrates for oxidation, which were oxidized by 15-LOX into ferroptosis signals, including PE-AdA-OH and PE-AA-OH, and participated in ferroptosis execution (D'Herde, et al., 2017).
The brain rich in PUFAs and iron is particularly vulnerable to lipid peroxidation since its low antioxidant defense capacities (Cobley et al., 2018). Arachidonate 15-lipoxygenase (ALOX15) is an enzyme that oxygenates PUFAs and bio-membranes, which can selectively and specifically induce the oxidation of AA/AdA-PE, resulting in the enzymatic production of AA/AdA-PE-OOHs (Han et al., 2014; Hinman et al., 2018). PUFAs are the major source of lipid peroxidation in ferroptosis, and ALOX15 can directly oxidize lipid membranes containing PUFAs (van Leyen et al., 2014). ALOX15 was strongly expressed in neurons and endothelial cells following localized ischemia, while its inhibition or deletion prevented the development of edema and preserved the BBB (Jin et al., 2008; Yigitkanli et al., 2017). Lipid peroxidation products serve as underlying biomarkers for Ischemia-associated CNS Diseases. Moreover, The accumulation of lipid peroxidation on the mitochondrial membrane plays a vital role in ferroptosis (Krainz et al., 2016).One clinical area where lipid peroxidation is especially significant is degenerative disease of the brain and Ischemia-associated CNS Diseases. The brain consumes a large volume of oxygen and triggers a high quantity of ROS as a via product of ATP synthesis. Membrane phospholipids in the CNS are highly enriched in PUFAs, incorporating them rapidly from free fatty acids (C. T. Chen et al., 2008)
3.2. GSH Depletion and GPX4 Inactivation
Ferroptosis is regulated by lipid peroxidation, which may be reversed by GSH production and GPX4 activation. GSH, a tripeptide consisting of glutamate, glycine, and cysteine with sulfhydryl groups, exerts an indispensable duty in free radical scavenging and detoxification through glutathione S-transferase and GPX4 (Aoyama et al., 2015). As a glutamate/cystine antiporter that produces GSH and GPX4, System xc− plays a beneficial role in preventing ferroptosis (L. Wang et al., 2020). However, extracellular glutamate levels will increase during a stroke, causing glutamate toxicity and system xc- impairment. As such, cystine-glutamate exchange will be blocked and the GSH production and GPX4 activation will be inhibited, which finally initiates ferroptosis (Lewerenz et al., 2013; Zhang et al., 2021). Additionally, the levels of GSH are decreased in cerebral patients and MCAO animal models, which was accompanied by elevated lipid peroxidation (Ahmad et al., 2014; J. H. Liu et al., 2020). GSH depletion increase ROS content, induce cell ferroptosis, and promote the degradation of dopaminergic neurons in PD (Asanuma et al., 2021). Temporarily, GSH supplementation or activation of Nrf2 pathway is expected to be an operative therapeutic process for neuroprotection in PD. In addition, GSH levels were significantly reduced in ischemic brain tissue of IS model rats, while cysteine supplementation to restore depleted GSH levels or administration of ferroptosis inhibitors significantly reduced neuronal injuries after IS (Lan et al., 2020).
GPX4, one type of lipid enzyme, transforms lipid hydroperoxides into nontoxic lipid alcohols, which prevent the accumulation of toxic lipid oxidation (S. Hao et al., 2018; Yang et al., 2014). Lethal ferroptosis and neurological dysfunction can be caused by the deletion or inactivation of the GPX4 (Brutsch et al., 2015; Imai et al., 2003), which mostly presents as progressive cognitive dysfunction and impaired behavior in the context of cerebral ischemia (Hambright et al., 2017; X. Jiang et al., 2018). It has been shown that the expression level of GPX4 was significantly reduced during the acute phase of ischemic stroke and that aggrandizing GPX4 levels can protect neurons from ferroptosis (Z. Zhang et al., 2018). In addition, supplementation with tetrahydroxy stilbene glycoside, γ-glutamylcysteine, etc, can restore the GSH-GPX4 antioxidant system and reduce ROS levels in AD patients, thereby reducing Aβ-induced brain injury (Y. Liu et al., 2021).Meanwhile, GPX4 expression was decreased, and lipid peroxidation appeared in the hippocampus of epileptic model rats, which could be reversed by Fer-1 (Mao et al., 2019).
3.3. Iron Overload
There are two main sources of iron: food-derived iron and iron generated by the hemoglobin of senescent erythrocytes (Sliwinska et al., 2018). The Fe2+ is absorbed by the small intestinal mucosa epithelial cells, and then, the absorbed Fe2+ is oxidized to Fe3+ by the ferroxidase hephaestin, eventually, Fe3+ enters the circulation by ferroportin (FPN) (Chifman et al., 2014; Dong et al., 2008; Ferris et al., 1999). Furthermore, virtually all circulating Fe3+ tightly combined with transferrin (TF) to form the complete TF, which can bind to the membrane protein transferrin receptor 1 (TFR1) to transport the Fe3+ into the acidified endosomes (Hamai et al., 2017). Finally, iron reductase reduces Fe3+ to Fe2+, which is transported into the labile iron pool in the cytoplasm by divalent metal transporter 1 (DMT1) (Garton et al., 2016). Most released Fe2+ is utilized in several physiological processes, including the Fenton reaction, DNA synthesis, and mitochondrial oxidative metabolism. Ferritin, a protein with two subunits, namely ferritin heavy chain 1 (FTH1) and ferritin light chain (FTL), can store part of the released Fe2+. At the same time, FPN allows other iron that is not utilized or stored to be exported to the extracellular environment. Subsequently, the ceruloplasmin (Cp) can oxidize the exported Fe2+, which is bound to serum TF again (Hentze et al., 2010). Under physiological conditions, iron homeostasis is kept through these intricate networks.
Prior to the concept of ferroptosis, it had already been acknowledged that the accumulation of iron in the brain was one of the causes of secondary brain damage (H. Liu et al., 2019; Tang et al., 2020). Physiologically, BBB can rescue the brain against undulations in systemic iron (Qian et al., 2019). However, the BBB is disrupted in acute ischemia, allowing free iron and ferritin to enter the brain parenchyma, and hydrogen peroxide is converted to hydroxyl radicals through the Fenton reaction (DeGregorio-Rocasolano et al., 2019; Y. Li et al., 2019). Besides, iron dissociation from TF is induced by the acidic environment of cerebral ischemia, which raises extracellular iron levels and transfers iron to neurons, increasing intracellular iron intake (Lipscomb et al., 1998; Palmer et al., 1999; Selim et al., 2004). Ferritin, TFR1, and DMT1 have been demonstrated to increase during cerebral ischemia, which may contribute to the intake of iron (Chi et al., 2000; Lan, et al., 2020). Meanwhile, FPN, the only protein that exports iron from cells, has been downregulated following cerebral ischemia, further aggravating intracellular iron overload by preventing its outflow (L. Li et al., 2009).
Iron overload has been identified as a significant provenience of oxidative stress in cerebral ischemia. Iron deposits severely in the basal ganglia, thalami, periventricular, and subcortical white matter regions following severe ischemic and hypoxic brain injury (Dietrich et al., 1988). This process sharply increases ROS production, directly damaging proteins, amino acids, nucleic acids and membrane lipids, ultimately mediating ferroptosis. Furthermore, the source of lipid peroxidation comes in three ways: lipid autoxidation catalyzed by iron, esterification and oxygenation of PUFAs, and production of lipid ROS associated with the Fenton reaction (Kaye et al., 2019; Yang et al., 2016). Iron participates in all three pathways, which are critical factors in triggering ferroptosis. It has been shown that multiple iron chelates or iron export compounds have indicated neuroprotective effects in ischemic strokes models(Hanafy et al., 2019), supporting iron accumulation as a therapeutic target in ischemic strokes.
4. The Interplay between Mitochondrial Dysfunction and Ferroptosis in Ischemia-Associated Central Nervous System Diseases
Mitochondria play a crucial role in cellular stress response and are dynamically involved in multiple types of regulated cell death mechanisms, including ferroptosis. Furthermore, there is mounting evidence from various sources, including structural, functional, and autophagic studies, indicating the existence of crosstalk between mitochondria and ferroptosis. This crosstalk is illustrated in Figure 2.
4.1. The Interplay between Mitochondrial Dysfunction and Ferroptosis in IS
In light of the existing animal studies, mitochondrial dysfunction was reported in ischemic stroke, which was correlated with oxygen and glucose deprivation (OGD) (T. Yang et al., 2020). Although a vast array of evidence suggested that the Fenton reaction was the primary source of ROS in ferroptosis (Shen et al., 2018; S. Wang et al., 2018), mitochondrial ROS production likely contributes to ferroptosis induction by promoting lipid peroxidation (Gan, 2021). In the ischemic brain, p53 rapidly accumulates and activates transcriptional-dependent and transcriptional-independent programs to cause neuronal apoptosis (Almeida et al., 2021). Recent studies revealed that p53 favors mitochondrial respiration and contributes to ROS-mediated ferroptosis, which has a direct impact on metabolic versatility (W. Zhang et al., 2018). Moreover, MitoROS may activate ER stress which triggers autophagy and ferroptosis (C. Zhao et al., 2021). ONOO−-mediated mitophagy is suspected t be excessive in stroke and could provoke uncontrolled degradation of mitochondria, resulting in stored iron releasing, which triggers iron overload and ferroptosis (Du et al., 2020; J. Feng et al., 2017). Additionally, the NOD LRR pyrin domain-containing protein 3 (NLRP3) inflammasome in microglia was activated immediately following cerebral I/R injury, and mitochondrial dysfunction played a key role via releasing mitochondrial ROS, mitochondrial DNA damage, and releasing phospholipid cardiolipin in this process (Gong et al., 2018; He et al., 2020). Exceptional activation of the NLRP3 inflammasome via mitochondrial dysfunction conduces to not merely pyroptosis but other kinds of cell death, including ferroptosis (Y. Huang et al., 2021).
The characteristic morphological changes of mitochondria in ferroptosis are the shrunken mitochondria, ruptured external membrane, reduced crista, a compressed internal membrane, and an electron lucent nucleus (H. Wang et al., 2020). These changes eventually lead to cell membrane rupture, mitochondrial membrane potential depolarization, and MPTP opening. MPTP is deeply involved in ROS production in the mitochondria of neuronal cells during ischemic stroke (Matsumoto, et al., 2018). Moreover, MPTP opening causes mitochondrial energetic dysfunction, organelle swelling, rupture as well as typically ferroptosis, thus creating a vicious circle (Basit et al., 2017; Kwong, et al., 2015). These studies suggest that MPTP is an important intersection point between mitochondrial dysfunction and ferroptosis. VDACs, a channel-forming protein situated at the mitochondrial outer membrane, allowed the metabolites and irons across the outer membrane (Colombini, 2012; Hodge et al., 1997). The partial closure of VDACs after global ischemia inhibits mitochondrial metabolism and reduces mitochondrial membrane potential (Maldonado et al., 2012; Yao et al., 2018). Interestingly, ferroptosis activator Erastin promotes VDAC2/3 opening, causing mitochondrial iron uptake and increasing the mitochondrial membrane potential. In the following hours, mitochondrial depolarization was identified, which is a manifestation of mitochondrial dysfunction (DeHart, Fang, et al., 2018; DeHart, Lemasters, et al., 2018; Gao, et al., 2019). Therefore, stabilizing VADCs can be used as an effective target in ferroptosis treatment.
Additionally, mitochondrial-dependent proapoptotic protein Bid is transactivated after IR, which may be caused by lipid peroxidation and the increase of ROS induced by ferroptosis. As a result, the mitochondrial membrane potential and integrity are lost, ATP synthesis is impaired, ROS production increase, and AIF is released from the mitochondria to the nucleus (Neitemeier et al., 2017; Yin et al., 2020).
4.2. The Interplay between Mitochondrial Dysfunction and Ferroptosis in AD
As a matter of fact, it is generally accepted that mitochondria participate in AD. Mitochondrial ferritin (FtMt) regulates iron metabolism by modulating the redistribution of iron from mitochondria to the cytoplasm, which protects mitochondria from iron-induced oxidative injury (Carocci et al., 2018). And lack of FtMt aggravates amyloid β-induced neurotoxicity. Therefore, we speculate that lack of FtMt may lead to iron overload by aggravating mitochondrial dysfunction via enhancing oxidative stress in AD (Fuhrmann et al., 2020; L. Wang et al., 2017). Iron regulatory protein 1 (IRP1) activation is secondary to mitochondrial dysfunction and is regulated by the mitochondrial ISC biogenesis pathway, it selectively regulates amyloid β precursor protein (APP) mRNA and causes iron accumulation (Calvelage et al., 2021; Rouault et al., 2017; Urrutia et al., 2017). To prevent excess cytoplasmic iron, mitoferrin1/2 allows iron to enter the mitochondria to maintain iron homeostasis. However, recent studies have shown that mitoferrin1/2 was unregulated in AD, causing cells to exhibit iron overload (J. Huang et al., 2018; H. Wang, et al., 2020). Additionally, the pathophysiology of AD has also been linked to the aberrant activation of the NLRP3 inflammasome induced by mitochondrial dysfunction. The NLRP3 inflammasome has been shown to be critical for the pathological drive of amyloid and tau proteins (Ising et al., 2019). Iron overload is the most apparent alteration of these pathological processes, which can promote ferroptosis by causing lipid peroxidation via intracellular ROS generated by the Fenton reaction. Mitophagy can reduce mitochondrial ROS production and degrade dysfunctional mitochondria (Guan et al., 2018). So far, Pink1-Parkin-mediated mitophagy is the most deeply understood mechanism of mitophagy in mammalian cells (Ni, et al., 2015). Pink1-Parkin-mediated mitophagy, as a particular type of autophagy, is discovered to be strongly activated at the onset of AD (Hajj Hussein et al., 2015). Cytosolic Parkin levels decrease during disease progression, resulting in increased mitochondrial dysfunction (Hajj Hussein, et al., 2015). Impaired mitophagy may lead to mitochondrial accumulation, increased oxygen consumption, and excessive formation of ROS, which is associated with lipid peroxidation (Evans et al., 2020).
