
Submitted:
21 July 2023
Posted:
21 July 2023
You are already at the latest version
Abstract
Photophysical properties of a series of bis(arylydene)cycloalkanone dyes with various donor substituentns are studied by quantum chemistry. Their capacity for luminescence and nonradiative relaxation through trans-cis isomerization is related to their structure, in particular, to the donor capacity of the substituents and the degree of conjugation due to the central cycloalkanone moiety. It is shown that cyclohexanone central moiety introduces distortions and disrupts the conjugation, thus leading to a nonmonotonic change in their properties. The increasing donor capacity of the substituents causes increase in the HOMO energy (raise of the oxidation potential) and decrease in the HOMO-LUMO gap, which results in the red shift of the absorption spectra. The ability of the excited dye to relax through fluorescence or through trans-cis isomerization is governed by the height of the barrier between the Franck–Condon and S1-S0 conical intersection regions on the potential energy surface of the lowest π-π* excited state. This barrier also correlates with the donor capacity of the substituents and the degree of conjugation between the central and donor moieties. The calculated fluorescence and trans-cis isomerization rates are in good agreement with the observed fluorescence quantum yields.
Keywords:
bis(arylydene)cycloalkanone dyes
; photophysical properties
; photochemical properties
; absorption
; luminescence
; trans-cis isomerization
; quantum chemistry
; density functional theory
; potential energy surface
; conical intersection
1. Introduction
Bis(arylidene)cycloalkanones (Figure 1), also known as symmetric cross-conjugated dienones, ketocyanine dyes, or diarylidene ketone derivatives, are D-π-A-π-D dyes with interesting photochemistry and photophysics. The electrochemistry, photophysics and photochemistry of bis(arylidene)cycloalkanone series have been studied extensively [1,2,3]. The compounds were found to exhibit efficient trans-cis photoisomerization reactions. This property makes bis(arylidene)cycloalkanones useful in the development of photoresponsive materials and devices, such as molecular switches and optical data storage. Bis(arylidene)cycloalkanone compounds display solvatochromic properties [4,5,6] and can be applied, for example, for determining the polarity of a medium [7,8]. Bis(arylidene)cycloalkanones modified with ionophoric fragments [9,10,11] form a basis for photocontrolled supramolecular devices.
Bis(arylidene)cycloalkanone compounds were found to exhibit strong two-photon absorption properties, which could be useful for two-photon microscopy and photodynamic therapy [12,13]. The studies found that the photophysical properties of the compounds were dependent on the size of the alicyclic ring, with larger rings leading to lower fluorescence quantum yields and shorter excited-state lifetimes. [11]. Ref. [14] discusses the recent advances on benzylidene cyclopentanones as photosensitizers for two-photon polymerization. The study focuses on the symmetrically substituted benzylidene cyclopentanones and their ability to initiate polymerization. In Ref. [15] discusses the molecular structure and vibrational spectra of 2,6-bis(benzylidene)cyclohexanone molecule. The paper presents the near-infrared Fourier transform (NIR-FT) Raman and Fourier transform infrared (FT-IR) spectra of 2,6-bis(benzylidene)cyclohexanone molecule along with the density functional calculations. Numerous papers [16,17] discuss the photoprocesses, kinetics, and photoproducts of bis(arylidene)cycloalkanones and their derivatives with electron-donating substituents. The studies analyze the spectral, luminescent, and time-resolved properties of the compounds and their derivatives.
The electrochemistry and positions of the absorption and emission bands are immediately related to the electronic structure of the molecules in study, while the luminescence quantum yields and excitation decay kinetics are governed by the potential energy surfaces. The goal of this paper is to provide a quantum chemical interpretation of the photochemical and photophysical properties of the cycloalkanone series as a function of their molecular structure and important features of their potential energy surfaces. This study will help one gain deeper understanding of the structure–property relationships in bis(arylidene)cycloalkanone derivatives and facilitate their targeted molecular design.
2. Results
We consider a series of bis(benzylidene)cycloalkanone derivatives (Figure 2) with electron-donating groups in the phenyl rings. In polar solvents, such as acetonitrile, the global energy minimum corresponds to the sterically unhindered (E,E) isomers, while the gas phase structure, as predicted by the calculations, is (Z,Z) form stabilized by intramolecular C-H…O hydrogen bonds. Therefore, all the calculations were performed taking solvent into account within Polarizable Continuum Model.
2.1. Ground state structures and molecular orbitals
The calculations demonstrated that the fractions of (E,Z) and (Z,Z) isomers is negligible in the ground state. This agrees with the experiment [1,2,3,17], where no electronic transitions corresponding to (E,Z) and (Z,Z) isomers were observed in the absorption spectra. The ground-state rotation barriers are ~50-60 kcal/mol, which indicates that trans-cis isomerization in the ground state is impossible at any reasonable temperature.
The depths of the local minima associated with the rotation of the phenyl rings (shown by arrows in Figure 2) differ by a fraction of kilocalorie, and these minima are separated by low barriers, which can easily be overcome at room temperature, ensuring slightly hindered rotation of these groups. The 1H NMR experiments indicate that dyes (1-3)c exist as mixtures of rotamers [1,2,3].
The planarity of the molecule reflects the degree of conjugation in it. In bis(benzylidene)cycloalkanones under study the central part is rather rigid, but rotation is possible around formally single bonds shown in Figure 2 by arrows. Table 1 shows the corresponding torsion angles characterizing the effect caused by the central alicyclic fragment on the conjugated chromophore chain.
One can see that cyclobutanone central moiety favors perfectly conjugated chromophore chain, the distortion introduced by cyclopentanone is also minor, while that introduced by cyclohexanone is noticeable. Donor substituents favor the conjugation diminishing the torsion from 7° in 2a to almost 0° in 2d,e. The distortion caused by cyclohexanone is so large that even donor substituents reduce it from 30° in 3a only to 21° in 3e. The same trend is observed in the available crystal structures: 0-2° in cyclobutanones, 1-8° in cyclopentanones, and 9-39° in cyclohexanones.
The chemical shifts (Table S1 of Supplementary Materials) show the following trend with the increase of the central alicycle: the aliphatic protons shift towards strong fields, while methine protons shift towards weak fields. This trend is observed both in the calculated and experimental chemical shifts.
The donor capacity of the substituents is reflected by the Mulliken charges induced by the substituents on the phenyl rings (Table 2). In more conjugated systems, the electron density can flow freely from the donors to the acceptor, thus causing positive charge on the donors and negative on the acceptor. Due to the distorted conjugation, the electron density remains on the donors, and the charge on the donor fragment slightly decreases in cyclohexanone relative to cyclobutanone and cyclopentanone. As for the donor capacity of the substituents, the charges suggest the following order: 4-H < 4-OMe ~ 3,4-OMe ~ 4-SMe < 4-NEt2.
