
Submitted:
12 July 2023
Posted:
13 July 2023
You are already at the latest version
Abstract
Coronary no-reflow (CNR) is a frequent phenomenon that develops in patients with ST-segment elevation myocardial infarction (STEMI) following reperfusion therapy. CNR is highly dynamic and develops gradually (over hours) and persists for days to weeks after reperfusion. Microvascular obstruction (MVO) developing as a consequence of myocardial ischemia, distal embolization and reperfusion-related injury is the main pathophysiological mechanism of CNR. The frequency of CNR or MVO after primary PCI differs widely depending on the sensitivity of the tools used for diagnosis and timing of examination. Coronary angiography is readily available and most convenient to diagnose CNR but it is highly conservative and underestimates the true frequency of CNR. Cardiac magnetic resonance (CMR) imaging is the most sensitive method to diagnose MVO and CNR that provides information on the presence, localization and extent of MVO. CMR imaging detects intramyocardial hemorrhage and accurately estimates the infract size. MVO and CNR markedly negate the benefits of reperfusion therapy and contribute to poor clinical outcomes including adverse remodeling of left ventricle, worsening or new congestive heart failure and reduced survival. Despite extensive research and the use of therapies that target almost all known pathophysiological mechanisms of CNR, no therapy has been found that prevents or reverses CNR and provides consistent clinical benefit in patients with STEMI undergoing reperfusion. Currently the prevention or alleviation of MVO and CNR remain unmet goals in the therapy of STEMI that continue to be under intense research.
Keywords:
Coronary no-reflow
; microcirculation steal syndrome
; microvascular obstruction
; pathophysiology
; therapy
Historical Perspective
Historical records of no-reflow are difficult to track because vascular events developing in early experimental ischemia/reperfusion models, later known as no-reflow, were described well before the no-reflow phenomenon was recognized. The term “no-reflow phenomenon was coined in 1967 by Guido Majno et al.1 in a Letter to the Editor in Lancet (September 9th issue, 1967) to describe the inability to reperfuse rabbit brain regions made ischemic by artery ligation despite restoration of blood flow. The work supportive of the proposal of the term “no-reflow phenomenon” was published approximately 6 months later in the February issue of American Journal of Pathology.2 Using electron microscopy, the authors described many morphological (cellular) characteristics of the no-reflow at the capillary level such as, capillary obstruction by cellular swelling, bleb formations originating from the endothelial cells, platelet and red cell aggregates and extravascular compression of the microcirculation.2-3 These histological findings are considered as hallmark characteristics of the no-reflow to this day. However, failure to restore tissue reperfusion following restoration of blood flow was described in a number of ischemia/reperfusion animal models before the term “no-reflow phenomenon” was coined. In 1948, Harman4 provided one of the best descriptions of the no-reflow in skeletal muscle in the right hind legs of albino male rabbits made ischemic by the application of tourniquets. Angiographic and dye studies assessing the rate of penetration and elimination of bromphenol blue from the ischemic muscle showed that blood circulation through the ischemic muscle after the release of occlusion was extremely sluggish. Of note, the study by Harman established a relationship between duration of ischemia and the speed of elimination of dye from the ischemic muscle, provided histological analysis of ischemic lesions such as tightly packed erythrocytes within the capillaries and interstitial fluid accumulation after release of occlusion and excluded an eventual role of thrombi in the genesis of the syndrome. In the subsequent years, no-reflow phenomenon was described in a number of experimental ischemia/reperfusion models in kidney,5-6 adrenal gland,7 brain,2-3 myocardium,8 and skin.9 Demonstration of no-reflow phenomenon in various animals and organs led Majno et al.1 to suggest that no-reflow after an ischemic insult may be a general phenomenon. In 1974, Kloner et al.10, while working in the laboratory of Robert Jennings, performed a specifically designed study to characterize coronary no-reflow (CNR) after coronary occlusion in anesthetized dogs subjected to 40 to 90-min ischemia (by clamping the circumflex coronary artery) followed by clamp release and reperfusion. Using electron microscopy, the authors offered the best description of ultrastructural alterations in the vasculature and working myocardium that stand at the pathophysiological basis of CNR to this day.10-11 The study by Kloner et al.10 was highly influential and often is considered as an inaugural study in the field of CNR.
