
Submitted:
26 July 2023
Posted:
28 July 2023
You are already at the latest version
Abstract
Immune checkpoint inhibitors (ICIs) have revolutionized cancer care and shown remarkable efficacy clinically. This efficacy is, however, limited to subsets of patients with significant infiltration of lymphocytes into the tumor microenvironment. To extend their efficacy to patients who fail to respond or achieve durable responses, it is now becoming evident that complex combinations of immunomodulatory agents may be required to extend efficacy to patients with immunologically “cold” tumours. Oncolytic viruses (OVs) have the capacity to selectively replicate within and kill tumour cells resulting in the induction of immunogenic cell death and the augmentation of anti-tumour immunity and have emerged as a promising modality for combination therapy to overcome the limitations seen with ICIs. Pre-clinical and clinical data has demonstrated that OVs can increase immune cell infiltration into the tumour and induce anti-tumour immunity, thus changing a “cold” tumour microenvironment that is commonly associated with poor response to ICIs, to a “hot” microenvironment which can render patients more susceptible to ICIs. Here, we review the major viral vector platforms used in OV clinical trials, their success when used as a monotherapy and when combined with adjuvant ICIs, as well as pre-clinical studies looking at the effectiveness of encoding OVs to deliver ICIs locally to the tumour microenvironment through transgene expression.
Keywords:
Oncolytic virus
; immune checkpoint inhibitors
; immunotherapy
1. Introduction
The development of immune checkpoint inhibitor (ICI) therapies, which target immunosuppressive signals and restore anti-tumour immunity, has revolutionised the immunotherapy field in recent years. Antibodies targeting cytotoxic T lymphocyte-associated protein 4 (CTLA-4), programmed cell death protein 1 (PD-1), and its ligand PD-L1 aim to disrupt these negative regulatory signals, which under physiological conditions protect the host from autoimmunity and chronic inflammation, to disrupt the ability of tumour cells to evade the host immune response. The goal of ICI therapy is to recruit and activate innate and adaptive immune cells within the tumour microenvironment (TME), reverse T cell exhaustion, and reinvigorate anti-tumour T cells to control tumour growth [1]. As ICI therapy primarily functions to reinvigorate existing tumour reactive T cells, rather than induce their formation, durable clinical responses are most commonly seen in cancers which demonstrate an immunologically “hot” TME, characterised by a high somatic tumour mutation burden (TMB) and highly infiltrated immune active TMEs [2]. However, a lack of therapeutic benefit has been observed in those tumours which possess an immunologically “cold” TME; these tumours demonstrate a low density of tumour infiltrating lymphocytes (TILs), lack of tumour antigen expression and/or loss of tumour antigen presentation machinery, and infiltration of immunosuppressive immune cells such as neutrophils, macrophages, myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs) [3]. Therefore, to effectively build on the recent successes of ICIs it is critical that to extend their efficacy to non-responders, combination strategies need to be generated which aim to “heat up” tumours to obtain durable clinical responses. To this end, oncolytic viruses (OVs), which preferentially infect and destroy cancer cells, thus inducing immunogenic cell death, are a compelling combination agent which possess the ability to increase immune cell infiltration and overcome immunosuppression within the TME [4]. The promise of this combinatorial approach has led to multiple clinical trials which aim to investigate the efficacy of adjuvant OV and ICI therapy in several cancers. In this review, we describe the clinical efficacy of OVs as monotherapies and when delivered as a neoadjuvant with systemic ICI therapy. Furthermore, we explore the pre-clinical studies of OVs engineered to encode antibodies against immune checkpoints which aim to locally target ICI expression to within the tumour to overcome the adverse events associated with systemic immunotherapy.
2. Oncolytic viruses
OVs are immunotherapies which exploit the ability of replication-competent viruses to infect and replicate in tumour cells, whilst leaving healthy cells intact, leading to tumour cell lysis and subsequent release of viral progeny. Upon infection of cells, viruses possess the ability to promote their replication and subsequent release of viral progeny by interacting with cellular proteins to avoid immune cell recognition and early host cell death. Viruses typically activate one or more cell death pathways during infection and replication. Some forms of cell death are intrinsically tolerogenic and result in the uptake of dead cells by phagocytic cells, conversely cell death can induce an innate and adaptive immune response termed immunogenic cell death (ICD). The induction of immunogenic tumour cell death results in local inflammation through the release of danger-associated molecular patterns (DAMPs) from dying infected cells, such as high mobility group box 1 (HMGB1), heat shock proteins (HSP), cell surface exposure of Calreticulin, and extracellular adenosine triphosphatase (ATP). Furthermore, virus replication and cell lysis leads to the release of pathogen-associated molecular patterns (PAMPs), such as viral proteins and nucleic acids which further contribute to intensifying the immune response [5] (Figure 1).
This therapeutic efficacy is dependent on a fine balance between viral immunogenicity and anti-tumour immunity in which OVs can persist and avoid immune clearance, at least temporarily, to allow sufficient time for OVs to infect and replicate within tumour cells and to initiate an anti-tumour immune response [6]. In addition to their immunogenicity, the size, pathogenicity, and transgene capacity of a virus all contribute towards the selection of the appropriate vector for use as an OV therapy (Table 1). Some OVs, such as those derived from strains of coxsackie virus, influenza A virus (IAV), newcastle disease virus (NDV), measles virus (MV), reovirus, vaccinia virus (VV) and vesicular stomatitis virus (VSV) demonstrate a natural tropism for tumours through exploitation of extracellular makers or dysregulated oncogenic intracellular pathways in tumour cells [7–12]. Alternatively, OVs such as those derived from adenovirus (Ad) and herpes simplex virus (HSV) can be genetically modified to increase tumour cell selectivity through deletion and modification of genes to alter the natural tropism of the virus and provide a replicative advantage in tumour cells [13,14]. In addition, OVs can be further engineered through the insertion of eukaryotic transgenes to promote replication competence, limit their pathogenicity, increase their immunogenicity, and deliver additional genetic “payloads” which can promote anti-tumour immunity or increase the extent of tumour cell death [15].
2.1. Turning cold tumours hot: the OV immune response
During tumour development, tumour cells undergoing continuous remodelling at the genetic, epigenetic, and metabolic levels generate the critical modifications necessary for these cells to escape both innate and adaptive immune control, thus leading to malignant progression and growth of the tumour in the face of a competent immune system. Tumour immune evasion can result from changes at the level of the tumour, through inhibition of immune cell recognition and the selection of tumour variants that are resistant to immune effectors, or through the induction and recruitment of distinctive immunosuppressive immune cells and cytokines within the TME, thus generating a “cold” immunosuppressive TME [16]. The aim of immunotherapies is to revert these “cold” TMEs into immune activated and infiltrated “hot” TMEs indicative of an active anti-tumour immune response taking place; therefore, OVs which induce immunogenic tumour cell death and induce innate and adaptive immune responses are an ideal therapeutic candidate [17].
Following OV infection of tumour cells and subsequent local inflammation, innate immune cells such as dendritic cells (DCs), natural killer (NK) cells and macrophages within the TME recognize the DAMPs, PAMPs and tumour antigens released by oncolysis, resulting in the secretion of inflammatory cytokines such as interferon-γ (IFN-γ), IFN-α, interleukin-6 (IL-6), IL-12 and tumour necrosis factor-α (TNF-α) which promote the maturation of DCs and further recruitment and activation of innate immune cells [18,19] (Figure 2). Antigen-loaded antigen-presenting cells (APCs) then migrate to draining lymph nodes where they initiate antigen-specific T cell priming and activation. In addition to T cell priming and activation, OV infection also elicits a potent type I IFN response, which stimulates the production of T cell-recruiting chemokines which increase TME T cell infiltration [20]. Furthermore, the induction of inflammatory cytokines such as TNF-α and IL-1β upregulates the expression of selectin on endothelial cells, allowing for enhanced extravasation of T cells into the tumour [17]. Upon entering the TME, TILs must contend with an often-dense network of stromal cells and extracellular matrix (ECM) which can prevent efficient T cell infiltration into the tumour. OV infection has been shown to alleviate these structural barriers through the recruitment of neutrophils which can secrete proteases, such as elastase and matrix metalloproteinases (MMPs), to degrade the ECM and increase immune cell infiltration [21,22]. In addition to the activation, priming, trafficking and infiltration of anti-tumour immune cells, OV infection can also overcome immunosuppressive signals within the TME through the stimulation of pro-inflammatory cytokine production and induction of potent pro-inflammatory M1 macrophages and type 1 helper (Th1) immune cell phenotypes [5,23,24].
3. Oncolytic virus monotherapy
To date, Ad, coxsackie virus, HSV, NDV, MV, VV and VSV OVs have all entered clinical trials for the treatment of several different cancers, with one adenovirus OV (H101) approved in China for the treatment of head and neck cancer, and two HSV OVs now approved for the treatment of metastatic melanoma in Europe and the US (T-VEC; Imlygic) and glioblastoma (GBM) in Japan (G47∆) (Table 2) [25,26]. Patients treated with OV monotherapy often demonstrate significant reductions in tumour burden, as demonstrated by decreased tumour size, partial response (PR), and complete response (CR) rates [8,27–40], or present with stable disease (SD) and progression free survival (PFS), suggesting an anti-tumour immune response is occurring to control and destroy the tumour [7,8,26–29,31,35–37,41–46]. Indeed, OV treated tumours have demonstrated an increase in tumour infiltrating CD8+ T cells and the systemic presence of tumour antigen specific CD8+ T cells, in addition to a decrease in immunosuppressive MDSCs and Tregs within the TME [26–28,30,32,38,39,41,43–45,47]. However, despite demonstrating disease control and the presence of an activated immune response, only a small proportion of these clinical trials are able to demonstrate a durable clinical response (≥ 6 months) in small subsets of patients [26–28,30,34,43,44,46,48].
Despite some promising examples of clinical efficacy, it is evident that OV monotherapies need to be enhanced for patient benefit. Several potential mechanisms, including the existence of neutralising antibodies, rapid anti-viral immune responses resulting in rapid and premature OV clearance, physical exclusion from the TME, and an immunosuppressive TME, may contribute to the modest activity seen with OV monotherapy and contribute to OV resistance [3]. Therefore, to enhance clinical efficacy, combined immunotherapeutic approaches comprising OV with adjuvant ICI therapy, have been developed and demonstrate improved clinical efficacy when compared to either therapy alone [49]
4. Combined OV and ICI therapy
4.1. Neoadjuvant therapies
Expression of immune checkpoint molecules by cancer cells is one of the major mechanisms by which tumours can induce immunosuppression and subsequent immune evasion. CTLA-4, lymphocyte-activation gene 3 (LAG-3), T cell immunoglobulin and mucin-domain containing-3 (TIM-3), and T cell immunoreceptor with Ig and ITIM domains (TIGIT) all primarily interact with their ligands during the T cell priming stage, thereby limiting T cell activation, whilst PD-1:PD-L1 interactions occur predominantly in the periphery to regulate activated T cells during the effector phase [50]. CTLA-4 was the first immune checkpoint to be clinically targeted; it is expressed exclusively on Tregs and activated effector T cells where it regulates the amplitude of T cell activation during priming; CTLA-4 expressing T cells often display tolerance towards tumours and CTLA-4 expressing Tregs contribute towards immunosuppression within the TME by further inhibiting the functions of other immune cells. Similar to CTLA-4 signalling, PD-1 binding with its ligands inhibits T cell function by reducing the intensity of IFN-γ, TNF and IL-2 production, reducing T cell survival through the inhibition of anti-apoptotic gene production, and suppressing T cell proliferation [51,52]. Such immune checkpoint molecule-mediated immunosuppression of the anti-tumour immune response facilitates the progression of cancer in the face of a competent immune system. Thus, ICI therapies aim to interrupt these immunosuppressive signals to restore anti-tumour immunity by exposing the tumour cells to a newly reinvigorated host immune response.
