Submitted:
27 July 2023
Posted:
28 July 2023
You are already at the latest version
Abstract
The Bcl-2 family plays a crucial role in regulating cell apoptosis, making it an attractive target for cancer therapy. In this study, a series of indole-based compounds, U1-6, were designed, synthesized, and evaluated for their anticancer activity against BCL-2-expressing cancer cell lines. The binding affinity, safety profile, cell cycle arrest, and apoptosis effects of the compounds were tested. The designed compounds exhibited potent inhibitory activity at sub-micromolar IC50 concentrations against MCF-7, MDA-MB-231, and A549 cell lines. Notably, U2 and U3 demonstrated the highest activity, particularly against MCF-7 cells. Respectively, both U2 and U3 showed potential Bcl-2 inhibition activity with IC50 values of 1.2 ± 0.02 and 11.10 ± 0.07 µM using an ELISA binding assay compared to 0.62 ± 0.01 µM for gossypol, employed as positive control. Molecular docking analysis suggested stable interactions of compound U2 at the Bcl-2 binding site through hydrogen bonding, pi-pi stacking, and hydrophobic interactions. Furthermore, U2 demonstrated significant induction of apoptosis and cell cycle arrest at G1/S phase. Importantly, U2 displayed a favorable safety profile on HDF human dermal normal fibroblast cells at 10-fold greater IC50 compared to MDA-MB-231 cells. These findings underscore the therapeutic potential of compound U2 as a Bcl-2 inhibitor and provide insights into its molecular mechanisms of action.
Keywords:
Bcl-2
; Anti-apoptotic protein
; Indole
; Cancer drug discovery
1. Introduction
The B-cell lymphoma 2 (Bcl-2) protein is a key regulator of apoptosis, a programmed cell death process required for tissue homeostasis and elimination of damaged cells [1]. Bcl-2 is a member of the Bcl-2 protein family, which is categorized into pro-apoptotic (such as Bax and Bak) and anti-apoptotic members (such as Bcl-2, Bcl-XL, and Mcl-1) [2]. Bcl-2 is as an anti-apoptotic protein that suppresses cell death by inhibiting the activation of pro-apoptotic proteins, hence favouring cell survival [3]. Bcl-2 expression or function dysregulation contributes to cancer development and progression [4,5]. Several studies have linked the overexpression levels of anti-apoptotic Bcl-2 protein with resistance to chemotherapy, radiation treatment, and targeted therapies in many cancer types [6,7]. This resistance permits cancer cell survival, resulting in therapeutic resistance and disease progression [8]. Furthermore, multiple Bcl-2 inhibitors are being tested in clinical studies against various cancer types [9], and activity is widely reported in pre-clinical models such as human breast cancer cell MCF-7 [10,11], MDA-MB-231 [12], and human lung cancer cell A549 [13,14]. These trials are assessing their safety, efficacy, and potential synergistic benefits when paired with existing medicines, offering promise for improved cancer treatment outcomes [15].
The BH3 domain (Bcl-2 homology 3) is a conserved region present in the pro-apoptotic Bcl-2 family proteins [16]. It facilitates the interaction of pro-apoptotic and anti-apoptotic proteins and is required for apoptosis regulation [17]. Small molecule inhibitors that mimic the BH3 domain and specifically bind to anti-apoptotic Bcl-2 proteins can disrupt this protein-protein interaction, resulting in apoptosis activation in cancer cells [18]. These inhibitors aim to sensitise cancer cells to apoptotic signals and enhance the effectiveness of cancer treatments [19]. Several BH3 mimetic molecules have been developed and tested as potential anticancer agents (Figure 1). Venetoclax (ABT-199) is an FDA-approved medication that targets Bcl-2[20]; it is the first-in-class Bcl-2 inhibitor that treats lymphoid cancers [21] and has shown remarkable clinical activity against certain types of haematological malignancies such as chronic lymphocytic leukaemia [22]. Venetoclax combined with azacitidine is used for the treatment of acute myeloid leukaemia [23]. ABT-737 was also one of the first developed Bcl-2 inhibitors that demonstrated efficient activity against lymphoma [24]. However, its major limitation is low oral bioavailability; therefore navitoclax (ABT-263) was developed [25]. Navitoclax (ABT-263) is a small molecule Bcl-2 inhibitor with a broader inhibition profile of Bcl-2 family proteins [26]. It induces apoptosis in small cell lung cancer and other solid tumour patients [27], and showed safety and efficacy when combined with erlotinib in the treatment of solid tumours [28]. Obatoclax mesylate (GX15-070) is a small molecule pan-Bcl-2 family inhibitor that is employed in the treatment of advanced chronic lymphocytic leukaemia [29].
Current Bcl-2 inhibitors, such as venetoclax, have limited application because of overexpression of anti-apoptotic proteins, particularly MCL-1, which leads to resistance and decrease in patient survival [30]. As a result, this study attempted to identify novel inhibitors of Bcl-2 protein that would provide enhanced therapeutic benefits through integrating distinct pharmacophoric moieties to improve potency, selectivity, and binding affinity towards the target proteins. Development of new Bcl-2 inhibitors represents a promising therapeutic approach in oncology, with the potential to overcome drug resistance and improve the efficacy of cancer treatments. Continued research and clinical trials can reveal the full potential of Bcl-2 inhibition in cancer treatment.
2. Experimental
2.1. Chemistry
The identity and purity of synthesised compounds were monitored on thin-layer chromatography (TLC) using pre-coated silica gel plates (Kieselgel 60F254, BDH, Taufkirchen, Germany) and visualised using UV light at 254 nm. A Gallenkamp melting point apparatus (London, UK) was used to calculate the melting points (mp). 1H NMR spectra were acquired at 500 MHz on a Bruker spectrometer. Chemical shifts were indicated in parts per million (ppm) relative to TMS, coupling constant (J) values were expressed in hertz (Hz), and the signals were labelled as s (singlet), d (doublet), t (triplet), and m (multiplet). Positive mode electrospray ionization (ESI) mass spectroscopy (Bruker Daltonics mass spectrometer, Bremen, Germany) was used to confirm molecular mass and formula.
2.1.1. Synthesis of 2-chloro-N-(4-nitrophenyl)acetamide (2)
Chloro-acetyl chloride (1 mL, 10 mmol) was added dropwise to a mixture of p-nitro aniline (1.4 g, 10 mmol) and potassium carbonate (20 mmol) in DCM (20 mL). The reaction mixture was refluxed for 4 h. The aqueous layer was extracted with dichloromethane, dried over anhydrous sodium sulphate and evaporated in vacuo until dryness. The crude product was re-crystallized from ethanol. Yield: 70%, mp: 158-159 °C. 1H-NMR (DMSO-d6) δ 4.32 (s, 2H, CH2), 7.83 (d, 2H, J = 9.15, ArH), 8.25 (d, 2H, J = 9.15, ArH), 10.38 (s, 1H, NH).
2.1.2. Synthesis of 2-morpholin-4-yl-N-(4-nitrophenyl)acetamide (3)
2-Chloro-N-(4-nitrophenyl) acetamide (2, 2 g, 10 mmol) in anhydrous THF (10 mL) was added dropwise to K2CO3 solution (2.8 g, 20 mmol), followed by addition of morpholine (1, 1 mL, 10 mmol). The reaction mixture was heated to 80 °C for 16 h. Water (30 mL) was added, and the resulting precipitate was collected by filtration, rinsed with water, and allowed to dry to obtain the crude product without further purification.
2.1.3. Synthesis of N-(4-amino-phenyl)-2-morpholin-4-yl-acetamide (4)
2-Morpholin-4-yl-N-(4-nitrophenyl)acetamide (3, 2.7 mL, 10 mmol) in 75% ethanol (30 mL) was added to a solution of reduced iron 5 gm in water (10 mL) and concentrated hydrochloric acid (0.5 mL). solution. The reaction mixture was heated to reflux for 1 h. Iron was removed through filtration and the filtrate was rinsed three times with ethanol. The filtrate was then run through a silica pad to eliminate any leftover iron residues. The filtrate was evaporated in vacuo until dryness and purified using preparative TLC. Yield: 92%, mp: 97-99°C. 1H-NMR (DMSO-d6) δ 2.49 (s, 4H, 2xCH2), 3.01 (s, 2H, CH2), 3.66 (m, 6H, 2xCH2, NH2), 6.54 (d, 2H, J = 8.65, ArH), 7.22 (d, 2H, J = 8.65, ArH), 8.78 (s, 1H, NH).
2.1.4. N-[4-(2-chloroacetylamino)phenyl]-2-morpholin-4-yl-acetamide (5)
Chloroacetyl chloride (1 mL, 10 mmol) in anhydrous THF (10 mL) was added dropwise to K2CO3 solution (2.8 g, 20 mmol), followed by N-(4-amino-phenyl)-2-morpholin-4-yl-acetamide (4, 2.4 mL, 10 mmol). The reaction mixture was heated at reflux for 16 h, crush ice was added, and the precipitate was collected by filtration and crystallized using ethanol. Yield: 78%, mp.: 97-99 °C. 1H-NMR (DMSO-d6) δ 2.50 (s, 4H, 2xCH2), 3.11 (s, 2H, CH2), 3.63 (s, 4H, 2xCH2), 4.23 (s, 2H, CH2), 7.52 (d, 2H, J = 8.69, ArH), 7.58 (d, 2H, J = 8.85, ArH), 9.71 (s, 2H, NH, NH).
2.1.5. General procedure for triazole thiol (7a-f) preparation
A solution of previously prepared indolyl-3-carbonyl-N-substituted phenyl thiosemicarbazides (6a-f, 10 mmol) in 2N NaOH (4 mL) was refluxed for 3h [31]. Water was added, and the solution was carefully neutralized with diluted HCl. The corresponding precipitate was filtered, dried, and recrystallized from ethanol.
2.1.5.1. 4-(4-Fluorophenyl)-5-(1H-indol-3-yl)-4H-1,2,4-triazole-3-thiol (7a). Yield: 62%, mp: 223-225 °C. 1H-NMR (DMSO-d6) δ 6.46 (d, 1H, J = 2.53, ArH), 7.18 (m, 2H, ArH), 7.42-7.48 (m, 3H, ArH), 7.51-7.55 (m, 2H, ArH), 8.06 (d, 1H, J = 7.5, ArH), 11.45 (s, 1H, NH), 13.94 (s, 1H, SH).
2.1.5.2. 4-(Methoxyphenyl)-5-(1H-indol-3-yl)-4H-1,2,4-triazole-3-thiol(7b). Yield 56%, mp: 210-212 °C. 1H-NMR (DMSO-d6) δ 3.86 (s, 3H, OCH3), 6.40 (d, 1H, J = 2.85, ArH), 7.12 -7.22 (m, 4H, ArH), 7.35 (d, 2H, J = 8.91, ArH), 7.43 (d, 1H, J = 7.84, ArH), 8.09 (d, 1H, J = 7.84, ArH), 11.52 (s, 1H, NH), 13.80 (s, 1H, SH).
2.1.5.3. 5-(1H-indol-3-yl)-4-methylphenyl-4H-1,2,4-triazole-3-thiol(7c). Yield 60%, mp: 208-210 °C. 1H-NMR (DMSO-d6) δ 2.45 (s, 3H, CH3), 6.38 (d, 1H, J = 2.84, ArH), 7.14 -7.23 (m, 2H, ArH), 7.32 (d, 2H, J = 8.18, ArH), 7.43 (d, 3H, J = 8.18, ArH), 8.08 (d, 1H, J = 8.18, ArH), 11.40 (s, 1H, NH), 13.93 (s, 1H, SH).