The early records of no-reflow in humans remain elusive or subject to interpretation. It is highly plausible that no-reflow could have played a role in the genesis of ischemic muscular contractures (called Volkman’s ischemic contracture) after surgical embolectomy of arterial thrombi. In 1934 Jefferson12 described a case of removal of a clot from the brachial artery two and a half hours after embolic artery occlusion. The clot was successfully removed but the patient rapidly developed a contracture of moderate severity in the forearm flexor muscles. Similar cases of contractures following clot removal from the acutely (embolic) occluded femoral artery as well as demonstration of this phenomenon in experiments with rabbits were reported by Griffiths13 in 1940. Thrombolytic studies opened the prospect of assessing the CNR in clinical setting. However, the interest on CNR in the thrombolytic era was low. In 1985, Schofer et al.14 were the first to demonstrate CNR by thallium-201 or technetium-99m scintigraphy in patients with acute myocardial infarction (AMI) of anterior wall after intracoronary thrombolysis. In the re-studied patients, the scintigraphic zone of CNR persisted for 2 to 4 weeks after intracoronary thrombolysis. In the subsequent years, CNR was described by angiography in case reports15-17 or in studies with a limited number of patients.18 In 1989, Wilson et al.18 described a syndrome characterized by angina, ST-segment elevation and a striking reduction of blood flow in the dilated artery immediately after balloon angioplasty in 5 patients with acute thrombotic coronary artery occlusions with no visible distal emboli or side branch occlusions. The syndrome lasted for 48 to 80 minutes and was not reversed by nitroglycerin or thrombolytic drugs. The condition was explained by microvascular constriction caused by release of potent vasoconstrictors from the clot. In 1992, Ito et al.19 demonstrated CNR in 39 patients undergoing thrombolysis (10 patients) or coronary angioplasty (29 patients) using myocardial contrast echocardiography (MCE). Of note, the study by Ito et al.19 showed that no-reflow was a predictor of poor functional recovery of the postischemic myocardium. A study of 1919 percutaneous coronary interventions (PCI) performed in early 1990s showed an overall frequency of CNR (defined as Thrombolysis in Myocardial Infarction [TIMI] blood flow <3) of 2%. However, the frequency of NCR was 11.5% in patients undergoing PCI for AMI and 4% in patients undergoing treatment of saphenous vein grafts.20 Morishima et al.21-22 demonstrated an association between angiographic CNR and in-hospital and long-term outcomes including cardiac death in patients with the first AMI treated with PCI. These studies suggested that the increased risk for long-term complications in patients with CNR may be related to adverse left ventricular remodeling associated with CNR. The purpose of this review is to provide an overview of frequency, diagnostic tools, pathophysiology, predisposing factors, clinical impact and principles of therapy of CNR in patients with ST-segment elevation myocardial infarction (STEMI) treated with primary PCI.
Diagnosis and Frequency of CNR
Thrombolytic and primary PCI studies in patients with AMI showed that CNR is a relevant clinical problem. CNR is highly dynamic in nature. It develops gradually (over hours) following coronary blood flow restoration and persists over days to weeks depending on severity, duration and extent of myocardial ischemia and application of therapeutic measures aiming to prevent or alleviate ischemia/reperfusion injury. The diagnostic yield of any method used to diagnose CNR depends on the extent and severity of CNR and the timing of examination. Transient slowing of restored myocardial blood flow or small under-reperfused myocardial segments may go undetected whereas fixed microvascular obstruction (MVO) developing over an extensive myocardial area/volume is more reliably detectable. Furthermore, the use of diagnostic methods early (before CNR has developed) or late (after CNR has resolved) after the restoration of epicardial blood flow may fail to detect (or underestimate) CNR. These factors as well as the sensitivity of the method per se used to diagnose CNR may explain the wide variations in the frequency of CNR across the studies. Before analyzing specific methods used to diagnose CNR in clinical setting, two concepts may need clarification. First, the term CNR is used to describe coronary blood flow stasis or MVO after all PCI procedures including elective PCI in patients with chronic coronary syndromes. However, since CNR is an ischemia/reperfusion syndrome resulting from the sequence of coronary artery occlusion (acute ischemia) and reopening (blood flow restoration-related reperfusion injury), blood slowing detected immediately after elective PCI in patients without an acute antecedent (or ongoing) myocardial ischemia may not be CNR. In this scenario, CNR may be caused by clogged microvasculature by distal embolization of atherosclerotic/thrombotic material in the course of PCI. Second, if the definition of CNR requires restoration of blood flow without flow-impeding obstacles at the large coronary artery level, massive and angiographically visible distal embolization of atherosclerotic/thrombotic material occurring during the primary PCI procedures simply shifts the mechanical obstacle to blood flow from the epicardial artery to a more distal location. Thus, it may not represent a true CNR phenomenon.
