Submitted:
28 July 2023
Posted:
31 July 2023
You are already at the latest version
Abstract
Small molecule kinase inhibitors (SMKIs) are one of the heightened concerns in the field of drug research and development. There are seventy-nine (up to July 2023) small molecule kinase inhibitors have been approved by FDA and hundreds kinase inhibitors candidates in clinical trials, which shed light on the treatment of some major diseases. As an important strategy of drug design, computer-aided drug design (CADD) plays an indispensable role in the discovery of SMKIs. CADD methods, such as docking, molecular dynamic, pharmacophore have been applied to the design and optimization of small molecule kinase inhibitors. In the review, we provide an overview of recent advances in CADD and SMKIs; and the application of CADD for discovery of SMKIs.
Keywords:
CADD
; SBDD
; LBDD
; kinase
; small molecule kinase inhibitors
1. Introduction
Drug marketing is a costly and lengthy process. According to statistics, ninety percent drugs that entered clinical trials while failed to get FDA approval, mainly because of various problems encountered in rational drug design.[1] With the rapid development of bioinformatics and computer technology, computer-aided drug design (CADD) has made great progress. CADD can be divided into structure-based drug design (SBDD) and ligand-based drug design (LBDD).[2] SBDD is based on the structure of receptor and ligand, analysis and evaluation the interaction between ligand and receptor, and then select or modify the structural of ligand to enhance the affinity with receptor.[3] LBDD can obtain information such as molecular structure, charge distribution and reactive site, which is helpful to understand the relationship between the structural properties of compounds and their pharmacological activities or druggability.
4] CADD not only reduces the cost of drug research and development, but also greatly shortens the time from discovery to market, so it plays an important role in drug research and development.[5]
Kinases play an important role in regulating cell metabolism, growth, exercise, differentiation and division, as well as signal pathways related to the formation and development of many human diseases, including cancer [6,7,8,9], vascular diseases [10,11,12,13], diabetes [14,15,16], inflammation [17,18,19,20,21] and degenerative diseases [22,23,24], which makes they are attractive targets for drug discovery. Since Imatinib was approved in 2002, the US Food and Drug Administration (FDA) has approved 79 small molecule kinase inhibitors (SMKIs), and there are currently several small molecule kinase inhibitor candidates in clinical trials. Most kinase inhibitors bind to kinase catalytic domain, that is, ATP binding site. Due to the high conservation of the ATP binding site in kinases, off-target effects (i.e., low selectivity) of kinase inhibitors can result in undesirable side effects. [25,26] Therefore, it is still an important challenge to find novel, effective and selective kinase inhibitors. CADD can analyze the subtle differences and structural characteristics of different kinase ATP binding site, and promote the discovery of kinase inhibitors. Up to July 2023, totally 26 kinase inhibitors discovered by CADD method have been approved by FDA.
In this review, we provide an overview of recent advances in CADD application examples, including the methods of structure-based drug design (SBDD) and ligand-based drug design (LBDD) for discovery drugs. Moreover, we focus on recent progress of kinase inhibitors and highlight representative investigations on kinase inhibitors by CADD.
2. Computer-Aided Drug Design
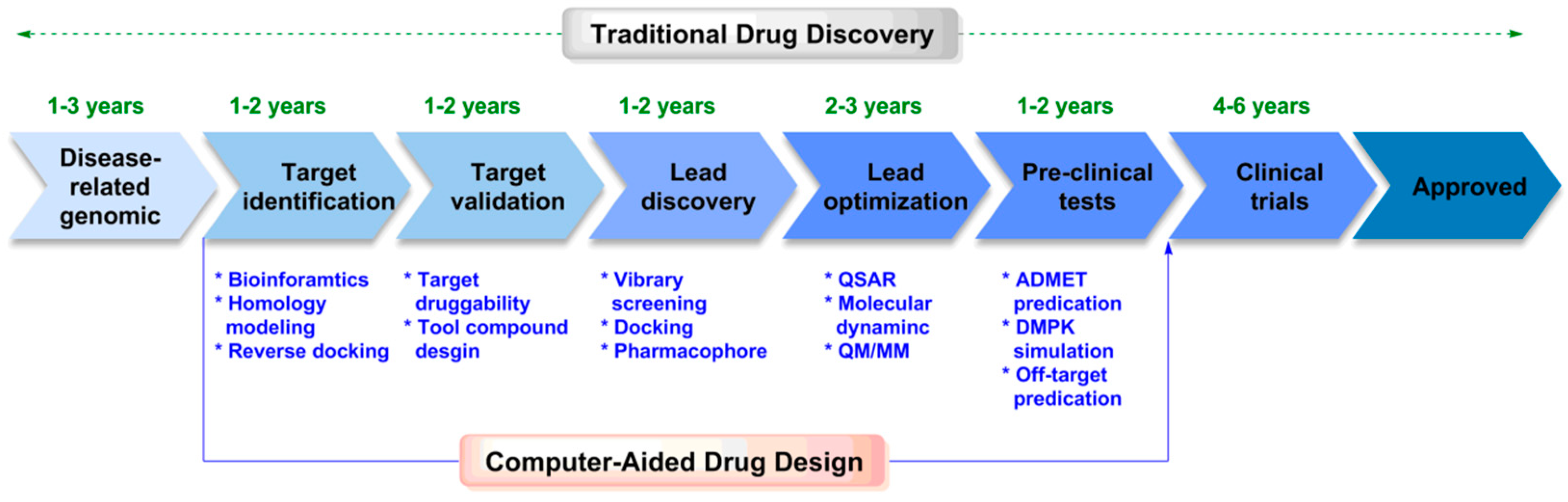
Traditional drug research and development (R&D) uses the method of random screening. According to data statistics, the average cost of a drug from laboratory research to market launch is about 2.558 billion US dollars, lasting for at least 13.5 years, and the success rate is only about 10.0%.[27,28] In order to meet the needs and accelerate the process of new drug R&D, the concept of drug molecular design has been proposed, which makes drug investigation into a new era. Drug molecular design is a scientific and efficient strategy which reasonably design new chemical entity molecules with expected pharmacological activities, safety, stability and quality controllability properties based on the structural characteristics of endogenous ligands and targets.[29]
Computer-aided drug design (CADD) provides new ideas for rational drug molecular design and promotes significant breakthroughs in drug molecular design. In 1981, the cover article of Fortune “Next Industrial Revolution: Designing Drugs by Computer at Merck” marked the formal entry of CADD into the field of pharmaceutical research.[30] Subsequently, the Nobel Prize in Chemistry in 1998 and 2013 was awarded for the related work in molecular simulation, demonstrating that experiments and theory are the twin pillars of chemistry. In the past few decades, scientists have developed a variety of CADD methods, such as sequence alignment [31], homologous modeling [32], molecular docking [33], pharmacophore [34], quantitative structure-activity relationship [35], molecular dynamics [36] and quantum mechanics [37], etc.; and these methods have been adopted to different stage of drug development. Compared to traditional drug R&D methods, CADD reduces the high cost, decreases the high-risk problems, shortens the cycle time and improves the efficiency of new drug development observably. With its remarkable advantages, CADD has successfully developed many drugs, such as Donepizil (treatment of Alzheimer disease), Zanamivir (anti-virus), Imatinib (anti-tumor), Saquinavir (treatment of immunodeficiency virus) and so on.[38,39,40]
Figure 1.
Comparison of traditional drug discovery and CADD in drug discovery process.
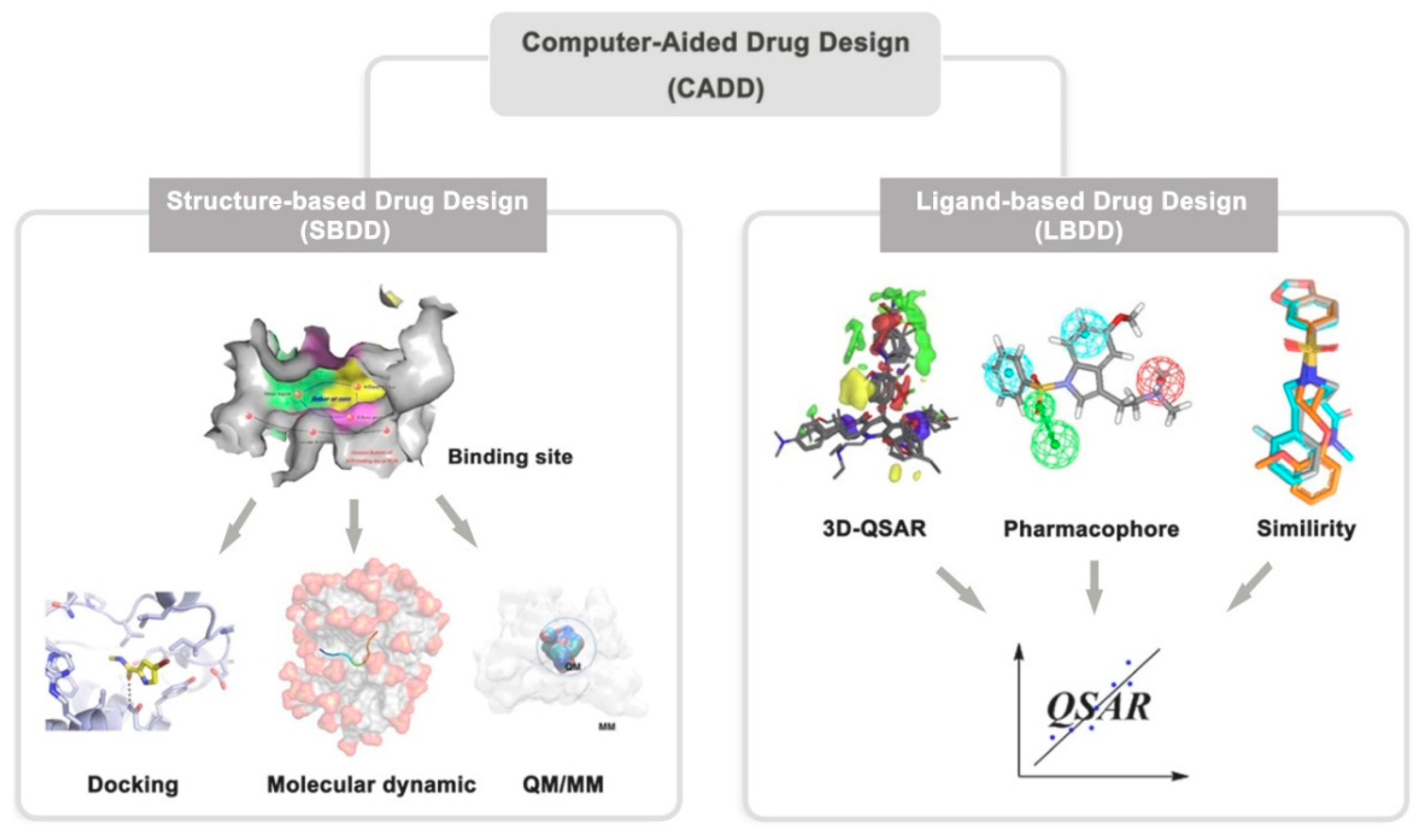
Computer-aided drug design [1,2,3] includes structure-based drug design (SBDD) and ligand-based drug design (LBDD). The prerequisite for the implementation of SBDD and LBDD is the 3D structure of targets and molecular structures of ligands, respectively. Structure-based drug design method is based on the three-dimensional structure of biological targets to find hit or lead compounds by using ab initio design, molecular docking or virtual screening methods with measuring the interaction modes and binding energy between ligand and receptor.[2] Ligand-based drug design preforms quantitative structure-activity relationship and pharmacophore model to establish the theoretical prediction model between molecular structure and biological activity which using for screening and optimizing active compounds.[3] Given the advantages and disadvantages of SBDD and LBDD, researchers typically use a combination of SBDD and LBDD methods for drug design in order to achieve better results.
Figure 2.
Description of computer-aided drug design.
2.1. Structure-Based Drug Design
2.1.1. Preparation of Target Structures
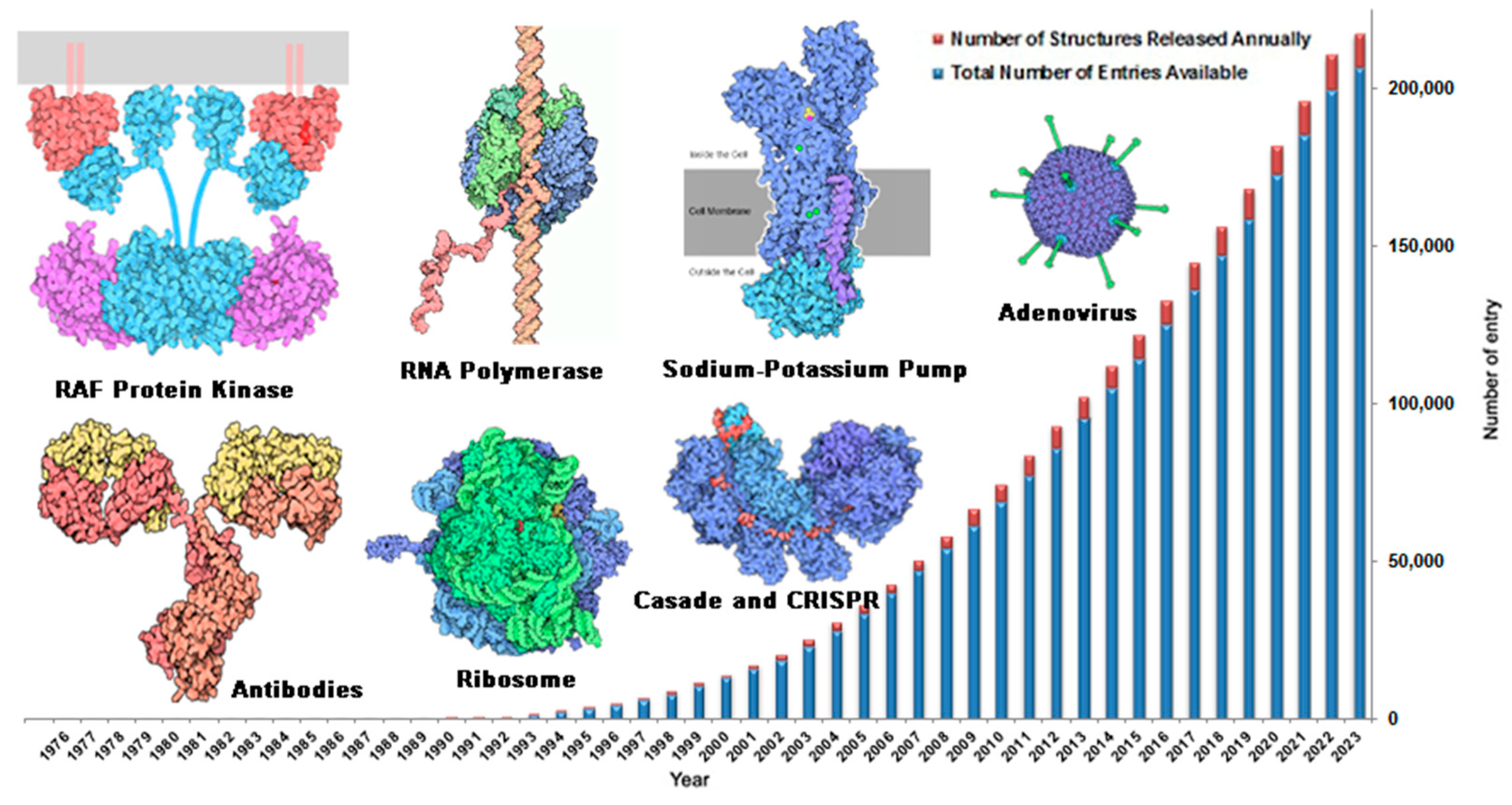
In order to achieve rational structure-based drug design, it is necessary to fully understand the three-dimensional structure of targets. Structural biology is devoted to study the complete, accurate and real-time three-dimensional structure of biological macromolecules by experimental means.[41,42] Nowadays, structure of a large number of biological macromolecules have been resolved by structural biology technology. The structural data of biological macromolecules are collected in the Protein Structure Database (PDB, http://www.rcsb.org). As of July 2023, 207,540 pieces of biological macromolecular structures information have been collected in PDB database, and the number of structures is increasing rapidly by nearly 10,000 pieces per year. Structural type of macromolecules involves a variety of drug targets associated with major human diseases, such as antitumor [43], antibacterial [44], antiviral [45], Alzheimer's disease [46] and diabetes [47], etc..
Figure 3.
PDB statistics: overall growth of released structures per year (https://www.rcsb.org).
Figure 3.
PDB statistics: overall growth of released structures per year (https://www.rcsb.org).
