
Submitted:
20 July 2023
Posted:
21 July 2023
You are already at the latest version
Abstract
A diagnosis of typical chronic lymphocytic leukemia (CLL) requires the presence of ≥5000 clonal B-lymphocytes/μL with the coexistence of CD19, CD20, CD5, and CD23 and the restriction of light chain immunoglobulin, and the lack of expression of antigens CD22 and CD79b. Atypical CLL (aCLL) can be distinguished from typical CLL morphologically and immunophenotypically. Atypical CLL cells are defined as prolymphocytes; morphologically, these present as deeply-clefted lymphocytes which are larger than those in typical CLL, with condensed chromatin and without prominent nucleoli. However, current aCLL diagnostics rely more on immunophenotypic characteristics rather than atypical morphology. Immunophenotypically, atypical CLL differs from classic CLL in the lack of expression of one or fewer surface antigens, most commonly CD5 and CD23, and the patient does not meet the criteria for a diagnosis of any other B-cell lymphoid malignancy. Morphologically atypical CLL has more aggressive clinical behavior and worse prognosis than classic CLL. Patients with aCLL are more likely to display markers associated with poor prognosis, including trisomy 12, unmutated IGVH, and CD38 expression compared with classic CLL. However, no standard or commonly-accepted criteria exist for differentiating aCLL from classic CLL and the clinical significance of aCLL is still under debate. This review summarizes the current state of knowledge on the morphological, immunophenotypic and genetic abnormalities of aCLL.
Keywords:
CLL
; atypical CLL
; CD5
; CD23
; CLL differential diagnosis.
1. Introduction
Chronic lymphocytic leukemia (CLL) is characterized by monoclonal B cell proliferation, as well as the accumulation of mature lymphocytes within various organs including the peripheral blood (PB), bone marrow (BM), lymph nodes and spleen [1,2]. It is the most common form of adult leukemia in the Western world, comprising 25 to 30 percent of all cases [3]. Its incidence has been estimated as 5.1 cases per 100,000 individuals [4], occuring about twice as often in men as in women. In the United States, an estimated 20,380 new cases (12,130 men and 6610 women), and 4490 deaths were reported for 2023 [5]. Most CLL patients are older, with a median age of 72 years at diagnosis [2] Only nine percent of newly diagnosed patients with CLL are younger than 45 years [6].
In most patients, CLL is diagnosed incidentally on routine blood analysis. However, in some patients, lymphadenopathy, splenomegaly, anemia or thrombocytopenia are observed. A diagnosis of CLL requires the presence of ≥5000 clonal B-lymphocytes/μL in the peripheral blood, together with CD5, CD19 and CD23 antigen expression and light-chain surface immunoglobulin (SIg) restriction, confirmed by flow cytometry, for a duration of at least three months [1,2].
In the recent International Consensus Classification (ICC) and the 5th edition of the World Health Organization Classification (WHO HAEM5) diagnostic criteria for CLL/small lymphocytic lymphoma (SLL) are the same and include coexistence of CD19, CD20, CD5, and CD23 antigens on the leukemic cells [7,8]. In some cases, CD10, CD43, CD49d, CD79b, CD81, CD200 and ROR1 can be useful for differentiating CLL from other lymphoproliferative disorders (LPD) [9]. The clonality of the circulating B-lymphocytes needs to be confirmed by flow cytometry. The WHO-HAEM5 classification of CLL includes prolymphocytic progression [8]. Moreover, it is important to distinguish accelerated CLL from diffuse large B-cell transformation (Richter syndrome) [10],
In 1989, by the French-American-British (FAB) Cooperative Group proposed subdividing CLL into typical and atypical subtypes [11]. However, the clinical significance of atypical CLL (aCLL) is still debated. In 1999, Criel et al proposed the concept of aCLL based on its unique morphology, immunophenotype, genetic abnormalities, clinical features and prognostic factors [12]. Since then, several studies have attempted to identify the causes of the poor prognosis in this group, such as abnormal lymphocyte morphology and karyotype (e.g., trisomy 12 and deletion 17p) [13]. However, atypical CLL is WHO HAEM5 nor the ICC [7,8]. Recent progress in elucidating the genetic characteristics of LPD has allowed a clearer understanding and more accurate definition of CLL and its related disorders [16]. Therefore, the present review summarizes the current state of knowledge regarding the morphology, immunophenotype and genetics of aCLL.
2. Atypical morphological feature
In blood smears from classic CLL, leukemic lymphocytes are characteristically small, mature lymphocytes with a narrow border of cytoplasm and a dense nucleus lacking discernible nucleoli, and having partially-aggregated chromatin (Figure 1A and B). However, there are currently no standard, commonly-accepted criteria for morphological differentiation between aCLL and classic CLL [17,18,19,20,21]. Morphologically-atypical CLL cells were defined mainly as large atypical forms (Figure 1 C and D)1C), prolymphocytes (Figure 2A and B), or cleaved cells (Figure 2C and D). Prolymphocytes, or larger atypical cells up to 55% of the blood lymphocytes do not exclude CLL diagnosis [22]. In some patients with atypical CLL, larger cells and/or cells with abundant cytoplasm are present. This subtype of CLL is commonly associated with trisomy of chromosome 12, a higher intensity of cell B markers, and a poor prognosis.
The FAB Cooperative Group defined three cytology-based subtypes of CLL. These comprise one classical form of CLL and two mixed-cell types: one CLL/prolymphocytic leukemia (PLL) form with 10%-55% prolymphocytes in the PB smear, and another form with larger cells characterized by lymphoplasmacytic features and more abundant cytoplasm cleaved cells. The latter was subsequently called aCLL [11,12]. In the FAB classification, patients with ≥55% prolymphocytes are diagnosed with B-cell prolymphocytic leukemia (B-PLL). Cases with 15–55% of the prolymphocytes previously called CLL/PLL are now known as aCLL [21,23]. However, before the era of multiparameter flow cytometry, aCLL probably included patients with other LPDs with similar morphological features, like mantle cell lymphoma (MCL) or follicular lymphoma (FL).
Criel et al. included cases with large lymphocytes, prolymphocytoid cells, and cells with nuclear clefts in morphologically atypical CLL [12]. Large cells in aCLL have similar morphology to tclasic CLL cells but they are typically at least twice the size of erythrocytes and are morphologically similar to typical CLL cells (Figure [1]). Melo et al. defined prolymphocytoid cells as being more pleomorphic and more heterogenous in size, with an irregular shape and large nucleolus, compared to prolymphocytes in PL [24]. Two types of CLL cells with nuclear clefts were identified: the first are small, while the second are twice as large as erythrocytes [25,26,27,28].
Several studies have indicated clinical, immunophenotypical and genetic differences between cytological aCLL and typical CLL [18,29,30]. Frater et al. compared the morphologic, clinical, and immunophenotypic characteristics between patients with atypical and typical morphologic features of CLL [18]. Morphologically atypical cells were defined as deeply clefted lymphocytes, larger than typical CLL with condensed chromatin and without prominent nucleoli. A minimum of 10% lymphocytes with clefted and folded nuclei in the peripheral blood were required for a diagnosis of aCLL. However, patients with circulating prolymphocytes or with increased numbers of prolymphocytoid cells were not included. Patients with a cytologically atypical CLL had a higher WBC count and a lower platelet count, and a significantly higher risk of disease progression. In addition, aCLL patients demontrated higher expression of CD23, but no significant difference was noted between aCLL and classic CLL regarding the expression of surface immunoglobulin (sIg), CD79b or CD5. In addition, 11 of patients with aCLL had trisomy 12. Morphologically atypical CLL had more aggressive clinical behavior and worse prognosis than classic CLL [12].
In other studies, however, a strong correlation was found between atypical morphology, trisomy 12 and an aberrant immunophenotype [31]. During a study of the validity and clinical impact of CLL on a group of 390 patients, Criel et al distinguished two closely-related but different entities with different prognostic parameters and different survival rates [31]. Typical CLL cases were mostly diagnosed in Binet stage A/Rai 0; these patients did not need treatment at diagnosis and expected a longer survival. In contrast, aCLL cases were mainly diagnosed at a higher risk stage (Binet B/Rai I-II), mostly required immediate treatment and were characterized by shorter survival. The most common chromosomal abnormalities were del(11q) (21% of those with an abnormal karyotype) in typical CLL, and trisomy 12 (65%) in aCLL. The authors concluded that this two types of CLL can be identified based on a specific clinical presentation, different cytogenetic abnormalities and prognostic parameters.
Finn et al. investigated the association between morphology, immunophenotype and karyotype of PB CLL lymphocytes in 26 patients with CLL [32]. Among the patients, seven of the eight (88%) with trisomy 12 had mixed cell morphology according to the FAB guidelines, compared to only three of 18 (17%) without trisomy 12 (P = 0.004). In contrast, only one patient (12%) with trisomy 12 had typical CLL morphology. The atypical immunophenotype comprising strong CD20 expression, strong surface light chain expression, or the absence of CD23 expression was noted in six of the eight (75%) patients with trisomy 12, compared to only two of the 18 patients (11%) without trisomy 12 (p= 0.005).
More recently, Marionneaux et al. used digital microscopy to classify CLL patients as morphologically aCLL or typical CLL on archived blood films of 97 CLL patients [33]. In this study, the CLL/PLL and mixed-type subgroups were classified as aCLL. Atypical CLL was identified in 26 of 97 (27%) CLL cases, including 11 patients (42.3%) with CLL/PLL and 15 patients (57.7%) with the mixed-cell type. All patients with aCLL had at least one prognostic negative cytogenetic abnormality associated with a poor prognosis, compared with 34% of typical CLL patients. The patients with aCLL were more likely to have trisomy 12, unmutated IGVH and CD38 expression compared with typical CLL.
