Submitted:
13 July 2023
Posted:
14 July 2023
You are already at the latest version
Abstract
Breast cancer (BC) is the most common malignancy and the second cause of cancer specific death in women from high-income countries. More than 70% of all breast cancer are hormone receptor-positive BC, and elevated estrogen circulating in blood has been proved to be a strong risk for BC development. This is due to its contribution to enhance proliferation of cancerous cells, angiogenesis and metastasis stimulation and therapy resistance. The estrogen (E) metabolism–gut microbiome axis is functional with underlying individual variations in E levels. It is reasonable that the estrobolome (bacterial genes whose products are capable of metabolizing E) could contribute to the risk of hormone-driven malignancies including BC and may serve as a potential biomarker and target. Gut microbial β -glucuronidase (GUS) enzymes have been suggested to be involved in the estrobolome. Furthermore, bacterial GUS enzymes within the gastrointestinal tract have been postulated to contribute to hormone breast cancer. In this review, we discuss the recent knowledge about the role of GUS enzyme in the pathogenesis of breast cancer. We focus on the role of GUS in (i)-the microbioma and estrogen metabolism, (ii) diet, estrobolome, and the BC development, (iii) other activities of the bacterial β-glucuronidase, and (iv) the new molecular target for BC therapeutic application.

Keywords:
breast cancer
; microbiota
; estrobolome
; β-glucuronidase
; dysbiosis
; inhibitors
; metabolomics
; personalized medicine
; review
1. Introduction
Breast cancer (BC) is the most frequent malignancy and the second most common cancer-specific cause of death in women in developed countries. According to the International Agency for Research on Cancer (IARC) global cancer statistics, more than 2 million new cases and 600 thousand deaths from BC were reported in 2018 [1]. The 5-year overall survival for BC is 98% for localized disease, 84% for regional disease, but only 23% for disease with metastases [2].
The most relevant known BC risk factors include lifestyle factors such as western diet, obesity, alcohol consumption, and a sedentary lifestyle, in addition to other equally relevant factors such as exposure to endogenous and exogenous estrogens, high breast density, a history of atypical hyperplasia, and genetic susceptibility [3]. However, less than 10% of breast cancer are attributable to genetic predisposition [4]. Thus, the mechanisms of the etiopathogenesis of breast cancer remain to be clarified.
Since 2010, the recognition of the gut microbiota on human health has been monumental, as demonstrated by the number of medical publications in well-respected, peer-reviewed journals. In recent years, a close relationship between breast cancer and the gastrointestinal tract (GI) microbiome has been suggested. The GI tract harbors more than 1000 different bacterial species and estimates of the number of bacteria we carry reach 1013 per gram of luminal content [5,6], 10 times the number of human cells in the body. Bacterial load and species diversity increase from the stomach to the colon, giving rise to a very complex microbial ecosystem [7,8,9]. The composition of the GI tract microbiota (archaea, protozoa, fungi, viruses, and bacteria) reflects host variables, such as delivery mode, genetics, diet, alcohol intake, environmental exposures, and drugs such as antibiotics and anticancer therapies.
Studies of bacterial microbiome function, composition, and assessment of the aggregate of its genes (the metagenome) are now possible via advances in 16S ribosomal RNA (rRNA) sequencing and bioinformatics [10,11]. Humans and microbes have co-evolved a complex intricate relationship to benefit the host while allowing the intestinal microbiota to live in a mutually advantageous equilibrium. Microbiome perturbation has been associated with inflammatory, autoimmune, and malignant diseases [12,13]. A pathological imbalance of the microbial community may promote oncogenesis and induce tumor progression, as well as influence responses to cancer therapies and the toxicity profiles of cytotoxic agents as antineoplastic agents [14,15,16]. In addition, the human gut microbiome is active and functional, exerting effects locally as well as over long distances that include metabolic, hormonal, and immunological messengers [17,18]. Therefore, host-microbe interactions may influence carcinogenesis through mechanisms such as the induction of genotoxic responses, alteration of the microenvironment, metabolism, and chronic inflammation [19,20].
In this review, we analyze the links between gut microbiota, estrogen metabolism, and breast cancer, and explore the possible implications of β-glucuronidase enzyme substrate metabolites that might impact breast cancer risk, prognosis, and possibly treatment options toward more personalized medicine. Finally, we put into perspective possible limitations and biases regarding current microbiota research and provide ideas for new and more robust studies in this promising and challenging field.
2. Gut microbiota, diversity and dysbiosis
The gut microbiota is composed of microorganisms (archaea, protozoa, fungi, viruses, bacteria) that colonize the gastrointestinal tract and other zones of the body. Microbiota is a collective term that refers to the group of microbes colonizing the human body, and the collection of genes they encode is known as our microbiome [21]. The human intestinal microbiota is estimated to contain 1013 bacterial cells, 10 times the number of human cells in the body. The bacterial load, along with species diversity, growing from the stomach to the colon, creating an intricate microbial ecosystem [7,8,9].
For the study of the microbiota-microbiome, we must know the main concepts that allow us to understand the differences between a homeostatic microbiota (eubiosis) and an altered microbiota (dysbiosis, term that describes a diseased state of intestinal microorganism communities, which conducts to an intestinal-microbial imbalance in the host). Therefore, α-diversity is the richness of microbes present in the gut and is determined by counting operational taxonomic units (meaning the number of different species in the gut) and the Shannon index (which measures the evenness of the distribution of microbes in the gut). The β-diversity is used to compare samples and assesses how different the microbial community is from one environment to another, like microbiological fingerprinting [7,8,9].
The microbiota has the ability of self-regeneration, known as resilience, which is explained by the ability to restore its homeostasis after an external disturbance (e.g. infections, antibiotic or antitumor treatments such as chemotherapy). These changes can be a pulse disturbance (a short period of time such as ingestion of a medication), or a press perturbation (continuous stimulus for a prolonged period of time, such as permanent changes in diet or changes in the environment) [7,8,9].
Furthermore, the resilience of the microbiota depends on three factors for dynamic stability: time (the same composition of the microbiota is maintained over time and in response to perturbations), taxonomic groups (group persistence across perturbations or over time), and functional groups (although species and taxonomic clusters may change, the functional characteristics of the microbiota remain the same). Only when a threshold of cumulative stress is exceeded is there a shift from homeostasis to a new equilibrium [7,8,9].
3. Microbioma and estrogen metabolism: the estrobolome
A major mechanism of action of the microbiome in the host is the synthesis of enzymes and, thus the production of their bacterial metabolites. These metabolites, synthesized by the microbiome, can enter the blood circulation at the site of their biosynthesis and then travel to distant organs, where they will exert their biological effects [17]. Bacterial metabolites behave like human hormones in the sense that they are synthesized by an “organ” (the microbiome) and are then transferred to the site of action by the circulation. The metabolism of estrogen, which includes hydroxylation and conjugation, takes place primarily in the liver and involves a pattern of enterohepatic circulation (Figure 1) [17]. Endogenous estrogen exists in three biologically active form, estradiol in premenopausal, estrone in postmenopausal and estriol in pregnant women [22,23]. Conjugated estrogens with their metabolites are excreted in the bile and subsequently into the GI, where they are deconjugated into wide array of estrogen metabolites and gain estrogenicity accordingly variable activity of microbial β-Glucuronidases (β-GUS, EC 3.2.1.31) [24].
Plottel and Baser refer to the existence of a group of genes present in some enteric bacteria that produce enzymes, such as β-Glucuronidases [22,24], capable of metabolizing estrogens; the sum total of these bacterial genes is known as “the estrobolome” (Figure 1). Therefore, these enzymes are responsible for estrogen deconjugation. Deconjugation of excreted estrogen is important in estrogen reuptake at the distal intestine and, thus, modulation of systemic estrogen (via portal vein) availability and the regulation of estrogen-associated pathways. In this scenario, it has been widely hypothesized that systemic estrogens and their metabolites (hydroxylated species from estrone or estradiol) can be modulated by gastrointestinal estrobolome. In human GI, the most important β-glucuronidase-encoding genes are the GUS genes. Mammalian UDP-glucosyltransferases bind the glucuronic acid moiety to complex compounds, including steroid hormones, marking them for excretion (more water soluble). Gut microbes possessing BGUS genes that encode β-glucuronidase enzyme activity can remove the glucuronic acid to be used as a carbon resource. The respective aglycones are either released into the GI for excretion or reabsorbed back into the circulation Most intestinal bacteria can express β-glucuronidase enzymatic activity, including Firmicutes and Bacteroidetes (Table 1). Therefore, these bacterial species would affect the levels of estrogens circulating in the blood and excreted in feces and urine. These reactivated estrogens increase their serum levels, which act through estrogen receptors (ERα and ERβ). Activation of these receptors modulates the expression of several genes, including mitochondrial genes. Elevated oxidative phosphorylation has been shown to promote metastasis [25,26], contribute to therapeutic failure and make tumors more aggressive [26,27]. Taken together, bacterial deconjugation of estrogens favors breast cancer progression and changes the risk of development and progression of estrogen-dependent cancers [26,27,28]. Currently, there is only one pilot study that directly links estrobolome, circulating and excreted estrogen levels to the presence of breast cancer. Goedert et al [29], (2015), in a pilot study of cases (N=48) and controls (N=48) in a population of postmenopausal women, demonstrated that women with BC had a statistically significant modified (β-diversity) composition, and that urine total estrogen positively correlated with α-diversity in healthy but not in women with BC, signifying lower microbial richness and diversity.
In addition, the enterohepatic circulation similarly metabolizes a number of complex molecules, such as neurotransmitters, anticancer drugs, nonsteroidal anti-inflammatory drugs (NSAIDs) and environmental carcinogens, however, it is the intestinal bacteria that largely determine whether they are excreted or reabsorbed into the circulation, where the bacterial β-glucuronidase enzyme plays an active role [30,31] (see section 5). Consequently, dysfunction of the estrogen metabolism-gut microbiota axis in combination with underlying individual variability in estrogen levels may contribute to an increased risk of hormone-mediated malignancies, including BC. In the future, interventions involving the use of prebiotics, probiotics, postbiotics, or antimicrobial agents should be considered to modulate gut bacterial populations with BGUS activity and decrease the risk of estrogen-related BC or, after cancer diagnosis, become adjunctive treatments [22,23].

Table 1.
Bacterial species can express β-glucuronidase.
| Genus | Species | Gene IDa | Estrogen deconjugationb | PDBc database (accession ID) |
Reference |
|---|---|---|---|---|---|
| Alistipes | EXC72_RS02090 ID: 78178623 |
[25,32] | |||
| Akkermansia | muciniphila | GOZ73_RS09295 ID: 60881251 |
[31,32] | ||
| Bacteroides | Fragilis | I6J55_RS13335 ID: 66330823 |
Yes | 3CMG | [31,32] |
| cellulosilyticus | INE78_RS14030 ID: 66307762 |
[32] | |||
| intestinalis | I1224_RS00440 ID: 69505108 |
[32] | |||
| uniformis | INE75_RS18175 ID: 66283800 |
6NZG, 6D1N, 6D41, 6D50, 6D6W, 6D7F, 6D89, 6D8G | [32,33,34] | ||
| Ovatus | Bovatus_RS21525 ID: 29455654 |
6D8K | [32,34] | ||
| Dorei | FYB91_RS01050 ID: 56614211 |
6ED1 | [32,35] | ||
| massiliensis | I6J55_RS13335 ID: 66330823 |
[32] | |||
| Vulgatus | GAIMETA21_RS00905 ID: 69838528 |
[32] | |||
| Bacillus |
thuringiensis |
A9498_RS29930 ID: 39691567 |
[32,36] | ||
| Bifidobacterium | Dentium | BIFDEN_RS03045 ID: 69535529 |
6LD0,6LD6, 6LDB, 6LDC, 6LDD | [32,37] | |
| Citrobacter | [32] | ||||
| Clostridium | perfringens | uidA [31] ID: 69447906 |
yes | 6CXS, 6JKM, | [32,38,39] |
| Collinsella | tanakaei YIT 12063 | uidA ID: 62759750 |
[32] | ||
| Dermabacter | [32] | ||||
| Edwardsiella |
piscicida |
uidA ID: 72529797 |
[32] | ||
| Ictaluri | uidA ID: 69540280 |
[32] | |||
| Escherichia | Coli | uidA ID: 946149 |
yes | 6LEG, 3K46, 3K4A, 3K4D, 3LPF, 3LPG, 4JHZ, 5CZK, 6LEG, 6LEJ, 6LEL, 6LEM, 7PR6 | [31,32,37,39,40,41,42] |
| Eubacterium | Eligens | uidA ID: 41357285 |
yes | 6BJW | [32,43] |
| Faecalibacterium | prausnitzii | uidA ID: 56863673 uidA ID: 34751772 |
yes | 6U7I, 6ED2 | [32,35] |
| Fusicatenibacter | saccharivorans | 6NCY, 6NCZ | [32,44] | ||
| Lactobacillus | rhamnosus | RHM_0050 ID: 12473125 |
yes | 6ECA | [32,35] |
| Gasseri | J3E66_RS04340 ID: 66468975 |
[32,45] | |||
| Marvinbryantia | [32] | ||||
| Propionibacterium | Acnes | uidA ID: 12534223 |
[32] | ||
| Parabacteroides | Merdae | DY317_RS05255 ID: 49202940 |
6DXU | [32] | |
| Johnsonii | HMPREF1077_RS04680 ID: 43351364 |
[32] | |||
| Roseburia | Hominis | uidA ID: 77458459 |
yes | 6MVH | [32] |
| intestinalis | uidA ID: 61434358 |
[32] | |||
| Ruminococcus | Gnavus | N769_RS0107715 ID: 35896210 |
yes | 6EC6 | [32,35] |
| Streptococcus | agalactiae | uidA ID: 66885601 |
yes | 4JKL, 4JKK, 4JKL, | [32,39] |
|
equisimilis |
GGS_1280 ID: 13799427 |
[32] | |||
| Tannerella | forsythia | BFO_RS10495 ID: 34759432 |
[32,39] |
a Accession ID are from NCBI. b Ervin S.M et al. 2019 JBC 294(49): 18586-18599. c AccessionID are from PDB database.