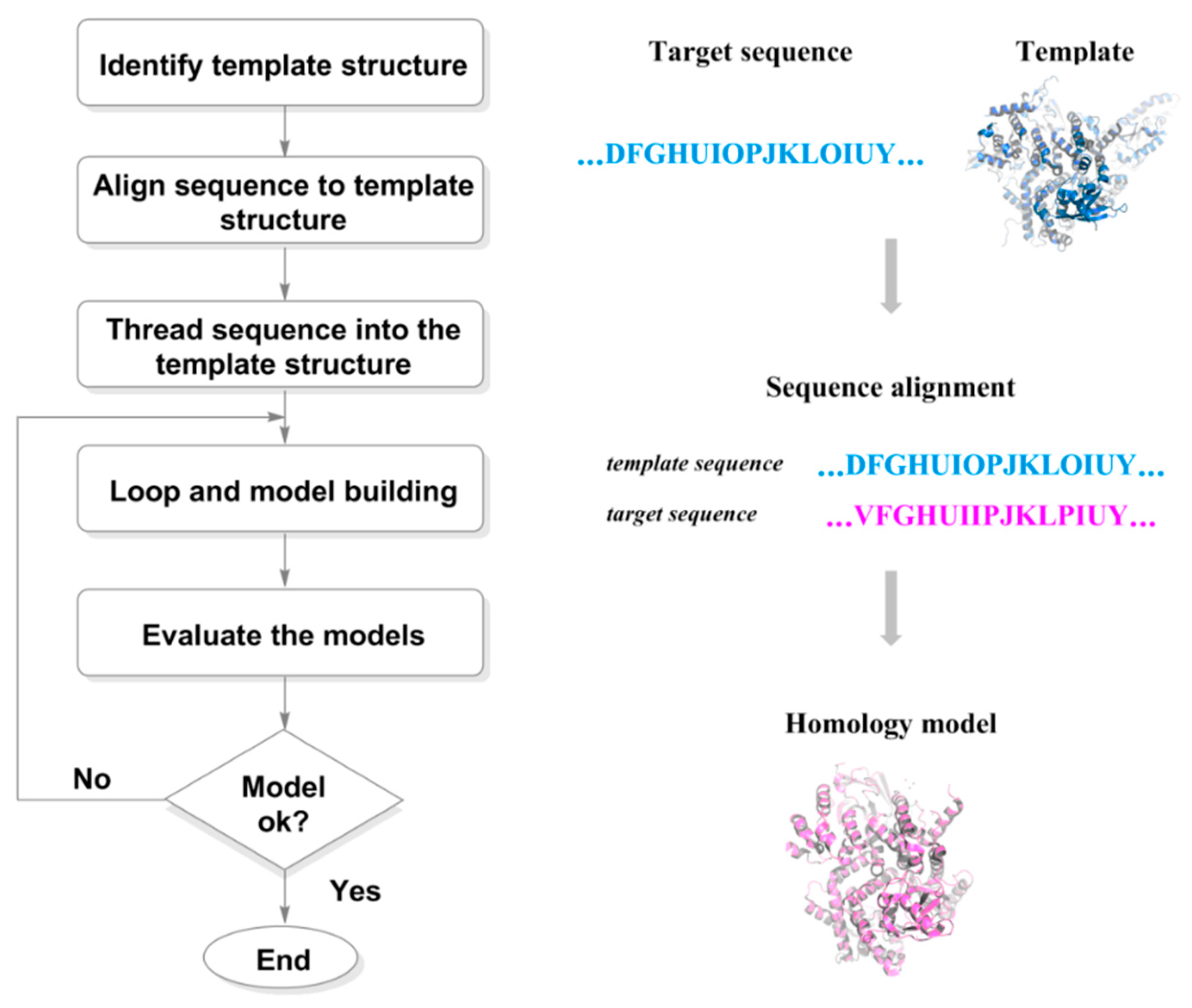
However, limited by the experimental techniques of structural analysis and the inherent properties of biological macromolecules, there are still a large number of drug target structures have not been accurately analyzed. With the help of computer simulation technology, scientists have developed a series of molecular structural prediction methods. The combination of homology modeling [32] and molecular dynamic (MD) [36] is the traditional method to simulate the structure of targets. Firstly, based on the principle that the primary structure of a protein determines the tertiary structure, homology modeling constructs the three-dimensional structure of protein through sequence alignment, structural construction (copying skeleton, constructing side chains, completing missing residues, and optimizing loop regions), and energy minimization. Then, molecular dynamics technology simulates the nearly real and accurate structure of protein structures obtained from homology modeling in solvent state. Moreover, a lot of software for protein prediction has also emerged in the field of artificial intelligence, such as AlphaFold and RosettaFold.
Figure 4.
Steps involved in homology model building process.
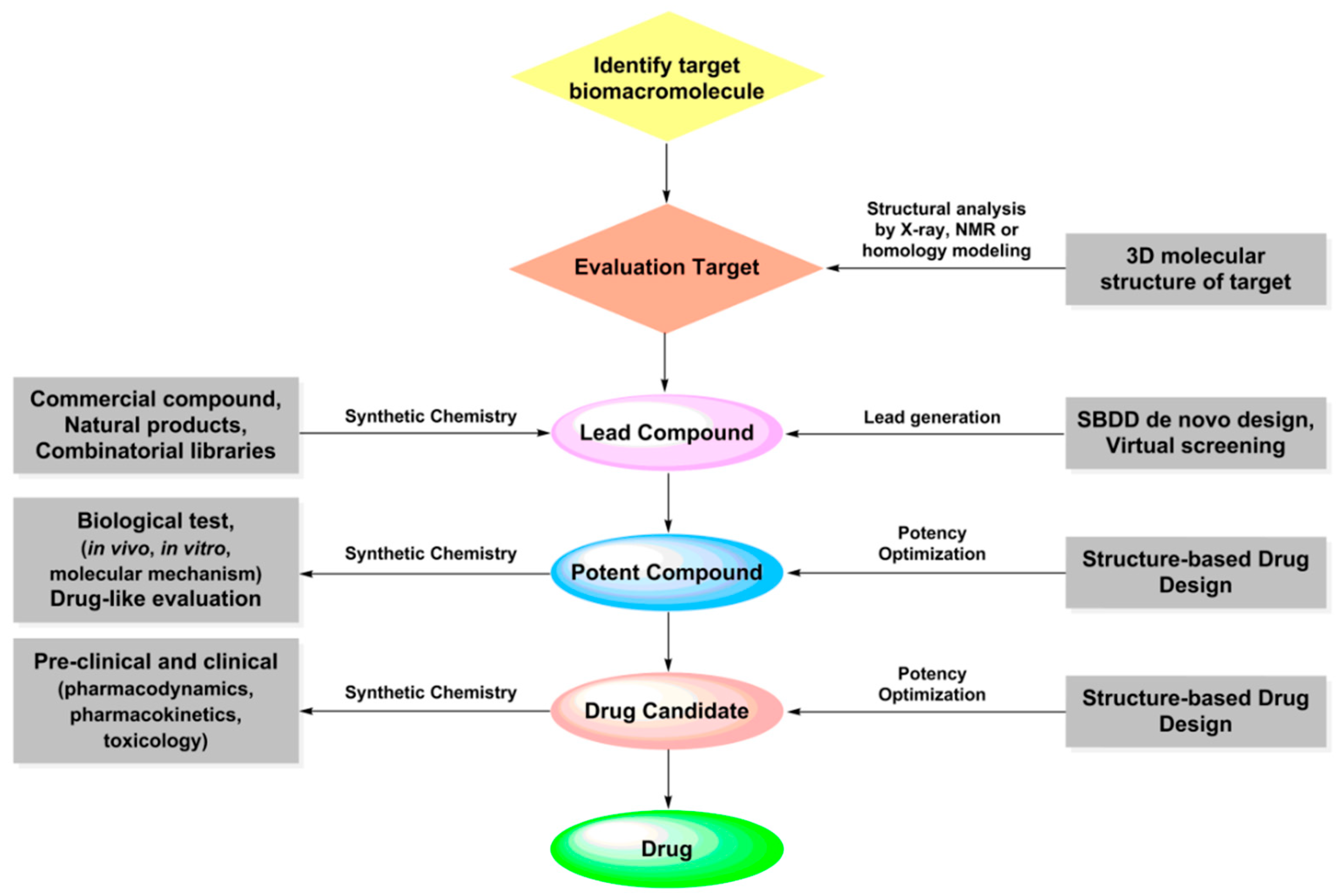
The widespread application of the above scientific and technological advancements contributes to deeply understand the molecular mechanism of the occurrence and development of related diseases, and provides new perspectives and research tools for rational structure-based drug design. The basic process of structure-based drug design: (1) according to the three-dimensional structural characteristics of the target, tailor-made ligand molecules with shape matching, electrical complementarity and clear intermolecular interaction are designed at molecular level; (2) the structure of ligand-receptor complex is established by molecular simulation to verify the completion of drug design; (3) a combined analysis of binding energy between ligand and receptor, physical and chemical properties of ligand and biological activity is preformed to evaluate the activity and druglikeness of designed compounds, which guides the subsequent drug design and optimization. Structure based drug design greatly reduces the blindness of drug design and screening, and accelerates the research and development process of lead compounds and candidate drugs. Among the methods of SBDD, molecular docking, molecular dynamic (MD) and quantum chemistry (QM) are commonly used to simulate and analyze the structure of ligand-receptor complexes, predict the binding strength between ligand and receptor and calculate the physical and chemical properties of compounds. The following will provide a detailed introduction to the above three methods.
Figure 5.
SBDD in drug discovery and design flow chart.
2.1.2. Molecular Docking
Since the early 1980s, molecular docking has gradually developed into the most frequently used method with high success rate in structure-based drug design.[48,49,50,51] Molecular docking predicts the orientation, binding pose and binding energy between small molecular ligands and the active sites of targets at atomic level; and the optimal binding pose will be screened by a comprehensively consideration of space matching and energy matching to clarify the ligand-receptor interaction.[52,53,54] Graphic analysis software can be conducted to visualize ligand-receptor binding mode and observe the fine structural differences between homologous targets, which offers key microscopic features for rational design of compounds with high target-compatibility and selectivity.
In the past three decades, scientific research achievements using molecular docking for drug design have increased year by year, covering almost all disease fields in pharmaceutical research, such as anti-tumor [55], anti-virus [56], anti-infection [57], diabetes [58], anti-hypertension [59], anti-depressant [60], anti-inflammatory [61], anti-bacterial [62] and so on. Undoubtedly, molecular docking has become one of the necessary tools for the research and development of targeted drugs, and it is one of the most widely used and effective means in the designing and screening of hit and lead compounds.[63,64,65]

Table 1.
Popular molecular docking software.
| Software | Algorithm | Scoring function | Website |
|---|---|---|---|
| Dock | Fragment growth | Force field, Surface matching score, Environment matching score | http://dock.compbio.ucsf.edu/DOCK_6/ |
| AutoDock | Genetic algorithm | Environment matching score | http://autodock.scripps.edu/ |
| GOLD | Genetic algorithm | Empirical | http://www.biosolveit.de/FlexX/ |
| FlexX | Fragment growth | Empirical | https://github.com/flexxui/flexx |
| Z-Dock | Geometric matching/Molecular dynamics | CAPRI+ | http://zdock.umassmed.edu/ |
| Hex | Geometric matching | CAPRI+ | http://www.csd.abdn.ac.uk/hex/ |
| SLIDE | Systematic | Force field, Empirical | http://www.bmb.msu.edu/~kuhn/software/slide/ |
| Fred | Systematic | Empirical | http://www.eyesopen.com/oedocking |
| LeDock | Annealing-Genetic algorithm | Physics/knowledge hybrid | http://www.lephar.com/software.htm |
| Glide | Systematic | XP/SP/HTVS | https://www.schrodinger.com |
| Surflex-Dock | Hammerhead | Empirical | http://www.tripos.com |
2.1.3. Molecular Dynamic
Drug-target binding is a competitive process among ligand, receptor and solvent. The solvent environment in which ligands and receptors are located plays an important role in molecular recognition and binding energy calculation. Solvent effect is not considered in molecular docking, which makes it impossible to provide microscopic information of ligand-receptor interaction accurately. Furthermore, the cell life activities are accompanied by protein conformational changes, so elucidating the details of drug target protein conformational changes is helpful to design efficient and highly selective compounds. However, at present, the dynamic process of protein conformational change cannot be measured by experimental methods.
Based on Newton's laws of motion, MD simulates the motion state of molecules and atoms in a certain period of time by computer, measuring the behavior of system properties and other parameters with time from a dynamic perspective.[63,64,65,66,67,68,69] Molecular dynamics can observe many details at the microscopic level which cannot be captured in real experiments, such as potential drug binding sites. The huge simulation system, long-time simulation and high-precision calculation method make the calculation cost of molecular dynamics simulation expensive. In order to reduce the calculation cost of MD, the sample count for MD is usually small. MD is often used to assist in the initial and post-processing stages of molecular docking, involving three aspects: protein flexibility, docking complex refinement and binding energy calculation.[70,71,72,73,74,75]
Table 2.
Popular molecular dynamic software.
| Software | Scoring function | Charge | Website |
|---|---|---|---|
| Amber | Mainly for biological system | AmberTools Free | http://ambermd.org/ |
| CPMD | Biological and chemical system | Free | http://www.cpmd.org/ |
| NAMD | Biological and chemical system | Free | http://www.ks.uiuc.edu/Research/namd |
| Lammps | Materials and solid state physical systems | Free | https://www.lammps.org/ |
| Gromacs | Mainly for biological system | Free | https://www.gromacs.org/ |
| Charmm | Mainly for biological system | Free | https://www.charmm.org/ |
| Tinker | Mainly for biological system | Free | http://dasher.wustl.edu/tinker/ |
Figure 6.
Three applications of MD in drug design. (a) Conformational simulation of flexible of protein. (b) Refine of ligand-target complex structure. (c) Calculation of binding energy. Figures are taken from ref.70, 75.
Figure 6.
Three applications of MD in drug design. (a) Conformational simulation of flexible of protein. (b) Refine of ligand-target complex structure. (c) Calculation of binding energy. Figures are taken from ref.70, 75.
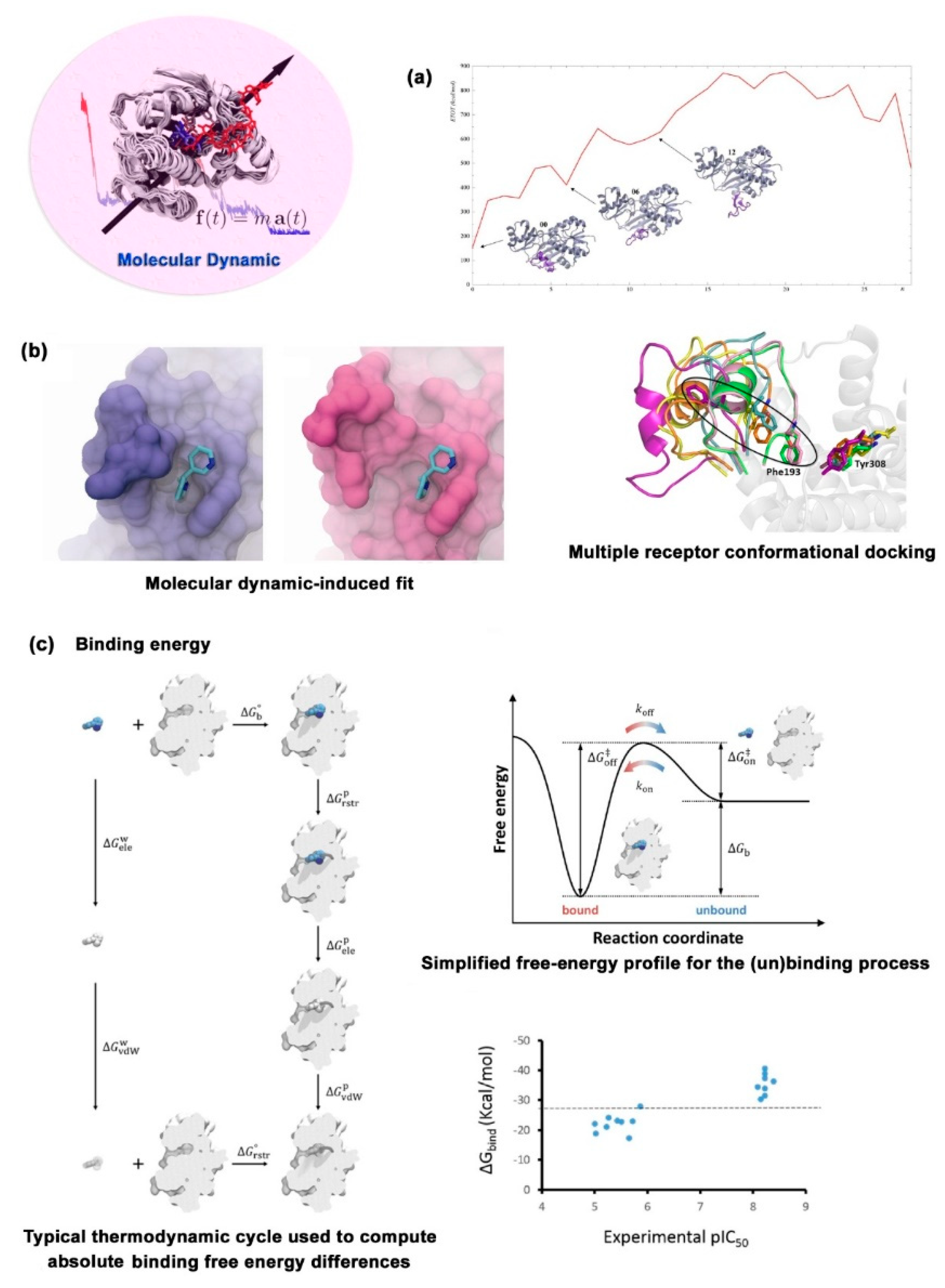
In the initial stage of molecular docking, molecular dynamics simulation mainly performs the work of “protein flexibility”. Different conformers of the target are extracted from the molecular dynamics simulation trajectory using “snapshots” as the initial structures for multiple receptor conformations (MRC) molecular docking, which nearly truthfully simulates the existing form of the target under physiological conditions. Merck company discovered the “mysterious binding pocket” of HIV integrase through MD simulation, and then discovered the highly potent antiviral drug Raltegravir by structure-based drug design targeting this site.[76] In the post-processing stage of docking, MD mainly carries out two aspects: “docking complex refinement” and “binding energy calculation”. The preferential conformational structure simulated by molecular docking was refined by considering the influence of solvent, complex flexibility and induced fit to obtain accurate ligand-receptor complex structure and interaction mode between ligand and receptor. Nowadays, molecular dynamic-induced fit (MD-IF) strategy has been widely used to refine the sub-binding pocket of active sites, find allosteric regulatory sites and improve the binding mode of ligand-receptor complexes.[70,71,72,73,74,75,77,78,79] The sophisticated intermolecular interactions between HIV-1 protease inhibitor Darunavir [80], Janus kinase 2 (JAK2) kinase inhibitor Ruxolitinib [81], TAF1 protein second bromine domain inhibitor acetylated lysine [82] and corresponding targets were predicted by MD-IF simulation. As for the ligand-receptor complex, MD simulation considers the influence of dominant solvents, especially the influence of water molecules in macromolecular structure and key water molecules near the binding site, which is critical for the accurate calculation of binding energy between receptor and ligand.[83] The structure of biomacromolecules and receptor-ligand interactions obtained by MD provides important clues for drug design and discovery; so MD has become one of the pivotal means in structure-based drug design.
2.1.4. Quantum Chemistry
Quantum chemistry (QC) is a method based on the basic principles of quantum mechanics (QM), reveals the interaction, transformation, structure and relationship between molecules in the chemical field by solving the Schrödinger equation and describing the motion behavior of microscopic particles.[84] At present, a relatively complete theoretical system and several calculation methods has been established in quantum chemistry, such as ab inito methods (ab initio, HF), density functional theory (DFT) and semi empirical methods (PM3, AM1, PM6, etc.).