A number of studies have found morphoclinically atypical CLL to demonstrate more aggressive clinical behavior and worse prognosis than classic CLL [23,27,34,35,38]. Ahn et al. evaluated the clinical and prognostic significance of 121 patients with CLL, nine patients with aCLL and 11 patients with B-PLL [28]. They found that lymphadenopathy was more common in CLL (42%), and aCLL (56%) than in B-PLL (0%). In contrast, splenomegaly was more common in B-PLL patients (100%) than in CLL (25%) or aCLL (33%). In addition, aCLL patients demonstrated more severe anemia, elevated lactate dehydrogenase, and β2-microglobulin than those with CLL or B-PLL. Importantly, patients with B-PLL commonly displayed CD5 or CD23 negativity, FMC7 positivity, and strong CD22 positivity. Patients with aCLL showed higher frequencies of FMC7 expression and strong expression of CD22 than those with classic CLL. In addition, the patients with aCLL or B-PLL had worse prognosis than those with classic CLL. The overall survival (OS) rates at 10 years were 22.2% in aCLL, 46.3 % in B-PLL, and 65.6% in classic CLL, respectively. These studies suggest that classic CLL and aCLL represent two closely-related but different entities.
3. Atypical immunophenotype
Currently, CLL is usually identified based on immunophenotypic characteristics. The typical immunophenotype includes the coexistence of CD19, CD20, CD5, and CD23 antigens on leukemic cells (Figure 3). While CLL can be accurately diagnosed by flow cytometry in the majority of patients, such diagnosis becomes more challenging when CD23 or CD5 is not expressed by the leukemic cells [18,29,36,37,38,39,40].
3.1. CD5-negative CLL
CD5-negative (Figure 4) and CD23-negative (Figure 5) variants of CLL are most commonly considered as aCLL. CD5 is expressed on the subset of normal T lymphocytes and in a subset of B-cells [41].
While most CLL patients demonstrate expression of CD5 on the surface of neoplastic cells, this is absent in 7 to 20% of cases [42]. The diagnostic criteria for CD5-negative CLL is an important issue. Several studies have established a diagnosis of CD5-negative CLL where fewer than 5% of leukemic cells demonstrated CD5 expression [42,43,44,45,46]. CD5-negative B-CLL seems to have a different clinical presentation to CD5-positive CLL (Table 1) [45,46,47]. Based on a study on 423 CLL patients, Friedman et al. propose that the mean fluorescence intensity of the CD5 antigen on CLL cells may correlate with the clinical course of the disease: high mean fluorescence intensity rate correlated with longer progression-free survival (PFS) [45]. Cartron et al. compared patients with CD5-negative CLL (n=42) and those with CD5-positive CLL (n=79). The CD5-negative group frequently expressed a higher level of surface immunoglobulin and were more likely to present with splenomegaly; however, the two groups demonstrated similar median OS [42]. In other study, CD5-negative patients were found to exhibit milder disease symptoms and longer survival than the CD5-positive patients [43].
Efstathiou et al. compared clinical and biological characteristics of 29 CD5-negative CLL patients with a control group of 29 sex- and age-matched, consecutive CD5-positive CLL patients [43]. In the CD5-negative group, splenomegaly, lymph node involvement and hemolytic anemia were significantly less common than in the CD5-positive group. The CD5-negative patients had also longer median OS (97.2 months) than the CD5-positive group (84.0 months, p = 0.0025). However, other studies have not confirmed that CD5-negative CLL has a better prognosis.
In a recent study, Demir et al compared 19 consecutive CD5-negative B-CLL cases observed from 2009 to 2015 with 105 CD5-positive CLL patients [44]. Lymphadenopathy was less common (31.5%) of the CD5-negative group than the CD5-positive group (51.4%; p=0.029) but splenomegaly was more common in the CD5-negative group (42.1%) than the CD5-positive group (16.1%, p=0.029). However, no difference in Binet staging or median neutrophil count was noted between the groups. The CD5-negative patients also exhibited a higher mean lymphocyte count (43.2×109/L) than the CD5-positive patients (36.7×109/L, p=0.001). The five-year survival rate was 84.2% in CD5-negative patients and 90.5% in the CD5-positive patients (p=0.393).
Romano et al. analyzed a cohort of 400 CLL patients, including 13 with a CD5-negative phenotype. No significant differences in clinical course and survival were observed between the CD5-positive and CD5-negative CLL cells [47]. Elsewhere, Kurec et al. report that CD5-negative CLL patients exhibited a lower hemoglobin level, higher disease stage (Rai’s classification) at diagnosis and worse survival: the five-year survival rate was 55% in CD5-negative patients >90% in CD5-positive patients [46]. 3.2. CD23-negative CLL
The CD23 surface glycoprotein is another important antigen in CLL diagnosis. It is involved in the activation and proliferation of normal B lymphocytes and plays a role in CLL pathogenesis [48]. In particular, elevated CD23 isotype expression appears to have a protective effect on neoplastic B-cells, with the CD23 isotype stimulating them [49]. In addition, while a diagnosis of CLL requires co-expression of CD23 with CD5 and CD19, MCL is based on the co-expression of CD5 and CD19 alone, without CD23 [50,51]. However, atypical forms of both diseases have been recorded with CD23-negative CLL (Figure 5) and CD23-positive MCL being reported [52]. Barna et al. investigated the cut-off levels of CD23 positivity and intensity in the differential diagnosis of CLL (84 patients) and MCL (26 patients) by flow cytometry analysis [52]. It was found that higher CD23 positivity (>92.5%) and/or mean fluorescence intensity (MFI) of CD23 greater than 44.5 correlated with a diagnosis of CLL. In contrast, CD23 positivity < 30% indicated a diagnosis of MCL. However, patients with CD23 positivity between 30 and 92.5% and intensity below 44.5 Median Fluorescence Intensity (MFI) can be seen both in CLL and MCL. In such cases, it is necessary to perform FISH for the translocation t(11;14) or immunohistochemical detection of cyclin D1 overexpression the differentiate CLL from MCL [52].
Some investigators indicate that the levels of CD23 may influence prognosis in CLL patients [53,54]. Jursic et al compared the level of expression of CD23 antigen and the clinical course of the disease in 77 previously-untreated patients with CLL [48]. The results indicated a correlation between a lower level of CD23 expression and the number of lymphocytes in PB, and that patients with CD23 expression over 40% had a longer PFS and OS.
4. Atypical genotype
As mentioned above, morphologically atypical CLL is often associated with genetic abnormalities, most frequently with trisomy 12 [23,31,53,54]. Some studies indicate a strong association between trisomy 12 and atypical morphology in CLL [53]. However, this abnormality is not limited to lymphocytes with atypical morphology, but can also occur in typical CLL cells. In addition, several mutations are known to render the leukemia more aggressive, such as TP53, ATM, SF3B1, FBXW7, POT1, CHD2, RPS15, IKZF3, ZNF292, ZMYM3, ARID1A, and PTPN11.7 mutations [22,55,56]. In total, 44 recurrently-mutated genes and 11 recurrent somatic copy number variations has been recognized so far, including NOTCH1 and MYD88. The (11;14)(q13;q32) translocation was previously considered to be the hallmark of MCL [57,58]. However, it can also be present in 2%-5% of patients with CLL. These patients have a poor prognosis due to an oncogenic IGH/CCND1 translocation and aberrant expression of cyclin D1 [57]. Trisomy 12 is one of the most frequent chromosomal abnormalities in CLL. Patents with CLL and t(11;14) commonly have an atypical morphology consisting of several small lymphocytes and some larger lymphocytes and prolymphocytes, and share some biological characteristics with MCL [59,60]. In addition, these patients have an atypical immunophenotype comprising CD5+, CD19+, CD23+, SIg+, FMC7+, and CD10−. FMC7 expression is not present in classic CLL, but has been observed in MCL. Moreover, surface Ig has variable expression in classic CLL.
Atypical CLL with t(11;14) has poorer prognosis than other CLL subtypes [53,60], and as these patients require prompt treatment, fast and accurate diagnosis is needed. However, the optimal management of aCLL with t(11;14) is not established yet, due to its low incidence [61,62].
Trisomy 12 was most frequently found in morphologically atypical CLL. One study examined trisomy 12 clones in CLL lymphocytes with atypical morphology based on MGG/FISH with standard cytomorphology [53]. Peripheral blood specimens was studied in four patients with aCLL using a DNA probe against the pericentromeric region of chromosome 12. Although trisomy 12 was identified in 10–34 % of the lymphocytes, the disorder is not confined to lymphocytes with atypical morphology, being also observed in typical CLL cells.
5. Scoring systems for CLL diagnosis
While several scoring systems have been developed for CLL diagnosis, Matutes et al. developed one on the basis of the most common CLL marker profile [62]. The system includes five markers known to be important for CLL diagnosis: CD5 positive, CD22 weak or negative, CD23 positive, FMC7 negative and Slg weak. For each of these five markers, a value of 1 or 0 is added according to whether it is typical or atypical for CLL, and the total score ranges from 0 in atypical CLL to 5 in typical CLL. Among 400 analyzed cases, 87% scored 5 and 4 and only 0.4% scored 0 or 1. In other B-cell leukemias, viz. prolymphocytic leukemia, hairy cell leukemia (HCL) and HCL variant, 89% of the patients scored 0 or 1. Moreover, higher scores were found in CLL cases with more typical morphology (p < 0.0015). Using this panel of five standard markers, the accuracy of the scoring system for distinguishing CLL from non-CLL LPD was 91.8%, using a cutoff of four points or higher.