ICI antibodies such as the anti-CTLA-4 Ipilimumab, anti-PD-1 Nivolumab and Pembrolizumab, and anti-PD-L1 Atezolizumab have been approved for the treatment of several solid and haematological malignancies and have shown durable clinical responses in a proportion of patients [4]. In those patients who do benefit, clinical responses correlate with high tumour mutational burden, a rich neoantigen repertoire and the presence of a pre-existing anti-tumour response, as evidenced by increased TILs [6]. Conversely, those patients with cold TMEs, characterised by low tumour mutational burdens, a lack of expression or presentation of neoantigens, and low infiltration of TILs do not demonstrate durable clinical responses following treatment. As ICI therapy functions to reactivate an exhausted and suppressed anti-tumour immune response, TILs are the most important component for a patient to derive durable responses from ICI therapy, with the presence of a prominent T cell infiltration prior to treatment associated with increased sensitivity and survival following ICI treatment [53,54]. While several combination therapy approaches are in development to reverse these deficiencies in non-responsive patients, OV therapy is a promising combination therapeutic as it induces tumour cell death in a highly immunogenic context, thereby triggering an “in situ” tumour vaccination through the release of tumour antigens in the presence of virus-induced inflammation. Combination therapies using ICI and OVs are therefore attractive and potentially synergistic, as the OV therapy can “heat up” the TME by recruiting TILs, promoting further immune cell activation, and triggering the release of tumour antigens [55]. Moreover, treatment with Ad and HSV OV monotherapies have demonstrated significant increases in tumour PD-L1 expression, thus sensitizing tumours to subsequent ICI therapy [32,38,45].
Patients treated with neoadjuvant oncolytic Ad, HSV and VV followed by ICIs targeting PD-1, PD-L1 and CTLA-4 have all demonstrated clinical benefit, with durable response rates observed in subsets of patients (Table 3) [56–58]. Local intra-tumoral injection of OVs prior to systemic ICI therapy resulted in both an increase in CD8+ and CD4+ TILs and increases in circulating CD8+ and CD4+ T cells, in addition to local inflammation and reductions in the size of non-injected tumours, suggesting the presence of a systemic anti-tumour immune response [30,53–60]. When compared to Pembrolizumab monotherapy in advanced stage immunotherapy-naïve melanoma patients, combination T-VEC and Pembrolizumab therapy demonstrated slightly increased response rates and PFS, although this did not reach significance [58]. Similarly, combination T-VEC and Ipilimumab therapy did not significantly increase PFS or OS when compared to Ipilimumab alone; however, the combination therapy did demonstrate a significant increase in ORR (CR/PR). Furthermore, combination therapy resulted in an increase in the reduction in size of visceral non-injected lesions, consistent with a systemic anti-tumour immune response [66].
4.1.2. Markers of response
As expected, greater persistence of viral DNA in the tumour is indicative of greater clinical responses, with the presence of ONCOS-102 DNA at week 9 post-injection detectable in responders but undetectable in patients with progressive disease, suggesting rapid OV clearance or less effective viral replication may prevent disease control [62]. In the same trial, the baseline presence of CD8+ and CD4+ TILs were also significantly greater in patients with disease control compared to those with progressive disease, with further increased tumour infiltration of CD4+ and CD8+ T cells following OV administration only seen in patients with disease control [57]. Pre-treatment presence of CD3+/CD8+ aggregates at the infiltrating tumour edge and a greater abundance of TILs were also indicators of responsive patients in patients treated with T-VEC and Pembrolizumab [64]. However, objective responses to DNX-2401 and Pembrolizumab were only observed in patients with moderately inflamed TMEs, with those presenting pre-treatment with highly inflamed tumours enriched with exhausted immune cells, characterised by high expression of immunosuppressive immune checkpoints, receiving no improvements in survival, suggesting that the immunosuppressive TME in these patients may suppress any immune response induced by OV or ICI therapy [57]. The correlation with pre-treatment immune infiltration and response to combination therapy raises some concerns, as although the neoadjuvant OV therapy aims to induce immunogenic cell death and increase immune infiltration into the tumour, it appears that the tumour must already have some evidence of an immune response in order to demonstrate a response. This suggests that, as with ICI therapy, those patients who present with immunologically cold tumours may not derive good clinical responses from these therapies.
4.1.3. Adverse events
Although all studies reported adverse effects in both the monotherapy and combination therapy arms none reported dose-limiting toxicities and all adverse events (AEs) were of those expected and observed with the single-agent use of either therapy. Combination treatments were not associated with an increase in the incidence or severity of AEs and most AEs such as fatigue, fever, chills, arthralgia, rash and nausea were of mild to moderate (Grade 1/2) severity. Grade 3/4 AEs occurred in subsets of patients in addition to some fatal AEs; however, when compared to monotherapies, again the incidence of these events was similar, suggesting that combination therapy is tolerable for patients [30,53–61]. ICI therapy functions to remove the inhibitory signals placed on effector immune cells, thus effectively removing the brakes on the anti-tumour immune response; however, although OVs are often locally delivered by intra-tumoural injection, ICIs are delivered systemically via the intravenous route. Under normal physiological conditions, immune checkpoints function to prevent over activation of the immune system and auto-immune responses, therefore systemic blocking of these signals often results in autoimmune AEs. These systemic toxicities associated with intravenous ICI therapy could potentially be reduced by targeted delivery of the antibodies directly into the TME, indeed low-dose intra-tumoural administration of ICIs has been shown to be comparable to systemic high-dose delivery [67,68]. Therefore, the use of OVs engineered to express ICIs, thus limiting ICI expression to areas of viral replication within the TME, is an attractive approach for local ICI delivery which may limit systemic AEs.
4.2. OVs encoding ICIs
Ad, HSV, IAV, NDV, MV, VV, VSV and chimeric poxviruses have all been engineered to express ICIs targeting CTLA-4, PD-1 and PD-L1 either as full-length IgG antibodies, single-chain fragment variables (scFV), or scFV-Fc fusion proteins, showing promising results in a variety of in vivo animal models (Table 4). In comparison to full length monoclonal antibodies (mAbs), scFvs are fragments of antibody consisting of variable regions of the light (VL) and heavy (VH) chains joined by a flexible linker peptide; this smaller size allows for greater penetration and efficient localisation into the tumour, and faster clearance from the blood. Furthermore, the reduced transgene size can allow for the addition of further transgenes in OVs with high loading capacity, thus further enhancing the immunotherapeutic viral payload [55]. Despite antibodies naturally being produced in highly specialized and differentiated plasma cells, it has been demonstrated that functional ICI antibodies can be detected in tumour cells in vitro [69–76] and in vivo [70] following infection with engineered OVs, and that these antibodies are therapeutically functional.
Intra-tumoural injection of ICI-encoding OVs (ICI-OVs) resulted in significant reductions in tumour volume compared to untreated [45,69,70,77–81] and parental OV treated tumours [72–76,82–86], with ICI-OVs also significantly increasing OS compared to untreated [78–81,87,88] and parental OV treated tumours [73,75,77,82,84–86,89], demonstrating that OVs encoding for antibodies against CLTA-4, PD-1 and PD-L1 significantly increase the anti-tumour immune response. This increase in therapeutic activity was associated with increased CD4+ and CD8+ T cell and decreased Treg tumour infiltration, and an increase in the proportion of activated and effector memory and central memory T cells [74–76,84,86,89,90]. Furthermore, local intra-tumoural administration of ICI-OVs elicited pronounced systemic increases in activated CD4+ and CD8+ T cells, increased effector memory and central memory T cells and decreased MDSC and Treg populations [73,77,81]. Moreover, an abscopal effect mediated by a systemic anti-tumour response was observed using bilateral tumour models with unilateral IT injection, wherein un-injected distant tumours demonstrated decreased tumour growth and increased immune infiltration and activation [74,75,90], with one melanoma model demonstrating delayed non-injected tumour growth and prolonged survival with IAV-CTLA4 compared to parental IAV treatment [72]. The delay in untreated tumour growth, in combination with the increased presence of memory T cells, is indicative of the induction of long-term immune memory. Indeed, when re-challenged with tumour cells in vivo, ICI-OV treated animals were able to successfully inhibit tumour growth [73,74,77,81,82], with one study demonstrating significant reductions in tumour growth and increased survival following VV-PDL1 treatment compared to parental VV treatment [90]. Taken together, these pre-clinical studies demonstrate that OVs encoding ICI antibodies can significantly decrease tumour growth and prolong survival when compared to parental OV treatment in multiple tumour models.
When compared to parental OV plus systemic ICI treatment, ICI-OVs demonstrate similar reductions in tumour volume and increased survival, suggesting that ICI-OVs could represent a promising new immunotherapeutic with reduced AEs compared to OV + systemic ICI [73,76,77,82,89–91]. Currently, research into IVI-OVs is predominantly limited to mouse models and data on AEs is limited at present. However, the safety profile of IV injection of an oncolytic HSV1 encoding a PD-1 scFv was assessed in a more clinically relevant non-human primate model and demonstrated no abnormal body weight or temperature changes, slight elevations in serum markers of renal and liver dysfunction which returned to normal after several days, no overt changes in leukocyte counts or increases in cytokine production, and no obvious pathological abnormalities in any organs [75]. These favorable safety outcomes in a non-human primate model, in combination with the toxicity studies in humanized mouse models, are an encouraging step in the translation of these ICI-OVs to the clinic, wherein the full therapeutic benefit of a human OV encoding for a human ICI can be studied.
4.2.1. Additional targets
Activation of an immune response following ICI therapy has been associated with the upregulation of additional immune checkpoint molecules such as CTLA-4, PD-1, PD-L1, TIGIT, LAG-3 and TIM-3 on immune cells [92]. This increase in immunosuppressive signals is one such mechanism by which patients can become resistant to ICI therapy; therefore combination therapies targeting multiple immune checkpoints are a promising mechanism to improve therapeutic outcomes, as by targeting multiple immunoregulatory pathways the likelihood of a successful and sustained anti-tumour immune response is increased [93]. Indeed, data from the ICI-OV pre-clinical studies has suggested that the therapeutic outcome of these ICI-OVs can be further improved with the addition of systemic ICIs targeting additional immune checkpoints. An oncolytic NDV encoding for an anti-PD-1 scFV demonstrated significant survival benefits over parental virus when combined with systemic CTLA-4 treatment, with the combination therapy targeting two immunoregulatory pathways at distinct yet synergistic stages in the immune response, namely the priming (CTLA-4) and effector (PD-1) phases of the adaptive immune response, inducing up to 50% CR rates [85]. Likewise, systemic targeting of TIM-3, an immune checkpoint that functions to suppress T cell responses, has been shown to more potently suppress tumour growth and improve the anti-tumour efficacy of an anti-PD1 scFV HSV OV [75]. In a different study, the anti-tumour efficacy of an anti-PD-1 scFV HSV OV was improved by the addition of systemic TIGIT ICI therapy, as evidenced by increased splenic tumour-specific CD8 T cells [74]. TIGIT is expressed on naïve T cells and activated NK cells and Tregs, and interacts with its two major ligands poliovirus receptor (PVR; CD155) and poliovirus receptor-related 2 (PVRL2; CD112), which are expressed on myeloid cells and tumour cells [94]. This enhanced therapeutic efficacy demonstrated with combinatorial PD-1 and CTLA-4/TIM-3/TIGIT blockade demonstrates that immune checkpoints which function in distinct yet synergistic stages in the immune response, namely the priming (CTLA-4/TIM-3/TIGIT) and effector (PD-1) phases of the adaptive immune response, can synergise to enhance anti-tumour immunity. In addition to systemic anti-TIGIT therapies, an oncolytic VV armed with an scFV against TIGIT has demonstrated enhanced anti-tumour efficacy and increased recruitment and activation of T cells within the TME compared to parental virus in several subcutaneous tumour models [95,96]. Research into additional novel immune checkpoints such as LAG-3, CD200, TIM-3, B7 homolog 3 protein (B7-H3) have shown promising results in pre-clinical and clinical models, suggesting that OVs could be engineered to express antibodies against these checkpoints in the future [97].