Another way of characterizing donor capacity of the substituents is their effect on the frontier orbital energies, mainly, HOMO. Figure 3 shows that the trend observed in the Mulliken charges is pronounced more clearly in the HOMO energy: 4-H < 4-OMe < 3,4-OMe ~ 4-SMe < 4-NEt2. The LUMO energy is mostly affected by the acceptor fragment, but the effect is much less pronounced. The HOMO-LUMO gap, which correlates with the absorption spectra, decreases in the series: 4-H > 4-OMe > 3,4-OMe ~ 4-SMe > 4-NEt2.
The HOMO-LUMO gap in cyclohexanone derivatives is in general higher than that in cyclobutanones and cyclopentanones owing to the distorted conjugation caused by the cyclohexanone moiety. This results in the blue-shifted spectra of the cyclohexanone derivatives. The HOMO and LUMO energies correlate with the oxidation and reduction potentials [1,2,3]. The HOMO-LUMO gaps also correlate with the experimental Eox-Ered gaps (see Figures S2 and S3 of Supplementary materials).
2.2. Absorption and emission spectra
The most interesting properties of bis(benzylidene)cycloalkanones are their absorption and emission spectra and their phototransformations. In this section we consider the energy of the lowest π-π* electronic transition as a function of the donor capacity of the substituent and the size of the central alicycle (see Table S2 of Supplementary Materials).
The trends in the energy of the lowest π-π* electronic transition (Figure 4) resemble those of the HOMO-LUMO gap, because these transitions are mainly contributed by HOMO->LUMO excitation. As before, cyclohexanone dyes have higher excitation energies due to the distorted conjugation.
Our calculations show that in addition to the intense π-π* and dark n-π* electronic transitions, the dyes exhibit one more π-π* electronic transition at ~400 nm resulting from HOMO-1->LUMO transition (Figure 5). The intensity of this transition decreases from cyclobutanone to cyclohexanone (Figure S4 of Supplementary Materials). This is supported by the experimental data [1,2,3,18,19]: in cyclobutanone 1e the long-wave absorption band consists of two distinct peaks that cannot be attributed to the vibronic progression, while in 2e and 3e the long-wave absorption band is only broadened. The position of this band changes only slightly, which can be explained by the fact that HOMO-1 is not affected either by the substituents or by the central moiety.
The trends in the order of excited states are shown in Figure S5 of Supplementary Materials. The nπ* state is the lowest in the unsubstituted dyes (1-3)a, and goes up as soon as donor substituents appear in the para position. At the same time, the two ππ* states go down. In (1-3)b, the nπ* state lies between the two ππ* states. In the dyes with more donor substituents, the order of stated depends on the central alicycle: in the most conjugated 1(c-e), the nπ* state lies above both ππ* states, while in the other cycloalkanones, the nπ* state goes above both ππ* states only in (2,3)e.
Frequently the changes taking place in excited dyes, in particular, the bond length alternation in the excited state, which facilitates trans-cis isomerization, are explained in terms of resonance structures (see, for example, [20]). In most cases, these features are reflected by the nodal structure of HOMO and LUMO. The latter, being populated in the excited state, correlates with the resonance structure with alternated bonds. In the case of benzylidene cycloalkanones, the nodal structure of LUMO also corresponds to the resonance structure with alternated bonds. The bond length in the first ππ* excited state change in agreement with the nodal structure of LUMO: appearance of the node in LUMO as compared to HOMO results in bond lengthening, while disappearance of the node results in bond shortening. The change is noticeable, especially in the (1-3)a, up to 0.03-0.04 A. In general, the largest alternations are in the exocyclic double bonds and in the adjacent single bonds shown by arrows in Figure 2. In some cases, the carbonyl bond also changes.
All cyclobutanone dyes except for 1a and 1b are fluorescent, with the quantum yields increasing from 1c to 1e. Cyclopentanones 2c-e are also fluorescent, while in cyclohexanones, only 3e is fluorescent. For (1-3)a, the lack of luminescence can be explained by the fact that their first electronic transition is dark n-π*; therefore, excitation to the bright S2 (ππ*) only causes relaxation to the dark S1(nπ*), which further relaxes nonradiatively thorough internal conversion or intersystem crossing.
The lack of luminescence of other dyes, whose first electronic transition is bright π-π*, is more difficult to explain. To do this, one needs to consider the potential energy surfaces of the ground and first excited states or, more specifically, their cross-sections (profiles) along certain internal coordinates (Figure 6a).
The internal coordinate involved in the relaxation of the S1 (ππ*) excited state of bis(arylidene)cycloalkanones is rotation along the formally double bond. This shape of the potential energy profile implies that the molecule excited from its global (E,E) minimum to the Franck-Condon region of the S1 state quickly relaxes to its nearest minimum. From this local minimum of the S1 state, the molecule can either emit light or further relax to the region of lower energy (here, it is the region of the S1-S0 conical intersection). If the barrier separating the local minimum from the CI region is not very high, this relaxation can successfully compete with the radiative relaxation. Near the CI point, the molecule quickly relaxes nonradiatively to the S0 state. From this point, the twisted chromophore can relax either back to the (E,E) region or forward to the (E,Z) region resulting in the trans-cis isomerization. Since the S1 state in the local minimum is rather short-lived (either owing to the radiative relaxation or to the structural relaxation to the CI), no equilibrium is possible on the S1 potential energy surface, and excited (E,Z)* states are not accessible directly from the excited (E,E)* states. All these processes are schematically shown in Figure 6b.
The right-hand part of the profile in Figure 6a represents the same process, but starting from the (E,Z) ground state structure. However, since the ground state is dominated by the (E,E) isomer, the right-hand part of the profile does not seem to be relevant.
Therefore, the luminescence quantum yield is governed by the competition between the radiative relaxation from the (E,E)* minimum and trans-cis photoisomerization. The triplet processes proceed on a microsecond timescale or even slower; therefore, we don’t need to include them in the scheme.
Figure 7 shows the trends in the activation energy and CI depth as a function of donor substituents and central ring size. Although the CI depth cannot be a driving force for the trans-cis isomerization process, it is also indicative of the dye properties. the CI depth decreases and the activation energy increases with the increasing donor capacity of the substituent. The barrier height also increases from cyclohexanone to cyclobutanone. This is in line with the observed trend in the fluorescence quantum yields.
