Submitted:
20 July 2023
Posted:
20 July 2023
You are already at the latest version
Abstract
Nonalcoholic fatty liver disease (NAFLD) is a liver metabolism-associated steatohepatitis caused by non-alcoholic factors. NAFLD is currently the most prevalent liver disease in the world, affecting one-fourth of the world's population, and its prevalence increases with age. There are no approved drugs specifically for the treatment of NAFLD, and one important reason hindering drug development is the lack of effective biomarkers. C-reactive protein (CRP), a marker of inflammation, has been linked to NAFLD and aging in recent studies. Coincidentally, hepatocytes are responsible for the major CRP production, and the levels of CRP increase with age. Therefore, CRP is not only a potential marker, but also acts as a key factor driving liver aging and NAFLD. Herein, we reviewed the biological function and production mechanism of CRP and the relationship between CRP and NAFLD. We also comprehensively described the potential molecular mechanisms of CRP-mediated signaling in aging-associated NAFLD. Finally, we proposed possible therapeutic approaches based on the mechanism of CRP signaling in the pathogenesis of NAFLD. We hope this study can provide new insights into the development of aging associated NAFLD biomarkers and suggest that modulation of CRP signaling is a potential therapeutic target.
Keywords:
NAFLD
; C-reactive Protein
; Aging
1. Introduction
The proportion of the global elderly population is gradually increasing[1]. The prevalence of chronic diseases, disabilities, and geriatric syndromes increases with age, leading to higher demand for wellness and long-term care services among older adults. A common feature of senescence at the cellular level is the decreased ability of cells to resume proliferation, which ultimately leads to dysfunction of organs and individuals[2]. Mammals undergo changes such as diminished physical activity, metabolic and cognitive capacity, impaired digestive function, and increased DNA damage[3].
As the largest metabolic organ in the body, the liver plays a key role in lipid synthesis and storage. However, liver disease is widespread, with non-alcoholic fatty liver disease (NAFLD) being the most common. NAFLD refers to a condition of excessive lipid deposition in the liver in the absence of significant alcohol intake[4]. However, no drug treatment is approved for NAFLD and NASH, and the main therapy is lifestyle modification, which is difficult to sustain[5]. The scarcity of reliable biomarkers and non-invasive diagnostics for assessing disease progression is a challenge for NAFLD and NASH drug development[6].
Aging is a major risk factor for NAFLD, evidenced by the rising prevalence of NAFLD with older age[7,8]. The main mechanisms of NAFLD pathogenesis include decreased fatty acid oxidation, increased de novo lipogenesis, disrupted lipid export through VLDL secretion, and augmented oxidative stress and inflammation susceptibility in the liver[9]. Extensive research has been conducted to identify biomarkers related to aging and NAFLD, and some biomarkers with correlations have been detected[10,11,12]. C-reactive protein (CRP), an indicator of inflammation, serves as a potential biomarker for both aging and NAFLD[13,14,15,16]. Furthermore, CRP is mainly synthesized by liver hepatocytes, suggesting a direct role of CRP in NAFLD[17].
In summary, CRP has clinical relevance with NAFLD, and the treatment for NAFLD based on the pathogenic mechanism of CRP is promising. Therefore, this review specifically focuses on the direct and the indirect regulation of aging associated NAFLD by CRP and provides reference for future research and treatment.
2. Aging is A Risk Factor for NAFLD
2.1. Aging and Liver Aging
Senescence is the process of biological aging, where cells gradually lose their ability to divide and function normally[18]. Senescence is characterized by several hallmarks, such as changes in gene expression and epigenetics, production of reactive oxygen species, secretion of pro-inflammatory factors, and alterations in metabolism[19]. These hallmarks contribute to the senescent phenotype and affect the surrounding cells and tissues. The liver’s unique ability to regenerate allows it to recover from surgical or chemical damage[20]. Interestingly, a recent study used retrospective radiocarbon (14C) birth dating of cells to determine the age of human liver samples and find that hepatocytes show continuous and lifelong turnover, allowing the liver to remain a young organ, which emphasizes the powerful self-renewal capacity of liver[21]. However, the living environment is not ideal and factors that come from outside the body. Pathogenic microorganisms, radiation damage and chemical contamination accelerate the aging of the liver and the dysfunction of liver is a common phenomenon in aging[22]. Liver aging attributes to the accumulation of senescent hepatocytes, where the number of hepatocytes decreases, the regenerative capacity of the liver impairs, and polyploid hepatocytes accumulate[23].
2.2. Aging-associated Impaired Lipid Metabolism and NAFLD
Aging leads to increased triglyceride levels, decreased ability to utilize and breakdown triglyceride (Figure 1) [24]. A population study showed that the incidence of fatty liver increased significantly from the age less than 30 to more than 60[25]. Another study pointed out that presence of diabetes, hypertension and triglycerides ≥150 mg/dl were associated with NAFLD in older adults[26]. Soberingly, a study showed that 88% of older adults with NAFLD had normal ALT levels, and nearly a quarter of older NAFLD patients were non-obese, which may lead to inaccurate diagnosis of NAFLD. For patients already diagnosed with NAFLD, aged people have a higher tendency for advanced fibrosis than younger people, and fibrosis may contribute to the lower degree of steatosis in patients by limiting fat accumulation[27]. Similar to the results of human studies, 22-month-old mice gained weight accompanied by lipid accumulation and glucose intolerance compared to 3-month-old mice[28]. Another rodent-based study compared high-fat, high-sugar, and high-cholesterol diets to model NAFLD in young and aged mice[29]. Their results showed that aged mice developed NASH, including hepatic steatosis, lobular inflammation, and hepatic ballooning, with a more severe phenotype than younger mice. A study using aged mice and reactive oxygen species (ROS) modeling of aged HepG2 cells (a hepatocellular carcinoma cell line) revealed increased lipid accumulation and increased cholesterol and glucose uptake in aged livers[30]. To examine the direct effect of cellular senescence on hepatic steatosis, a study of middle-aged mice and human liver biopsies showed that the accumulation of senescent cells induced hepatic lipid accumulation and steatosis[31]. Moreover, they also revealed that the removal of these senescent hepatocytes reversed the progression of steatohepatitis.
Taken together, current research supports that aging leads to increased incidence and poorer prognosis of NAFLD. Aging-induced changes in the liver include direct effects of reduced lipid metabolism and indirect effects through decreased clearance in senescent cells. Both directly or indirectly, aging and NAFLD are associated with the generation of oxidative stress and inflammation, which is to be discussed in detail in the next section.
3. CRP is A Potential Biomarker for Aging associated NAFLD
3.1. CRP definition, expression regulation and function
In 1930, Tillett and Francis identified a serum protein that formed a precipitate with the C-polysaccharide of pneumococcus and they called it C-reactive protein (CRP)[32]. CRP was originally identified as an acute phase protein because a rapid increase of CRP is found at sites of infection or inflammation.
Hepatocytes are the main parenchymal cells in the liver, making up about 80% of total liver cells[33]. Hepatocytes participate in liver immunity as antigen-presenting cells and activate innate immunity through secreting immune molecules such as CRP[34,35]. The production of CRP by hepatocytes is mainly regulated by cytokines, especially interleukin-6 (IL-6)[36,37]. IL-6 binds to IL-6 receptor on hepatocytes and activates the JAK/STAT signaling pathway, which leads to the transcription of the CRP gene. IL-6 can also induce the expression of CCAAT/enhancer-binding protein β (C/EBPβ), a transcription factor that binds to the CRP promoter and enhances its activity[38]. Interleukin-1β (IL-1β) can enhance the effect of IL-6 on CRP synthesis by enhancing the expression of IL-6 receptor and C/EBPβ[39]. Interleukin-10 (IL-10) can inhibit the CRP-induced tissue factor genes expression in peripheral blood mononuclear cells[40]. CRP mRNA can be translated into a polypeptide chain of 206 amino acids, which contains a signal peptide of 18 amino acids at the N-terminus[41]. CRP then undergoes processing and folding in the endoplasmic reticulum and moves to the Golgi apparatus, where it is sorted into vesicles and secreted into the blood circulation[42].