2.1.5.4. 4-(3-Chlorophenyl)-5-(1H-indol-3-yl)-4H-1,2,4-triazole-3-thiol(7d). Yield: 71%, mp: 170-172 °C. 1H-NMR (DMSO-d6) δ 6.49 (d, 1H, J = 2.49, ArH), 7.19 (m, 2H, ArH), 7.45 (t, J = 7.5, 2H, ArH), 7.64 (t, J =6.50, 1H, ArH), 7.68 (d, 1H, J = 1.25, ArH), 7.7 (d, 1H, J = 1.25, ArH), 8.0 (d, 1H, J = 7.97, ArH), 11.49 (s, 1H, NH), 13.9 (s, 1H, SH).
2.1.5.5. 4-(4-Chlorophenyl)-5-(1H-indol-3-yl)-4H-1,2,4-triazole-3-thiol (7e). Yield: 67%, mp: 165-167 °C. 1H-NMR (DMSO-d6) δ 6.51 (d, 1H, J = 2.48, ArH), 7.19 (m, 2H, ArH), 7.45 (d, 1H, J = 8.14, ArH), 7.51 (d, 2H, J = 8.50, ArH) 7.68 (d, 2H, J = 8.14, ArH), 8.04 (d, 1H, J = 7.79, ArH), 11.40 (s, 1H, NH), 13.96 (s, 1H, SH).
2.1.5.6. 4-(3,4-Dichlorophenyl)-5-(1H-indol-3-yl)-4H-1,2,4-triazole-3-thiol(7f). Yield: 63%, mp: 198-200 °C. 1H-NMR (DMSO-d6) δ 6.62 (d, 1H, J = 2.85, ArH), 7.17 (m, 2H, ArH), 7.46 (d, 1H, J = 8.05, ArH), 7.5 (d, 1H, J = 8.56, ArH), 7.87 (d, 1H, J = 8.67, ArH), 7.92 (s, 1H, ArH), 8.0 (d, 1H , J =7.97, ArH), 11.63 (s, 1H , NH), 14.0 (s, 1H , SH).
2.1.6. General procedure of S alkylation of triazole thiol (U1-6)
N-[4-(2-chloroacetylamino)phenyl]-2-morpholin-4-yl-acetamide (5, 10 mmol) was added to a mixture of 4-(substituted-phenyl)-5-(1H-indol-3-yl)-4H-1,2,4-triazole-3-thiol (7a-f, 10 mmol) in ethanol and KOH (10 mmol) and stirred for 16 h at room temperature. The produced precipitate was then filtered, dried, and recrystallized from ethanol to obtain the corresponding S-alkyl triazole thiol U1-6.
2.1.6.1. 2-(4-(4-Fluorophenyl)-5-(1H-indol-3-yl)-4H-1,2,4-triazol-3-ylthio)-N-(4-(2 morpholinoacetamido)phenyl)acetamide(U1). Yield: 65%, mp: 208-210 °C. 1H-NMR (DMSO-d6) δ 2.14 (s, 4H, 2xCH2), 3.14 (s, 4H, 2xCH2), 3.63 (s, 2H, CH2), 4.15 (s, 2H, CH2), 6.60 (d, 1H, J = 2.12, ArH), 7.19 (m, 2H, ArH), 7.45 (d, 1H, J = 6.37, ArH), 7.50-7.56 (m, 4H, ArH), 7.60-7.66 (m, 4H, ArH), 8.2 (d, 1H, J = 7.79, ArH), 9.73 (s, 1H, NH), 10.32 (s, 1H, NH), 11.35 (s, 1H, NH). MS analysis for C30H28FN7O3S: Calcd mass: 585.65, found (m/z, M+): 586.16.
2.1.6.2. 2-(5-(1H-indol-3-yl)-4-(4-methoxyphenyl)-4H-1,2,4-triazol-3-ylthio)-N- (4-(2-morpholinoacetamido)phenyl)acetamide (U2). Yield: 65%, mp: 173-175 °C. 1H-NMR (DMSO-d6) δ 2.12 (s, 4H, 2xCH2), 3.11 (s, 4H, 2xCH2), 3.60 (s, 2H, CH2), 3.86 (s, 3H, OCH3), 4.11 (s, 2H, CH2), 6.54 (d, 1H, J = 2.21, ArH), 7.15 (m, 4H, ArH), 7.41-7.51 (d, 4H, J = 8.50, ArH), 7.60 (d, 3H, J = 8.50, ArH), 8.20 (d, 1H, J =5.66, ArH), 9.68 (s, 1H , NH), 10.31 (s, 1H, NH), 11.30 (s, 1H, NH). MS analysis for C31H31FN7O4S: Calcd mass 597.69, found (m/z, M+): 598.22.
2.1.6.3. 2-(5-(1H-indol-3-yl)-4-methylphenyl-4H-1,2,4-triazol-3-ylthio)-N-(4-(2- morpholinoacetamido)phenyl)acetamide(U3). Yield: 67%, mp: 250-252 °C. 1H-NMR (DMSO-d6) δ 2.10 (s, 4H, 2xCH2), 2.41 (s, 3H, CH3), 3.09 (s, 4H, 2xCH2), 3.57 (s, 2H, CH2), 4.09 (s, 2H, CH2), 6.50 (d, 1H, J = 2.12, ArH), 7.19 (m, 2H, ArH), 7.45 (dd, 5H, J = 6.37, 7.15 ArH), 7.52 (d, 2H, J = 6.90, ArH), 7.64 (d, 2H, J = 6.90, ArH), 8.2 (d, 1H, J = 7.79, ArH), 9.73 (s, 1H, NH), 10.39 (s, 1H, NH), 11.43 (s, 1H, NH). MS analysis for C31H31FN7O3S: Calcd mass 581.69, found (m/z, M+): 582.18.
2.1.6.4. 2-(4-(3-Chlorophenyl)-5-(1H-indol-3-yl)-4H-1,2,4-triazol-3-ylthio)-N-(4-(2 morpholinoacetamido)phenyl) acetamide U4. Yield: 45%, mp: 220-222 °C. 1H-NMR (DMSO-d6) δ 2.16 (s, 4H, 2xCH2), 3.17 (s, 4H, 2xCH2), 3.68 (s, 2H, CH2), 4.18 (s, 2H, CH2), 6.50 (d, 1H, J = 2.30, ArH), 7.15 (m, 2H, ArH), 7.45 (d, 1H, J = 6.37, ArH), 7.54-7.57 (m, 4H, ArH), 7.62-7.66 (m, 3H, ArH), 7.74 (t, J =6.75, 1H, ArH), 8.2 (d, 1H, J = 7.79, ArH), 9.63 (s, 1H, NH), 10.52 (s, 1H, NH), 11.34 (s, 1H, NH). MS analysis for C30H28ClN7O3S: Calcd mass: 601.17, found (m/z, M+): 602.20.
2.1.6.5. 2-(4-(4-Chlorophenyl)-5-(1H-indol-3-yl)-4H-1,2,4-triazol-3-ylthio)-N-(4-(2 morpholinoacetamido)phenyl)acetamide(U5). Yield 65%, mp: 208-210 °C. 1H-NMR (DMSO-d6) δ 2.15 (s, 4H, 2xCH2), 3.16 (s, 4H, 2xCH2), 3.64 (s, 2H, CH2), 4.18 (s, 2H, CH2), 6.58 (d, 1H, J = 2.42, ArH), 7.14 (m, 2H, ArH), 7.40 (d, 1H, J = 8.37, ArH), 7.52-7.54 (m, 4H, ArH), 7.68 (d, 2H, J = 7.50, ArH), 7.62-7.64 (m, 2H, ArH), 8.2 (d, 1H, J = 7.79, ArH), 9.73 (s, 1H, NH), 10.30 (s, 1H, NH), 11.20 (s, 1H, NH). MS analysis for C30H28ClN7O3S: Calcd mass: 601.17, found (m/z, M+): 601.98.
2.1.6.6. 2-[4-(3,4-Dichlorophenyl)-5-(1H-indol-3-yl)-4H-1,2,4-triazol-3-ylthio-N-[4-(2-morpholinoacetamido)phenyl acetamide (U6). Yield: 57%, mp: 218-220 °C. 1H-NMR (DMSO-d6) δ 2.10 (s, 4H, 2xCH2), 3.04 (s, 4H, 2xCH2), 3.12 (s, 2H, CH2), 4.07 (s, 2H, CH2), 6.58 (d, 1H, J = 2.12, ArH), 7.18-7.21 (m, 2H, ArH), 7.46 (d, 1H, J = 8.05, ArH), 7.64 (t, J = 7.1, 1H, ArH), 7.70-7.74 (m, 3H, ArH), 7.82-7.84 (m, 2H, ArH), 7.92 (s, 1H, ArH), 8.0 (d, 1H, J =7.97, ArH), 9.73 (s, 1H, NH), 10.34 (s, 1H, NH), 11.63 (s, 1H, NH). MS analysis for C30H27Cl2N7O3S: Calcd mass: 635.13, found (m/z, M+): 636.78.
2.2. Biology
2.2.1. Cell culture and maintenance
MDA-231 triple-negative breast cancer cells, MCF-7 breast cancer cells, and A549 adenocarcinomic human alveolar basal epithelial cells were maintained in Roswell Park Memorial Institute media (RPMI 1640, Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% foetal bovine serum and 1% penicillin/streptomycin. HDF human dermal normal fibroblast cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM, Sigma-Aldrich, Germany). MCF-7 breast cancer cell line was obtained from cell lines service (CLS; Germany). MDA-MB-231 breast cancer cell line and A549 cancer cells were obtained from European Collection of Authenticated Cell Cultures (ECACC; UK). Cell lines were maintained at 37° C in a humidified incubator with 5% CO2.
2.2.2. Cytotoxicity assay
The antiproliferative activity of compounds U1-6 was assessed using 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide MTT assay as previously described [32,33]. Cancer cell lines were seeded in 96-well flat bottom plates at a density of 104 cells/ well for 24 h. The cells were subsequently treated with U1-6 compounds at a screening concentration of 50 µM and incubated at 37°C for 48 h in a humidified incubator containing 5% CO2. Serial dilution concentrations ranged from 1, 10, 25, 50, 100 µM were used for IC50 calculation of each compound on the three cancer cell lines. DMSO was used as a negative vehicle control. The culture media was then removed and incubated for 2 h with fresh 200 µl culture media containing 5 mg/ml MTT. After removing the culture media and dissolving the produced formazan crystals in 100 µl DMSO and then incubated for another 30 min at 37 °C. The produced colour was measured at 570 nm using a microplate reader (Thermo-Scientific, Vantaa, Finland). GraphPad Prism 9.1 software was used to generate plots of absorbance versus the compound’s concentration. Three independent repeat experiments were performed for each concentration to ensure reproducibility. IC50 values were obtained from nonlinear regression plots of absorbance versus log concentration of the tested compounds using GraphPad Prism 5 software (San Diego, CA, USA).
2.2.3. Cell cycle assay
Most active compound U2 was applied to MCF7 cells for 24 h at its IC50 concentration. DMSO vehicle was used as negative control. The cells were collected, centrifuged, and fixed in 70% ethanol on ice for 20 min. The fixed cells were incubated for 1 h at room temperature with staining solution (50 mg/mL propidium iodide PI, 0.05% Triton X-100, 0.1 mg/mL RNaseA). Gallios flow cytometer (Beckman Coulter, Brea, CA, USA) was used to measure cell cycle proportion [34].