Table 3 shows the radiative lifetimes calculated using the oscillator strengths of the S1-S0 transition, and characteristic trans-cis isomerization times calculated using the Arrhenius equation on the S1 potential energy surface of the trans isomer. One can see that emission can proceed in the nanosecond timescale, while trans-cis isomerization time can range from fractions of nanosecond to microseconds or longer. Fast isomerization successfully competes with the radiative decay channel, thus causing fluorescence quenching. One can see that trans-cis isomerization in 1(c-e) is several orders of magnitude slower than in 1(a,b), and isomerization of 3b competes with the radiative relaxation. In 3(c,d), relaxation via triplet state adds to the relaxation via trans-cis isomerization, The calculated trans-cis isomerization times correlate with the observed fluorescence of the cycloalkanones (Table S2 of Supplementary materials).
4. Materials and Methods
The structures and energies of the molecules were calculated using the density functional theory (DFT) with the PBE0 functional and 6-31+G(d,p) basis set by the FireFly program [21], partially based on GAMESS code [22]. The solvent (MeCN) effects were taken into account using the dielectric polarizable continuum model (D-PCM) [23]. Previously [10], we have shown that for dienones, solvent effects are important to properly reproduce the structures and conformation energies.
The vertical absorption and emission spectra, energy profiles of (E,E)-(E,Z) isomerization and rotation of the aromatic ring around C(β)-C(γ) bond were calculated by the time-dependent DFT (TDDFT) with the same functional, basis set, and solvent model. The vertical absorption spectra were calculated by the TDDFT after DFT optimization of the ground state geometry. A similar method was applied for vertical emission spectra calculations after geometry optimization of the π-π* lowest excited state using the TDDFT and D-PCM. The radiative lifetimes were calculated according to the formula:
where f 0i and ν0i are oscillator strength and the frequency of the electronic transition of the ith isomer respectively; kr is the radiation constant. The isomerization periods were calculated according to the formula:
where νi is the vibrational mode frequency of the ith isomer, and EAi is the activation barrier of this isomer.
kr = (⅔)f0iν2i0; τr = 1/kr
ktc = cνi·exp(-EAi/RT); ttc = 1/ktc
The main channel of the structural relaxation of the S1 state of (E,E) bis(benzylidene)cycloalkanones is rotation around formally double bond leading to trans-cis isomerization. To construct the profiles of (E,E)-(E,Z) isomerization and an alternative channel, rotation of the aromatic ring around formally single bond, we used a simple (unrelaxed) scan of the potential energy surface along the corresponding dihedral angles. The energy values correspond to the non-optimized structures obtained by twisting of the initial isomer or rotamer. In the case of (E,E)-(E,Z) isomerization the left-hand rotation barriers were estimated by the energy difference of the maximum on the S1 profile (corresponding to the left transition state) and stable structures of the (E,E) isomers in the S1 state (left minimum).
We understand that phototransformation, which proceeds via a conical intersection, requires multireference quantum chemistry for an adequate description of the potential energy profiles [24]. Nevertheless, our semi-quantitative description gives insights into the mechanism of phototransformations in organic dyes [25].
The vertical ionization potentials (IP) and electron affinities (EA) were calculated by restricted-open-shell DFT (RO-DFT) for the corresponding monocation and monoanion of each dye. The functional, basis set, and solvation model were the same.
5. Conclusions
Photophysical properties of a series of bis(arylydene)cycloalkanone dyes with various donor substituentns are studied by quantum chemistry. Their capacity for luminescence and nonradiative relaxation through trans-cis isomerization is related to their structure, in particular, to the donor capacity of the substituents and the degree of conjugation due to the central cycloalkanone moiety. It is shown that cyclohexanone central moiety introduces distortions and disrupts the conjugation, thus leading to a nonmonotonic change in their properties. The donor capacity of the substituents is found to increase in the series 4-H < 4-OMe < 3,4-OMe ~ 4-SMe < 4-NEt2, which causes increase in the HOMO energy (raise of the oxidation potential) and decrease in the HOMO-LUMO gap (decrease in the excitation energy and a red shift of the absorption spectra). The ability of the excited dye to relax through fluorescence or through the trans-cis isomerization is governed by the height of the barrier between the Franck–Condon and S1-S0 conical intersection regions on the potential energy surface of the lowest π-π* excited state. This barrier also correlates with the donor capacity of the substituents and the degree of conjugation between the central and donor moieties. The calculated fluorescence and trans-cis isomerization rates are in good agreement with the observed fluorescence quantum yields.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Calculated and experimental chemical shifts in 4-H, 4-OMe, and 3,4-OMe; Figure S2: Correlations between the calculated HOMO and LUMO energies and experimental oxidation and reduction potentials; Figure S3: Correlations between the calculated HOMO-LUMO gap and experimental gap between the oxidation and reduction potentials; Figure S4: Oscillator strengths of the first and second ππ* transitions; Figure S5: Energy diagrams of the excited states of (E,E) isomer in cyclobutanone, cyclopentanone, and cyclopentanone series; Table S1: Comparison of experimental and calculated chemical shifts; Table S2: Experimental and calculated electronic transitions.
Author Contributions
Conceptualization, A.F.; methodology, A.F.; formal analysis, R.S.; investigation, A.F. and R.S.; writing—original draft preparation, A.F. and R.S.; writing—review and editing, A.F. and R.S.; visualization, A.F. and R.S.; supervision, S.G.; project administration, S.G.; funding acquisition, S.G. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Russian Science Foundation (project No. 22-13-00064).
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
The calculations were performed using the computational facilities of the Joint Supercomputer Center of the Russian Academy of Sciences.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Fomina, M.V.; Vatsadze, S.Z.; Freidzon, A.Y.; Kuz’mina, L.G.; Moiseeva, A.A.; Starostin, R.O.; Nuriev, V.N.; Gromov, S.P. Structure–Property Relationships of dibenzylidenecyclohexanones. ACS Omega 2022, 7, 10087–10099. [Google Scholar] [CrossRef]
- Fomina, M.V.; Freidzon, A.Y.; Kuz’mina, L.G.; Moiseeva, A.A.; Starostin, R.O.; Kurchavov, N.A.; Nuriev, V.N.; Gromov, S.P. Synthesis, Structure and Photochemistry of Dibenzylidenecyclobutanones. Molecules 2022, 27, 7602. [Google Scholar] [CrossRef] [PubMed]
- Vatsadze, S.Z.; Gavrilova, G.V.; Zyuz’kevich, F.S.; Nuriev, V.N.; Krut’ko, D.P.; Moiseeva, A.A.; Shumyantsev, A.V.; Vedernikov, A.I.; Churakov, A.V.; Kuz’mina, L.G.; et al. Synthesis, structure, electrochemistry, and photophysics of 2,5- dibenzylidenecyclopentanones containing in benzene rings substituents different in polarity. Russ. Chem. Bull. 2016, 65, 1761–1772. [Google Scholar] [CrossRef]
- Doroshenko, A.O.; Pivovarenko, V.G. Fluorescence quenching of the ketocyanine dyes in polar solvents: anti-TICT behavior. J. Photochem. Photobiol. A. 2003, 156, 55–64. [Google Scholar] [CrossRef]
- Gutrov, V.N.; Zakharova, G.V.; Fomina, M.V.; Nuriev, V.N.; Gromov, S.P.; Chibisov, A.K. Molecular photonics of dienones based on cycloalkanones and their derivatives. J. Photochem. Photobiol. A 2022, 425, 113678. [Google Scholar] [CrossRef]
- Homocianu, M.; Serbezeanu, D.; Tachita, V.B. Solvatochromism, Acidochromism and Photochromism of the 2,6-Bis(4-hydroxybenzylidene) Cyclohexanone Derivative. Int. J. Mol. Sci. 2023, 24, 5286. [Google Scholar] [CrossRef]
- Kedia, N.; Sarkar, A.; Shannigrahi, M.; Bagchi, S. Photophysics of representative ketocyanine dyes: Dependence on molecular structure. Spectrochim. Acta A 2011, 81, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Kessler, M.A.; Wolfbeis, O.S. Spectrochim. Acta A 1991, 47A, 187–192.