A plethora of evidence showed that lipid peroxidation and GSH depletion, the primary signature of ferroptosis, have been described in AD (N. Yan et al., 2019). And iron-induced oxidative stress also is an important pathological mechanism of AD that can directly cause neuronal damage and cell death. The ferroptosis pathway may contribute to our understanding of how iron enhances the neurotoxicity of other major pathological features of AD, including those associated with Aβ and tau (Lane et al., 2018). Anna et al. found a significant decrease in the activity of GPX in Alzheimer's disease (Zalewska et al., 2021). Consistent with this, adult mice with conditional GPX4 ablation in their forebrain neurons have cognitive deficits and hippocampus degeneration resembling Alzheimer's disease. It is noteworthy that lipid peroxidation and mitochondrial impairments related to ferroptosis are found in this model, which can be partly rescued by inhibitors of ferroptosis, confirming ferroptosis involvement (Hambright, et al., 2017). Moreover, under iron overload conditions, mtDNA is damaged by ROS produced by the labile iron pool (Matsuo-Tezuka et al., 2019). MtDNA double-strand breaks, as well as progressive loss of intact mtDNA, reduced mtDNA transcription and decreased expression of respiratory chain subunits encoded by the mitochondrial genome were reported in AD cells, which were also observed in ferroptosis mitochondrial. Therefore, we speculated that one of the main culprits of mtDNA damage in AD cells was ferroptosis (W. Hao et al., 2021; Tonnies et al., 2017).
4.3. The Interplay between Mitochondrial Dysfunction and Ferroptosis in PD
Mitochondrial dysfunction is a key pathophysiological change in PD and is an essential initiator of dopaminergic neuron loss (Malpartida et al., 2021). Excessive ROS leads to mitophagy in PD patients, a negative feedback mechanism that plays a crucial role in eliminating ROS by inhibiting oxidative stress. Hence, mitophagy is regarded to be neuroprotective by scavenging ROS generation (Amro et al., 2018; Orellana-Urzua et al., 2020; Wible et al., 2018). However, mitophagy dysfunction has been observed in several PD models (J. Liu et al., 2019), which causes ROS could not be effectively scavenged. Excessive ROS expedites oxidative stress to cell components and causes extensive lipid peroxidation, leading to ferroptosis (Gutteridge et al., 1979). MitoNEET, an iron-containing outer mitochondrial membrane protein, participates in exporting iron from mitochondria (Mittler et al., 2019). MitoNEET KO mice exhibit multiple characteristics of early neurodegeneration in PD, including mitochondrial dysfunction, loss of striatal dopamine and tyrosine hydroxylase (Geldenhuys et al., 2017). Knockout of MitoNEET will increase the mitochondrial iron content and lipid peroxidation (H. Yuan et al., 2016), aggravating the adverse effects of erastin-induced ferroptosis. NEET proteins were identified as a newly discovered iron-sulfur protein (2Fe-2S), which mediated the export of sulfur ions and iron between the cytosol and the mitochondria when the synthesis of Fe-S clusters was disturbed (Mittler, et al., 2019). Deletion of one of the mitoNEET isoforms, CDGSH iron sulfur domain 1 (CISD1), led to ferroptosis by causing mitochondrial iron accumulation and the production of mitochondrial lipid peroxides (H. Yuan, et al., 2016). Interestingly, CISD1 knockout mice exhibit many features of Parkinson’s disease. In addition, recent studies also highlight that Mfrn1/2 is impaired not only in AD but also in PD, which are all linked to ferroptosis. These findings illustrate mitochondria play a crucial role in cellular iron homeostasis, and the regulation of iron storage in mitochondria will be an effective strategy to inhibit ferroptosis in PD.
Intracellular calcium overload interacts with iron overload, which is characteristic of ferroptosis, and each ion disorderliness occurs in PD and AD (Maher et al., 2018). Increased cytoplasmic Ca2+ promotes 12/15-LOX activation (Brinckmann et al., 1998). Furthermore, excess Ca2+ entering mitochondria produces mitochondrial dysfunction and damage, whereas iron overload induces oxidative stress, which increases the flow of excessive calcium signals (Do Van et al., 2016). In addition, in the experimental model of arsenite-induced neuronal cell death that has been shown to be associated with PD and AD, it was shown that the loss of neurons is caused by the generation of ROS, the imbalance of GSH and GSSG, and ferroptosis-related signaling pathways. Of note, in this model, VADCs and mitogen-activated protein kinases were activated, leading to mitochondrial dysfunction, which may be associated with ferroptosis. Despite the clinical distinctions between AD and PD, these disorders share partially similar clinicopathologic features in ferroptosis, including unregulated mitoferrin1/2, calcium overload, iron overload and VADCs opening.
4.4. The Interplay between Mitochondrial Dysfunction and Ferroptosis in Epilepsy
Mitochondrial oxidative stress induced by ROS is emerging as a crucial element involved in the onset of epilepsy (N. Yang et al., 2020). Dysregulation of astrocyte glutamate metabolism due to mitochondrial oxidative stress results in high extracellular levels of glutamate occurring in the brain during epileptic seizures, which can cause recurrent seizures (Albrecht et al., 2017; Boison et al., 2018; Fulton et al., 2021). Moreover, excessive glutamate induces neuronal over-excitation and neuronal excitotoxicity and triggers ferroptosis (Dixon, et al., 2012). Additionally, previous studies have discovered that autophagy induced by mitochondrial oxidative stress is crosstalk with ferroptosis. Mitochondrial antioxidants Mito-TEMPO can reduce GSH depletion and autophagic response, which induces autophagy inhibition to block the ferroptosis process (Wei et al., 2020).
GSH serves as imperative free radical scavenger compounds, which play an imperative role in alleviating oxidative stress and preventing neuronal death (Mueller et al., 2001). It was found to reverse mitochondrial dysfunction (Diao et al., 2020). Patients with epilepsy have lower GPX4 and GSH levels compared to normal controls, which have been linked to ferroptosis (Y. Cai et al., 2021). The persistently low GPX4 and GSH result in neuronal excitability changes, hippocampal neuron loss, and astrocyte proliferation, which may induce mitochondrial dysfunction (Kurzatkowski et al., 2013). Meanwhile, in rats with temporal lobe epilepsy (TLE) induced by kainic acid, a considerable accumulation of lipid peroxidation and consumption of GSH have been observed, combined with a decrease in the mitochondrial region of hippocampal neurons (Ye et al., 2019). Collectively, these findings imply that mitochondrial dysfunction due to low GPX4 and GSH levels during ferroptosis is a nonnegligible reason for cell damage in epilepsy. Particularly, the p53 pathway, which can adjust necrosis and autophagic activity, including mitophagy, directly alters the metabolic versatility by promoting mitochondrial respiration and contributing to the ROS-mediated ferroptosis in Ischemia-associated CNS Diseases (Lu et al., 2020; Morrison et al., 2000; Shi et al., 2020; P. Zhang et al., 2020; W. Zhang, et al., 2018).
5. Targeting Intervention of Mitochondrial Dysfunction and Ferroptosis for the Treatment against Ischemia-Associated Central Nervous System Diseases
Recent studies have revealed that mitochondrial dysfunction and ferroptosis are not only associated with mechanisms of neuronal death but also have significant clinical applications in the diagnosis and treatment of ischemia-associated CNS diseases. As a result, numerous studies have focused on this aspect and have suggested that the potential mechanism for preventing and treating ischemia-associated CNS diseases may involve the intervention of mitochondrial dysfunction and ferroptosis. This relationship is illustrated in Table 1.
Mitochondria are double-membrane organelles with functionally and structurally distinct outer and inner membranes, separating the intermembrane space from the matrix1. Continuous mitochondrial damage results in the dysfunction of energy metabolism; consequently, this leads to decreased ATP production, increased ROS burden, and reduced calcium buffering, all of which lead to neuronal loss characteristic of ischemia-associated CNS diseases. It has been reported that α-Lipoic acid (LA) supplementation effectively inhibited the high phosphorylation of Tau in several AD-related sites and reversed the cognitive decline of P301S Tau transgenic mice. When studying the death patterns of various neural cells in the brain tissues of mice treated with LA, it was found that Tau-induced iron overload, lipid peroxidation, and inflammation related to ferroptosis were significantly blocked by LA administration, suggesting that LA plays a role in inhibiting Tau hyperphosphorylation and ferroptosis (Y. H. Zhang, et al., 2018). Besides, LA, as a "mitochondrial nutraceutical", cannot only improve the antioxidant system and stimulate mitochondrial biogenesis but also directly prevent ROS production using the thiol group for redox regulation. LA also can be inducing the overexpression of Nrf2, which promotes improved mitochondrial function in neurons (Dos Santos, et al., 2019). Idebenone, an analog of the well-known antioxidant compound coenzyme Q10 (CoQ10), rescues cells against ferroptosis in PD by decreasing the striatal levels of the lipid peroxidation products and increasing the expression of GPX4 (Avci, et al., 2021). Meanwhile, idebenone can regulate the expression of the mitophagy-related outer membrane proteins VDAC1 and BNIP3, activate Parkin/PINK1 mitophagy, promote the degradation of damaged mitochondria, and reduce dopaminergic neuron damage, which improves behavioral disorders in PD mice (J. Yan, et al., 2022). Clinical phase therapeutic vatiquinone (EPI-743, α-tocotrienol quinone) has decreased the frequency and associated morbidity of mitochondrial disease. Further research indicates that EPI-743 reduces the incidence of epilepsy by protecting from ferroptosis under GSH depletion and iron overload conditions (Kahn-Kirby et al., 2019). In addition, RTA 408 activates Nrf2 via blocking Keap1, which prevents mitochondrial depolarization, ferroptosis, and lipid peroxidation in an in vitro model of seizure-like activity (Shekh-Ahmad, et al., 2019).
BID-mediated mitochondrial damage is a prerequisite for death signaling in neurons in severe oxidative stress paradigms. Recent studies have found that erastin-induced ferroptosis in neuronal cells was accompanied by BID transactivation to mitochondria, loss of mitochondrial membrane potential, enhanced mitochondrial fragmentation and decreased ATP levels. Bid knockout can prevent mitochondrial dysfunction in paradigms of ferroptosis. Vice versa, the ferroptosis inhibitor ferrostatin-1 blocked BID transactivation, mitochondrial fragmentation and the loss of mitochondrial membrane potential after glutamate exposure in neuronal cells, confirming that ferroptosis shares detrimental mechanisms of ROS formation upstream of BID-dependent mitochondrial demise (Neitemeier, et al., 2017). These findings show that mitochondrial transactivation of BID links ferroptosis to mitochondrial dysfunction as the final execution step in this paradigm of oxidative cell death. Nrf2 is involved in modulating ferroptosis related genes, including genes for GSH regulation (synthesis, cysteine supply via system xc−, GSH reductase, GPX4), NADPH regeneration which is crucial for Gpx4 activity (glucose 6-phosphate dehydrogenase, phosphogluconate dehydrogenase, malic enzyme) and iron regulation (iron export and reserve, heme synthesis and catabolism) (Kerins et al., 2018; J. M. Lee et al., 2003; Sasaki et al., 2002; K. C. Wu et al., 2011). Peroxisome proliferator-activated receptor gamma (PPARγ), a major regulator of lipid metabolism (W. Cai et al., 2018), can be activated by oxidized lipids relevant to the initiation of ferroptosis (Itoh et al., 2008), which reciprocally regulated with Nrf2, participating in the regulation of lipids (W. Cai, et al., 2018; C. Lee, 2017). Additionally, Nrf2 also plays an important role in modulating the mitochondrial quality control system including biogenesis via interaction with PGC-1α and mitophagy via a P62-dependent, PINK1/Parkin-independent mechanism (East et al., 2014; Gureev et al., 2019). Impaired mitochondrial function was observed in Nrf2 knockout mice, whereas activation of Nrf2 enhances mitochondrial function and resistance to stressors (Greco et al., 2010; Holmstrom et al., 2016; Neymotin et al., 2011). Although BID and Nrf2 have not been thoroughly studied, current evidence suggests that BID and Nrf2 are effective targets for intervention in ferroptosis and mitochondrial dysfunction. Further studies to determine the neuroprotective effects of BID and Nrf2 may be a beacon for future treatment of Ischemia-associated CNS diseases.