5. Conclusions
Monoclonal antibodies targeting the immune checkpoints CTLA-4, PD-1 and PD-L1 have demonstrated promising clinical efficacy in several cancers, with subsets of patients deriving durable clinical responses. However, as PD1/PD-L1 ICI therapy works to re-invigorate tumour reactive T cells, its success is based on the presence of a pre-existing anti-tumour immune response and an immunologically “hot” TME. Thus, overall response rates are 47-63%, with non-responder patients demonstrating both cell intrinsic and extrinsic primary resistance mechanisms. Similarly, response rates with CTLA-4 ICI therapy are 10-20% [98,99]. Furthermore, of those that do show an initial or sustained response, disease relapse and progression occur in most cases due to acquired secondary resistance mechanisms within the TME. Therefore, therapeutic combinations which can turn an immunologically “cold” tumour into a highly inflamed “hot” tumour which is primed for subsequent ICI therapy are a promising mechanism to overcome ICI resistance. OVs which selectively replicate within and kill tumour cells, resulting in the induction of immunogenic cell death and the augmentation of anti-tumour immunity, have emerged as a promising modality for combination therapy.
OV monotherapies have demonstrated moderate clinical efficacy in several clinical trials and, when used as a neoadjuvant with systemic ICIs, have been shown to increase the anti-tumour immune response. There are currently several ongoing clinical trials in a range of cancers looking at novel neoadjuvant OV and ICI combination therapies with the aim of achieving durable clinical responses in patients who would often not benefit from systemic ICI treatment alone. In addition, pre-clinical animal models of OVs engineered to encode for ICI antibodies have shown promising results, with tumour growth control and overall survival similar to that seen with OV and systemic ICI therapy. A clinical trial looking at the efficacy of an oncolytic adenoviral vector encoding an anti-CD40 antibody in advanced tumours, alone and in combination with systemic Pembrolizumab, has completed and is currently awaiting results (NCT03852511). This targeted and local expression of ICI antibodies could represent a mechanism by which the AEs associated with systemic alleviation of immunosuppression, a major drawback to ICI therapy, can be overcome; therefore, these results are eagerly anticipated.
Funding
CL is funded by a Pancreatic Cancer Research Fund project award to ALP, reference 522761. ALP is also supported by a Cancer Research UK Biotherapeutic Programme grant (reference C52915/A29104).
Acknowledgements
All figures were created with biorender.com
Conflicts of Interest
ALP is Founder and Chief Scientific Officer of Trocept Therapeutics Ltd.
References
- Dyck, L.; Mills, K.H.G. Immune Checkpoints and Their Inhibition in Cancer and Infectious Diseases. European Journal of Immunology 2017, 47, 765–779. [CrossRef]
- Maleki Vareki, S. High and Low Mutational Burden Tumors versus Immunologically Hot and Cold Tumors and Response to Immune Checkpoint Inhibitors. Journal for ImmunoTherapy of Cancer 2018, 6, 4–8. [CrossRef]
- Harrington, K.; Freeman, D.J.; Kelly, B.; Harper, J.; Soria, J.C. Optimizing Oncolytic Virotherapy in Cancer Treatment. Nature Reviews Drug Discovery 2019, 18, 689–706. [CrossRef]
- Hwang, J.K.; Hong, J.; Yun, C.O. Oncolytic Viruses and Immune Checkpoint Inhibitors: Preclinical Developments to Clinical Trials. International Journal of Molecular Sciences 2020, 21, 1–26. [CrossRef]
- Ma, J.; Ramachandran, M.; Jin, C.; Quijano-Rubio, C.; Martikainen, M.; Yu, D.; Essand, M. Characterization of Virus-Mediated Immunogenic Cancer Cell Death and the Consequences for Oncolytic Virus-Based Immunotherapy of Cancer. Cell Death Dis 2020, 11, 1–15. [CrossRef]
- Bommareddy, P.K.; Shettigar, M.; Kaufman, H.L. Integrating Oncolytic Viruses in Combination Cancer Immunotherapy. Nature Reviews Immunology 2018, 18, 498–513. [CrossRef]
- Hotte, S.J.; Lorence, R.M.; Hirte, H.W.; Polawski, S.R.; Bamat, M.K.; O’Neil, J.D.; Roberts, M.S.; Groene, W.S.; Major, P.P. An Optimized Clinical Regimen for the Oncolytic Virus PV701. Clinical Cancer Research 2007, 13, 977–985. [CrossRef]
- Heinzerling, L.; Künzi, V.; Oberholzer, P.A.; Kündig, T.; Naim, H.; Dummer, R. Oncolytic Measles Virus in Cutaneous T-Cell Lymphomas Mounts Antitumor Immune Responses in Vivo and Targets Interferon-Resistant Tumor Cells. Blood 2005, 106, 2287–2294. [CrossRef]
- Zhang, Q.; Yu, Y.A.; Wang, E.; Chen, N.; Danner, R.L.; Munson, P.J.; Marincola, F.M.; Szalay, A.A. Eradication of Solid Human Breast Tumors in Nude Mice with an Intravenously Injected Light-Emitting Oncolytic Vaccinia Virus. Cancer Research 2007, 67, 10038–10046. [CrossRef]
- Felt, S.A.; Grdzelishvili, V.Z. Recent Advances in Vesicular Stomatitis Virus-Based Oncolytic Virotherapy: A 5-Year Update. J Gen Virol 2017, 98, 2895–2911. [CrossRef]
- Kemp, V.; Hoeben, R.C.; van den Wollenberg, D.J.M. Exploring Reovirus Plasticity for Improving Its Use as Oncolytic Virus. Viruses 2015, 8, 4. [CrossRef]
- Geisler, A.; Hazini, A.; Heimann, L.; Kurreck, J.; Fechner, H. Coxsackievirus B3—Its Potential as an Oncolytic Virus. Viruses 2021, 13, 718. [CrossRef]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic Viruses: A New Class of Immunotherapy Drugs. Nature Reviews Drug Discovery 2015, 14, 642–662. [CrossRef]
- Cunliffe, T.G.; Bates, E.A.; Parker, A.L. Hitting the Target but Missing the Point: Recent Progress towards Adenovirus-Based Precision Virotherapies. Cancers 2020, 12, 1–24. [CrossRef]
- Shalhout, S.Z.; Miller, D.M.; Emerick, K.S.; Kaufman, H.L. Therapy with Oncolytic Viruses: Progress and Challenges. Nature Reviews Clinical Oncology 2023, 20, 160–177. [CrossRef]
- Vesely, M.D.; Kershaw, M.H.; Schreiber, R.D.; Smyth, M.J. Natural Innate and Adaptive Immunity to Cancer. Annual Review of Immunology 2011, 29, 235–271. [CrossRef]
- Twumasi-Boateng, K.; Pettigrew, J.L.; Kwok, Y.Y.E.; Bell, J.C.; Nelson, B.H. Oncolytic Viruses as Engineering Platforms for Combination Immunotherapy. Nature Reviews Cancer 2018, 18, 419–432. [CrossRef]
- Shi, T.; Song, X.; Wang, Y.; Liu, F.; Wei, J. Combining Oncolytic Viruses With Cancer Immunotherapy: Establishing a New Generation of Cancer Treatment. Frontiers in Immunology 2020, 11, 1–13. [CrossRef]
- Ren, Y.; Miao, J.M.; Wang, Y.Y.; Fan, Z.; Kong, X.B.; Yang, L.; Cheng, G. Oncolytic Viruses Combined with Immune Checkpoint Therapy for Colorectal Cancer Is a Promising Treatment Option. Frontiers in Immunology 2022, 13, 1–13. [CrossRef]
- Li, Q.; Tan, F.; Wang, Y.; Liu, X.; Kong, X.; Meng, J.; Yang, L.; Cen, S. The Gamble between Oncolytic Virus Therapy and IFN. Frontiers in Immunology 2022, 13.