CRP is a pentameric protein (pCRP) belonging to the pentraxin family of proteins, which have a characteristic ring-shaped structure composed of five identical monomeric human CRP (mCRP)[43]. Each subunit has a molecular weight of about 23 kDa and contains a phosphocholine (PCh)-binding site that can bind to certain molecules on the surface of dead or dying cells and some types of bacteria[44]. In human inflammatory tissues, pCRP undergoes structural changes through association with cell-produced vesicles, but still maintains the structural symmetry of the pentamer called pCRP*-microvesicle complexes, which enhance immune cell recruitment to the site of inflammation[45]. In recent decades, CRP was recognized as an acute-phase reactant that increased in response to various inflammatory stimuli, such as infections, injuries, and autoimmune diseases[46]. CRP binds to phosphocholine moieties on microbial and necrotic cells, initiates the complement cascade, engages Fc receptors to induce pro-inflammatory cytokine secretion, and regulates apoptosis, phagocytosis, nitric oxide synthesis, and coagulation[47,48]. CRP activates macrophages to secrete tissue factor, which plays a procoagulant function[49]. Tissue factor contributes to disseminated intravascular coagulation and thrombus formation under inflammatory conditions. CRP also increases the uptake of low-density lipoprotein (LDL) by macrophages and the ability to form foam cells[50]. Evidence also suggest that CRP may also play an anti-inflammatory role. CRP binding to histone H4 can be a protective response to excessive inflammation and downregulates the neutrophil respiratory burst response[51]. CRP also reduced the excessive activation of complement to prevent drug-induced liver damage in mice[52]. Therefore, CRP should be viewed dialectically, and further mechanistic studies are needed.
Recently, high-sensitivity C-reactive protein (hs-CRP) is proposed as an independent clinical feature of the severity of NASH and the fibrosis caused by NASH[53]. Coincidentally, the close relationship between CRP and aging was recognized earlier[54,55]. Increased CRP was associated with an increased risk of adverse aging outcomes in a large, nationally representative follow-up of older adults in England for up to 10 years[16]. A study of 6060 healthy people showed that serum hs-CRP increased significantly with age and people over 45 years old was one of the risk factors[56]. It is worth mentioning that with increasing age and development of NAFLD, an increase in CRP strongly predicted poor prognosis[57,58].
Since CRP is produced by mainly hepatocytes, which allows CRP to directly cause liver inflammation. Therefore, it is reasonable to speculate that CRP increases with age to be an important biomarker and driver of aging-associated NAFLD. Although the mechanism is not fully understood, insulin signaling, mitochondrial metabolism, and NF-κB pathway inclusive of CRP are the potential factors that modulate aging-associated NAFLD (Figure 2).
3.2. CRP, Insulin signaling pathway and aging-associated NAFLD
Insulin plays an important role in metabolic syndrome of aging and defects in the regulation of insulin signaling lead to a variety of metabolic diseases including NAFLD[59,60]. Elevated CRP is associated with insulin resistance[61]. Insulin resistance and CRP have been used together as indicators of NAFLD[62]. CRP has shown to inhibit insulin signaling through Fcγ, and genetic elimination of CRP confers resistance to obesity and insulin resistance in rats[63,64]. Insulin-like growth factor 1 (IGF-1) is shown to be inversely correlated with CRP in previous studies[65,66,67]. IGF-1 mRNA levels in the liver and brain decline with age[68]. Low serum IGF-1 levels are associated with increased histologic severity of NAFLD in a diagnosis-based study[69]. Reductions in IGF-1 and IGF-binding protein were associated with increased NAFLD severity[70]. Insulin initiates phosphorylation events that activate phosphoinositide 3-kinase (PI3K), a lipid kinase that coordinates glucose uptake and utilization[71]. IGF-1 was proved to block CRP by activating the PI3K/Akt pathway and inhibiting the JNK/c-Jun signaling pathways [72]. Therefore, an antagonistic relationship between CRP and IGF-1 needs further verifications.
AMP-activated protein kinase (AMPK) signaling suppresses insulin resistance, promotes glucose metabolism and uptake, and has been shown to alleviate diabetes[73]. In NAFLD treatment, AMPK inhibits fatty acid synthesis by downregulating the expression of fatty acid production genes and increases the expression of genes involved in fatty acid oxidation[74]. Studies in obese mice also proved that hepatic AMPK activation is effective in preventing steatosis and inflammation [75]. However, high levels of CRP inhibits AMPK signaling[76], and AMPK activation decreases with age, causing impaired metabolism, increased oxidative stress and reduced autophagic clearance[77]. Therefore, in addition to directly affecting the insulin signaling pathway, CRP can also indirectly reduce the function of insulin by inhibiting AMPK signaling.
The hypothesis that CRP affects NAFLD by affecting the insulin pathway was further substantiated in the mammalian target of rapamycin (mTOR), one of the downstream of IGF-1 and AMPK. mTOR regulates cell growth and metabolism in response to nutrients, growth factors and cellular energy[78]. mTOR has been identified as a central node in a network regulating hepatic lipid metabolism, but its specific impact on NAFLD is unclear[79]. A mouse model of spontaneous HCC reveals that mTOR crosstalk with STAT5 promotes de novo lipid synthesis and induces HCC[80]. Rapamycin inhibitors, which block mTOR signaling, have been used to treat NAFLD-associated HCC and other cancers[81]. A study using CRP transgenic mice have shown that CRP activates Smad3/mTOR signaling through TGF-β and ERK/mitogen-activated protein kinase (MAPK)[82]. Collectively, CRP shows inhibitory effects on the insulin signaling pathway, and increased CRP with age is a potential biomarker and cause of insulin resistance and NAFLD.
3.3. CRP, mitochondria dysfunction and aging-associated NAFLD
Mitochondria are the factories of cellular energy synthesis and oxidative phosphorylation. Insulin resistance underlies the development of type 2 diabetes mellitus (T2DM), where mitochondrial dysfunction is one of the main mechanisms[83]. Patients with T2DM are prone to develop NAFLD and NASH[84]. Reactive oxygen species (ROS) are byproducts of mitochondrial aerobic respiration, causing oxidative damage to DNA, proteins and lipids[85]. Cellular senescence leads to decreased mitochondrial ATP synthesis and ROS accumulation, which leads to an increase in ROS with age[86]. ROS damage mitochondrial membranes, leading to MPTP formation and subsequent release of mtDNA as danger-associated molecular patterns (DAMPs), which activate the NLRP3 inflammasome to trigger inflammation through toll like receptor (TLR) signaling [87]. CRP upregulates ROS production in target cells through Fcγ receptors[88]. CRP enhances ROS formation and innate killer mechanism phagocytosis in a complement-dependent manner in patients with sepsis[88]. Radiation-induced CRP proved to interact with the STAT3/Ref-1 complex through ROS, resulting in further mitochondrial dysfunction[89]. ROS overproduction suppresses the ability of antioxidant defense systems in NAFLD, leading to further oxidative damage[90]. Low mitochondrial DNA (mtDNA), important for cellular homeostasis, was associated with higher CRP levels in healthy adults[91]. Another population-based study also showed that participants with higher levels of CRP and IL-6 had lower blood mtDNA levels[92]. ROS-induced mitochondrial damage is an important cause of mitochondrial DNA release, exacerbating mitochondrial dysfunction and chronic inflammation[93].
3.4. CRP, NF-κB pathway and aging-associated NAFLD
Nuclear factor kappa-light-chain-enhancer of activated B cells(NF-κB) acts as a transcription factor that regulates multiple aspects of immune function and plays a central role in inflammatory responses[94]. NF-κB transcription increases with age and is associated with age-related degenerative diseases, and pathways such as insulin/IGF-1 FoxO and mTOR are all correlated with NF-κB signaling[95]. Activation of NF-κB promotes the expression of inflammatory cytokines, such as TNF-α, IL-1 and IL-6, which in turn enhance NF-κB activity[96]. NF-κB activation is shown to be increased in the liver of NAFLD patients and has a central role in the regulation of hepatitis fibrosis and carcinogenesis[97]. CRP activates the NF-κB signaling pathway in endothelial cells through the degradation of IκB-α[98]. Studies in ARPE-19 cells revealed that CRP mediates IL-8 production and triggers inflammation via Fcγ/NF-kB and MAPK pathways[99]. NF-kB is critical for intracellular signaling induced by CRP pro-inflammatory and catabolic mediators[100]. NF-κB activation and CRP expression are region-specific in response to endotoxemia, and the NF-κB transcription factor subunit p50 interacts with consensus sequence elements of CRP promoter, which implies a cyclic relationship between CRP and NF-κB [101].
4. CRP as a potential therapeutic target for aging associated NAFLD (Table 1)

Table 1.