6. Conclusion and Perspectives
Cerebral ischemia, which includes inadequate blood flow to the brain and thus insufficient oxygen delivery (Levchenkova, et al., 2021), may initiate massive pathological processes, eventually result in different sorts of neuronal cell death, such as apoptosis, necrosis, and autophagy (G. C. Zhao et al., 2017).These processes impede essential sensory, cognitive functions, and motor functions (Stradecki-Cohan et al., 2017). Neurodegenerative diseases of CNS such as IS, AD, PD and epilepsy are pathological conditions that involve the loss of neuronal function or structure in the brain or spinal cord.
Mitochondria, intracellular organelles that regulate metabolism and produce ATP, undergo dysfunction during cerebral ischemia. Ferroptosis is a novel form of cell death, and its associated mechanisms, including iron overload, GSH depletion, and GPX4 inactivation, can exacerbate the damage caused by age-related ischemia-associated CNS diseases. Relevant research has demonstrated that mitochondrial dysfunction and ferroptosis interact with each other in the context of ischemia-associated CNS diseases (Abdalkader et al., 2018; Johnson et al., 2021; Saralkar et al., 2020). During cerebral ischemia, mitochondrial dysfunction leads to impaired mitophagy, resulting in the release of high amounts of reactive oxygen species (ROS) by damaged mitochondria and abnormal mitochondrial iron metabolism, which contributes to iron overload. These processes are closely related to the mechanism of ferroptosis. Ferroptosis is characterized by inspissated membrane densities, ruptured outer membrane, diminished or vanished mitochondrial inner membrane, mitochondrial oxidative stress, and mitochondrial depolarization. Furthermore, ferroptosis can also interfere with mitophagy via the P35 pathway. GSH plays a role in rescuing mitochondria from oxidative stress, but its depletion is a key factor in the failure of mitochondrial function recovery during ferroptosis. The interplay between mitochondrial dysfunction and ferroptosis often occurs in a vicious cycle, causing further injury. Considering the complex pathogenesis of Ischemia-associated CNS Diseases and the lack of treatment options, there is an instant need to develop new therapeutic approaches including traditional Chinese medicine formulations, to affect multiple targets and prevent neuronal death. Therefore, simultaneously pharmacological regulation of mitochondrial dysfunction and ferroptosis may be an effective strategy for treating Ischemia-associated CNS Diseases. However, several challenges need to be addressed before exploiting this interplay in a clinical setting. For instance, the roles of common targets in mitochondrial dysfunction and ferroptosis in the interplay require further investigation. Additionally, the exact mechanisms by which certain drugs, especially those from traditional Chinese medicine, modulate mitochondrial dysfunction and ferroptosis in Ischemia-associated CNS Diseases, remain to be fully elucidated. Addressing these issues will enhance our understanding of the molecular mechanisms underlying the interplay between ferroptosis and mitochondrial dysfunction and provide critical insights for intervention in Ischemia-associated CNS Diseases. Although there are still many gaps in the study of mitochondrial dysfunction and ferroptosis in Ischemia-associated CNS Diseases, it no denying that a complete analysis of the underlying mechanism of this interplay may provide new therapeutic opportunities for the treatment of Ischemia-associated CNS Diseases.
Author Contributions
Z. M. conceived the review, drafted partly and revised the manuscript. Z. M. and S. C. reviewed all the literature and draft the manuscript, S. C. and Z. M. gave some positive suggestions for the English language, J. G. and Z. M. provided valuable critical revisions of the manuscript. All authors approved the final version of the manuscript.
Acknowledgments
Thanks to Bo-yang Huang for technique assistance in figure production and beneficial suggestions in discussions. This work was supported by the National Natural Science Foundation of China (81774033, 81774174); Hunan Provincial Natural Science Foundation (2022JJ40303).
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Abbreviations
| AA | Arachidonoyl |
| ACSL4 | Acyl-CoA synthetase long-chain family member 4 |
| AdA | Adrenoyl |
| AD | Alzheimer’s disease |
| ADP | Adenosine diphosphate |
| AIF | Apoptosis-inducing factor |
| ALOX15 | Arachidonate 15-lipoxygenase |
| AMPA | Amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid |
| ANT | Adenine nucleotide translocator |
| APP | Amyloid β precursor protein |
| ATP | Adenosine triphosphate |
| Aβ | Amyloid β |
| BACE1 | Beta-site amyloid precursor protein cleaving enzyme 1 |
| BBB | Blood-brain barrier |
| CNS | Central Nervous System |
| CAC | Citric acid cycle |
| CISD1 | CDGSH iron sulfur domain 1 |
| Cp | Ceruloplasmin |
| CoQ10 | Coenzyme Q10 |
| CypD | Cyclophilin D |
| DAMPs | Damage-associated molecular patterns |
| DMT1 | Divalent metal transporter 1 |
| DNA | Deoxyribonucleic Acid |
| Endo G | Endonuclease G |
| FPN | Ferroportin |
| FTH1 | Ferritin heavy chain 1 |
| FTL | Ferritin light chain |
| FtMt | Mitochondrial ferritin |
| FXN | Frataxin |
| GSH | Glutathione |
| GSSG | Oxidized glutathione |
| GPX4 | Glutathione peroxidase 4 |
| IRP1 | Iron regulatory protein 1 |
| IS | Ischemic stroke |
| LA | α-Lipoic acid |
| LAMP1 | Lysosomal-associated membrane protein 1 |
| LPCAT3 | Lysophosphatidylcholine acyltransferase 3 |
| MDA | Malondialdehyde |
| MPTP | Mitochondrial permeability transition pore |
| MSCs | Mesenchymal stem cells |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NCOA4 | Nuclear receptor coactivator 4 |
| NLRP3 | The NOD LRR pyrin domain-containing protein 3 |
| NO | Nitric oxide |
| Nrf2 | Nuclear factor E2-related factor 2 |
| PD | Parkinson’s disease |
| PEs | Phosphatidylethanolamines |
| PGC-1 | Peroxisome proliferator-activated receptor-coactivator |
| Pi | Inorganic phosphate |
| PKC | Protein kinase C |
| PPARγ | Peroxisome proliferator-activated receptor gamma |
| PUFAs | Polyunsaturated fatty acids |
| RCD | Regulated cell death |
| RET | Reverse electron transport |
| ROS | Reactive oxygen species |
| Se | Selenium |
| SOD | Superoxide dismutase |
| Sp1 | Specificity protein 1 |
| SNpc | Substantia nigra pars compacta |
| TFAP2C | Transcription factor activating protein 2 gamma |
| tPA | Tissue plasminogen activator |
| TF | Transferrin |
| TFR1 | Transferrin receptor 1 |
| TLE | Temporal lobe epilepsy |
| TSG | Tetrahydroxy stilbene glycoside |
| VDACs | Voltage-dependent anion channels |
| 2Fe-2S | Iron-sulfur protein |
| 4-HNE | 4-hydroxynonenal |
| 8OHdG | 8 hydroxy 2V deoxyguanosine |
References
- Abdalkader, M.; Lampinen, R.; Kanninen, K.M.; Malm, T.M.; Liddell, J.R. Targeting Nrf2 to Suppress Ferroptosis and Mitochondrial Dysfunction in Neurodegeneration. Front. Neurosci. 2018, 12, 466. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Elsherbiny, N.M.; Haque, R.; Khan, M.B.; Ishrat, T.; Shah, Z.A.; Ali, M.; Jamal, A.; Katare, D.P.; Liou, G.I.; et al. Sesamin attenuates neurotoxicity in mouse model of ischemic brain stroke. NeuroToxicology 2014, 45, 100–110. [Google Scholar] [CrossRef]
- Aizenman, E.; Hartnett, K.A.; Reynoldst, I.J. Oxygen free radicals regulate NMDA receptor function via a redox modulatory site. Neuron 1990, 5, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, J.; Zielińska, M. Mechanisms of Excessive Extracellular Glutamate Accumulation in Temporal Lobe Epilepsy. Neurochem. Res. 2016, 42, 1724–1734. [Google Scholar] [CrossRef]
- Almeida, A.; Sánchez-Morán, I.; Rodríguez, C. Mitochondrial–nuclear p53 trafficking controls neuronal susceptibility in stroke. IUBMB Life 2021, 73, 582–591. [Google Scholar] [CrossRef] [PubMed]
- Amro, M.S.; Teoh, S.L.; Norzana, A.G.; Srijit, D. The potential role of herbal products in the treatment of Parkinson's disease. Clin Ter. 2018, 169, e23–e33. [Google Scholar] [CrossRef]
- Aoyama, K.; Nakaki, T. Glutathione in Cellular Redox Homeostasis: Association with the Excitatory Amino Acid Carrier 1 (EAAC1). Molecules 2015, 20, 8742–8758. [Google Scholar] [CrossRef]
- Asanuma, M.; Miyazaki, I. Glutathione and Related Molecules in Parkinsonism. Int. J. Mol. Sci. 2021, 22, 8689. [Google Scholar] [CrossRef]
- Avcı, B.; Günaydın, C.; Güvenç, T.; Yavuz, C.K.; Kuruca, N.; Bilge, S.S. Idebenone Ameliorates Rotenone-Induced Parkinson’s Disease in Rats Through Decreasing Lipid Peroxidation. Neurochem. Res. 2020, 46, 513–522. [Google Scholar] [CrossRef]
- Babaei, P. NMDA and AMPA receptors dysregulation in Alzheimer's disease. Eur. J. Pharmacol. 2021, 908, 174310. [Google Scholar] [CrossRef]
- Basit, F.; Van Oppen, L.M.P.E.; Schöckel, L.; Bossenbroek, H.M.; Van Emst-de Vries, S.E.; Hermeling, J.C.W.; Grefte, S.; Kopitz, C.; Heroult, M.; Willems, P.H.; et al. Mitochondrial complex I inhibition triggers a mitophagy-dependent ROS increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death Dis. 2017, 8, e2716. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, D.; Choi, M.E. The Emerging Role of Mitophagy in Kidney Diseases. JoLS, J. Life Sci. 2019, 1, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Boczek, T.; Lisek, M.; Ferenc, B.; Zylinska, L. Plasma membrane Ca2+-ATPase is a novel target for ketamine action. Biochem. Biophys. Res. Commun. 2015, 465, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Boison, D.; Steinhäuser, C. Epilepsy and astrocyte energy metabolism. Glia 2018, 66, 1235–1243. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; Pinton, P. The Mitochondrial Permeability Transition Pore and Cancer: Molecular Mechanisms Involved in Cell Death. Front. Oncol. 2014, 4, 302. [Google Scholar] [CrossRef]