4. Axis diet, estrobolome and breast cancer
Diet plays an integral role in the complex interrelationship between the human gut microbiota, estrogen metabolism, and its influence on breast cancer recurrence as well as metastatic potential. The standard Western diet results in the increased propagation of unhealthy bacteria, which contain high levels of β-glucuronidase (estrobolome composition). In addition, the bacterial composition of the estrobolome is influenced by host-specific factors (e.g., age and ethnicity), as well as by environmental influences throughout life, such as alcohol, hormonal treatments, and antibiotic use. All of these factors would exert selective pressures on the bacterial populations of the estrobolome. Although the correlations between BC risk and dietary intake have been intensively studied, the underlying associations or effector mechanisms remain poorly understood. Historically, increased risk of BC has been tied to high intake of red meat and animal fat, with decreased risk being concurrently linked to fruit and vegetables consumption, associated with high and low levels of β-glucuronidase, respectively [46,47], Western diet (ie, those that are high in processed meat, sugar and fat) effects, for example, are only significant in postmenopausal patients with hormone receptor-positive (HR+) tumors, while “healthy” diet (ie, high fresh fruit, vegetables and fish) effects are only significant in premenopausal women, but across receptor-positive and receptor-negative tumours [48]. What has been referred to as the standard western diet results in obesity, insulin resistance, dysbiosis, and inflammation [49].
The synthesis of insulin-like growth factor-1 (IGF-1), related to tumor growth and metastasis, is stimulated by insulin. In addition, this pancreatic hormone binds to steroid hormone-binding globulin (SHBG), increasing estrogen availability, promoting higher estrogen levels, and contributing to mammary carcinogenesis [50]. In turn, adiponectin levels decrease, leading to insulin resistance and increased IGF-1 levels, which promote cell proliferation. Both estrogen and IGF-1-mediated signaling are increased in obese postmenopausal women “Cross-talk” between such pathways represents an important link to tumor progression [50]. Obesity, affects more than half of postmenopausal women, is a risk factor for BC [51]. A meta-analysis of 50 prospective observational studies confirmed a relationship between adult weight gain in women and hormone-dependent cancer risk; for every 5-kilogram increase in weight was associated with increases in postmenopausal breast (+11%), ovarian (+13%), and endometrial (+39%) cancers [52]. Adipose tissue is known to be metabolically active, with elevated levels of the enzyme aromatase that converts androgens to estrogens, the main source of estrogens in postmenopausal women. Therefore, excessive estrogen biosynthesis from expanded adipose tissue is associated with adverse disease outcomes in obese women with hormone-sensitive and hormone-resistant cancers [53]. No less important, related between obesity-breast cancer is the chronic inflammatory process that the obese state originates. This leads to the activation of a large number of metabolic pathways such as JAK2/STAT3, MAPK, EGFR, etc., which are a regulators of lipid metabolism, promote chemoresistance, or have mitogenic activity, respectively [54]. However, the biological basis for the differences in the natural history of breast cancer in obese women is not entirely clear. In addition, there are numerous prominent studies and reviews that clearly associate obesity with gut dysbiosis and its health effects, such as cancer, including breast cancer [55,56,57]. Although, this is not the subject of this review, we should not fail to mention them.
Alcohol consumption is also an important risk factor, as high alcohol consumption is associated with disease recurrence and poorer survival, especially in estrogen receptor-positive breast cancer in postmenopausal women [58]. The evidence is consistent that alcohol consumption, even ingestion of less than 10-50 g per day, carries an increased risk of this disease. In the European Prospective Investigation into Cancer and Nutrition (EPIC) study, which recruited a cohort of more than 360,000 from 10 countries in Europe, a strong association was demonstrated between alcohol consumption and the risk of breast cancer in estrogen receptor-positive tumors [59,60,61,62]. Consuming ethanol increases levels of endogenous estrogens-notably estradiol and estrone [61,63]. Ethanol likely affects estrogen levels multifactorially including upregulation of the estrogen receptor (ER), the steroid hormone signaling pathway and ER alfa ligand [63,64,65]. In MCF-7 cell models of human breast cancer, ethanol increased cell proliferation, ERα expression, and aromatase enzyme activity. Ethanol has been observed to stimulate the proliferation of ER+ but not ER- human breast cancer cells in vitro [66]. Furthermore, in hormone-positive MCF-7 and T47D cells, the increase in ER-α ligand activity was dependent on the dose of ethanol delivered and resulted in inhibition of the expression of the tumor suppressor gene, BRCA1 [67].
Alcohol consumption can lead to small intestinal bacterial overgrowth (SIBO). Anaerobic and aerobic bacteria are elevated in individuals with chronic alcohol consumption and alcoholic cirrhosis compared with healthy controls [68]. SIBO and altered microbiome composition have been observed in rats with alcoholic liver disease [69].
Ethanol may affect the metabolism of intestinal bacteria, although this has not been well studied. In animal models, 4-hydroxyestrone, an estrogenic metabolite of catechol, had biologically significant estrogenic activity, and it has been shown that estrogen induces tumorigenesis [70]. The interactions between alcohol, estrogens, the estrobolome, and mammary carcinogenesis in humans need further definition.
Another dietary pattern linked to BC is the Mediterranean Diet, with recent studies showing an inverse relationship, particularly in the context of triple-negative disease [48]. A whole-food, plant-based diet (especially one high in fiber) results in the promotion of “healthy” microbiota. A decrease in β-glucuronidase activity minimizes circulating estrogen levels and increases SGBH along with the fecal estrogen excretion. It is known that intestinal bacteria are involved in this process; however, what is not known is which bacteria are high producers of β-glucuronidase, a topic that needs to be investigate [19,49,50]. Firmicutes and Bacteroidetes are the main colonic phyla responsible for the metabolism of fiber and polyphenols. A plant-based diet contributes to a favorable Firmicutes/Bacteroidetes ratio. Conversely, an intake rich in fats and dairy increases Bacteroidetes, while the consumption of vegetables and fiber increases Prevotella, Akkermansia and other beneficial bacteria [71,72]. The gut microbiota, including the estrobolome, may be our most potent endocrine regulator, because it acts on almost all distant organs and their proper functions [73]. Therefore, a more nuanced microbiota and defined diet approach may represent a more realistic avenue to improve cancer outcomes. This highlights the importance of the relationship between the composition of the microbiota and the production of its metabolites, and that both factors must be taken into account if we are to realistically improve cancer outcomes [47,48,74,75,76].
5. Other activities of the bacterial estrobolome
The bacterial estrobolome also acts on other substrates (androgens, anticarcinogens, polyphenols, phytoestrogens, hetrocyclic amines, etc) that directly or indirectly would negatively or positively affect the development of hormone-dependent breast cancer.
In normal body, drugs and other xenobiotics are detoxified via glucuronidation in liver (phase II metabolism) by UDP-glucuronosyltransferases (UGTs). This glucuronide molecules are less active, more soluble and excreted by renal clearance and faeces [77]. However, elevated levels of GUS activity revert this process by deglucuronidation and regenerating the active form. In this way the estrobolome has been implicated in genotoxicity, toxicity, and resistance to therapies [78]. Pharmacomicrobiomics is defined as the effect of microbiome variations on drug disposition, action, and toxicity [79,80].
Irinotecan (CPT-11) treats a range of solid tumors, the prodrug is active in vivo to SN38, a potent topoisomerase I inhibitor that retards the growth of rapidly proliferating cells in tumors and intestinal epithelium [81,82]. SN38 is detoxified through the addition of glucuronic acid in the liver, to form SN38-glucuronic (SN38-G), this molecule is eliminated via the GI tract. The estrobolome remove the glucuronic acid through the enzyme B-glucuronidase, again releasing the active molecule SN38. Reactived SN38 inflicts epithelial damage, causing toxicities (diarrhea and weight loss) in the treated patient [82]. Thus, treating irinotecan induced diarrhea is a significant clinical need. Wallace et al. have reported several selective inhibitors that block the action of the β-glucuronidase enzyme. Irinotecan induces shifts in gut microbial composition, including increases in Proteobacteria [83,84]. Proteobacteria, specifically the Enterobacteriaceae, are unique among gut microbial taxa in encoding an operon of genes encoding GUS enzymes that are up regulated in response to the presence of glucuronidated compounds, allowing these bacterial species to use Glucuronic acid for growth [85]. By inhibiting Enterobacteriaceae GUS enzymes and blocking access to Glucuronic acid, GUS inhibitors alone seems capable of blunting the growth of Proteobacteria in the mouse GI tract. The utility of GUS inhibition also extends to drugs beyond irinotecan and would be relevant in the preventive treatment of postmenopausal women with elevated levels of β-glucuronidase activity (see section 7) [38,39,86,87].
In contrast, a recent novel study by An. J et al. proposes that the estrobolome can be used in the treatment of hormone-dependent breast cancer [88]. These authors analyzed the composition of the blood microbiome of healthy controls (192 women) and patients with stage 0-III breast cancer (96 women). In particular, they investigated the estrobolome bacteria (producers of β-glucuronidase and/or β-galactosidase), which are involved in estrogen metabolism. They found that Staphylococcus species were more abundant in healthy controls than in breast cancer patients, so these bacteria were selected for further studies. Next, they analyzed the effect of Staphylococcus aureus on endocrine therapy combined with tamoxifen. Analysis of the microbiome of blood samples showed that β-glucuronidase-producing species were more abundant in breast cancer patients than in healthy controls. Further experiments in MCF7 and BT474 breast cancer cells culture confirmed that the efficacy of tamoxifen increased when administered in conjugation with S. aureus extracellular vesicles (EVs). The authors propose that the combination treatment of S. aureus EVs could potentially be used as adjuvants for breast cancer treatment in the future [88].
Testosterone therapy is being increasingly used in the management of postmenopausal women. However, as clinical trials have demonstrated a significantly increased risk of breast cancer with oral combined estrogen-progestin therapy [89]. Testosterone exhibits high variability in pharmacokinetics and glucuronidation after oral administration. Although testosterone metabolism has been studied for decades, the role of gut bacterial β-glucuronidases on its disposition are not well characterized. 5β-dihydrotestosterone (5β-DHT) and 3α,5β-tetrahydrotestosterone (3α,5β-THT), synthetized in the liver, also appeared in human hepatocytes treated with testosterone and in human serum collected after oral testosterone dosing in men. Basit et al. showed that 5β-DHT and 3α, 5β-THT are then eliminated through hepatic glucuronidation. They evaluated the potential reactivation of testosterone glucuronide (TG) after its secretion into the intestinal lumen. Incubation of TG with purified gut microbial β-glucuronidase enzymes and with human fecal extracts confirmed testosterone reactivation into testosterone by gut bacterial enzymes. Both testosterone metabolic switching and variable testosterone activation by gut microbial enzymes are important mechanisms for explaining the disposition of orally administered testosterone and appear essential to unraveling the molecular mechanisms underlying to hepatic glucuronidation-associated pathophysiological conditions [90].
Consuming red and processed meat has been associated with increased risk of cancer, this action is attributed to exposure to carcinogens molecules such as heterocyclic amines (HCA) formed during cooking and conservation processes. The major form of HCA in the colon are glucuronides (HCA-G), Zhang et al. [91], such as Faecalibacterium prausnitzii, have the ability to hydrolyze G-HCA, releasing free HCA. Interestingly, this deglucuronidation reaction coupled with bacterial glycerol/diol dehydratase activity from Flavonifractor plautii, Blautia obeum and Lactobacillus reuteri, produces metabolites (heterocyclic amines to glycerol conjugates, HCA-M1) with lower mutagenic potential, this study suggests a potential target to modulate estrobolome activities to mitigate the risk of HCA carcinogenic activity.