CRP-lowering therapy for aging associated NAFLD.
| CRP-lowering Strategies | Drugs/Methods | Mechanisms | Effectiveness | Limitations |
|---|---|---|---|---|
| Caloric restriction and Antioxidation | MedDiet, Dietary restriction, Astaxanthin and β-Cryptoxanthin |
Energy intake reduction, Anti-oxidative stress, Inflammatory factors reduction |
Weight Loss, Increased Lifespan, Reduced risk of NAFLD |
lacking long-term clinical evidence, Requiring long-time maintenance |
| CRP-lowering drugs | Pioglitazone, Statins, Vitamin E, MitoQ |
PPARγ agonists, HMG CoA reductase inhibitors, Anti-oxidation |
Reducing steatosis, inflammation and fibrosis, Anti-inflammation |
Drug inefficiency, Nonspecific binding, Immunosuppression |
| CPR Adsorption Technology | CPR Apheresis | Using adsorbent specifically binding CRP in serum | Effectively reducing CRP acute infection, Recyclable and no side effects |
Safety and efficacy evaluations needed in the treatment of NAFLD |
4.1. Caloric restriction and Antioxidation
Excessive intake of lipids and other energy sources are underlying factors of NAFLD[102]. Numerous studies used dietary restriction to reduce the absorption of lipid[103]. In the 1930s, Clive McCay made the astonishing discovery that mice fed a strictly restricted diet lived 33% longer than previously known[104]. A diet that is restrictive but maintains basic nutrients has been proved to be effective in extending the lifespan of some organisms[105,106]. Whether dietary restriction is effective in human beings needs further investigations, although reduced energy intake has been reported to be beneficial in alleviating inflammation and chronic disease[107]. The Traditional Mediterranean Diet (MedDiet), advocating less carbohydrate intake and more antioxidant polyphenols, has shown to be effective in reducing hs-CRP concentrations by 24%, IL-6 concentrations by 16%, and E-selectin concentrations by 13%[108]. Evidence also supports that better adherence to the MedDiet, but not others, is strongly associated with a lower risk of major cardiovascular disease[109]. Dietary restriction also leads to significant reductions in body weight and CRP, especially in severely obese individuals[110]. Astaxanthin and β-Cryptoxanthin, two strong antioxidants belonging to carotenoids, has been proved to treat NAFLD and aging by resisting oxidative stress, inhibiting inflammation, and promoting M2 macrophage polarization[111]. A meta-analysis showed that more astaxanthin intake is associated with a decrease in CRP, and no significant associations were observed for other indicators[112]. A study of the link between vitamins and CRP found that only carotene is inversely associated with increased CRP level[113].
4.2. CRP-lowering drugs for NAFLD treatment
Although there is no specific drug for the treatment of NAFLD, some potential NAFLD drugs have been shown to reduce CRP. Pioglitazone is a class of PPARγ agonists that alleviate NAFLD by reducing insulin resistance[114]. Early studies have demonstrated the efficacy of pioglitazone in reducing CRP, which may be mediated through PPARγ-NF-κB signaling[115,116]. As a class of HMG CoA reductase inhibitors, statins are widely used lipid-lowering drugs[117]. Statins reduce the production of IL-6 and reduce the number of LDL particles, thereby reducing the production of inflammatory mediators produced by CRP[118]. Limited cross-sectional studies show that statins are effective in reducing steatosis, inflammation and fibrosis in NAFLD or NASH[117]. Vitamin E, either in the alpha-tocopherol or gamma-tocopherol form, has been shown to lower serum CRP levels[119]. The mechanism of vitamin E in anti-inflammation and reducing CRP production is also mediated through PPARγ[120]. Vitamin E improves biochemical parameters in NASH patients with histological abnormalities by inhibiting inflammation and reducing ROS[121]. Furthermore, mitochondrial quinone (MitoQ), a mitochondria-targeted antioxidant, can reduce ROS production through the action of NF-κB and lower the risk of cardiovascular disease in type 2 diabetes[122].
4.3. CPR Adsorption Technology
A more efficient way to reduce CRP is needed, due to the inefficiency to reduce through diet and habits. Pharmaceutical companies have tried a variety of approaches targeting CRP lowering, including: inhibition of CRP synthesis, CRP receptor antagonists, and complement activation inhibitors. Pharmaceutical companies have tried a variety of approaches targeting CRP, including CRP synthesis inhibitors, CRP receptor antagonists, and complement activation inhibitors[123]. However, these methods are limited by low efficiency and side effects caused by non-specific binding. An exciting technology has been published and put into use, that is, CRP Apheresis[124]. This technology uses a novel adsorbent (PentraSorb CRP) for CRP apheresis in human plasma, specifically binding CRP in human plasma and can regenerate up to 200 times without losing its binding ability or affect biocompatibility[124]. Although there are no examples of CRP apheresis applied to the treatment of NAFLD, it has been used in the treatment of other acute diseases. A clinical study based on myocardial infarction patients showed that CRP concentration can be effectively reduced by CRP apheresis, and no side effects were observed during the 12-month operation[125]. CRP apheresis used in acute infections including sepsis and coronavirus showed to be effective in reducing CRP in plasma, as well[126,127]. After further confirmation of safety and necessity evaluation, CRP apheresis is worthy of becoming one of the potential methods for the treatment of NAFLD and other chronic diseases.
5. Conclusions
In summary, as an indicator of inflammation that increases with age, CRP can contribute to the development of aging-associated NAFLD by mediating insulin signaling, mitochondrial metabolism, and NF-κB signaling. From growth and development, metabolism and apoptosis, CRP is so "faithful" that CRP in different conditions are mostly consistent, making it an important bridge connecting chronic inflammation, aging and NAFLD. Treatment of NAFLD by lowering CRP is promising, but current strategies are limited. This review is the first to propose CRP as both a biomarker and regulator for aging-associated NAFLD. Future research is needed to explore the deeper mechanism of CRP in aging-associated NAFLD, and develop new drugs or treatments based on CRP signaling.
Author Contributions
Conceptualization, Z.D. and J.S.; methodology, Z.D.; investigation, Z.D.; writing—original draft preparation, Z.D.; writing—review and editing, Z.D., J.S., J.P., Y.W., S.W. and G.C.; visualization, Z.D.; supervision, J.S.; project administration, J.S.; funding acquisition, J.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- He, W.; Goodkind, D.; Kowal, P. R., An aging world: 2015. United States Census Bureau Washington, DC: 2016.
- López-Otín, C.; Blasco, M. A.; Partridge, L.; Serrano, M.; Kroemer, G. , The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M. A.; Partridge, L.; Serrano, M.; Kroemer, G. , Hallmarks of aging: An expanding universe. Cell 2023. [Google Scholar] [CrossRef] [PubMed]
- Lazarus, J. V.; Mark, H. E.; Anstee, Q. M.; Arab, J. P.; Batterham, R. L.; Castera, L.; Cortez-Pinto, H.; Crespo, J.; Cusi, K.; Dirac, M. A. , Advancing the global public health agenda for NAFLD: a consensus statement. Nature Reviews Gastroenterology & Hepatology 2022, 19, 60–78. [Google Scholar]
- Harvey, B. E. , NASH: regulatory considerations for clinical drug development and US FDA approval. Acta pharmacologica Sinica 2022, 43, 1210–1214. [Google Scholar] [CrossRef] [PubMed]
- Nassir, F. , NAFLD: Mechanisms, treatments, and biomarkers. Biomolecules 2022, 12, 824. [Google Scholar] [CrossRef]
- Hu, X.; Huang, Y.; Bao, Z.; Wang, Y.; Shi, D.; Liu, F.; Gao, Z.; Yu, X. , Prevalence and factors associated with nonalcoholic fatty liver disease in Shanghai work-units. BMC gastroenterology 2012, 12, 1–9. [Google Scholar] [CrossRef]
- Li, W.; Ng, C. H.; Quek, J.; Chan, K. E.; Tan, C.; Zeng, R. W.; Yong, J. N.; Tay, H.; Tan, D. J. H.; Lim, W. H. , The growing prevalence of nonalcoholic fatty liver disease (NAFLD), determined by fatty liver index, amongst young adults in the United States. A 20-year experience. Metabol Target Organ Damage 2022, 2, 19. [Google Scholar] [CrossRef]
- Alqahtani, S. A.; Schattenberg, J. M. , NAFLD in the Elderly. Clinical interventions in aging 2021, 1633–1649. [Google Scholar] [CrossRef]
- Drescher, H. K.; Weiskirchen, S.; Weiskirchen, R. , Current status in testing for nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH). Cells 2019, 8, 845. [Google Scholar] [CrossRef]
- Green, S.; Hillersdal, L. , Aging biomarkers and the measurement of health and risk. History and Philosophy of the Life Sciences 2021, 43, 1–23. [Google Scholar] [CrossRef]
- Galkin, F.; Mamoshina, P.; Aliper, A.; de Magalhães, J. P.; Gladyshev, V. N.; Zhavoronkov, A. , Biohorology and biomarkers of aging: Current state-of-the-art, challenges and opportunities. Ageing Research Reviews 2020, 60, 101050. [Google Scholar] [CrossRef] [PubMed]
- Oruc, N.; Ozutemiz, O.; Yuce, G.; Akarca, U. S.; Ersoz, G.; Gunsar, F.; Batur, Y. , Serum procalcitonin and CRP levels in non-alcoholic fatty liver disease: a case control study. BMC gastroenterology 2009, 9, 1–5. [Google Scholar] [CrossRef]
- Kumar, R.; Porwal, Y. C.; Dev, N.; Kumar, P.; Chakravarthy, S.; Kumawat, A. , Association of high-sensitivity C-reactive protein (hs-CRP) with non-alcoholic fatty liver disease (NAFLD) in Asian Indians: A cross-sectional study. Journal of Family Medicine and Primary Care 2020, 9, 390. [Google Scholar]
- Tang, Y.; Fung, E.; Xu, A.; Lan, H. Y. , C-reactive protein and ageing. Clinical and Experimental Pharmacology and Physiology 2017, 44, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Lassale, C.; Batty, G. D.; Steptoe, A.; Cadar, D.; Akbaraly, T. N.; Kivimäki, M.; Zaninotto, P. , Association of 10-year C-reactive protein trajectories with markers of healthy aging: findings from the English longitudinal study of aging. The Journals of Gerontology: Series A 2019, 74, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Sproston, N. R.; Ashworth, J. J. , Role of C-reactive protein at sites of inflammation and infection. Frontiers in immunology 2018, 9, 754. [Google Scholar] [CrossRef] [PubMed]
- Sławińska, N.; Krupa, R. , Molecular aspects of senescence and organismal ageing—DNA damage response, telomeres, inflammation and chromatin. International journal of molecular sciences 2021, 22, 590. [Google Scholar] [CrossRef]
- Dodig, S.; Čepelak, I.; Pavić, I. , Hallmarks of senescence and aging. Biochemia medica 2019, 29, 483–497. [Google Scholar] [CrossRef]
- Michalopoulos, G. K., Liver regeneration. The Liver: Biology and Pathobiology 2020, 566-584.