- Bonora, M.; Pinton, P. A New Current for the Mitochondrial Permeability Transition. Trends Biochem. Sci. 2019, 44, 559–561. [Google Scholar] [CrossRef]
- Brinckmann, R.; Schnurr, K.; Heydeck, D.; Rosenbach, T.; Kolde, G.; Kuhn, H. Membrane translocation of 15-lipoxygenase in hematopoietic cells is calcium-dependent and activates the oxygenase activity of the enzyme. Blood 1998, 91, 64–74. [Google Scholar] [CrossRef]
- Bromont, C.; Marie, C.; Bralet, J. Increased lipid peroxidation in vulnerable brain regions after transient forebrain ischemia in rats. Stroke 1989, 20, 918–924. [Google Scholar] [CrossRef]
- Brütsch, S.H.; Wang, C.C.; Li, L.; Stender, H.; Neziroglu, N.; Richter, C.; Kuhn, H.; Borchert, A.; Maiorino, M.; Conrad, M.; et al. Expression of Inactive Glutathione Peroxidase 4 Leads to Embryonic Lethality, and Inactivation of theAlox15Gene Does Not Rescue Such Knock-In Mice. Antioxidants Redox Signal. 2015, 22, 281–293. [Google Scholar] [CrossRef]
- Cai, W. , Yang, T., Liu, H., Han, L., Zhang, K., Hu, X. Peroxisome proliferator-activated receptor gamma (PPARgamma): A master gatekeeper in CNS injury and repair. Prog Neurobiol. 2018, 163–164, 27–58. [Google Scholar] [CrossRef]
- Cai, Y.; Yang, Z. Ferroptosis and Its Role in Epilepsy. Front. Cell. Neurosci. 2021, 15, 696889. [Google Scholar] [CrossRef] [PubMed]
- Calvelage, S.; Tammiranta, N.; Nokireki, T.; Gadd, T.; Eggerbauer, E.; Zaeck, L.M.; Potratz, M.; Wylezich, C.; Höper, D.; Müller, T.; et al. Genetic and Antigenetic Characterization of the Novel Kotalahti Bat Lyssavirus (KBLV). Viruses 2021, 13, 69. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.Y.; Dixon, S.J. Mechanisms of ferroptosis. Cell Mol. Life Sci. 2016, 73, 2195–2209. [Google Scholar] [CrossRef]
- Carocci, A.; Catalano, A.; Sinicropi, M.S.; Genchi, G. Oxidative stress and neurodegeneration: The involvement of iron. BioMetals 2018, 31, 715–735. [Google Scholar] [CrossRef]
- Chen, C.T.; Green, J.T.; Orr, S.K.; Bazinet, R.P. Regulation of brain polyunsaturated fatty acid uptake and turnover. Prostaglandins, Leukot. Essent. Fat. Acids 2008, 79, 85–91. [Google Scholar] [CrossRef]
- Chen, S.D. , Lin, T. K., Lin, J.W., Yang, D.I., Lee, S.Y., Shaw, F.Z. Activation of calcium/calmodulin-dependent protein kinase IV and peroxisome proliferator-activated receptor gamma coactivator-1alpha signaling pathway protects against neuronal injury and promotes mitochondrial biogenesis in the hippocampal CA1 subfield after transient global ischemia. J. Neurosci. Res. 2010, 88, 3144–3154. [Google Scholar] [CrossRef]
- Cheng, A.; Wan, R.; Yang, J.-L.; Kamimura, N.; Son, T.G.; Ouyang, X.; Luo, Y.; Okun, E.; Mattson, M.P. Involvement of PGC-1α in the formation and maintenance of neuronal dendritic spines. Nat. Commun. 2012, 3, 1250. [Google Scholar] [CrossRef]
- Cheng, J.; Wang, F.; Yu, D.-F.; Wu, P.-F.; Chen, J.-G. The cytotoxic mechanism of malondialdehyde and protective effect of carnosine via protein cross-linking/mitochondrial dysfunction/reactive oxygen species/MAPK pathway in neurons. Eur. J. Pharmacol. 2011, 650, 184–194. [Google Scholar] [CrossRef]
- Chi, S.I.; Wang, C.K.; Chen, J.J.; Chau, L.Y.; Lin, T.N. Differential regulation of H- and L-ferritin messenger RNA subunits, ferritin protein and iron following focal cerebral ischemia-reperfusion. Neuroscience 2000, 100, 475–484. [Google Scholar] [CrossRef]
- Chifman, J.; Laubenbacher, R.; Torti, S.V. A Systems Biology Approach to Iron Metabolism. 2014, 844, 201–225. [Google Scholar] [CrossRef]
- Chung, K.; Chen, Y.; Juan, Y.; Hsu, C.; Nakahira, K.; Huang, Y.; Lin, M.; Wu, S.; Shih, J.; Chang, Y.; et al. Multi-kinase framework promotes proliferation and invasion of lung adenocarcinoma through activation of dynamin-related protein 1. Mol. Oncol. 2020, 15, 560–578. [Google Scholar] [CrossRef] [PubMed]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Colombini, M. VDAC structure, selectivity, and dynamics. Biochim. Biophys. Acta 2012, 1818, 1457–1465. [Google Scholar] [CrossRef] [PubMed]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J.; Siggens, L.; Figg, N.; Bennett, M.; Foo, R.; Chastain, P.D.; Nakamura, J.; et al. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef]
- Cui, J.; Holmes, E.H.; Greene, T.G.; Liu, P.K. Oxidative DNA damage precedes DNA fragmentation after experimental stroke in rat brain. FASEB J. 2000, 14, 955–967. [Google Scholar] [CrossRef]
- D'Herde, K.; Krysko, D.V. Ferroptosis: Oxidized PEs trigger death. Nat. Chem. Biol. 2017, 13, 4–5. [Google Scholar] [CrossRef]
- DeGregorio-Rocasolano, N.; Martí-Sistac, O.; Gasull, T. Deciphering the Iron Side of Stroke: Neurodegeneration at the Crossroads Between Iron Dyshomeostasis, Excitotoxicity, and Ferroptosis. Front. Neurosci. 2019, 13, 85. [Google Scholar] [CrossRef]
- DeHart, D.N.; Fang, D.; Heslop, K.; Li, L.; Lemasters, J.J.; Maldonado, E.N. Opening of voltage dependent anion channels promotes reactive oxygen species generation, mitochondrial dysfunction and cell death in cancer cells. Biochem. Pharmacol. 2017, 148, 155–162. [Google Scholar] [CrossRef]
- DeHart, D.N.; Lemasters, J.J.; Maldonado, E.N. Erastin-Like Anti-Warburg Agents Prevent Mitochondrial Depolarization Induced by Free Tubulin and Decrease Lactate Formation in Cancer Cells. SLAS Discov. Adv. Sci. Drug Discov. 2017, 23, 23–33. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Elmoselhi, A.B.; Hata, T.; Makino, N. Status of myocardial antioxidants in ischemia–reperfusion injury. Cardiovasc. Res. 2000, 47, 446–456. [Google Scholar] [CrossRef]
- Diao, X.; Zhou, Z.; Xiang, W.; Jiang, Y.; Tian, N.; Tang, X.; Chen, S.; Wen, J.; Chen, M.; Liu, K.; et al. Glutathione alleviates acute intracerebral hemorrhage injury via reversing mitochondrial dysfunction. Brain Res. 2020, 1727, 146514. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, R.B.; Bradley, W.G., Jr. Iron accumulation in the basal ganglia following severe ischemic-anoxic insults in children. Radiology 1988, 168, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Do Van, B. , Gouel, F., Jonneaux, A., Timmerman, K., Gele, P., Petrault, M. Ferroptosis, a newly characterized form of cell death in Parkinson's disease that is regulated by PKC. Neurobiol Dis. 2016, 94, 169–178. [Google Scholar] [CrossRef]
- Domínguez, C.; Delgado, P.; Vilches, A.; Martín-Gallán, P.; Ribó, M.; Santamarina, E.; Molina, C.; Corbeto, N.; Rodríguez-Sureda, V.; Rosell, A.; et al. Oxidative Stress After Thrombolysis-Induced Reperfusion in Human Stroke. Stroke 2010, 41, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.-P.; Cheng, X.; Mills, E.; Delling, M.; Wang, F.; Kurz, T.; Xu, H. The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel. Nature 2008, 455, 992–996. [Google Scholar] [CrossRef]
- Dos Santos, S.M.; Romeiro, C.F.R.; Rodrigues, C.A.; Cerqueira, A.R.L.; Monteiro, M.C. Mitochondrial Dysfunction and Alpha-Lipoic Acid: Beneficial or Harmful in Alzheimer's Disease? Oxid. Med. Cell Longev. 2019, 2019, 8409329. [Google Scholar] [CrossRef]
- Doyle, K.P.; Simon, R.P.; Stenzel-Poore, M.P. Mechanisms of ischemic brain damage. Neuropharmacology 2008, 55, 310–318. [Google Scholar] [CrossRef]
- Dröge, W. Free Radicals in the Physiological Control of Cell Function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef]
- Du, J.; Zhou, Y.; Li, Y.; Xia, J.; Chen, Y.; Chen, S.; Wang, X.; Sun, W.; Wang, T.; Ren, X.; et al. Identification of Frataxin as a regulator of ferroptosis. Redox Biol. 2020, 32, 101483. [Google Scholar] [CrossRef]
- East, D.A.; Fagiani, F.; Crosby, J.; Georgakopoulos, N.D.; Bertrand, H.; Schaap, M. PMI: A DeltaPsim independent pharmacological regulator of mitophagy. Chem Biol. 2014, 21, 1585–1596. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.S.; Holzbaur, E.L. Quality Control in Neurons: Mitophagy and Other Selective Autophagy Mechanisms. J. Mol. Biol. 2020, 432, 240–260. [Google Scholar] [CrossRef]
- Feng, J.; Chen, X.; Guan, B.; Li, C.; Qiu, J.; Shen, J. Inhibition of Peroxynitrite-Induced Mitophagy Activation Attenuates Cerebral Ischemia-Reperfusion Injury. Mol. Neurobiol. 2018, 55, 6369–6386. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Chen, X.; Shen, J. Reactive nitrogen species as therapeutic targets for autophagy: Implication for ischemic stroke. Expert Opin. Ther. Targets 2017, 21, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Hu, W.; Marnett, L.J.; Tang, M.-S. Malondialdehyde, a major endogenous lipid peroxidation product, sensitizes human cells to UV- and BPDE-induced killing and mutagenesis through inhibition of nucleotide excision repair. Mutat. Res. Mol. Mech. Mutagen. 2006, 601, 125–136. [Google Scholar] [CrossRef]
- Ferris, C.D.; Jaffrey, S.R.; Sawa, A.; Takahashi, M.; Brady, S.D.; Barrow, R.K.; Tysoe, S.A.; Wolosker, H.; Barañano, D.E.; Doré, S.; et al. Haem oxygenase-1 prevents cell death by regulating cellular iron. Nature 1999, 1, 152–157. [Google Scholar] [CrossRef]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef]
- Forcina, G.C.; Dixon, S.J. GPX4 at the Crossroads of Lipid Homeostasis and Ferroptosis. PROTEOMICS 2019, 19, e1800311. [Google Scholar] [CrossRef]
- Franco-Iborra, S.; Vila, M.; Perier, C. Mitochondrial Quality Control in Neurodegenerative Diseases: Focus on Parkinson's Disease and Huntington's Disease. Front. Neurosci. 2018, 12, 342. [Google Scholar] [CrossRef]