Subsequently, Moreau et al investigated whether the Matutes scoring system could be improved with the monoclonal antibody SN8 (CD79b) [63]. Briefly, PB samples were taken from 298 patients with CLL and 166 patients with non-CLL and analysed using the five standard markers (CD5, CD22, CD23, FMC7, Slg), together with the SN8 monoclonal antibody, by flow cytometry. SN8 recognizes an extracellular epitope on the B29 protein of the B-cell antigen receptor and has been clustered as CD79b. The addition of SN8 and a cut off of 4 points or higher, as in the original Matutes scoring system, increased the accuracy of the scoring system from 91.8% to 96.6%. However, a similar accuracy (96.8%) was observed if CD22 was excluded and a cutoff of 3 points or higher was used. Thus, the replacement of CD22 by SN8 antibody in the original Matutes scoring system significantly increases the possibility of discrimination between CLL and other B-cell LPD. Some patients with CLL show an immunophenotype that overlaps with other LPD, especially those scoring 3 of 5 points in the Moreau system [63]. However, these classifications cannot define aCLL [17].
In other studies, the name aCLL has been used for CLL cases with uncommon laboratory features. Sandes et al. characterized patients with aCLL as having a Moreau score of 3 or less, or lacking CD23 expression [37]. However, even now, the definition of aCLL remains controversial.
The most recent immunophenotypic European Research Initiative on CLL (ERIC) criteria for diagnosis of CLL does not include any definition of aCLL [9]. The ERIC and European Society for Clinical Cell Analysis (ESCCA) Harmonization project study examined the diagnostic criteria for CLL based on morphology and immunophenotype. The authors classified 14 of 35 potential markers as "required" or "recommended" for CLL diagnosis. It was agreed that the following diagnostic markers were required: CD19, CD5, CD20, CD23,and SIg Kappa, and Lambda. These markers are consistent with current diagnostic criteria and are used in clinical practice. The markers CD43, CD79b, CD81, CD200, CD10, and ROR1 appear to be potentially useful for differential diagnosis and are recommended as such. Immunophenotypically, atypical CLL correlates with higher Sig expression, FMC7 positivity and, typically, a lower Matutes score [31,54,62].
A new scoring system for CLL diagnosis in Chinese patients was developed recently by Li et al. [64]. In this system CD5 and CD23 antigens were replaced with CD43 and CD180. Samples from 237 patients with diagnosis of mature B-cell LPD were randomly included into an exploratory and a validation group. The expression of CD5, CD19, CD20, CD23, CD43, CD79b, CD180, CD200, FMC7, and SIg were analyzed in all the samples. The sensitivity of 91.8% and specificity of 83.1% was calculated based on the receiver operating characteristic curves (ROC). These results were confirmed in a validation cohort (sensitivity of 90.5% (p = 0.808) and a specificity of 79.5% (p = 0.639)). This new CLL score improved sensitivity of 79.4% in CD5-negative or CD23 negative CLL group compared to Moreau score (41.2%) and CLL flow score (47.1%). In aCLL group, the new CLL improved sensitivity of 84.2% compared to Moreau score (61.4%) and CLL flow score (64.9%). This proposed atypical CLL score helped to offer an accurate differentiation of CLL from non-CLL together with morphological and molecular methods, particularly in Chinese patients with atypical immunophenotype [64].
6. Differential diagnosis of atypical CLL with other lymphoproliferative disorders
In patients with aCLL, additional markers should be used for differentiating CLL from other chronic B-cell neoplasms, especially MCL. Classic MCL cases are characterized by a homogeneous population of small- to medium-sized lymphocytes with irregular nuclear contours [66]. However, both CLL and MCL are characterized by the presence of CD5-positive neoplastic cells. In such overlapping cases, an accurate diagnosis can be facilitated by including a cytogenetic study for t(11;14), which is characteristic for MCL. In MCL, t(11;14), (q13;q32) and cyclin D1 overexpression is observed in >95% of cases. However, in some patients, the differential diagnosis of CLL/SLL from other CD5-positive small B-cell lymphomas can be challenging due to the presence of overlapping morphologic and immunophenotypic features [65].
Classic MCL cells are positive for cyclin D1 and SOX11 and negative for CD23 and CD200. However, some MCL patients express CD23 and CD200 but lack SOX11. These patients morphologically and immunophenotypically resemble to CLL, and can be referred to as CLL-like MCL. Qiu et al. compared the clinic-pathological features of 14 cases with CLL-like MCL and 33 classic CLL cases [66]. Patients with CLL-like MCL were found to have lower numbers of neoplastic cells in the PB and lower BM involvement. Moreover more patients with CLL-like MCL had mutated IGHV. CLL-like MCL was more likely to display moderate to high expression of B-cell antigens and Sig light chain. However, patients with CLL-like MCL have distinctive immunophenotypic features that are useful for distinguishing MCL from CLL. The OS of patients with CLL-like MCL was similar to that of CLL patients.
In the majority of patients, CLL can be differentiated from MCL and other lymphoid malignancies by flow cytometry [67]. However, the differential diagnosis may be challenging if CD23 is not expressed by the CLL lymphocytes, or where CD23 is expressed by MCL, as the two entites share many similarities [68]. In such cases, MCL diagnosis should be confirmed by immunohistochemical cyclin D1 detection, together with other cytofluorimetric parameters, cytogenetics or molecular tests.
CD200 is a particularly useful marker for differentiating between aCLL and MCL. It is a membrane glycoprotein belonging to the immunoglobulin superfamily expressed on a subset of T and B lymphocytes. CD200 is expressed on myeloma, plasma cells, and in most patients with CLL. However, in MCL it is totally absent or expressed by a small minority of CD5-positive cells [67,68,69,70]. In a recent study on patients with aCLL, Ting et al. investigated the expression of CD200 on mature B cell neoplasms by eight-color flow cytometry in combination with a conventional panel of flow cytometry markers [70]. The study included 63 samples with CLL or an atypical CLL phenotype, six samples of MCL, and 40 samples of other mature B cell neoplasms. CD200 was expressed in all CLL samples whereas MCL samples were dim or negative for CD200. Among seven aCLL identified by conventional flow cytometry, with Matutes scores ≤3 all were strongly positive for CD200 and negative for t(11;14) translocation.
Mehrpouri et al. attempted to distinguish patients with various lymphoproliferative disorders, including 91 CLL, 15 atypical CLL, 14 MCL and 11 CD5-/CD10-lymphoma, using a panel of specific markers and flow cytometry [71]. They found that CD22, CD23, FMC-7 and CD5 expression could be used to diagnose CLL, MCL, and CD5-/CD10- lymphoma, but could not differentiate MCL from atypical CLL. However, the expression patterns of CD38 and immunoglobulin light chain were found to differ between aCLL and MCL: CD38 was expressed in 92.8% of MCL and 1.1% aCLL patients, and lambda light chain was expressed in 85% of MCL and 0% aCLL. Moreover, all of the patients with aCLL expressed kappa light chain.
In other study, Ho et al. attempted to distinguish between MCL and phenotypically atypical CLL patients presenting with typical MCL phenotypes; the method was based on flow cytometric analysis and a FISH panel for the detection of t(11;14) and other cytogenetic abnormalities characteristic for CLL [66].
Another marker used to differentiate the typical CLL from other B cell chronic lymphoproliferative disorders, including aCLL, is CD45. In a study of the expression of CD45 in typical CLL, aCLL, CLL/PLL, HCL, B-PLL, and B cell-non Hodgkin's lymphoma (B-NHL), Maljaei et al. found lower CD45 density in typical CLL than the other conditions including aCLL [72]. The diagnostic value of CD45 in CLL has been confirmed by other authors [73].
Another antigen that may be useful in the differentiation of CLL from other lymphoproliferative disorders is CD43 [74]. CD43 is a transmembrane sialogclyoprotein expressed on a subset of B-cells, T-cells, monocytes and granulocytes but not on follicular lymphoma or MCL. A recent study showed that CD43 and CD200 can differentiate CLL from non-CLL LPD with higher accuracy than the Matutes scoring system [74]. In addition, no difference in CD43 and CD200 expression has been observed between classic CLL and aCLL patients. In other study, CD81, CD5, CD23 and CD200 were found to be useful markers for distinguishing CLL from other LPDs [75]. Lower lymphoid enhancer-binding factor (LEF-1) expression was found in CLL patients with atypical immunophenotypic or morphological findings compared to classic CLL [76].
7. Conclusions
Atypical CLL (aCLL) is characterized by morphologic, phenotypic and cytogenetic differences compared to classic CLL. However, aCLL is not formally defined and is not included in the 2022 WHO list of hematological neoplasms, nor in the recent International Consensus Classification. Moreover, other LPDs exhibit overlapping expression of some antigens present on CLL cells. Furthermore, the biological relationship between phenotypically standard CLL, CLL with a minor alteration and non-CLL leukemic LPD with some CLL-like findings, is unknown. In the future, molecular studies will play a key role in characterizing aCLL and related diseases. Today, a diagnosis of aCLL has no therapeutic implications, as it requires a similar treatment to classic CLL. However, following the recent introduction of targeted drugs designed on the basis of biological mechanisms, a better definition of aCLL is needed to provide optimal treatment for each group of patients.
Author Contributions
T.R. wrote the paper. All authors examined the available data, reviewed, and revised the manuscript and provided their approval of the final version of the manuscript. All authors agree to be accountable for all aspects of the work. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the grants from the Medical University of Lodz, Poland(No. 503/1-093-01/503-11-004 and 503/1093-1/503-11-003).