2.2.4. Apoptosis Assay
The Annexin-V-FITC apoptosis detection kit (Catalog # k101-25, Biovision, USA) was used for the apoptosis experiment according to previously published data. MCF-7 cells were treated with U2 compound for 24 h. The negative control was 0.1% DMSO. The cells were collected by centrifugation and resuspended in 500 µl of 1X Binding Buffer. Treated and control cells were stained with 5 µl Annexin V-FITC and 5 µl propidium iodide (PI 50 mg/ml) and then incubated at room temperature for 5 min in dark. Fluorescence-activated cell sorting (FACS) was quantified by flow cytometry. Flow cytometry (Excision at 488 nm; Emission at 530 nm) was used to examine Annexin V-FITC binding and PI staining using the FITC signal detector and the phycoerythrin emission signal detector [35].
2.2.5. Bim Enzyme-Linked Immunosorbent Assay (ELISA)
Bim binding assay (Thermo scientific, catalogue no # BMS244-3, Austria) was performed according to previously published data [31,36]. PBS solution containing 0.05% Tween-20 was used to wash a streptavidin-coated 96-well plate. For immobilisation, biotinylated Bim peptide (residues 81–106) was diluted in superblock blocking solution and applied to each well. After incubation, the plate was washed with 0.5% BSA in PBS with Tween-20 solution. In PBS, test compounds were treated with His-tagged Bcl-2 protein and incubated for 1h after that transferred to the wells containing the immobilised Bim peptide. After another wash, each well received anti-His antibody with horseradish peroxidase enzyme. Following incubation and washing, O-phenylenediamine and hydrogen peroxide solution was added for color production. A plate reader (Thermo-Scientific, Vantaa, Finland) was used to measure the optical density at 450 nm. The reduction in Bim affinity was measured using non-linear regression curve (GraphPad Prism 5), which was also utilized to generate the IC50 value for Bcl-2 inhibition.
2.3. Computational modeling
All computational work was carried out using Schrödinger suite 12.7 available at www. Schrödinger.com and using Maestro graphical user interface software.
2.3.1. Protein and ligand preparation
The protein data bank (https://www.rcsb.org) was used to obtain the 3D crystal structure of human Bcl-2 (PDB ID: 4AQ3). The protein was prepared and refined using the Protein Preparation Wizard Maestro. Water molecules that were crystallographically larger than 5 A° were eliminated. At pH 7.3, all the missing hydrogen atoms were added to the protein for correct ionization using EPIC module [37,38]. The tautomerization state and bond order of amino acid residues were assigned. Water molecules having three hydrogen bonds to non-waters were eliminated. Finally, to alleviate steric conflicts, energy was minimized using OPLS4 [39]. Ligand compounds were built, and energy minimized using LigPrep tool [40], and the ligand 2D structure was transformed to 3D structure [41].
2.3.2. Grid generation and molecular docking
In grid generation, the ligand having the crystal structure of human Bcl-2 (PDB ID: 4AQ3) was utilized. For docking investigations, a grid box was constructed at the active site's centroid, and the active site was defined around the native ligand of Bcl-2 (PDB ID: 4AQ3) crystal structure. The prepared ligands were docked within the grid-generated Bcl-2 (PDB: 4AQ3) binding site using the standard precision (SP) mode of Glide without any limitations [42,43]. The visual inspection of the interaction indicated the affinity of docked ligands to the binding site.
2.3.3. Pharmacokinetics predication
Using the Swiss-ADME website (http://www.swissadme.ch/), the bioavailability radar of the best active substance U2 was projected versus Bcl-2 small molecule inhibitor ABT-263. Structure sketches were converted to SMILES format [44]. The bioavailability radar indicates desired features such as size, solubility, saturation, polarity, lipophilicity, and flexibility [45].
2.4. Statistical analysis
GraphPad prism (version 9.1.0) was used for data analysis using one-way analysis of variance (ANOVA) and Multiple Comparison Test. The data were represented by the mean SEM of three independent replicates. P< 0.05 was used as the statistical significance level. * Reveals that P-value <0.05, ** reveals that P-value <0.01, *** reveals that P-value <0.001, **** reveals that P-value <0.0001.
3. Results
3.1. Rational design
The rational design of Bcl-2 inhibitors has been explored using various small molecule inhibitors with core heterocyclic scaffolds, such as isoquinoline [2,46], and indole [47,48], along with different substitutions to enhance their activity and physiochemical properties. We previously designed series of indole-based compounds with triazole [36], oxadiazole [3,31], and quinoline-fused triazolothiadiazoles [2] scaffolds. The inclusion of a triazole ring was beneficial in targeting anti-apoptotic proteins Bcl-2 and Mcl-1 [49]. Morpholino substitution has demonstrated efficacy in targeting Bcl-2 proteins as in the case of ABT-263 [50,51]. Additionally, acetamide derivatives have been investigated for their ability to inhibit anti-apoptotic Bcl-2 proteins [52]. Various substitutions of electron donating or withdrawing groups have been introduced to the aforementioned compounds to alter the electronic properties and physicochemical characteristics of the molecule, potentially impacting its interactions with the target protein or modulating its biological activity. The rational construction of a series of molecules involves incorporating various functional groups and substitution patterns including an indole ring, a 1,2,4-triazole ring, a thioether linkage, an acetamide linker, and a morpholino moiety. The arrangement and connectivity of these structural elements can contribute to the compound's functional activity. It is worth noting that the discovery of small molecule inhibitors targeting the Bim binding site of anti-apoptotic Bcl-2 is an active area of research (Figure 2). The rational design of these compounds aims to optimize their potency, selectivity, and other pharmacological properties, with the ultimate goal of developing effective therapeutic agents for the treatment of specific disease.
3.2. Synthesis of the title compounds U1-6
A multistep procedure was used to synthesize compounds U1-6. First, 2-chloro-N-(4-nitrophenyl)acetamide 2 was synthesised by refluxing chloroacetyl chloride in a dichloromethane solution of p-nitroaniline and potassium carbonate. Then 2-morpholin-4-yl-N-(4-nitrophenyl)acetamide 3 was prepared by heating compound 2 in anhydrous THF with morpholine. Following that, N-(4-amino-phenyl)-2-morpholin-4-yl-acetamide 4 was synthesized by refluxing compound 3 in ethanol and HCl with reduced iron. Further, reaction of compound 4 with 2 chloroacetyl chloride produced N-[4-(2-chloro-acetylamino)-phenyl]-2-morpholin-4-yl-acetamide 5. The substituted-phenyl-5-(1H-indol-3-yl)-4H-1,2,4-triazole-3-thiol (7a-f) were synthesized by treatment of previously reported indolyl-3-carbonyl-N-substituted phenyl thiosemicarbazides (6a-f, 10 mmol) in refluxing sodium hydroxide solution [31]. Finally, compound 5 was mixed in ethanol and KOH with substituted-phenyl-5-(1H-indol-3-yl)-4H-1,2,4-triazole-3-thiol (7a-f) to effect S-alkylation of the triazole thiol and yield the desired product, U1-6 as illustrated in Figure 3.
3.3. Compounds U2 and U3 showed potent inhibitory activity towards Bcl-2- expressing cancer cells
The designed compounds demonstrated inhibitory activity against Bcl-2 expressing human cell lines including breast cancer lines MCF-7 and MDA-MB-231; and A549 lung cancer cells at sub-micromolar concentrations indicating their potency (Table 1, Figure 4). The most potent activity was observed by U2, followed by U3, U1 and U4. Overall, the antiproliferative activity of all compounds was the highest against the MCF-7 cancer cell line. Compounds U2, U3 showed IC50 values of 0.83 ± 0.11 and 1.17 ± 0.10 µM against MCF-7; 0.73 ± 0.07 and 2.98 ± 0.19 µM against A549; and 5.22 ± 0.55, 4.07 ± 0.35 µM against the metastatic and treatment-refractory triple-negative breast cancer cell line MDA-MB-231, respectively. The physicochemical characteristics of various substitution patterns and the potent activity of U2, U3, led to their selection for further investigation.
3.4. ELISA indicated the superior activity of compound U2 against Bcl-2 protein
The ability of compounds U2 and U3 to bind to the Bcl-2 binding pocket as BH3 mimetics was tested by the ELISA binding assay, as previously described by our group [31,36] The different attributes of the substitution such as size, shape, and electrostatic interaction influence their effectiveness as Bcl-2 competitive inhibitors. Compound U2 exhibited an IC50 = 1.2±0.02 µM, which is 2-fold less potent than gossypol (IC50 0.62 ± 0.01µM), while U3 exhibited lower binding affinity with IC50 = 11.10 ± 0.07 µM (Table 2, Figure 5). These results indicated the potential activity of compound U2 as a BCL-2 inhibitor warranting further investigation.
3.5. Compounds U1-6 showed an excellent safety prolife on human normal cells.
All tested compounds displayed minimal inhibitory activity against the HDF human dermal normal fibroblast cells at a concentration of 50 µM (over 24 hrs), indicative of a high safety profile. In particular, compounds U1, U3, U4, and U5 showed no significant change compared to the negative control. U6 and U2 exhibited limited toxicity on fibroblasts with P values of < 0.0001and 0.01, respectively (Figure 6).
3.6. Compound U2 induced apoptosis and cell cycle arrest at G1/S phase.
Compound U2 effectively inhibited cell growth through apoptosis, and significantly increased early apoptosis, displaying a 43-fold increase compared to control untreated cells (Figure 7A, B, C), while inducing a remarkable 111-fold increase in late apoptosis (Figure 7A, B, C). Additionally, the compound caused cell cycle arrest at G1/S phase as indicted in Figure 7D, E, F. These findings highlight the ability of compound U2 to induce programmed cell death and disrupt cell cycle progression.
3.7. Molecular docking revealed efficacy and selectivity of U2 compound against Bcl-2
Molecular docking analysis revealed that all U1-6 compounds formed stable interactions at Bcl-2 binding site (PDB: 4AQ3). The carbonyl group of U2 showed H-bond interactions with Arg-60, the phenyl ring showed pi-pi staking interaction with Phe-63 alongside hydrophobic interaction with Ala-108, Arg-105, Glu-95, Tyr-67 and Phe-63 that further contributed to the binding stability (Table 3, Figure 8A). Carbonyl group of compound U3 showed H-bond interactions with Arg-105, in addition to aromatic H-bond interaction of the phenyl ring with Val-92, and hydrophobic interaction with Arg-105, Glu-95, Asp-70, Leu-96, Tyr-67, and Phe-63 (Table 3, Figure 8B). On the other hand, carbonyl group of compound U1 showed H- bond interactions with Arg-105 and aromatic H-bond interaction with Val-92, hydrophobic interaction with Asp-70, Phe-63, and Try-67, and Arg-105 within the binding site (Table 3, Figure 8C). The collective interactions resulted in an overall glide score of -5.8 kcal/mole, suggesting a strong binding affinity.
3.8. Compound U2 showed improved properties compared to small molecule inhibitor ABT263.
The bioavailability radars (Swiss-ADME) [53] of the newly designed and highly potent compound U2 were compared to those of ABT-263 compound (Figure 9). The pink areas in the figure represent the optimal range of six properties, namely unstauration, insolubility, lipophilicity, flexibility, polarity, and size. It was observed that U2 compound fell within the desired range and exhibited acceptable parameters when compared to ABT-236. This suggests that U2 exhibited favorable bioavailability characteristics, making it a promising candidate for further development.