- Heinke, P.; Rost, F.; Rode, J.; Trus, P.; Simonova, I.; Lázár, E.; Feddema, J.; Welsch, T.; Alkass, K.; Salehpour, M. , Diploid hepatocytes drive physiological liver renewal in adult humans. Cell Systems 2022, 13, 499–507. [Google Scholar] [CrossRef]
- Cieslak, K. P.; Baur, O.; Verheij, J.; Bennink, R. J.; van Gulik, T. M. , Liver function declines with increased age. Hpb 2016, 18, 691–696. [Google Scholar] [CrossRef]
- Zhao, Y.; Yang, Y.; Li, Q.; Li, J. , Understanding the unique microenvironment in the aging liver. Frontiers in Medicine 2022, 9. [Google Scholar] [CrossRef]
- Chung, K. W. , Advances in understanding of the role of lipid metabolism in aging. Cells 2021, 10, 880. [Google Scholar] [CrossRef]
- Wong, V. W.-S.; Wong, G. L.-H.; Woo, J.; Abrigo, J. M.; Chan, C. K.-M.; Shu, S. S.-T.; Leung, J. K.-Y.; Chim, A. M.-L.; Kong, A. P.-S.; Lui, G. C.-Y. , Impact of the new definition of metabolic associated fatty liver disease on the epidemiology of the disease. Clinical Gastroenterology and Hepatology 2021, 19, 2161–2171. [Google Scholar] [CrossRef] [PubMed]
- Koehler, E. M.; Schouten, J. N. L.; Hansen, B. E.; van Rooij, F. J. A.; Hofman, A.; Stricker, B. H.; Janssen, H. L. A. , Prevalence and risk factors of non-alcoholic fatty liver disease in the elderly: results from the Rotterdam study. Journal of hepatology 2012, 57, 1305–1311. [Google Scholar] [CrossRef] [PubMed]
- Noureddin, M.; Yates, K. P.; Vaughn, I. A.; Neuschwander-Tetri, B. A.; Sanyal, A. J.; McCullough, A.; Merriman, R.; Hameed, B.; Doo, E.; Kleiner, D. E. , Clinical and histological determinants of nonalcoholic steatohepatitis and advanced fibrosis in elderly patients. Hepatology 2013, 58, 1644–1654. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R. H.; Argmann, C.; Houten, S. M.; Cantó, C.; Jeninga, E. H.; Andreux, P. A.; Thomas, C.; Doenlen, R.; Schoonjans, K.; Auwerx, J. , The metabolic footprint of aging in mice. Sci Rep 2011, 1, 134. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lu, Y.; Liang, X.; Zhou, X.; Li, D.; Zhang, Z.; Niu, Y.; Liu, S.; Ye, L.; Zhang, R. , A new NASH model in aged mice with rapid progression of steatohepatitis and fibrosis. Plos one 2023, 18, e0286257. [Google Scholar] [CrossRef]
- Seo, E.; Kang, H.; Choi, H.; Choi, W.; Jun, H. S. , Reactive oxygen species-induced changes in glucose and lipid metabolism contribute to the accumulation of cholesterol in the liver during aging. Aging Cell 2019, 18, e12895. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Miwa, S.; Tchkonia, T.; Tiniakos, D.; Wilson, C. L.; Lahat, A.; Day, C. P.; Burt, A.; Palmer, A.; Anstee, Q. M. , Cellular senescence drives age-dependent hepatic steatosis. Nature communications 2017, 8, 15691. [Google Scholar] [CrossRef]
- Tillett, W. S.; Francis Jr, T. , Serological reactions in pneumonia with a non-protein somatic fraction of pneumococcus. The Journal of experimental medicine 1930, 52, 561. [Google Scholar] [CrossRef]
- Schulze, R. J.; Schott, M. B.; Casey, C. A.; Tuma, P. L.; McNiven, M. A. , The cell biology of the hepatocyte: A membrane trafficking machine. Journal of Cell Biology 2019, 218, 2096–2112. [Google Scholar] [CrossRef] [PubMed]
- Knolle, P. A. , Staying local—Antigen presentation in the liver. Current opinion in immunology 2016, 40, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Xu, M.-J.; Gao, B. , Hepatocytes: a key cell type for innate immunity. Cellular & molecular immunology 2016, 13, 301–315. [Google Scholar]
- Zhang, D.; Sun, M.; Samols, D.; Kushner, I. , STAT3 Participates in Transcriptional Activation of the C-reactive Protein Gene by Interleukin-6 (∗). Journal of Biological Chemistry 1996, 271, 9503–9509. [Google Scholar] [CrossRef]
- Ngwa, D. N.; Pathak, A.; Agrawal, A. , IL-6 regulates induction of C-reactive protein gene expression by activating STAT3 isoforms. Molecular immunology 2022, 146, 50–56. [Google Scholar] [CrossRef]
- Agrawal, A.; Cha-Molstad, H.; Samols, D.; Kushner, I. , Transactivation of C-reactive protein by IL-6 requires synergistic interaction of CCAAT/enhancer binding protein β (C/EBPβ) and Rel p50. The Journal of Immunology 2001, 166, 2378–2384. [Google Scholar] [CrossRef]
- Kramer, F.; Torzewski, J.; Kamenz, J.; Veit, K.; Hombach, V.; Dedio, J.; Ivashchenko, Y. , Interleukin-1β stimulates acute phase response and C-reactive protein synthesis by inducing an NFκB-and C/EBPβ-dependent autocrine interleukin-6 loop. Molecular immunology 2008, 45, 2678–2689. [Google Scholar] [CrossRef]
- Ramani, M.; Khechai, F.; Ollivier, V.; Ternisien, C.; Bridey, F.; Hakim, J.; de Prost, D. , Interleukin-10 and pentoxifylline inhibit C-reactive protein-induced tissue factor gene expression in peripheral human blood monocytes. FEBS Lett 1994, 356, 86–8. [Google Scholar] [CrossRef]
- Pathak, A.; Agrawal, A. , Evolution of C-reactive protein. Frontiers in immunology 2019, 10, 943. [Google Scholar] [CrossRef]
- Macintyre, S.; Samols, D.; Dailey, P. , Two carboxylesterases bind C-reactive protein within the endoplasmic reticulum and regulate its secretion during the acute phase response. Journal of Biological Chemistry 1994, 269, 24496–24503. [Google Scholar] [CrossRef]
- Ansar, W., Multiple Faces of C-Reactive Protein: Structure–Function Relationships. Clinical Significance of C-reactive Protein 2020, 1-34.