Acknowledgments
We thank Edward Lowczowski from the Medical University of Lodz, Poland for editorial assistance.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; interpretation of data, writing of the manuscript, or in the decision to publish the review
References
- Hallek, M.; Cheson, B.D.; Catovsky, D.; Caligaris-Cappio, F.; Dighiero, G.; Dohner, H.; Hillmen, P.; Keating, M.; Montserrat, E.; Chiorazzi, N.; et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood. 2018, 131, 2745–2760 / Blood 2018 Jun21;131(25):2745. [Google Scholar] [CrossRef] [PubMed]
- Eichhorst, B.; Robak, T.; Montserrat, E.; Ghia, P.; Niemann, C.U.; Kater, A.P.; Gregor, M.; Cymbalista, F.; Buske, C.; Hillmen, P.; Hallek, M.; Mey, U. ESMO Guidelines Committee. Chronic lymphocytic leukaemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2021, 32, 23–33. [Google Scholar] [CrossRef]
- Scarfò L, Ferreri AJM, Ghia P. Chronic lymphocytic leukaemia. Crit Rev Oncol Hematol. 2016, 104, 169–182. [CrossRef] [PubMed]
- Teras L.R., DeSantis C.E., Cerhan J.R., Morton L.M., Jemal A., Flowers C.R. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J Clin. 2016, 66, 443–459.
- Siegel R.L., Miller K.D., Wagle N.S, Jemal A. Cancer statistics, 2023. CA Cancer J Clin. 2023, 73, 17–48.
- The Surveillance, Epidemiology, and End Results (SEER) Program of the National Cancer Institute. Cancer Stat Facts: Leukemia—Chronic Lymphocytic Leukemia (CLL). https://seer.cancer.gov/statfacts/html/clyl.html; 2021.
- Campo E., Jaffe E.S., Cook J.R., Quintanilla-Martinez L., Swerdlow S.H., Anderson K.C., Brousset P., Cerroni L., de Leval L., Dirnhofer S., et al. The International Consensus Classification of Mature Lymphoid Neoplasms: a report from the Clinical Advisory Committee. Blood. 2022, 140, 1229–1253.
- Alaggio R., Amador C., Anagnostopoulos I., Attygalle A.D., Araujo IBO, Berti E., Bhagat G., Borges A.B. , Boyer D., Calaminici M., et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia. 2022, 36, 1720–1748. [CrossRef]
- Rawstron A.C, Kreuzer K.A., Soosapilla A., Spacek M., Stehlikova O., Gambell P., McIver-Brown N., Villamor N., Psarra K., Arroz M., et al. Reproducible diagnosis of chronic lymphocytic leukemia by flow cytometry: an European Research Initiative on CLL (ERIC) & European Society for Clinical Cell Analysis (ESCCA) Harmonisation project. Cytom B Clin Cytom. 2018, 94, 121–128.
- Giné E., Martinez A., Villamor N., López-Guillermo A., Camos M., Martinez D., Esteve J., Calvo X., Muntañola A., Abrisqueta P., et al. Expanded and highly active proliferation centers identify a histological subtype of chronic lymphocytic leukemia (“accelerated” chronic lymphocytic leukemia) with aggressive clinical behavior. Haematologica. 2010, 95, 1526–1533.
- Bennett J.M., Catovsky D., Daniel M.T., Flandrin G., Galton D.A., Gralnick H.R., Sultan C. Proposals for the classification of chronic (mature) B and T lymphoid leukaemias. French-American-British (FAB) Cooperative Group. J Clin Pathol. 1989, 42, 567–584. [CrossRef]
- Criel A., Michaux L., De Wolf-Peeters C. The concept of typical and atypical chronic lymphocytic leukaemia. Leuk Lymphoma. 1999, 33, 33–45. [CrossRef] [PubMed]
- Landau D.A., Tausch E., Taylor-Weiner A.N., Stewart C., Reiter J.G., Bahlo J., Kluth S., Bozic I., Lawrence M., Böttcher S., et al. Mutations driving CLL and their evolution in progression and relapse. Nature. 2015, 526, 525–535.
- Jaffe E.S., Harris N.L., Stein H., Vardiman J.W., eds. Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: WHO Press; 2001. Swerdlow S.H, Campo E., Harris N.L., et al., eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: WHO Press; 2008.
- Swerdlow, S.H., Campo E., Harris N.L., et al., eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: WHO Press; 2017.
- Navarro A., Clot G., Martínez-Trillos A., Pinyol M., Jares P., González-Farré B., Martínez D., Trim N., Fernández V., Villamor N., et al. Improved classification of leukemic B-cell lymphoproliferative disorders using a transcriptional and genetic classifier. Haematologica. 2017, 102, e360–e363.
- Sorigue M., Junca J. Atypical chronic lymphocytic leukemia: Brief historical overview and current usage of an equivocal concept. Int J Lab Hematol. 2019, 41, e17–e19.
- Frater J.L., McCarron K.F., Hammel J.P., Shapiro J.L., Miller M.L., Tubbs R.R., Pettay J., Hsi E.D. Typical and atypical chronic lymphocytic leukemia differ clinically and immunophenotypically. Am J Clin Pathol. 2001, 116, 655–66. [CrossRef] [PubMed]
- O’Connor S.J., Su’ut L., Morgan G.J., Jack A.S. The relationship between typical and atypical B-cell chronic lymphocytic leukemia. A comparative genomic hybridization-based study. Am J Clin Pathol. 2000, 114, 448–458. [CrossRef]
- Marionneaux S., Maslak P., Keohane E.M. Morphologic identification of atypical chronic lymphocytic leukemia by digital microscopy. Int J Lab Hematol. 2014, 36, 459–464. [CrossRef]
- Matutes E, Attygalle A, Wotherspoon A, Catovsky D. Diagnostic issues in chronic lymphocytic leukaemia (CLL). Best Pract Res Clin Haematol. 2010, 23, 3–20. [CrossRef]
- Hallek M, Al-Sawaf O. Chronic lymphocytic leukemia: 2022 update on diagnostic and therapeutic procedures. Am J Hematol. 2021, 96, 1679–1705. [CrossRef]
- Oscier DG, Matutes E, Copplestone A, Pickering RM, Chapman R, Gillingham R, Catovsky D, Hamblin TJ. Atypical lymphocyte morphology: an adverse prognostic factor for disease progression in stage A CLL independent of trisomy 12. Br J Haematol 1997, 98, 934–939. [CrossRef]
- Melo JV, Catovsky D, Galton DA. The relationship between chronic lymphocytic leukaemia and prolymphocytic leukaemia. I. Clinical and laboratory features of 300 patients and characterization of an intermediate group. Br J Haematol. 1986, 63, 377–387.
- Ghani AM, Krause JR. Investigation of cell size and nuclear clefts as prognostic parameters in chronic lymphocytic leukemia. Cancer. 1986, 58, 2233–2238. [CrossRef]
- Molica S, Alberti A. Investigation of nuclear clefts as a prognostic parameter in chronic lymphocytic leukemia. Eur J Haematol. 1988, 41, 62–5;
- Vallespí T, Montserrat E, Sanz MA. Chronic lymphocytic leukaemia: prognostic value of lymphocyte morphological subtypes. A multivariate survival analysis in 146 patients. Br J Haematol. 1991, 77, 478–485.
- Ahn A, Kim M S, Park C J, Seo E J, Jang S, Cho Y U, Ji M, Choi YM, Lee K H, Lee J Het al. Atypical chronic lymphocytic leukemia has a worse prognosis than CLL and shows different clinical and laboratory features from B-cell prolymphocytic leukemia. HemaSphere 2019, 3, 864. [CrossRef]
- D'Arena G, Dell'Olio M, Musto P, Cascavilla N, Perla G, Savino L, Greco MM. Morphologically typical and atypical B-cell chronic lymphocytic leukemias display a different pattern of surface antigenic density. Leuk Lymphoma. 2001, 42, 649–54. [CrossRef]
- Cro L, Ferrario A, Lionetti M, Bertoni F, Zucal N N, Nobili L, Fabris S, Todoerti K, Cortelezzi A, Guffanti A, et al. The clinical and biological features of a series of immunophenotypic variant of B-CLL. Eur J Haematol. 2010, 85, 120–129.
- Criel A, Verhoef G, Vlietinck R, et al. Further characterization of morphologically defined typical and atypical CLL: a clinical, immunophenotypic, cytogenetic, and prognostic study on 390 cases. Br J Haematol. 1997, 97, 383–391. [CrossRef]
- Finn WG, Thangavelu M, Yelavarthi KK, et al. Karyotype correlates with peripheral blood morphology and immunophenotype in chronic lymphocytic leukemia. Am J Clin Pathol. 1996, 105, 458–467. [CrossRef]
- Marionneaux S, Maslak P, Keohane EM. Morphologic identification of atypical chronic lymphocytic leukemia by digital microscopy. Int J Lab Hematol. 2014 Aug;36(4):459-64. [CrossRef]
- Peterson LC, Bloomfield CD, Brunning RD. Relationship of clinical staging and lymphocyte morphology to survival in chronic lymphocytic leukaemia. Br J Haematol. 1980, 45, 563–567.
- Ralfkiær E, Geisler C, Hansen MM, et al. Nuclear clefts in chronic lymphocytic leukemia: a light microscopic and ultrastructural study of a new prognostic parameter. Scand J Haematol. 1983, 30, 5–12.