4. Discussion
Venetoclax, an FDA licensed Bcl-2 inhibitor is quickly evolving as the standard care for acute myeloid leukaemia and chronic lymphocytic leukaemia. Because of the concomitant upregulation of anti-apoptotic proteins, particularly MCL-1 following prolonged exposure to venetoclax [54], which provides an escape route leading to venetoclax resistance, its use is limited and hence it has become part of a combination therapy [55]. To overcome this limitation, researchers have studied the dual inhibition of MCL-1 and Bcl-2 [56]. This includes the use of an indole-based lead compound that exhibits potent inhibition of MCL-1 and moderate inhibition of Bcl-2 [57].
Indole-based compounds showed potent anticancer activity through dual inhibition activity of both Bcl-2/Mcl-1 proteins [58]. Indole-based derivatives designed by incorporation of amide, biphenyl and sulfonamide pharmacophoric moieties aimed to enhance the inhibitory activity against Bcl-2 and Mcl-1 [59], potentially leading to more effective dual inhibitors [58]. Furthermore, indole-based coumarin compounds were reported as a potent anticancer agent by targeting Bcl-2, and halogen substitution showed the highest activity [60]. We previously designed and synthesized a series of indole-based compounds by incorporating various heterocyclic rings such as oxadiazole [61], triazole [62], thiazole [2,63], quinoline [2], and fused triazolo-thiazole ring [32]. Our previous studies highlight the chloro-substitution of indole-based oxadiazole amine as a promising inhibitor of Bcl-2 with similar binding affinity to gossypol; triazolo-thiazole and the dimethoxy derivatives showed 2-fold less potent binding affinity to Bcl-2 compared to gossypol employed as positive control [32].
The structure modifications aimed to optimize the binding interactions between the inhibitor and the protein. The extension to occupy more of the binding pocket of Bcl-2 played a vital role in enhancing the inhibitory activity against both proteins. The design of new molecular features that enhance potency and specificity was achieved by studying the pharmacophoric features of potent inhibitors and structural characteristics to make favorable interactions. A molecular docking study indicated the binding affinity of the designed compounds with Bcl-2 active site key amino acid residues due to the amide carbonyl moiety incorporated with H-bond interactions. The structure activity relationship study indicated that electron-donating groups substitution at the para position such as p-methoxy U2 and p-methyl substituted U3 showed the highest activity, followed by the electron-withdrawing functional groups such as p-fluoro substituted U1. These modifications can impact the compound's pharmacokinetic profile and potentially improve its efficacy and safety.
U2 compound showed cytotoxic effects mainly by inducing the apoptosis and G1/S cell cycle arrest. This inference is consistent with earlier research highlighting the involvement of Bcl-2 inhibitors in cell cycle arrest, specifically in G0/G1 and S phases. For example, TW-37, a small molecule Bcl-2 inhibitor, induces S phase arrest in tumours [64]. Obatoclax inhibited the G0/G1 cell cycle in human oesophageal cancer cells [65]. Other studies have found that Bcl-2 has the most pronounced effect on the cell cycle by delaying passage from G0/G1 to S phases [66], and that direct inhibition of Bcl-2 by obatoclax improves G1/G0 to S phase transition rather than causing G1/G0 phase arrest [67]. Gossypol also limited cell cycle progression by triggering S phase arrest [68]. These findings contribute to continuing attempts to find more effective BCL-2 inhibitors, perhaps leading to new therapeutic alternatives for cancer treatment. More study and optimization of these compounds are required to increase their potency and selectivity.
5. Conclusion
This study investigated the effects of designed indole-based derivatives U1-6 on Bcl2-expressing cancer cell growth, apoptosis, and cell cycle progression. Out of the designed compounds, compound U2 with a 4-methoxy substitution demonstrated potent inhibitory effects on cell growth through the induction of apoptosis. In comparison to untreated control cells, U2 compound exhibited a significant 43-fold increase in early apoptosis and a remarkable 111-fold increase in late apoptosis. Moreover, the compound induced cell cycle arrest at G1/S phase. These findings highlight the promising anticancer activity of U2. Further research and development of U2 can lead to novel strategies to combating cell proliferation-related diseases.
Author Contributions
Conceptualization, R.H. and A.D.W; methodology, A.M, A.S., R.H, S.S and A.D.W.; investigation, R.H, A.M and A.D.W.; resources, A.M, SS and A.D.W.; writing, R.H., A.M. and SS. All authors have read and agreed to the published version of the manuscript.
Funding
This study was funded by the University of Sharjah (Grant No: 2301110176) to SSMS.
Conflicts of interest
The authors declare no conflict of interest.
References
- Aniogo, E.C., B.P.A. George, and H. Abrahamse, Role of Bcl-2 family proteins in photodynamic therapy mediated cell survival and regulation. Molecules, 2020. 25(22): p. 5308. [CrossRef]
- Hamdy, R., et al., New quinoline-based heterocycles as anticancer agents targeting bcl-2. Molecules, 2019. 24(7): p. 1274. [CrossRef]
- Hamdy, R., et al., Design, synthesis and evaluation of new bioactive oxadiazole derivatives as anticancer agents targeting bcl-2. International Journal of Molecular Sciences, 2020. 21(23): p. 8980. [CrossRef]
- Kaloni, D. , et al., BCL-2 protein family: Attractive targets for cancer therapy. Apoptosis, 2023. 28(1-2): p. 20-38. [CrossRef]
- Keller, M.A. , et al., Bcl-x short-isoform is essential for maintaining homeostasis of multiple tissues. Iscience, 2023. 26(4): p. 106409. [CrossRef]
- Kang, M.H. and C.P. Reynolds, Bcl-2 inhibitors: targeting mitochondrial apoptotic pathways in cancer therapy. Clinical cancer research, 2009. 15(4): p. 1126-1132. [CrossRef]
- Longley, D. and P. Johnston, Molecular mechanisms of drug resistance. The Journal of Pathology: A Journal of the Pathological Society of Great Britain and Ireland, 2005. 205(2): p. 275-292.
- Mohammad, R.M., et al. Broad targeting of resistance to apoptosis in cancer. in Seminars in cancer biology. 2015. Elsevier. [CrossRef]
- Thomas, S. , et al., Targeting the Bcl-2 family for cancer therapy. Expert opinion on therapeutic targets, 2013. 17(1): p. 61-75. [CrossRef]
- Oh, S. , et al., Downregulation of autophagy by Bcl-2 promotes MCF7 breast cancer cell growth independent of its inhibition of apoptosis. Cell Death & Differentiation, 2011. 18(3): p. 452-464. [CrossRef]
- Akar, U. , et al., Silencing of Bcl-2 expression by small interfering RNA induces autophagic cell death in MCF-7 breast cancer cells. Autophagy, 2008. 4(5): p. 669-679. [CrossRef]
- Hudson, S.G. , et al., Microarray determination of Bcl-2 family protein inhibition sensitivity in breast cancer cells. Experimental Biology and Medicine, 2013. 238(2): p. 248-256. [CrossRef]
- Li, Y. , et al., Curcumin inhibits human non-small cell lung cancer A549 cell proliferation through regulation of Bcl-2/Bax and cytochrome C. Asian Pacific Journal of Cancer Prevention, 2013. 14(8): p. 4599-4602.
- Xiong, S. , et al., MicroRNA-7 inhibits the growth of human non-small cell lung cancer A549 cells through targeting BCL-2. International journal of biological sciences, 2011. 7(6): p. 805. [CrossRef]
- Lima, K. , et al., Obatoclax reduces cell viability of acute myeloid leukemia cell lines independently of their sensitivity to venetoclax. Hematology, Transfusion and Cell Therapy, 2022. 44: p. 124-127. [CrossRef]
- Daniel, P.T. , et al., Guardians of cell death: the Bcl-2 family proteins. Essays in biochemistry, 2003. 39: p. 73-88. [CrossRef]
- Warren, C.F., M.W. Wong-Brown, and N.A. Bowden, BCL-2 family isoforms in apoptosis and cancer. Cell death & disease, 2019. 10(3): p. 177. [CrossRef]
- Bajwa, N., C. Liao, and Z. Nikolovska-Coleska, Inhibitors of the anti-apoptotic Bcl-2 proteins: a patent review. Expert opinion on therapeutic patents, 2012. 22(1): p. 37-55. [CrossRef]
- Carneiro, B.A. and W.S. El-Deiry, Targeting apoptosis in cancer therapy. Nature reviews Clinical oncology, 2020. 17(7): p. 395-417. [CrossRef]
- Mullard, A. , Pioneering apoptosis-targeted cancer drug poised for FDA approval: AbbVie's BCL-2 inhibitor venetoclax--the leading small-molecule protein-protein interaction inhibitor--could soon become the first marketed drug to directly target the ability of cancer cells to evade apoptosis. Nature Reviews Drug Discovery, 2016. 15(3): p. 147-150.
- King, A.C. , et al., Venetoclax: a first-in-class oral BCL-2 inhibitor for the management of lymphoid malignancies. Annals of Pharmacotherapy, 2017. 51(5): p. 410-416. [CrossRef]
- Korycka-Wolowiec, A. , et al., Venetoclax in the treatment of chronic lymphocytic leukemia. Expert Opinion on Drug Metabolism & Toxicology, 2019. 15(5): p. 353-366. [CrossRef]
- DiNardo, C.D. , et al., Azacitidine and venetoclax in previously untreated acute myeloid leukemia. New England Journal of Medicine, 2020. 383(7): p. 617-629.
- Mason, K.D. , et al., In vivo efficacy of the Bcl-2 antagonist ABT-737 against aggressive Myc-driven lymphomas. Proceedings of the National Academy of Sciences, 2008. 105(46): p. 17961-17966. [CrossRef]
- Tse, C. , et al., ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer research, 2008. 68(9): p. 3421-3428. [CrossRef]
- Chen, J. , et al., The Bcl-2/Bcl-XL/Bcl-w Inhibitor, Navitoclax, Enhances the Activity of Chemotherapeutic Agents In Vitro and In VivoNavitoclax Enhances the Activity of Chemotherapeutic Agents. Molecular cancer therapeutics, 2011. 10(12): p. 2340-2349.
- Gandhi, L. , et al., Phase I study of Navitoclax (ABT-263), a novel Bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. Journal of clinical oncology, 2011. 29(7): p. 909. [CrossRef]
- Tolcher, A.W. , et al., Safety, efficacy, and pharmacokinetics of navitoclax (ABT-263) in combination with erlotinib in patients with advanced solid tumors. Cancer chemotherapy and pharmacology, 2015. 76: p. 1025-1032. [CrossRef]
- O'Brien, S.M. , et al., Phase I study of obatoclax mesylate (GX15-070), a small molecule pan–Bcl-2 family antagonist, in patients with advanced chronic lymphocytic leukemia. Blood, The Journal of the American Society of Hematology, 2009. 113(2): p. 299-305. [CrossRef]
- Bose, P., V. Gandhi, and M. Konopleva, Pathways and mechanisms of venetoclax resistance. Leukemia & lymphoma, 2017. 58(9): p. 2026-2039. [CrossRef]
- Hamdy, R. , et al., Synthesis and evaluation of 5-(1H-indol-3-yl)-N-aryl-1, 3, 4-oxadiazol-2-amines as Bcl-2 inhibitory anticancer agents. Bioorganic & Medicinal Chemistry Letters, 2017. 27(4): p. 1037-1040. [CrossRef]
- Hamdy, R. , et al., New bioactive fused triazolothiadiazoles as Bcl-2-targeted anticancer agents. International Journal of Molecular Sciences, 2021. 22(22): p. 12272. [CrossRef]
- Ziedan, N.I. , et al., Virtual screening, SAR, and discovery of 5-(indole-3-yl)-2-[(2-nitrophenyl) amino][1, 3, 4]-oxadiazole as a novel Bcl-2 inhibitor. Chemical biology & drug design, 2017. 90(1): p. 147-155.