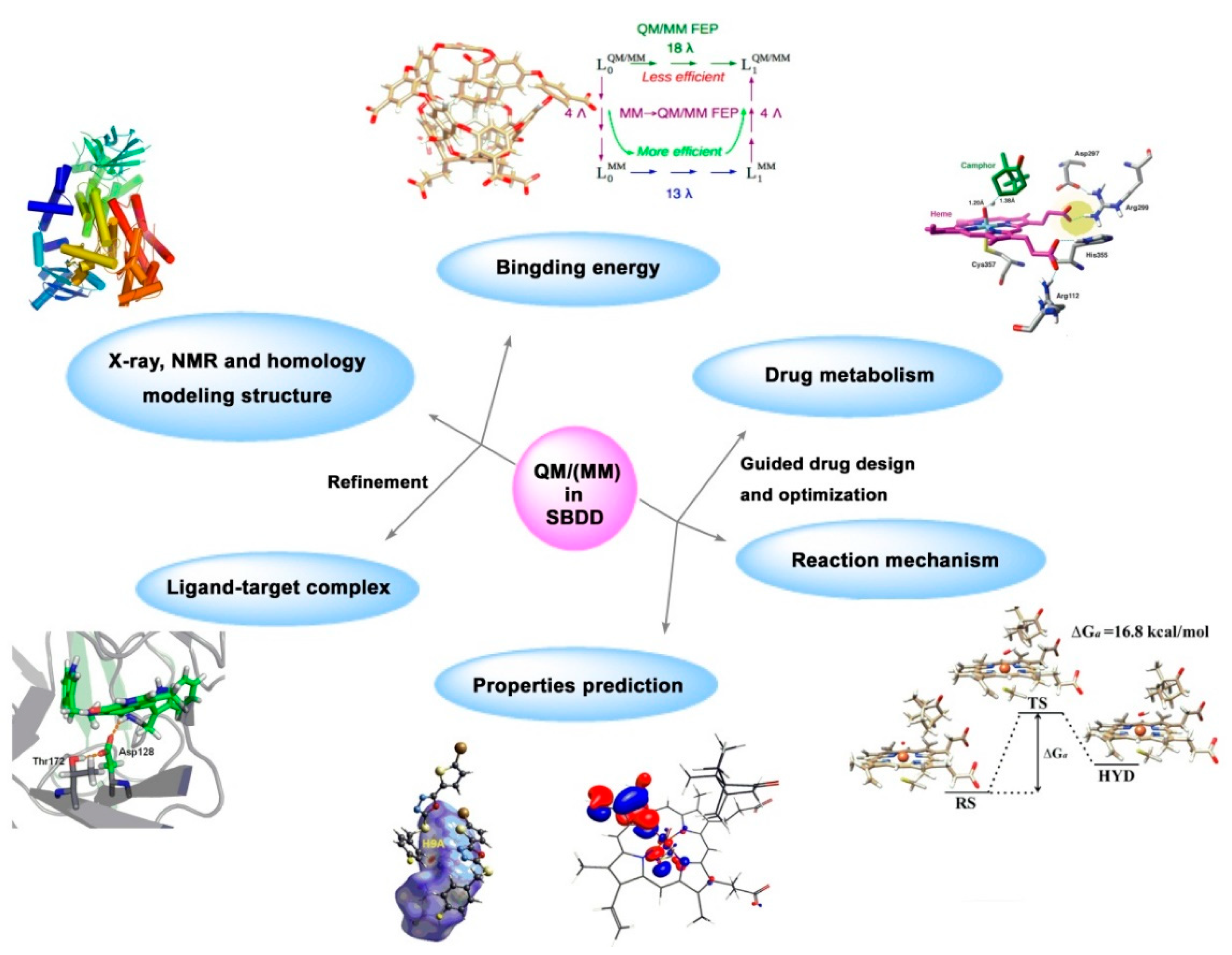
Since quantum chemistry calculation involves the interaction between electrons, it plays an irreplaceable role in many fields such as life science, medical science, material science and so on.[85,86,87,88,89] It breaks the boundaries between different disciplines and promotes interdisciplinary penetration. For example, quantum medicinal chemistry [90] plays a vital role in the discovery and development of innovative drugs, mainly used to explore the structures and properties of drug molecules, the interaction between biological macromolecules and ligands, and the chemical reaction mechanism. The drug targets are usually macromolecules, such as protein, receptors, nucleic acids, etc. For such large systems, the ab initio method and DFT need a lot of calculation time when dealing with a large number of electron correlations, and the semi-empirical method has poor accuracy. In order to overcome the above shortcomings, Martin Karplus et al. proposed the hybrid strategy of quantum mechanics and molecular mechanics (QM/MM) for the simulation of macromolecular systems in the 1990s, which has the characteristics of high calculation accuracy and fast calculation speed. Three scientists, including Martin Karplus, were awarded the 2013 Nobel Prize in Chemistry for their contribution on the development of QM/MM.[91,92,93,94,95] The basic algorithm of QM/MM method is to divide the whole molecular system into two layers. The core region of the study system (such as the active cavity in the ligand-receptor complex) is treated by QM method, while the peripheral region (such as the inactive cavity in the ligand-receptor complex and the solvent environment) is treated by MM method. QM/MM simulation model leads the calculated results are similar to those obtained by the QM calculation method for the entire system, owning to the consideration of the changes in electronic structure accurately.[96,97,98,99,100]
At present, QM/MM has gradually become a powerful tool in biology, crystal engineering, material science and supramolecule fields. In drug design, QM/MM is conducted to refine the structure of ligand-receptor complex, calculate the binding energy between ligand and receptor, predict physical and chemical properties (charge density, pKa, water solubility, etc.) of ligands, simulate the action of endogenous ligand, drug metabolism pathway and drug resistance mechanism.[101,102,103,104] In summary, QM/MM provides more refined structural and property parameters for structure-based drug design; and has been used to assist in the study of drug targets, such as tubulin [105], human carbonic anhydrase (hCAII) [106], cyclin-dependent kinase 2 (CDK2) [107] and lipoxygenase [108].
Figure 7.
Application of QM or QM/MM in structure-based drug design.
2.1.5. Molecular Docking-Molecular Dynamic-Quantum Chemistry
Molecular docking, molecular dynamics and quantum chemistry are the three commonly used methods in structure-based drug design; these methods have their own advantages and disadvantages. Generally speaking, molecular docking can quickly obtain the dominant conformer of ligand-receptor complex, and analyze the interaction between them preliminarily. Although the calculation speed of docking is the fastest among these three methods, while the calculation accuracy is the lowest. Molecular dynamics can provide the dynamic process of molecular motion over time-evolution at microscopic level. Although it can simulate the dynamic process of macromolecular systems in solution realistically, however, the molecular interaction cannot be accurately described due to the use of molecular force fields in MD algorithm. QM/MM hybrid method not only has high computational speed, but also ensures high computational accuracy. QM/MM can be used to predict the structure, properties, binding energy and interaction of ligand-acceptor complexes accurately, except for obtaining the global dynamic conformer of ligand-acceptor complexes. The combination of the three above-mentioned computation methods can enhance the strengths, circumvent the weaknesses and complement each other. A combined of docking, MD and QM/MM generally used to simulate structures and properties of receptors and ligands for SBDD. In terms of structure, it is possible to distinguish subtle differences between homologous receptors, obtain the structure of ligand receptor complexes accurately, and analyze the fine interactions between the ligand and receptor reasonably by the combined calculation method. As for properties, the combined method (docking-MD-QM/MM) can estimate the activity of small molecular drugs by calculating ligand-receptor binding energy; evaluate druglinkeness by predicting the metabolic site, lipid-water partition coefficient, atomic charge and other properties of the given molecule. At present, a variety of drug screening platforms have been established based on the combination of multi-molecular simulation technologies, among which the MD-high throughput screening-SBDD-MD-activity testing module built by Caflisch’s research group [109] has been applied to design and screen many targeted drugs efficiently and accurately.
2.1.6. Virtual Screening
Virtual screening (VS) technology is a method to screen potential active compounds by evaluating the binding energy between compounds and targets in small molecule databases.[117] Molecular docking is the ordinary used in VS process, so VS is a rapid and cheap drug screening method. The commonly used databases of small molecular compounds are Zinc (https://zinc.docking.org/), DrugBank (https://www.drugbank.com/), PubChem (https://pubchem.ncbi.nlm.nih.gov/), ChEMBL (https://pubchem.ncbi.nlm.nih.gov/source/ChEMBL), BindingDB (https://www.bindingdb.org/). Virtual screening has become a common method in drug screening, especially in screening of hit or lead with novel skeleton. Its great potential and value have been confirmed by the discovery of various drugs, such as cholinesterase inhibitors [118], Topoisomerase I inhibitors [119] and anti-coronavirus SARA-CoV-2 compounds[120].
As a rational, scientific and efficient drug design method, structure-based drug design plays an important role in the discovery of hit compounds, optimization of lead compounds and development of innovative drugs. Since Captopril, the first drug discovered by SBDD, came into the market, anti-HIV drugs Raltegravir and Amprenavir, anti-tumor drugs Imatinib and Ponatinib, anti-coagulant drugs Rivaroxaban and Dabigatran etexilate, anti-viral drugs Oseltamivir and Boceprevir and many other blockbuster drugs have come out one after another. As of July 2023, a total of over seventy drugs discovered by SBDD have been marketed, and a large number of excellent candidate drugs in clinical and preclinical research. Structure-based drug design has become a powerful means of designing, researching and developing innovative drugs in the field of medicinal chemistry. In addition, diversity oriented synthesis (DOS) [110], combinatorial chemistry [111], computer aided synthesis planning (CASP) [112], artificial intelligence (AI) [113,114,115,116] robot systems and other technologies were used to explore novel chemical reactions, which have increased the synthetic blocks in organic synthesis, broadened the space of the chemical universe and improved the possibility for pharmaceutical chemists to design a variety of ligand molecules. Therefore, we are bound to believe that SBDD will play an important role in the stage of drug research and development, and continue to promote the process of novel drugs discovery.
2.2. Ligand-Based Drug Design
2.2.1. Quantitative Structure-Activity Relationship
In 2002, at the 70th anniversary of the founding of the Chinese Chemical Society, Academician Guangxian Xu proposed “the quantitative relationship between molecular structure and its properties” as one of the “four century puzzles in chemistry” in the 21st century. According to a basic law of chemistry, “structure determines performance, and performance reflects structure”, so there must be a close relationship between the structure of a substance and its performance, and exploring the relationship between structure and performance has become one of the hot topics in chemical research.
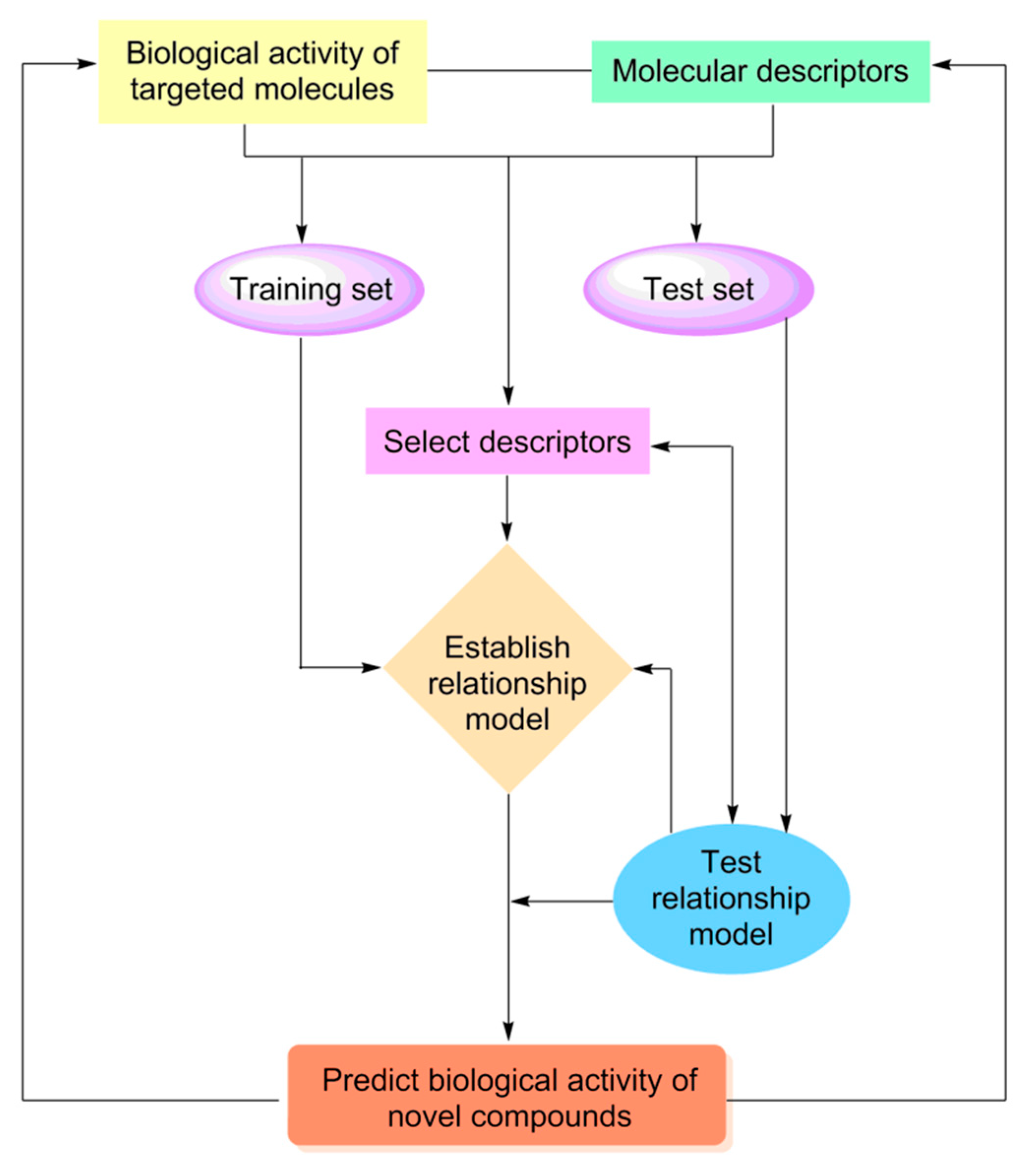
Quantitative structure–activity relationship (QSAR) is the mainstream method to explore the relationship between the structure and properties of compounds. The theoretical basis of this method is that the structural characteristics of a series of compounds with similar structures and the same mechanism of action are interrelated with their activities/properties.[121,122,123] In a broad sense, the activity in QSAR includes physiological and biochemical indexes (e.g. pharmacological activity, enzyme inhibition toxicity and neurotoxicity) and physical and chemical properties (e.g. solubility, retention time, lipid-water partition coefficient, reaction rate constant, boiling point and melting point).[122] In pharmaceutical research, the research thought of QSAR is: firstly, a mathematical model between the structures and activities/properties of known or assumed drug molecules or active compounds by statistical method to reveal the mechanism and mode of action of drug molecules; then, the obtained mathematical model used to quantitatively explain and predict the activities and properties of unknown compounds, so as to optimize and design drug molecules rationally and effectively. At present, QSAR is widely used in pharmaceutical field in the prediction of pharmacological activity of compounds, discovery and optimization of lead compounds, and evaluation absorption, distribution, metabolism, excretion and toxicity (ADMET) and other properties of drugs.[123,124,125,126,127,128]
QSAR is one of the earliest and most widely used strategies in ligand-based drug design filed. It has the characteristics of small amount of calculation, high accuracy and short calculation period; and plays an important role in predicting and screening compounds, especially suitable for rational drug design when the target structure is unknown. Quinolone antibiotics, monoamine oxidase (MAO) inhibitors, HIV-1 integrase inhibitors, proteolytic enzyme inhibitors and tyrosinase inhibitors are discovered by QSAR method.[129,130]
Figure 8.
The flow diagram of quantitative structure-activity relationship investigation.
2.2.2. DFT-Based Quantitative Structure-Activity Relationship
The primary work of QSAR research is to select appropriate and sufficient molecular structure descriptors which are crucial for establish a good QSAR model. The ideal descriptors can accurately and perfectly exhibit the structural information that affects the biological activity, so that the established QASR model not only has high predictive ability, but also has clear physical meaning which can be used to explain the mechanism of action of compounds.
Commonly used molecular descriptors include molecular surface area, molecular volume, lipid-water partition coefficient, Hammett electrical parameters, thermodynamic parameters and stereoscopic effect parameters, etc. The above descriptors are measured experimentally or obtained by empirical parameters fitting, which usually show the shortcomings of large experimental workload and inaccurate parameter data. Nowadays, the quantum chemistry parameters calculated by quantum chemistry calculations became an important approach to obtain molecular structure descriptors in QSAR research. Compared with traditional molecular descriptors, quantum chemical parameters demonstrate three advantages; firstly, quantum chemical parameters are more accurate and have clear physical and chemical significance; secondly, quantum chemical parameters can be fully simulated theoretically without experimental measured, which is efficient; thirdly, quantum chemistry parameters are not limited to obtained compounds, but also can evaluate the designed compounds in advance.[131,132] Thence, quantum chemical parameters have gradually penetrated into the field of QSAR research, especially, density functional theory-based quantitative structure-activity relationship (DFT-based QSAR) [133]. DFT-based QSAR has been widely used in the development stage of various types of drugs, such as adrenergic receptor inhibitors [134], calcium ion channel blockers [135], atty acid synthase inhibitors [136], protoporphyrinogen oxidase inhibitors [137], melanin-concentrating hormone receptor 1 inhibitors [138] and so on. In the study of kinase inhibitors, DFT-QSAR is applied to mammalian target of rapamycin (mTOR) kinase inhibitors [139], phosphatidylinositol 3 kinase (PI3K) inhibitors [140] and cyclin-dependent kinases (CDK) inhibitors [141].
Table 3.
Quantum-chemical descriptions.[133].
Table 3.
Quantum-chemical descriptions.[133].
| Definition | Name |
|---|---|
| Charges | |
| QA | net atomic charge on atom A |
| Qmin, Qmax | net charges of the most negative and most positive atoms |
| QAB | net group charge on atoms A, B |
| QT, QA | sum of absolute values of the charges of all the atoms in a given molecule |
| QT2, QA2 | sum of squares of the charges of all the atoms in a given molecule or functional group |
| Qm | mean absolute atomic charge (i.e. the average of the absolute values of the charges on all atoms) |
| HOMO and LUMO Energies | |
| EHOMO, ELUMO | energies of the highest occupied (HOMO) and (LUMO) molecular orbitals lowest unoccupied |
| ∆ELUMO-HOMO | HOMO and LUMO orbital energy difference |
| η = (ELUMO - EHOMO)/2 | hardness |
| S = 1/(ELUMO - EHOMO). | softness |
| ∆η = ηR - ηT | activation hardness, R and T stand for reactant and transition state |
| Molecular Polarizabilities | |
| α | molecular polarizability |
| α = (αxx + αyy + αzz)/3 | mean polarizability of the molecule |
| β2 = [(αxx - αyy)2 + (αyy - αzz)2 + (αzz - αxx)2] | anisotropy of the polarizability |
| Dipole Moments and Polarity Indices | |
| µ | molecular dipole moment |
| µchar, µ | charge and hybridization components of the dipole moment |
| µ2 | square of the molecular dipole moment |
| DX, DY, DZ | components of dipole moment along inertia axes |
| ∆ | submolecular polarity parameter (largest difference in electron charges between two atoms) |
| τ | quadrupole moment tensor |
| Energies | |
| E | total energy |
| H | Enthalpy |
| G | Gibbs free energy |
| S | entropy |
| IP | ionization potential |
| EA | electron affinity, difference in total energy between the neutral and anion radical species |
| Orbital Electron Densities | |
| qA, σ, qA, π | σ- and π-electron densities of the atom A |
| QA,H, QA,L | HOMO/LUMO electron densities on the atom A |
| FrE = frE/EHOMO | electrophilic atomic frontier electron densities |
| FrN = frN/ELUMO | |
| Atom-Atom Polarizabilities | |
| πAA, πAB | self-atom polarizabilities and atom-atom polarizabilities |
| Superdelocalizabilities | |
| SE, A, SN, A | electrophilic and nucleophilic superdel ocalizabilities |
2.2.3. Pharmacophore Modeling
In 1909, Ehrlich put forward the concept of pharmacophore, that is, the key structural features of compounds determine their biological activity. The earliest developed pharmacophore method is ligand-based pharmacophore, which aims to identify the common structural characteristics of compounds with similar pharmacological activities and different structures.[142,143,144,145] Pharmacophore is an important strategy for CADD research when the target structure is unknown. The commonly used automatic pharmacophore generation programs include Discovery Studio, PHASE, LigandScout and MOE.