- Zhang, Y.; Li, Y.; Chen, K.; Qian, L.; Wang, P. Oncolytic Virotherapy Reverses the Immunosuppressive Tumor Microenvironment and Its Potential in Combination with Immunotherapy. Cancer Cell International 2021, 21, 1–17. [CrossRef]
- Coffelt, S.B.; Wellenstein, M.D.; De Visser, K.E. Neutrophils in Cancer: Neutral No More. Nature Reviews Cancer 2016, 16, 431–446. [CrossRef]
- Hofman, L.; Lawler, S.E.; Lamfers, M.L.M. The Multifaceted Role of Macrophages in Oncolytic Virotherapy. Viruses 2021, 13, 1570. [CrossRef]
- Prestwich, R.; Errington, F.; Ilett, E.; Morgan, R.; Scott, K.; Kottke, T.; Thompson, J.; Morrison, E.; Harrington, K.; Pandha, H.; et al. Tumor Infection by Oncolytic Reovirus Primes Adaptive Anti-Tumor Immunity. Clin Cancer Res 2008, 14, 7358–7366. [CrossRef]
- Andtbacka, R.H.I.; Collichio, F.; Harrington, K.J.; Middleton, M.R.; Downey, G.; Öhrling, K.; Kaufman, H.L. Final Analyses of OPTiM: A Randomized Phase III Trial of Talimogene Laherparepvec versus Granulocyte-Macrophage Colony-Stimulating Factor in Unresectable Stage III-IV Melanoma. Journal for ImmunoTherapy of Cancer 2019, 7, 1–11. [CrossRef]
- Todo, T.; Ito, H.; Ino, Y.; Ohtsu, H.; Ota, Y.; Shibahara, J.; Tanaka, M. Intratumoral Oncolytic Herpes Virus G47∆ for Residual or Recurrent Glioblastoma: A Phase 2 Trial. Nature Medicine 2022, 28, 1630–1639. [CrossRef]
- Heo, J.; Reid, T.; Ruo, L.; Breitbach, C.J.; Rose, S.; Bloomston, M.; Cho, M.; Lim, H.Y.; Chung, H.C.; Kim, C.W.; et al. Randomized Dose-Finding Clinical Trial of Oncolytic Immunotherapeutic Vaccinia JX-594 in Liver Cancer. Nature Medicine 2013, 19, 329–336. [CrossRef]
- Holloway, R.W.; Kendrick, J.E.; Stephens, A.; Kennard, J.; Burt, J.; LeBlanc, J.; Sellers, K.; Smith, J.; Coakley, S. Phase 1b Study of Oncolytic Vaccinia Virus GL-ONC1 in Recurrent Ovarian Cancer (ROC). JCO 2018, 36, 5577–5577. [CrossRef]
- Streby, K.A.; Currier, M.A.; Triplet, M.; Ott, K.; Dishman, D.J.; Vaughan, M.R.; Ranalli, M.A.; Setty, B.; Skeens, M.A.; Whiteside, S.; et al. First-in-Human Intravenous Seprehvir in Young Cancer Patients: A Phase 1 Clinical Trial. Mol Ther 2019, 27, 1930–1938. [CrossRef]
- Lang, F.F.; Conrad, C.; Gomez-Manzano, C.; Alfred Yung, W.K.; Sawaya, R.; Weinberg, J.S.; Prabhu, S.S.; Rao, G.; Fuller, G.N.; Aldape, K.D.; et al. Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. Journal of Clinical Oncology 2018, 36, 1419–1427. [CrossRef]
- Galanis, E.; Hartmann, L.C.; Cliby, W.A.; Long, H.J.; Prema, P.; Barrette, B.A.; Kaur, J.S.; Jr, P.J.H.; Aderca, I.; Zollman, P.J.; et al. Phase I Trial of Intraperitoneal Administration of an Oncolytic Measles Virus Strain Engineered to Express Carcinoembryonic Antigen for Recurrent Ovarian Cancer. Cancer Research 2010, 70, 875–882. [CrossRef]
- Ranki, T.; Pesonen, S.; Hemminki, A.; Partanen, K.; Kairemo, K.; Alanko, T.; Lundin, J.; Linder, N.; Turkki, R.; Ristimäki, A.; et al. Phase I Study with ONCOS-102 for the Treatment of Solid Tumors - an Evaluation of Clinical Response and Exploratory Analyses of Immune Markers. Journal for ImmunoTherapy of Cancer 2016, 4, 1–18. [CrossRef]
- Andtbacka, R.H.I.; Ross, M.I.; Agarwala, S.S.; Taylor, M.H.; Vetto, J.T.; Neves, R.I.; Daud, A.; Khong, H.T.; Ungerleider, R.S.; Tanaka, M. Efficacy and Genetic Analysis for a Phase II Multicenter Trial of HF10, a Replication-Competent HSV-1 Oncolytic Immunotherapy, and Ipilimumab Combination Treatment in Patients with Stage IIIb-IV Unresectable or Metastatic Melanoma. Journal of Clinical Oncology 2018, 36, 9541–9541. [CrossRef]
- Cook, J.; Peng, K.W.; Witzig, T.E.; Broski, S.M.; Villasboas, J.C.; Paludo, J.; Patnaik, M.; Rajkumar, V.; Dispenzieri, A.; Leung, N.; et al. Clinical Activity of Single-Dose Systemic Oncolytic VSV Virotherapy in Patients with Relapsed Refractory T-Cell Lymphoma. Blood Advances 2022, 6, 3268–3279. [CrossRef]
- Ferris, R.L.; Gross, N.D.; Nemunaitis, J.J.; Andtbacka, R.H.I.; Argiris, A.; Ohr, J.; Vetto, J.T.; Senzer, N.N.; Bedell, C.; Ungerleider, R.S.; et al. Phase I Trial of Intratumoral Therapy Using HF10, an Oncolytic HSV-1, Demonstrates Safety in HSV+/HSV- Patients with Refractory and Superficial Cancers. Journal of Clinical Oncology 2014, 32, 6082–6082. [CrossRef]
- Markert, J.M.; Razdan, S.N.; Kuo, H.C.; Cantor, A.; Knoll, A.; Karrasch, M.; Nabors, L.B.; Markiewicz, M.; Agee, B.S.; Coleman, J.M.; et al. A Phase 1 Trial of Oncolytic HSV-1, G207, given in Combination with Radiation for Recurrent GBM Demonstrates Safety and Radiographic Responses. Molecular Therapy 2014, 22, 1048–1055. [CrossRef]
- Cui, C.L.; Wang, X.; Lian, B.; Ji, Q.; Zhou, L.; Chi, Z.; Si, L.; Sheng, X.; Kong, Y.; Yu, J.; et al. OrienX010, an Oncolytic Virus, in Patients with Unresectable Stage IIIC-IV Melanoma: A Phase Ib Study. Journal for ImmunoTherapy of Cancer 2022, 10. [CrossRef]
- Shirakawa, Y.; Tazawa, H.; Tanabe, S.; Kanaya, N.; Noma, K.; Koujima, T.; Kashima, H.; Kato, T.; Kuroda, S.; Kikuchi, S.; et al. Phase I Dose-Escalation Study of Endoscopic Intratumoral Injection of OBP-301 (Telomelysin) with Radiotherapy in Oesophageal Cancer Patients Unfit for Standard Treatments. European Journal of Cancer 2021, 153, 98–108. [CrossRef]
- Musher, B.L.; Smaglo, B.G.; Abidi, W.; Othman, M.; Patel, K.; Jawaid, S.; Jing, J.; Brisco, A.; Wenthe, J.; Eriksson, E.; et al. A Phase I/II Study of LOAd703, a TMZ-CD40L/4-1BBL-Armed Oncolytic Adenovirus, Combined with Nab-Paclitaxel and Gemcitabine in Advanced Pancreatic Cancer. Journal of Clinical Oncology 2022, 40, 4138–4138. [CrossRef]
- Annels, N.E.; Mansfield, D.; Arif, M.; Ballesteros-Merino, C.; Simpson, G.R.; Denyer, M.; Sandhu, S.S.; Melcher, A.A.; Harrington, K.J.; Davies, B.; et al. Phase I Trial of an ICAM-1-Targeted Immunotherapeutic-Coxsackievirus A21 (CVA21) as an Oncolytic Agent Against Non Muscle-Invasive Bladder Cancer. Clinical Cancer Research 2019, 25, 5818–5831. [CrossRef]
- Moreno, V.; Barretina-Ginesta, M.P.; García-Donas, J.; Jayson, G.C.; Roxburgh, P.; Vázquez, R.M.; Michael, A.; Antón-Torres, A.; Brown, R.; Krige, D.; et al. Safety and Efficacy of the Tumor-Selective Adenovirus Enadenotucirev with or without Paclitaxel in Platinum-Resistant Ovarian Cancer: A Phase 1 Clinical Trial. Journal for ImmunoTherapy of Cancer 2021, 9, 1–14. [CrossRef]
- Bazan-Peregrino, M.; Garcia-Carbonero, R.; Laquente, B.; Álvarez, R.; Mato-Berciano, A.; Gimenez-Alejandre, M.; Morgado, S.; Rodríguez-García, A.; Maliandi, M.V.; Riesco, M.C.; et al. VCN-01 Disrupts Pancreatic Cancer Stroma and Exerts Antitumor Effects. Journal for ImmunoTherapy of Cancer 2021, 9, 1–12. [CrossRef]
- Friedman, G.K.; Johnston, J.M.; Bag, A.K.; Bernstock, J.D.; Li, R.; Aban, I.; Kachurak, K.; Nan, L.; Kang, K.-D.; Totsch, S.; et al. Oncolytic HSV-1 G207 Immunovirotherapy for Pediatric High-Grade Gliomas. New England Journal of Medicine 2021, 384, 1613–1622. [CrossRef]
- Hirooka, Y.; Kasuya, H.; Ishikawa, T.; Kawashima, H.; Ohno, E.; Villalobos, I.B.; Naoe, Y.; Ichinose, T.; Koyama, N.; Tanaka, M.; et al. A Phase I Clinical Trial of EUS-Guided Intratumoral Injection of the Oncolytic Virus, HF10 for Unresectable Locally Advanced Pancreatic Cancer. BMC Cancer 2018, 18, 1–9. [CrossRef]
- Zhang, B.; Huang, J.; Tang, J.; Hu, S.; Luo, S.; Luo, Z.; Zhou, F.; Tan, S.; Ying, J.; Chang, Q.; et al. Intratumoral OH2, an Oncolytic Herpes Simplex Virus 2, in Patients with Advanced Solid Tumors: A Multicenter, Phase I/II Clinical Trial. Journal for ImmunoTherapy of Cancer 2021, 9, 1–10. [CrossRef]
- Mayo Clinic Phase I Trial of a Measles Virus Derivative Producing CEA (MV-CEA) in Patients With Recurrent Glioblastoma Multiforme (GBM); clinicaltrials.gov, 2019;
- Garcia-Carbonero, R.; Gil Martín, M.; Alvarez Gallego, R.; Macarulla Mercade, T.; Riesco Martinez, M.C.; Guillen-Ponce, C.; Vidal, N.; Real, F.X.; Moreno, R.; Maliandi, V.; et al. Systemic Administration of the Hyaluronidase-Expressing Oncolytic Adenovirus VCN-01 in Patients with Advanced or Metastatic Pancreatic Cancer: First-in-Human Clinical Trial. Annals of Oncology 2019, 30, v271–v272. [CrossRef]
- Packiam, V.T.; Lamm, D.L.; Barocas, D.A.; Trainer, A.; Fand, B.; Davis, R.L.; Clark, W.; Kroeger, M.; Dumbadze, I.; Chamie, K.; et al. An Open Label, Single-Arm, Phase II Multicenter Study of the Safety and Efficacy of CG0070 Oncolytic Vector Regimen in Patients with BCG-Unresponsive Non–Muscle-Invasive Bladder Cancer: Interim Results. Urologic Oncology: Seminars and Original Investigations 2018, 36, 440–447. [CrossRef]
- Li, S.J.; Sun, Z.J. Fueling Immune Checkpoint Blockade with Oncolytic Viruses: Current Paradigms and Challenges Ahead. Cancer Letters 2022, 550, 215937–215937. [CrossRef]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways Similarities, Differences, and Implications of Their Inhibition. American Journal of Clinical Oncology: Cancer Clinical Trials 2016, 39, 98–106. [CrossRef]
- De Silva, P.; Aiello, M.; Gu-Trantien, C.; Migliori, E.; Willard-Gallo, K.; Solinas, C. Targeting CTLA-4 in Cancer: Is It the Ideal Companion for PD-1 Blockade Immunotherapy Combinations? International Journal of Cancer 2021, 149, 31–41. [CrossRef]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and Its Ligands in Tolerance and Immunity. Annual Review of Immunology 2008, 26, 677–704. [CrossRef]
- Schalper, K.A.; Kaftan, E.; Herbst, R.S. Predictive Biomarkers for PD-1 Axis Therapies: The Hidden Treasure or a Call for Research. Clinical Cancer Research 2016, 22, 2102–2104.
- Doroshow, D.B.; Sanmamed, M.F.; Hastings, K.; Politi, K.; Rimm, D.L.; Chen, L.; Melero, I.; Schalper, K.A.; Herbst, R.S. Immunotherapy in Non–Small Cell Lung Cancer: Facts and Hopes. Clinical Cancer Research 2019, 25, 4592–4602. [CrossRef]
- Kontermann, R.E.; Ungerechts, G.; Nettelbeck, D.M. Viro-Antibody Therapy: Engineering Oncolytic Viruses for Genetic Delivery of Diverse Antibody-Based Biotherapeutics. mAbs 2021, 13. [CrossRef]
- Li, R.; Steinberg, G.; Uchio, E.; Lamm, D.; Paras, S.; Kamat, A.; Bivalacqua, T.; Packiam, V.; Chisamore, M.; McAdory, J.; et al. 666 Phase 2, Single Arm Study of CG0070 Combined with Pembrolizumab in Patients with Non-Muscle Invasive Bladder Cancer (NMIBC) Unresponsive to Bacillus Calmette-Guerin (BCG).; BMJ Specialist Journals 2022.