- Chan, G.K.Y. , et al., A simple high-content cell cycle assay reveals frequent discrepancies between cell number and ATP and MTS proliferation assays. PloS one, 2013. 8(5): p. e63583. [CrossRef]
- Vertrees, R.A. , et al., Synergistic interaction of hyperthermia and gemcitabine in lung cancer. Cancer biology & therapy, 2005. 4(10): p. 1144-1153. [CrossRef]
- Hamdy, R. , et al., Synthesis and evaluation of 3-(benzylthio)-5-(1H-indol-3-yl)-1, 2, 4-triazol-4-amines as Bcl-2 inhibitory anticancer agents. Bioorganic & medicinal chemistry letters, 2013. 23(8): p. 2391-2394. [CrossRef]
- Shelley, J.C. , et al., Epik: a software program for pK a prediction and protonation state generation for drug-like molecules. Journal of computer-aided molecular design, 2007. 21(12): p. 681-691. [CrossRef]
- Greenwood, J.R. , et al., Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. Journal of computer-aided molecular design, 2010. 24(6): p. 591-604. [CrossRef]
- Hamdy, R. , et al., Iterated virtual screening-assisted antiviral and enzyme inhibition assays reveal the discovery of novel promising anti-SARS-CoV-2 with dual activity. International Journal of Molecular Sciences, 2021. 22(16): p. 9057.
- Giardina, S.F. , et al., Novel, Self-Assembling Dimeric Inhibitors of Human β Tryptase. Journal of medicinal chemistry, 2020. 63(6): p. 3004-3027.
- Hamdy, R. , et al., Efficient selective targeting of Candida CYP51 by oxadiazole derivatives designed from plant cuminaldehyde. RSC Medicinal Chemistry, 2022. 13(11): p. 1322-1340. [CrossRef]
- Halgren, T.A. , et al., Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. Journal of medicinal chemistry, 2004. 47(7): p. 1750-1759.
- Friesner, R.A. , et al., Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. Journal of medicinal chemistry, 2004. 47(7): p. 1739-1749.
- Daina, A., O. Michielin, and V. Zoete, SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Scientific reports, 2017. 7: p. 42717. [CrossRef]
- Alzaabi, M.M. , et al., Flavonoids are promising safe therapy against COVID-19. Phytochemistry Reviews, 2021: p. 1-22. [CrossRef]
- Bang, S. , et al., Azaphilones from an endophytic Penicillium sp. prevent neuronal cell death via inhibition of MAPKs and reduction of Bax/Bcl-2 ratio. Journal of Natural Products, 2021. 84(8): p. 2226-2237.
- Kamath, P.R. , et al., Indole-coumarin-thiadiazole hybrids: An appraisal of their MCF-7 cell growth inhibition, apoptotic, antimetastatic and computational Bcl-2 binding potential. European Journal of Medicinal Chemistry, 2017. 136: p. 442-451. [CrossRef]
- Nagy, M.I. , et al., Design, synthesis, anticancer activity, and solid lipid nanoparticle formulation of indole-and benzimidazole-based compounds as pro-apoptotic agents targeting bcl-2 protein. Pharmaceuticals, 2021. 14(2): p. 113.
- Liu, T. , et al., Single and dual target inhibitors based on Bcl-2: Promising anti-tumor agents for cancer therapy. European Journal of Medicinal Chemistry, 2020. 201: p. 112446. [CrossRef]
- Dwivedi, A.R. , et al., Morpholine substituted quinazoline derivatives as anticancer agents against MCF-7, A549 and SHSY-5Y cancer cell lines and mechanistic studies. RSC Medicinal Chemistry, 2022. 13(5): p. 599-609. [CrossRef]
- Ono, Y. , et al., Design and synthesis of quinoxaline-1, 3, 4-oxadiazole hybrid derivatives as potent inhibitors of the anti-apoptotic Bcl-2 protein. Bioorganic Chemistry, 2020. 104: p. 104245. [CrossRef]
- Kulabaş, N. , et al., Synthesis and antiproliferative evaluation of novel 2-(4H-1, 2, 4-triazole-3-ylthio) acetamide derivatives as inducers of apoptosis in cancer cells. European Journal of Medicinal Chemistry, 2016. 121: p. 58-70.
- Daina, A., O. Michielin, and V. Zoete, SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Scientific reports, 2017. 7(1): p. 42717. [CrossRef]
- Liu, J. , et al., Mechanisms of venetoclax resistance and solutions. Frontiers in Oncology, 2022. 12: p. 1005659. [CrossRef]
- Janssen, M. , et al., Venetoclax synergizes with gilteritinib in FLT3 wild-type high-risk acute myeloid leukemia by suppressing MCL-1. Blood, The Journal of the American Society of Hematology, 2022. 140(24): p. 2594-2610. [CrossRef]
- Timucin, A.C., H. Basaga, and O. Kutuk, Selective targeting of antiapoptotic BCL-2 proteins in cancer. Medicinal research reviews, 2019. 39(1): p. 146-175. [CrossRef]
- Wan, Y. , et al., Small-molecule Mcl-1 inhibitors: Emerging anti-tumor agents. European Journal of Medicinal Chemistry, 2018. 146: p. 471-482. [CrossRef]
- Liu, T. , et al., Design, synthesis and preliminary biological evaluation of indole-3-carboxylic acid-based skeleton of Bcl-2/Mcl-1 dual inhibitors. Bioorganic & Medicinal Chemistry, 2017. 25(6): p. 1939-1948.
- Xu, G. , et al., 1-Phenyl-1H-indole derivatives as a new class of Bcl-2/Mcl-1 dual inhibitors: Design, synthesis, and preliminary biological evaluation. Bioorganic & medicinal chemistry, 2017. 25(20): p. 5548-5556.
- Kamath, P.R. , et al., Some new indole–coumarin hybrids; Synthesis, anticancer and Bcl-2 docking studies. Bioorganic Chemistry, 2015. 63: p. 101-109.
- Nayak, S. , et al., 1, 3, 4-Oxadiazole-containing hybrids as potential anticancer agents: Recent developments, mechanism of action and structure-activity relationships. Journal of Saudi Chemical Society, 2021. 25(8): p. 101284. [CrossRef]
- Xu, Z., S. -J. Zhao, and Y. Liu, 1, 2, 3-Triazole-containing hybrids as potential anticancer agents: Current developments, action mechanisms and structure-activity relationships. European journal of medicinal chemistry, 2019. 183: p. 111700. [CrossRef]
- Sharma, P.C. , et al., Thiazole-containing compounds as therapeutic targets for cancer therapy. European journal of medicinal chemistry, 2020. 188: p. 112016. [CrossRef]
- Ashimori, N. , et al., TW-37, a small-molecule inhibitor of Bcl-2, mediates S-phase cell cycle arrest and suppresses head and neck tumor angiogenesis. Molecular cancer therapeutics, 2009. 8(4): p. 893-903. [CrossRef]
- Zhong, D. , et al., Obatoclax Induces G1/G0-Phase Arrest via p38/p21waf1/Cip1 Signaling Pathway in Human Esophageal Cancer Cells. Journal of Cellular Biochemistry, 2014. 115(9): p. 1624-1635.
- Gupta, S., F. Afaq, and H. Mukhtar, Involvement of nuclear factor-kappa B, Bax and Bcl-2 in induction of cell cycle arrest and apoptosis by apigenin in human prostate carcinoma cells. Oncogene, 2002. 21(23): p. 3727-3738. [CrossRef]
- Opydo-Chanek, M. , et al., The pan-Bcl-2 inhibitor obatoclax promotes differentiation and apoptosis of acute myeloid leukemia cells. Investigational New Drugs, 2020. 38: p. 1664-1676. [CrossRef]
- Wang, Y.-c. and P.N. Rao, Effect of gossypol on DNA synthesis and cell cycle progression of mammalian cells in vitro. Cancer research, 1984. 44(1): p. 35-38.
Figure 1.
Reported small molecule inhibitors of Bcl-2.
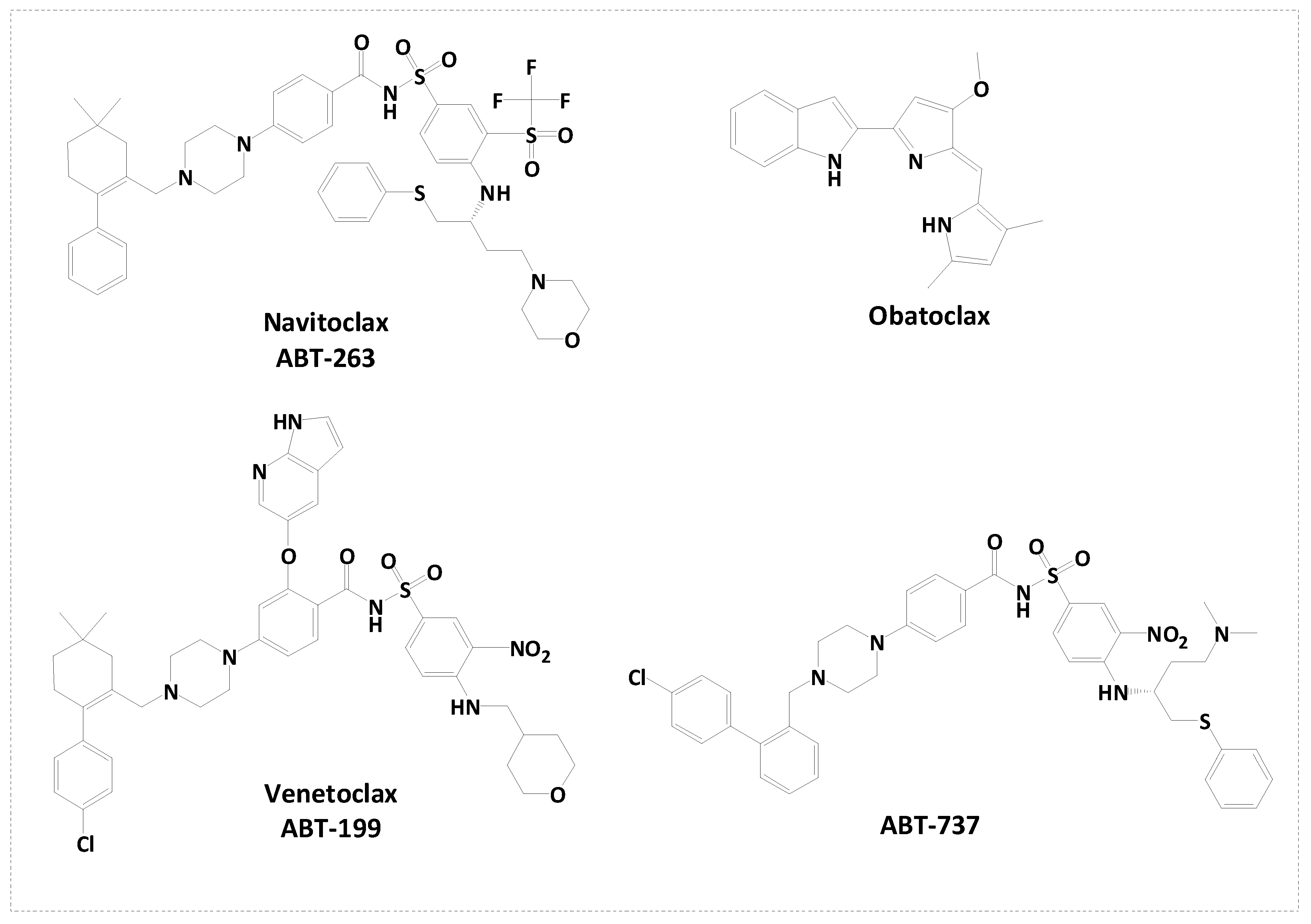
Figure 2.