- Vatsadze, S.Z.; Gromov, S.P. Novel Linear Bis-Crown Receptors with Cross-Conjugated and Conjugated Central Cores. Macroheterocycles 2017, 10, 432–445. [Google Scholar] [CrossRef]
- Fomina, M.V.; Kurchavov, N.A.; Freidzon, A.Y.; Nuriev, V.N.; Vedernikov, A.I.; Strelenko, Y.A.; Gromov, S.P. Self-assembly involving hydrogen bonds. Spectral properties and structure of supramolecular complexes of bis-aza-18-crown-6-containing dienones with alkanediammonium salts. J. Photochem. Photobiol. A 2020, 402, 112801. [Google Scholar] [CrossRef]
- Volchkov, V.V.; Khimich, M.N.; Melnikov, M.Y.; Egorov, A.E.; Starostin, R.O.; Freidzon, A.Ya.; Dmitrieva, S.N.; Gromov, S.P. Hydrogen-Bonded Self-assembly of Supramolecular Donor–Acceptor Complexes of (E)-Bis(18-crown-6)azobenzene with Bis(ammoniopropyl) Derivatives of Bipyridine and Dipyridylethylene in Acetonitrile. J. Solution Chem. 2023. [Google Scholar] [CrossRef]
- Zou, Q.; Zhao, Y.; Makarov, N.S.; Campo, J.; Yuan, H.; Fang, D.C.; Perry, J.W.; Wu, F. Effect of alicyclic ring size on the photophysical and photochemical properties of bis(arylidene)cycloalkanone compounds. Phys. Chem. Chem. Phys 2012, 14, 11743–11752. [Google Scholar] [CrossRef]
- Zou, Q.; Zhao, H.; Zhao, Y.; Wang, Y.; Gu, Y.; Wu, F. Polyethylene glycol-functionalized bis(arylidene)cycloalkanone photosensitizers for two-photon excited photodynamic therapy. Proc. SPIE 2012, Optics in Health Care and Biomedical Optics V, 85530J. [Google Scholar]
- Dumur, F. Recent advances on benzylidene cyclopentanones as visible light photoinitiators of polymerization. Eur. Polym. J. 2022, 181, 111639. [Google Scholar] [CrossRef]
- Sajan, D.; Lakshmi, K.U.; Erdogdu, Y.; Joe, I.H. Molecular structure and vibrational spectra of 2,6-bis(benzylidene)cyclohexanone: A density functional theoretical study. Spectrochim. Acta A 2011, 78, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Gutrov, V.N.; Zakharova, G.V.; Fomina, M.V.; Gromov, S.P.; Chibisov, A.K. Intermediates of the Photoinduced 2,4-Bis(4-Diethylaminobenzylidene)cyclobutanone Redox Reaction in Methanol. High Energy Chemistry 2020, 54, 436–440. [Google Scholar] [CrossRef]
- Zakharova, G.V.; Zyuz’kevich, F.S.; Nuriev, V.N.; Vatsadze, S.Z.; Plotnikov, V.G.; Gromov, S.P.; Chibisov, A.K. Photonics of Bis(diethylaminobenzylidene)cyclopentanone and Its Analogue with the Bisazacrown Moiety in Acetonitrile. High Energy Chemistry 2016, 50, 27–31. [Google Scholar] [CrossRef]
- Zakharova, G.V.; Zyuz’kevich, F.S.; Gutrov, V.N., Gavrilova G.V., Nuriev, V.N.; Vatsadze, S.Z.; Plotnikov, V.G.; Gromov, S.P.; Chibisov, A.K. Effect of Substituents on Spectral, Luminescent and Time-Resolved Char-acteristics of 2,5-Diarylidene Derivatives of Cyclopentanone. High Energy Chemistry 2017, 51, 113–117.
- Zakharova, G.V.; Gutrov, V.N.; Nuriev, V.N.; Zyuz’kevich, F.S.; Vatsadze, S.Z.; Gromov, S.P.; Chibisov, A.K. Effect of Substituents on Spectral, Luminescent, and Time-Resolved Spectral Properties of 2,6-Diarylidene De-rivatives of Cyclohexanone. High Energy Chemistry 2017, 51, 424–426. [Google Scholar] [CrossRef]
- L.A. Huck, W.J. Leigh. A Better Sunscreen: Structural Effects on Spectral Properties. Journal of Chemical Education 2010, 87, 1384–1387. [CrossRef]
- Granovsky, A.A. Firefly Version 8.2.0. Available online: http://classic.chem.msu.su/gran/firefly/index.html (accessed on 28 October 2022).
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.J.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Freidzon, A.Y.; Safonov, A.A.; Bagaturyants, A.A.; Alfimov, M.V. Solvatofluorochromism and Twisted Intramolecular ChargeTransfer State of the Nile Red Dye. Int. J. Quantum Chem. 2012, 112, 3059–3067. [Google Scholar] [CrossRef]
- Quentin, C.; Gerasimaite, R.; Freidzon, A.; Atabekyan, L.S.; Lukinaviˇcius, G.; Belov, V.N.; Mitronova, G.Y. Direct Visualization of ˙ Amlodipine Intervention into Living Cells by Means of Fluorescence Microscopy. Molecules 2021, 26, 2997. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic structure of ketocyanine dyes.