- Nassiri, F.; Patil, V.; Yefet, L.S.; Singh, O.; Liu, J.; Dang, R.M.A.; Yamaguchi, T.N.; Daras, M.; Cloughesy, T.F.; Colman, H.; et al. Oncolytic DNX-2401 Virotherapy plus Pembrolizumab in Recurrent Glioblastoma: A Phase 1/2 Trial. Nat Med 2023, 1–9. [CrossRef]
- Chesney, J.A.; Ribas, A.; Long, G.V.; Kirkwood, J.M.; Dummer, R.; Puzanov, I.; Hoeller, C.; Gajewski, T.F.; Gutzmer, R.; Rutkowski, P.; et al. Randomized, Double-Blind, Placebo-Controlled, Global Phase III Trial of Talimogene Laherparepvec Combined with Pembrolizumab for Advanced Melanoma. Journal of Clinical Oncology 2023, 41, 528–540. [CrossRef]
- Fakih, M.G.; Wang, D.; Harb, W.; Rosen, L.; Mahadevan, D.; Berlin, J.D.; Basciano, P.; Brown, R.; Arogundade, O.; Cox, C.; et al. SPICE, a Phase I Study of Enadenotucirev in Combination with Nivolumab in Tumours of Epithelial Origin: Analysis of the Metastatic Colorectal Cancer Patients in the Dose Escalation Phase. Annals of Oncology 2019, 30, v231–v231. [CrossRef]
- Lillie, T.; Stone, A.; Lockwood, S.; Brown, R.; Fox, A.; Bournazou, E.; Beadle, J. 329 Prolonged Overall Survival (OS) in Patients with Metastatic Colorectal Cancer (MCRC) in SPICE, a Phase I Study of Enadenotucirev in Combination with Nivolumab.; 2020; Vol. 8, p. A202.1-A202.
- Krige, D.; Fakih, M.; Rosen, L.; Wang, D.; Harb, W.; Babiker, H.; Berlin, J.; Di Genova, G.; Miles, D.; Mark, P.; et al. Combining enadenotucirev and nivolumab increased tumour immune cell infiltration/ activation in patients with microsatellite- stable/instability-low metastatic colorectal cancer in a phase 1 study. 2021; Vol. 5, pp. 2021–2021.
- Shoushtari, A.N.; Olszanski, A.J.; Nyakas, M.; Hornyak, T.J.; Wolchok, J.D.; Levitsky, V.; Kuryk, L.; Hansen, T.B.; Jäderberg, M. Pilot Study of ONCOS-102 and Pembrolizumab: Remodeling of the Tumor Microenvironment and Clinical Outcomes in Anti-PD-1-Resistant Advanced Melanoma. Clinical cancer research : an official journal of the American Association for Cancer Research 2023, 29, 100–109. [CrossRef]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.I.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017, 170, 1109-1119.e10. [CrossRef]
- Kelly, C.M.; Antonescu, C.R.; Bowler, T.; Munhoz, R.; Chi, P.; Dickson, M.A.; Gounder, M.M.; Keohan, M.L.; Movva, S.; Dholakia, R.; et al. Objective Response Rate Among Patients With Locally Advanced or Metastatic Sarcoma Treated With Talimogene Laherparepvec in Combination With Pembrolizumab: A Phase 2 Clinical Trial. JAMA Oncology 2020, 6, 402–408. [CrossRef]
- Monge, C.; Xie, C.; Myojin, Y.; Coffman, K.; Hrones, D.M.; Wang, S.; Hernandez, J.M.; Wood, B.J.; Levy, E.B.; Juburi, I.; et al. Phase I/II Study of PexaVec in Combination with Immune Checkpoint Inhibition in Refractory Metastatic Colorectal Cancer. Journal for ImmunoTherapy of Cancer 2023, 11, 1–14. [CrossRef]
- Chesney, J.; Puzanov, I.; Collichio, F.; Singh, P.; Milhem, M.M.; Glaspy, J.; Hamid, O.; Ross, M.; Friedlander, P.; Garbe, C.; et al. Randomized, Open-Label Phase II Study Evaluating the Efficacy and Safety of Talimogene Laherparepvec in Combination with Ipilimumab versus Ipilimumab Alone in Patients with Advanced, Unresectable Melanoma. Journal of Clinical Oncology 2018, 36, 1658–1667. [CrossRef]
- Fransen, M.F.; Van Der Sluis, T.C.; Ossendorp, F.; Arens, R.; Melief, C.J.M. Controlled Local Delivery of CTLA-4 Blocking Antibody Induces CD8 + T-Cell-Dependent Tumor Eradication and Decreases Risk of Toxic Side Effects. Clinical Cancer Research 2013, 19, 5381–5389. [CrossRef]
- Han, X.; Li, H.; Zhou, D.; Chen, Z.; Gu, Z. Local and Targeted Delivery of Immune Checkpoint Blockade Therapeutics. Acc Chem Res 2020, 53, 2521–2533. [CrossRef]
- Du, T.; Shi, G.; Li, Y.M.; Zhang, J.F.; Tian, H.W.; Wei, Y.Q.; Deng, H.; Yu, D.C. Tumor-Specific Oncolytic Adenoviruses Expressing Granulocyte Macrophage Colony-Stimulating Factor or Anti-CTLA4 Antibody for the Treatment of Cancers. Cancer Gene Therapy 2014, 21, 340–348. [CrossRef]
- Dias, J.D.; Hemminki, O.; Diaconu, I.; Hirvinen, M.; Bonetti, A.; Guse, K.; Escutenaire, S.; Kanerva, A.; Pesonen, S.; Löskog, A.; et al. Targeted Cancer Immunotherapy with Oncolytic Adenovirus Coding for a Fully Human Monoclonal Antibody Specific for CTLA-4. Gene Therapy 2012, 19, 988–998. [CrossRef]
- Thomas, S.; Kuncheria, L.; Roulstone, V.; Kyula, J.N.; Mansfield, D.; Bommareddy, P.K.; Smith, H.; Kaufman, H.L.; Harrington, K.J.; Coffin, R.S. Development of a New Fusion-Enhanced Oncolytic Immunotherapy Platform Based on Herpes Simplex Virus Type 1. Journal for ImmunoTherapy of Cancer 2019, 7, 1–17. [CrossRef]
- Hamilton, J.R.; Vijayakumar, G.; Palese, P. A Recombinant Antibody-Expressing Influenza Virus Delays Tumor Growth in a Mouse Model. Cell Reports 2018, 22, 1–7. [CrossRef]
- Zhou, P.; Wang, X.; Xing, M.; Yang, X.; Wu, M.; Shi, H.; Zhu, C.; Wang, X.; Guo, Y.; Tang, S.; et al. Intratumoral Delivery of a Novel Oncolytic Adenovirus Encoding Human Antibody against PD-1 Elicits Enhanced Antitumor Efficacy. Molecular Therapy - Oncolytics 2022, 25, 236–248. [CrossRef]
- Lin, C.; Ren, W.; Luo, Y.; Li, S.; Chang, Y.; Li, L.; Xiong, D.; Huang, X.; Xu, Z.; Yu, Z.; et al. Intratumoral Delivery of a PD-1-Blocking ScFv Encoded in Oncolytic HSV-1 Promotes Antitumor Immunity and Synergizes with TIGIT Blockade. Cancer Immunology Research 2020, 8, 632–648. [CrossRef]
- Ju, F.; Luo, Y.; Lin, C.; Jia, X.; Xu, Z.; Tian, R.; Lin, Y.; Zhao, M.; Chang, Y.; Huang, X.; et al. Oncolytic Virus Expressing PD-1 Inhibitors Activates a Collaborative Intratumoral Immune Response to Control Tumor and Synergizes with CTLA-4 or TIM-3 Blockade. Journal for ImmunoTherapy of Cancer 2022, 10, 1–17. [CrossRef]
- Tian, C.; Liu, J.; Zhou, H.; Li, J.; Sun, C.; Zhu, W.; Yin, Y.; Li, X. Enhanced Anti-Tumor Response Elicited by a Novel Oncolytic HSV-1 Engineered with an Anti-PD-1 Antibody. Cancer Letters 2021, 518, 49–58. [CrossRef]
- Zhu, Y.; Hu, X.; Feng, L.; Yang, Z.; Zhou, L.; Duan, X.; Cheng, S.; Zhang, W.; Liu, B.; Zhang, K. Enhanced Therapeutic Efficacy of a Novel Oncolytic Herpes Simplex Virus Type 2 Encoding an Antibody Against Programmed Cell Death 1. Molecular Therapy - Oncolytics 2019, 15, 201–213. [CrossRef]
- Woo, Y.; Zhang, Z.; Yang, A.; Chaurasiya, S.; Park, A.K.; Lu, J.; Kim, S.I.; Warner, S.G.; Von Hoff, D.; Fong, Y. Novel Chimeric Immuno-Oncolytic Virus CF33-HNIS-AntiPDL1 for the Treatment of Pancreatic Cancer. Journal of the American College of Surgeons 2020, 230, 709–717. [CrossRef]
- Chaurasiya, S.; Yang, A.; Zhang, Z.; Lu, J.; Valencia, H.; Kim, S.I.; Woo, Y.; Warner, S.G.; Olafsen, T.; Zhao, Y.; et al. A Comprehensive Preclinical Study Supporting Clinical Trial of Oncolytic Chimeric Poxvirus CF33-HNIS-Anti-PD-L1 to Treat Breast Cancer. Molecular Therapy - Methods and Clinical Development 2022, 24, 102–116. [CrossRef]