- Fucci, L.; Oliver, C.N.; Coon, M.J.; Stadtman, E.R. Inactivation of key metabolic enzymes by mixed-function oxidation reactions: Possible implication in protein turnover and ageing. Proc. Natl. Acad. Sci. USA 1983, 80, 1521–1525. [Google Scholar] [CrossRef]
- Fuhrmann, D.C.; Mondorf, A.; Beifuß, J.; Jung, M.; Brüne, B. Hypoxia inhibits ferritinophagy, increases mitochondrial ferritin, and protects from ferroptosis. Redox Biol. 2020, 36, 101670. [Google Scholar] [CrossRef]
- Fulton, R.E.; Pearson-Smith, J.N.; Huynh, C.Q.; Fabisiak, T.; Liang, L.-P.; Aivazidis, S.; High, B.A.; Buscaglia, G.; Corrigan, T.; Valdez, R.; et al. Neuron-specific mitochondrial oxidative stress results in epilepsy, glucose dysregulation and a striking astrocyte response. Neurobiol. Dis. 2021, 158, 105470. [Google Scholar] [CrossRef] [PubMed]
- Gan, B. Mitochondrial regulation of ferroptosis. J. Cell Biol. 2021, 220, e202105043. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of Mitochondria in Ferroptosis. Mol. Cell 2018, 73, 354–363. [Google Scholar] [CrossRef]
- Garton, T.; Keep, R.F.; Hua, Y.; Xi, G. Brain iron overload following intracranial haemorrhage. Stroke Vasc. Neurol. 2016, 1, 172–184. [Google Scholar] [CrossRef]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochemical and Biophysical Research Communications. 2017, 482, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Geldenhuys, W.J.; Benkovic, S.A.; Lin, L.; Yonutas, H.M.; Crish, S.D.; Sullivan, P.G.; Darvesh, A.S.; Brown, C.M.; Richardson, J.R. MitoNEET (CISD1) Knockout Mice Show Signs of Striatal Mitochondrial Dysfunction and a Parkinson’s Disease Phenotype. ACS Chem. Neurosci. 2017, 8, 2759–2765. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.; Pan, J.; Shen, Q.; Li, M.; Peng, Y. Mitochondrial dysfunction induces NLRP3 inflammasome activation during cerebral ischemia/reperfusion injury. J. Neuroinflam. 2018, 15, 1–17. [Google Scholar] [CrossRef]
- Goulay, R.; Romo, L.M.; Hol, E.M.; Dijkhuizen, R.M. From Stroke to Dementia: A Comprehensive Review Exposing Tight Interactions Between Stroke and Amyloid-β Formation. Transl. Stroke Res. 2019, 11, 601–614. [Google Scholar] [CrossRef]
- Gray, W.A.; Feiring, A.; Cioppi, M.; Hibbard, R.; Gray, B.; Khatib, Y.; Jessup, D.; Bachinsky, W.; Rivera, E.; Tauth, J.; et al. Self-Expanding Nitinol Stent for the Treatment of Atherosclerotic Lesions in the Superficial Femoral Artery (STROLL): 1-Year Outcomes. J. Vasc. Interv. Radiol. 2014, 26, 21–28. [Google Scholar] [CrossRef]
- Greco, T.; Fiskum, G. Brain mitochondria from rats treated with sulforaphane are resistant to redox-regulated permeability transition. J. Bioenerg. Biomembr. 2010, 42, 491–497. [Google Scholar] [CrossRef]
- Grune, T.; Reinheckel, T.; Joshi, M.; Davies, K.J.A. Proteolysis in Cultured Liver Epithelial Cells during Oxidative Stress. Perspect. Surg. 1995, 270, 2344–2351. [Google Scholar] [CrossRef]
- Guan, R.; Zou, W.; Dai, X.; Yu, X.; Liu, H.; Chen, Q.; Teng, W. Mitophagy, a potential therapeutic target for stroke. J. Biomed. Sci. 2018, 25, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Guéraud, F.; Peiro, G.; Bernard, H.; Alary, J.; Créminon, C.; Debrauwer, L.; Rathahao, E.; Drumare, M.-F.; Canlet, C.; Wal, J.-M.; et al. Enzyme immunoassay for a urinary metabolite of 4-hydroxynonenal as a marker of lipid peroxidation. Free. Radic. Biol. Med. 2006, 40, 54–62. [Google Scholar] [CrossRef]
- Guo, C.; Ma, Y.-Y. Calcium Permeable-AMPA Receptors and Excitotoxicity in Neurological Disorders. Front. Neural Circuits 2021, 15, 711564. [Google Scholar] [CrossRef] [PubMed]
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N. Regulation of Mitochondrial Biogenesis as a Way for Active Longevity: Interaction Between the Nrf2 and PGC-1α Signaling Pathways. Front. Genet. 2019, 10, 435. [Google Scholar] [CrossRef]
- Gustafsson, Å.B.; Gottlieb, R.A. Heart mitochondria: Gates of life and death. Cardiovasc. Res. 2007, 77, 334–343. [Google Scholar] [CrossRef]
- Gutteridge, J.M.C.; Richmond, R.; Halliwell, B. Inhibition of the iron-catalysed formation of hydroxyl radicals from superoxide and of lipid peroxidation by desferrioxamine. Biochem. J. 1979, 184, 469–472. [Google Scholar] [CrossRef] [PubMed]
- Hussein, I.H.; Chams, N.; Chams, S.; El Sayegh, S.; Badran, R.; Raad, M.; Gerges-Geagea, A.; Leone, A.; Jurjus, A. Vaccines Through Centuries: Major Cornerstones of Global Health. Front. Public Health 2015, 3, 269. [Google Scholar] [CrossRef]
- Hamai, A.; Mehrpour, M. [Autophagy and iron homeostasis]. Med. Sci. 2017, 33, 260–267. [Google Scholar] [CrossRef]
- Hambright, W.S.; Fonseca, R.S.; Chen, L.; Na, R.; Ran, Q. Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol. 2017, 12, 8–17. [Google Scholar] [CrossRef]
- Han, H.; Xu, D.; Liu, C.; Claesson, H.-E.; Björkholm, M.; Sjöberg, J. Interleukin-4-Mediated 15-Lipoxygenase-1 Trans-Activation Requires UTX Recruitment and H3K27me3 Demethylation at the Promoter in A549 Cells. PLoS ONE 2014, 9, e85085. [Google Scholar] [CrossRef] [PubMed]
- Hanada, T. Ionotropic Glutamate Receptors in Epilepsy: A Review Focusing on AMPA and NMDA Receptors. Biomolecules 2020, 10, 464. [Google Scholar] [CrossRef] [PubMed]
- Hanafy, K.A.; Gomes, J.A.; Selim, M. Rationale and Current Evidence for Testing Iron Chelators for Treating Stroke. Curr. Cardiol. Rep. 2019, 21, 20. [Google Scholar] [CrossRef] [PubMed]
- Hao, S.; Liang, B.; Huang, Q.; Dong, S.; Wu, Z.; He, W.; Shi, M. Metabolic networks in ferroptosis (Review). Oncol. Lett. 2018, 15, 5405–5411. [Google Scholar] [CrossRef]
- Hao, W.; Fengli, W.; Na, T.; Ting, Z.; Weihua, G. The Multifaceted Regulation of Mitochondria in Ferroptosis. Life 2021, 11, 222. [Google Scholar] [CrossRef]
- He, Z.; Ning, N.; Zhou, Q.; Khoshnam, S.E.; Farzaneh, M. Mitochondria as a therapeutic target for ischemic stroke. Free. Radic. Biol. Med. 2019, 146, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to Tango: Regulation of Mammalian Iron Metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Hinman, A.; Holst, C.R.; Latham, J.C.; Bruegger, J.J.; Ulas, G.; McCusker, K.P.; Amagata, A.; Davis, D.; Hoff, K.G.; Kahn-Kirby, A.H.; et al. Vitamin E hydroquinone is an endogenous regulator of ferroptosis via redox control of 15-lipoxygenase. PLoS ONE 2018, 13, e0201369. [Google Scholar] [CrossRef] [PubMed]
- Hodge, T.; Colombini, M. Regulation of Metabolite Flux through Voltage-Gating of VDAC Channels. J. Membr. Biol. 1997, 157, 271–279. [Google Scholar] [CrossRef]
- Holmström, K.M.; Kostov, R.V.; Dinkova-Kostova, A.T. The multifaceted role of Nrf2 in mitochondrial function. Curr. Opin. Toxicol. 2016, 1, 80–91. [Google Scholar] [CrossRef]
- Huang, J.; Chen, S.; Hu, L.; Niu, H.; Sun, Q.; Li, W.; Tan, G.; Li, J.; Jin, L.; Lyu, J.; et al. Mitoferrin-1 is Involved in the Progression of Alzheimer’s Disease Through Targeting Mitochondrial Iron Metabolism in a Caenorhabditis elegans Model of Alzheimer’s Disease. Neuroscience 2018, 385, 90–101. [Google Scholar] [CrossRef]
- Huang, Y.; Xu, W.; Zhou, R. NLRP3 inflammasome activation and cell death. Cell. Mol. Immunol. 2021, 18, 2114–2127. [Google Scholar] [CrossRef]
- Imai, H.; Hirao, F.; Sakamoto, T.; Sekine, K.; Mizukura, Y.; Saito, M.; Kitamoto, T.; Hayasaka, M.; Hanaoka, K.; Nakagawa, Y. Early embryonic lethality caused by targeted disruption of the mouse PHGPx gene. Biochem. Biophys. Res. Commun. 2003, 305, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef] [PubMed]
- Itoh, T.; Fairall, L.; Amin, K.; Inaba, Y.; Szanto, A.; Balint, B.L. Structural basis for the activation of PPARgamma by oxidized fatty acids. Nat Struct Mol Biol. 2008, 15, 924–931. [Google Scholar] [CrossRef]
- Jena, N.R. DNA damage by reactive species: Mechanisms, mutation and repair. J. Biosci. 2012, 37, 503–517. [Google Scholar] [CrossRef]
- Jiang, L.; Hu, X. Positive Effect of α-Asaronol on the Incidence of Post-Stroke Epilepsy for Rat with Cerebral Ischemia-Reperfusion Injury. Molecules 2022, 27, 1984. [Google Scholar] [CrossRef]
- Jiang, X.; Andjelkovic, A.V.; Zhu, L.; Yang, T.; Bennett, M.V.L.; Chen, J.; Keep, R.F.; Shi, Y. Blood-brain barrier dysfunction and recovery after ischemic stroke. Prog. Neurobiol. 2018, 163–164, 144–171. [Google Scholar] [CrossRef]
- Jin, G.; Arai, K.; Murata, Y.; Wang, S.; Stins, M.F.; Lo, E.H. Protecting against cerebrovascular injury: Contributions of 12/15-lipoxygenase to edema formation after transient focal ischemia. Stroke 2008, 39, 2538–2543. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.; Mercado-Ayon, E.; Mercado-Ayon, Y.; Na Dong, Y.; Halawani, S.; Ngaba, L.; Lynch, D.R. Mitochondrial dysfunction in the development and progression of neurodegenerative diseases. Arch. Biochem. Biophys. 2020, 702, 108698. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.F.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized Arachidonic and Adrenic PEs Navigate Cells to Ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Kahn-Kirby, A.H.; Amagata, A.; Maeder, C.I.; Mei, J.J.; Sideris, S.; Kosaka, Y.; Hinman, A.; Malone, S.A.; Bruegger, J.J.; Wang, L.; et al. Targeting ferroptosis: A novel therapeutic strategy for the treatment of mitochondrial disease-related epilepsy. PLoS ONE 2019, 14, e0214250. [Google Scholar] [CrossRef]
- Kalani, K.; Yan, S.F.; Yan, S.S. Mitochondrial permeability transition pore is a potential drug target for neurodegeneration. Drug Discovery Today 2018, 23, 1983–1989. [Google Scholar] [CrossRef] [PubMed]