- Urbaniak M, Iskierka-Jażdżewska E, Majchrzak A, Robak T. Atypical immunophenotype of chronic lymphocytic leukemia. Acta Haematol Pol. 2022, 53, 48–52. [CrossRef]
- Sandes AF, de Lourdes Chauffaille M, Oliveira CR, Maekawa Y, Tamashiro N, Takao TT, Ritter EC, Rizzatti EG. CD200 has an important role in the differential diagnosis of mature B-cell neoplasms by multiparameter flow cytometry. Cytometry B Clin Cytom. 2014, 86, 98–105. [CrossRef]
- Ting YS, Smith SABC, Brown DA, Dodds AJ, Fay KC, Ma DDF, Milliken S, Moore JJ, Sewell WA. CD200 is a useful diagnostic marker for identifying atypical chronic lymphocytic leukemia by flow cytometry. Int J Lab Hematol. 2018, 40, 533–539. [CrossRef]
- Sheikh SS, Kallakury BV, Al-Kuraya KA, Meck J, Hartmann DP, Bagg A. CD5-negative, CD10-negative small B-cell leukemia: variant of chronic lymphocytic leukemia or a distinct entity? Am J Hematol. 2002, 71, 306–310 25. [CrossRef] [PubMed]
- Huang JC, Finn WG, Goolsby CL, Variakojis D, Peterson LC. CD5-small B-cell leukemias are rarely classifiable as chronic lymphocytic leukemia. Am J Clin Pathol. 1999, 111, 123–130. [CrossRef] [PubMed]
- Delia D, Bonati A, Giardini R, Villa S, De Braud F, Cattoretti G, Rilke F. Expression of the T1 (CD5, p67) surface antigen in B-CLL and B-NHL and its correlation with other B-cell differentiation markers. Hematol Oncol. 1986, 4, 237–248. [CrossRef]
- Cartron G, Linassier C, Bremond JL, Desablens B, Georget MT, Fimbel B, Luthier F, Dutel JL, Lamagnere JP, Colombat P. CD5 negative B-cell chronic lymphocytic leukemia: clinical and biological features of 42 cases. Leuk Lymphoma. 1998, 31, 209–216. [CrossRef] [PubMed]
- Efstathiou S, Tsioulos D, Zacharos I, Tsiakou A, Mastorantonakis S, Salgami E, Katirtzoglou N, Psarra A, Roussou P. The prognostic role of CD5 negativity in B-cell chronic lymphocytic leukaemia: a case-control study. Haematologia (Budap). 2002, 32, 209–218. [CrossRef]
- Demir C, Kara E, Ekinci Ö, Ebinç S. Clinical and Laboratory Features of CD5-Negative Chronic Lymphocytic Leukemia. Med Sci Monit. 2017, 23, 2137–2142. [CrossRef]
- Friedman DR, Guadalupe E, Volkheimer A, Moore JO, Weinberg JB. Clinical outcomes in chronic lymphocytic leukaemia associated with expression of CD5, a negative regulator of B-cell receptor signalling. Br J Haematol. 2018, 183, 747–754. [CrossRef] [PubMed]
- Kurec AS, Threatte GA, Gottlieb AJ, Smith JR, Anderson J, Davey FR. Immunophenotypic subclassification of chronic lymphocytic leukaemia (CLL). Br J Haematol. 1992, 81, 45–51. [CrossRef] [PubMed]
- Romano C, Sellitto A, Chiurazzi F, Simeone L, De Fanis U, Raia M, Del Vecchio L, Lucivero G. Clinical and phenotypic features of CD5-negative B cell chronic lymphoproliferative disease resembling chronic lymphocytic leukemia. Int J Hematol. 2015, 101, 67–74. [CrossRef]
- Jurisic V, Colovic N, Kraguljac N, Atkinson HD, Colovic M. Analysis of CD23 antigen expression in B-chronic lymphocytic leukaemia and its correlation with clinical parameters. Med Oncol. 2008, 25, 315–322. [CrossRef] [PubMed]
- Fournier S, Yang LP, Delespesse G, Rubio M, Biron G, Sarfati M. The two CD23 isoforms display differential regulation in chronic lymphocytic leukaemia. Br J Haematol. 1995, 89, 373–379. [CrossRef] [PubMed]
- Kilo MN, Dorfman DM. The utility of flow cytometric immunophenotypic analysis in the distinction of small lymphocytic lymphoma/chronic lymphocytic leukemia from mantle cell lymphoma. Am J Clin Pathol. 1996, 105, 451–457. [CrossRef]
- Gong JZ, Lagoo AS, Peters D, Horvatinovich JM, Benz P, Buckley PJ. Value of CD23 determination by flow cytometry in differentiating mantle cell lymphoma from chronic lymphocytic leukemia/small lymphocytic lymphoma. Am J clin Pathol. 2001, 116, 893–897. [CrossRef]
- Barna G, Reiniger L, Tátrai P, Kopper L, Matolcsy A. The cut-off levels of CD23 expression in the differential diagnosis of MCL and CLL. Hematol Oncol. 2008, 26, 167–170. [CrossRef]
- Hjalmar V, Kimby E, Matutes E, Sundström C, Wallvik J, Hast R. Atypical lymphocytes in B-cell chronic lymphocytic leukemia and trisomy 12 studied by conventional staining combined with fluorescence in situ hybridization. Leuk Lymphoma. 2000, 37, 571–576. [CrossRef]
- Matutes E, Oscier D, Garcia-Marco J, Ellis J, Copplestone A, Gillingham R, Hamblin T, Lens D, Swansbury GJ, Catovsky D. Trisomy 12 defines a group of CLL with atypical morphology: correlation between cytogenetic, clinical and laboratory features in 544 patients. Br J Haematol. 1996, 92, 382–388.
- Puente XS, Pinyol M, Quesada V, Conde L, Ordóñez GR, Villamor N, Escaramis G, Jares P, Beà S, González-Díaz M, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011, 475, 101–105. [CrossRef] [PubMed]
- Puente XS, Beà S, Valdés-Mas R, Villamor N, Gutiérrez-Abril J, Martín-Subero JI, Munar M, Rubio-Pérez C, Jares P, Aymerich M, et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature. 2015, 526, 519–524.
- Huret, J. t(11;14)(q13;q32). Atlas Genet Cytogenet Oncol Haematol. 1998, 2, 129–131. [Google Scholar] [CrossRef]
- Muddasani R, Talwar N, Suarez-Londono JA, Braunstein M. Management of atypical chronic lymphocytic leukemia presenting with extreme leukocytosis. Clin Case Rep. 2020, 8, 877–882. [CrossRef] [PubMed]
- Cuneo A, Balboni M, Piva N, Rigolin GM, Roberti MG, Mejak C, Moretti S, Bigoni R, Balsamo R, Cavazzini P, et al. Atypical chronic lymphocytic leukaemia with t(11;14)(q13;q32): karyotype evolution and prolymphocytic transformation. Br J Haematol. 1995, 90, 409–416. [CrossRef]
- Gómez Pescie M, Denninghoff V, García A, Rescia C, Avagnina A, Elsner B. Linfoma del manto vs. leucemia linfatica crónica atipica. Utilización de inmunohistoquímica, citometría de flujo y biología molecular para su correcta tipificación [Mantle cell lymphoma vs. atypical chronic lymphocytic leukemia. Use of immunohistochemistry, flow cytometry and molecular biology for their adequate typing]. Medicina (B Aires). 2005, 65, 419–424.
- DE Braekeleer M, Tous C, Guéganic N, LE Bris MJ, Basinko A, Morel F, Douet-Guilbert N. Immunoglobulin gene translocations in chronic lymphocytic leukemia: A report of 35 patients and review of the literature. Mol Clin Oncol. 2016, 4, 682–694. [CrossRef]
- Matutes E, Owusu-Ankomah K, Morilla R, Garcia Marco J, Houlihan A, Que TH, Catovsky D. The immunological profile of B-cell disorders and proposal of a scoring system for the diagnosis of CLL. Leukemia. 1994, 8, 1640–1645.
- Moreau EJ, Matutes E, A'Hern RP, Morilla AM, Morilla RM, Owusu-Ankomah KA, Seon BK, Catovsky D. Improvement of the chronic lymphocytic leukemia scoring system with the monoclonal antibody SN8 (CD79b). Am J Clin Pathol. 1997, 108, 378–382. [CrossRef]
- Li Y, Tong X, Huang L, Li L, Wang C, He C, Liu S, Wang Z, Xiao M, Mao X, Zhang D. A new score including CD43 and CD180: Increased diagnostic value for atypical chronic lymphocytic leukemia. Cancer Med. 2022, 10, 4387–4396.