- Torzewski, M. , C-reactive protein: friend or foe? Phylogeny from heavy metals to modified lipoproteins and SARS-CoV-2. Frontiers in Cardiovascular Medicine 2022, 9. [Google Scholar] [CrossRef] [PubMed]
- Braig, D.; Nero, T. L.; Koch, H.-G.; Kaiser, B.; Wang, X.; Thiele, J. R.; Morton, C. J.; Zeller, J.; Kiefer, J.; Potempa, L. A. , Transitional changes in the CRP structure lead to the exposure of proinflammatory binding sites. Nature communications 2017, 8, 14188. [Google Scholar] [CrossRef] [PubMed]
- Pepys, M. B.; Hirschfield, G. M. , C-reactive protein: a critical update. The Journal of clinical investigation 2003, 111, 1805–1812. [Google Scholar] [CrossRef] [PubMed]
- Forte, L.; Cimmino, G.; Loffredo, F.; De Palma, R.; Abbate, G.; Calabrò, P.; Ingrosso, D.; Galletti, P.; Carangio, C.; Casillo, B. , C-reactive protein is released in the coronary circulation and causes endothelial dysfunction in patients with acute coronary syndromes. International journal of cardiology 2011, 152, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Zhang, Y.; Wu, H. , Regulation of C-reactive protein conformation in inflammation. Inflammation Research 2019, 68, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Luan, Y.-y.; Yao, Y.-m. , The clinical significance and potential role of C-reactive protein in chronic inflammatory and neurodegenerative diseases. Frontiers in Immunology 2018, 9, 1302. [Google Scholar] [CrossRef]
- Zwaka, T. P.; Hombach, V.; Torzewski, J. , C-reactive protein–mediated low density lipoprotein uptake by macrophages: implications for atherosclerosis. Circulation 2001, 103, 1194–1197. [Google Scholar] [CrossRef]
- Hsieh, I. N.; White, M.; Hoeksema, M.; Deluna, X.; Hartshorn, K. , Histone H4 potentiates neutrophil inflammatory responses to influenza A virus: Down-modulation by H4 binding to C-reactive protein and Surfactant protein D. PloS One 2021, 16, e0247605. [Google Scholar] [CrossRef]
- Li, H.-Y.; Tang, Z.-M.; Wang, Z.; Lv, J.-M.; Liu, X.-L.; Liang, Y.-L.; Cheng, B.; Gao, N.; Ji, S.-R.; Wu, Y. , C-reactive protein protects against acetaminophen-induced liver injury by preventing complement overactivation. Cellular and Molecular Gastroenterology and Hepatology 2022, 13, 289–307. [Google Scholar] [CrossRef]
- Yoneda, M.; Mawatari, H.; Fujita, K.; Iida, H.; Yonemitsu, K.; Kato, S.; Takahashi, H.; Kirikoshi, H.; Inamori, M.; Nozaki, Y.; Abe, Y.; Kubota, K.; Saito, S.; Iwasaki, T.; Terauchi, Y.; Togo, S.; Maeyama, S.; Nakajima, A. , High-sensitivity C-reactive protein is an independent clinical feature of nonalcoholic steatohepatitis (NASH) and also of the severity of fibrosis in NASH. Journal of Gastroenterology 2007, 42, 573–582. [Google Scholar] [CrossRef]
- Yeh, E. T. H.; Willerson, J. T. , Coming of age of C-reactive protein: using inflammation markers in cardiology. Circulation 2003, 107, 370–371. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Szmitko, P. E.; Ridker, P. M. , C-reactive protein comes of age. Nature Clinical Practice Cardiovascular Medicine 2005, 2, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Liang, P.; Chen, J.; Fu, S.; Liu, B.; Feng, M.; Lin, B.; Lee, B.; Xu, A.; Lan, H. Y. , The baseline levels and risk factors for high-sensitive C-reactive protein in Chinese healthy population. Immunity & Ageing 2018, 15, 21. [Google Scholar]
- Kravitz, B. A.; Corrada, M. M.; Kawas, C. H. , High levels of serum C-reactive protein are associated with greater risk of all-cause mortality, but not dementia, in the oldest-old: results from The 90+ Study. J Am Geriatr Soc 2009, 57, 641–6. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wang, M.; Wu, Y.; Kumar, R.; Lin, S. , Serum high-sensitive C-reactive protein is a simple indicator for all-cause among individuals with MAFLD. Frontiers in Physiology 2022, 13. [Google Scholar] [CrossRef]
- Marušić, M.; Paić, M.; Knobloch, M.; Liberati Pršo, A.-M. , NAFLD, insulin resistance, and diabetes mellitus type 2. Canadian Journal of Gastroenterology and Hepatology 2021, 2021. [Google Scholar] [CrossRef]
- Barzilai, N.; Ferrucci, L. , Insulin Resistance and Aging: A Cause or a Protective Response? The Journals of Gerontology: Series A 2012, 67, 1329–1331. [Google Scholar] [CrossRef]
- Alissa, E. M.; Algarni, S. A.; Khaffji, A. J.; Al Mansouri, N. M. , Role of inflammatory markers in polycystic ovaries syndrome: In relation to insulin resistance. Journal of Obstetrics and Gynaecology Research 2021, 47, 1409–1415. [Google Scholar] [CrossRef]
- Cetin, E. G.; Demir, N.; Sen, I. , The Relationship between Insulin Resistance and Liver Damage in non-alcoholic Fatty Liver Patients. Sisli Etfal Hastan Tip Bul 2020, 54, 411–415. [Google Scholar]
- Tanigaki, K.; Mineo, C.; Yuhanna, I. S.; Chambliss, K. L.; Quon, M. J.; Bonvini, E.; Shaul, P. W. , C-reactive protein inhibits insulin activation of endothelial nitric oxide synthase via the immunoreceptor tyrosine-based inhibition motif of FcgammaRIIB and SHIP-1. Circ Res 2009, 104, 1275–82. [Google Scholar] [CrossRef]
- Yang, M.; Qiu, S.; He, Y.; Li, L.; Wu, T.; Ding, N.; Li, F.; Zhao, A. Z.; Yang, G. , Genetic ablation of C-reactive protein gene confers resistance to obesity and insulin resistance in rats. Diabetologia 2021, 64, 1169–1183. [Google Scholar] [CrossRef] [PubMed]
- Dąbrowski, R.; Szczubiał, M.; Kostro, K.; Wawron, W.; Ceron, J. J.; Tvarijonaviciute, A. , Serum insulin-like growth factor-1 and C-reactive protein concentrations before and after ovariohysterectomy in bitches with pyometra. Theriogenology 2015, 83, 474–477. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Nilsson, E.; Lindholm, B.; Heimbürger, O.; Barany, P.; Stenvinkel, P.; Qureshi, A. R.; Chen, J. , Low-plasma insulin-like growth factor-1 associates with increased mortality in chronic kidney disease patients with reduced muscle strength. Journal of Renal Nutrition 2023, 33, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y.; Sukhanov, S.; Anwar, A.; Shai, S.-Y.; Delafontaine, P. , Aging, Atherosclerosis, and IGF-1. The Journals of Gerontology: Series A 2012, 67A, 626–639. [Google Scholar] [CrossRef]
- Ashpole, N. M.; Sanders, J. E.; Hodges, E. L.; Yan, H.; Sonntag, W. E. , Growth hormone, insulin-like growth factor-1 and the aging brain. Exp Gerontol 2015, 68, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Dichtel, L. E.; Corey, K. E.; Misdraji, J.; Bredella, M. A.; Schorr, M.; Osganian, S. A.; Young, B. J.; Sung, J. C.; Miller, K. K. , The Association Between IGF-1 Levels and the Histologic Severity of Nonalcoholic Fatty Liver Disease. Clin Transl Gastroenterol 2017, 8, e217. [Google Scholar] [CrossRef]
- Stanley, T. L.; Fourman, L. T.; Zheng, I.; McClure, C. M.; Feldpausch, M. N.; Torriani, M.; Corey, K. E.; Chung, R. T.; Lee, H.; Kleiner, D. E.; Hadigan, C. M.; Grinspoon, S. K. , Relationship of IGF-1 and IGF-Binding Proteins to Disease Severity and Glycemia in Nonalcoholic Fatty Liver Disease. The Journal of Clinical Endocrinology & Metabolism 2020, 106, e520–e533. [Google Scholar]
- Hopkins, B. D.; Goncalves, M. D.; Cantley, L. C. , Insulin–PI3K signalling: an evolutionarily insulated metabolic driver of cancer. Nature Reviews Endocrinology 2020, 16, 276–283. [Google Scholar] [CrossRef]
- Liu, S.-J.; Zhong, Y.; You, X.-Y.; Liu, W.-H.; Li, A.-Q.; Liu, S.-M. , Insulin-like growth factor 1 opposes the effects of C-reactive protein on endothelial cell activation. Molecular and Cellular Biochemistry 2014, 385, 199–205. [Google Scholar] [CrossRef]
- Entezari, M.; Hashemi, D.; Taheriazam, A.; Zabolian, A.; Mohammadi, S.; Fakhri, F.; Hashemi, M.; Hushmandi, K.; Ashrafizadeh, M.; Zarrabi, A.; Ertas, Y. N.; Mirzaei, S.; Samarghandian, S. , AMPK signaling in diabetes mellitus, insulin resistance and diabetic complications: A pre-clinical and clinical investigation. Biomedicine & Pharmacotherapy 2022, 146, 112563. [Google Scholar]
- Fang, C.; Pan, J.; Qu, N.; Lei, Y.; Han, J.; Zhang, J.; Han, D. , The AMPK pathway in fatty liver disease. Frontiers in Physiology 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.; Hellberg, K.; Chaix, A.; Wallace, M.; Herzig, S.; Badur, M. G.; Lin, T.; Shokhirev, M. N.; Pinto, A. F. M.; Ross, D. S.; Saghatelian, A.; Panda, S.; Dow, L. E.; Metallo, C. M.; Shaw, R. J. , Genetic Liver-Specific AMPK Activation Protects against Diet-Induced Obesity and NAFLD. Cell Reports 2019, 26, 192–208e6. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Wang, L.; Guo, M.; He, J.; Deng, Y.; Liu, J.; Wei, Y.; Wang, C.; Zhou, J.; Ma, L. , Clinically relevant high levels of human C-reactive protein induces endothelial dysfunction and hypertension by inhibiting the AMPK-eNOS axis. Clinical Science 2020, 134, 1805–1819. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K. , AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Research Reviews 2012, 11, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D. M. , mTOR signaling at a glance. Journal of cell science 2009, 122, 3589–3594. [Google Scholar] [CrossRef]