2.2.4. Molecular Similarity
Molecular similarity refers to the structural similarity of two compounds, which is an important application of chemoinformatics method in CADD. The basic idea is that compounds with similar structures may have similar pharmaceutical activity. Drug screening based on molecular similarity is to screen out compounds with structural similarity from compound database by using active compounds as templates. Molecular similarity is not only an approach to screen hit or lead compound, but also guide for optimization of compounds.[146,147] For example, the principle of electron isosteric substitution drug design strategy is molecular similarity.
3. Kinase
Kinases participate in many important physiological processes of organisms; and an increasing number of signaling pathways have been confirmed to be closely related to substrate phosphorylation. The 1989 Nobel Prize was awarded to Bishop and Varmus for their discovery of protein kinases associated with tumors [148]; the 1992 Nobel Prize in Medicine was awarded to Fischer and Krebs for the discovery of reversibility protein phosphorylation process is a biological self-regulation mechanism, and the imbalance of intracellular substances can lead to the occurrence of diseases [149]; the 2001 Nobel Prize was awarded to Nurse and Hunt for their discovery of the important regulatory role of cell cycle dependent protein kinases in the cell cycle [150]. Numerous investigations have shown that substrate phosphorylation, kinases, and their regulatory mechanisms play an important role in the field of life sciences.
Figure 9.
Phosphorylation and dephosphorylation processes are catalyzed by kinase and protein phosphatase.
Figure 9.
Phosphorylation and dephosphorylation processes are catalyzed by kinase and protein phosphatase.
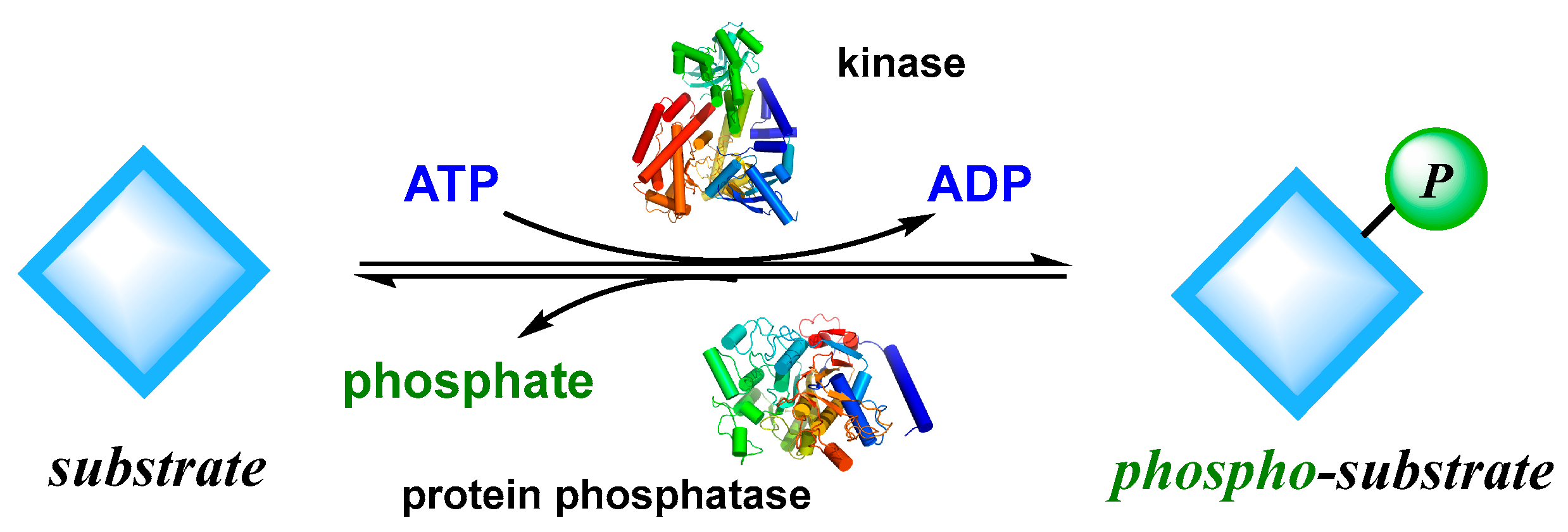
Reversible phosphorylation reactions can affect and regulate various functional processes of cells by regulating and balancing the activity of substrates, such as protein synthesis, gene expression, signal factor release, cell metabolism, morphological changes, apoptosis and so on. Abnormal phosphorylation level and function are closely related to many diseases such as tumor, inflammation, immunity, cardiovascular disease, neurodegenerative disease, and diabetes. Kinases have become one of the important targets in drug research and development; and kinase inhibitors have also become drugs for treating various important diseases. According to literature report, about one-third of drugs in research or development projects worldwide are related to kinases.
3.1. Structure and Function of Kinase
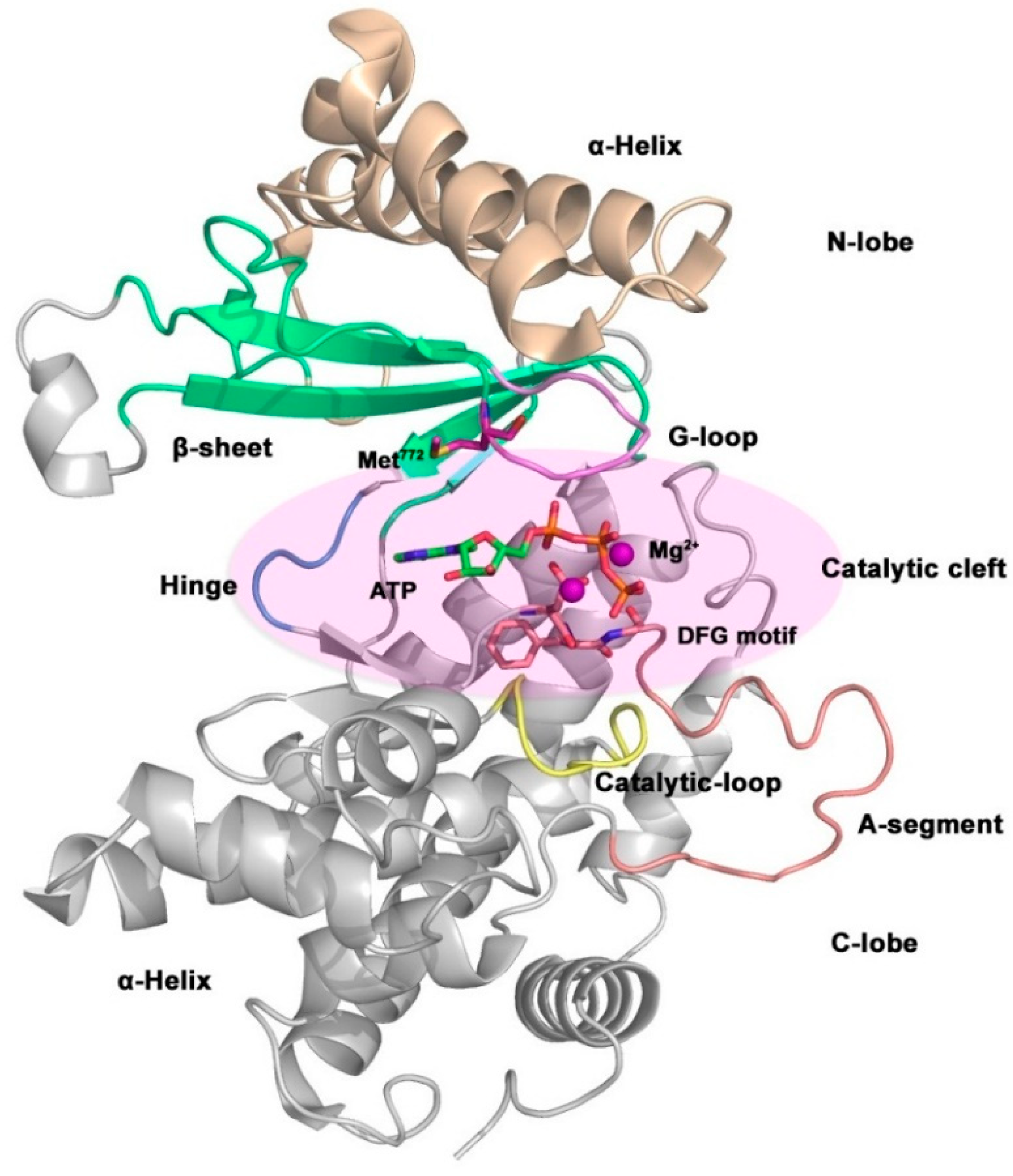
The general structure of kinase is shown in Figure 10, which mainly includes a catalytic domain, a helical domain, and multiple binding domains. The function of kinase is to realize the phosphorylation of substrate, so its catalytic domain is highly conserved and the three-dimensional structures of different kinases are very similar. The catalytic domain is composed of a bilobate region which linked by a hinge region, a G-loop region (also known as P-loop), a catalytic loop, and an activation loop (A-segment). Bilobate region located at the C and N-terminal of kinase, called C-lobe and N-lobe fragments, respectively. N-lobe is composed of five β-sheet and a α-helix; while the C-lobe is mainly composed of α-helix structures. The catalytic cleft between the bilobate region is ATP binding site and it’s also the binding site of most ATP competitive kinase inhibitors.[151,152,153]
Figure 10.
Overall crystal structure of the catalytic structure in p110α subunit (PDB code: 1IRK).
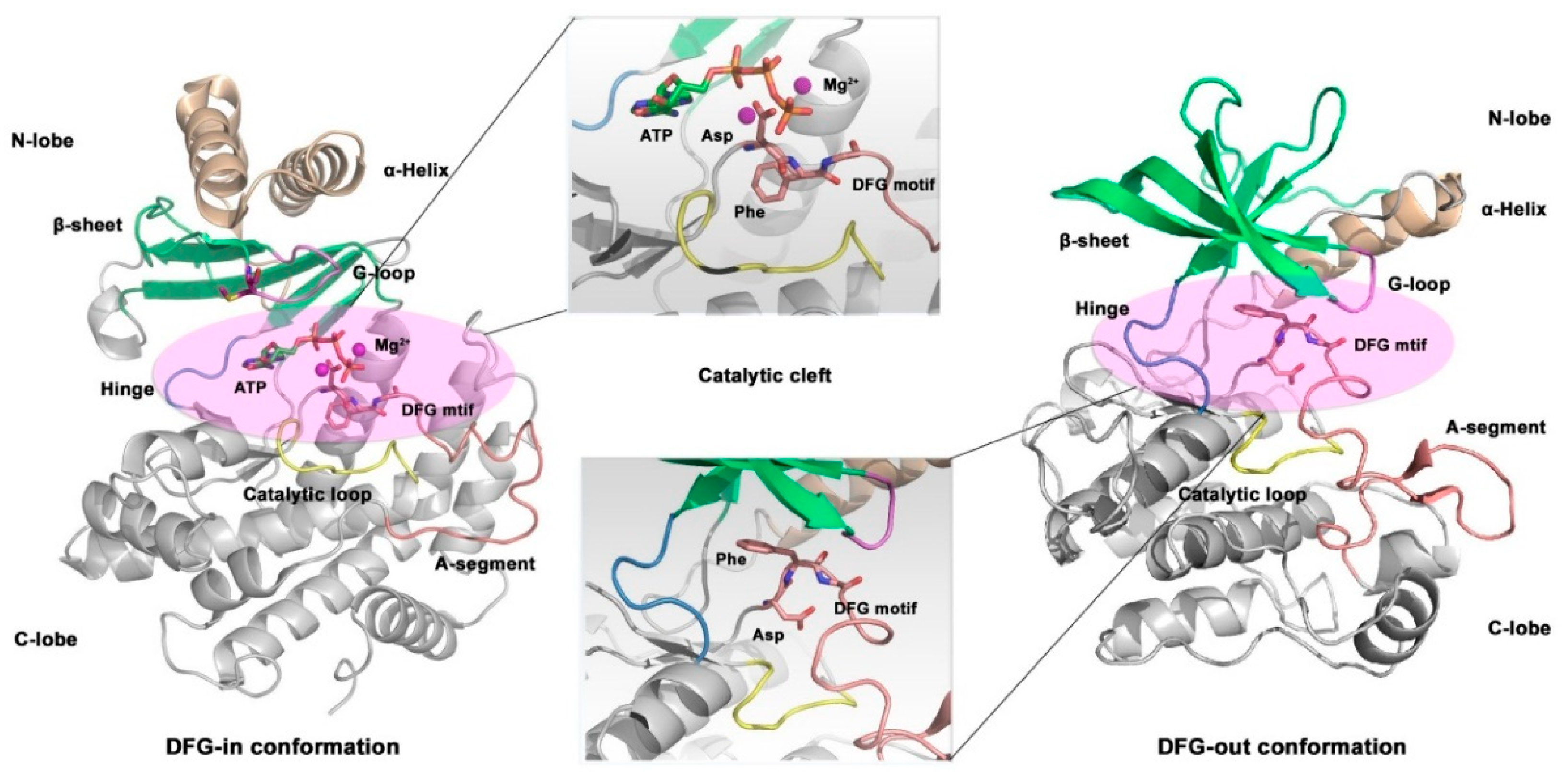
Kinase exists in two states, active and in-active states, acting as switches during signal transduction process. Under normal physiological conditions, most kinases are in the inactive state; and when kinases activated by upstream signals, they start the reaction of substrate phosphorylation. Asp-Phe-Gly motif (DFG motif) located at the N-terminal of the activation loop is recognized as playing an important role in regulating the kinase activity, the conformation of amino acid residues in DFG motif characterizes different states of kinase.[154,155,156] When kinase in the in-active state, the aspartate residue in the DFG motif is facing away from the ATP binding site, leading to the activation loop prevents the substrate from contacting with the active site, that is, the “DFG out” conformation. When kinase is in the active state, the conformation of DFG motif is reversed, and the aspartic acid residue faces the ATP binding site, so that the kinase can function normally, namely, the “DFG-in” conformation.[157]
Figure 11.
Overall crystal structure of the catalytic structure in p110α subunit (PDB code: 1IRK).
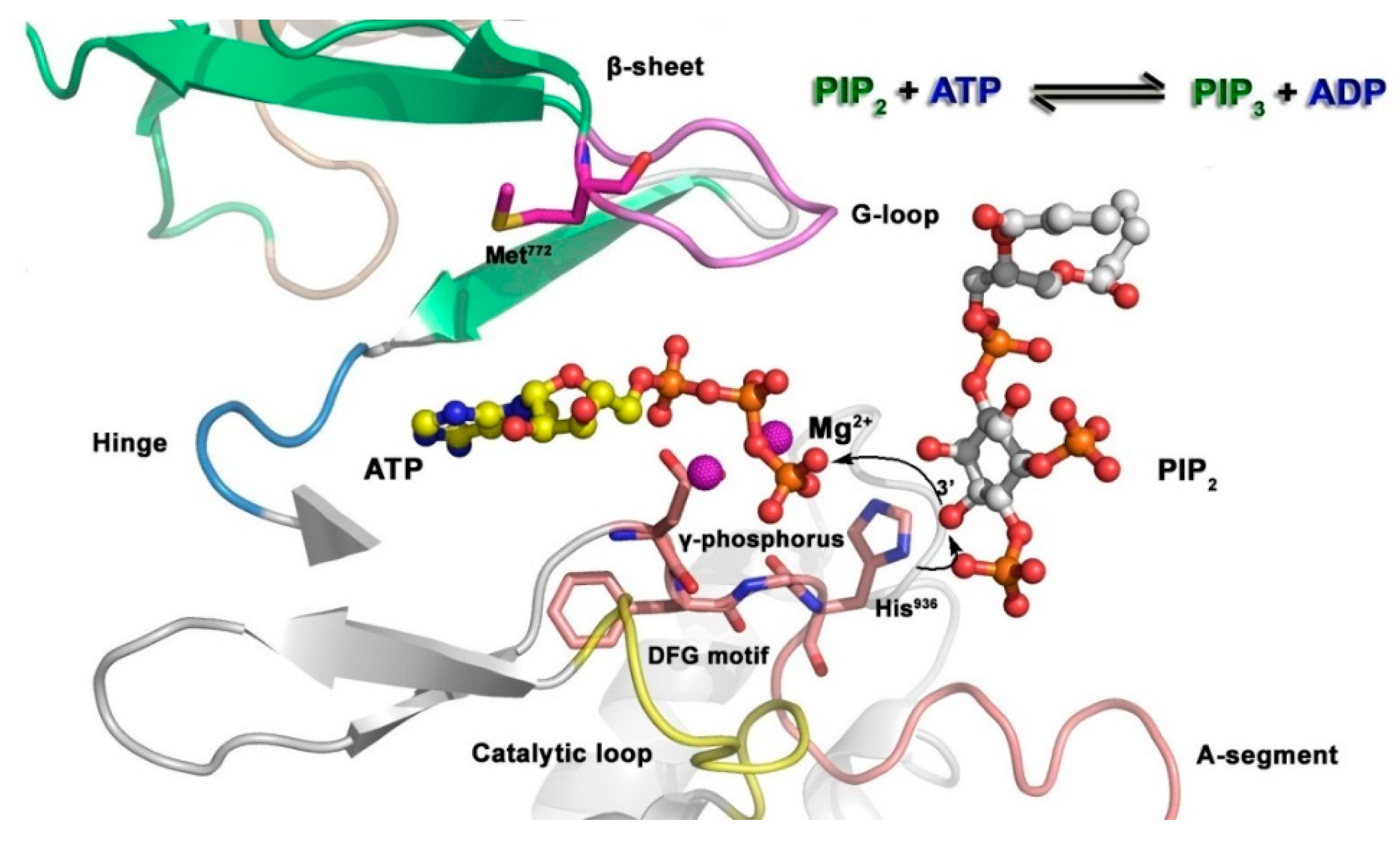
The mechanism of substrate phosphorylation catalyzed by kinases[158] is shown in Figure 12. Phosphatidylinositol 3 kinase (PI3K) is taken as the example to describe the phosphorylation of substrate: firstly, ATP and substrate phosphatidylinositol 4,5-biphosphate (PIP2) bind to the catalytic domain of PI3K, and the activation loop, phosphate groups of ATP and Mg2+ ions forming molecular interaction; then, His936 captures the protons from PIP2, allowing PIP2 as a nucleophile to attack γ-phosphoryl group; finally, the phosphoryl group transfers to PIP2 to generate phosphatidylinositol-3,4,5-triphosphate (PIP3), thus the whole phosphorylation process finished.