Polyphenols, which are widely distributed in plants and the human diets, they can be found in wine [92], coffee [93], tea [94], fruits, and vegetables [95], are known to have numerous biological activities and can modulate the composition of the gut microbiota, and hence indirectly influence their own metabolism and biovailability [96]. However, humans have a low absorption of these beneficial molecules, and this is largely due to the fact that their absorption is mediated by coupled metabolic pathways between intestinal bacteria and humans (esterase, glucosidase, demethylation, etc.). The study of polyphenols has become of great interest in the prevention of chronic diseases since epidemiological studies have shown that most of these compounds from the diet have numerous benefits for human health, such as reducing the incidence of cancer [97], cardiovascular disease [98], stroke [99], and type 2 diabetes [100].
One of the best studied types of polyphenol metabolites are nonsteroidal estrogens. Some intestinal bacteria, such as Eggerthella spp. strain YY7918, Eggerthella spp. Julong 732, Enterococcus faecium, Adlercreutzia equolifaciens, Slackia equolifaciens, Lactobacillus mucosae, Bifidobacterium spp, Slackia isoflavoniconvertens and Bacteroides ovatus [101,102,103,104,105,106,107], are able to metabolize the soybean isoflavone daidzein to equol and/or and/or O-desmethylangolensin (O-DMA). The rate of equol formation depends on dietary habits, the composition of the intestinal microflora, the degree of intestinal bacterial fermentation, intestinal transit time and alterations of the redox level in the intestine [96]. Equol exerts many different effects, the most important being endocrine. Its high binding affinity to the estrogen receptor, preferentially activating ERβ [106,107], has been used to alleviate menopausal symptoms [105]. Anti-androgenic effects and inhibition of osteoclast formation have also been observed [108,109]. In vitro studies have revealed anticancer activities, mainly by inhibition of cancer cell migration and invasion, as well as induction of apoptosis. Other estrogen receptor ligands include enterolactone, enterodiol, urolithin A and 8-prenylnaringenin. These are polyphenolic metabolites generated by various bacteria, e.g. Bacteroides spp, Clostridium spp, Eubacterium limosum and E. lenta. All of them can bind to estrogen receptors and inhibit cancer development by inhibiting tumor proliferation and invasion, and inactivating angiogenesis [110,111,112].
In the liver, dietary polyphenols are glucuronidated by UGTs enzymes and released into the intestine where they are hydrolyzed by enzymes of the estrobolome, such as β-glucuronidases and possibly sulfatases, and returned to the circulation via the portal vein [96]. However, this circuit is very complex since it involves a network of enzymes and transporters that make the absorption of these beneficial molecules less effective. The combined actions of UGTs, efflux transporters, and β-glucuronidase derived from the intestinal/enterocyte microflora play a crucial role in triple recycling (local, enteric, and enterohepatic recycling), thereby increasing the residence time of polyphenols and their glucuronides in the local intestine and liver [113]. Further studies on these recycling mechanisms and the gut estrobolome would provide in-depth insights into the disposition of polyphenols in relation to their benefits to human health.
6. Gut microbiota β-glucuronidase structure
Given the important impact of microbial β-glucuronidase activity on different forms of cancer, including breast cancer, GUS enzymes have been the target of intensive structural analysis by a significant number of researchers. The first β-glucuronidase (GUS) structure, corresponding to the human enzyme, was determined in 1996 [114], nevertheless, it was not until nearly 15 years later when the first microbial enzyme structure was characterized corresponding to the Escherichia coli GUS (EcGUS) (Table 1) [40]. Up to date more than 40 crystallographic structures of GUS belonging to gut microbiota have been deposited at the Protein Data Bank database (Table 1), and many others not related with the digestive tract [115]. Microbial GUS enzymes (EC 3.2.1.31) are a broad structural and functional group of enzymes encoded by the uidA gene and included in the Glycoside Hydrolase families 2 (GH2) of the Carbohydrate Active enZymes (CAZy) classification, possessing exo β-D-glucuronidase activity. The quaternary structure of bacterial GUS enzymes plays an important role to explain its biological function; in fact, the majority of the GUS enzymes show a homotetrameric structure (Figure 1a) with minimal exceptions, as is the case of Bacteroides uniformis (BuGUS) and Fusicatenibacter saccharivorans GUS (FsGUS) that reveals dimeric or hexameric architecture [34,44]. Furthermore, the quaternary structure is essential for the deconjugating activity of this enzyme since in the majority of the cases the N-terminus of the polypeptide chain contributes to the active center formation of the adjacent subunit with an important role in the recognition of the aglycone portion of the conjugated estrogens or related substrates [44].
In humans, in addition to GUS proteins, another type of enzymes with β-glucuronidase activity are the heparanases (HPSE), enzymes with endo β-D-glucuronidase activity responsible for the cleavage of heparan sulphate, an important component of the extracellular matrix, but not involved in estrogen metabolism [116]. These enzymes, although sharing a similar activity with GUS proteins, are structurally different and it is important not to confuse them, since they exhibit a dimeric architecture and belongs to the GH79 family [117].
Microbial GUS enzymes are large enzymes with a protomer of around 600 aminoacids comprising an N-terminal jelly roll β-sandwich domain followed by an immunoglobulin-constant-chain-like domain. The C-terminal sequence includes a core (β/α)8 TIM-barrel fold domain containing the glucuronic acid binding site with the catalytic residues located at the C-terminal end of the central-barrel [34,78]. In spite the variability found in the active center of microbial GUS enzymes, there are specific sequence features that are essential for GUS activity over a wide range of glucuronidated substrates, including estrogen glucuronides, as is the case of the key catalytic glutamates, the N-K motif, and the Y loop (Figure 1b). The catalytic glutamate residue, located nearby the loop 2 (E413 in EcGUS) (Figure 1b), interacts with the anomeric hydroxyl group of the glucuronic acid moiety of estrogen-conjugated substrates and promotes proton transfer, acting as a general acid/base, while the second glutamate residue (E504 in EcGUS) (Figure 1b) which interacts with the anomeric and O2 hydroxyl groups being the residue in charge of the nucleophilic attack [44]. The lysine and asparagine residues belonging to the N-K sequence motif (N566 and K568 in EcGUS) (Figure 1b) recognizes the characteristic carboxylic acid moiety of the glucuronic acid conjugate, representing a crucial signature to differentiate the glucuronic acid moiety relative to galactose [39]. Residues N412 and H330 hydrogen bonds the 2-hydroxyl and 3-hydroxyl groups, respectively, while D163 and W549 interacts with the 4-hydroxyl group, collaborating to the adequate positioning of the glucuronic acid sugar (EcGUS numbering). In addition, the Y loop in GUS enzymes is made up by three tyrosine residues (Y468, Y469 and Y472 in EcGUS) (Figure 1b) nearby the N-K motif, revealing important structural adaptability and facilitating, through pi-stacking interactions, the binding of estrogen glucuronides [32,39]. Residue Y468 (EcGUS numbering) collaborates in the proper orientation of the nucleophilic glutamate for catalysis while residue Y472 contributes to the recognition of the carboxylate moiety of the glucuronic acid belonging to an aromatic cage critically involved in the binding of estrogen-glucuronides (Figure 2a) [32,34].
The GH2 family includes, in addition to GUS enzymes, β-galacturonidases (GalAses), enzymes capable to cleave the sugar conjugates of the epimer galacturonate (GalA), instead of glucuronic acid, and also hybrid GUS/GalAses, enzymes with a catalytic machinery capable to differentiate and process both epimeric substrates [44]. In the case of GalAses, an arginine residue (R337 in Eisenbergiella tayi GalAse (EtGalAse)) is the responsible for the GalA epimer selectivity. In fact, a mutation of this residue abolished GalAse activity while conferring GUS activity, as occurs in EtGalAse [44]. In hybrid GUS/GalAse enzymes, including BuGUS-1 and FsGUS, the specific arginine residue of GalAse is replaced by a tyrosine (YW motif), that occupies the same position, recognizing the 3-hydroxyl group of both epimers and the axial 4-hydroxyl of GalA [34,44]. In GUS enzymes the YW motif is occupied by small residues, hence, these sequences are target signatures to differentiate gut microbial GUS. Interestingly, some hybrid GUS/GalAses, such as BuGUS-1, that combine both activities, are competent to metabolize estrogen conjugates showing similar catalytic efficiency for either estrone-3-glucuronide or estradiol-17-glucuronide [44].
In addition, the active center of the GUS enzymes is coordinated by a network of water molecules. Seven water molecules were found in the Bifidobacterium dentium GUS interacting with the inhibitor and surrounding residues. Some of these water molecules (W2 and W3) are also found in similar positions in the active center of microbial EcGUS (PDB 3K4D) interacting with the 3-hydroxyl group of the inhibitor and the catalytic residue E413, and hence are key players in the binding and deconjugation of estrogen-glucuronides (Figure 1b and Figure 2c) [37]. Interestingly, we have performed active site comparison of these GUS enzymes revealing that both water molecules are absent in hybrid GUS/GalAses, such as BuGUS1 (PDB 6D6W) (Figure 2c), and its interactions are mimicked by residues W383 and Y382 (BuGUS-1 numbering), belonging to the YW motif, highlighting the relevance of these interactions for the organization of the active center in GUS enzymes. We have observed that a water molecule (W1) (Figure 1b) interacting with H296 in EcGUS helps to bind the 3-hydroxyl group of both epimers being present in either GUS, hybrid GUS/GalAses and GalAses, thereby demonstrating its relevance for substrate binding including estrogen-glucuronides.
Another key element for the binding of glucuronic acid containing substrates to GUS enzymes is the presence of flexible loops surrounding the active center (Table 2). As a consequence of the wide variability in the length of the amino acid chain of loop 1 and loop 2 (residues 356-380 and 416-419 in EcGUS, respectively), a classification has been created including 7 categories: Loop 1 (bacterial loop) (>15 residues), mini-Loop 1 (10-15 residues), Loop 2 (≥12 residues), mini-Loop 2 (9-11 residues), No loop and no coverage. The latter category is used in case that sequence information of one of the loops is missing [34]. As a general rule, those enzymes whose active center is surrounded by longer loops are specialized for the binding and processing of smaller molecule glucuronides, such as p-nitrophenol glucuronide, while those containing smaller loops, which possess a more open active site, allow for the accommodation and processing of larger substrate-glucuronides, as is the case of a heparosan nonasaccharide substrate [34]. Furthermore, those GUS enzymes specialized in processing smaller substrate-glucuronides, as is the case of the Loop 1 group, tend to have an intracellular location, while the vast majority of them, with the capacity to deconjugate larger substrates, are located in the periplasmic space, as indicated by the fact that 78% of GUS enzymes found in human gut microbiota belongs to Mini-Loop 1, Mini-Loop 2, Mini-Loop 1,2 or No Loop categories [31]. Nevertheless, a few Loop 1 enzymes reveal low affinity towards small substrates, as found in Ruminococcus gnavus GUS, likely due to an alpha helix conformation observed at Loop 1 [35]. The combination of GUS enzymes belonging to different loop categories permits these enzymes to process a wide variability of glucuronic acid conjugated substrates in the intestinal tract [31]. Those GUS enzymes capable to process glucuronides of estrogen (estrone-3-glucuronide or estradiol-17-glucuronide) are included mainly in the Loop 1, as is the case of EcGUS or Clostridium perfringens GUS (CpGUS), and mini-Loop 1 categories which also includes Bacteroides fragilis and Roseburia hominis GUS (Figure 3b) [32]. However, Loop 1 GUS enzymes represent those with the highest catalytic efficiency towards estrogen glucuronides due to its high content in aromatic residues that facilitate its binding, showing a clear preference towards estrone-3-glucuronide likely due to the presence of an extra planar aromatic ring not present in estradiol-17-glucuronide and also to the different position of the methyl group. Interestingly, GUS enzymes that possess an active site able to accommodate flavin-mononucleotide (FMN) cofactors are also capable of processing glucuronides of estrogen [32]. Since FMN-binding GUS enzymes possess a wider active center they are able to process both estradiol-conjugates with the same efficiency. The steric occlusion caused by a novel 25 residues long loop nearby the active site in Faecalibacterium prausnitzii GUS, a mini-Loop 1 enzyme, appears responsible for its surprisingly low processing activity on estrogen-glucuronide substrates [32].
7. Inhibitors of β-glucuronidase as potential anti-cancer treatment
As mentioned before, gut microbiome GUS enzymes, as an active member of the estrobolome, are capable of metabolizing estrogens and other toxic compounds masked with glucuronic acid, as is the case of the colon carcinogen azoxymethane, releasing them into the gut and causing adverse effects that might sometimes be severe including tumorigenesis [118]. However, blocking these enzymes in the gut by developing specific inhibitors could prevent these adverse effects. An extensive review on GUS enzyme inhibitors has been published by Paul Awolade el al. in 2020 [78].
So far there are a number of selective inhibitors targeting human gut microbiota GUS enzymes which mostly affect GUS activity, even taking into account that druggability studies performed previously reflected the limited predisposition of their active sites as a drug target [45]. Among them are included some examples of prenylflavonoids (Sanggenon C and Kuwanon G), índole-based compounds (Bazedoxifene), piperazines (Amoxapine) or phenoxy thiophene sulphonamides (BRITE-355252) showing high inhibitory potency in the low micromolar or even low nanomolar range [78].