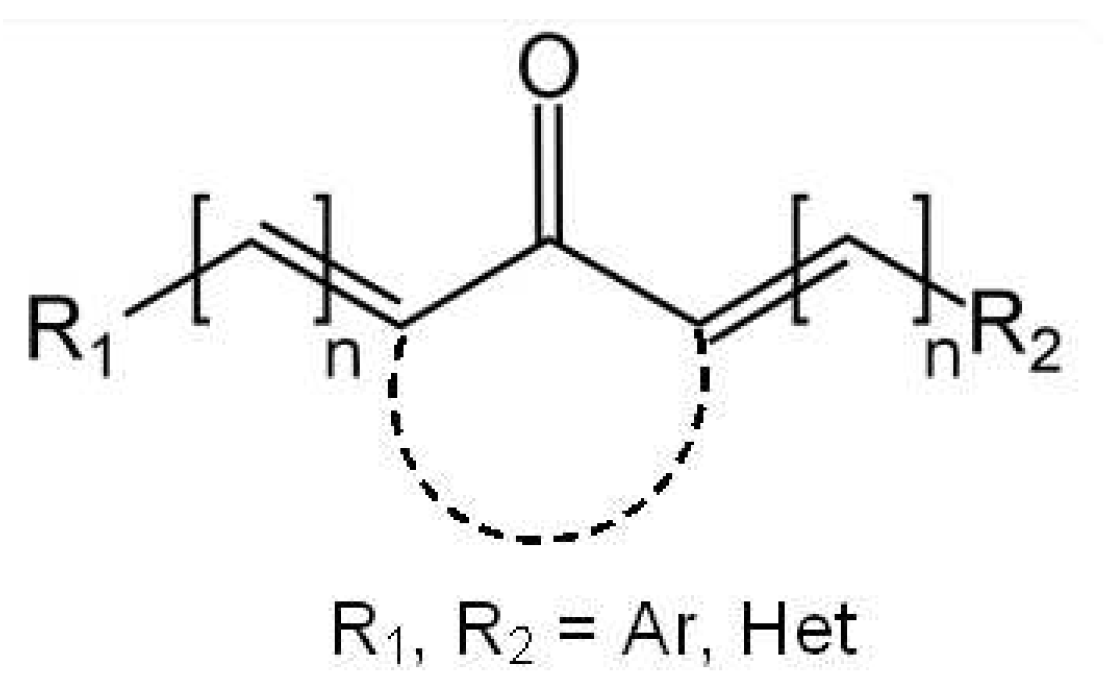
Figure 2.
Dyes under study with important torsion angles shown by arrows.
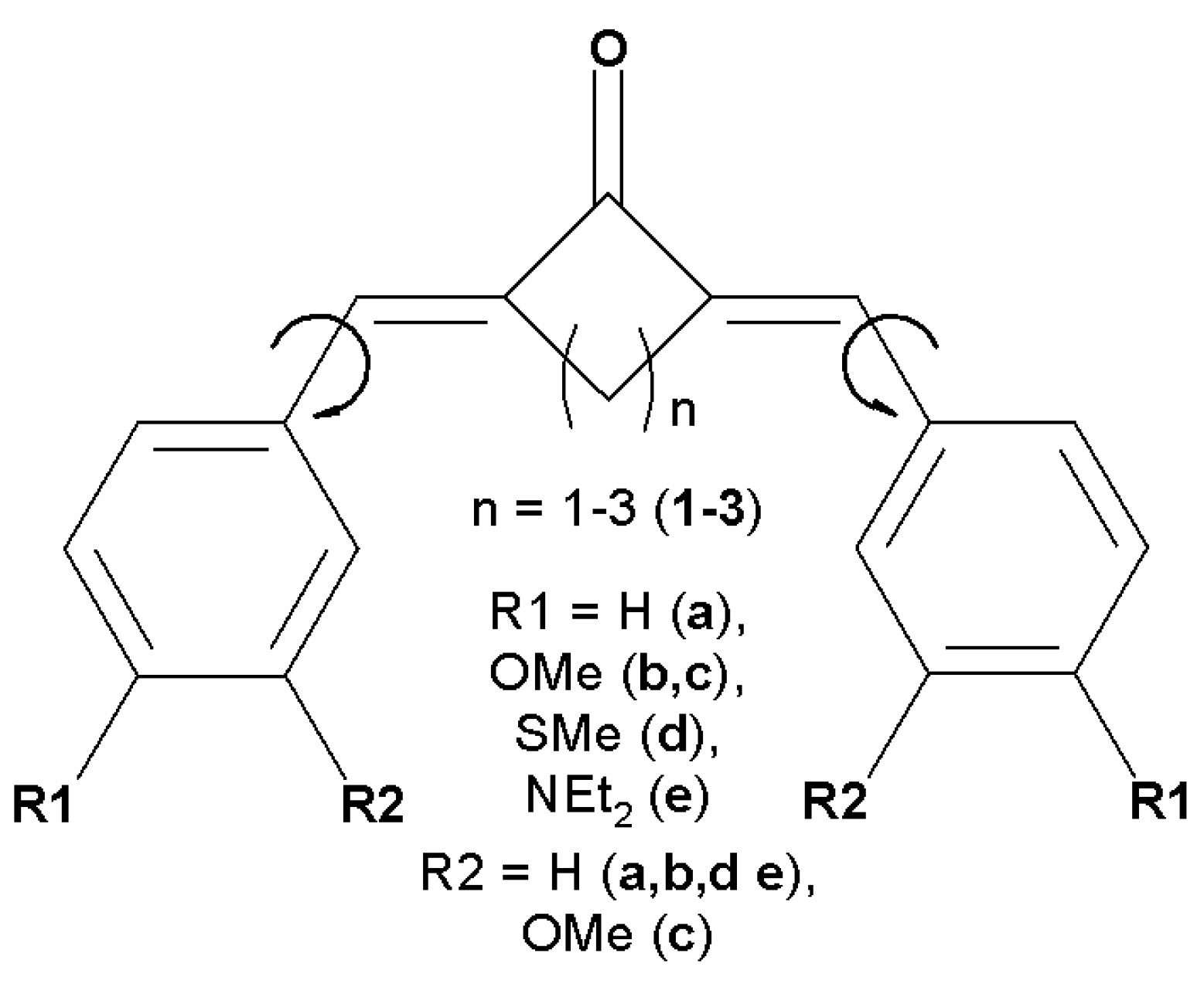
Figure 3.
(a) HOMO, (b) LUMO energies and (c) HOMO-LUMO gap as functions of the donor substituents.