- Ho AK, Hill S, Preobrazhensky SN, Miller ME, Chen Z, Bahler DW. Small B-cell neoplasms with typical mantle cell lymphoma immunophenotypes often include chronic lymphocytic leukemias. Am J Clin Pathol. 2009, 131, 27–32. [CrossRef]
- Qiu L, Xu J, Tang G, Wang SA, Lin P, Ok CY, Garces S, Yin CC, Khanlari M, Vega F, Medeiros LJ, Li S. Mantle cell lymphoma with chronic lymphocytic leukemia-like features: a diagnostic mimic and pitfall. Hum Pathol. 2022, 119, 59–68. [CrossRef] [PubMed]
- El-Sewefy DA, Khattab DA, Sallam MT, Elsalakawy WA, Dahlia A. Flow cytometric evaluation of CD200 as a tool for differentiation between chronic lymphocytic leukemia and mantle cell lymphoma. Egypt J Haematol. 2014, 39, 42–46. [CrossRef]
- Palumbo GA, Parrinello NL, Fargione G, Cardillo K, Chiarenza A, Berretta S, Conticello C, Villari L, Di Raimondo F. CD200 expression may help in differential diagnosis between mantle cell lymphoma and B-cell chronic lymphocytic leukemia. Leuk. Res. 2009, 33, 1212–1216. [CrossRef] [PubMed]
- Palumbo GA, Parrinello N, Fargione G, Cardillo K, Chiarenza A, Berretta S, Conticello C, Villari L, Di Raimondo F. CD200 expression may help in differential diagnosis between mantle cell lymphoma and B-cell chronic lymphocytic leukemia. Leuk Res. 2009, 33, 1212–1216. [CrossRef]
- Ting YS, Smith SABC, Brown DA, Dodds AJ, Fay KC, Ma DDF, Milliken S, Moore JJ, Sewell WA. CD200 is a useful diagnostic marker for identifying atypical chronic lymphocytic leukemia by flow cytometry. Int J Lab Hematol. 2018, 40, 533–539. [CrossRef]
- Mehrpouri M, Sadat Hosseini M, Jafari L, Mosleh M, Shahabi Satlsar E. A Flow Cytometry Panel for Differential Diagnosis of Mantle Cell Lymphoma from Atypical B-Chronic Lymphocytic Leukaemia. Iran Biomed J. 2023, 27, 15–22. [CrossRef]
- Maljaei SH, Asvadi-E-Kermani I, Eivazi-E-Ziaei J, Nikanfar A, Vaez J. Usefulness of CD45 density in the diagnosis of B-cell chronic lymphoproliferative disorders. Indian J Med Sci. 2005, 59, 187–94. [CrossRef]
- Ramalingam TR, Mohanraj S, Muthu A, Prabhakar V, Ramakrishnan B, Vaidhyanathan L, Easow J, Raja T. Independent diagnostic utility of CD20, CD200, CD43 and CD45 in chronic lymphocytic leukaemia. Leuk Lymphoma. 2022, 63, 377–384. [CrossRef] [PubMed]
- Hoffmann J, Rother M, Kaiser U, Thrun MC, Wilhelm C, Gruen A, Niebergall U, Meissauer U, Neubauer A, Brendel C. Determination of CD43 and CD200 surface expression improves accuracy of B-cell lymphoma immunophenotyping. Cytometry B Clin Cytom. 2020, 98, 476–482. [CrossRef]
- Ozdemir ZN, Falay M, Parmaksiz A, Genc E, Beyler O, Gunes AK, Ceran F, Dagdas S, Ozet G. A novel differential diagnosis algorithm for chronic lymphocytic leukemia using immunophenotyping with flow cytometry. Hematol Transfus Cell Ther. 2023, 45, 176–181. [CrossRef] [PubMed]
- Soliman DS, Al-Kuwari E, Siveen KS, Al-Abdulla R, Chandra P, Yassin M, Nashwan A, Hilmi FA, Taha RY, Nawaz Z, El-Omri H, Mateo JM, Al-Sabbagh A. Downregulation of Lymphoid enhancer-binding factor 1 (LEF-1) expression (by immunohistochemistry and/ flow cytometry) in chronic Lymphocytic Leukemia with atypical immunophenotypic and cytologic features. Int J Lab Hematol. 2021, 43, 515–525.
Figure 1.
Morphological features of classic (A and B) large (C and D) CLL cells. Mature CLL cells are lymphocytes with a narrow border of cytoplasm and partially aggregated chromatin in a dense nucleus (A-peripheral blood, B- bone marrow). Large atypical CLL cells (C- peripheral blood, D- bone marrow) ((magnification 63x).
Figure 1.
Morphological features of classic (A and B) large (C and D) CLL cells. Mature CLL cells are lymphocytes with a narrow border of cytoplasm and partially aggregated chromatin in a dense nucleus (A-peripheral blood, B- bone marrow). Large atypical CLL cells (C- peripheral blood, D- bone marrow) ((magnification 63x).
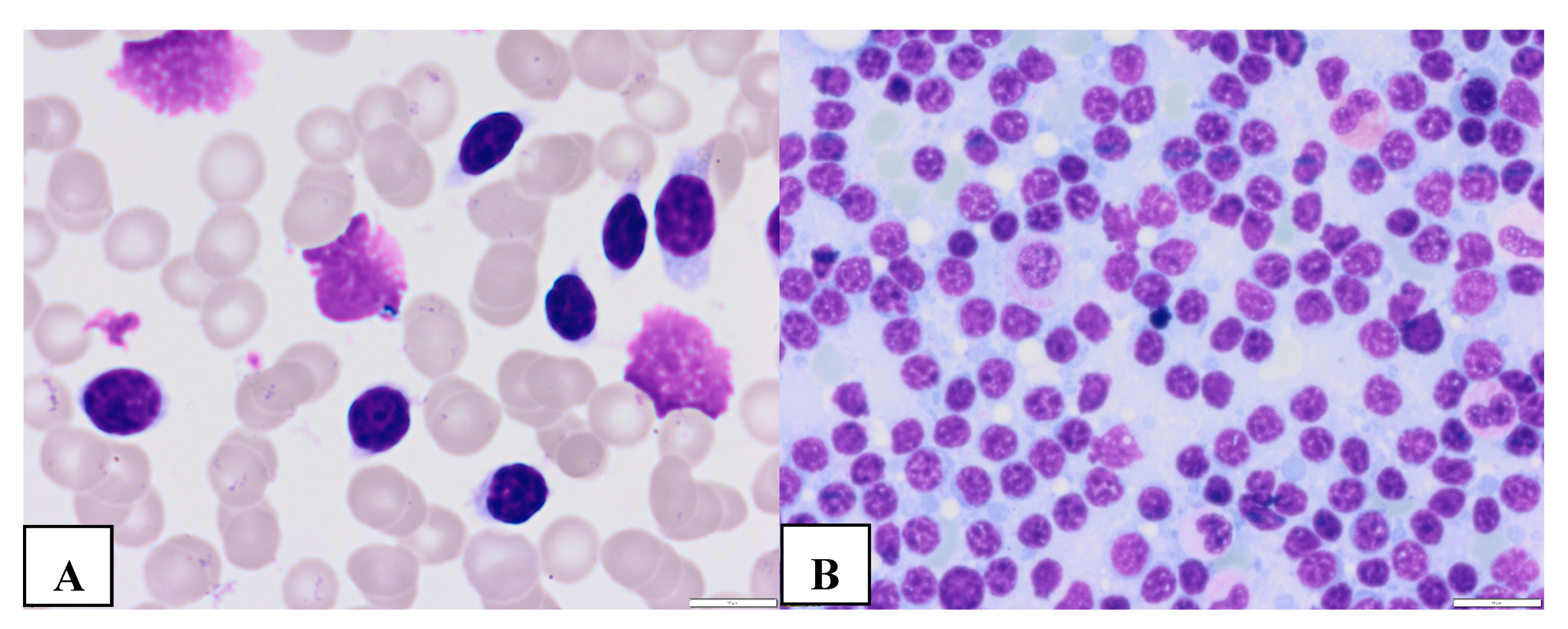
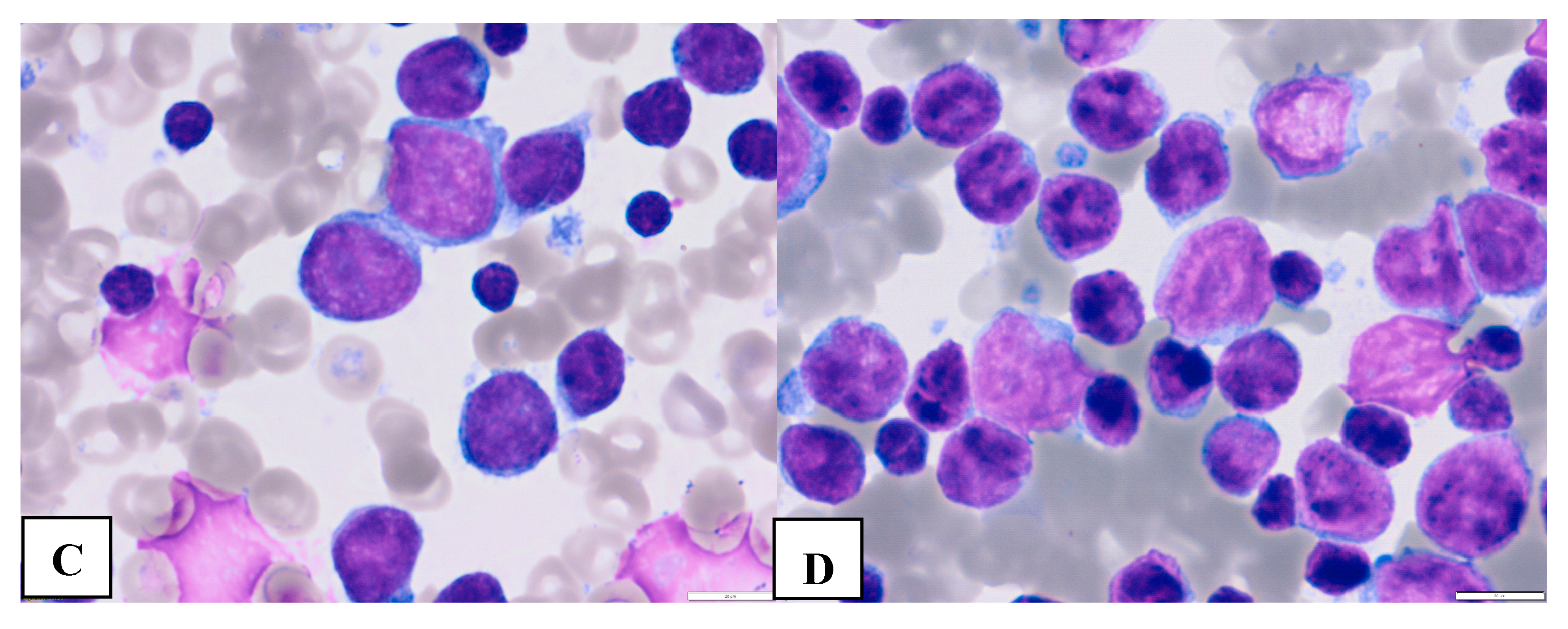
Figure 2.