- Feng, J.; Qiu, S.; Zhou, S.; Tan, Y.; Bai, Y.; Cao, H.; Guo, J.; Su, Z. , mTOR: A potential new target in nonalcoholic fatty liver disease. International Journal of Molecular Sciences 2022, 23, 9196. [Google Scholar] [CrossRef]
- Li, T.; Weng, J.; Zhang, Y.; Liang, K.; Fu, G.; Li, Y.; Bai, X.; Gao, Y. , mTOR direct crosstalk with STAT5 promotes de novo lipid synthesis and induces hepatocellular carcinoma. Cell Death & Disease 2019, 10, 619. [Google Scholar]
- Ling, S.; Zhan, Q.; Jiang, G.; Shan, Q.; Yin, L.; Wang, R.; Que, Q.; Wei, X.; Xu, S.; Yu, J. , E2F7 promotes mammalian target of rapamycin inhibitor resistance in hepatocellular carcinoma after liver transplantation. American Journal of Transplantation 2022, 22, 2323–2336. [Google Scholar] [CrossRef]
- You, Y.-K.; Huang, X.-R.; Chen, H.-Y.; Lyu, X.-F.; Liu, H.-F.; Lan, H. Y. , C-reactive protein promotes diabetic kidney disease in db/db mice via the CD32b-Smad3-mTOR signaling pathway. Scientific reports 2016, 6, 26740. [Google Scholar] [CrossRef]
- Sangwung, P.; Petersen, K. F.; Shulman, G. I.; Knowles, J. W. , Mitochondrial Dysfunction, Insulin Resistance, and Potential Genetic Implications: Potential Role of Alterations in Mitochondrial Function in the Pathogenesis of Insulin Resistance and Type 2 Diabetes. Endocrinology 2020, 161. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A. R.; Roden, M. , NAFLD and diabetes mellitus. Nature Reviews Gastroenterology & Hepatology 2017, 14, 32–42. [Google Scholar]
- Cheung, E. C.; Vousden, K. H. , The role of ROS in tumour development and progression. Nature Reviews Cancer 2022, 22, 280–297. [Google Scholar] [CrossRef] [PubMed]
- Stefanatos, R.; Sanz, A. , The role of mitochondrial ROS in the aging brain. FEBS letters 2018, 592, 743–758. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, D.; Hu, D.; Zhou, X.; Zhou, Y. , The role of mitochondria in NLRP3 inflammasome activation. Molecular immunology 2018, 103, 115–124. [Google Scholar] [CrossRef]
- Zeller, J.; Bogner, B.; Kiefer, J.; Braig, D.; Winninger, O.; Fricke, M.; Karasu, E.; Peter, K.; Huber-Lang, M.; Eisenhardt, S. U. , CRP Enhances the Innate Killing Mechanisms Phagocytosis and ROS Formation in a Conformation and Complement-Dependent Manner. Front Immunol 2021, 12, 721887. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.-W.; Jung, I.-H.; Park, E.-Y.; Kim, K.-H.; Kim, K.; Yeom, J.; Jung, J.; Lee, S.-w. , Radiation-induced C-reactive protein triggers apoptosis of vascular smooth muscle cells through ROS interfering with the STAT3/Ref-1 complex. Journal of Cellular and Molecular Medicine 2022, 26, 2104–2118. [Google Scholar] [CrossRef] [PubMed]
- Farzanegi, P.; Dana, A.; Ebrahimpoor, Z.; Asadi, M.; Azarbayjani, M. A. , Mechanisms of beneficial effects of exercise training on non-alcoholic fatty liver disease (NAFLD): Roles of oxidative stress and inflammation. European journal of sport science 2019, 19, 994–1003. [Google Scholar] [CrossRef]
- Wu, I. C.; Liu, C.-S.; Cheng, W.-L.; Lin, T.-T.; Chen, H.-L.; Chen, P.-F.; Wu, R.-C.; Huang, C.-W.; Hsiung, C. A.; Hsu, C.-C. , Association of leukocyte mitochondrial DNA copy number with longitudinal C-reactive protein levels and survival in older adults: a cohort study. Immunity & Ageing 2022, 19, 62. [Google Scholar]
- Knez, J.; Marrachelli, V. G.; Cauwenberghs, N.; Winckelmans, E.; Zhang, Z.; Thijs, L.; Brguljan-Hitij, J.; Plusquin, M.; Delles, C.; Monleon, D. , Peripheral blood mitochondrial DNA content in relation to circulating metabolites and inflammatory markers: A population study. Plos one 2017, 12, e0181036. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, B.; Xu, L.; Yu, S.; Fu, J.; Wang, J.; Yan, X.; Su, J. , ROS-Induced mtDNA Release: The Emerging Messenger for Communication between Neurons and Innate Immune Cells during Neurodegenerative Disorder Progression. Antioxidants (Basel) 2021, 10. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. , NF-κB signaling in inflammation. Signal transduction and targeted therapy 2017, 2, 1–9. [Google Scholar] [CrossRef]
- Tilstra, J. S.; Clauson, C. L.; Niedernhofer, L. J.; Robbins, P. D. , NF-κB in Aging and Disease. Aging Dis 2011, 2, 449–65. [Google Scholar] [PubMed]
- Kunnumakkara, A.; Shabnam, B.; Girisa, S.; Harsha, C.; Banik, K.; Devi, T. B.; Choudhury, R.; Sahu, H.; Parama, D.; Sailo, B. L. , Inflammation, NF-κB, and chronic diseases: how are they linked? Critical Reviews™ in Immunology 2020, 40. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yu, K.; Cao, Y.; Luo, Y.; Liu, Y.; Zhao, C. , miR-125b promotes the NF-κB-mediated inflammatory response in NAFLD via directly targeting TNFAIP3. Life Sciences 2021, 270, 119071. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Badiwala, M. V.; Weisel, R. D.; Li, S.-H.; Wang, C.-H.; Fedak, P. W. M.; Li, R.-K.; Mickle, D. A. G. , C-reactive protein activates the nuclear factor-κB signal transduction pathway in saphenous vein endothelial cells: implications for atherosclerosis and restenosis. The Journal of Thoracic and Cardiovascular Surgery 2003, 126, 1886–1891. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Bian, Z.-M.; Yu, W.-Z.; Yan, Z.; Chen, W.-C.; Li, X.-X. , Induction of interleukin-8 gene expression and protein secretion by C-reactive protein in ARPE-19 cells. Experimental Eye Research 2010, 91, 135–142. [Google Scholar] [CrossRef]
- Ruiz-Fernández, C.; Gonzalez-Rodríguez, M.; Francisco, V.; Rajab, I. M.; Gómez, R.; Conde, J.; Lago, F.; Pino, J.; Mobasheri, A.; Gonzalez-Gay, M. A. , Monomeric C reactive protein (mCRP) regulates inflammatory responses in human and mouse chondrocytes. Laboratory Investigation 2021, 101, 1550–1560. [Google Scholar] [CrossRef]
- McCarthy, W. C.; Sherlock, L. G.; Grayck, M. R.; Zheng, L.; Lacayo, O. A.; Solar, M.; Orlicky, D. J.; Dobrinskikh, E.; Wright, C. J. , Innate Immune Zonation in the Liver: NF-κB (p50) Activation and C-Reactive Protein Expression in Response to Endotoxemia Are Zone Specific. The Journal of Immunology 2023, 210, 1372–1385. [Google Scholar] [CrossRef]
- Im, Y. R.; Hunter, H.; de Gracia Hahn, D.; Duret, A.; Cheah, Q.; Dong, J.; Fairey, M.; Hjalmarsson, C.; Li, A.; Lim, H. K. , A systematic review of animal models of NAFLD finds high-fat, high-fructose diets most closely resemble human NAFLD. Hepatology 2021, 74, 1884–1901. [Google Scholar] [CrossRef]
- Mair, W.; Dillin, A. , Aging and survival: the genetics of life span extension by dietary restriction. Annu. Rev. Biochem. 2008, 77, 727–754. [Google Scholar] [CrossRef]
- McDonald, R. B.; Ramsey, J. J. , Honoring Clive McCay and 75 years of calorie restriction research. The Journal of nutrition 2010, 140, 1205–1210. [Google Scholar] [CrossRef]
- Crawford, D.; Libina, N.; Kenyon, C. , Caenorhabditis elegans integrates food and reproductive signals in lifespan determination. Aging cell 2007, 6, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Moatt, J. P.; Savola, E.; Regan, J. C.; Nussey, D. H.; Walling, C. A. , Lifespan extension via dietary restriction: time to reconsider the evolutionary mechanisms? BioEssays 2020, 42, 1900241. [Google Scholar] [CrossRef] [PubMed]
- Green, C. L.; Lamming, D. W.; Fontana, L. , Molecular mechanisms of dietary restriction promoting health and longevity. Nature Reviews Molecular Cell Biology 2022, 23, 56–73. [Google Scholar] [CrossRef] [PubMed]
- Fung, T. T.; McCullough, M. L.; Newby, P.; Manson, J. E.; Meigs, J. B.; Rifai, N.; Willett, W. C.; Hu, F. B. , Diet-quality scores and plasma concentrations of markers of inflammation and endothelial dysfunction–. The American journal of clinical nutrition 2005, 82, 163–173. [Google Scholar] [CrossRef]
- Martínez-González, M. A.; Gea, A.; Ruiz-Canela, M. , The Mediterranean diet and cardiovascular health: A critical review. Circulation research 2019, 124, 779–798. [Google Scholar] [CrossRef]
- Kemalasari, I.; Fitri, N. A.; Sinto, R.; Tahapary, D. L.; Harbuwono, D. S. , Effect of calorie restriction diet on levels of C reactive protein (CRP) in obesity: A systematic review and meta-analysis of randomized controlled trials. Diabetes & Metabolic Syndrome: Clinical Research & Reviews 2022, 16, 102388. [Google Scholar]
- Ota, T., Prevention of NAFLD/NASH by Astaxanthin and β-Cryptoxanthin. Carotenoids: Biosynthetic and Biofunctional Approaches 2021, 231-238.