Figure 12.
Mechanism of the phosphorylation for p110α (PDB code: 1E8X). The oxygen of the PIP2 substrate attacks the γ-phosphorus of ATP. His936 serves as a catalytic base by removing the proton from the hydroxyl group of PIP2.
Figure 12.
Mechanism of the phosphorylation for p110α (PDB code: 1E8X). The oxygen of the PIP2 substrate attacks the γ-phosphorus of ATP. His936 serves as a catalytic base by removing the proton from the hydroxyl group of PIP2.
According to the substrate of the kinase, kinases can be divided into protein kinase, lipid kinase, carbohydrate kinase and other kinases. Protein kinases include eight categories, such as AGC (e.g. PKA, PKC, PKG and etc.), CMGC (e.g. CDK, CDKL, MAPK, CLK and etc.), CAMK (e.g. CaMKI, MLCK, eEF2K and etc.), CK1 (e.g. CK1, TTBK, VRK and etc.), STE (e.g. STE20, STE11, MAP4K and etc.), TK (e.g. RTK, CTK and etc.), TKL (e.g. MLK, MLKL, RAF and etc.) and atypical kinase (e.g. ATR, mTOR, ADCK1 and etc.). The hottest lipid kinase is PI3K. Up to data, over one million kinases related literature published, over 500 kinase related protein structures and over 900 kinase sequence information analyzed, and over four fifths of human kinase group related kinase determination methods determined.[159]
3.2. Small Molecule Kinase Inhibitors
Studies have shown that kinases are closely related to human diseases, so more and more pharmaceutical companies and research institutions are focusing on the development of kinase inhibitors. Kinase inhibitors include macromolecule antibody inhibitor and small molecule kinase inhibitor (SMKI). SMKI has advantages in oral administration, easy operation and low cost than antibody inhibitors, leading to SMKIs have become one of the hot spots in targeting drug research and development.[160]
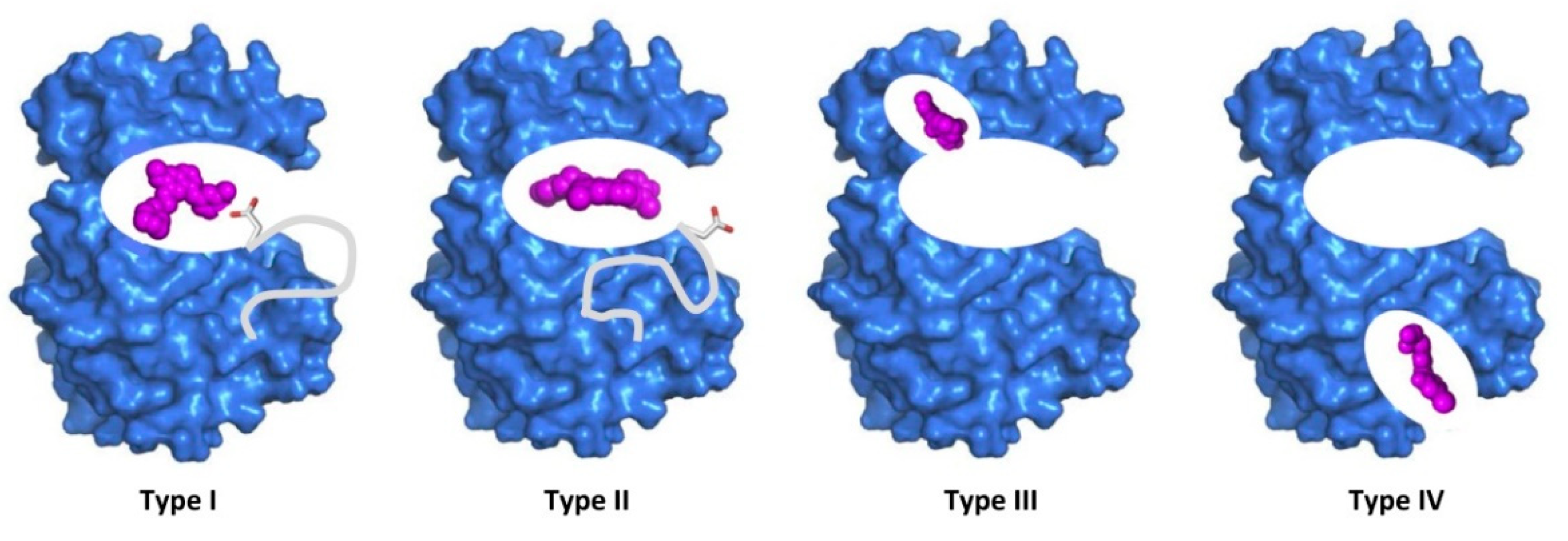
According to the binding mode of small molecule kinase inhibitor with target, SMKIs can be divided into reversible SMKIs and irreversible SMKIs. Irreversible SMKIs occupy the ATP-binding site by forming covalent bonds with residues; and then sealing the ATP-binding pocket, blocking kinase function. Reversible SMKIs can be divided into four types based on ligand binding sites and the conformation of DFG motif. Type I inhibitors ATP-competitive inhibitors that bind to the active conformation of the kinase with the aspartate residue (white backbone) of the DFG motif pointing into the ATP-binding pocket [161]; type II inhibitors bind and stabilize the inactive conformation of the kinase with the flipped aspartate residue facing outward of the binding pocket [162]; type III inhibitors occupy an allosteric pocket that is adjacent to the ATP-binding pocket but does not overlap with it [163]; type IV inhibitors bind to an allosteric pocket remote from the ATP-binding pocket [164].
Figure 13.
Four types of reversible binding mode. Type I inhibitors bind to the active conformation of the kinase with the aspartate residue (white backbone) of the DFG motif pointing into the ATP-binding pocket; type II inhibitors bind and stabilize the inactive conformation of the kinase with the flipped aspartate residue facing outward of the binding pocket; type III inhibitors occupy an allosteric pocket that is adjacent to the ATP-binding pocket but does not overlap with it; type IV inhibitors bind to an allosteric pocket remote from the ATP-binding pocket. Figure was taken from ref.160.
Figure 13.
Four types of reversible binding mode. Type I inhibitors bind to the active conformation of the kinase with the aspartate residue (white backbone) of the DFG motif pointing into the ATP-binding pocket; type II inhibitors bind and stabilize the inactive conformation of the kinase with the flipped aspartate residue facing outward of the binding pocket; type III inhibitors occupy an allosteric pocket that is adjacent to the ATP-binding pocket but does not overlap with it; type IV inhibitors bind to an allosteric pocket remote from the ATP-binding pocket. Figure was taken from ref.160.
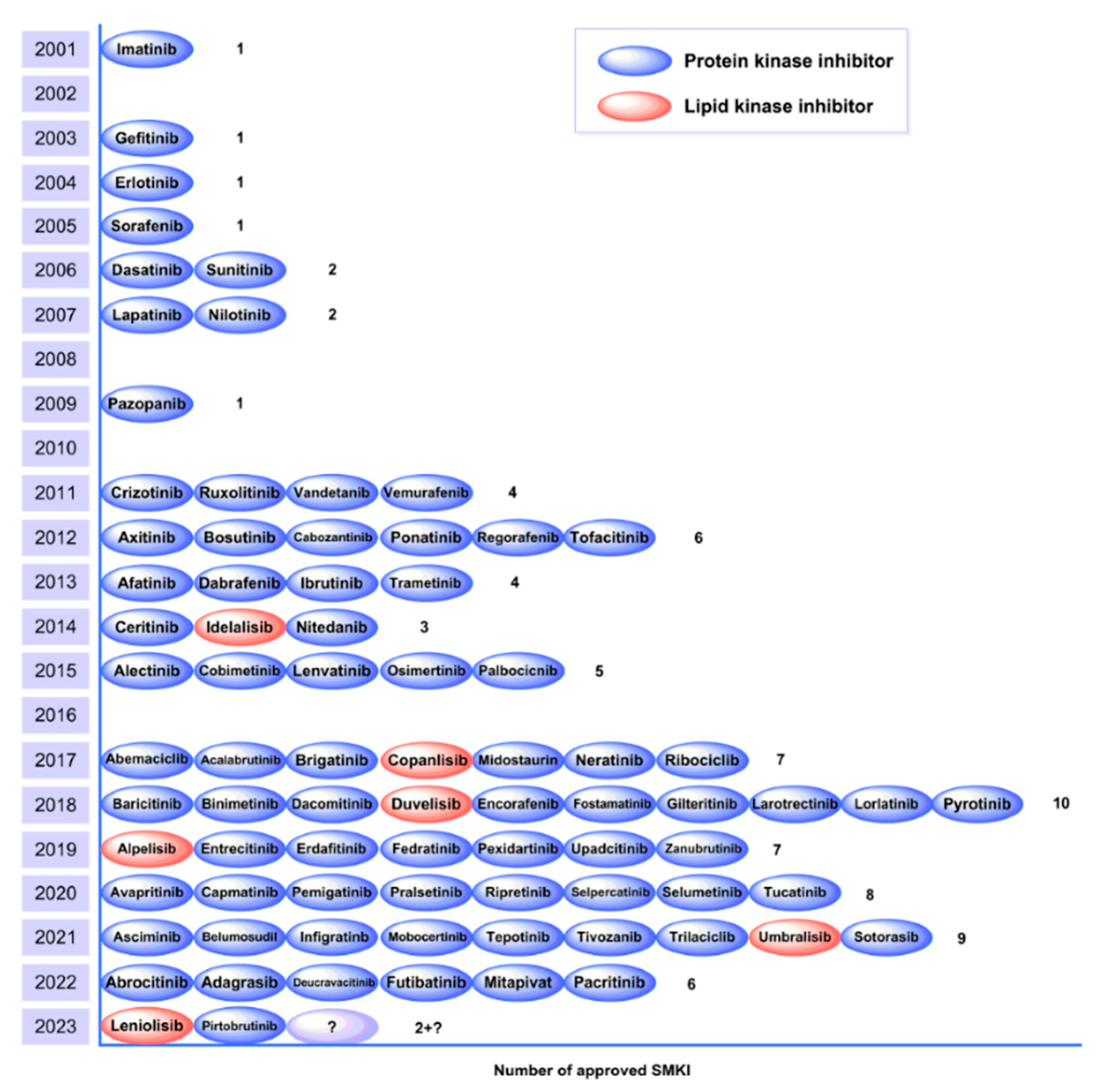
In 1950s, researchers realized that kinase inhibitors played a crucial role in signal transduction and then started the theoretical investigation of kinase inhibitors. Until the 1980s, the studies and application of epidermal growth factor receptor (EGFR) inhibitors opened a new chapter in the study of kinase inhibitors.[165,166] In 2001, Imatinib (Gleevc®), the first kinase inhibitor, was approved by the US Food and Drug Administration (FDA), which becoming a milestone in the research history of kinase inhibitors.[167] After the following twenty years, over seventy kinase inhibitors have been approved by FDA,[168] and hundreds of SMKIs are in preclinical and clinical research stage for the treatment of tumor, rheumatoid diseases, diabetes and so on. Among the marketed SMKIs, the sales of over ten drugs have reached or exceeded $1 billion in 2022, indicating that small molecule kinase inhibitors have become and will continue to be an important component of drug research and development.
Figure 14.
Number of approved SMKIs from 2001 to July 2023.
4. Small Molecule Kinase Inhibitors Discovered by CADD
Figure 15.
SMKIs discovered by CADD.
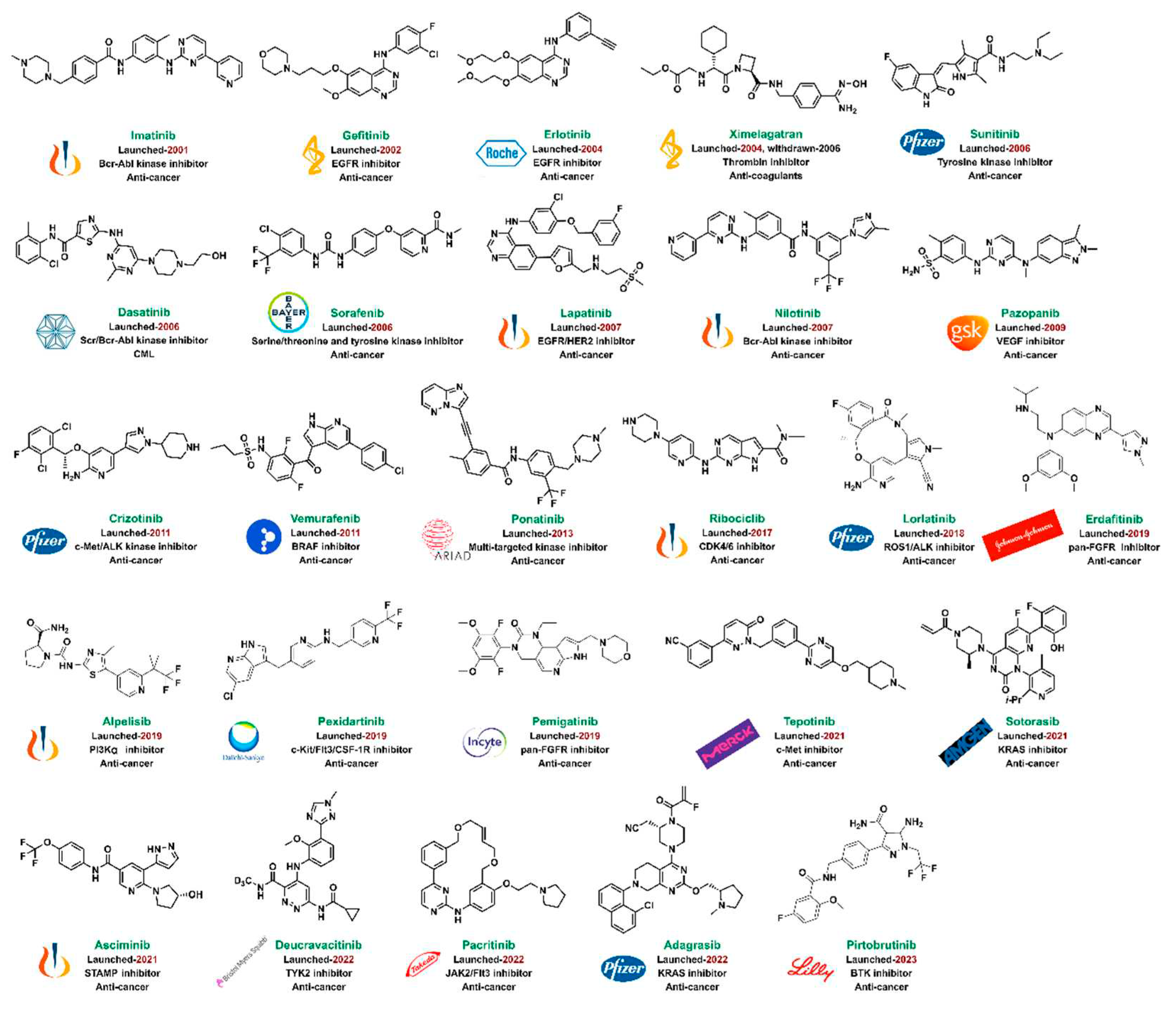
Kinase plays a key regulatory role in biological process, involving cell growth, differentiation, infiltration, angiogenesis and metastasis. There are at least 500 different kinases in human body, and it was initially believed that kinases may not be good drug targets, due to the highly conserved ATP-binding pocket of kinases, so it is a challenge to design specific kinase inhibitor.[25,26] Computer-aided drug design strategy can distinguish the subtle differences and structural characteristics of different kinase ATP binding site, and promote the discovery of kinase inhibitors. Imatinib, Abl inhibitor, was the first kinase inhibitor discovery by SBDD in 2001. Up to July 2023, totally 26 kinase inhibitors approved by FDA and hundreds compounds in clinic investigation were discovered by CADD method. Furthermore, the proportion of marketed kinase inhibitors guided by CADD has increased year by year. An overview of FDA-approved kinase inhibitors using the CADD approach since 2021 is provided below (Figure 16, Figure 17, Figure 18 and Figure 19).[169,170,171,172,173,174,175]
In order to have a better understanding of the CADD in the discovery of kinase inhibitors, Deucravacitinib, Adagrasib and Pirtobrutinib were selected to offer a detail description.
Deucravacitinib, a TYK2 inhibitor, was discovered by Bristol-Myers Squibb Company and approved by FDA in 2022. In the design process of Deucravacitinib, firstly, the sulfone group of lead compound was instead of amide group to break the interaction with conserved water, and then form H-bond with the target; secondly, triazole fragment introduced to the structure to improve potency and metabolic stability; then, the modification of aromatic ring to from hydrogen bondings with the target; finally, TYK2 inhibitor Deucravacitinib was obtained.
Adagrasib, a KRAS inhibitor, was approved by FDA in 2022. The investigator discovered the lead compound in-house which showed moderate inhibition against KRAS with poor pharmacokinetic properties. In order to improve the pharmacokinetic properties of lead compound, the investigator remove the hydroxyl group from the naphthalene ring to weaken O-atom metabolism. Then, the cyanide group is introduced into the piperazine ring in order to form hydrogen bondings with the amino acid residue of KRAS. Subsequently, chlorine atom was added into naphthalene ring in order to embed in hydrophobic cleft. Finally, electrophile is introduced to the olefinic bond to attenuate reactivity.
The discovery process of Pirtobrutinib is shown in Figure 18. Ibrtinib was selected as the lead compound, which contains three pharmacophore fragments, hinge region, H3 pocket (hydrophobic pocket) and covalent binding region (covalently binding with cysteine at position 481). The optimization process is carried out for the above three regions. The pyrazolopyrimidine of Ibtinib was broken to obtain a primary amide with an amino group in the ortho position of pyrazole, and the two substituent groups formed a pseudo-bicyclic ring through hydrogen bonding; the benzene ring connected by oxygen in H3 pocket was changed into an aromatic ring connected by amide methylene; and the covalent binding region was changed into a non-covalently bonded CF3-substituted ethyl group; and finally Pirtobrutinib was optimized.