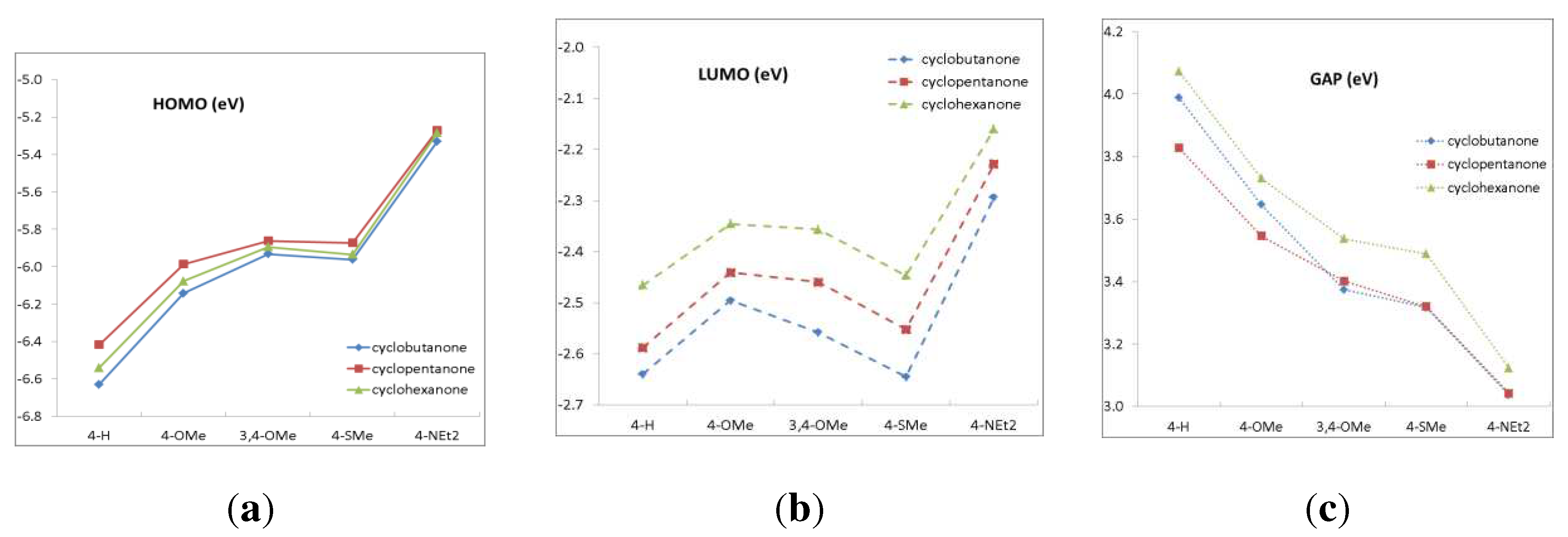
Figure 4.
(a) Experimental and (b) calculated energy of the lowest π-π* electronic transition as a function of the donor substituent.
Figure 4.
(a) Experimental and (b) calculated energy of the lowest π-π* electronic transition as a function of the donor substituent.
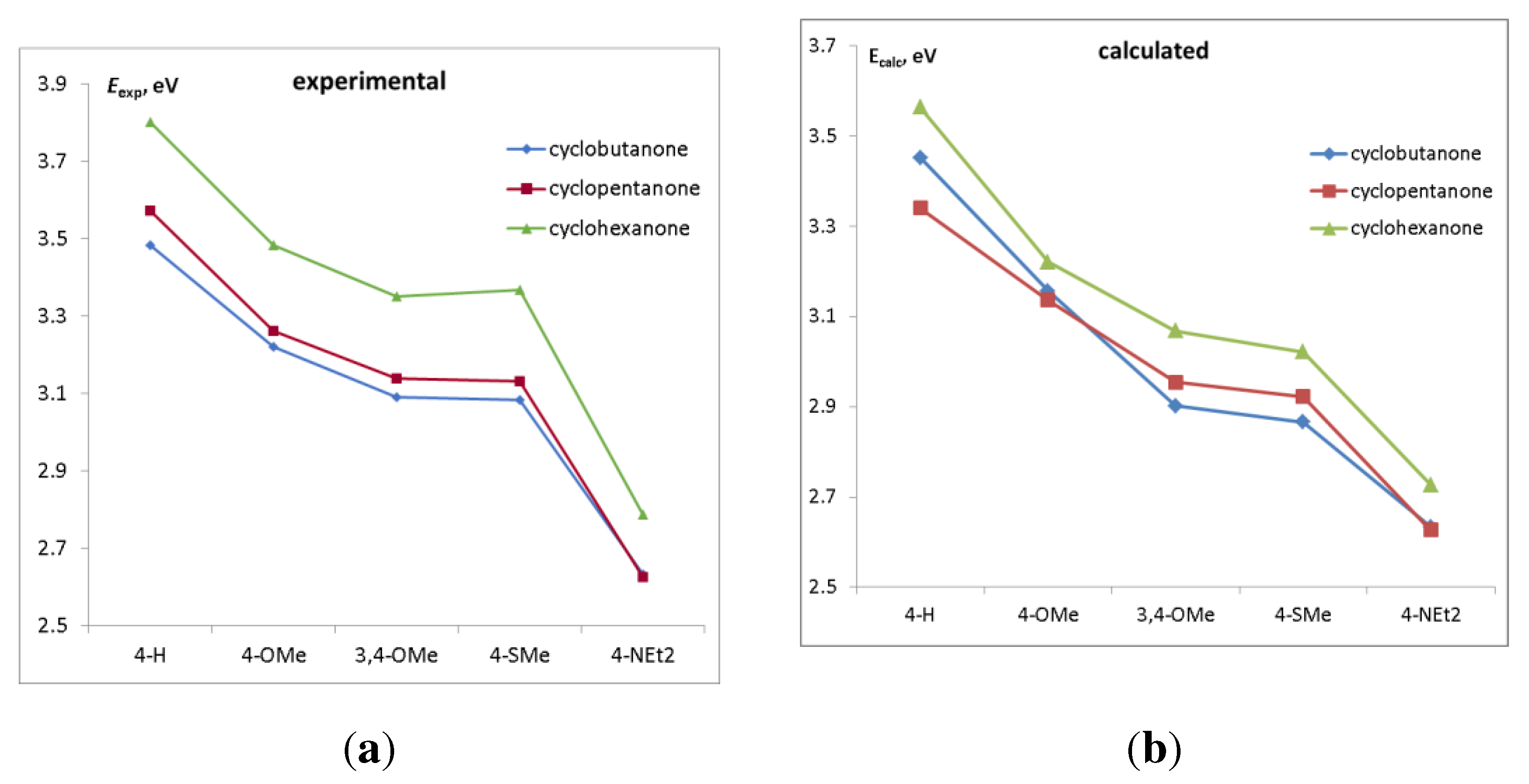
Figure 5.
Frontier orbitals of (1-3)a.