It has been proven that most of the compounds that exhibits high potency towards EcGUS specifically bind to Loop 1 (Inhibitors 2, 3, R1) [40]. Interestingly, some of these compounds (R1, R3, 7, and 8) show no inhibition towards other Loop 1 members like the Firmicute enzymes CpGUS and Streptococcus agalactiae GUS [39]. The same authors tested the capacity to reduce Irinotecan-induced diarrhea with Inhibitor 1 and R1 in mice, observing an important reduction of the symptoms with Inhibitor 1 relative to Inhibitor R1 [119]. Inhibitor 1 was also proven effective in the protection against the adverse effects caused by nonsteroidal anti-inflammatory drugs [86]. Similar effects were obtained with other chemotypes including pyrazolo [4,3-c]quinolines or amoxapine which showed comparable efficacy as Inhibitor 1 in the reduction of the side effects of Irinotecan and, in addition, reduced tumour growth in mice [119,120]. Also probiotic lactic acid bacteria have beneficial effects by reducing GUS activity in colorectal cancer [118].
At the moment only one inhibitor of gut microbiome GUS enzymes, UNC10201652 (4-(8-(piperazin-1-yl)-1,2,3,4-tetrahydro- [1,2,3]triazino [4',5':4,5]thieno [2,3-c]isoquinolin-5-yl)morpholine) or its derivatives, capable of potently inhibiting deconjugation of estrone or estradiol glucuronides, including on fecal samples, has been tested with potency in the low nM range [32,43]. This compound, which contains a piperazine ring, is a slow–binding inhibitor that targets a catalytic intermediate showing effect, preferentially, on those GUS belonging to the Loop 1 group and it has been crystallized bound to CpGUS (PDB 6CXS) (Figure 4) [32,38,43]. Other piperazine-containing compounds have been designed but with less potency [43]. The fact that the enzymes belonging to the Loop 1 group are relatively rare (~5.5% of the GUS enzymes identified in the human intestinal microbiome up to date) [31], explains why the UNC10201652 inhibitor alone is not capable of inhibiting all GUS enzymes belonging to the estrobolome [32]. Nevertheless, a weak inhibition was found in No Loop enzymes (Bacteroides dorei GUS) indicating that other interactions different from those of Loop 1 are involved [35]. It has been hypothesized that this inhibitor could be an effective candidate to prevent tumour growth in an HR+ breast cancer model, and despite the fact that it has been found to be effective in preventing estrogen-glucuronide deconjugation in living E. coli cells, it has not been proven effective in transgenic mouse models that exhibit a progression similar to human breast cancer [32]. A study carried out by Bhatt and co-workers compared the in vitro efficacy of the GUS inhibitors UNC10201652 and Inhibitor 1, regarding irinotecan-induced gut toxicity, concluding that the first one exhibits higher potency and efficacy towards Loop 1 enzymes as a result of a stronger interaction network in the active site [38]. In this study the UNC10201652 inhibitor was tested in vivo observing an improvement in tumour regression, a survival increase and a mitigation of the side effects using a breast cancer mouse model, without significantly affecting the overall metabolism of the host [38,121].
8. Future perspective
The term estrobolome first entered the scientific vocabulary in 2011 and defines the set of all enteric bacteria capable of metabolizing estrogens (Figure 1). These bacteria modulate the enterohepatic circulation of estrogens, thus affecting their plasma levels. Much of our knowledge of the enterohepatic circulation and estrogen metabolism is based on research conducted during the 1970s. Recent advances in analytical methods (NGS and mass spectrometry), including accurate and sensitive measurement of different conjugated and unconjugated estrogens in serum and urine, should provide tools for meaningful comparisons between subjects. Animal and cellular models serve to test hypotheses under controlled conditions. However, more large-scale observational studies in humans are needed to identify and confirm associations while controlling for other variables (genetic, epigenetic, dietary, and environmental) that affect the estrobolome and confound cancer risk.
Several authors, as discussed throughout this review, suggest that intestinal microbial β-glucuronidase enzymes are especially important in total estrogen circulation; therefore, it has been hypothesized that a estrobolome rich in deconjugating GUS enzymes would be a risk factor in breast cancer (BC). Characterization of the structural and enzymological properties of these estrogen-reactivating proteins, the GUS enzymes, in bacteria found in normal individuals and in breast cancer patients could provide us with multiple pieces of information for the modification or modulation of these enzymes. GUS enzymes from the intestinal microbiota share mostly the same structural architecture and an active center with wide variability but with well-defined characteristic sequences that allow their rapid identification. The processing of a wide variety of substrate sizes in the intestinal tract is made possible by the wide variability of existing GUS that show important differences in the length of the loops surrounding the active center, with those possessing loop 1 being the most efficient processors of estrogenic glucuronides and, at the same time, the best targets for current inhibitors. Although there is a wide variety of compounds capable of inhibiting the GUS enzyme, so far few of them have demonstrated their efficacy in reducing symptoms and tumor regression, and only one of them has shown high potency in the deconjugation of estrone or estradiol glucuronides. The active sites explored in very few studies should be further inspected for their possible role in the glucuronidation of glucuronidated estrogens. The study of these catalytic sites could help modify GUS enzymes to avoid their estrogen deconjugation potential. These modifications could include alterations to the structure of the active sites involved in deglucuronidation by inducing point mutations in the GUS gene and the deletion of conserved protein motifs, thereby inhibiting their estrogenic reactivation potential. However, genetic manipulation of the human microbiome is not a readily feasible approach. In this scenario, manipulations at the dietary level, such as probiotic or postbiotic supply, are very easy to perform, therefore, modification of the estrobolome at the dietary level could be useful to reduce the risk of breast cancer by inhibiting the reactivation of this estrogen-associated protein.
9. Conclusions
In the near future, induced changes in the estrobolome could be considered a non-hereditary risk factor for breast cancer, mainly estrogen receptor positive, and potentially modulable by a personalized preventive and therapeutic approach. Likewise, in already established BC, the microbiota could be a prognostic and predictive factor for treatment response (resistance) and/or its side effects and toxicities. Furthermore, modification of the microbiota could be used to improve the outcomes of therapies in patients with BC.
Thus, metabolic disturbances of the gut estrogen-microbiota axis (estrobolome) combined with variations in individual-specific estrogen levels could contribute to an increased risk of hormone-mediated malignancies, including BC. In the future, interventions involving the use of prebiotics, probiotics, postbiotics, synbiotics, and/or antimicrobial agents could be considered. These therapeutic strategies, easy to implement in clinical practice, can be specifically designed to modulate gut bacterial populations with BGUS activity and decrease the risk of estrogen-related BC or, after cancer diagnosis, become adjuvant treatments.
Ethics Statement
This manuscript does not contain any studies with human participants or animals performed by any of the authors. The study did not require ethical approval.
Author Contributions
The present manuscript is the result of original work by the authors. MLFM and FGO: conception and design, development of methodology, acquisition, analysis, and interpretation of data. MLFM, FGO and LSG: writing, review, and/or revision of the manuscript. ALLC: manuscript supervision. All authors contributed to the article and approved the submitted version.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. All the authors declare no conflict of interest.
References
- Ferlay, J.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory Today.
- Saraiva, D.; Guadalupe Cabral, M.; Jacinto, A.; Braga, S. How Many Diseases Is Triple Negative Breast Cancer: The Protagonism of the Immune Microenvironment. ESMO Open 2017, 2, e000208. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F.; Kyriakides, S.; Ohno, S.; Penault-Llorca, F.; Poortmans, P.; Rubio, I.T.; Zackrisson, S.; Senkus, E. Early Breast Cancer: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Annals of Oncology 2019, 30, 1194–1220. [Google Scholar] [CrossRef] [PubMed]
- Saarinen, N.M.; Wärri, A.; Airio, M.; Smeds, A.; Mäkelä, S. Role of Dietary Lignans in the Reduction of Breast Cancer Risk. Mol Nutr Food Res 2007, 51, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Macpherson, A.J.; Harris, N.L. Interactions between Commensal Intestinal Bacteria and the Immune System. Nat Rev Immunol 2004, 4, 478–485. [Google Scholar] [CrossRef]
- Hooper, L.V.; Gordon, J.I. Commensal Host-Bacterial Relationships in the Gut. Science (1979) 2001, 292, 1115–1118. [Google Scholar] [CrossRef]
- Roy, S.; Trinchieri, G. Microbiota: A Key Orchestrator of Cancer Therapy. Nat Rev Cancer 2017, 17, 271–285. [Google Scholar] [CrossRef]
- Sommer, F.; Anderson, J.M.; Bharti, R.; Raes, J.; Rosenstiel, P. The Resilience of the Intestinal Microbiota Influences Health and Disease. Nat Rev Microbiol 2017, 15, 630–638. [Google Scholar] [CrossRef]
- Stewart, C.J.; Ajami, N.J.; O’Brien, J.L.; Hutchinson, D.S.; Smith, D.P.; Wong, M.C.; Ross, M.C.; Lloyd, R.E.; Doddapaneni, H.; Metcalf, G.A.; et al. Temporal Development of the Gut Microbiome in Early Childhood from the TEDDY Study. Nature 2018, 562, 583–588. [Google Scholar] [CrossRef]
- Clarridge, J.E. Impact of 16S RRNA Gene Sequence Analysis for Identification of Bacteria on Clinical Microbiology and Infectious Diseases. Clin Microbiol Rev 2004, 17, 840–862. [Google Scholar] [CrossRef]
- Thomas, T.; Gilbert, J.; Meyer, F. Metagenomics - a Guide from Sampling to Data Analysis. Microb Inform Exp 2012, 2, 3. [Google Scholar] [CrossRef]
- Zitvogel, L.; Galluzzi, L.; Viaud, S.; Vétizou, M.; Daillère, R.; Merad, M.; Kroemer, G. Cancer and the Gut Microbiota: An Unexpected Link. Sci Transl Med 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- YANG J BREAST CANCER. 2016.