Diagnostic methods used to detect CNR in clinical setting differ widely with respect to their sensitivity to detect the condition. Furthermore, the most optimal timing of their use to diagnose CNR remains unknown. Historically, thallium-201 and technetium-99m scintigraphy14 and MCE19 have demonstrated the presence of CNR in clinical setting. Although nuclear imaging methods (single photon emission tomography and positron emission tomography) can detect CNR, they are not commonly used to diagnose CNR in current practice. MCE uses gas-filled microbubbles, which are very effective in scattering of ultrasound and an ideal tracer of microcirculation. Human left ventricular myocardium has more than 2200 capillaries per mm2 in cross-sectional view23, and 90% of blood in microcirculation (approximately 8% of ventricular mass) is present in capillaries.24 Microbubbles have rheological properties similar to the red blood cells and their size (<5 μm) allows them to pass through capillaries without blocking them. Microbubbles remain entirely within the vascular space and myocardial contrast intensity following intravenous or intracoronary microbubble injection reflects their concentration in the microvascular compartment of myocardium.25-26 The microbubble lingering inside the myocardium or the lack of contrast opacification in the echocardiograms obtained following intravenous or intracoronary microbubble injection indicate CNR and MVO. MCE can localize the zone of MVO and quantify its extent within the infarcted myocardium.27 The prevalence of CNR diagnosed by MCE varies between 37%19 and 66%.28 MCE is a validated method to assess myocardial reperfusion29-30 and the CNR diagnosed by this technique correlates closely with adverse left ventricular remodeling and poor prognosis after AMI.19 MCE is limited by moderate spatial resolution and dependence of operator’s experience31, poor echocardiographic window preventing reliable measurements in 8% of patients32 and inability to detect vasodilatation or vasoconstriction within a previously ischemic myocardial area.27 MCE is not frequently used to diagnose CNR in contemporary clinical practice.
Coronary angiography is routinely used to diagnose CNR following PCI. There are at least 4 angiographic metrics that have been used to diagnose CNR: Thrombolysis in Myocardial Infarction (TIMI) flow grade, corrected TIMI frame count, myocardial blush grade and TIMI myocardial perfusion grade. TIMI flow grade assesses blood flow in the epicardial coronary arteries. It is quantified using a scale between 0 and 3 and a TIMI flow grade of <3 is used to diagnose CNR. TIMI flow grade has prognostic value and failure to restore a TIMI flow grade <3 was independently associated with increased risk of mortality after PCI in patients with acute coronary syndromes (ACS).33 Although, the assessment of CNR using TIMI flow grade is convenient and simple, it does not reflect tissue reperfusion (or microvascular function) and thus, the method lacks sensitivity to diagnose CNR.34-35 A study from our group showed that tissue reperfusion assessed by myocardial perfusion grade was not fully restored (myocardial perfusion grade ≤2) in 34% of patients with a TIMI flow grade of 3 after primary PCI.36 Thus, a definition of CNR as a TIMI flow grade of <3 is highly conservative and leads to underestimation of CNR in a high proportion of patients undergoing primary PCI. Corrected TIMI flow count represents the number of frames required for the dye to reach a standardized distal landmark after correction for the vessel (epicardial artery) length.37 Faster (lower) corrected TIMI frame TFC count was associated with improved in-hospital and one-month outcomes after thrombolysis38 and better functional recovery after successful primary angioplasty in patients with AMI.39 Corrected TIMI frame count correlated inversely with peak blood flow velocity by intracoronary Doppler but not with the degree of microvascular injury after primary coronary angioplasty in patients with the first STEMI of anterior wall.40 These data suggest that corrected TIMI frame count reflects coronary epicardial blood flow but not microcirculation.