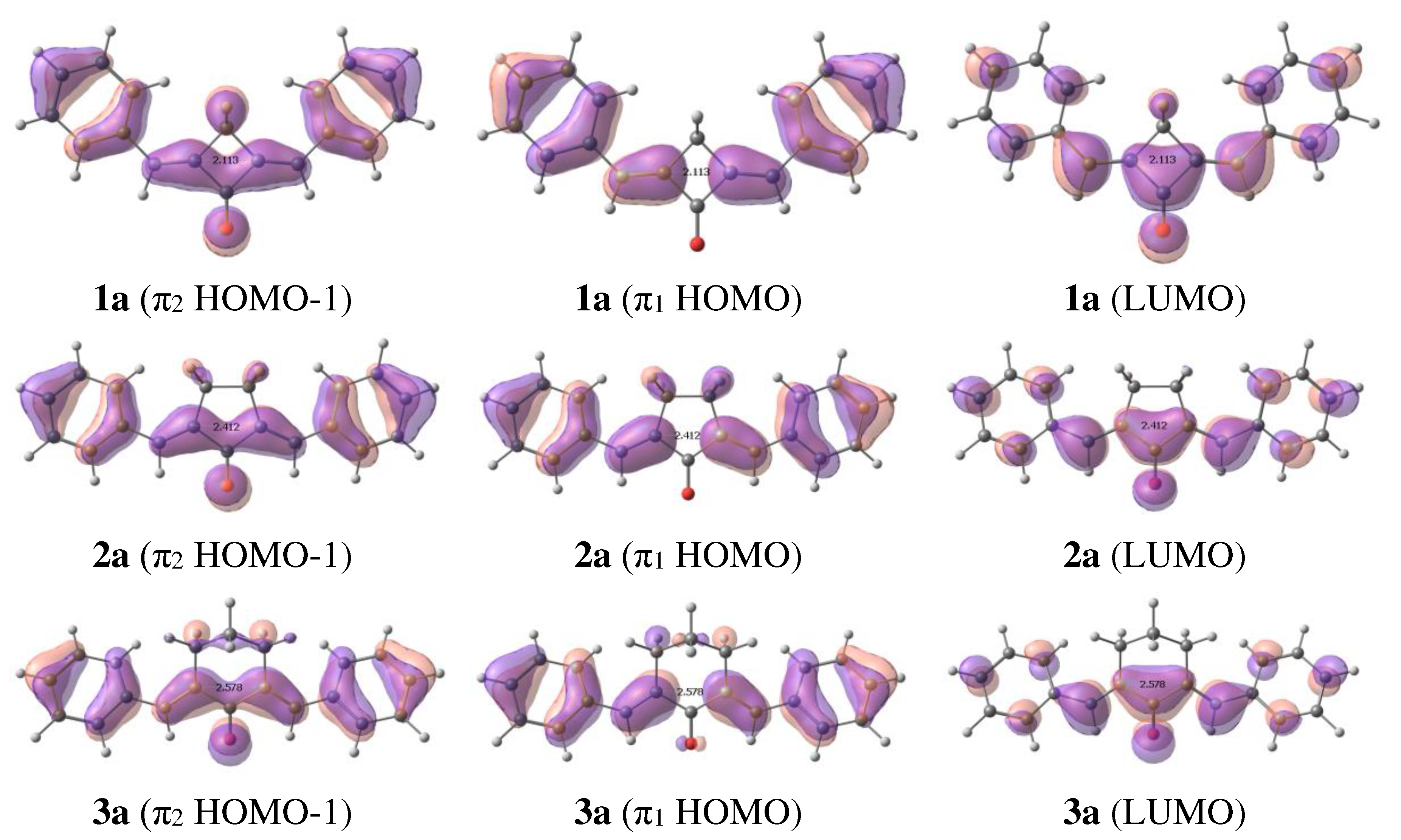
Figure 6.
(a) Typical potential energy profile of the ground S0 and lowest excited S1 (ππ*) and S2 (nπ*) states. (b) Relaxation processes resulting in the trans-cis isomerization.
Figure 6.
(a) Typical potential energy profile of the ground S0 and lowest excited S1 (ππ*) and S2 (nπ*) states. (b) Relaxation processes resulting in the trans-cis isomerization.
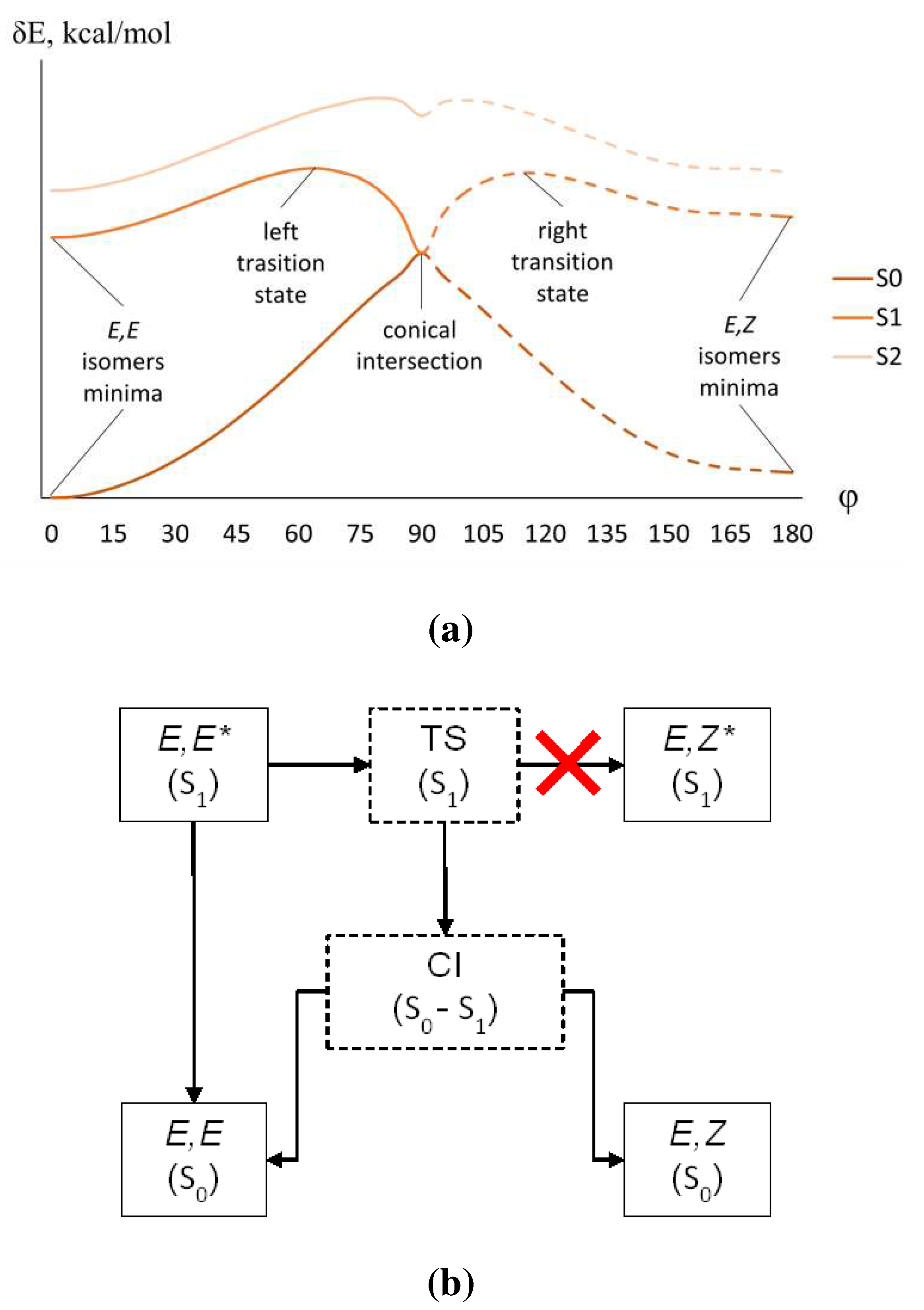
Figure 7.