- Kaye, D.M.; Petrie, M.C.; McKenzie, S.; Hasenfuβ, G.; Malek, F.; Post, M.; Doughty, R.N.; Trochu, J.-N.; Gustafsson, F.; Lang, I.; et al. Impact of an interatrial shunt device on survival and heart failure hospitalization in patients with preserved ejection fraction. ESC Hearth Fail. 2018, 6, 62–69. [Google Scholar] [CrossRef]
- Kerins, M.J.; Ooi, A. The Roles of NRF2 in Modulating Cellular Iron Homeostasis. Antioxid. Redox Signal. 2018, 29, 1756–1773. [Google Scholar] [CrossRef]
- Khalifa, A.R.M.; Abdel-Rahman, E.A.; Mahmoud, A.M.; Ali, M.H.; Noureldin, M.; Saber, S.H.; Mohsen, M.; Ali, S.S. Sex-specific differences in mitochondria biogenesis, morphology, respiratory function, and ROS homeostasis in young mouse heart and brain. Physiol. Rep. 2017, 5, e13125. [Google Scholar] [CrossRef]
- Khoshnam, S.E.; Winlow, W.; Farzaneh, M. The Interplay of MicroRNAs in the Inflammatory Mechanisms Following Ischemic Stroke. J. Neuropathol. Exp. Neurol. 2017, 76, 548–561. [Google Scholar] [CrossRef]
- Khoshnam, S.E.; Winlow, W.; Farzaneh, M.; Farbood, Y.; Moghaddam, H.F. Pathogenic mechanisms following ischemic stroke. Neurol. Sci. 2017, 38, 1167–1186. [Google Scholar] [CrossRef]
- Koch, R.E.; Josefson, C.C.; Hill, G.E. Mitochondrial function, ornamentation, and immunocompetence. Biol. Rev. Camb. Philos Soc. 2017, 92, 1459–1474. [Google Scholar] [CrossRef] [PubMed]
- Krainz, T.; Gaschler, M.M.; Lim, C.; Sacher, J.R.; Stockwell, B.R.; Wipf, P. A Mitochondrial-Targeted Nitroxide Is a Potent Inhibitor of Ferroptosis. ACS Central Sci. 2016, 2, 653–659. [Google Scholar] [CrossRef]
- Kurzatkowski, D.M.; Trombetta, L.D. Maneb causes pro-oxidant effects in the hippocampus of Nrf2 knockout mice. Environ. Toxicol. Pharmacol. 2013, 36, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Kwong, J.Q.; Molkentin, J.D. Physiological and Pathological Roles of the Mitochondrial Permeability Transition Pore in the Heart. Cell Metab. 2015, 21, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Lai, T.K.Y.; Zhai, D.; Su, P.; Jiang, A.; Boychuk, J.; Liu, F. The receptor-receptor interaction between mGluR1 receptor and NMDA receptor: A potential therapeutic target for protection against ischemic stroke. FASEB J. 2019, 33, 14423–14439. [Google Scholar] [CrossRef] [PubMed]
- Lan, B.; Ge, J.-W.; Cheng, S.-W.; Zheng, X.-L.; Liao, J.; He, C.; Rao, Z.-Q.; Wang, G.-Z. Extract of Naotaifang, a compound Chinese herbal medicine, protects neuron ferroptosis induced by acute cerebral ischemia in rats. J. Integr. Med. 2020, 18, 344–350. [Google Scholar] [CrossRef]
- Lane, D.J.R.; Ayton, S.; Bush, A.I. Iron and Alzheimer’s Disease: An Update on Emerging Mechanisms. J. Alzheimer Dis. 2018, 64, S379–S395. [Google Scholar] [CrossRef] [PubMed]
- Lee, C. Collaborative Power of Nrf2 and PPARgamma Activators against Metabolic and Drug-Induced Oxidative Injury. Oxid. Med. Cell Longev. 2017, 2017, 1378175. [Google Scholar] [CrossRef]
- Lee, J.-M.; Calkins, M.J.; Chan, K.; Kan, Y.W.; Johnson, J.A. Identification of the NF-E2-related Factor-2-dependent Genes Conferring Protection against Oxidative Stress in Primary Cortical Astrocytes Using Oligonucleotide Microarray Analysis. PEDIATRICS 2003, 278, 12029–12038. [Google Scholar] [CrossRef]
- Lei, P.; Bai, T.; Sun, Y. Mechanisms of Ferroptosis and Relations With Regulated Cell Death: A Review. Front. Physiol. 2019, 10, 139. [Google Scholar] [CrossRef]
- Levchenkova, O.S.; Novikov, V.E.; Korneva, Y.S.; Dorosevich, A.E.; Parfenov, E.A. Combined Preconditioning Reduces the Negative Influence of Cerebral Ischemia on the Morphofunctional Condition of CNS. Bull. Exp. Biol. Med. 2021, 171, 489–493. [Google Scholar] [CrossRef]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W. The cystine/glutamate antiporter system x(c)(-) in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox. Signal. 2013, 18, 522–555. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, Y.-W.; Zhao, J.-Y.; Liu, Y.-Z.; Holscher, C. Quantitative analysis of iron concentration and expression of ferroportin 1 in the cortex and hippocampus of rats induced by cerebral ischemia. J. Clin. Neurosci. 2009, 16, 1466–1472. [Google Scholar] [CrossRef]
- Yu, W.-F.; Li, P.-Y.; Wang, X.; Stetler, R.; Chen, J. Anti-inflammatory signaling: The point of convergence for medical gases in neuroprotection against ischemic stroke. Med. Gas Res. 2016, 6, 227–231. [Google Scholar] [CrossRef]
- Li, S.; Zheng, J.; Carmichael, S.T. Increased oxidative protein and DNA damage but decreased stress response in the aged brain following experimental stroke. Neurobiol. Dis. 2005, 18, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Li, W. Targeting oxidative stress for the treatment of ischemic stroke: Upstream and downstream therapeutic strategies. Brain Circ. 2016, 2, 153–163. [Google Scholar] [CrossRef]
- Li, Y.; Zhong, W.; Jiang, Z.; Tang, X. New progress in the approaches for blood–brain barrier protection in acute ischemic stroke. Brain Res. Bull. 2019, 144, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Lipscomb, D.C.; Gorman, L.G.; Traystman, R.J.; Hurn, P.D. Low Molecular Weight Iron in Cerebral Ischemic Acidosis In Vivo. Stroke 1998, 29, 487–493. [Google Scholar] [CrossRef]
- Liu, H.; Hua, Y.; Keep, R.F.; Xi, G. Brain Ceruloplasmin Expression After Experimental Intracerebral Hemorrhage and Protection Against Iron-Induced Brain Injury. Transl. Stroke Res. 2018, 10, 112–119. [Google Scholar] [CrossRef]
- Liu, J.; Liu, W.; Li, R.; Yang, H. Mitophagy in Parkinson's Disease: From Pathogenesis to Treatment. Cells 2019, 8, 712. [Google Scholar] [CrossRef]
- Liu, J.-H.; Wang, T.-W.; Lin, Y.-Y.; Ho, W.-C.; Tsai, H.-C.; Chen, S.-P.; Lin, A.M.-Y.; Liu, T.-Y.; Wang, H.-T. Acrolein is involved in ischemic stroke-induced neurotoxicity through spermidine/spermine-N1-acetyltransferase activation. Exp. Neurol. 2019, 323, 113066. [Google Scholar] [CrossRef]
- Liu, X.; Feng, Z.; Du, L.; Huang, Y.; Ge, J.; Deng, Y.; Mei, Z. The Potential Role of MicroRNA-124 in Cerebral Ischemia Injury. Int. J. Mol. Sci. 2019, 21, 120. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, Z.; Li, B.; Yao, H.; Zarka, M.; Welch, J.; Sachdev, P.; Bridge, W.; Braidy, N. Supplementation with γ-glutamylcysteine (γ-GC) lessens oxidative stress, brain inflammation and amyloid pathology and improves spatial memory in a murine model of AD. Neurochem. Int. 2020, 144, 104931. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, S.; Grigoletto, J.; Bernis, M.E.; Pesch, V.; Ma, L.; Reithofer, S.; Tamgüney, G. Ischemic stroke causes Parkinson’s disease-like pathology and symptoms in transgenic mice overexpressing alpha-synuclein. Acta Neuropathol. Commun. 2022, 10, 1–17. [Google Scholar] [CrossRef]
- Lu, J.; Xu, F.; Lu, H. LncRNA PVT1 regulates ferroptosis through miR-214-mediated TFR1 and p53. Life Sci. 2020, 260, 118305. [Google Scholar] [CrossRef]
- Luo, T.; Park, Y.; Sun, X.; Liu, C.; Hu, B. Protein Misfolding, Aggregation, and Autophagy After Brain Ischemia. Transl. Stroke Res. 2013, 4, 581–588. [Google Scholar] [CrossRef] [PubMed]
- MacManus, J.P.; Fliss, H.; Preston, E.; Rasquinha, I.; Tuor, U. Cerebral Ischemia Produces Laddered DNA Fragments Distinct from Cardiac Ischemia and Archetypal Apoptosis. J. Cereb. Blood Flow Metab. 1999, 19, 502–510. [Google Scholar] [CrossRef]
- Maher, P.; van Leyen, K.; Dey, P.N.; Honrath, B.; Dolga, A.; Methner, A. The role of Ca2+ in cell death caused by oxidative glutamate toxicity and ferroptosis. Cell Calcium 2018, 70, 47–55. [Google Scholar] [CrossRef]
- Maldonado, E.N.; Lemasters, J.J. Warburg Revisited: Regulation of Mitochondrial Metabolism by Voltage-Dependent Anion Channels in Cancer Cells. Experiment 2012, 342, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Malpartida, A.B.; Williamson, M.; Narendra, D.P.; Wade-Martins, R.; Ryan, B.J. Mitochondrial Dysfunction and Mitophagy in Parkinson’s Disease: From Mechanism to Therapy. Trends Biochem. Sci. 2021, 46, 329–343. [Google Scholar] [CrossRef]
- Mao, X.-Y.; Zhou, H.-H.; Jin, W.-L. Ferroptosis Induction in Pentylenetetrazole Kindling and Pilocarpine-Induced Epileptic Seizures in Mice. Front. Neurosci. 2019, 13, 721. [Google Scholar] [CrossRef]
- Matsumoto, S.; Murozono, M.; Kanazawa, M.; Nara, T.; Ozawa, T.; Watanabe, Y. Edaravone and cyclosporine A as neuroprotective agents for acute ischemic stroke. Acute Med. Surg. 2018, 5, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Matsuo-Tezuka, Y.; Sasaki, Y.; Iwai, T.; Kurasawa, M.; Yorozu, K.; Tashiro, Y. T2 ( *) Relaxation Time Obtained from Magnetic Resonance Imaging of the Liver Is a Useful Parameter for Use in the Construction of a Murine Model of Iron Overload. Contrast Media Mol. Imaging 2019, 2019, 7463047. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S.L.; Kumari, S.; Mendelev, N.; Li, P.A. Selenium preserves mitochondrial function, stimulates mitochondrial biogenesis, and reduces infarct volume after focal cerebral ischemia. BMC Neurosci. 2012, 13, 79–79. [Google Scholar] [CrossRef] [PubMed]
- Mittler, R.; Darash-Yahana, M.; Sohn, Y.S.; Bai, F.; Song, L.; Cabantchik, I.Z.; Jennings, P.A.; Onuchic, J.N.; Nechushtai, R. NEET Proteins: A New Link Between Iron Metabolism, Reactive Oxygen Species, and Cancer. Antioxidants Redox Signal. 2019, 30, 1083–1095. [Google Scholar] [CrossRef]
- Liu, K.-Y.; Mo, Y.; Sun, Y.-Y. Autophagy and inflammation in ischemic stroke. Neural Regen. Res. 2020, 15, 1388–1396. [Google Scholar] [CrossRef]
- Morrison, R.S.; Kinoshita, Y. The role of p53 in neuronal cell death. Cell Death Differ. 2000, 7, 868–879. [Google Scholar] [CrossRef]