5. Future Perspectives
Despite the impressive achievements of small molecule kinase inhibitors, the research on kinases and kinase inhibitors still needs to be further deepened. In terms of kinases, firstly, the detailed function of many kinases needs to be clarified; secondly, only one-fifth of human kinase targets have been reported with corresponding small-molecule kinase inhibitors, the druggability of other kinases need to be investigated. For the SMKIs concept, the selectivity and resistance of SMKIs are the key concerns; the launched SMKIs are mainly focus on type I and II inhibitors, and most of them are protein kinase inhibitors, the development of novel SMKIs cannot be underestimated. Structure-based drug design can be based on target structure for rational drug design; ligand-based drug design can extract the structural characteristics of active molecules. Computer-aided drug design, comprehensive consideration of ligand and receptor information, greatly reduce the blindness of the experiment, improve the screening efficiency of lead compounds and candidates, thus saving manpower, material and financial resources, shorten the drug development cycle effect. Given the widespread use of CADD in the development of kinase inhibitors, we believe that CADD will continue to promote kinase inhibitor research.
Author Contributions
Conceptualization, P.L. and Y.C.; data curation, L.L., S.L., Y.L., B.W. and S.X.; writing—original draft preparation, L.L., and S.L.; writing—review and editing, S.X., F.L. and Y.C.; visualization, L.L., S.L, Y.L. and B.W.; supervision, P.L. and Y. C.; project administration, L.L, P.L. and Y.C.; funding acquisition, L.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was financially supported by the Natural Science Foundation of Jiangsu Province (BK20200287).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zhao, L.; Ciallella, H.L.; Aleksunes, L.M.; et al. Advancing computer-aided drug discovery (CADD) by big data and data-driven machine learning modeling. Drug discovery today 2020, 25, 1624–1638. [Google Scholar] [CrossRef]
- Gomeni, R.; Bani, M.; D'angeli, C.; et al. Computer-assisted drug development (CADD): an emerging technology for designing first-time-in-man and proof-of-concept studies from preclinical experiments. European Journal of Pharmaceutical Sciences 2001, 13, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Finn. Application of SBDD to the discovery of new antibacterial drugs. In <i>Structure-Based Drug, Discovery</i>; Tari, L. (Eds.) Finn. Application of SBDD to the discovery of new antibacterial drugs. In Structure-Based Drug Discovery; Tari, L., Eds.; Humana Press: 2012.
- Acharya, C.; Coop, A.; Polli, J.E.; et al. Recent Advances in Ligand-Based Drug Design: Relevance and Utility of the Conformationally Sampled Pharmacophore Approach. Curr Comput Aided Drug Des 2011, 7, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Sabe, V.T.; Ntombela, T.; Jhamba, L.A.; et al. Current trends in computer aided drug design and a highlight of drugs discovered via computational techniques: A review. European Journal of Medicinal Chemistry 2021, 224, 113705. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.S.; Wimalasena, J. Estrogen regulates activity of cyclin-dependent kinases and retinoblastoma protein phosphorylation in breast cancer cells. Molecular Endocrinology 1996, 10, 488–498. [Google Scholar]
- Haldar, S.; Chintapalli, J.; Croce, C.M. Taxol Induces bcl-2 Phosphorylation and Death of Prostate Cancer Cells. Cancer Research 1996, 56, 1253–1255. [Google Scholar]
- Itoh, N.; Semba, S.; Ito, M.; et al. Phosphorylation of Akt/PKB is required for suppression of cancer cell apoptosis and tumor progression in human colorectal carcinoma. Cancer 2010, 94, 3127–3134. [Google Scholar] [CrossRef]
- Mcdonald, P.C.; Oloumi, A.; Mills, J.; et al. Rictor and integrin-linked kinase interact and regulate Akt phosphorylation and cancer cell survival. Cancer Research 2008, 68, 1618–1624. [Google Scholar] [CrossRef]
- Cordwell, S.J.; White, M.Y. Targeted proteomics for determining phosphorylation site-specific associations in cardiovascular disease. Circulation 2012, 126, 1803–1807. [Google Scholar] [CrossRef]
- Nishida, M.; Saiki, S.; Kitajima, N.; et al. ChemInform Abstract: Regulation of Cardiovascular Functions by the Phosphorylation of TRPC Channels. Cheminform 2011, 42, no. [Google Scholar] [CrossRef]
- Streeter, J.; Schickling, B.; Thiel, W.; et al. Nox1 Phosphorylation in Cardiovascular Disease. Free Radical Biology & Medicine 2012, 53, S175–S175. [Google Scholar]
- Wieland, T.; Attwood, P.V. Alterations in reversible protein histidine phosphorylation as intracellular signals in cardiovascular disease. Frontiers in Pharmacology 2015, 6, 173–173. [Google Scholar] [CrossRef] [PubMed]
- Ja, P.; et al. Targeted Deletion of AIF Decreases Mitochondrial Oxidative Phosphorylation and Protects from Obesity and Diabetes. Cell 2007, 131, 476–491. [Google Scholar]
- Liu, Y.; Liu, F.; Grundkeiqbal, I.; et al. Brain glucose transporters, O-GlcNAcylation and phosphorylation of tau in diabetes and Alzheimer's disease. Journal of Neurochemistry 2010, 111, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Szendroedi, J.; Schmid, A.I.; Chmelik, M.; et al. Muscle mitochondrial ATP synthesis and glucose transport/phosphorylation in type 2 diabetes. Plos Medicine 2007, 4, 154–154. [Google Scholar] [CrossRef]
- Liu, B.; Yang, Y.; Chernishof, V.; et al. Proinflammatory stimuli induce IKKalpha-mediated phosphorylation of PIAS1 to restrict inflammation and immunity. Cell 2007, 129, 903–914. [Google Scholar] [CrossRef]
- Caudle, R.M.; Perez, F.M.; Vallepinero AY, D.; et al. Spinal cord NR1 serine phosphorylation and NR2B subunit suppression following peripheral inflammation. Molecular Pain 2005, 1, 25–25. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Mimuro, H.; Kiga, K.; et al. Helicobacter pylori CagA Phosphorylation-Independent Function in Epithelial Proliferation and Inflammation. Cell Host & Microbe 2009, 5, 23–34. [Google Scholar]
- Wertz, I.E.; Newton, K.; Seshasayee, D.; et al. Phosphorylation and linear ubiquitin direct A20 inhibition of inflammation. Nature 2015, 528, 370–375. [Google Scholar] [CrossRef]
- Zhang, Y.; Qiu, J.; Wang, X.; et al. AMP-activated protein kinase suppresses endothelial cell inflammation through phosphorylation of transcriptional coactivator p300. Arterioscler Thromb Vasc Biol 2011, 31, 2897–2908. [Google Scholar] [CrossRef]
- BuãE, L.; BussiãRe, T.; BuãE-Scherrer V, et al. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev 2000, 33, 95–130. [Google Scholar] [CrossRef] [PubMed]
- Hanger, D.P.; Anderton, B.H.; Noble, W. Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends in Molecular Medicine 2009, 15, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Mp, M.; Pm, F. Untangling tau hyperphosphorylation in drug design for neurodegenerative diseases. Nature Reviews Drug Discovery 2007, 6, 464–479. [Google Scholar]
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; et al. Comprehensive analysis of kinase inhibitor selectivity. Nature Biotechnology 2011, 29, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Attwood, M.M.; Fabbro, D.; Sokolov, A.V.; et al. Trends in kinase drug discovery: targets, indications and inhibitor design. Nature Reviews Drug Discovery 2021, 20, 839–861. [Google Scholar] [CrossRef]
- Macalino, S.Y.; Vijayakumar, G.; Sunhye, H.; et al. Role of computer-aided drug design in modern drug discovery. Archives of Pharmacal Research 2015, 38, 1686–1701. [Google Scholar] [CrossRef]
- Dimasi, J.A.; Grabowski, H.G.; Hansen, R.W. Innovation in the pharmaceutical industry: New estimates of R&D costs. Journal of Health Economics 2016, 47, 20–33. [Google Scholar]
- Patani, G.A.; Lavoie, E. Bioisosterism: A Rational Approach in Drug Design. Chemical Reviews 1996, 96, 3147–3176. [Google Scholar] [CrossRef]
- Brown, F.K.; Sherer, E.C.; Johnson, S.A.; et al. The evolution of drug design at Merck Research Laboratories. Journal of Computer-Aided Molecular Design 2017, 31, 1–12. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- Cavasotto, C.N.; Phatak, S.S. Homology modeling in drug discovery: current trends and applications. Drug Discovery Today 2009, 14, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Lim-Wilby, M. Molecular docking. Methods in Molecular Biology 2008, 443, 365–382. [Google Scholar] [PubMed]
- Jones, G.; Willett, P.; Glen, R.C. A genetic algorithm for flexible molecular overlay and pharmacophore elucidation. Journal of Computer-Aided Molecular Design 1995, 9, 532–532. [Google Scholar] [CrossRef] [PubMed]
- Hopfinger, A.J.; Wang, S.; Tokarski, J.S.; Jin, B.; Albuquerque, M.; Madhav, P.J.; Duraiswami, C. Construction of 3D-QSAR Models Using the 4D-QSAR Analysis Formalism. Journal of the American Chemical Society 1997, 119, 10509–10524. [Google Scholar] [CrossRef]
- Durrant, J.D.; Andrew, M.C. Molecular dynamics simulations and drug discovery. BMC Biology 2011, 9, 71–71. [Google Scholar] [CrossRef] [PubMed]
- Keseru, G.M.; Kolossváry, I. Molecular mechanics and conformational analysis in drug design[M]. 1999.
- Gravenstein, S.; Johnston, S.L.; Loeschel, E.; et al. Zanamivir. Drug Safety 2001, 24, 1113–1125. [Google Scholar] [CrossRef]
- Noble, S.; Faulds, D. Saquinavir. Drugs 1996, 52, 93–112. [Google Scholar] [CrossRef]
- Silver, R.T. Imatinib mesylate (Gleevec (TM)) reduces phlebotomy requirements in polycythemia vera. Leukemia 2003, 17, 1186–1187. [Google Scholar] [CrossRef]
- Chirikjian, G.S. Conformational Modeling of Continuum Structures in Robotics and Structural Biology: A Review. Advanced Robotics the International Journal of the Robotics Society of Japan 2015, 29, 817–829. [Google Scholar] [CrossRef]
- Congreve, M.; Murray, C.W.; Blundell, T.L. Keynote review: Structural biology and drug discovery. Drug Discovery Today 2005, 10, 895–907. [Google Scholar] [CrossRef]
- Garratt, R. Structural biology and cancer. Bmc Proceedings 2013, 7, 1–1. [Google Scholar] [CrossRef]
- Holler, T.P.; Evdokimov, A.G.; Narasimhan, L. Structural biology approaches to antibacterial drug discovery. Expert Opinion on Drug Discovery 2007, 2, 1085–1101. [Google Scholar] [CrossRef] [PubMed]
- Penin, F.; Dubuisson, J.; Rey, F.A.; et al. Structural biology of hepatitis C virus. Clinics in Liver Disease 2003, 7, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Tomita, T.; Iwatsubo, T. Structural Biology of Presenilins and Signal Peptide Peptidases. Journal of Biological Chemistry 2013, 288, 14673–14680. [Google Scholar] [CrossRef]
- Fenalti, G.; Buckle, A.M. Structural biology of the GAD autoantigen. Autoimmunity Reviews 2010, 9, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; et al. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- Meng, X.Y.; Zhang, H.X.; Mezei, M.; et al. Molecular docking: a powerful approach for structure-based drug discovery. Current Computer-Aided Drug Design 2016, 7, 146–157. [Google Scholar] [CrossRef]
- Yuriev, E.; Holien, J.; Ramsland, P.A. Improvements, trends, and new ideas in molecular docking: 2012-2013 in review. Journal of Molecular Recognition 2015, 28, 581–604. [Google Scholar] [CrossRef] [PubMed]
- Yuriev, E.; Ramsland, P.A. Latest developments in molecular docking: 2010-2011 in review. Journal of Molecular Recognition Jmr 2013, 26, 215–239. [Google Scholar] [CrossRef]
- Chen, H.; Lyne, P.D.; Giordanetto, F.; et al. On evaluating molecular-docking methods for pose prediction and enrichment factors. Journal of Chemical Information & Modeling 2006, 46, 401–415. [Google Scholar]
- Dias, R.; Timmers, L.F.; Caceres, R.A.; et al. Evaluation of molecular docking using polynomial empirical scoring functions. Current Drug Targets 2008, 9, 1062–1070. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.D.; Jewsbury, P.J.; Essex, J.W. A review of protein-small molecule docking methods. Journal of Computer-Aided Molecular Design 2002, 16, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Yadav, D.K.; Kumar, S.; et al. Molecular docking, QSAR and ADMET studies of withanolide analogs against breast cancer. Drug Design Development & Therapy 2017, 11, 1859–1870. [Google Scholar]
- Nair, V.; Ma, X.; Shu, Q.; Zhang, F.; Uchil, V.; Cherukupalli, G.R. Impdh As A Biological Probe For Rna Antiviral Drug Discovery: Synthesis, Enzymology, Molecular Docking, And Antiviral Activity Of New Ribonucleosides With Surrogate Bases. Cheminform 2007, 26, 651–654. [Google Scholar]
- Ding, W.; Gu, J.; Cao, L.; et al. Traditional Chinese herbs as chemical resource library for drug discovery of anti-infective and anti-inflammatory. Journal of Ethnopharmacology 2014, 155, 589–598. [Google Scholar] [CrossRef]
- Balamurugan, R.; Stalin, A.; Ignacimuthu, S. Molecular docking of γ-sitosterol with some targets related to diabetes. European Journal of Medicinal Chemistry 2012, 47, 38–43. [Google Scholar] [CrossRef]
- Rayalu, D.J.; Selvaraj, C.; Singh, S.K.; et al. Homology modeling, active site prediction, and targeting the anti hypertension activity through molecular docking on endothelin-B receptor domain. Bioinformation 2012, 8, 81–86. [Google Scholar] [CrossRef]
- Mathew, B. Molecular Docking Studies of Some Novel Antidepressant 5-Substituted Phenyl-3-(Thiophen-2-yl)-4, 5-Dihydro-1h-Pyrazole-1-Carboxamides against Monoamine Oxidase Isoforms. Central Nervous System Agents in Medicinal Chemistry(Formerly Current Medicinal 2015, 16, 75–80. [Google Scholar] [CrossRef]
- Abdel-Aziz, A.A.; Eltahir, K.E.; Asiri, Y.A. Synthesis, anti-inflammatory activity and COX-1/COX-2 inhibition of novel substituted cyclic imides. Part 1: Molecular docking study. European Journal of Medicinal Chemistry 2011, 46, 1648–1655. [Google Scholar] [CrossRef]
- Cheng, K.; Zheng, Q.Z.; Qian, Y.; et al. Synthesis, antibacterial activities and molecular docking studies of peptide and Schiff bases as targeted antibiotics. Bioorg Med Chem 2009, 17, 7861–7871. [Google Scholar] [CrossRef]
- Ma, D.L.; Chan, S.H.; Leung, C.H. Molecular docking for virtual screening of natural product databases. Chemical Science 2011, 2, 1656–1665. [Google Scholar] [CrossRef]
- Ruyck, J.D.; Brysbaert, G.; Blossey, R.; et al. Molecular docking as a popular tool in drug design, an in silico travel. Advances & Applications in Bioinformatics & Chemistry Aabc 2016, 9, 1–11. [Google Scholar]
- Stark, J.L.; Powers, R. Application of NMR and molecular docking in structure-based drug discovery. Topics in Current Chemistry 2012, 326, 1–34. [Google Scholar] [PubMed]
- Stark, J.L.; Powers, R. Application of NMR and molecular docking in structure-based drug discovery. Topics in Current Chemistry 2012, 326, 1–34. [Google Scholar]
- Foroutan, M.; Fatemi, S.M.; Esmaeilian, F. A review of the structure and dynamics of nanoconfined water and ionic liquids via molecular dynamics simulation. European Physical Journal E 2017, 40, 19. [Google Scholar] [CrossRef]
- Komanduri; Raff, M. L. A review on the molecular dynamics simulation of machining at the atomic scale. Proceedings of the Institution of Mechanical Engineers Part B Journal of Engineering Manufacture 2001, 215, 1639–1672. [Google Scholar] [CrossRef]
- Car, R.; Parrinello, M. Unified approach for molecular dynamics and density-functional theory. Physical Review Letters 1985, 55, 2471. [Google Scholar] [CrossRef]
- Vivo, M.D.; Masetti, M.; Bottegoni, G.; et al. Role of Molecular Dynamics and Related Methods in Drug Discovery. Journal of Medicinal Chemistry 2016, 59, 4035–4061. [Google Scholar] [CrossRef]
- Galeazzi, R. Molecular Dynamics as a Tool in Rational Drug Design: Current Status and Some Major Applications. Current Computer-Aided Drug Design 2009, 5, 225–240. [Google Scholar] [CrossRef]
- Mortier, J.; Rakers, C.; Bermudez, M.; et al. The impact of molecular dynamics on drug design: applications for the characterization of ligand–macromolecule complexes. Drug Discovery Today 2015, 20, 686–702. [Google Scholar] [CrossRef]
- Perryman, A.L.; Lin, J.H.; Mccammon, J.A. Restrained molecular dynamics simulations of HIV-1 protease: the first step in validating a new target for drug design. Biopolymers 2010, 82, 272–284. [Google Scholar] [CrossRef]
- Durrant, J.D.; MxCammon, J.A. Molecular dynamic simulations and drug discovery. BMC Biology.