- Dzutsev, A.; Goldszmid, R.S.; Viaud, S.; Zitvogel, L.; Trinchieri, G. The Role of the Microbiota in Inflammation, Carcinogenesis, and Cancer Therapy. Eur J Immunol 2015, 45, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Lobionda, S.; Sittipo, P.; Kwon, H.Y.; Lee, Y.K. The Role of Gut Microbiota in Intestinal Inflammation with Respect to Diet and Extrinsic Stressors. Microorganisms 2019, 7, 271. [Google Scholar] [CrossRef]
- Nogueira, A.R.; Shoenfeld, Y. Microbiome and Autoimmune Diseases: Cause and Effect Relationship. Curr Opin Rheumatol 2019, 31, 471–474. [Google Scholar] [CrossRef] [PubMed]
- Mikó, E.; Kovács, T.; Sebő, É.; Tóth, J.; Csonka, T.; Ujlaki, G.; Sipos, A.; Szabó, J.; Méhes, G.; Bai, P. Microbiome—Microbial Metabolome—Cancer Cell Interactions in Breast Cancer—Familiar, but Unexplored. Cells 2019, 8, 293. [Google Scholar] [CrossRef]
- Maynard, C.L.; Elson, C.O.; Hatton, R.D.; Weaver, C.T. Reciprocal Interactions of the Intestinal Microbiota and Immune System. Nature 2012, 489, 231–241. [Google Scholar] [CrossRef]
- Shapira, I.; Sultan, K.; Lee, A.; Taioli, E. Evolving Concepts: How Diet and the Intestinal Microbiome Act as Modulators of Breast Malignancy. ISRN Oncol 2013, 2013, 1–10. [Google Scholar] [CrossRef]
- Hullar, M.A.J.; Fu, B.C. Diet, the Gut Microbiome, and Epigenetics. The Cancer Journal 2014, 20, 170–175. [Google Scholar] [CrossRef]
- García-Castillo, V.; Sanhueza, E.; McNerney, E.; Onate, S.A.; García, A. Microbiota Dysbiosis: A New Piece in the Understanding of the Carcinogenesis Puzzle. J Med Microbiol 2016, 65, 1347–1362. [Google Scholar] [CrossRef]
- Plottel, C.S.; Blaser, M.J. Microbiome and Malignancy. Cell Host Microbe 2011, 10, 324–335. [Google Scholar] [CrossRef]
- Kwa, M.P.C.B.M.A.S. The Intestinal Microbiome and Estrogen Receptor–Positive Female Breast Cancer. JNCI: Journal of the National Cancer Institute 2016. [Google Scholar] [CrossRef]
- Zhu, B.T.; Han, G.-Z.; Shim, J.-Y.; Wen, Y.; Jiang, X.-R. Quantitative Structure-Activity Relationship of Various Endogenous Estrogen Metabolites for Human Estrogen Receptor α and β Subtypes: Insights into the Structural Determinants Favoring a Differential Subtype Binding. Endocrinology 2006, 147, 4132–4150. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, N.; Das, G. Metabolic Reprogramming in Breast Cancer and Its Therapeutic Implications. Cells 2019, 8, 89. [Google Scholar] [CrossRef] [PubMed]
- Kovács, T.; Mikó, E.; Ujlaki, G.; Sári, Z.; Bai, P. The Microbiome as a Component of the Tumor Microenvironment. 2020; 137–153. [Google Scholar]
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and Transfer of Mitochondrial DNA via Exosomes Regulate Escape from Dormancy in Hormonal Therapy-Resistant Breast Cancer. Proceedings of the National Academy of Sciences 2017, 114. [Google Scholar] [CrossRef]
- Iván, J.; Major, E.; Sipos, A.; Kovács, K.; Horváth, D.; Tamás, I.; Bay, P.; Dombrádi, V.; Lontay, B. The Short-Chain Fatty Acid Propionate Inhibits Adipogenic Differentiation of Human Chorion-Derived Mesenchymal Stem Cells Through the Free Fatty Acid Receptor 2. Stem Cells Dev 2017, 26, 1724–1733. [Google Scholar] [CrossRef]
- Goedert, J.J.; Jones, G.; Hua, X.; Xu, X.; Yu, G.; Flores, R.; Falk, R.T.; Gail, M.H.; Shi, J.; Ravel, J.; et al. Investigation of the Association Between the Fecal Microbiota and Breast Cancer in Postmenopausal Women: A Population-Based Case-Control Pilot Study. JNCI: Journal of the National Cancer Institute 2015, 107. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A Human Gut Microbial Gene Catalogue Established by Metagenomic Sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef]
- Pollet, R.M.; D’Agostino, E.H.; Walton, W.G.; Xu, Y.; Little, M.S.; Biernat, K.A.; Pellock, S.J.; Patterson, L.M.; Creekmore, B.C.; Isenberg, H.N.; et al. An Atlas of β-Glucuronidases in the Human Intestinal Microbiome. Structure 2017, 25, 967–977. [Google Scholar] [CrossRef]
- Ervin, S.M.; Li, H.; Lim, L.; Roberts, L.R.; Liang, X.; Mani, S.; Redinbo, M.R. Gut Microbial β-Glucuronidases Reactivate Estrogens as Components of the Estrobolome That Reactivate Estrogens. J Biol Chem 2019, 294, 18586–18599. [Google Scholar] [CrossRef]
- Jariwala, P.B.; Pellock, S.J.; Goldfarb, D.; Cloer, E.W.; Artola, M.; Simpson, J.B.; Bhatt, A.P.; Walton, W.G.; Roberts, L.R.; Major, M.B.; et al. Discovering the Microbial Enzymes Driving Drug Toxicity with Activity-Based Protein Profiling. ACS Chem Biol 2020, 15, 217–225. [Google Scholar] [CrossRef]
- Pellock, S.J.; Walton, W.G.; Biernat, K.A.; Torres-Rivera, D.; Creekmore, B.C.; Xu, Y.; Liu, J.; Tripathy, A.; Stewart, L.J.; Redinbo, M.R. Three Structurally and Functionally Distinct β-Glucuronidases from the Human Gut Microbe Bacteroides Uniformis. Journal of Biological Chemistry 2018, 293, 18559–18573. [Google Scholar] [CrossRef] [PubMed]
- Biernat, K.A.; Pellock, S.J.; Bhatt, A.P.; Bivins, M.M.; Walton, W.G.; Tran, B.N.T.; Wei, L.; Snider, M.C.; Cesmat, A.P.; Tripathy, A.; et al. Structure, Function, and Inhibition of Drug Reactivating Human Gut Microbial β-Glucuronidases. Sci Rep 2019, 9, 825. [Google Scholar] [CrossRef] [PubMed]
- Bang, W.Y.; Ban, O.-H.; Lee, B.S.; Oh, S.; Park, C.; Park, M.-K.; Jung, S.K.; Yang, J.; Jung, Y.H. Genomic-, Phenotypic-, and Toxicity-Based Safety Assessment and Probiotic Potency of Bacillus Coagulans IDCC 1201 Isolated from Green Malt. J Ind Microbiol Biotechnol 2021. [Google Scholar] [CrossRef]
- Lin, H.-Y.; Chen, C.-Y.; Lin, T.-C.; Yeh, L.-F.; Hsieh, W.-C.; Gao, S.; Burnouf, P.-A.; Chen, B.-M.; Hsieh, T.-J.; Dashnyam, P.; et al. Entropy-Driven Binding of Gut Bacterial β-Glucuronidase Inhibitors Ameliorates Irinotecan-Induced Toxicity. Commun Biol 2021, 4, 280. [Google Scholar] [CrossRef]
- Bhatt, A.P.; Pellock, S.J.; Biernat, K.A.; Walton, W.G.; Wallace, B.D.; Creekmore, B.C.; Letertre, M.M.; Swann, J.R.; Wilson, I.D.; Roques, J.R.; et al. Targeted Inhibition of Gut Bacterial β-Glucuronidase Activity Enhances Anticancer Drug Efficacy. Proceedings of the National Academy of Sciences 2020, 117, 7374–7381. [Google Scholar] [CrossRef]
- Wallace, B.D.; Roberts, A.B.; Pollet, R.M.; Ingle, J.D.; Biernat, K.A.; Pellock, S.J.; Venkatesh, M.K.; Guthrie, L.; O’Neal, S.K.; Robinson, S.J.; et al. Structure and Inhibition of Microbiome β-Glucuronidases Essential to the Alleviation of Cancer Drug Toxicity. Chem Biol 2015, 22, 1238–1249. [Google Scholar] [CrossRef]
- Wallace, B.D.; Wang, H.; Lane, K.T.; Scott, J.E.; Orans, J.; Koo, J.S.; Venkatesh, M.; Jobin, C.; Yeh, L.-A.; Mani, S.; et al. Alleviating Cancer Drug Toxicity by Inhibiting a Bacterial Enzyme. Science (1979) 2010, 330, 831–835. [Google Scholar] [CrossRef]
- Roberts, A.B.; Wallace, B.D.; Venkatesh, M.K.; Mani, S.; Redinbo, M.R. Molecular Insights into Microbial β -Glucuronidase Inhibition to Abrogate CPT-11 Toxicity. Mol Pharmacol 2013, 84, 208–217. [Google Scholar] [CrossRef]
- de Boer, C.; Armstrong, Z.; Lit, V.A.J.; Barash, U.; Ruijgrok, G.; Boyango, I.; Weitzenberg, M.M.; Schröder, S.P.; Sarris, A.J.C.; Meeuwenoord, N.J.; et al. Mechanism-Based Heparanase Inhibitors Reduce Cancer Metastasis in Vivo. Proceedings of the National Academy of Sciences 2022, 119. [Google Scholar] [CrossRef]
- Pellock, S.J.; Creekmore, B.C.; Walton, W.G.; Mehta, N.; Biernat, K.A.; Cesmat, A.P.; Ariyarathna, Y.; Dunn, Z.D.; Li, B.; Jin, J.; et al. Gut Microbial β-Glucuronidase Inhibition via Catalytic Cycle Interception. ACS Cent Sci 2018, 4, 868–879. [Google Scholar] [CrossRef]
- Pellock, S.J.; Walton, W.G.; Redinbo, M.R. Selecting a Single Stereocenter: The Molecular Nuances That Differentiate β-Hexuronidases in the Human Gut Microbiome. Biochemistry 2019, 58, 1311–1317. [Google Scholar] [CrossRef] [PubMed]
- Muccee, F.; Ghazanfar, S.; Ajmal, W.; Al-Zahrani, M. In-Silico Characterization of Estrogen Reactivating β-Glucuronidase Enzyme in GIT Associated Microbiota of Normal Human and Breast Cancer Patients. Genes (Basel) 2022, 13, 1545. [Google Scholar] [CrossRef] [PubMed]
- Komorowski, A.S.; Pezo, R.C. Untapped “-Omics”: The Microbial Metagenome, Estrobolome, and Their Influence on the Development of Breast Cancer and Response to Treatment. Breast Cancer Res Treat 2020, 179, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Leeming, E.R.; Johnson, A.J.; Spector, T.D.; Le Roy, C.I. Effect of Diet on the Gut Microbiota: Rethinking Intervention Duration. Nutrients 2019, 11, 2862. [Google Scholar] [CrossRef] [PubMed]
- Teng, N.M.Y.; Price, C.A.; McKee, A.M.; Hall, L.J.; Robinson, S.D. Exploring the Impact of Gut Microbiota and Diet on Breast Cancer Risk and Progression. Int J Cancer 2021, 149, 494–504. [Google Scholar] [CrossRef]
- Bodai, B.I.; Nakata, T.E.; Wong, W.T.; Clark, D.R.; Lawenda, S.; Tsou, C.; Liu, R.; Shiue, L.; Cooper, N.; Rehbein, M.; et al. Lifestyle Medicine: A Brief Review of Its Dramatic Impact on Health and Survival. Perm J 2018, 22. [Google Scholar] [CrossRef]
- Sinicrope, F.A.; Dannenberg, A.J. Obesity and Breast Cancer Prognosis: Weight of the Evidence. Journal of Clinical Oncology 2011, 29, 4–7. [Google Scholar] [CrossRef]
- Lee, K.; Kruper, L.; Dieli-Conwright, C.M.; Mortimer, J.E. The Impact of Obesity on Breast Cancer Diagnosis and Treatment. Curr Oncol Rep 2019, 21, 41. [Google Scholar] [CrossRef]
- Keum, N.; Greenwood, D.C.; Lee, D.H.; Kim, R.; Aune, D.; Ju, W.; Hu, F.B.; Giovannucci, E.L. Adult Weight Gain and Adiposity-Related Cancers: A Dose-Response Meta-Analysis of Prospective Observational Studies. JNCI: Journal of the National Cancer Institute 2015, 107. [Google Scholar] [CrossRef]