- Mueller, S.G.; Trabesinger, A.H.; Boesiger, P.; Wieser, H.G. Brain glutathione levels in patients with epilepsy measured by in vivo 1H-MRS. Neurology 2001, 57, 1422–1427. [Google Scholar] [CrossRef]
- Neitemeier, S.; Jelinek, A.; Laino, V.; Hoffmann, L.; Eisenbach, I.; Eying, R.; Ganjam, G.K.; Dolga, A.M.; Oppermann, S.; Culmsee, C. BID links ferroptosis to mitochondrial cell death pathways. Redox Biol. 2017, 12, 558–570. [Google Scholar] [CrossRef]
- Neymotin, A.; Calingasan, N.Y.; Wille, E.; Naseri, N.; Petri, S.; Damiano, M. Neuroprotective effect of Nrf2/ARE activators, CDDO ethylamide and CDDO trifluoroethylamide, in a mouse model of amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2011, 51, 88–96. [Google Scholar] [CrossRef]
- Nguyen, D.; Alavi, M.V.; Kim, K.-Y.; Kang, T.; Scott, R.T.; Noh, Y.H.; Lindsey, J.D.; Wissinger, B.; Ellisman, M.H.; Weinreb, R.N.; et al. A new vicious cycle involving glutamate excitotoxicity, oxidative stress and mitochondrial dynamics. Cell Death Dis. 2011, 2, e240–e240. [Google Scholar] [CrossRef]
- Ni, H.-M.; Williams, J.A.; Ding, W.-X. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 2015, 4, 6–13. [Google Scholar] [CrossRef]
- Niedernhofer, L.J.; Daniels, J.S.; Rouzer, C.A.; Greene, R.E.; Marnett, L.J. Malondialdehyde, a product of lipid peroxidation, is mutagenic in human cells. J Biol Chem. 2003, 278, 31426–31433. [Google Scholar] [CrossRef] [PubMed]
- Niizuma, K.; Yoshioka, H.; Chen, H.; Kim, G.S.; Jung, J.E.; Katsu, M.; Okami, N.; Chan, P.H. Mitochondrial and apoptotic neuronal death signaling pathways in cerebral ischemia. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2010, 1802, 92–99. [Google Scholar] [CrossRef]
- Oliver, C.N.; E Starke-Reed, P.; Stadtman, E.R.; Liu, G.J.; Carney, J.M.; A Floyd, R. Oxidative damage to brain proteins, loss of glutamine synthetase activity, and production of free radicals during ischemia/reperfusion-induced injury to gerbil brain. Proc. Natl. Acad. Sci. USA 1990, 87, 5144–5147. [Google Scholar] [CrossRef]
- Orellana-Urzúa, S.; Rojas, I.; Líbano, L.; Rodrigo, R. Pathophysiology of Ischemic Stroke: Role of Oxidative Stress. Curr. Pharm. Des. 2020, 26, 4246–4260. [Google Scholar] [CrossRef] [PubMed]
- Orrenius, S.; Gogvadze, V.; Zhivotovsky, B. Mitochondrial Oxidative Stress: Implications for Cell Death. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 143–183. [Google Scholar] [CrossRef]
- Palmer, C.; Menzies, S.L.; Roberts, R.L.; Pavlick, G.; Connor, J.R. Changes in iron histochemistry after hypoxic-ischemic brain injury in the neonatal rat. J. Neurosci. Res. 1999, 56, 60–71. [Google Scholar] [CrossRef]
- Picaud, J.C.; Steghens, J.P.; Auxenfans, C.; Barbieux, A.; Laborie, S.; Claris, O. Lipid peroxidation assessment by malondialdehyde measurement in parenteral nutrition solutions for newborn infants: A pilot study. Acta Paediatr. 2004, 93, 241–245. [Google Scholar] [CrossRef]
- Epizzimenti, S.; Ciamporcero, E.S.; Edaga, M.; Epettazzoni, P.; Earcaro, A.; Ecetrangolo, G.; Eminelli, R.; Edianzani, C.; Elepore, A.; Egentile, F.; et al. Interaction of aldehydes derived from lipid peroxidation and membrane proteins. Front. Physiol. 2013, 4, 242. [Google Scholar] [CrossRef]
- Pluta, R.; Januszewski, S.; Czuczwar, S.J. Brain Ischemia as a Prelude to Alzheimer's Disease. Front. Aging Neurosci. 2021, 13. [Google Scholar] [CrossRef]
- Qian, Z.M.; Ke, Y. Brain iron transport. Biol. Rev. Camb. Philos Soc. 2019, 94, 1672–1684. [Google Scholar] [CrossRef]
- Qiu, Z.; Wei, Y.; Song, Q.; Du, B.; Wang, H.; Chu, Y.; Hu, Y. The Role of Myocardial Mitochondrial Quality Control in Heart Failure. Front. Pharmacol. 2019, 10, 1404. [Google Scholar] [CrossRef] [PubMed]
- Radak, D.; Resanovic, I.; Isenovic, E.R. Link Between Oxidative Stress and Acute Brain Ischemia. Angiology 2013, 65, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Rouault, T.A.; Maio, N. Biogenesis and functions of mammalian iron-sulfur proteins in the regulation of iron homeostasis and pivotal metabolic pathways. J. Biol. Chem. 2017, 292, 12744–12753. [Google Scholar] [CrossRef]
- Sanderson, T.H.; Reynolds, C.A.; Kumar, R.; Przyklenk, K.; Hüttemann, M. Molecular Mechanisms of Ischemia–Reperfusion Injury in Brain: Pivotal Role of the Mitochondrial Membrane Potential in Reactive Oxygen Species Generation. Mol. Neurobiol. 2012, 47, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Saralkar, P.; Arsiwala, T.; Geldenhuys, W.J. Nanoparticle formulation and in vitro efficacy testing of the mitoNEET ligand NL-1 for drug delivery in a brain endothelial model of ischemic reperfusion-injury. Int. J. Pharm. 2020, 578, 119090–119090. [Google Scholar] [CrossRef]
- Sasaki, H.; Sato, H.; Kuriyama-Matsumura, K.; Sato, K.; Maebara, K.; Wang, H.; Tamba, M.; Itoh, K.; Yamamoto, M.; Bannai, S. Electrophile Response Element-mediated Induction of the Cystine/Glutamate Exchange Transporter Gene Expression. PEDIATRICS 2002, 277, 44765–44771. [Google Scholar] [CrossRef] [PubMed]
- Scarpulla, R.C. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 2011, 1813, 1269–1278. [Google Scholar] [CrossRef]
- Selim, M.H.; Ratan, R.R. The role of iron neurotoxicity in ischemic stroke. Ageing Res. Rev. 2004, 3, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Serteser, M.; Ozben, T.; Gumuslu, S.; Balkan, S.; Balkan, E. Lipid Peroxidation in Rat Brain During Focal Cerebral Ischemia: Prevention of Malondialdehyde and Lipid Conjugated Diene Production by a Novel Antiepileptic, Lamotrigine. NeuroToxicology 2002, 23, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Sesso, A.; Belizário, J.; Marques, M.; Higuchi, M.; Schumacher, R.; Colquhoun, A.; Ito, E.; Kawakami, J. Mitochondrial Swelling and Incipient Outer Membrane Rupture in Preapoptotic and Apoptotic Cells. Anat. Rec. 2012, 295, 1647–1659. [Google Scholar] [CrossRef]
- Shekh-Ahmad, T.; Lieb, A.; Kovac, S.; Gola, L.; Wigley, W.C.; Abramov, A.Y.; Walker, M.C. Combination antioxidant therapy prevents epileptogenesis and modifies chronic epilepsy. Redox Biol. 2019, 26, 101278. [Google Scholar] [CrossRef]
- Shen, Y.; Li, X.; Dong, D.; Zhang, B.; Xue, Y.; Shang, P. Transferrin receptor 1 in cancer: A new sight for cancer therapy. Am. J. Cancer Res. 2018, 8, 916–931. [Google Scholar]
- Shi, Z.; Zhang, K.; Zhou, H.; Jiang, L.; Xie, B.; Wang, R. Increased miR-34c mediates synaptic deficits by targeting synaptotagmin 1 through ROS-JNK-p53 pathway in Alzheimer's Disease. Aging Cell. 2020, 19, e13125. [Google Scholar] [CrossRef]
- Simmons, E.C.; Scholpa, N.E.; Schnellmann, R.G. Mitochondrial biogenesis as a therapeutic target for traumatic and neurodegenerative CNS diseases. Exp. Neurol. 2020, 329, 113309–113309. [Google Scholar] [CrossRef]
- Slatter, D.A.; Avery, N.C.; Bailey, A.J. Identification of a New Cross-link and Unique Histidine Adduct from Bovine Serum Albumin Incubated with Malondialdehyde. J. Biol. Chem. 2004, 279, 61–69. [Google Scholar] [CrossRef]
- Śliwińska, A.; Luty, J.; Aleksandrowicz-Wrona, E.; Małgorzewicz, S. Iron status and dietary iron intake in vegetarians. Adv. Clin. Exp. Med. 2018, 27, 1383–1389. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Angeli, J.P.F.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Jiang, X.; Gu, W. Emerging Mechanisms and Disease Relevance of Ferroptosis. Trends Cell Biol. 2020, 30, 478–490. [Google Scholar] [CrossRef]
- Stradecki-Cohan, H.M.; Cohan, C.H.; Raval, A.P.; Dave, K.R.; Reginensi, D.; Gittens, R.A.; Youbi, M.; Perez-Pinzon, M.A. Cognitive Deficits after Cerebral Ischemia and Underlying Dysfunctional Plasticity: Potential Targets for Recovery of Cognition. J. Alzheimer's Dis. 2017, 60, S87–S105. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Liu, J.; Yan, X.-Y.; Zhang, Y.; Zhang, J.-J.; Zhang, L.-C.; Sun, L.-K. Cytoprotective Effect of the UCP2-SIRT3 Signaling Pathway by Decreasing Mitochondrial Oxidative Stress on Cerebral Ischemia–Reperfusion Injury. Int. J. Mol. Sci. 2017, 18, 1599. [Google Scholar] [CrossRef] [PubMed]
- Suárez, I.; Bodega, G.; Fernández, B. Glutamine synthetase in brain: Effect of ammonia. Neurochem. Int. 2002, 41, 123–142. [Google Scholar] [CrossRef]
- Sun, Y.; Zheng, Y.; Wang, C.; Liu, Y. Glutathione depletion induces ferroptosis, autophagy, and premature cell senescence in retinal pigment epithelial cells. Cell Death Dis. 2018, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Sveinsson, O.A.; Kjartansson, O.; Valdimarsson, E.M. [Cerebral ischemia/infarction—Epidemiology, causes and symptoms]. Laeknabladid 2014, 100, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Gao, P.; Chen, H.; Zhou, X.; Ou, Y.; He, Y. The Role of Iron, Its Metabolism and Ferroptosis in Traumatic Brain Injury. Front. Cell. Neurosci. 2020, 14. [Google Scholar] [CrossRef]
- Tonnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef]
- Toyokuni, S.; Ito, F.; Yamashita, K.; Okazaki, Y.; Akatsuka, S. Iron and thiol redox signaling in cancer: An exquisite balance to escape ferroptosis. Free. Radic. Biol. Med. 2017, 108, 610–626. [Google Scholar] [CrossRef]
- Tuo, Q.-Z.; Lei, P.; A Jackman, K.; Li, X.-L.; Xiong, H.; Liuyang, Z.-Y.; Roisman, L.C.; Zhang, S.-T.; Ayton, S.; Wang, Q.; et al. Tau-mediated iron export prevents ferroptotic damage after ischemic stroke. Mol. Psychiatry 2017, 22, 1520–1530. [Google Scholar] [CrossRef]
- Urrutia, P.J.; Aguirre, P.; Tapia, V.; Carrasco, C.M.; Mena, N.P.; Núñez, M.T. Cell death induced by mitochondrial complex I inhibition is mediated by Iron Regulatory Protein 1. Biochim. et Biophys. Acta (BBA) - Mol. Basis Dis. 2017, 1863, 2202–2209. [Google Scholar] [CrossRef]