Myocardial blush grade or the degree of myocardial contrast staining following intracoronary contrast injection is also used to assess myocardial microvasculature and tissue reperfusion after primary PCI.41 Myocardial blush grade is scaled between 0 (no contrast staining) to 3 (contrast staining similar to that of contralateral non-infarcted myocardium) and a myocardial blush grade of <3 indicates CNR. However, myocardial blush grade represents a low contrast-to-nose ratio imaging, depends on operator’s experience which may lead to unacceptably high interobserver variability and suffers from the same limitations as the TIMI flow grade method.42-43 These characteristics limit the usefulness of myocardial blush grade to assess CNR after primary PCI. Post-procedural TIMI flow grade or myocardial blush grade do not necessarily correlate with the presence of MVO detected by cardiac magnetic resonance (CMR) imaging in patients with STEMI undergoing successful primary PCI.44-45 Although myocardial perfusion grade appears to correlate with one-year mortality even in patients with TIMI flow grade of 346, it was discordant with ST-segment resolution in up to 40% of patients after primary PCI.47 One study has shown that MVO assessed by CMR but not TIMI flow grade or myocardial blush grade correlated with the left ventricular function following primary PCI.48 TIMI myocardial perfusion grade is also used to assess myocardial reperfusion based on the densitometry of contrast entry, duration, and clearance from the ischemic myocardium.43 It is scaled between 0 (no tissue reperfusion) and 3 (minimally persistent myocardial contrast staining following 3 cardiac cycles of washout) and a TIMI myocardial perfusion grade of ≤2 indicates impaired tissue reperfusion and CNR. TIMI myocardial perfusion grade appears to be superior to other angiographic indices with respect to the assessment of tissue reperfusion in reperfused patients with STEMI. A combination of TIMI flow grade with TIMI myocardial perfusion grade (a TIMI flow grade of ≤2 or a TIMI flow grade of 3 with a TIMI myocardial perfusion grade of 0-1) diagnosed CNR in 29% of patients with STEMI treated successfully with primary PCI.49 Impaired TIMI myocardial perfusion grade correlated with MVO assessed by CMR imaging at 3 to 4 days50-51, infarct size at 7 days and 3 months52 and left ventricular ejection fraction (LVEF) at 90 days51 after STEMI. Impaired TIMI myocardial perfusion grade (≤2) correlated with biochemical markers of myocardial necrosis and increased risk of death, myocardial infarction or ischemic events on Holter monitoring by 48 hours in patients with non-ST-segment elevation ACS undergoing PCI.53 In aggregate, although achieving favorable coronary angiography indices are associated with an improvement of prognosis after primary PCI and should be strived for them, with exception of TIMI myocardial perfusion grade, these indices are poor correlates of MVO and CNR assessed by sensitive CMR imaging. One reason of the low sensitivity of angiographic methods to detect CNR may be related to the timing of coronary angiography, which may be well ahead of development of CNR. Since CNR is a dynamic process that develops hours to days following blood flow restoration to previously ischemic myocardium, angiographic indices obtained at the end of primary PCI procedure reflect ischemia and distal embolization related factors but not the most important factor of CNR, that is reperfusion-related injury (discussed later in this review).
CMR imaging is the most sensitive technique to detect MVO and diagnose CNR in clinical setting. By providing multi-slice views, with a high spatial resolution, CMR imaging enables an accurate quantification and localization of MVO within the infarcted area as well as transmural extent of CNR and necrosis (infarct size) within the infarcted region. CMR imaging detects also the hemorrhagic transformation and extravasation of erythrocytes within the infarct core, which represent a frequent component of CNR that portends a poor prognosis.26 Contrast-enhanced CMR is based on the differences in the distribution of the contrast agent (gadolinium chelate injected intravenously) depending on the status (degree of injury or obstruction) of microcirculation within the injured versus healthy myocardium.42 CMR imaging-based detection of MVO (and consequently CNR) is defined as the lack of contrast uptake during the first pass of contrast agent (imaging obtained within <one minute after injection), the lack of early gadolinium enhancement (imaging obtained within <2-3 minutes after injection) or the lack of late gadolinium enhancement (imaging 10-15 minutes after contrast injection).54 On the first pass imaging, MVO typically appears as a central dark zone within an area of early enhanced myocardium indicating a focal absence of contrast enhancement within the infarcted area.55 Since gadolinium may diffuse slowly over time from the CNR zone, the area of MVO may become smaller in late gadolinium enhancement imaging, which may explain the diagnosis of MVO in a higher proportion of patients on the first pass imaging compared with late gadolinium enhancement imaging.55 However, it has been suggested that MVO on late gadolinium enhancement imaging is a better correlate (prognostic marker) of subsequent left ventricular remodeling and adverse cardiovascular events than MVO seen on the first pass gadolinium imaging.56-57 Using CMR imaging, MVO was diagnosed in up to 95% of patients with STEMI and restored TIMI flow grade of 3 during first pass contrast enhanced imaging58 and 57% of patients with STEMI within 7 days after primary PCI by late gadolinium enhancement imaging.59 Using CMR imaging, MVO was diagnosed in 13.8% of patients with non-ST-segment elevation myocardial infarction.60 CMR imaging typically is performed hours to days after reperfusion of patients with STEMI. Consequently, early events developing after epicardial blood restoration and the relationship between CNR and infarct size (or myocardial salvage) cannot be assessed by this technique.