- Xia, W.; Tang, N.; Kord-Varkaneh, H.; Low, T. Y.; Tan, S. C.; Wu, X.; Zhu, Y. , The effects of astaxanthin supplementation on obesity, blood pressure, CRP, glycemic biomarkers, and lipid profile: A meta-analysis of randomized controlled trials. Pharmacological Research 2020, 161, 105113. [Google Scholar] [CrossRef]
- Julia, C.; Galan, P.; Touvier, M.; Meunier, N.; Papet, I.; Sapin, V.; Cano, N.; Faure, P.; Hercberg, S.; Kesse-Guyot, E. , Antioxidant Status and the Risk of Elevated C-Reactive Protein 12 Years Later. Annals of Nutrition and Metabolism 2014, 65, 289–298. [Google Scholar] [CrossRef]
- Liss, K. H. H.; Finck, B. N. , PPARs and nonalcoholic fatty liver disease. Biochimie 2017, 136, 65–74. [Google Scholar] [CrossRef]
- Satoh, N.; Ogawa, Y.; Usui, T.; Tagami, T.; Kono, S.; Uesugi, H.; Sugiyama, H.; Sugawara, A.; Yamada, K.; Shimatsu, A. , Antiatherogenic effect of pioglitazone in type 2 diabetic patients irrespective of the responsiveness to its antidiabetic effect. Diabetes Care 2003, 26, 2493–2499. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Liu, J.-t.; Li, K.; Wang, S.-y.; Xu, S. , Genistein inhibits Ang II-induced CRP and MMP-9 generations via the ER-p38/ERK1/2-PPARγ-NF-κB signaling pathway in rat vascular smooth muscle cells. Life sciences 2019, 216, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Nascimbeni, F.; Pellegrini, E.; Lugari, S.; Mondelli, A.; Bursi, S.; Onfiani, G.; Carubbi, F.; Lonardo, A. , Statins and nonalcoholic fatty liver disease in the era of precision medicine: More friends than foes. Atherosclerosis 2019, 284, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Kandelouei, T.; Abbasifard, M.; Imani, D.; Aslani, S.; Razi, B.; Fasihi, M.; Shafiekhani, S.; Mohammadi, K.; Jamialahmadi, T.; Reiner, Ž. , Effect of statins on serum level of hs-CRP and CRP in patients with cardiovascular diseases: a systematic review and meta-analysis of randomized controlled trials. Mediators of Inflammation 2022, 2022. [Google Scholar] [CrossRef]
- Saboori, S.; Shab-Bidar, S.; Speakman, J. R.; Yousefi Rad, E.; Djafarian, K. , Effect of vitamin E supplementation on serum C-reactive protein level: a meta-analysis of randomized controlled trials. European journal of clinical nutrition 2015, 69, 867–873. [Google Scholar] [CrossRef]
- Nagashimada, M.; Ota, T. , Role of vitamin E in nonalcoholic fatty liver disease. IUBMB life 2019, 71, 516–522. [Google Scholar] [CrossRef]
- Perumpail, B. J.; Li, A. A.; John, N.; Sallam, S.; Shah, N. D.; Kwong, W.; Cholankeril, G.; Kim, D.; Ahmed, A. , The Role of Vitamin E in the Treatment of NAFLD. Diseases 2018, 6, 86. [Google Scholar] [CrossRef]
- Escribano-Lopez, I.; Diaz-Morales, N.; Rovira-Llopis, S.; de Marañon, A. M.; Orden, S.; Alvarez, A.; Bañuls, C.; Rocha, M.; Murphy, M. P.; Hernandez-Mijares, A.; Victor, V. M. , The mitochondria-targeted antioxidant MitoQ modulates oxidative stress, inflammation and leukocyte-endothelium interactions in leukocytes isolated from type 2 diabetic patients. Redox Biology 2016, 10, 200–205. [Google Scholar] [CrossRef]
- Torzewski, J.; Brunner, P.; Ries, W.; Garlichs, C. D.; Kayser, S.; Heigl, F.; Sheriff, A. , Targeting C-reactive protein by selective apheresis in humans: pros and cons. Journal of Clinical Medicine 2022, 11, 1771. [Google Scholar] [CrossRef]
- Mattecka, S.; Brunner, P.; Hähnel, B.; Kunze, R.; Vogt, B.; Sheriff, A. , PentraSorb C-Reactive Protein: Characterization of the Selective C-Reactive Protein Adsorber Resin. Therapeutic Apheresis and Dialysis 2019, 23, 474–481. [Google Scholar] [CrossRef]
- Ries, W.; Torzewski, J.; Heigl, F.; Pfluecke, C.; Kelle, S.; Darius, H.; Ince, H.; Mitzner, S.; Nordbeck, P.; Butter, C. , C-reactive protein apheresis as anti-inflammatory therapy in acute myocardial infarction: results of the CAMI-1 study. Frontiers in cardiovascular medicine 2021, 8, 591714. [Google Scholar] [CrossRef] [PubMed]
- Esposito, F.; Matthes, H.; Schad, F. , Seven COVID-19 patients treated with C-reactive protein (CRP) apheresis. Journal of clinical medicine 2022, 11, 1956. [Google Scholar] [CrossRef] [PubMed]
- Fendl, B.; Weiss, R.; Eichhorn, T.; Linsberger, I.; Afonyushkin, T.; Puhm, F.; Binder, C. J.; Fischer, M. B.; Weber, V. , Extracellular vesicles are associated with C-reactive protein in sepsis. Scientific Reports 2021, 11, 6996. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Aging and NAFLD. Impairment of lipid metabolism and liver regenerative capacity with age lead to hepatic lipid accumulation and steatosis.
Figure 1.
Aging and NAFLD. Impairment of lipid metabolism and liver regenerative capacity with age lead to hepatic lipid accumulation and steatosis.