- Yang, A.; Zhang, Z.; Chaurasiya, S.; Park, A.K.; Kim, I.; Priceman, S.; Fong, Y.; Jung, A.; Lu, J. Development of the Oncolytic Virus , CF33 , and Its Derivatives for Peritoneal- Directed Treatment of Gastric Cancer Peritoneal Metastases. Journal for ImmunoTherapy of Cancer 2023, 11, e006280–e006280. [CrossRef]
- Wu, C.; Wu, M.; Liang, M.; Xiong, S.; Dong, C. A Novel Oncolytic Virus Engineered with PD-L1 ScFv Effectively Inhibits Tumor Growth in a Mouse Model. Cellular and Molecular Immunology 2019, 16, 780–782. [CrossRef]
- Veinalde, R.; Pidelaserra-Martí, G.; Moulin, C.; Jeworowski, L.M.; Küther, L.; Buchholz, C.J.; Jäger, D.; Ungerechts, G.; Engeland, C.E. Oncolytic Measles Vaccines Encoding PD-1 and PD-L1 Checkpoint Blocking Antibodies to Increase Tumor-Specific T Cell Memory. Molecular Therapy - Oncolytics 2022, 24, 43–58. [CrossRef]
- Lei, G.L.; Wang, L.P.; Dong, S.H.; Sun, F.; Cheng, J.X.; Yang, X.L.; Zhang, S.G.; Wang, X.L.; Wang, X.X.; Yang, P.H. A Recombinant Influenza Virus with a CTLA4-Specific ScFv Inhibits Tumor Growth in a Mouse Model. Cell Biology International 2021, 45, 1202–1210. [CrossRef]
- Kleinpeter, P.; Fend, L.; Thioudellet, C.; Geist, M.; Sfrontato, N.; Koerper, V.; Fahrner, C.; Schmitt, D.; Gantzer, M.; Remy-Ziller, C.; et al. Vectorization in an Oncolytic Vaccinia Virus of an Antibody, a Fab and a ScFv against Programmed Cell Death -1 (PD-1) Allows Their Intratumoral Delivery and an Improved Tumor-Growth Inhibition. OncoImmunology 2016, 5, 1–14. [CrossRef]
- Vijayakumar, G.; McCroskery, S.; Palese, P. Engineering Newcastle Disease Virus as an Oncolytic Vector for Intratumoral Delivery of Immune Checkpoint Inhibitors and Immunocytokines. Journal of Virology 2020, 94. [CrossRef]
- Vitale, M.; Scialò, F.; Passariello, M.; Leggiero, E.; D’Agostino, A.; Tripodi, L.; Gentile, L.; Bianco, A.; Castaldo, G.; Cerullo, V.; et al. Oncolytic Adenoviral Vector-Mediated Expression of an Anti-PD-L1-ScFv Improves Anti-Tumoral Efficacy in a Melanoma Mouse Model. Frontiers in Oncology 2022, 12, 1–12. [CrossRef]
- Passaro, C.; Alayo, Q.; De Laura, I.; McNulty, J.; Grauwet, K.; Ito, H.; Bhaskaran, V.; Mineo, M.; Lawler, S.E.; Shah, K.; et al. Arming an Oncolytic Herpes Simplex Virus Type 1 with a Single-Chain Fragment Variable Antibody against PD-1 for Experimental Glioblastoma Therapy. Clinical Cancer Research 2019, 25, 290–299. [CrossRef]
- Zhang, Z.; Yang, A.; Chaurasiya, S.; Park, A.K.; Lu, J.; Kim, S.I.; Warner, S.G.; Yuan, Y.C.; Liu, Z.; Han, H.; et al. CF33-HNIS-AntiPDL1 Virus Primes Pancreatic Ductal Adenocarcinoma for Enhanced Anti-PD-L1 Therapy. Cancer Gene Therapy 2022, 29, 722–733. [CrossRef]
- Engeland, C.E.; Grossardt, C.; Veinalde, R.; Bossow, S.; Lutz, D.; Kaufmann, J.K.; Shevchenko, I.; Umansky, V.; Nettelbeck, D.M.; Weichert, W.; et al. CTLA-4 and PD-L1 Checkpoint Blockade Enhances Oncolytic Measles Virus Therapy. Molecular Therapy 2014, 22, 1949–1959. [CrossRef]
- Wang, G.; Kang, X.; Chen, K.S.; Jehng, T.; Jones, L.; Chen, J.; Huang, X.F.; Chen, S.Y. An Engineered Oncolytic Virus Expressing PD-L1 Inhibitors Activates Tumor Neoantigen-Specific T Cell Responses. Nature Communications 2020, 11. [CrossRef]
- Vijayakumar, G.; Palese, P.; Goff, P.H. Oncolytic Newcastle Disease Virus Expressing a Checkpoint Inhibitor as a Radioenhancing Agent for Murine Melanoma. EBioMedicine 2019, 49, 96–105. [CrossRef]
- Barrueto, L.; Caminero, F.; Cash, L.; Makris, C.; Lamichhane, P.; Deshmukh, R.R. Resistance to Checkpoint Inhibition in Cancer Immunotherapy. Transl Oncol 2020, 13, 100738. [CrossRef]
- Das, R.; Verma, R.; Sznol, M.; Boddupalli, C.S.; Gettinger, S.N.; Kluger, H.; Callahan, M.; Wolchok, J.D.; Halaban, R.; Dhodapkar, M.V. Combination Therapy with Anti–CTLA-4 and Anti–PD-1 Leads to Distinct Immunologic Changes in Vivo. The Journal of Immunology 2015, 194, 950–959. [CrossRef]
- Chauvin, J.-M.; Zarour, H.M. TIGIT in Cancer Immunotherapy. J Immunother Cancer 2020, 8, e000957. [CrossRef]
- Zuo, S.; Wei, M.; He, B.; Chen, A.; Wang, S.; Kong, L.; Zhang, Y.; Meng, G.; Xu, T.; Wu, J.; et al. Enhanced Antitumor Efficacy of a Novel Oncolytic Vaccinia Virus Encoding a Fully Monoclonal Antibody against T-Cell Immunoglobulin and ITIM Domain (TIGIT). EBioMedicine 2021, 64, 103240–103240. [CrossRef]
- Zuo, S.; Wei, M.; Xu, T.; Kong, L.; He, B.; Wang, S.; Wang, S.; Wu, J.; Dong, J.; Wei, J. An Engineered Oncolytic Vaccinia Virus Encoding a Single-Chain Variable Fragment against TIGIT Induces Effective Antitumor Immunity and Synergizes with PD-1 or LAG-3 Blockade. J Immunother Cancer 2021, 9, e002843. [CrossRef]
- Lee, J.B.; Ha, S.-J.; Kim, H.R. Clinical Insights Into Novel Immune Checkpoint Inhibitors. Frontiers in Pharmacology 2021, 12.
- Popat, S.; Grohé, C.; Corral, J.; Reck, M.; Novello, S.; Gottfried, M.; Radonjic, D.; Kaiser, R. Anti-Angiogenic Agents in the Age of Resistance to Immune Checkpoint Inhibitors: Do They Have a Role in Non-Oncogene-Addicted Non-Small Cell Lung Cancer? Lung Cancer 2020, 144, 76–84. [CrossRef]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front Oncol 2018, 8, 86. [CrossRef]
Figure 1.
OV anti-tumour mechanism of action. OVs selectively infect, replicate within, and lyse tumour cells whilst leaving healthy cells intact. Upon infection of tumour cells, OVs replicate and lyse the tumour cell, resulting in tumour cell death and release of viral progeny. This tumour cell lysis results in destruction of the local tumour microenvironment and induction of an anti-tumour immune response through local immune infiltration and release of tumour antigens.
Figure 1.
OV anti-tumour mechanism of action. OVs selectively infect, replicate within, and lyse tumour cells whilst leaving healthy cells intact. Upon infection of tumour cells, OVs replicate and lyse the tumour cell, resulting in tumour cell death and release of viral progeny. This tumour cell lysis results in destruction of the local tumour microenvironment and induction of an anti-tumour immune response through local immune infiltration and release of tumour antigens.
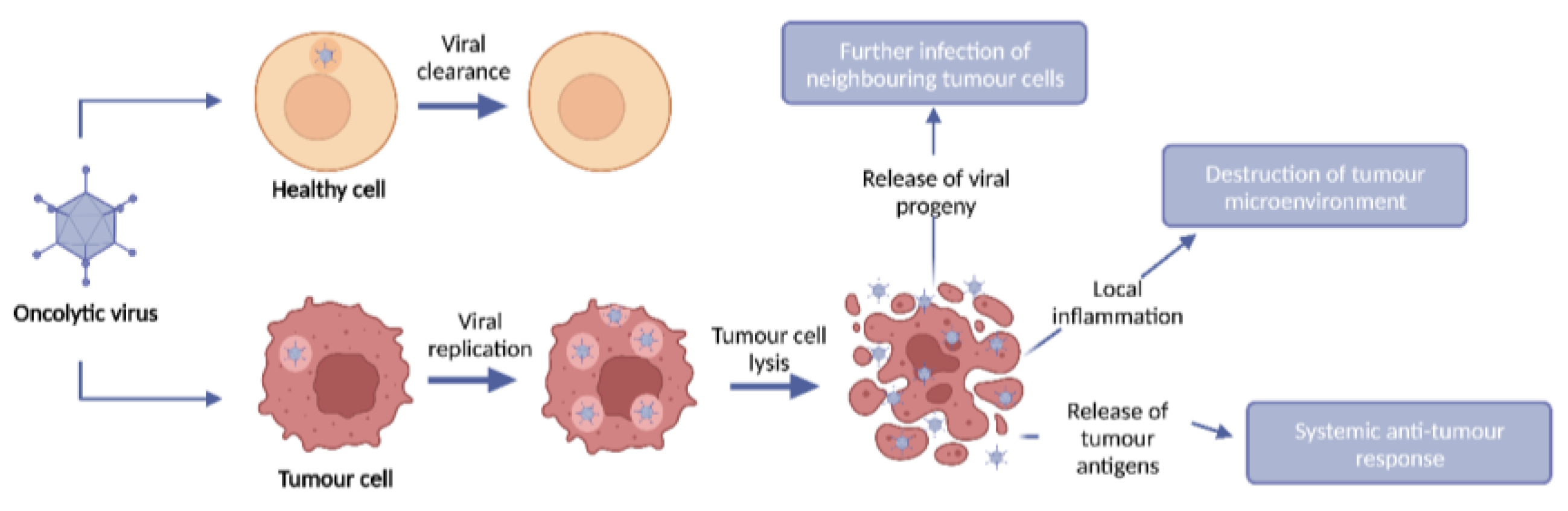
Figure 2.
OV infection can turn immunologically “cold” tumours, which do not respond well to ICI therapy, to immunologically “hot” tumours through the induction of local and systemic anti-tumour immune responses.
Figure 2.
OV infection can turn immunologically “cold” tumours, which do not respond well to ICI therapy, to immunologically “hot” tumours through the induction of local and systemic anti-tumour immune responses.
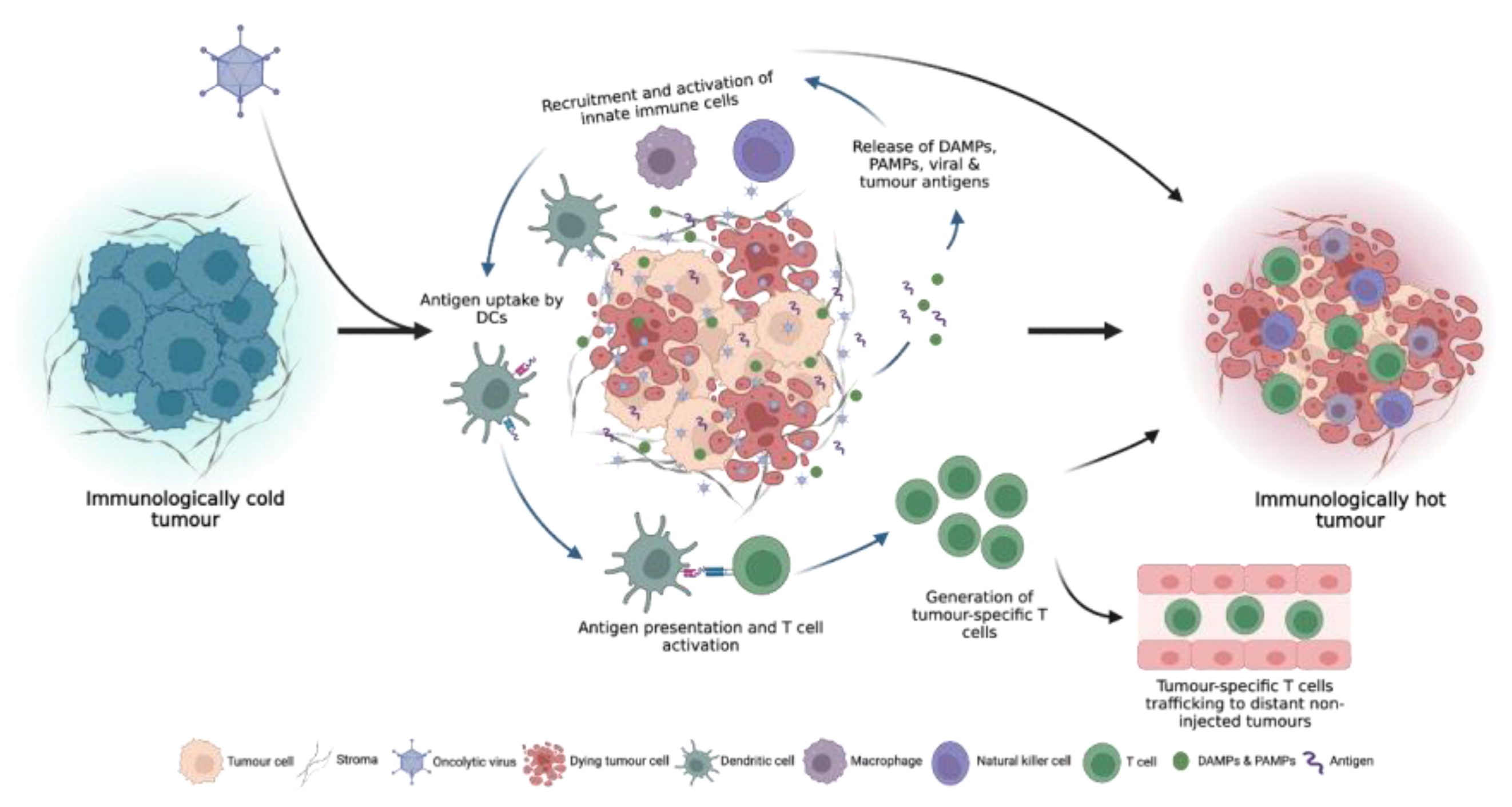

Table 1.