(a) The activation energy for the transition from (E,E) to CI and (b) CI depth in the S1 state.
Figure 7.
(a) The activation energy for the transition from (E,E) to CI and (b) CI depth in the S1 state.
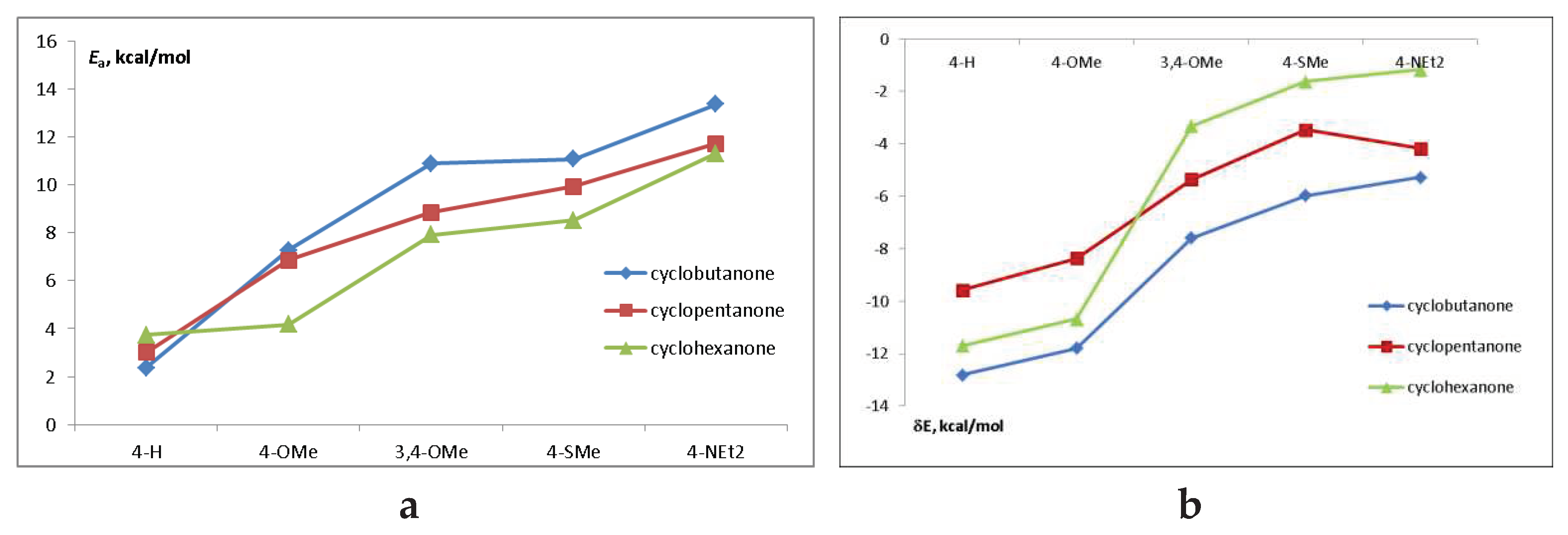

Table 1.
Torsion angle (°) characterizing sterical distortion of π system.
| a | b | c | d | e | |
|---|---|---|---|---|---|
| 1 | 0.1 | 0.0 | 0.0 | 0.0 | 0.4 |
| 2 | 7.0 | 1.0 | 0.1 | 0.5 | 0.2 |
| 3 | 30.3 | 26.9 | 23.9 | 26.6 | 21.0 |
Table 2.
Mulliken charges on donor fragments.
| a | b | c | d | e | |
|---|---|---|---|---|---|
| 1 | 0.11 | 0.14 | 0.14 | 0.13 | 0.20 |
| 2 | 0.10 | 0.13 | 0.12 | 0.11 | 0.19 |
| 3 | 0.07 | 0.10 | 0.10 | 0.09 | 0.16 |
Table 3.
Calculated radiative lifetime (τr) of E,E isomer of cycloalkanones, (E,E)-(E,Z) isomerization rate constant (k) and isomerization time (ttc).
Table 3.
Calculated radiative lifetime (τr) of E,E isomer of cycloalkanones, (E,E)-(E,Z) isomerization rate constant (k) and isomerization time (ttc).
| Cyclobutanone | |||
| Dienone | τr, ns | k, s-1 | ttc, ns |
| 1a | - | 1.12·1010 | 0.09 |
| 1b | 2.28 | 4.97·106 | 201 |
| 1c | 3.05 | 4.40·103 | 227400 |
| 1d | 2.62 | 4.06·103 | 2465000 |
| 1e | 3.07 | 1.22·102 | 8182600 |
| Cyclopentanone | |||
| 2a | - | 4.66·109 | 0.21 |
| 2b | 1.83 | 8.28·106 | 121 |
| 2c | 2.23 | 8.13·104 | 12300 |
| 2d | 1.97 | 3.34·104 | 29900 |
| 2e | 1.90 | 2.55·104 | 39200 |
| Cyclohexanone | |||
| 3a | - | 1.41·109 | 0.71 |
| 3b | 2.90 | 5.47·108 | 1.83 |
| 3c | 2.26 | 7.12·105 | 1404 |
| 3d | 2.02 | 4.92·105 | 2033 |
| 3e | 2.46 | 3.40·103 | 294000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