Rational design of novel Bcl-2 inhibitors.
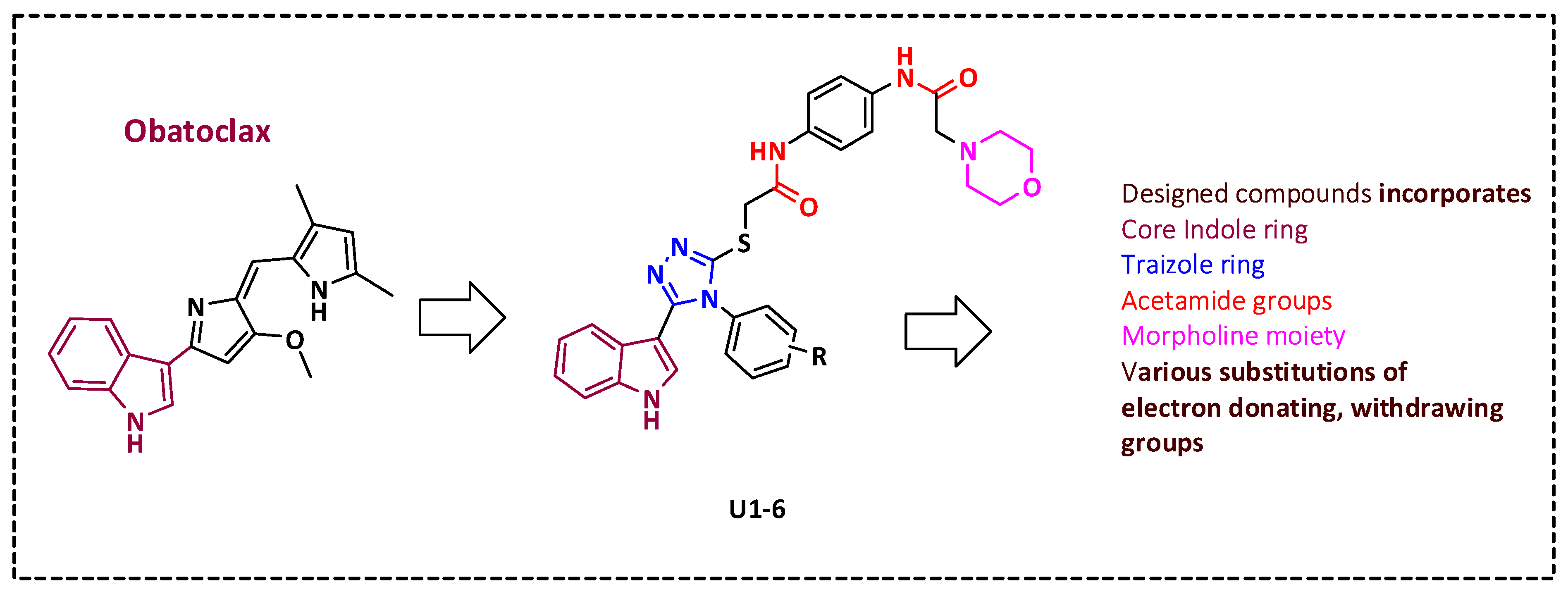
Figure 3.
(Scheme 1). Reagent and condition, (i) THF, K2CO3, r.t, overnight, (ii) Fe, HCl, ethanol, 3hrs reflux, (iii) Chloroacetyl chloride, K2CO3, DCM, 60 °C, 4h, (iv) a- 2 N NaOH, reflux, 3 h ; b- HCl, H2O , (v) Ethanol 70 %, KOH, overnight stir at room temperature (RT).
Figure 3.
(Scheme 1). Reagent and condition, (i) THF, K2CO3, r.t, overnight, (ii) Fe, HCl, ethanol, 3hrs reflux, (iii) Chloroacetyl chloride, K2CO3, DCM, 60 °C, 4h, (iv) a- 2 N NaOH, reflux, 3 h ; b- HCl, H2O , (v) Ethanol 70 %, KOH, overnight stir at room temperature (RT).
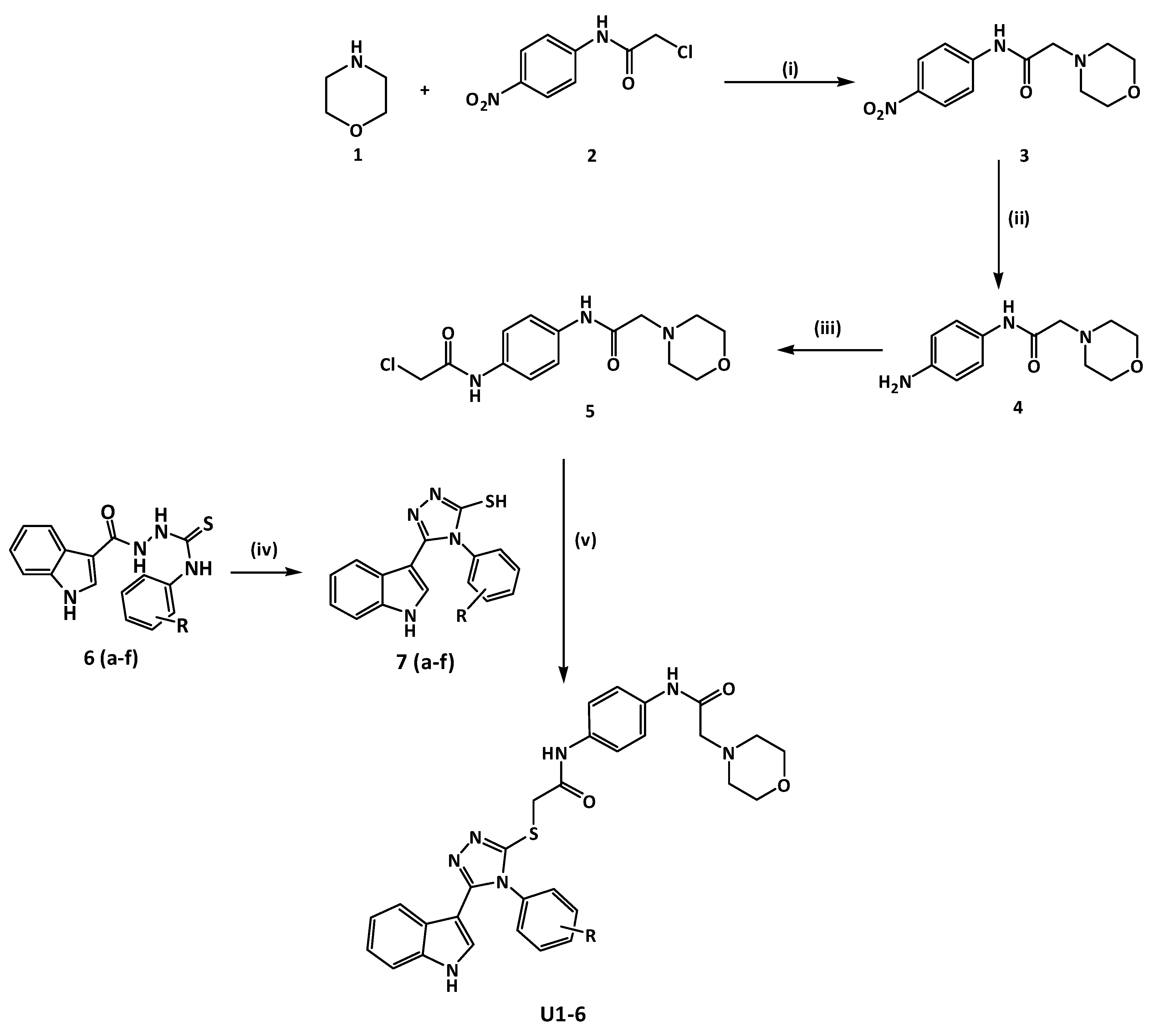
Figure 4.
Measurement of IC50 values of U1-6 compounds against different cancer cell lines. (A) MDA-MB-231 cells. (2) MCF7 cells. (C) A549 cells. The data were analyzed using GraphPad prism and were calculated with nonlinear retrogression curve analysis. The data display the mean ± standard error (SEM) of three replicates.
Figure 4.
Measurement of IC50 values of U1-6 compounds against different cancer cell lines. (A) MDA-MB-231 cells. (2) MCF7 cells. (C) A549 cells. The data were analyzed using GraphPad prism and were calculated with nonlinear retrogression curve analysis. The data display the mean ± standard error (SEM) of three replicates.
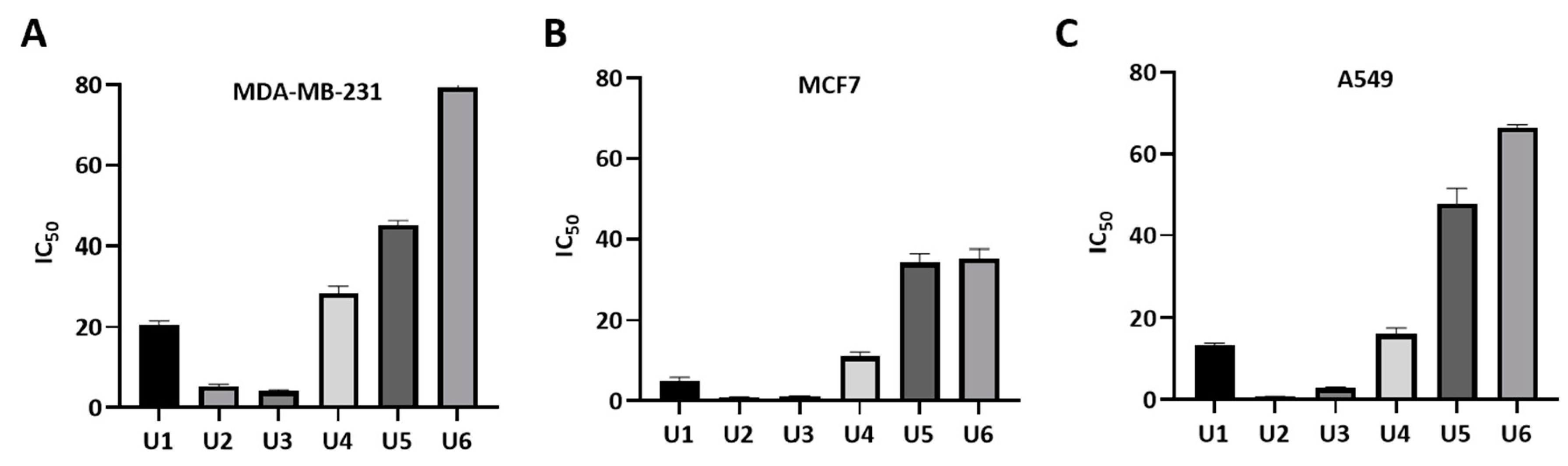
Figure 5.