- van der Worp, H.B. , Macleod, M.R., Kollmar, R., & European Stroke Research Network for, H. Therapeutic hypothermia for acute ischemic stroke: Ready to start large randomized trials? J. Cereb. Blood. Flow Metab. 2010, 30, 1079–1093. [Google Scholar] [CrossRef]
- van Leyen, K.; Holman, T.R.; Maloney, D.J. The potential of 12/15-lipoxygenase inhibitors in stroke therapy. Future Med. Chem. 2014, 6, 1853–1855. [Google Scholar] [CrossRef] [PubMed]
- Varanyuwatana, P.; Halestrap, A.P. The roles of phosphate and the phosphate carrier in the mitochondrial permeability transition pore. Mitochondrion 2012, 12, 120–125. [Google Scholar] [CrossRef]
- Vuckovic, A.M.; Bosello Travain, V.; Bordin, L.; Cozza, G.; Miotto, G.; Rossetto, M. Inactivation of the glutathione peroxidase GPx4 by the ferroptosis-inducing molecule RSL3 requires the adaptor protein 14-3-3epsilon. FEBS Lett. 2020, 594, 611–624. [Google Scholar] [CrossRef]
- Wang, H.; Liu, C.; Zhao, Y.; Gao, G. Mitochondria regulation in ferroptosis. Eur. J. Cell Biol. 2019, 99, 151058. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, Y.; Du, T.; Yang, H.; Lei, L.; Guo, M.; Ding, H.-F.; Zhang, J.; Wang, H.; Chen, X.; et al. ATF3 promotes erastin-induced ferroptosis by suppressing system Xc–. Cell Death Differ. 2019, 27, 662–675. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, L.; Dai, Z.; Wu, P.; Shi, H.; Zhao, S. Lack of mitochondrial ferritin aggravated neurological deficits via enhancing oxidative stress in a traumatic brain injury murine model. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef]
- Wang, R.; Liu, Y.-Y.; Liu, X.-Y.; Jia, S.-W.; Zhao, J.; Cui, D.; Wang, L. Resveratrol Protects Neurons and the Myocardium by Reducing Oxidative Stress and Ameliorating Mitochondria Damage in a Cerebral Ischemia Rat Model. Cell. Physiol. Biochem. 2014, 34, 854–864. [Google Scholar] [CrossRef]
- Wang, S.; Li, F.; Qiao, R.; Hu, X.; Liao, H.; Chen, L.; Wu, J.; Wu, H.; Zhao, M.; Liu, J.; et al. Arginine-Rich Manganese Silicate Nanobubbles as a Ferroptosis-Inducing Agent for Tumor-Targeted Theranostics. ACS Nano 2018, 12, 12380–12392. [Google Scholar] [CrossRef]
- Wei, S.; Qiu, T.; Yao, X.; Wang, N.; Jiang, L.; Jia, X.; Tao, Y.; Wang, Z.; Pei, P.; Zhang, J.; et al. Arsenic induces pancreatic dysfunction and ferroptosis via mitochondrial ROS-autophagy-lysosomal pathway. J. Hazard. Mater. 2019, 384, 121390. [Google Scholar] [CrossRef]
- Weiss, J.N.; Korge, P.; Honda, H.M.; Ping, P. Role of the Mitochondrial Permeability Transition in Myocardial Disease. Circ. Res. 2003, 93, 292–301. [Google Scholar] [CrossRef]
- Wible, D.J.; Bratton, S.B. Reciprocity in ROS and autophagic signaling. Curr. Opin. Toxicol. 2017, 7, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Luo, H.; Zhou, X.; Cheng, C.-Y.; Lin, L.; Liu, B.-L.; Liu, K.; Li, P.; Yang, H. Succinate-induced neuronal mitochondrial fission and hexokinase II malfunction in ischemic stroke: Therapeutical effects of kaempferol. Biochim. et Biophys. Acta (BBA) Mol. Basis Dis. 2017, 1863, 2307–2318. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.C.; Cui, J.Y.; Klaassen, C.D. Beneficial Role of Nrf2 in Regulating NADPH Generation and Consumption. Toxicol. Sci. 2011, 123, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zheng, Y.; Liu, M.; Li, Y.; Ma, S.; Tang, W.; Yan, W.; Cao, M.; Zheng, W.; Jiang, L.; et al. BNIP3L/NIX degradation leads to mitophagy deficiency in ischemic brains. Autophagy 2020, 17, 1934–1946. [Google Scholar] [CrossRef]
- Yamamoto, M.; Shima, T.; Uozumi, T.; Sogabe, T.; Yamada, K.; Kawasaki, T. A possible role of lipid peroxidation in cellular damages caused by cerebral ischemia and the protective effect of alpha-tocopherol administration. Stroke 1983, 14, 977–982. [Google Scholar] [CrossRef]
- Yan, J.; Sun, W.; Shen, M.; Zhang, Y.; Jiang, M.; Liu, A.; Ma, H.; Lai, X.; Wu, J. Idebenone improves motor dysfunction, learning and memory by regulating mitophagy in MPTP-treated mice. Cell Death Discov. 2022, 8, 1–8. [Google Scholar] [CrossRef]
- Yan, N.; Zhang, J. Iron Metabolism, Ferroptosis, and the Links With Alzheimer's Disease. Front. Neurosci. 2019, 13, 1443. [Google Scholar] [CrossRef]
- Yang, N.; Guan, Q.-W.; Chen, F.-H.; Xia, Q.-X.; Yin, X.-X.; Zhou, H.-H.; Mao, X.-Y. Antioxidants Targeting Mitochondrial Oxidative Stress: Promising Neuroprotectants for Epilepsy. Oxidative Med. Cell. Longev. 2020, 2020, 1–14. [Google Scholar] [CrossRef]
- Yang, T.; Sun, Y.; Li, Q.; Li, S.; Shi, Y.; Leak, R.K.; Chen, J.; Zhang, F. Ischemic preconditioning provides long-lasting neuroprotection against ischemic stroke: The role of Nrf2. Exp. Neurol. 2019, 325, 113142–113142. [Google Scholar] [CrossRef]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Yao, G.-Y.; Zhu, Q.; Xia, J.; Chen, F.-J.; Huang, M.; Liu, J.; Zhou, T.-T.; Wei, J.-F.; Cui, G.-Y.; Zheng, K.-Y.; et al. Ischemic postconditioning confers cerebroprotection by stabilizing VDACs after brain ischemia. Cell Death Dis. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Ye, Q.; Zeng, C.; Dong, L.; Wu, Y.; Huang, Q.; Wu, Y. Inhibition of ferroptosis processes ameliorates cognitive impairment in kainic acid-induced temporal lobe epilepsy in rats. Am. J. Transl. Res. 2019, 11, 875–884. [Google Scholar] [PubMed]
- Yigitkanli, K.; Zheng, Y.; Pekcec, A.; Lo, E.H.; van Leyen, K. Increased 12/15-Lipoxygenase Leads to Widespread Brain Injury Following Global Cerebral Ischemia. Transl. Stroke Res. 2017, 8, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.; Zhou, H.; Fang, Y.; Li, C.; He, Y.; Yu, L.; Wan, H.; Yang, J. Astragaloside IV alleviates ischemia reperfusion-induced apoptosis by inhibiting the activation of key factors in death receptor pathway and mitochondrial pathway. J. Ethnopharmacol. 2019, 248, 112319. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef]
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation. Biochem. Biophys. Res. Commun. 2016, 478, 838–844. [Google Scholar] [CrossRef]
- Yuan, Y.; Zhai, Y.; Chen, J.; Xu, X.; Wang, H. Kaempferol Ameliorates Oxygen-Glucose Deprivation/Reoxygenation-Induced Neuronal Ferroptosis by Activating Nrf2/SLC7A11/GPX4 Axis. Biomolecules 2021, 11, 923. [Google Scholar] [CrossRef]
- Zalewska, A.; Klimiuk, A.; Zięba, S.; Wnorowska, O.; Rusak, M.; Waszkiewicz, N.; Szarmach, I.; Dzierżanowski, K.; Maciejczyk, M. Salivary gland dysfunction and salivary redox imbalance in patients with Alzheimer’s disease. Sci. Rep. 2021, 11, 1–16. [Google Scholar] [CrossRef]
- Zhang, P.; Chen, L.; Zhao, Q.; Du, X.; Bi, M.; Li, Y. Ferroptosis was more initial in cell death caused by iron overload and its underlying mechanism in Parkinson's disease. Free Radic. Biol. Med. 2020, 152, 227–234. [Google Scholar] [CrossRef]
- Zhang, W.; Gai, C.; Ding, D.; Wang, F.; Li, W. Targeted p53 on Small-Molecules-Induced Ferroptosis in Cancers. Front. Oncol. 2018, 8, 507. [Google Scholar] [CrossRef]
- Zhang, Y.; Lu, X.; Tai, B.; Li, W.; Li, T. Ferroptosis and Its Multifaceted Roles in Cerebral Stroke. Front. Cell. Neurosci. 2021, 15. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, R.; Li, X.; Fang, W. Porcine Circovirus 2 Induction of ROS Is Responsible for Mitophagy in PK-15 Cells via Activation of Drp1 Phosphorylation. Viruses 2020, 12, 289. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.H.; Wang, D.W.; Xu, S.F.; Zhang, S.; Fan, Y.G.; Yang, Y.Y. alpha-Lipoic acid improves abnormal behavior by mitigation of oxidative stress, inflammation, ferroptosis, and tauopathy in P301S Tau transgenic mice. Redox. Biol. 2018, 14, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wu, Y.; Yuan, S.; Zhang, P.; Zhang, J.; Li, H.; Li, X.; Shen, H.; Wang, Z.; Chen, G. Glutathione peroxidase 4 participates in secondary brain injury through mediating ferroptosis in a rat model of intracerebral hemorrhage. Brain Res. 2018, 1701, 112–125. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Yu, D.; He, Z.; Bao, L.; Feng, L.; Chen, L.; Liu, Z.; Hu, X.; Zhang, N.; Wang, T.; et al. Endoplasmic reticulum stress-mediated autophagy activation is involved in cadmium-induced ferroptosis of renal tubular epithelial cells. Free. Radic. Biol. Med. 2021, 175, 236–248. [Google Scholar] [CrossRef]
- Zhao, G.-C.; Yuan, Y.-L.; Chai, F.-R.; Ji, F.-J. Effect of Melilotus officinalis extract on the apoptosis of brain tissues by altering cerebral thrombosis and inflammatory mediators in acute cerebral ischemia. BioMedicine 2017, 89, 1346–1352. [Google Scholar] [CrossRef]
- Zhou, R.-P.; Chen, Y.; Wei, X.; Yu, B.; Xiong, Z.-G.; Lu, C.; Hu, W. Novel insights into ferroptosis: Implications for age-related diseases. Theranostics 2020, 10, 11976–11997. [Google Scholar] [CrossRef]
Figure 1.
Overview of the pathophysiology of cerebral ischemia.
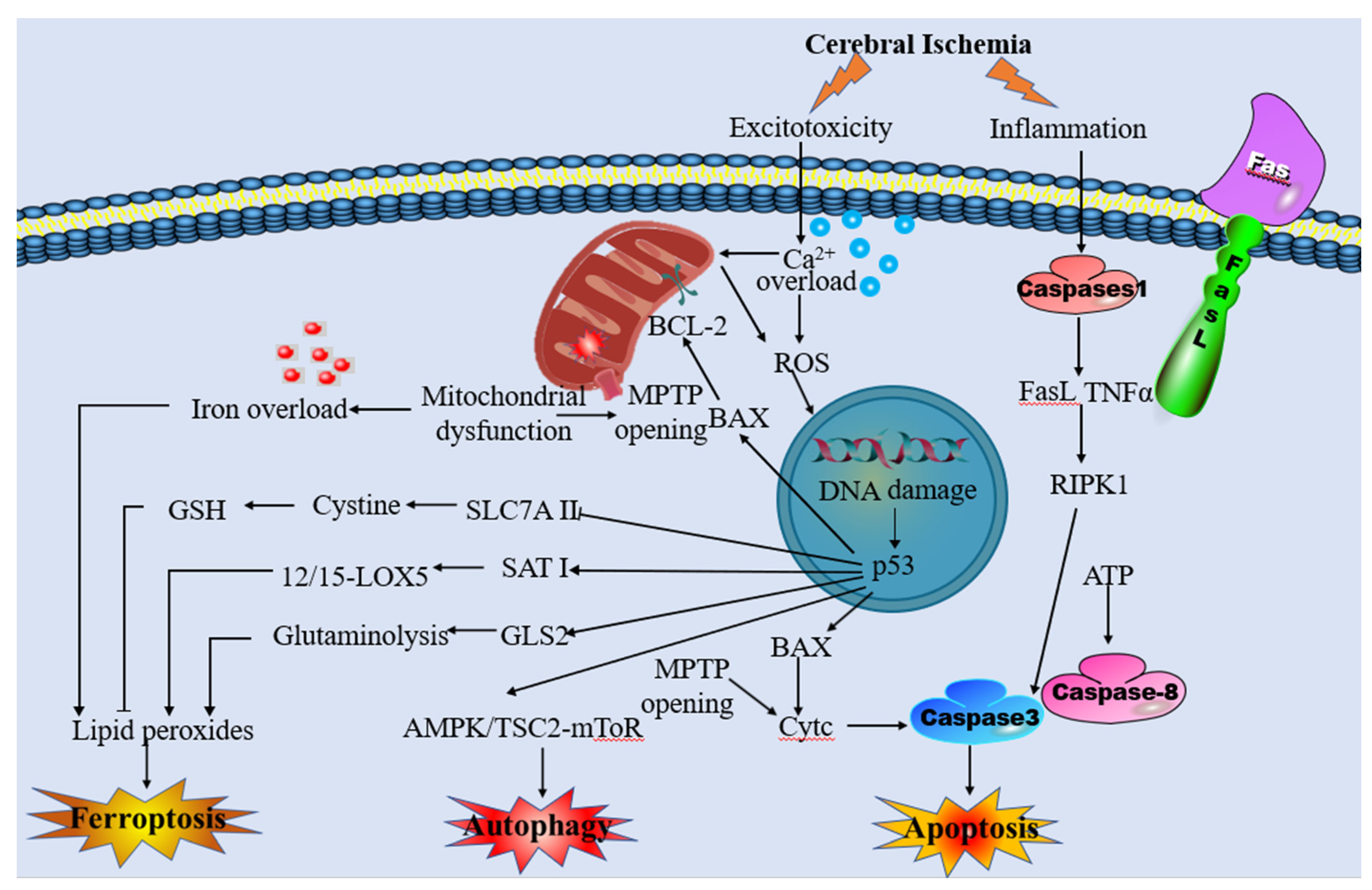
Figure 2.
The Interplay between Mitochondrial Dysfunction and Ferroptosis in ischemia-associated CNS diseases.
Figure 2.
The Interplay between Mitochondrial Dysfunction and Ferroptosis in ischemia-associated CNS diseases.
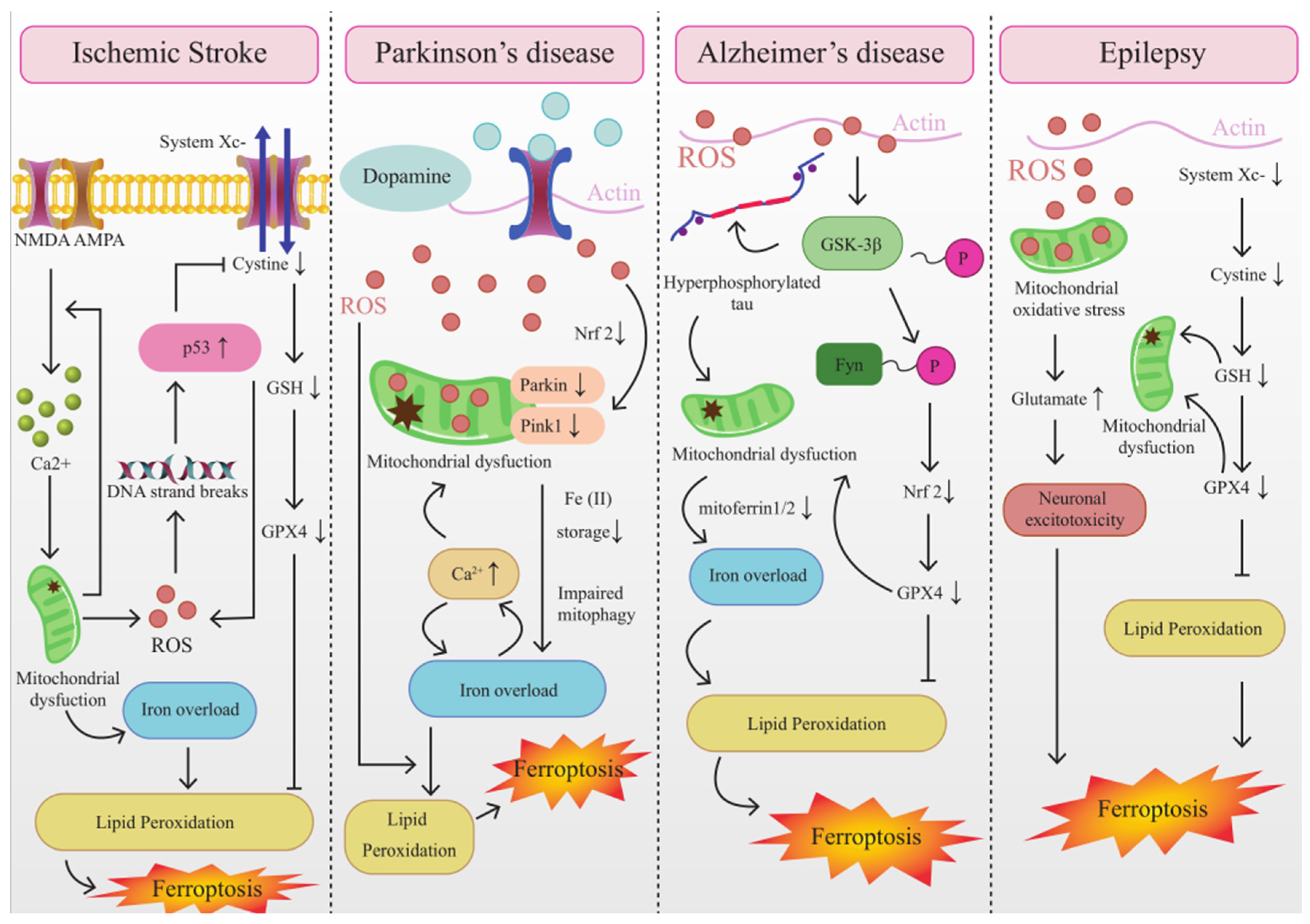

Table 1.
Study on ischemic encephalopathy treatment about mitochondrial dysfunction and ferroptosis.
Table 1.
Study on ischemic encephalopathy treatment about mitochondrial dysfunction and ferroptosis.
| Interventions | Mechanism for Mitochondrial Dysfunction | Mechanism for Ferroptosis | References |
|---|---|---|---|
| Kaempferol |
|
|
(B. Wu et al., 2017; Y. Yuan et al., 2021) |
| α-Lipoic acid (LA) |
|
|
(Dos Santos et al., 2019; Y. H. Zhang et al., 2018) |
| Idebenone |
|
|
(Avci et al., 2021; J. Yan et al., 2022) |
| RTA 408 |
|
|
(Shekh-Ahmad et al., 2019) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