- Ewertz, M.; Jensen, M.-B.; Gunnarsdóttir, K.Á.; Højris, I.; Jakobsen, E.H.; Nielsen, D.; Stenbygaard, L.E.; Tange, U.B.; Cold, S. Effect of Obesity on Prognosis After Early-Stage Breast Cancer. Journal of Clinical Oncology 2011, 29, 25–31. [Google Scholar] [CrossRef]
- Abenavoli, L.; Scarpellini, E.; Colica, C.; Boccuto, L.; Salehi, B.; Sharifi-Rad, J.; Aiello, V.; Romano, B.; De Lorenzo, A.; Izzo, A.A.; et al. Gut Microbiota and Obesity: A Role for Probiotics. Nutrients 2019, 11, 2690. [Google Scholar] [CrossRef] [PubMed]
- Bergom, C.; Kelly, T.; Bedi, M.; Saeed, H.; Prior, P.; Currey, A.D.; Wilson, J.; White, J. Does Size Matter: Examining the Association of BMI with Breast Cancer Recurrence and Survival in an Early Stage Breast Cancer Cohort with a High Median BMI. International Journal of Radiation Oncology*Biology*Physics 2014, 90, S47–S48. [Google Scholar] [CrossRef]
- Naaman, S.C.; Shen, S.; Zeytinoglu, M.; Iyengar, N.M. Obesity and Breast Cancer Risk: The Oncogenic Implications of Metabolic Dysregulation. J Clin Endocrinol Metab 2022, 107, 2154–2166. [Google Scholar] [CrossRef] [PubMed]
- Picon-Ruiz, M.; Morata-Tarifa, C.; Valle-Goffin, J.J.; Friedman, E.R.; Slingerland, J.M. Obesity and Adverse Breast Cancer Risk and Outcome: Mechanistic Insights and Strategies for Intervention. CA Cancer J Clin 2017, 67, 378–397. [Google Scholar] [CrossRef] [PubMed]
- Starek-Świechowicz, B.; Budziszewska, B.; Starek, A. Alcohol and Breast Cancer. Pharmacological Reports 2023, 75, 69–84. [Google Scholar] [CrossRef]
- Assi, N.; Rinaldi, S.; Viallon, V.; Dashti, S.G.; Dossus, L.; Fournier, A.; Cervenka, I.; Kvaskoff, M.; Turzanski-Fortner, R.; Bergmann, M.; et al. Mediation Analysis of the Alcohol-postmenopausal Breast Cancer Relationship by Sex Hormones in the EPIC Cohort. Int J Cancer 2020, 146, 759–768. [Google Scholar] [CrossRef]
- Onland-Moret, N.C.; Peeters, P.H.M.; van der Schouw, Y.T.; Grobbee, D.E.; van Gils, C.H. Alcohol and Endogenous Sex Steroid Levels in Postmenopausal Women: A Cross-Sectional Study. J Clin Endocrinol Metab 2005, 90, 1414–1419. [Google Scholar] [CrossRef]
- Dorgan, J.F.; Baer, D.J.; Albert, P.S.; Judd, J.T.; Brown, E.D.; Corle, D.K.; Campbell, W.S.; Hartman, T.J.; Tejpar, A.A.; Clevidence, B.A.; et al. Serum Hormones and the Alcohol-Breast Cancer Association in Postmenopausal Women. JNCI Journal of the National Cancer Institute 2001, 93, 710–715. [Google Scholar] [CrossRef]
- Mahabir, S.; Baer, D.J.; Johnson, L.L.; Dorgan, J.F.; Campbell, W.; Brown, E.; Hartman, T.J.; Clevidence, B.; Albanes, D.; Judd, J.T.; et al. The Effects of Moderate Alcohol Supplementation on Estrone Sulfate and DHEAS in Postmenopausal Women in a Controlled Feeding Study. Nutr J 2004, 3, 11. [Google Scholar] [CrossRef]
- Seitz, H.K.; Maurer, B. The Relationship between Alcohol Metabolism, Estrogen Levels, and Breast Cancer Risk. Alcohol Res Health 2007, 30, 42–43. [Google Scholar]
- Liu, Y.; Nguyen, N.; Colditz, G.A. Links between Alcohol Consumption and Breast Cancer: A Look at the Evidence. Women’s Health 2015, 11, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Freudenheim, J.L. Alcohols Effects on Breast Cancer in Women. Alcohol Res 2020, 40. [Google Scholar] [CrossRef] [PubMed]
- Etique, N.; Chardard, D.; Chesnel, A.; Merlin, J.-L.; Flament, S.; Grillier-Vuissoz, I. Ethanol Stimulates Proliferation, ERalpha and Aromatase Expression in MCF-7 Human Breast Cancer Cells. Int J Mol Med 2004, 13, 149–155. [Google Scholar] [PubMed]
- Fan, S.; Meng, Q.; Gao, B.; Grossman, J.; Yadegari, M.; Goldberg, I.D.; Rosen, E.M. Alcohol Stimulates Estrogen Receptor Signaling in Human Breast Cancer Cell Lines. Cancer Res 2000, 60, 5635–5639. [Google Scholar]
- Bode, J.C.; Bode, C.; Heidelbach, R.; Dürr, H.K.; Martini, G.A. Jejunal Microflora in Patients with Chronic Alcohol Abuse. Hepatogastroenterology 1984, 31, 30–34. [Google Scholar] [PubMed]
- Mutlu, E.; Keshavarzian, A.; Engen, P.; Forsyth, C.B.; Sikaroodi, M.; Gillevet, P. Intestinal Dysbiosis: A Possible Mechanism of Alcohol-Induced Endotoxemia and Alcoholic Steatohepatitis in Rats. Alcohol Clin Exp Res 2009, 33, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.T.; Bui, Q.D.; Weisz, J.; Liehr, J.G. Conversion of Estrone to 2- and 4-Hydroxyestrone by Hamster Kidney and Liver Microsomes: Implications for the Mechanism of Estrogen-Induced Carcinogenesis. Endocrinology 1994, 135, 1772–1779. [Google Scholar] [CrossRef]
- Dao, M.C.; Everard, A.; Aron-Wisnewsky, J.; Sokolovska, N.; Prifti, E.; Verger, E.O.; Kayser, B.D.; Levenez, F.; Chilloux, J.; Hoyles, L.; et al. Akkermansia Muciniphila and Improved Metabolic Health during a Dietary Intervention in Obesity: Relationship with Gut Microbiome Richness and Ecology. Gut 2016, 65, 426–436. [Google Scholar] [CrossRef]
- Geerlings, S.; Kostopoulos, I.; de Vos, W.; Belzer, C. Akkermansia Muciniphila in the Human Gastrointestinal Tract: When, Where, and How? Microorganisms 2018, 6, 75. [Google Scholar] [CrossRef]
- Evans, J.M.; Morris, L.S.; Marchesi, J.R. The Gut Microbiome: The Role of a Virtual Organ in the Endocrinology of the Host. Journal of Endocrinology 2013, 218, R37–R47. [Google Scholar] [CrossRef]
- Laborda-Illanes, A.; Sanchez-Alcoholado, L.; Dominguez-Recio, M.E.; Jimenez-Rodriguez, B.; Lavado, R.; Comino-Méndez, I.; Alba, E.; Queipo-Ortuño, M.I. Breast and Gut Microbiota Action Mechanisms in Breast Cancer Pathogenesis and Treatment. Cancers (Basel) 2020, 12, 2465. [Google Scholar] [CrossRef] [PubMed]
- Klement, R.; Pazienza, V. Impact of Different Types of Diet on Gut Microbiota Profiles and Cancer Prevention and Treatment. Medicina (B Aires) 2019, 55, 84. [Google Scholar] [CrossRef]
- Ostan, R.; Lanzarini, C.; Pini, E.; Scurti, M.; Vianello, D.; Bertarelli, C.; Fabbri, C.; Izzi, M.; Palmas, G.; Biondi, F.; et al. Inflammaging and Cancer: A Challenge for the Mediterranean Diet. Nutrients 2015, 7, 2589–2621. [Google Scholar] [CrossRef] [PubMed]
- Caira, M.R.; Lonescu, C. Drug Metabolism: Current Concepts; 1st ed.; Springer Dordrecht: Midtown Manhattan, New York City, USA, 2010; Vol. 7. [Google Scholar]
- Awolade, P.; Cele, N.; Kerru, N.; Gummidi, L.; Oluwakemi, E.; Singh, P. Therapeutic Significance of β-Glucuronidase Activity and Its Inhibitors: A Review. Eur J Med Chem 2020, 187, 111921. [Google Scholar] [CrossRef] [PubMed]
- ElRakaiby, M.; Dutilh, B.E.; Rizkallah, M.R.; Boleij, A.; Cole, J.N.; Aziz, R.K. Pharmacomicrobiomics: The Impact of Human Microbiome Variations on Systems Pharmacology and Personalized Therapeutics. OMICS 2014, 18, 402–414. [Google Scholar] [CrossRef] [PubMed]
- Panebianco, C.; Andriulli, A.; Pazienza, V. Pharmacomicrobiomics: Exploiting the Drug-Microbiota Interactions in Anticancer Therapies. Microbiome 2018, 6, 92. [Google Scholar] [CrossRef]
- Smith, N.F.; Figg, W.D.; Sparreboom, A. Pharmacogenetics of Irinotecan Metabolism and Transport: An Update. Toxicology in Vitro 2006, 20, 163–175. [Google Scholar] [CrossRef]
- Parvez, M.M.; Basit, A.; Jariwala, P.B.; Gáborik, Z.; Kis, E.; Heyward, S.; Redinbo, M.R.; Prasad, B. Quantitative Investigation of Irinotecan Metabolism, Transport, and Gut Microbiome Activation. Drug Metabolism and Disposition 2021, 49, 683–693. [Google Scholar] [CrossRef]
- Lin, X.B.; Dieleman, L.A.; Ketabi, A.; Bibova, I.; Sawyer, M.B.; Xue, H.; Field, C.J.; Baracos, V.E.; Gänzle, M.G. Irinotecan (CPT-11) Chemotherapy Alters Intestinal Microbiota in Tumour Bearing Rats. PLoS One 2012, 7, e39764. [Google Scholar] [CrossRef]
- Stringer, A.M.; Gibson, R.J.; Bowen, J.M.; Logan, R.M.; Ashton, K.; Yeoh, A.S.J.; Al-Dasooqi, N.; Keefe, D.M.K. Irinotecan-Induced Mucositis Manifesting as Diarrhoea Corresponds with an Amended Intestinal Flora and Mucin Profile. Int J Exp Pathol 2009, 90, 489–499. [Google Scholar] [CrossRef]
- Little, M.S.; Pellock, S.J.; Walton, W.G.; Tripathy, A.; Redinbo, M.R. Structural Basis for the Regulation of β-Glucuronidase Expression by Human Gut Enterobacteriaceae. Proceedings of the National Academy of Sciences 2018, 115. [Google Scholar] [CrossRef] [PubMed]
- LoGuidice, A.; Wallace, B.D.; Bendel, L.; Redinbo, M.R.; Boelsterli, U.A. Pharmacologic Targeting of Bacterial β-Glucuronidase Alleviates Nonsteroidal Anti-Inflammatory Drug-Induced Enteropathy in Mice. Journal of Pharmacology and Experimental Therapeutics 2012, 341, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Saitta, K.S.; Zhang, C.; Lee, K.K.; Fujimoto, K.; Redinbo, M.R.; Boelsterli, U.A. Bacterial β-Glucuronidase Inhibition Protects Mice against Enteropathy Induced by Indomethacin, Ketoprofen or Diclofenac: Mode of Action and Pharmacokinetics. Xenobiotica 2014, 44, 28–35. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Kwon, H.; Lim, W.; Moon, B.-I. Staphylococcus Aureus-Derived Extracellular Vesicles Enhance the Efficacy of Endocrine Therapy in Breast Cancer Cells. J Clin Med 2022, 11, 2030. [Google Scholar] [CrossRef] [PubMed]
- Somboonporn, W.; Davis, S.R. Postmenopausal Testosterone Therapy and Breast Cancer Risk. Maturitas 2004, 49, 267–275. [Google Scholar] [CrossRef]
- Basit, A.; Amory, J.K.; Mettu, V.S.; Li, C.Y.; Heyward, S.; Jariwala, P.B.; Redinbo, M.R.; Prasad, B. Relevance of Human Aldoketoreductases and Microbial β -Glucuronidases in Testosterone Disposition. Drug Metabolism and Disposition 2023, 51, 427–435. [Google Scholar] [CrossRef]