Inhibition activity of the most active compounds against Bcl-2-Bim binding. (A). IC50 calculation of U2. (B) IC50 calculation of U3. (C) IC50 calculation of gossypol as positive control. The data were analyzed using GraphPad prism and were calculated with nonlinear retrogression curve analysis. DMSO was used as a negative control. The data display the mean ± standard error (SEM) of three replicates. .
Figure 5.
Inhibition activity of the most active compounds against Bcl-2-Bim binding. (A). IC50 calculation of U2. (B) IC50 calculation of U3. (C) IC50 calculation of gossypol as positive control. The data were analyzed using GraphPad prism and were calculated with nonlinear retrogression curve analysis. DMSO was used as a negative control. The data display the mean ± standard error (SEM) of three replicates. .
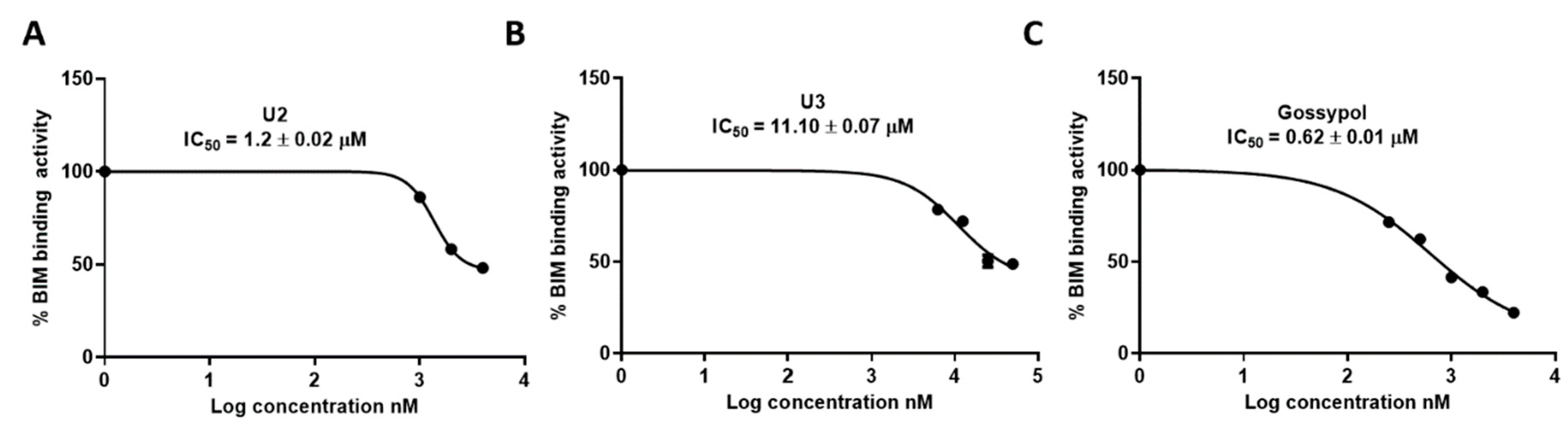
Figure 6.
Safety profile of the designed compounds against HDF (normal cell fibroblasts). The data were analyzed using one-way ANOVA and were calculated with multiple comparisons test. The significance level indicated by asterisks (*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001). The data display the mean ± standard error (SEM) of three replicates. .
Figure 6.
Safety profile of the designed compounds against HDF (normal cell fibroblasts). The data were analyzed using one-way ANOVA and were calculated with multiple comparisons test. The significance level indicated by asterisks (*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001). The data display the mean ± standard error (SEM) of three replicates. .
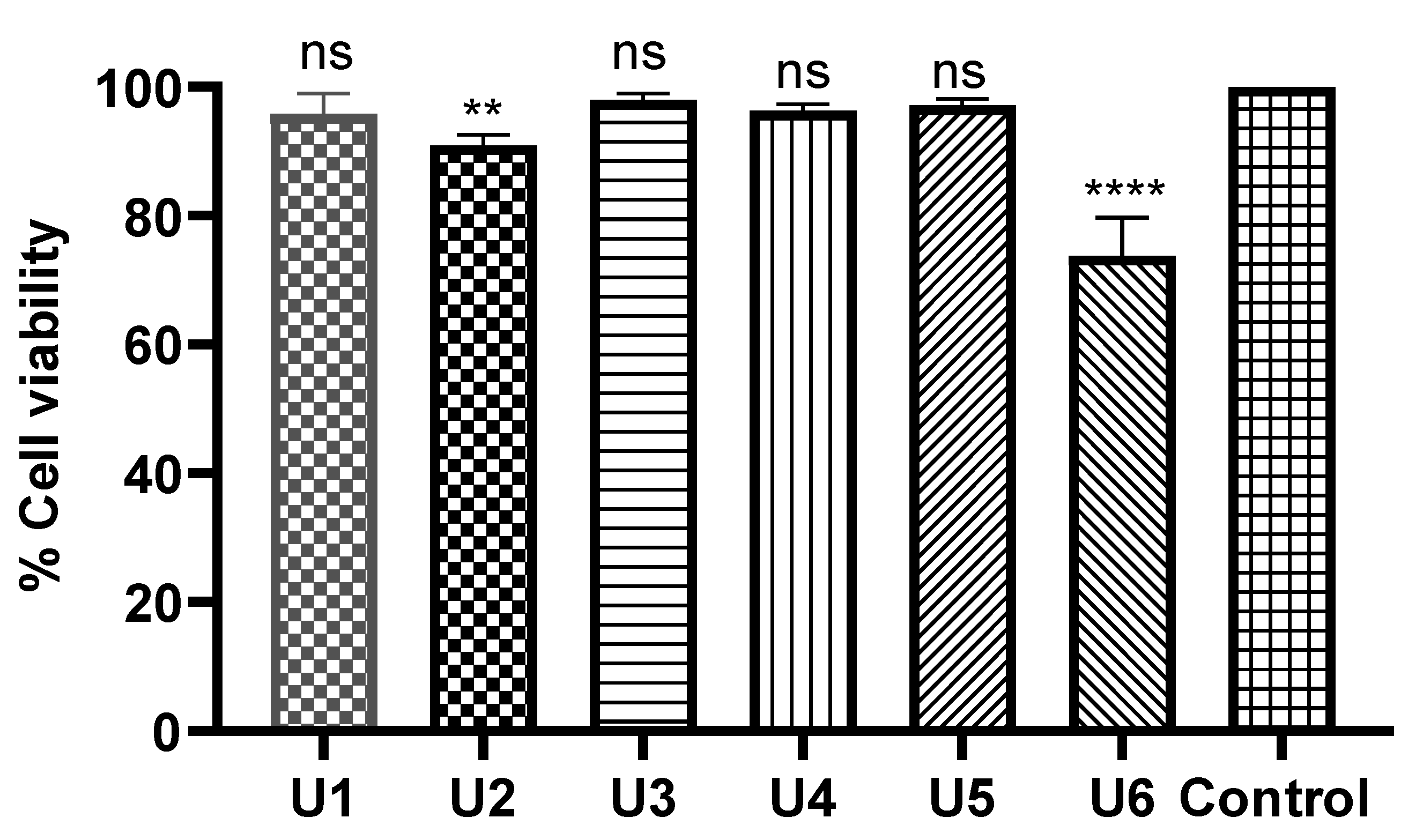
Figure 7.
Cell cycle analysis and detection of apoptosis. (A) Percentage of cells showing the increase in apoptosis because of U2. (B) MCF-7 treated with compound U2 for 24 h showed an increase in early and late apoptosis. (C) MCF-7 treated with vehicle as negative control. (D) Percentages of cells in different phases indicatingg G1/S cell cycle arrest. (E) Cell cycle analysis of U2-treated MCF-7 cells. (F) Cell cycle analysis of vehicle-treated MCF-7 cells.
Figure 7.
Cell cycle analysis and detection of apoptosis. (A) Percentage of cells showing the increase in apoptosis because of U2. (B) MCF-7 treated with compound U2 for 24 h showed an increase in early and late apoptosis. (C) MCF-7 treated with vehicle as negative control. (D) Percentages of cells in different phases indicatingg G1/S cell cycle arrest. (E) Cell cycle analysis of U2-treated MCF-7 cells. (F) Cell cycle analysis of vehicle-treated MCF-7 cells.
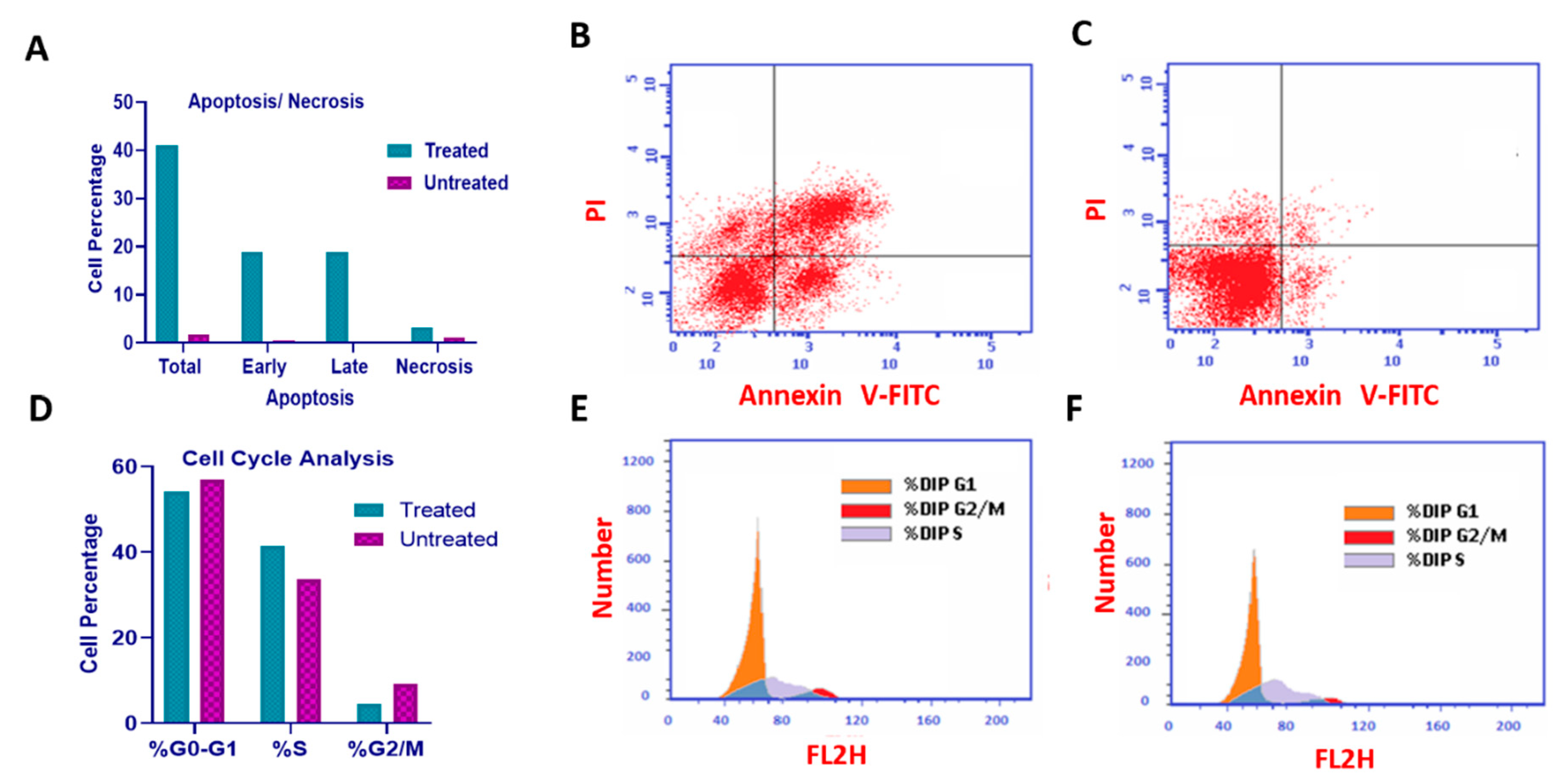
Figure 8.