Morphologic features of atypical CLL cells. Prolymphocytes in peripheral blood (A,B) and cleaved cells ( C- in peripheral blood, D- bone marrow) (magnification 63x).
Figure 2.
Morphologic features of atypical CLL cells. Prolymphocytes in peripheral blood (A,B) and cleaved cells ( C- in peripheral blood, D- bone marrow) (magnification 63x).
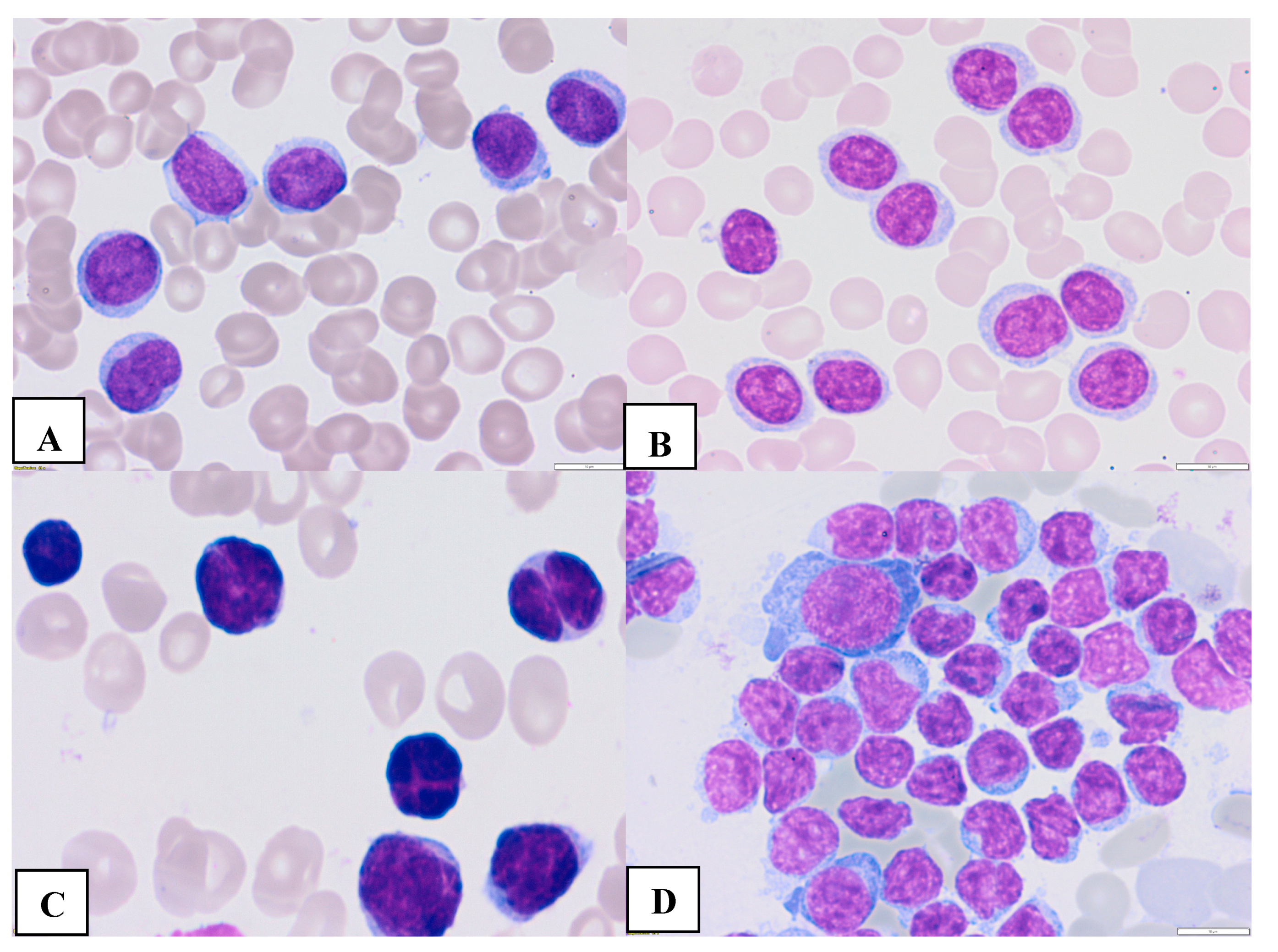
Figure 3.
Representative flow cytometry classic CLL cells showed positive expression of CD5, CD19 and CD23.
Figure 3.
Representative flow cytometry classic CLL cells showed positive expression of CD5, CD19 and CD23.
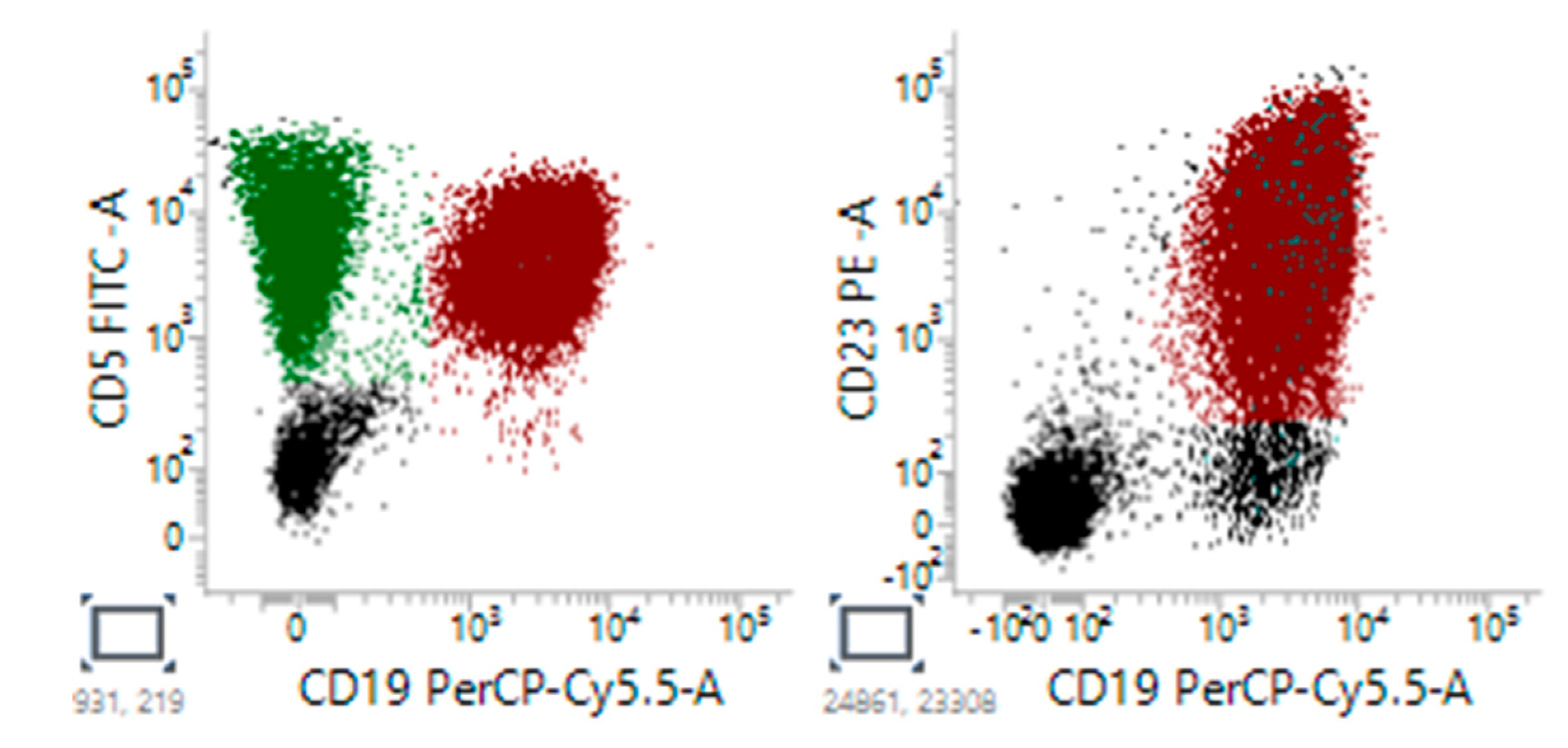
Figure 4.
Representative flow cytomety of atypical, CD5 negative CLL cells showed positive expression of CD19, CD23, and no expression of CD5.
Figure 4.
Representative flow cytomety of atypical, CD5 negative CLL cells showed positive expression of CD19, CD23, and no expression of CD5.