- Zhang, J.; Lacroix, C.; Wortmann, E.; Ruscheweyh, H.-J.; Sunagawa, S.; Sturla, S.J.; Schwab, C. Gut Microbial Beta-Glucuronidase and Glycerol/Diol Dehydratase Activity Contribute to Dietary Heterocyclic Amine Biotransformation. BMC Microbiol 2019, 19, 99. [Google Scholar] [CrossRef]
- Landeka Jurčević, I.; Dora, M.; Guberović, I.; Petras, M.; Rimac Brnčić, S.; Đikić, D. Wine Lees Polyphenols as a Novel Functional Bioactive Compound in the Protection against Oxidative Stress and Hyperlipidemia. Food Technol Biotechnol 2017, 55. [Google Scholar] [CrossRef]
- Miranda, A.; Steluti, J.; Fisberg, R.; Marchioni, D. Association between Coffee Consumption and Its Polyphenols with Cardiovascular Risk Factors: A Population-Based Study. Nutrients 2017, 9, 276. [Google Scholar] [CrossRef]
- Rothenberg, D.; Zhou, C.; Zhang, L. A Review on the Weight-Loss Effects of Oxidized Tea Polyphenols. Molecules 2018, 23, 1176. [Google Scholar] [CrossRef]
- Pérez-Jiménez, J.; Saura-Calixto, F. Macromolecular Antioxidants or Non-Extractable Polyphenols in Fruit and Vegetables: Intake in Four European Countries. Food Research International 2015, 74, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Duda-Chodak, A.; Tarko, T.; Satora, P.; Sroka, P. Interaction of Dietary Compounds, Especially Polyphenols, with the Intestinal Microbiota: A Review. Eur J Nutr 2015, 54, 325–341. [Google Scholar] [CrossRef] [PubMed]
- Moga, M.; Dimienescu, O.; Arvatescu, C.; Mironescu, A.; Dracea, L.; Ples, L. The Role of Natural Polyphenols in the Prevention and Treatment of Cervical Cancer—An Overview. Molecules 2016, 21, 1055. [Google Scholar] [CrossRef] [PubMed]
- Goszcz, K.; Duthie, G.G.; Stewart, D.; Leslie, S.J.; Megson, I.L. Bioactive Polyphenols and Cardiovascular Disease: Chemical Antagonists, Pharmacological Agents or Xenobiotics That Drive an Adaptive Response? Br J Pharmacol 2017, 174, 1209–1225. [Google Scholar] [CrossRef] [PubMed]
- Nabavi, S.; Dean, O.; Turner, A.; Sureda, A.; Daglia, M.; Nabavi, S. Oxidative Stress and Post-Stroke Depression: Possible Therapeutic Role of Polyphenols? Curr Med Chem 2014, 22, 343–351. [Google Scholar] [CrossRef]
- Leonidas, D.; Hayes, J.; Kato, A.; Skamnaki, V.; Chatzileontiadou, D.; Kantsadi, A.; Kyriakis, E.; Chetter, B.; Stravodimos, G. Phytogenic Polyphenols as Glycogen Phosphorylase Inhibitors: The Potential of Triterpenes and Flavonoids for Glycaemic Control in Type 2 Diabetes. Curr Med Chem 2017, 24, 384–403. [Google Scholar] [CrossRef]
- Wang, X.-L.; Kim, H.-J.; Kang, S.-I.; Kim, S.-I.; Hur, H.-G. Production of Phytoestrogen S-Equol from Daidzein in Mixed Culture of Two Anaerobic Bacteria. Arch Microbiol 2007, 187, 155–160. [Google Scholar] [CrossRef]
- Yokoyama, S.; Niwa, T.; Osawa, T.; Suzuki, T. Characterization of an O-Desmethylangolensin-Producing Bacterium Isolated from Human Feces. Arch Microbiol 2010, 192, 15–22. [Google Scholar] [CrossRef]
- YOKOYAMA, S.; SUZUKI, T. Isolation and Characterization of a Novel Equol-Producing Bacterium from Human Feces. Biosci Biotechnol Biochem 2008, 72, 2660–2666. [Google Scholar] [CrossRef]
- Matthies, A.; Blaut, M.; Braune, A. Isolation of a Human Intestinal Bacterium Capable of Daidzein and Genistein Conversion. Appl Environ Microbiol 2009, 75, 1740–1744. [Google Scholar] [CrossRef]
- Raimondi, S.; Roncaglia, L.; De Lucia, M.; Amaretti, A.; Leonardi, A.; Pagnoni, U.M.; Rossi, M. Bioconversion of Soy Isoflavones Daidzin and Daidzein by Bifidobacterium Strains. Appl Microbiol Biotechnol 2009, 81, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Setchell, K.D.R.; Clerici, C. Equol: History, Chemistry, and Formation, J Nutr 2010, 140, 1355S–1362S. [Google Scholar] [CrossRef] [PubMed]
- Setchell, K.D.R.; Clerici, C. Equol: Pharmacokinetics and Biological Actions, J Nutr 2010, 140, 1363S–1368S. [Google Scholar] [CrossRef]
- Setchell, K.D.; Clerici, C.; Lephart, E.D.; Cole, S.J.; Heenan, C.; Castellani, D.; Wolfe, B.E.; Nechemias-Zimmer, L.; Brown, N.M.; Lund, T.D.; et al. S-Equol, a Potent Ligand for Estrogen Receptor β, Is the Exclusive Enantiomeric Form of the Soy Isoflavone Metabolite Produced by Human Intestinal Bacterial Flora1–4, Am J Clin Nutr 2005, 81, 1072–1079. [Google Scholar] [CrossRef]
- Hwang, C.S.; Kwak, H.S.; Lim, H.J.; Lee, S.H.; Kang, Y.S.; Choe, T.B.; Hur, H.G.; Han, K.O. Isoflavone Metabolites and Their in Vitro Dual Functions: They Can Act as an Estrogenic Agonist or Antagonist Depending on the Estrogen Concentration. J Steroid Biochem Mol Biol 2006, 101, 246–253. [Google Scholar] [CrossRef]
- Högger, P. Nutrition-Derived Bioactive Metabolites Produced by Gut Microbiota and Their Potential Impact on Human Health. Nutr Med 2013, 122. [Google Scholar]
- Brunelli, E.; Minassi, A.; Appendino, G.; Moro, L. 8-Prenylnaringenin, Inhibits Estrogen Receptor-α Mediated Cell Growth and Induces Apoptosis in MCF-7 Breast Cancer Cells. J Steroid Biochem Mol Biol 2007, 107, 140–148. [Google Scholar] [CrossRef]
- Wang, L.-Q. Mammalian Phytoestrogens: Enterodiol and Enterolactone. Journal of Chromatography B 2002, 777, 289–309. [Google Scholar] [CrossRef]
- Wang, L.; Sun, R.; Zhang, Q.; Luo, Q.; Zeng, S.; Li, X.; Gong, X.; Li, Y.; Lu, L.; Hu, M.; et al. An Update on Polyphenol Disposition via Coupled Metabolic Pathways. Expert Opin Drug Metab Toxicol 2019, 15, 151–165. [Google Scholar] [CrossRef]
- Jain, S.; Drendel, W.B.; Chen, Z.W.; Mathews, F.S.; Sly, W.S.; Grubb, J.H. Structure of Human Beta-Glucuronidase Reveals Candidate Lysosomal Targeting and Active-Site Motifs. Nat Struct Biol 1996, 3, 375–381. [Google Scholar] [CrossRef]
- Michikawa, M.; Ichinose, H.; Momma, M.; Biely, P.; Jongkees, S.; Yoshida, M.; Kotake, T.; Tsumuraya, Y.; Withers, S.G.; Fujimoto, Z.; et al. Structural and Biochemical Characterization of Glycoside Hydrolase Family 79 β-Glucuronidase from Acidobacterium Capsulatum. J Biol Chem 2012, 287, 14069–14077. [Google Scholar] [CrossRef] [PubMed]
- Jayatilleke, K.M.; Hulett, M.D. Heparanase and the Hallmarks of Cancer. J Transl Med 2020, 18, 453. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Jiang, J.; Jin, Y.; Kallemeijn, W.W.; Kuo, C.-L.; Artola, M.; Dai, W.; van Elk, C.; van Eijk, M.; van der Marel, G.A.; et al. Activity-Based Probes for Functional Interrogation of Retaining β-Glucuronidases. Nat Chem Biol 2017, 13, 867–873. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.C.; Jobin, C. The Struggle within: Microbial Influences on Colorectal Cancer. Inflamm Bowel Dis 2011, 17, 396–409. [Google Scholar] [CrossRef]
- Kong, R.; Liu, T.; Zhu, X.; Ahmad, S.; Williams, A.L.; Phan, A.T.; Zhao, H.; Scott, J.E.; Yeh, L.-A.; Wong, S.T.C. Old Drug New Use--Amoxapine and Its Metabolites as Potent Bacterial β-Glucuronidase Inhibitors for Alleviating Cancer Drug Toxicity. Clin Cancer Res 2014, 20, 3521–3530. [Google Scholar] [CrossRef]
- Cheng, K.-W.; Tseng, C.-H.; Yang, C.-N.; Tzeng, C.-C.; Cheng, T.-C.; Leu, Y.-L.; Chuang, Y.-C.; Wang, J.-Y.; Lu, Y.-C.; Chen, Y.-L.; et al. Specific Inhibition of Bacterial β-Glucuronidase by Pyrazolo [4,3-c]Quinoline Derivatives via a PH-Dependent Manner To Suppress Chemotherapy-Induced Intestinal Toxicity. J Med Chem 2017, 60, 9222–9238. [Google Scholar] [CrossRef]
- Letertre, M.P.M.; Bhatt, A.P.; Harvey, M.; Nicholson, J.K.; Wilson, I.D.; Redinbo, M.R.; Swann, J.R. Characterizing the Metabolic Effects of the Selective Inhibition of Gut Microbial β-Glucuronidases in Mice. Sci Rep 2022, 12, 17435. [Google Scholar] [CrossRef]
Figure 1.
!-glucuronidase enzymes from the intestinal bacteria that make up the stroboloma release glucuronidated estrogens in the liver by the enzyme UDP-glucuronosyltransferases (UGTs). This reactivation allows estrogens to be recirculated through the portal vein, possibly contributing to hormonal disorders including breast tumor development.
Figure 1.
!-glucuronidase enzymes from the intestinal bacteria that make up the stroboloma release glucuronidated estrogens in the liver by the enzyme UDP-glucuronosyltransferases (UGTs). This reactivation allows estrogens to be recirculated through the portal vein, possibly contributing to hormonal disorders including breast tumor development.
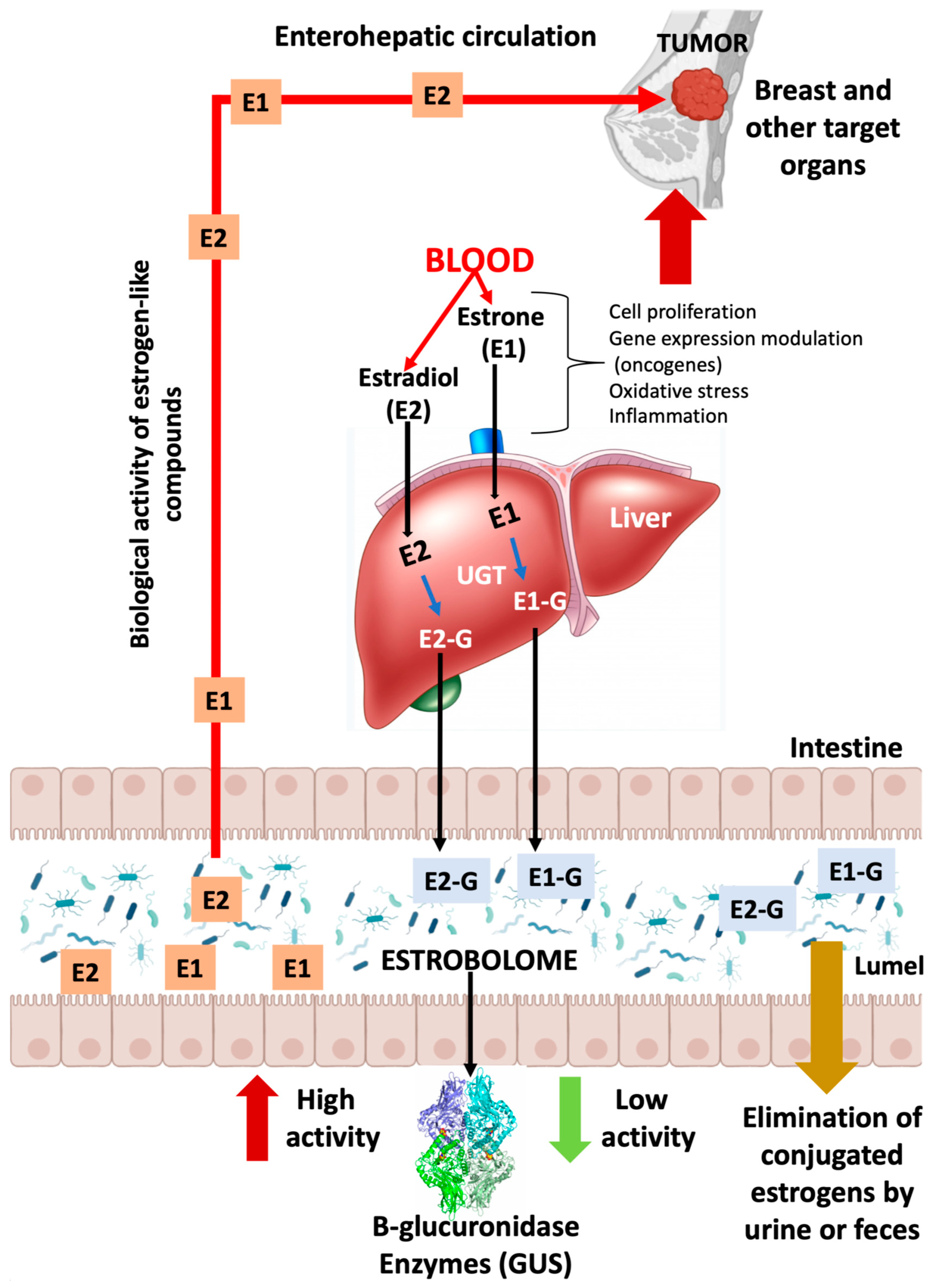
Figure 2.
Gut microbial GUS structure. A) Quaternary architecture of GH2 family GUS. The PDB 3K4D corresponding to EcGUS bound to the glucaro-d-lactam inhibitor is shown. Each dimer is represented in different shades of red and blue and the inhibitor in sphere representation. B) Detail of the active center of one of the subunits showing some structural elements involved in conjugated estrogen binding in GUS enzymes including Loop 1 (magenta), Loop 2 (salmon), N-K motif (pink) and Y loop (dark green). Also, those residues and water molecules involved in substrate binding and catalysis are labelled.
Figure 2.
Gut microbial GUS structure. A) Quaternary architecture of GH2 family GUS. The PDB 3K4D corresponding to EcGUS bound to the glucaro-d-lactam inhibitor is shown. Each dimer is represented in different shades of red and blue and the inhibitor in sphere representation. B) Detail of the active center of one of the subunits showing some structural elements involved in conjugated estrogen binding in GUS enzymes including Loop 1 (magenta), Loop 2 (salmon), N-K motif (pink) and Y loop (dark green). Also, those residues and water molecules involved in substrate binding and catalysis are labelled.