Molecular docking of U1-U3 compounds within Bcl-2 binding site (PDB 4AQ3). (A) Interaction of U2 within the active site of Bcl-2. (B) Interaction of U3 within the active site of Bcl-2. (C) Interaction of U1 within the active site of Bcl-2.
Figure 8.
Molecular docking of U1-U3 compounds within Bcl-2 binding site (PDB 4AQ3). (A) Interaction of U2 within the active site of Bcl-2. (B) Interaction of U3 within the active site of Bcl-2. (C) Interaction of U1 within the active site of Bcl-2.
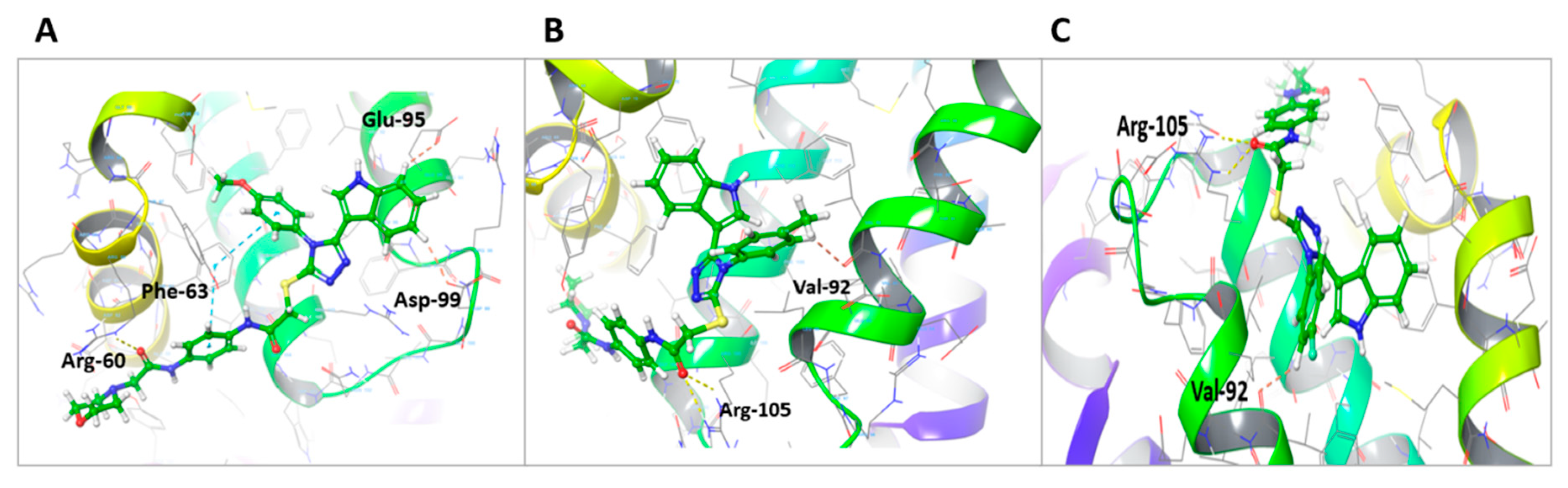
Figure 9.
(A) Bioavailability radar of U2. (B) Bioavailability radar of BCL-2 inhibitor ABT-263. The pink area indicates preferred properties range.
Figure 9.
(A) Bioavailability radar of U2. (B) Bioavailability radar of BCL-2 inhibitor ABT-263. The pink area indicates preferred properties range.
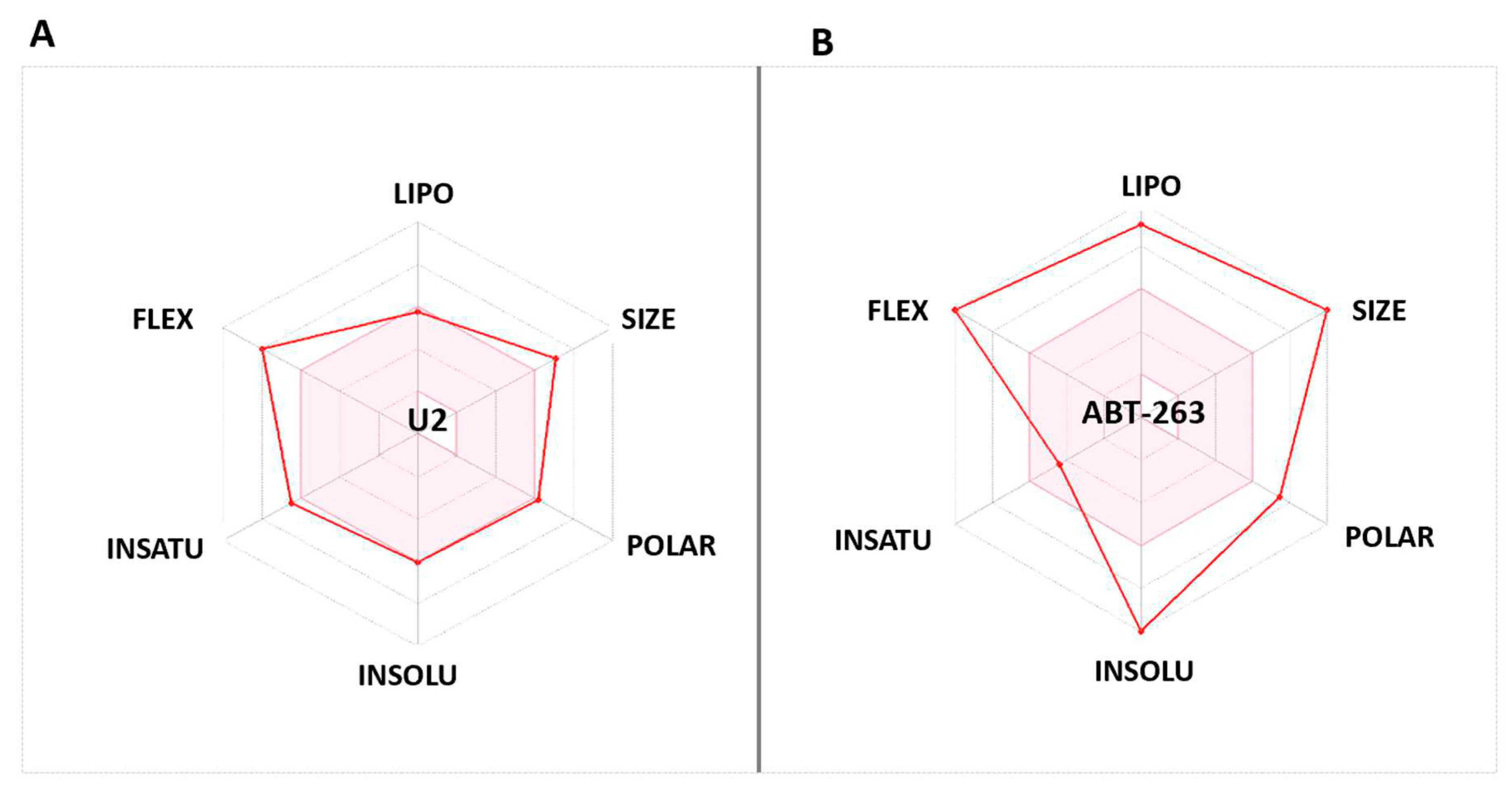

Table 1.
IC50 of U1-6 compounds against MDA-MB-231, MCF-7, and A549 human cancer cells.
| Compound | R | IC50 * | ||
|---|---|---|---|---|
| MDA-MB-231 | MCF-7 | A549 | ||
| U1 | 4-F | 20.66 ± 0.85 | 5.05 ± 0.80 | 13.34 ± 0.58 |
| U2 | 4-OCH3 | 5.22 ± 0.55 | 0.83 ± 0.11 | 0.73 ± 0.07 |
| U3 | 4-CH3 | 4.07 ± 0.35 | 1.17 ± 0.10 | 2.98 ± 0.19 |
| U4 | 3-Cl | 28.38 ± 1.79 | 11.0 ± 1.2 | 16.11 ± 1.4 |
| U5 | 4-Cl | 45.1 ± 1.15 | 34.43 ± 2.20 | 47.77 ± 3.7 |
| U6 | 3,4-Dichloro | 79.4 ± 4.04 | 35.45 ± 2.14 | 66.5 ± 0.62 |
| Gossypol | 5.5 ± 0.35 | 4.43 ± 0.54 | 3.45 ± 0.40 | |
*The results are presented as triplicate mean values ±STD, from testing on three independent occasions.
Table 2.
IC50 of the selected compounds using ELISA against Bcl-2.
| Compound | IC50 * |
|---|---|
| U2 | 1.2 ± 0.02 |
| U3 | 11.10 ± 0.07 |
| Gossypol | 0.62 ± 0.01 |
*Mean value and SEM score of three independent measurements.
Table 3.
Molecular docking of U1-3 within the binding site of Bcl-2 (PDB: 4AQ3).
| Compound | Moiety | Interaction | Amino acid |
|---|---|---|---|
| U1 | Carbonyl group Phenyl ring Phenyl ring Phenyl ring Phenyl ring Sulfanyl group |
2 H-bond interaction Aromatic H-bond Hydrophobic interaction Hydrophobic interaction Hydrophobic interaction Hydrophobic interaction |
Arg-105 Val-92 Tyr-67 Phe-63 Glu-95 Leu- 96 |
| U2 | Carbonyl group Phenyl ring Phenyl ring Phenyl ring Phenyl ring Sulfanyl group Phenyl ring Phenyl ring Phenyl ring |
H-bond interaction Pi-pi staking Aromatic H-bond Pi-pi staking Aromatic H-bond Hydrophobic interaction Hydrophobic interaction Hydrophobic interaction Hydrophobic interaction |
Arg-60 Phe-63 Asp-99 Phe-63 Glu-95 Arg-105, Tyr-63 and Ala-108 Ala-108 Tyr-67 Glu-95 |
| U3 | Carbonyl group Phenyl ring CH3 Sulfanyl group Carbonyl group Phenyl ring Phenyl ring Phenyl ring |
2 H-bond interaction Aromatic H-bond Hydrophobic interaction Hydrophobic interaction Hydrophobic interaction Hydrophobic interaction Hydrophobic interaction Hydrophobic interaction |
Arg-105 Val-92 Glu-95 Leu-96 Arg-105 Asp-70 Tyr-67 and Glu-95 Val-92 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