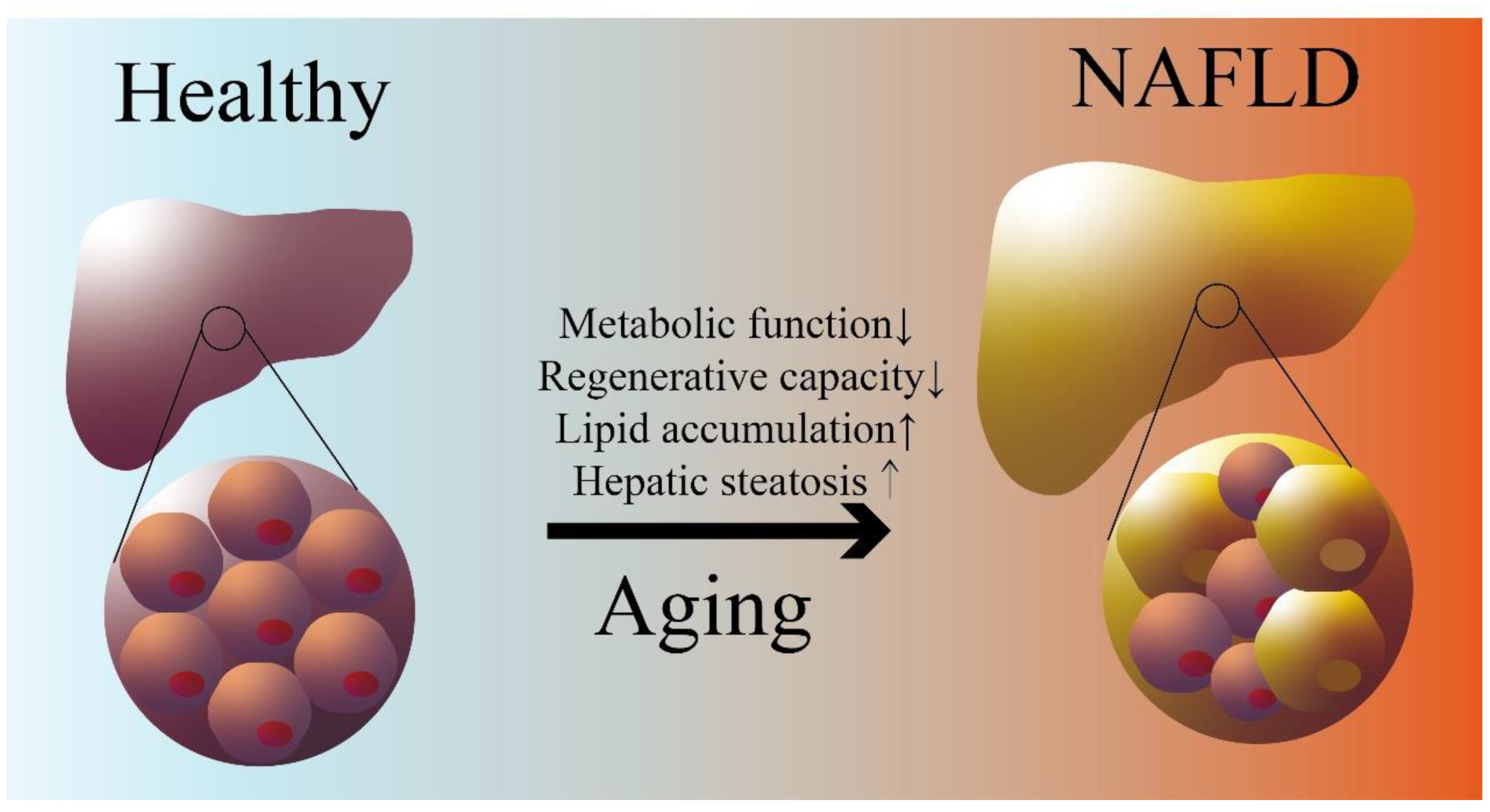
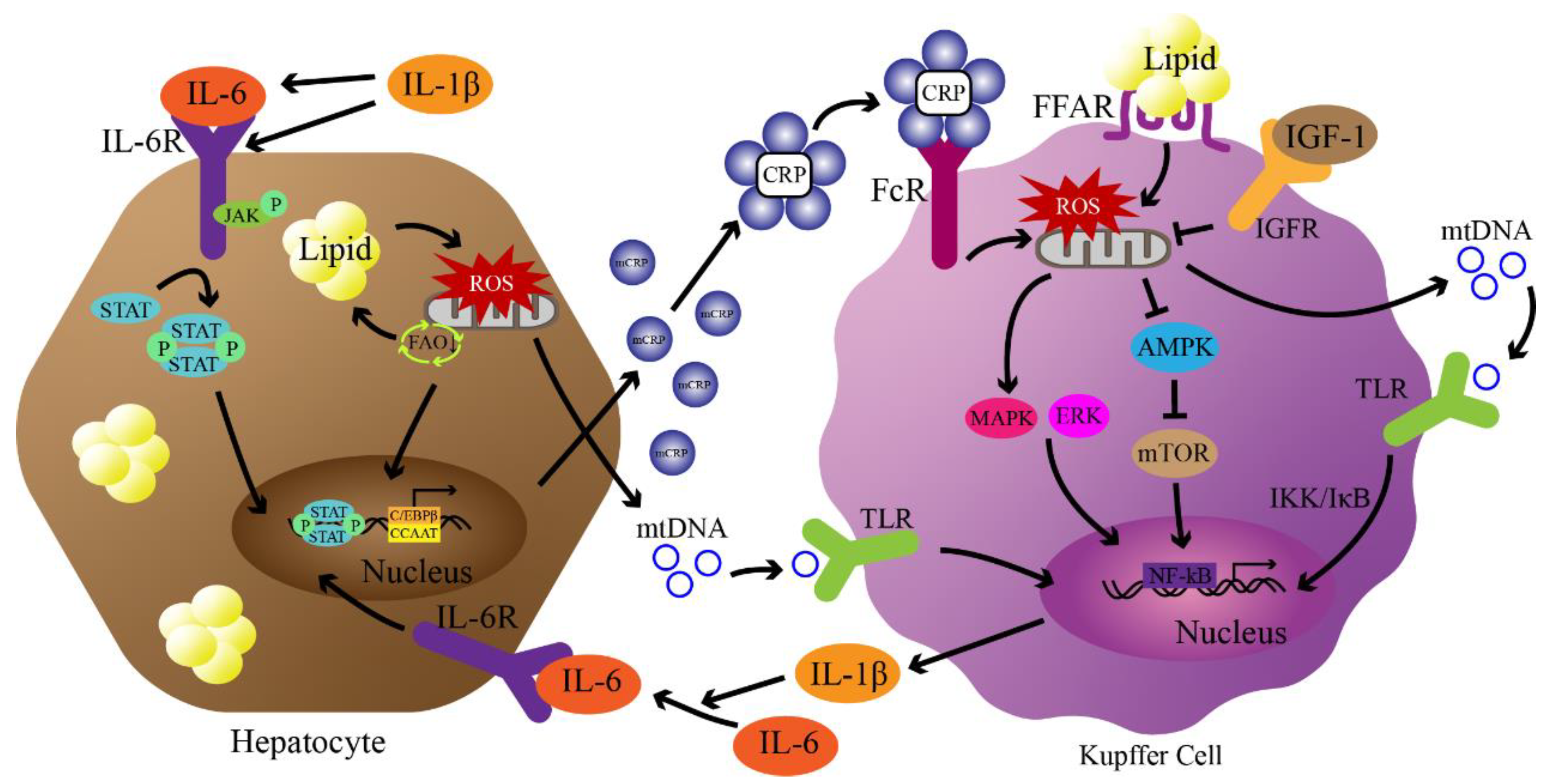
Figure 2.
Potential pathways of CRP-mediated NAFLD pathogenesis. First, lipid accumulation in hepatocytes exceeds mitochondrial metabolic capacity, leading to ROS production. ROS damage mitochondria leading to increased lipid accumulation, and mitochondrial DNA (mtDNA) release activates Kupffer cells through Toll-like receptors (TLRs). Activation of TLRs leads to nuclear transfer of NF-κB through IKK/IκB to produce IL-6 and IL-1β. Under the stimulation of IL-6 and the promotion of IL-1β, the JAK/STAT pathway is activated and mediates the production of monomeric CRP (mCRP) through C/EBPβ with enhancer CCAAT. Assembly of mCRP into CRP occurs at sites of inflammation and activates Kupffer cells via Fcγ receptors. Subsequently, activation of Fcγ receptors leads to the production of mitochondrial ROS in concert with fatty acid accumulation. ROS activates MAPK/ERK signaling and inhibits AMPK signaling to further mediate the production of inflammatory cytokines. IGF-1 blocks the regulation of CRP through PI3K/AKT signaling (not shown in the figure). Finally, inflammatory factors further lead to steatosis and damage of hepatocytes, triggering NAFLD and NASH.
Figure 2.
Potential pathways of CRP-mediated NAFLD pathogenesis. First, lipid accumulation in hepatocytes exceeds mitochondrial metabolic capacity, leading to ROS production. ROS damage mitochondria leading to increased lipid accumulation, and mitochondrial DNA (mtDNA) release activates Kupffer cells through Toll-like receptors (TLRs). Activation of TLRs leads to nuclear transfer of NF-κB through IKK/IκB to produce IL-6 and IL-1β. Under the stimulation of IL-6 and the promotion of IL-1β, the JAK/STAT pathway is activated and mediates the production of monomeric CRP (mCRP) through C/EBPβ with enhancer CCAAT. Assembly of mCRP into CRP occurs at sites of inflammation and activates Kupffer cells via Fcγ receptors. Subsequently, activation of Fcγ receptors leads to the production of mitochondrial ROS in concert with fatty acid accumulation. ROS activates MAPK/ERK signaling and inhibits AMPK signaling to further mediate the production of inflammatory cytokines. IGF-1 blocks the regulation of CRP through PI3K/AKT signaling (not shown in the figure). Finally, inflammatory factors further lead to steatosis and damage of hepatocytes, triggering NAFLD and NASH.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