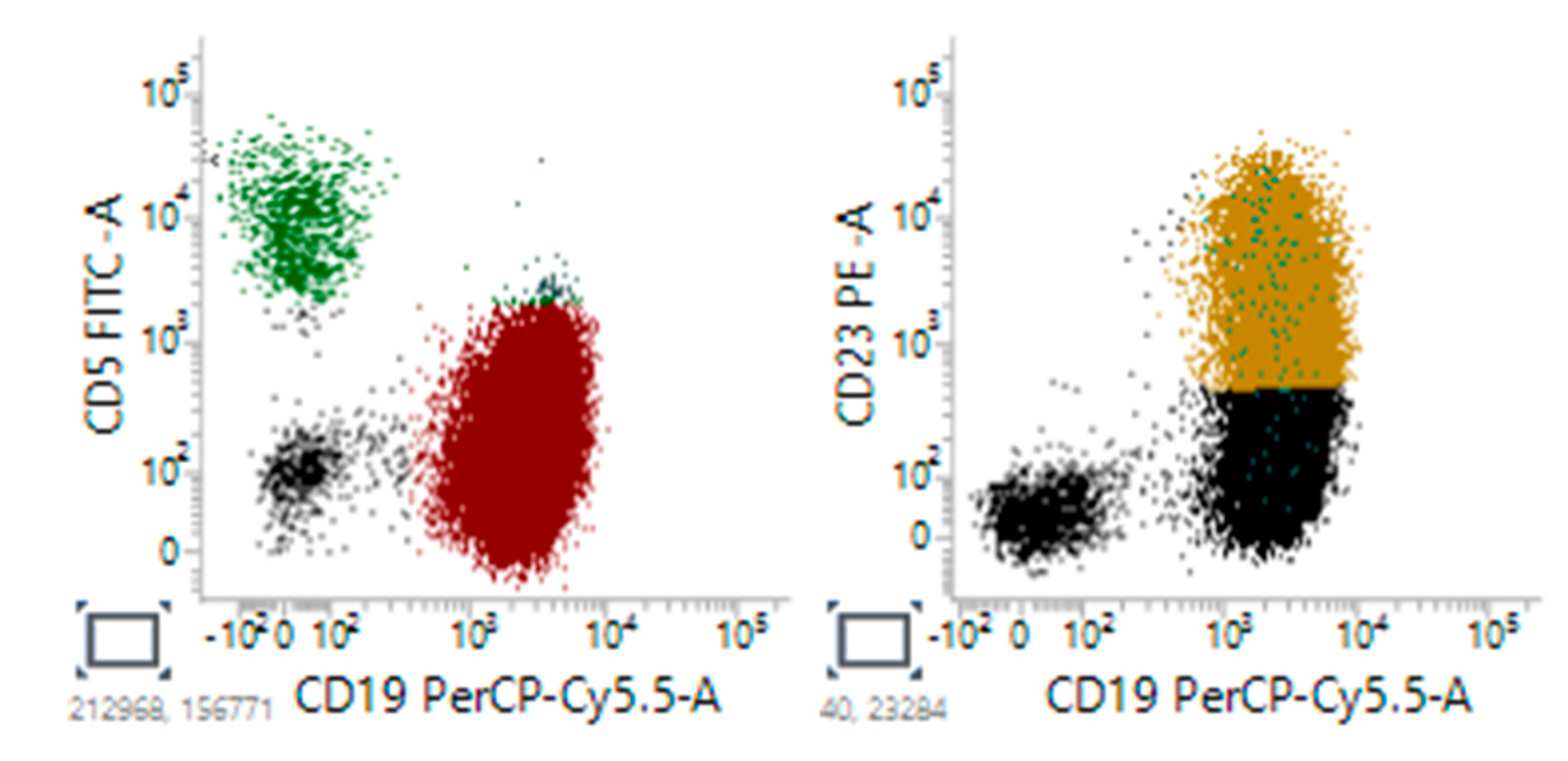
Figure 5.
Representative flow cytomety CLL patient showed positive expression of CD5 and CD19, and negative expression of CD23.
Figure 5.
Representative flow cytomety CLL patient showed positive expression of CD5 and CD19, and negative expression of CD23.
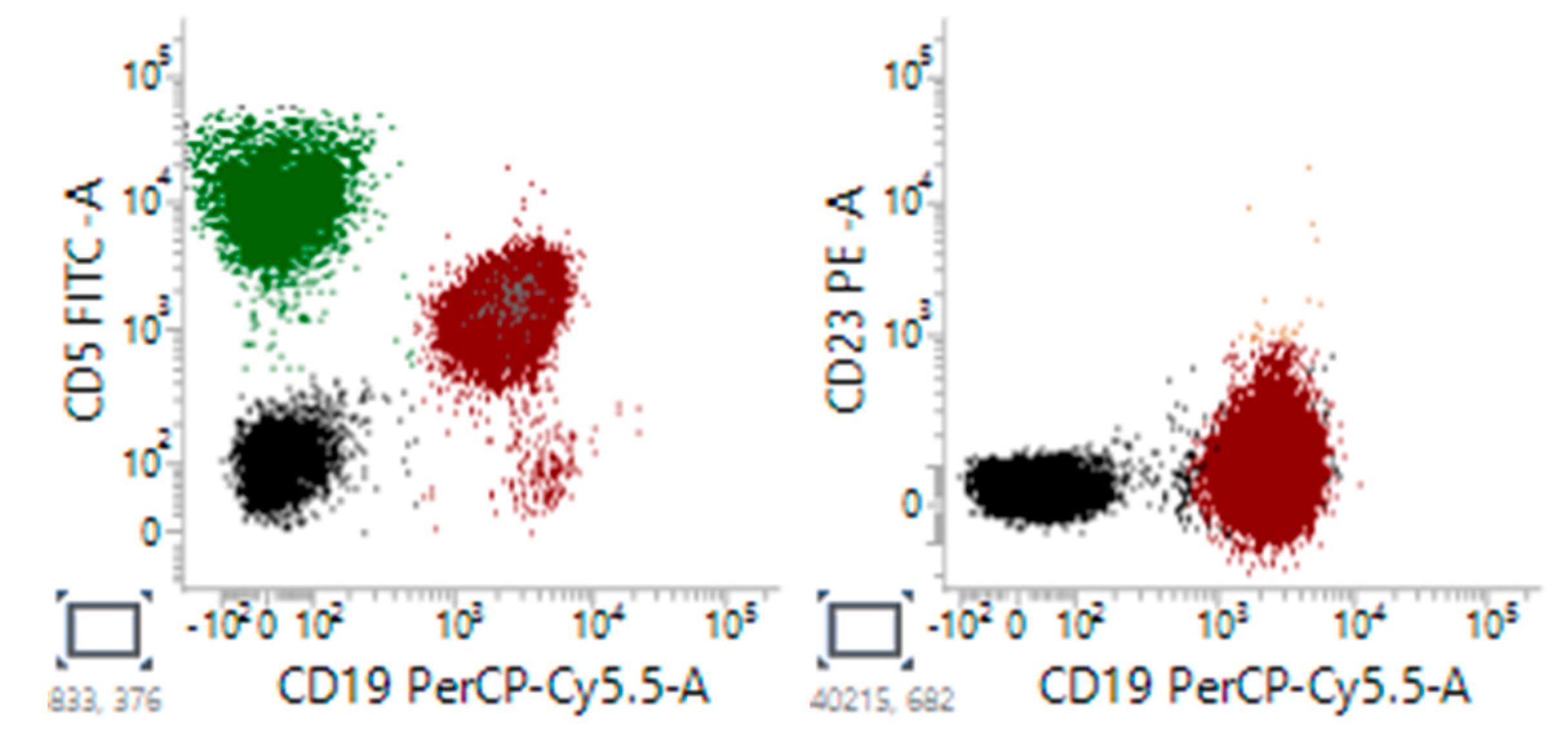

Table 1.
Larger studies comparing patients with CD5 negative and CLL positive CLL.
| Authors/ Reference |
No of pts CD5- vs CD5+ | Median age: CD5- vs CD5+ | Definition of CD5- CLL | PB CD5- vs CD5+ |
Treatment CD5- vs CD5+ |
Survival CD5- vs CD5+ |
Commentary |
|---|---|---|---|---|---|---|---|
| Cartron et al 1998 [42] | 42 vs 79 | 68/64.8 | <5% of mononuclear cells | Hb: 126/137 G/L(p=ns) PLT: 216/200 x 109/L (p=ns) Lymphocytes:27.3/32,7 x 109/L x 109/L (p=ns) |
No initially treated:64.3% vs 29.1% | 52% at 120 m vs 6% at 90 m (p=0.97) | CD5- CLL expressed a higher level of surface of immunoglobulin and had more frequently isolated splenomegaly. |
| Efstathiou et al 2002 [43] | 29 vs 29 | 68.8/68.4 | <5% of mononuclear cells | Hb: 131/10.5 G/L(p=ns) (p<0.05) PLT: 211/198 x 109/L(p=ns) Lymphocytes:38.2/39.6 x 109/L (p=ns) |
No initially treated:72.4% vs 24.1% | Median:97.2m vs 84.0m (p=0.0025) |
Splenomegaly, lymph node involvement, and haemolytic anemia less common in CD5- CLL. CD5- CLL patients had a more favourable prognosis compared with CD5+ patients |
| Demir et al. 2017 [44] | 19 vs 105 | 65.8/66.5 | <20% of mononuclear cells | HB: 133/127g/L (p=0.180) PLT: 144/ 160×6 x 109/L (p=0.044) Neutrophils: 3.5/3.36 x 109/L (p=0.169) Lymphocytes: 43.2/ 36.7 x 109/L (p=0.001). |
NR | 84.2% vs 90.5% at 5 yr (p=0.393) | Lymphadenopathy less frequent in CD5- (p=0.029). Splenomegaly more frequent in CD5- (p=0.029). No difference in clinical stage, autoimmune phenomena, hemo- globin and neutrophil count, and survival |
| Kurec et al 1992 [46] | 12 vs 27 | 66/67 | <20% of lymphoid cells | Hb: 11.2/13.7g/L PLT:172/175 x 109/L WBC:88 x 109/L /60 x109/L |
NR | 55% vs 90 % at 5 yr | Lack of CD5 antigen was with more advanced stage of disease and poor patient survival. |
Abbreviations: CLL – chronic lymphocytic leukemia;Hb-hemoglobin; ND -not differ; NR – not reported; PLT – platelets; WBC – white biood cells.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