Viruses commonly used as OV vectors and their features.
| Virus | Diameter | Genome | Genome size | Transgene capacity |
|---|---|---|---|---|
| Adenovirus | 90-100 nm | dsDNA | 30-36 kb | ~2.5 kb |
| Herpes simplex virus | 200 nm | dsDNA | ~152 kb | ~30 kb |
| Vaccinia virus | 350 nm | dsDNA | ~192 kb | ~25 kb |
| Influenza A virus | 80-120 nm | ss(-)RNA | ~13.5 kb | ~2.4 kb |
| Newcastle disease virus | 100-500 nm | ss(-)RNA | ~15 kb | ~4.5 kb |
| Measles virus | 100-200 nm | ss(-)RNA | ~16 kb | ~6 kb |
| Vesicular stomatitis virus | 70 nm | ss(-)RNA | ~11.1 kb | ~4.5 kb |
| Coxsackie virus | 22-30 nm | ss(+)RNA | ~7.5 kb | < 1 kb |
| Reovirus | 80 nm | dsRNA | 24 kb | ~1.5 kb |
ds, double stranded; ss, single stranded; kb, kilobase.
Table 2.
OV clinical trials and their key findings.
| Virus | OV | Engineered specificity | Transgene | Indication | Delivery | Key findings | Ref. |
|---|---|---|---|---|---|---|---|
| Adenovirus | CG0070 | Ad5 with E1a under E2F-1 promoter | GM-CSF | NMIBC Phase II |
IVS | 47% 6-month CR; 29% 12-month CR | [48] |
| DNX-2401 | Ad5 with 24 bp E1a deletion; RGD integrin-binding motif | GBM Phase I |
IT | 20% >3-year survival; 12% demonstrated >95% tumour reduction; increased tumour CD8+ and T-bet+ cells; decreased TIM-3+ cells | [30] | ||
| EnAd | Ad11p/3 chimera generated through directed evolution | Ovarian Phase I |
IV | 64% PFS; 10% ORR; 35% achieved stable disease; 65% saw reduction in tumour burden; 83.3% demonstrated increased CD8+ TILs | [41] | ||
| LoAd-703 | Ad5 with 24 bp E1a deletion; pseudotyped Ad35 knob |
TMZ-CD40L; 4-1BBL |
PDAC Phase I/II |
IT | 44% ORR; 94% DCR; OS 8.7 months; increased effector memory T cells; decreased Tregs and MDSCs; | [39] | |
| ONCOS-102 | Ad5 with 24 bp E1a deletion; pseudotyped Ad3 knob |
GM-CSF | Solid tumours Phase I |
IT | Increase in TILs; increase in systemic tumour-specific CD8+ T cells; increased tumour PD-L1 expression | [32] | |
| Telomelysin | Ad5 with E1a under hTERT promoter | Oesophageal Phase I |
IT | 91.7% ORR; 83.3% Stage I and 60% Stage II/III CRR; increased tumour CD8+ T cells; increased tumour PD-L1 expression | [38] | ||
| VCN-01 | Ad5 with 24 bp E1a deletion; E2F1 promoter insertion; RGDK integrin-binding motif | Hyaluronidase | PDAC Phase I |
IT | Injected tumours reduced in size or remained stable; reduction in tumour stiffness | [42] | |
| IV | 40-45% ORR including 1 complete response; CD8+ T cell tumour infiltration and IDO upregulation in 64% of patients | [47] | |||||
| Coxsackie virus | CVA21 | NMIBC Phase I |
IVS | 1/15 demonstrated CR; CR patient demonstrated increased immune infiltration; viral protein detected in 86% of tumours with no viral protein seen in stroma; RNA-seq demonstrated increased intrinsic apoptotic cell death pathway and PD-L1, LAG-3 and IDO within the TME | [40] | ||
| Herpes simplex virus |
T-VEC | HSV1 with ICP34.5 deletion; US11 deletion | GM-CSF | Melanoma Phase III |
IT | Median OS 23.3 months; 19% DRR; 31.5% ORR; 50% demonstrated CR of which 88.5% were estimated to survive at 5-years; median time to CR 8.6 months Approved for the local treatment of unresectable metastatic stage IIIB/C–IVM1a melanoma in Europe and US |
[25] |
| G207 | HSV1 with ICP34.5 deletion; UL39 deletion; | GBM Phase I (+Rad) |
IT | Median OS 7.5 months; median PFS 2.5 months; 67% demonstrated stable or partial response at ≥ 1 time point | [36] | ||
| Paediatric glioma Phase I |
IT | Median OS 12.2 months; 18% demonstrated stable disease at 12 months; 36% still alive at 18 months; increased CD4+ and CD8+ T cell tumour infiltration | [43] | ||||
| G47∆ | G207 with additional α47 deletion; US11 promoter deletion | GBM Phase II |
IT | Median OS 20.2 months; median PFS 4.7 months; 84.2% survival at 12 months; stable disease in 18 patients at 2 years; increased CD4+, CD8+ and decreased Foxp3+ TIL Approvd for treatment of GBM in Japan |
[26] | ||
| HF10 | HSV1 with UL43, UL49.5, UL55 & UL56 deletions; Latency-associated transcripts deletions; UL53 & UL54 overexpression | Pancreatic cancer Phase I |
IT | Median OS 15.5 months; median PFS 6.3 months; 33.3% PR; 44.4% SD; 2 patients demonstrated surgical CR; 2 patients were alive at 3 year follow up; increased CD4+, CD8+ TILs | [44] | ||
| Superficial solid tumours Phase II |
IT | 33.3% SD; 1 patient demonstrated pathological CR after 4 months; 30-61% reduction in tumour size in those demonstrating responses | [35] | ||||
| Seprehvir | HSV1 with ICP34.5 deletion | Paediatric solid tumours Phase I |
IT | Median OS 7 months; 80% demonstrated SD at 14 days; 43% SD at 28 days | [29] | ||
| OrienX010 | HSV1 with ICP34.5 deletion; US12 deletion | GM-CSF | Melanoma Phase I |
IT | Median OS 19.2 months; median PFS 2.9 months; 54.6% of injected tumours regressed, 25.8% of which regressed by ≥30%; 54.1% of non-injected regional tumours regressed, 32.8% of which regressed by ≥30%; 1 distant non-injected metastases regressed by 58% | [37] | |
| OH2 | HSV2 with ICP34.5 & ICP47 deletion; | GM-CSF | Solid tumours Phase I/II |
IT | 1 PR; 33% stable disease; 79% saw increased CD8+ TILs; 86% increased CD3+ TILs; 71.4% increased PD-L1+ cells | [45] | |
| Newcastle disease virus | PV701 | Solid tumours Phase I |
IV | 61% PFS at 4 months; 33% OR; 1 CR cervical cancer; 2 PRs colorectal; 1 PR melanoma | [7] | ||
| Measles virus | MV-CEA | Carcinoembryonic antigen | Ovarian cancer Phase I/II |
IP | Median OS 12.15 months; 67% SD; 36% demonstrated >30% tumour reduction | [8] | |
| GBM Phase I |
IT | Median OS 11.6 months; 59% 3-month PFS; 23% 6-month PFS | [46] | ||||
| MV-NIS | Sodium iodide symporter | Ovarian cancer Phase I |
IP | Median OS 26.2 months; 81% SD | [31] | ||
| Vaccinia virus | GL-ONC1 | Β-galactosidase; β-glucuronidase | Ovarian cancer Phase I |
IP | Median PFS 11.6 months; 78% 6-month PFS; 63% ORR; 52% CR; increased CD4+ & CD8+ TILs | [28] | |
| JX-594 | TK1 deletion | GM-CSF | HCC Phase II |
IT | Median OS 9 months; ~35% alive at 2 years; 46% demonstrated tumour control at 8 weeks; average 32.2% decrease in tumour size; increased tumour specific CD8+ TILs | [27] | |
| Vesicular stomatitis virus | VSV-IFNβ-NIS | IFN-β; sodium iodide symporter | TCL Phase I |
IV | 1 6-month PR; 1 20-month CR; 71.4% reduction in ≥1 tumour | [34] |
Abbreviations: Ad, adenovirus; CR, complete response; CRR, complete response rate; DCR, disease control rate; DRR, disease response rate; GBM, glioblastoma; GM-CSF, granulocyte-macrophage colony-stimulating factor 2; HCC, hepatocellular carcinoma; HSV, herpes simplex virus; IDO, indoleamine 2,3-dioxygenase; IP, intraperitoneal; IT, intratumoural; IV, intravenous; IVS, intravesicular;LAG-3, lymphocyte activation gene 3; MDSC, myeloid-derived suppressor cell; NDV, Newcastle disease virus; NMIBC, non-muuscle-invasive bladder cancer; MV, measels virus; ORR, overall response rate; OS, overall survival; PDAC, pancreatic ductal adenocarcinoma; PD-L1, programmed death-ligand 1; PFS, progression free survival; PR, partial response; SD, stable disease; TCL, T cell lymphoma; TIL, tumour-infiltrating lymphocyte; TIM-3, T-cell immunoglobulin and mucin domain 3; Treg, regulatory T cell; VSV, vesicular stomatitis virus; VV, vaccinia virus.
Table 3.