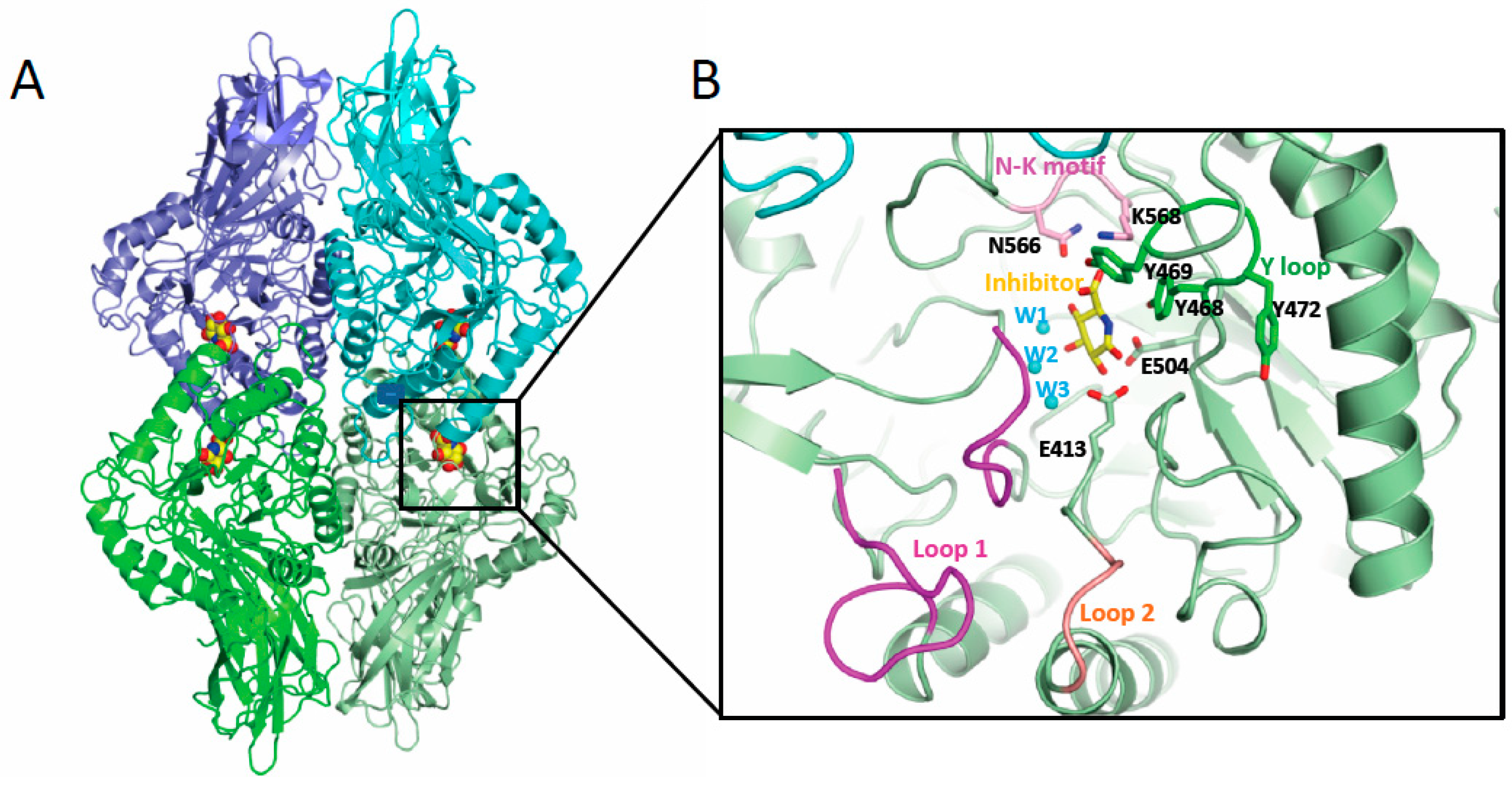
Figure 3.
Structural elements involved in the binding of glucuronides. Representation of the active center of GUS enzymes showing in A) EcGUS (PDB 3K4D) (green) with the Y-loop and aromatic cage highlighted in orange and a molecule of estrone-3-glucuronide (yellow) modelled; B) Mini-Loop 1 GUS of Bacteroides fragilis (PDB 3CMG) (green) highlighting the location of the mini-Loop 1 (magenta) involved in the binding of estrogen-glucuronides. C) Detail of the interactions of the water molecules 2 and 3 (cyan) in the active centre of EcGUS (PDB 3K4D) (green) (left panel) compared with those observed in BuGUS-1 (pink) (right panel) involving residues Y382 and W383.
Figure 3.
Structural elements involved in the binding of glucuronides. Representation of the active center of GUS enzymes showing in A) EcGUS (PDB 3K4D) (green) with the Y-loop and aromatic cage highlighted in orange and a molecule of estrone-3-glucuronide (yellow) modelled; B) Mini-Loop 1 GUS of Bacteroides fragilis (PDB 3CMG) (green) highlighting the location of the mini-Loop 1 (magenta) involved in the binding of estrogen-glucuronides. C) Detail of the interactions of the water molecules 2 and 3 (cyan) in the active centre of EcGUS (PDB 3K4D) (green) (left panel) compared with those observed in BuGUS-1 (pink) (right panel) involving residues Y382 and W383.
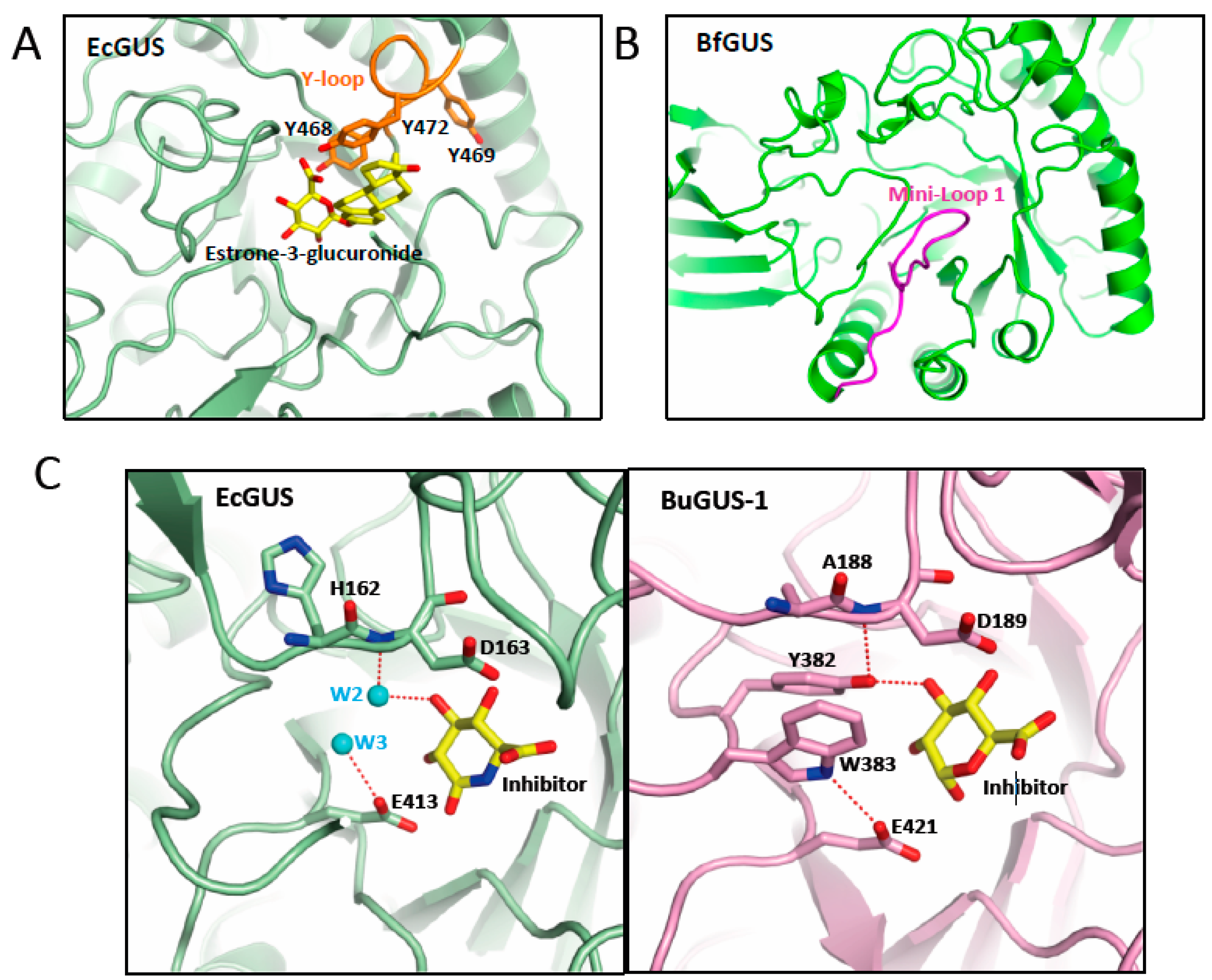
Figure 4.
Inhibitor binding to GUS enzymes. UNC10201652 inhibitor represented in sticks is shown bound to the active center of the Loop 1 enzyme CpGUS (PDB 6CXS) (blue ribbon). Loop 1 is coloured in magenta.
Figure 4.
Inhibitor binding to GUS enzymes. UNC10201652 inhibitor represented in sticks is shown bound to the active center of the Loop 1 enzyme CpGUS (PDB 6CXS) (blue ribbon). Loop 1 is coloured in magenta.
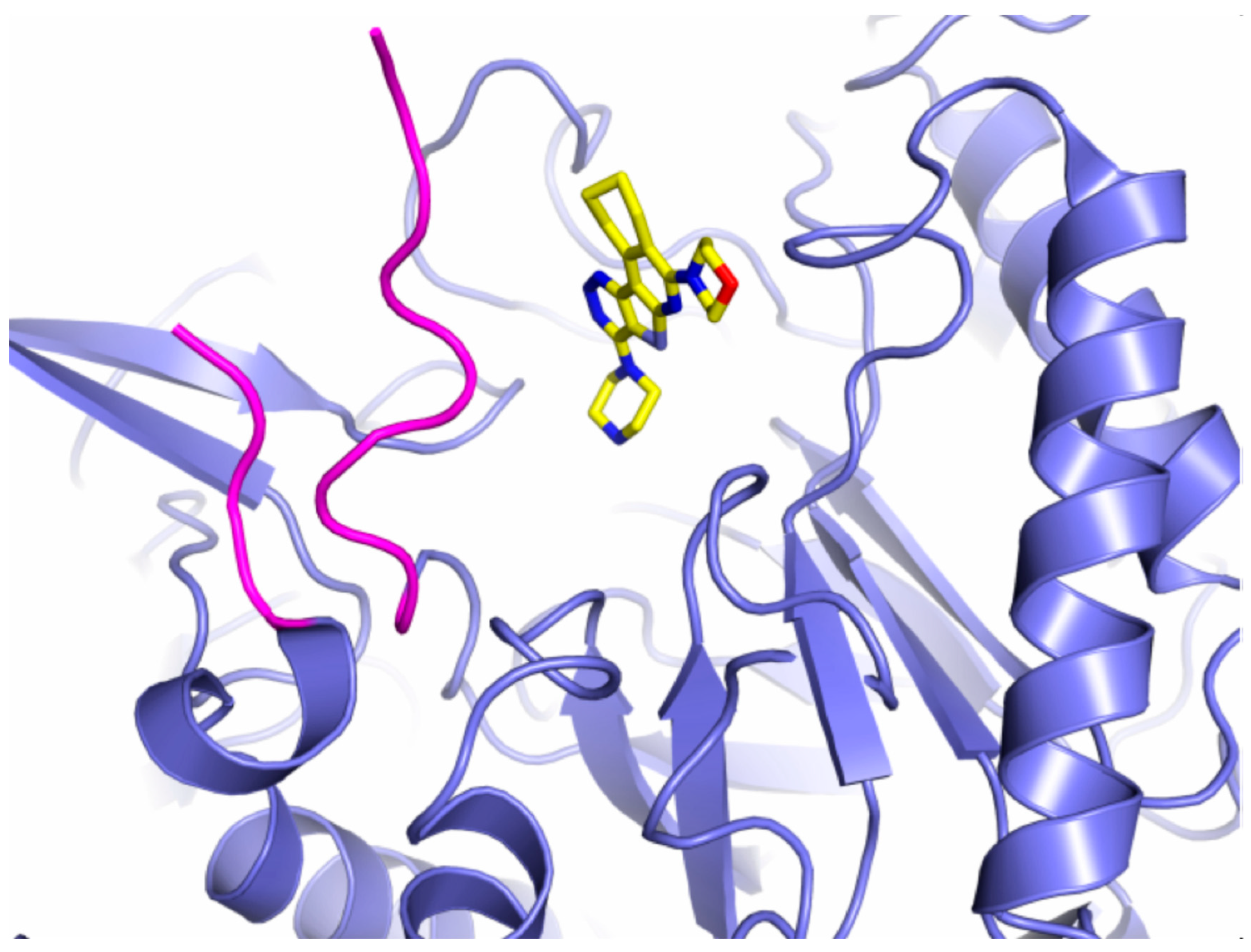
Table 2.
Distinct GUS enzymes architectures and cellular localization.
| Phylum (GUS abundance %) |
GUS Loop classification | Localization | References |
|---|---|---|---|
| Bacteroidetes (52%) | L2 mL1 mL2 NL rare mL1,2 |
Transported across the inner microbial membrane Periplasmic space Transported across the inner microbial membrane Periplasmic space Transported across the inner microbial membrane |
[31,38,39] |
| Firmicutes (41%) | L1 L2 NL mL1 |
Intracellular Transported across the inner microbial membrane Periplasmic space Periplasmic space |
[31,38,39] |
| Verrucomicrobia (1.5%) | mL2 | Transported across the inner microbial membrane | [31,38,39] |
| Proteobacteria (4%) | L1 | Intracellular | [31,38,39] |
NL: no loop < 10 residues in Loop 1 region; < 9 residues in Loop 2 region. Loop 1 (L1) >15 residue in Loop 1 region of E.coli GUS; < 9 residues in Loop 2 region. Mini-loop 1 (mL1) contain a loop of 10-15 residues in Loop 1 region; < 9 residues in Loop 2 region. Loop 2 (L2) < 10 residue in Loop 1 region; > 12 residues in Loop 2 region. Mini-loop 2 (mL2) < 10 residues in Loop 1 region; >9 and < 12 residues in Loop 2 region. Mini-loop 1,2 (mL1,2) contain a loop of 10-15 residues in Loop 1 region; >9 and < 12 residues in Loop 2 region.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