- Wereszczynski, J.; Mccammon, J.A. Accelerated molecular dynamics in computational drug design. Methods in Molecular Biology 2012, 819, 515. [Google Scholar]
- Zhao, H.; Caflisch, A. Molecular dynamics in drug design. European Journal of Medicinal Chemistry 2015, 91, 4–14. [Google Scholar] [CrossRef]
- Lin, J.H.; Perryman, A.L.; Schames, J.R.; et al. Computational drug design accommodating receptor flexibility: the relaxed complex scheme. Journal of the American Chemical Society 2002, 124, 5632. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.H.; De Groot, B.L. Ubiquitin dynamics in complexes reveal molecular recognition mechanisms beyond induced fit and conformational selection. Plos Computational Biology 2012, 8, e1002704. [Google Scholar] [CrossRef]
- Sotriffer, C.A.; Krämer, O.; Klebe, G. Probing flexibility and "induced-fit" phenomena in aldose reductase by comparative crystal structure analysis and molecular dynamics simulations. Proteins Structure Function & Bioinformatics 2010, 56, 52–66. [Google Scholar]
- Biswa Ranjan, M.; Yixuan, W. Interaction of I50V mutant and I50L/A71V double mutant HIV-protease with inhibitor TMC114 (darunavir): molecular dynamics simulation and binding free energy studies. Journal of Physical Chemistry B 2012, 116, 1884–1900. [Google Scholar]
- Zhou, T.; Georgeon, S.; Moser, R.; et al. Specificity and mechanism-of-action of the JAK2 tyrosine kinase inhibitors ruxolitinib and SAR302503 (TG101348). Leukemia 2014, 28, 404–407. [Google Scholar] [CrossRef]
- Spiliotopoulos, D.; Caflisch, A. Molecular Dynamics Simulations of Bromodomains Reveal Binding-Site Flexibility and Multiple Binding Modes of the Natural Ligand Acetyl-Lysine. Israel Journal of Chemistry 2015, 54, 1084–1092. [Google Scholar] [CrossRef]
- De, V.M.; Masetti, M.; Bottegoni, G.; et al. The Role of Molecular Dynamics and Related Methods in Drug Discovery. Journal of Medicinal Chemistry 2016, 59, 4035–406. [Google Scholar]
- Johnson, K.H. Quantum Chemistry. Annual Review of Physical Chemistry 1975, 26, 39–57. [Google Scholar] [CrossRef]
- Arnold, A.; Weigend, F.; Evers, F. Quantum chemistry calculations for molecules coupled to reservoirs: formalism, implementation, and application to benzenedithiol. Journal of Chemical Physics 2007, 126, 174101–174101. [Google Scholar] [CrossRef] [PubMed]
- Chernev, P.; Zaharieva, I.; Rossini, E.; et al. Merging Structural Information from X-ray Crystallography, Quantum Chemistry and EXAFS Spectra: The Oxygen Evolving Complex in PSII. Journal of Physical Chemistry B 2016, 120, 10899–10922. [Google Scholar] [CrossRef]
- Hernándezvaldés, D.; Alberto, R.; Jáureguihaza, U. Quantum chemistry calculations of technetium and rhenium compounds with application in radiopharmacy: review. Rsc Advances 2016, 6, 107127–107140. [Google Scholar] [CrossRef]
- Kim, W.Y.; Choi, Y.C.; Min, S.K.; et al. Application of quantum chemistry to nanotechnology: electron and spin transport in molecular devices. Cheminform 2009, 38, 2319–2333. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Min, G.; Zheng, X.; et al. Application of Quantum Chemistry Method in the Performance Evaluation and Mechanism Study of Corrosion Inhibitors. Corrosion & Protection 2017, 38, 829–833. [Google Scholar]
- Mannhold, R.; Kubinyi, H.; Folkers, G. Quantum Medicinal Chemistry[M]. 2005.
- Lyne, P.D.; Hodoscek, M.; Karplus, M. A Hybrid QM-MM Potential Employing Hartree-Fock or Density Functional Methods in the Quantum Region. Journal of Physical Chemistry A 1999, 103, 3462–3471. [Google Scholar] [CrossRef]
- Dinner, A.R.; Lopez, X.; Karplus, M. A charge-scaling method to treat solvent in QM/MM simulations. Theoretical Chemistry Accounts 2003, 109, 118–124. [Google Scholar] [CrossRef]
- Reuter, N.; Dejaegere, A.; Maigret, B.; Karplus, M. Frontier Bonds in QM/MM Methods: A Comparison of Different Approaches. Journal of Physical Chemistry A 2011, 104, 1720–1735. [Google Scholar] [CrossRef]
- Pezeshki, S.; Lin, H. Adaptive-Partitioning QM/MM for Molecular Dynamics Simulations: 4. Proton Hopping in Bulk Water. Journal of Chemical Theory & Computation 2015, 11, 2398–2411. [Google Scholar]
- Zhou, Y.Q. Martin Karplus Feeling of winning. Journal of Seeking Knowledge Guide 2013, 150–151. [Google Scholar]
- Cui, Q.; Karplus, M. Quantum Mechanical/Molecular Mechanical Studies of the Triosephosphate Isomerase-Catalyzed Reaction: Verification of Methodology and Analysis of Reaction Mechanisms. Journal of Physical Chemistry B 2002, 106, 1768–1798. [Google Scholar] [CrossRef]
- Chung, L.W.; Sameera, W.M.; Ramozzi, R.; et al. The ONIOM Method and Its Applications. Chemical Reviews 2015, 115, 5678–5796. [Google Scholar] [CrossRef]
- Svensson, M.; Humbel, S.; Froese RD, J.; et al. ONIOM: A Multilayered Integrated MO+MM Method for Geometry Optimizations and Single Point Energy Predictions. A Test for Diels-Alder Reactions and Pt(P(t-Bu)3)2+H2 Oxidative Addition. Journal of Physical Chemistry 1996, 100, 174–186. [Google Scholar] [CrossRef]
- Vreven, T.; Byun, K.S.; Komáromi, I.; et al. Combining Quantum Mechanics Methods with Molecular Mechanics Methods in, O.N.I.O.M. Journal of Chemical Theory & Computation 2006, 2, 815–826. [Google Scholar]
- Vreven, T.; Morokuma, K.; Farkas, O.; et al Geometry optimization with, Q.M.; et al. /.M.M.; ONIOM; other combined methods, I. Microiterations and constraints. Journal of Computational Chemistry 2003, 24, 760–769. [Google Scholar] [CrossRef] [PubMed]
- Adeniyi, A.A.; Soliman, M.E.S. Implementing QM in docking calculations: is it a waste of computational time? Drug Discovery Today 2017, 22, 1216–1223. [Google Scholar] [CrossRef] [PubMed]
- Chaskar, P.; Zoete, V.; Röhrig, U.F. On-the-fly QM/MM Docking with Attracting Cavities. Journal of Chemical Information & Modeling 2016, 57, 73–84. [Google Scholar]
- Lu, J.; Zhang, Z.; Zhong, N.; et al. QM/MM–PB/SA scoring of the interaction strength between Akt kinase and apigenin analogues. Computational Biology & Chemistry 2014, 52, 25–33. [Google Scholar]
- Raha K, Peters M B, Wang B, et al. The role of quantum mechanics in structure-based drug design. Drug Discovery Today 2007, 12, 725–731. [Google Scholar] [CrossRef]
- Kelly, E.B.; Tuszynski, J.A.; Klobukowski, M. QM and QM/MD simulations of the Vinca alkaloids docked to tubulin. Journal of Molecular Graphics & Modelling 2011, 30, 54–66. [Google Scholar]
- Adam, P.; Martin, L.; Jan, E.; et al. QM/MM calculations reveal the different nature of the interaction of two carborane-based sulfamide inhibitors of human carbonic anhydrase, I.I. Journal of Physical Chemistry B 2013, 117, 16096–16104. [Google Scholar]
- Alzate-Morales, J. Caballero Computational study of the interactions between guanine derivatives cyclin-dependent kinase 2 (CDK2) by, C.o.M.F.A.; QM/MM. Journal of Chemical Information & Modeling 2010, 50, 110–122. [Google Scholar]
- Cebriã, N.-P.A.; Rovira, T.; Saura, P.; et al. On the Inhibition of Mammalian 15-Lipoxygenase by Three Ebselen-like Drugs. A QM/MM and MM/PBSA Comparative Study. Journal of Physical Chemistry A 2017, 121, 9752–9763. [Google Scholar]
- Jung-Hsin, L.; Perryman, A.L.; Schames, J.R.; et al. Computational drug design accommodating receptor flexibility: the relaxed complex scheme. Journal of the American Chemical Society 2002, 124, 5632–5633. [Google Scholar]
- Nie, F.; Kunciw, D.L.; Wilcke, D.; et al. A Multidimensional Diversity-Oriented Synthesis Strategy for Structurally Diverse and Complex Macrocycles. Angew Chem Int Ed Engl 2016, 55, 11139–11143. [Google Scholar] [CrossRef]
- Lehn, J.M. Dynamic Combinatorial Chemistry and Virtual Combinatorial Libraries. Chemistry – A European Journal 2015, 5, 2455–2463. [Google Scholar] [CrossRef]
- Szymkuć, S.; Gajewska, E.P.; Klucznik, T.; et al. Computer-Assisted Synthetic Planning: The End of the Beginning. Angewandte Chemie 2016, 55, 5904–5937. [Google Scholar] [CrossRef]
- Shah, S.; Reddy, S.; Sardeshmukh, A.; et al. Application of Machine Learning Techniques for Inverse Prediction in Manufacturing Process Chains[M]. Springer International Publishing, 2015.
- Butler, K.T.; Davies, D.W.; Cartwright, H.; et al. Machine learning for molecular and materials science. Nature 2018, 559, 547–555. [Google Scholar] [CrossRef]
- Granda, J.M.; Donina, L.; Dragone, V.; et al. Controlling an organic synthesis robot with machine learning to search for new reactivity. Nature 2018, 559, 377–381. [Google Scholar] [CrossRef]
- Segler, M.H.S.; Preuss, M.; Waller, M.P. Planning chemical syntheses with deep neural networks and symbolic AI. Nature 2018, 555, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Schneider, G. Virtual screening: an endless staircase? Nature Reviews Drug Discovery 2010, 9, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Miles, J.A.; Ross, B.P. Recent Advances in Virtual Screening for Cholinesterase Inhibitors. Acs Chemical Neuroscience 2021, 12, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Kuma, V.; Kundu, B.; et al. Ligand-based Pharmacophore Modeling, Virtual Screening and Molecular Docking Studies for Discovery of Potential Topoisomerase I Inhibitors. Computational and Structural Biotechnology Journal 2019, 17, 291–310. [Google Scholar] [CrossRef]
- Gahlawat, A.; Kumar, N.; Kumar, R.; et al. Structure-Based Virtual Screening to Discover Potential Lead Molecules for the SARS-CoV-2 Main Protease. Journal of Chemical Information and Modeling 2020, 60, 5781–5793. [Google Scholar] [CrossRef]
- Dudek, A.Z.; Tomasz, A.; Jorge, G. Computational methods in developing quantitative structure-activity relationships (QSAR): a review. Combinatorial Chemistry & High Throughput Screening 2006, 9, 213–228. [Google Scholar]
- Chen, B. Development of quantitative structure activity relationship (QSAR) model for disinfection byproduct (DBP) research: A review of methods and resources. Journal of Hazardous Materials.
- Lill, M.A. Multi-dimensional QSAR in drug discovery. Drug Discovery Today 2007, 12, 1013–1017. [Google Scholar] [CrossRef]
- Ghafourian, T.; Zandasrar, P.; Hamishekar, H.; et al. The effect of penetration enhancers on drug delivery through skin: a QSAR study. Journal of Controlled Release 2004, 99, 113–125. [Google Scholar] [CrossRef]
- Vilar, S.; Cozza, G. ; S Medicinal chemistry and the molecular operating environment (MOE): application of QSAR and molecular docking to drug discovery. Current Topics in Medicinal Chemistry 2008, 8, 1555–1572. [Google Scholar] [CrossRef]
- Devillers. Neural Networks in QSAR and Drug Design[M]. City: Harcourt Brace, 1996.
- Low, Y.; Uehara, T.; Minowa, Y.; et al. Predicting Drug-induced Hepatotoxicity Using QSAR and Toxicogenomics Approaches. Chemical Research in Toxicology 2011, 24, 1251–1262. [Google Scholar] [CrossRef]
- Yoshida, F.; Topliss, J.G. QSAR model for drug human oral bioavailability. Journal of Medicinal Chemistry 2000, 43, 2575–2585. [Google Scholar] [CrossRef]
- Viskupicova, J.; Danihelova, M.; Majekova, M.; et al. Polyphenol fatty acid esters as serine protease inhibitors: a quantum-chemical QSAR analysis. J Enzyme Inhib Med Chem 2012, 27, 800–809. [Google Scholar] [CrossRef] [PubMed]
- Nongonierma, A.; Fitzgerald, D. Learnings from quantitative structure activity relationship (QSAR) studies with respect to food protein-derived bioactive peptides: A review. Rsc Advances 2016, 6, 75400–75413. [Google Scholar] [CrossRef]
- Mccoy, E.F.; Sykes, M. Quantum-mechanical QSAR/QSPR descriptors from momentum-space wave functions. Journal of Chemical Information & Computer Sciences 2003, 43, 545–553. [Google Scholar]
- Gozalbes, R.; Doucet, J.P.; Derouin, F. Application of topological descriptors in QSAR and drug design: history and new trends. Current Drug Targets - Infectious Disorders 2002, 2, 93–102. [Google Scholar] [CrossRef]
- Karelson, M.; Lobanov, V.S.; Katritzky, A.R. Quantum-Chemical Descriptors in QSAR/QSPR Studies. Chemical Reviews 1996, 96, 1027–1044. [Google Scholar] [CrossRef] [PubMed]
- Borota A, Mracec M, Gruia A, et al. A QSAR study using MTD method and Dragon descriptors for a series of selective ligands of αC adrenoceptor. European Journal of Medicinal Chemistry 2011, 46, 877–884. [Google Scholar] [CrossRef]
- Davood, A.; Nematollahi, A.; Iman, M.; et al. Computational studies of new 1,4-dihydropyridines containing 4-(5)-chloro-2-ethyl-5-(4)-imidazolyl substituent: QSAR and docking. Medicinal Chemistry Research 2010, 19, 58–70. [Google Scholar] [CrossRef]
- Milan, M.; Mirjana, M.; Desanka, B.; et al. In VitroAntioxidant Activity of Selected 4-Hydroxy-chromene-2-one Derivatives-SAR, QSAR and DFT Studies. International Journal of Molecular Sciences 2011, 12, 2822–2841. [Google Scholar]
- Zhang, L.; Wan, J.; Yang, G. A DFT-based QSARs study of protoporphyrinogen oxidase inhibitors: phenyl triazolinones. Bioorganic & Medicinal Chemistry 2004, 12, 6183–6191. [Google Scholar]
- Morales-Bayuelo, A.; Matute, R.A.; Caballero, J. Understanding the comparative molecular field analysis (CoMFA) in terms of molecular quantum similarity and DFT-based reactivity descriptors. Journal of Molecular Modeling 2015, 21, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ran, T.; Lu, T.; Yuan, H.; et al. A selectivity study on mTOR/PI3Kα inhibitors by homology modeling, 3.D.-Q.S.A.R. Journal of Molecular Modeling 2012, 18, 171–186. [Google Scholar] [CrossRef] [PubMed]
- Bharate, S.B.; Singh, B.; Bharate, J.B.; et al. QSAR and Pharmacophore Modeling of N-Acetyl-2-aminobenzothiazole Class of Phosphoinositide-3-kinase-α Inhibitors. Medicinal Chemistry Research 2012, 22, 890–899. [Google Scholar] [CrossRef]
- Wrã³bel, T.M.; Kieå‚Bus, M.; Kaczor, A.A.; et al. Discovery of nitroaryl urea derivatives with antiproliferative properties. J Enzyme Inhib Med Chem 2016, 31, 608–618. [Google Scholar] [CrossRef]
- Yang, S.Y. Pharmacophore modeling and applications in drug discovery: challenges and recent advances. Drug Discovery Today 2010, 15, 444–450. [Google Scholar] [CrossRef]
- Schaller, D.; Šribar, D.; Noonan, T.; et al. Next generation 3D pharmacophore modeling. Wiley Interdisciplinary Reviews 2020, 10, e1468. [Google Scholar] [CrossRef]
- Giordano, D.; Biancaniello, C.; Argenio, M.A.; et al. Drug Design by Pharmacophore and Virtual Screening Approach. Pharmaceuticals 2022, 15, 646. [Google Scholar] [CrossRef]
- Seidel, T.; Wieder, O.; Garon, A.; et al. Applications of the Pharmacophore Concept in Natural Product inspired Drug Design. Molecular Informatics 2020, 39, 2000059. [Google Scholar] [CrossRef]
- Muhammed, M.T.; Akl-Yalcin, E. Pharmacophore Modeling in Drug Discovery: Methodology and Current Status. Journal of the Turkish Chemical Society Section A; Chemistry 2021, 8, 748–762. [Google Scholar] [CrossRef]
- Seidel, T.; Schuetz, D.A.; Garon, A.; et al. The Pharmacophore Concept and Its Applications in Computer-Aided Drug Design. Progress in the Chemistry of Organic Natural Products 2019, 110, 99–141. [Google Scholar]
- Levinson, A.D.; Oppermann, H.; Levintow, L.; et al. Evidence that the transforming gene of avian sarcoma virus encodes a protein kinase associated with a phosphoprotein. Cell 1978, 15, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Lerea, K.M.; Tonks, N.K.; Krebs, E.G.; et al. Vanadate and molybdate increase tyrosine phosphorylation in a 50-kilodalton protein and stimulate secretion in electropermeabilized platelets. Biochemistry 1989, 28, 9286–9292. [Google Scholar] [CrossRef] [PubMed]
- Erickson, A.K.; Payne, D.M.; Martino, P.A.; et al. Identification by mass spectrometry of threonine 97 in bovine myelin basic protein as a specific phosphorylation site for mitogen-activated protein kinase. Journal of Biological Chemistry 1990, 265, 19728–19735. [Google Scholar] [CrossRef] [PubMed]
- Endicott, J.A.; Johnson, L.N. Protein Kinase Inhibitors: Insights into Drug Design from Structure. Science 2004, 303, 1800–1805. [Google Scholar]
- Hiles, I.D.; Otsu, M.; Volinia, S.; et al. Phosphatidylinositol 3-kinase: structure and expression of the 110 kd catalytic subunit. Cell 1992, 70, 419–429. [Google Scholar] [CrossRef]
- Wymann, M.P.; Pirola, L. Structure and function of phosphoinositide 3-kinases. Biochim Biophys Acta 1998, 1436, 127–150. [Google Scholar] [CrossRef]
- Liu, Y.; Gray, N.S. Liu, Y. & Gray, N.S. Rational design of inhibitors that bind to inactive kinase conformations. Nat. Chem. Biol. 2, 358-364. Nature Chemical Biology 2006, 2, 358–364. [Google Scholar]
- Tengholm, A.; Meyer, T. A PI3-Kinase Signaling Code for Insulin-Triggered Insertion of Glucose Transporters into the Plasma Membrane. Current Biology 2002, 12, 1871–1876. [Google Scholar] [CrossRef]
- Ung, P.M.; Schlessinger, A. DFGmodel: predicting protein kinase structures in inactive states for structure-based discovery of type-II inhibitors. Acs Chemical Biology 2015, 10, 269–278. [Google Scholar] [CrossRef]
- Mk, A.; Jd, V. Small molecule inhibitors of phosphoinositide 3-kinase (PI3K) delta and gamma. Current Topics in Medicinal Chemistry 2009, 9, 738–753. [Google Scholar]
- Johnson, L.N.; Lewis, R. Structural basis for control by phosphorylation. Chemical Reviews 2001, 101, 2209–2209. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Properties of FDA-approved small molecule protein kinase inhibitors. Pharmacological Research 2023, 187, 106552. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Nielsen, T.E.; Clausen, M.H. FDA-approved small-molecule kinase inhibitors. Trends in Pharmacological Sciences 2015, 36, 422–439. [Google Scholar]
- Noble ME, M.; Endicott, J.A.; Johnson, L.N. Protein kinase inhibitors: insights into drug design from structure. Science 2004, 303, 1800–1805. [Google Scholar] [CrossRef]
- Norman, R.A.; Toader, D.; Ferguson, A.D. Structural approaches to obtain kinase selectivity. Trends in Pharmacological Sciences 2012, 33, 273–278. [Google Scholar] [CrossRef]
- Cox, K.J.; Shomin, C.D.; Ghosh, I. Tinkering outside the kinase ATP box: allosteric (type IV) and bivalent (type V) inhibitors of protein kinases. Future Medicinal Chemistry 2011, 3, 29–43. [Google Scholar] [CrossRef]
- Lamba, V.; Ghosh, I. New directions in targeting protein kinases: focusing upon true allosteric and bivalent inhibitors. Current Pharmaceutical Design 2012, 18, 2936–2945. [Google Scholar] [CrossRef]
- Gazit, A.; Yaish, P.; Gilon, C.; et al. Tyrphostins I: synthesis and biological activity of protein tyrosine kinase inhibitors. Journal of Medicinal Chemistry 1989, 32, 2344–2352. [Google Scholar] [CrossRef]
- Yaish, P.; Gazit, A.; Gilon, C.; et al. Blocking of EGF-dependent cell proliferation by EGF receptor kinase inhibitors. Science 1988, 242, 933–935. [Google Scholar] [CrossRef]
- Capdeville, R.; Buchdunger, E.; Zimmermann, J.; et al. Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nature Reviews Drug Discovery 2002, 1, 493–502. [Google Scholar] [CrossRef]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. Small-molecule kinase inhibitors: an analysis of FDA-approved drugs. Drug Discovery Today 2016, 21, 5–10. [Google Scholar] [CrossRef]
- Dorsch, D.; Schadt, O.; Stieber, F.; et al. Identification and optimization of pyridazinones as potent and selective c-Met kinase inhibitors. Bioorganic & Medicinal Chemistry Letters 2015, 25, 1597–1602. [Google Scholar]
- Lanman, B.A.; Allen, J.R.; Allen, J.G.; et al. Discovery of a covalent inhibitor of KRASG12C (AMG 510) for the treatment of solid tumors. Journal of Medicinal Chemistry 2020, 63, 52–65. [Google Scholar] [CrossRef] [PubMed]
- Schoepfer, J.; Jahnke, W.; Berellini, G.; et al. Discovery of asciminib (ABL001), an allosteric inhibitor of the tyrosine kinase activity of BCR-ABL1. Journal of Medicinal Chemistry 2018, 61, 8120–8135. [Google Scholar] [CrossRef] [PubMed]
- William, A.D.; Lee AC, H.; Blanchard, S.; et al. Discovery of the Macrocycle 11-(2-Pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (SB1518), a Potent Janus Kinase 2/Fms-Like Tyrosine Kinase-3 (JAK2/FLT3) Inhibitor for the Treatment of Myelofibrosis and Lymphoma. Journal of Medicinal Chemistry 2011, 54, 4638–4658. [Google Scholar]
- Wrobleski, S.T.; Moslin, R.; Lin, S.; et al. Highly Selective Inhibition of Tyrosine Kinase 2 (TYK2) for the Treatment of Autoimmune Diseases: Discovery of the Allosteric Inhibitor BMS-986165. Journal of Medicinal Chemistry 2019, 62, 8973–8995. [Google Scholar] [CrossRef]
- Fell, J.B.; Fischer, J.P.; Baer, B.R.; et al. Identification of the Clinical Development Candidate MRTX849, a Covalent KRASG12C Inhibitor for the Treatment of Cancer. Journal of Medicinal Chemistry 2020, 63, 6679–6693. [Google Scholar] [CrossRef]
- Nivolas, G. Compounds useful as kinase inhibitors. WO201710 3611.