Neoadjuvant OV and ICI therapy trials and their key finding.
| Virus | OV | ICI | Indication | Key findings | Ref. |
|---|---|---|---|---|---|
| Ad | CG0070 (IVS) |
PD-1: Pembrolizumab |
NMIBC Phase II |
82% 6-month CR; 81% 9-month CR; 68% 12-month CR | |
| DNX-2401 (IT) |
GBM Phase II |
ORR 10.4%; 42.9% SD; 4.8% CR; 7.1% PR; 52.7% 12-month survival; 12.5 months median OS; 3 patients alive > 45 months | [57] | ||
| EnAd (IT) |
PD-1: Nivolumab |
mCRC Phase I |
Median OS 15.4 months (5 months placebo); median PFS 2.8 months; 85% demonstrated increased CD8+ TILs; 77% increased CD4+ TILs; 62% increased PD-L1+ TILs | [59–61] | |
| ONCOS-102 (IT) |
PD-1: Pembrolizumab |
Melanoma progressing post-PD-1 blockade Pilot |
35% ORR; 64% SD; 27% demonstrated CR in injected tumour 53% demonstrated reduction in ≥1 non-injected tumour; increased CD4+ & CD8+ TILs | [62] | |
| HSV | T-VEC (IT) |
PD-1: Pembrolizumab |
Melanoma Phase Ib |
82% demonstrated >50% reduction of injected tumours; 43% in non-injected tumours; 67% demonstrated increased CD8+ TILs; demonstrated increased systemic proliferating CD8+ T cells | [63] |
| Melanoma Phase III T-VEC+Pemb vs Pemb |
T+P: 17.9% CR; 48.6% ORR (CR/PR); 14.3 months PFS P: 11.6% CR; 41.3% ORR; 8.5 months PFS |
[58] | |||
| Sarcoma Phase II |
21% PR; 47% SD; median PFS 17.1 months; responders saw increased CD8+ TILs and CD8+ aggregates at tumour edge; non-responders saw no increase in CD8+ TILs or aggregates | [64] | |||
| CTLA-4: Ipilimumab |
Melanoma Phase II TVEC+Ipi vs Ipi |
T+I: 13% CR; 26% PR; 39% ORR (CR/PR); 8.2 months median PFS; 52% non-injected visceral tumour reduction I: 7% CR; 11% PR; 18% ORR; 6.4 months median PFS; 23% non-injected visceral tumour reduction |
[66] | ||
| HF10 (IT) |
CTLA-4: Ipilimumab |
Melanoma Phase II |
Median OS 26 months; median PFS 19 months; 68% SD; increased CD8+ and decreased CD4+ TILs | [33] | |
| VV | JX-594 (IT) |
CTLA-4: Tremelumab PD-L1: Durvalumab |
ICI refractory CRC Phase I/II |
J+D: Median OS 7.5 months; median PFS 2.3 months; 12.5% DCR J+D+T: Median OS 5.2 months; median PFS 2.1 months; 16.7% DCR Increased proliferating CD3+ TILs after OV treatment and again after ICI treatment; increased M1 macrophages in tumours |
[65] |
Abbreviations: Ad, adenovirus; CRC, colorectal cancer; DRC, disease control rate; GBM, glioblastoma;HSV, herpes simplex virus; ICI, immune checkpoint inhibitor; IT, intratumoural; IVS, intravesicular; mCRC, metastatic colorectal cancer; NMIBC non-muscle-invasive bladder cancer; ORR, overall response rate; OS, overall survival; OV, oncolytic virus; PD-1, programmed cell death protein 1; PD-L1, programmed death-ligand 1; PFS, progression free survival; PR, partial response; SD, stable disease; TIL, tumour infiltrating lymphocyte; VV, vaccinia virus.
Table 4.
Pre-clinical trials of OVs engineered to express ICI antibodies and their key findings.
| OV | Target | ICI format | Indication | Key findings | Ref. |
|---|---|---|---|---|---|
| Ad5 | CTLA-4 mouse |
IgG2 | Melanoma NSCLC SCLC |
Subcutaneous mouse xenograft model with intravenous OV injection: significant 72% reduction in tumour growth compared to untreated tumours Subcutaneous mouse xenograft model with intratumoural OV injection: significant 3-fold decrease in tumour growth compared to untreated tumours |
[69] |
| Ad5/3 | CTLA-4 human |
IgG2 | NSCLC Prostate |
Subcutaneous T-cell-deficient mouse xenograft model with intratumoural OV injection: significantly decreased tumour growth compared to untreated; 43-fold increase in tumour anti-CTLA-4 antibody concentrations compared to systemic plasma In vitro human T cell activation assay: PBMCs from advanced solid cancer patients cultured in the presence of supernatant from OV-infected cells saw increase in T cell IL-2 and IFN-γ production |
[70] |
| HSV-1 | CTLA-4 & GM-CSF mouse |
scFv fused to mouse IgG1 | Lymphoma | Bilateral subcutaneous mouse xenograft model with single-sided intratumoural OV injection: decreased tumour growth in both injected and non-injected tumours (not significant) | [71] |
| IAV | CTLA-4 mouse |
scFV | Melanoma | Bilateral subcutaneous mouse xenograft model with single-sided intratumoural OV injection: significantly decreased tumour growth in both injected and non-injected tumours and prolonged survival compared to parental virus | [72] |
| IAV | CTLA-4 mouse | scFV | HCC | Spontaneous homograft model with intratumoural OV injection: significantly decreased tumour growth and prolonged survival compared to parental OV | [83] |
| NDV | CTLA-4 mouse |
scFV | Melanoma | Intradermal mouse tumour model with intratumoural OV injection: demonstrated the same efficacy as systemic CTLA-4 treatment plus parental NDV, with comparable tumour growth inhibition and prolonged survival | [91] |
| MV | CTLA-4 mouse | scFV-IgG1 Fc fusion | Melanoma | Subcutaneous synergic mouse tumour model with intratumoural OV injection: significantly decreased tumour growth compared to parental virus and untreated; significant increase in tumour T cell infiltration and a decrease in Treg infiltration compared to parental OV and untreated; increased splenocyte IFN-γ release upon re-stimulation with tumour cells in vitro compared to parental OV and untreated | [89] |
| Ad68 | PD-1 | IgG4 | Colorectal | Bilateral subcutaneous humanised PD-1 transgenic mouse tumour model with single-sided intratumoural OV injection: significantly decreased tumour growth and prolonged survival compared to parental OV and untreated, with successful tumour rejection upon rechallenge; significantly increased systemic CD8+ T cell and effector and central memory T cell proportions; significantly decreased PD-1+ CD4+ and CD8+ T cell proportions | [73] |
| HSV-1 | PD-1 mouse | scFv | HCC | Bilateral subcutaneous synergic mouse tumour model with single-sided intratumoural OV injection: significantly decreased tumour growth in both injected and non-injected tumours and greater long-term tumour growth inhibition compared to parental OV and untreated; successful tumour rejection upon rechallenge; significantly increased activated CD4+ and CD8+ cell tumour infiltration compared to parental OV; however, also saw significantly greater MDSC infiltration compared to parental OV | [74] |
| HSV-1 | PD-1 human | scFv | HCC | Orthotopic HCC xenograft tumour model with intravenous OV injection in humanised PD-1 transgenic mice: significantly decreased tumour growth and increased overall survival compared to parental OV and untreated mice, with all anti-PD-1 OV treated mice tumour free at 12 weeks Bilateral subcutaneous mouse xenograft tumour model with single-sided intratumoural OV injection in humanised PD-1 transgenic mice: significantly decreased tumour growth in both injected and non-injected tumours compared to parental OV and untreated; anti-PD-1 OV treated tumours demonstrated significantly reduced proportions of exhausted CD8+ T cell populations and increased effector memory CD8+ T cell populations compared to parental OV and untreated |
[75] |
| HSV-1 | PD-1 human | scFv | Melanoma | Bilateral subcutaneous mouse xenograft tumour model with intratumoural OV injection in humanised PD-1 transgenic mice: significantly decreased tumour growth compared to untreated and parental OV; significantly increased tumour CD4+ and CD8+ T cell infiltration compared to untreated; RNA-seq analysis demonstrated significant enrichment in anti-viral, IFN and antigen presentation and processing pathways compared to untreated | [76] |
| HSV-1 | PD-1 human | scFV | GBM | Orthoptic GBM synergic mouse tumour model with intratumoural OV injection: increased median survival time compared to untreated (significant) and parental OV (not significant); successful tumour rejection following rechallenge | [87] |
| HSV-2 | PD-1 human | IgG | Melanoma | Subcutaneous mouse xenograft tumour model with intratumoural OV injection in humanised PD-1 transgenic mice: significantly decreased tumour growth and prolonged survival compared to untreated; improved tumour-free survival compared to parental OV and untreated; successful tumour rejection following rechallenge; increased systemic percentages of CD4+, CD8+ and CD3+ T cells and significant increase in T cell activation markers compared to parental OV and untreated; significant reduction in Tregs and MDSCs compared to untreated | [77] |
| VV | PD-1 mouse | IgG & scFV | Fibrosarcoma Melanoma |
Subcutaneous synergic mouse tumour model with intratumoural OV injection: significantly decreased tumour growth, and prolonged survival (IgG significant; scFV not significant) compared to parental OV and untreated; IgG-OV significantly increased tumour infiltration of CD4+ and CD8+ T cells, the proportion of activated CD8+ T cells, and the CD8+/Foxp3+ T cell ratio compared to systemic anti-PD-L1 treatment, but to a lesser extent than parental OV alone | [84] |
| NDV | PD-1 and PD-L1 mouse & IL-2 | scFV | Melanoma | Unilateral subcutaneous synergic mouse tumour model with intratumoural OV injection: significantly decreased tumour growth and prolonged survival compared to parental OV Bilateral subcutaneous synergic mouse tumour model with single-sided intratumoural OV injection: when combined with systemic anti-CTLA-4 treatment, PD-1 and PD-L1 OV demonstrated significantly prolonged survival and inhibited tumour growth in non-injected tumours compared to parental OV |
[85] |
| MV | PD-1 & PD-L1 mouse | scFV-IgG1 Fc fusion | Melanoma | Subcutaneous synergic mouse tumour model with intratumoural OV injection: significantly decreased tumour growth and prolonged survival compared to parental OV and untreated; successful tumour rejection following rechallenge; significantly increased activated CD8+ T cell and reduced Foxp3+ Treg tumour infiltration; higher effector memory T cell: central memory T cell ratio for PD-1 (significant) and PD-L1 (not significant) OVs compared to untreated | [82,89] |
| Ad5/24 | PD-L1 mouse |
scFV | Colorectal | Bilateral subcutaneous synergic mouse tumour model with intratumoural OV injection: significantly decreased tumour growth and prolonged survival compared to parental OV and untreated; significantly increased tumour CD8+ T cell infiltration compared to parental OV | [86] |
| Chimeric poxvirus | PD-L1 human | scFv | Breast cancer Gastric cancer PDAC |
Orthotopic synergic mouse breast cancer model with intratumoural or intravenous OV injection: significantly decreased tumour growth and prolonged survival compared to untreated Orthotopic mouse breast cancer xenograft model with intratumoural OV injection: significantly decreased tumour growth and prolonged survival compared to untreated Peritoneal mouse GC and PDAC xenograft tumour model with intraperitoneal OV injection: significantly decreased tumour growth and prolonged survival compared to untreated |
[78–80,88] |
| VSV | PD-L1 human | scFV | Lung carcinoma | Subcutaneous mouse hPD-L1 knock-in synergic tumour model with intratumoural OV injection: significantly decreased tumour growth and prolonged survival compared to untreated; successful tumour rejection following rechallenge; significant systemic increase in total number of CD8+ effector memory and CD8/CD4+ central memory T cells | [81] |
| VV | PD-L1 & GM-CSF human | Soluble PD-1 ED fused to IgG1 Fc | Melanoma | Bilateral subcutaneous synergic mouse tumour models with intratumoural OV injection: decreased tumour growth in 3 solid tumour models; significantly decreased tumour growth and prolonged survival upon tumour rechallenge compared to untreated and parental OV; significantly increased CD45+, DC, CD4+ and CD8+ T cell, and decreased MDSC and Treg tumour infiltration in injected tumours; untreated distant tumours also demonstrated increased infiltration and activation of lymphocytes and other immune cells | [90] |
Abbreviations: Ad, adenovirus; CTLA-4, cytotoxic T-lymphocyte associated protein 4; DC, dendritic cell; GBM, glioblastoma; GM-CSF, granulocyte-macrophage colony-stimulating 2; HCC, hepatocellular carcinoma; HSV, herpes simplex virus ; IAV, influenza A virus; IFN, interferon; IL-2, interleukin-2; MDSC, myeloid-derived suppressor cell; MV, measles virus; NDV, Newcastle disease virus; NSCLC, non-small cell lung carcinoma; OV, oncolytic virus; PBMC, peripheral blood mononuclear cell; PD-1, programmed cell death protein 1; PD-L1, programmed death-ligand 1; PDAC, pancreatic ductal adenocarcinoma; scFV, single chain variable fragment; SCLC, small cell lung carcinoma; treg, regulatory T cell; VSV, vesicular stomatitis virus; VV, vaccinia virus.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