Figure 16.
The discovery process of Deucravacitinib by computer-aided drug design strategy.
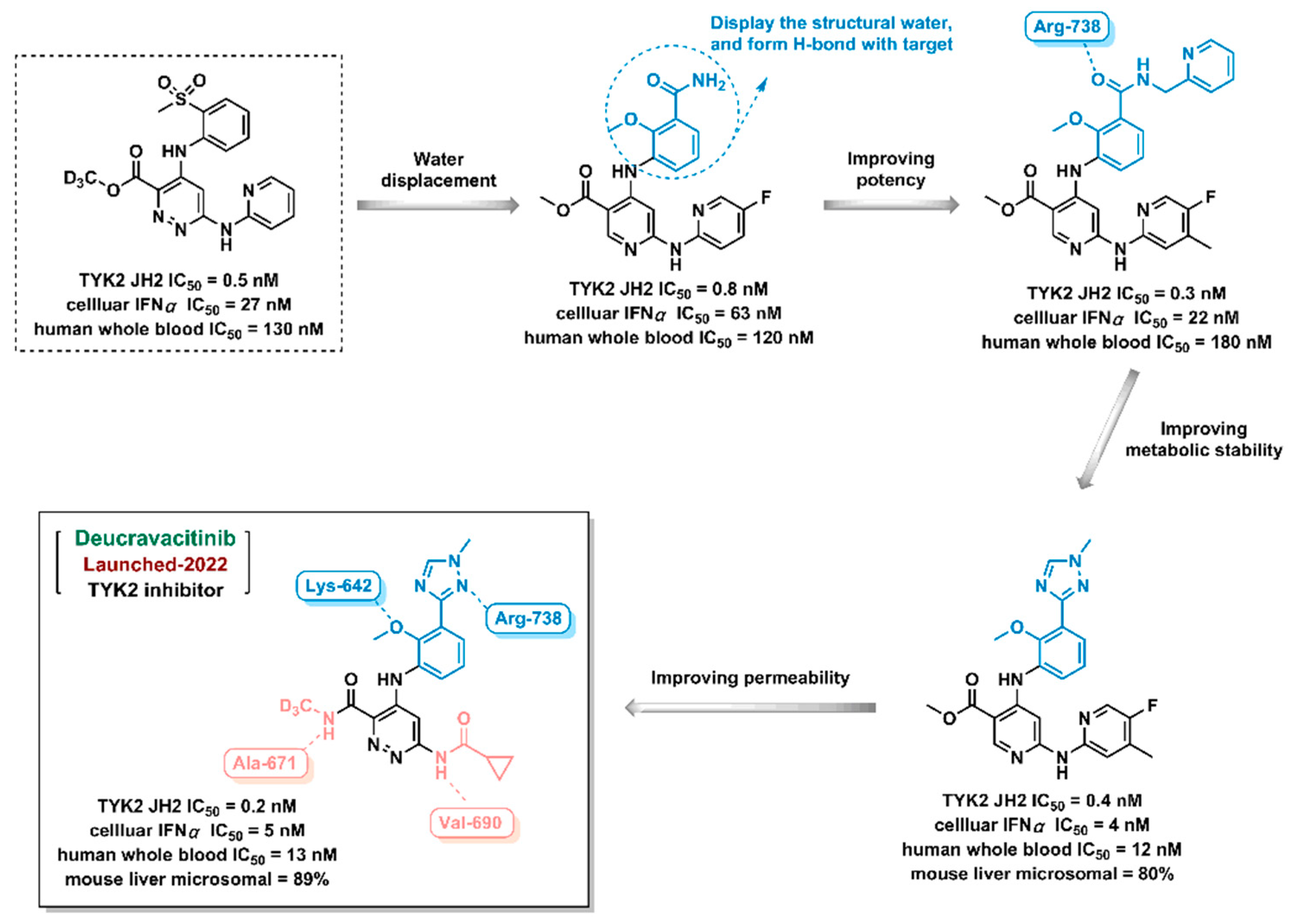
Figure 17.
The discovery process of Adagrasib by computer-aided drug design strategy.
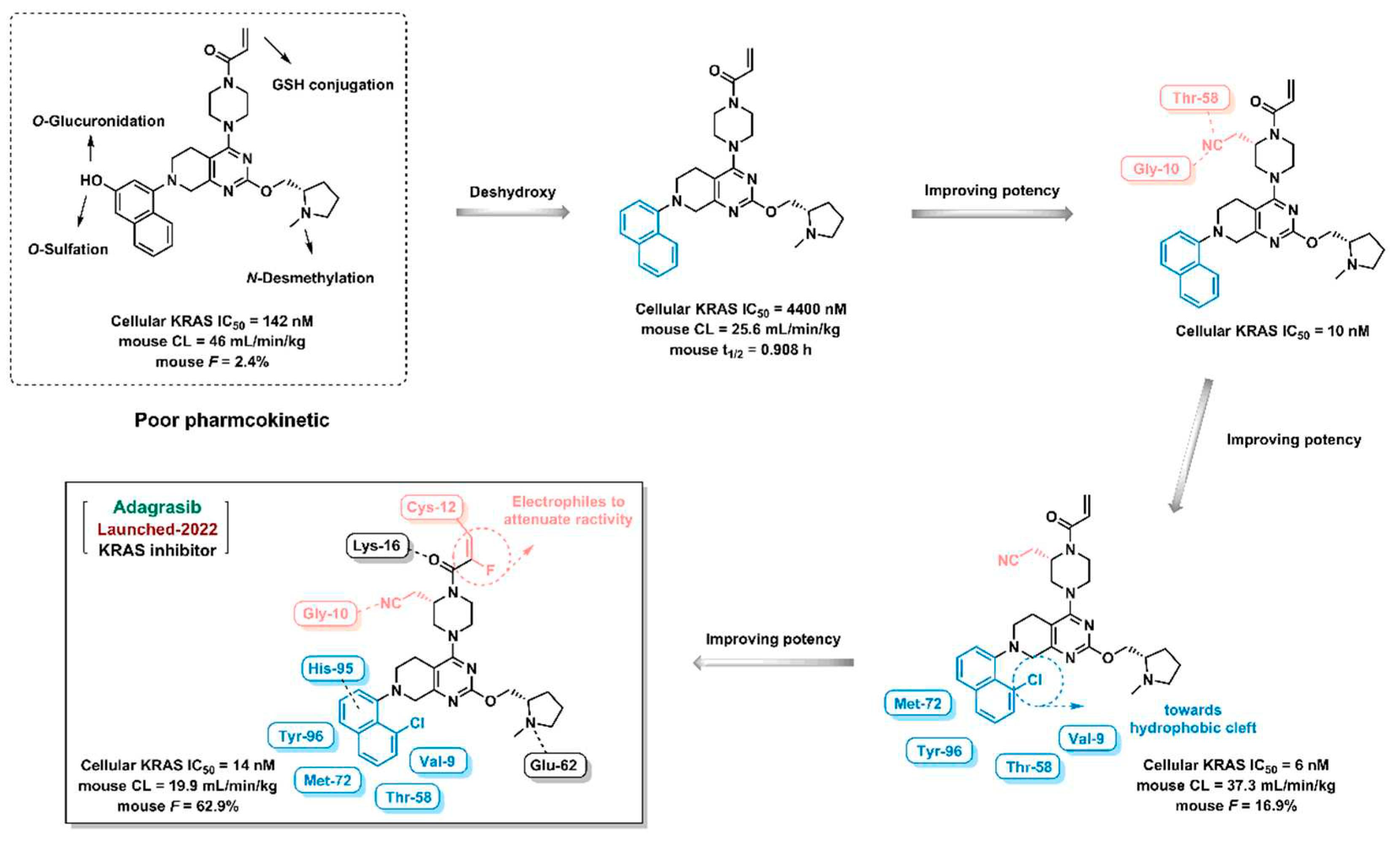
Figure 18.
The discovery process of Pitobrutinib by computer-aided drug design strategy.
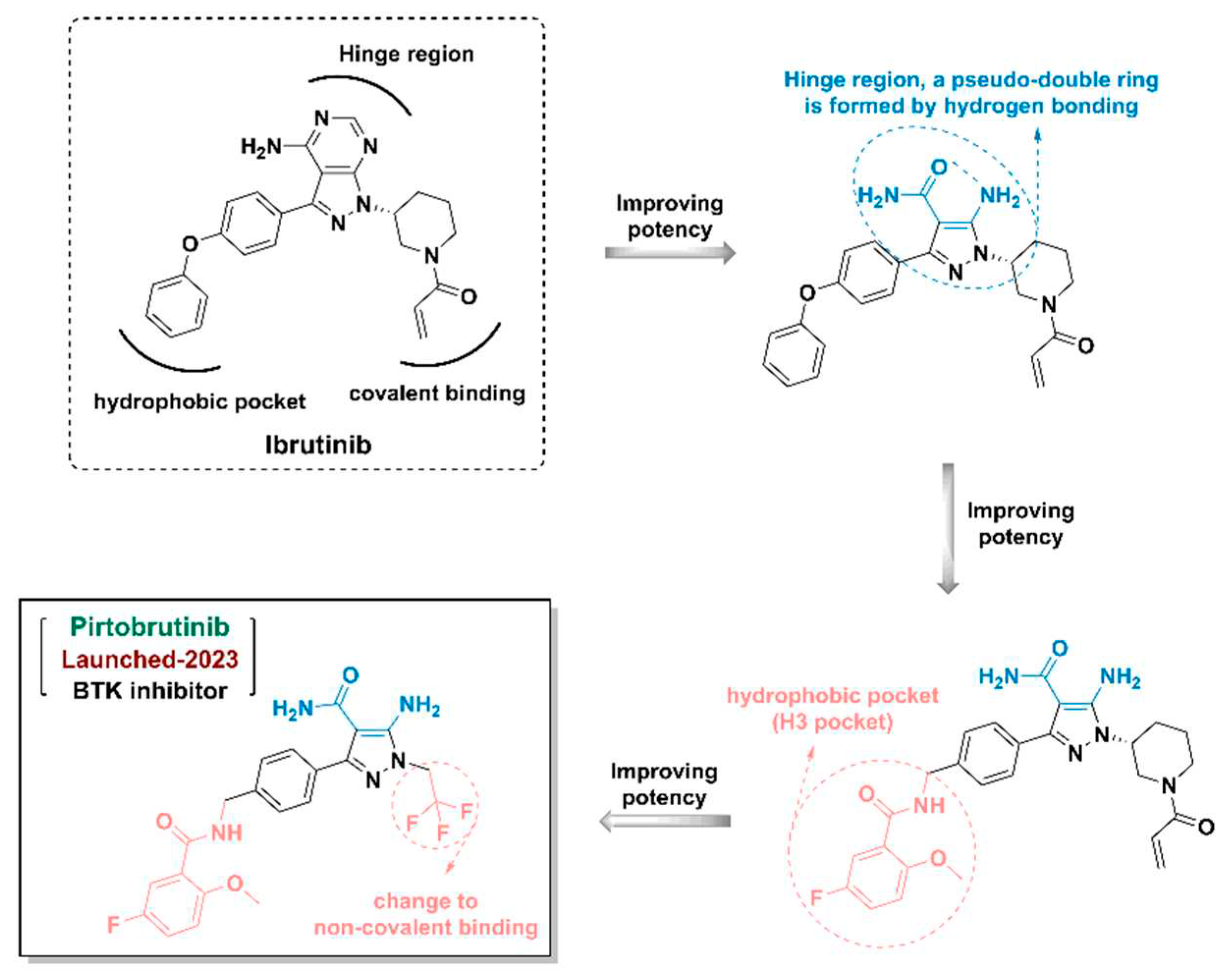
Figure 19.
FDA-approved kinase inhibitors discovered by computer-aided drug design strategy (2021-2023).
Figure 19.
FDA-approved kinase inhibitors discovered by computer-aided drug design strategy (2021-2023).
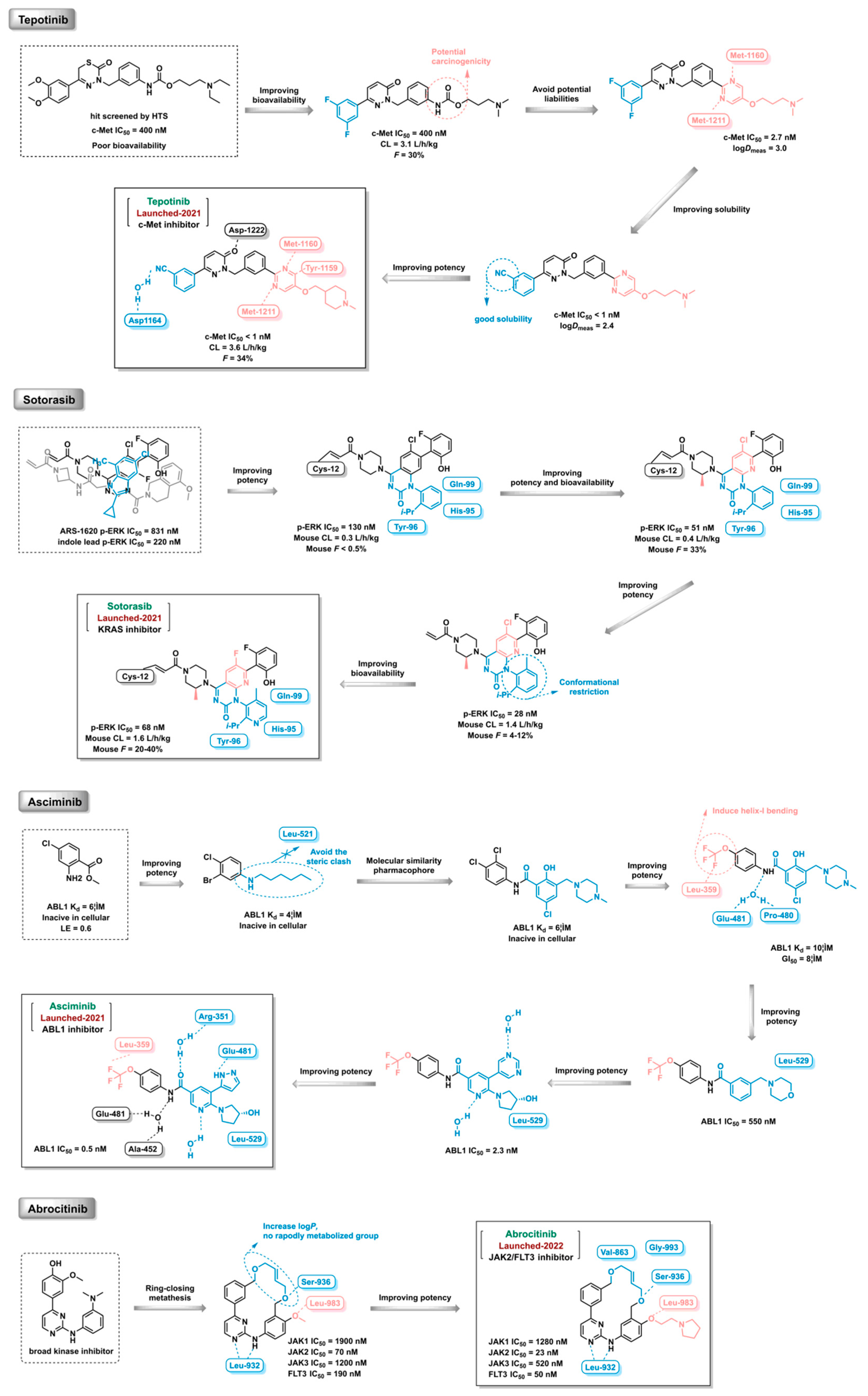
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