Other diagnostic methods have been used to assess CNR in clinical setting. The degree and speed of ST-segment elevation resolution in the post-reperfusion electrocardiogram correlates closely with tissue reperfusion and MVO after primary PCI. A rapid and complete ST-segment elevation resolution after primary PCI indicates prompt (and complete) restoration of tissue reperfusion61, which is associated with markedly reduced frequency of MVO or CNR on CME62 or CMR imaging.45 In a study by Nijveldt et al.45 residual ST-segment elevation was the only independent predictor of microvascular injury after adjustment in multivariable analysis. ST-segment resolution in the intracoronary electrocardiogram recorded at the time of procedure correlated with CMR-assessed MVO 4 days after reperfusion in 64 patients with STEMI undergoing primary PCI.51 Although standard electrocardiogram is cheap and readily available, it lacks sensitivity to diagnose CNR and there is no consensus on the most appropriate electrocardiographic leads or timing of electrocardiogram recording after reperfusion. MVO and CNR can be assessed using invasive (catheter-based) coronary physiology indices. Several physiological indices that characterize coronary microcirculation may be obtained such as coronary flow velocity patters, coronary flow reserve, index of microvascular resistance, hyperemic microvascular resistance, resistive reserve ratio, instantaneous hyperemic diastolic flow velocity-pressure slope and coronary zero flow pressure.63 In brief, microvascular injury (and consequently MVO) is characterized by shortening of diastolic deceleration time and presence of systolic retrograde flow, decrease in coronary flow reserve, an increase in hyperemic microvascular resistance index, an increase in the index of microcirculatory resistance, a decrease in the coronary conductance by the instantaneous hyperemic diastolic velocity pressure slope index and increase in zero-flow pressure26, 63 (for details see Lanzer et al.26 and Konijnenberg et al.63). Although these indices offer a good characterization of microvascular injury (and MVO) after reperfusion and may be applied in the catheterization laboratory, they mostly remain as research tools and not routinely used to diagnose MVO and CNR in patients with STEMI. Although myocardial scintigraphy offered the first evidence on the existence of CNR in humans,14 nuclear imaging techniques - single photon emission tomography (SPECT) and positron emission tomography (PET) with various perfusion tracers - have limited use to assess CNR in current practice due to problems related to technical difficulties, costs, patient’s radiation, and availability of other highly sensitive imaging techniques to detect CNR, particularly CMR imaging.
In aggregate, due to prognostic implications, CNR following primary reperfusion of patients with AMI should be searched for and diagnosed. Coronary angiography-based techniques are most convenient, but they are performed too early after reperfusion (before full-scale CNR has developed) and consequently they underestimate CNR. CMR imaging is the most sensitive technique to diagnose CNR that provides information on its extent and location within the infarcted myocardium in clinical setting. Apart from CNR diagnosis and characterization, CMR imaging detects also intramyocardial hemorrhage and accurately estimates infarct size further improving the risk stratification of patients with AMI. Other techniques (electrocardiogram, MCE, and nuclear imaging) have limited use for the diagnosis of CNR in current clinical practice.
Pathophysiology of CNR
MVO is the underlying pathophysiological mechanism of CNR following reperfusion of an occluded coronary artery. MVO and CNR after reperfusion of an occluded coronary artery are explained by a joint action of at least 4 factors: myocardial ischemia, spontaneous or iatrogenic distal embolization, reperfusion-related injury and individual susceptibility (predisposing conditions that increase the odds of developing MVO and CNR). Pathophysiological mechanisms of MVO and CNR are shown in Figure 1.
A Short Description of Myocardial Microcirculation
Coronary microcirculation refers to blood circulation in vessels <200 µm in diameter that are not visualized on coronary angiography. It consists of arterioles, capillaries and venules. All 3 structures participate in the MVO and CNR following ischemia and reperfusion, albeit with different roles. Coronary arterioles have a relatively thick smooth muscle wall, act as resistance vessels and are responsible for keeping a constant pre-capillary pressure of ~45 mmHg in the setting of autoregulation. In the setting of ischemia/reperfusion injury arterioles contribute to MVO and CNR through impaired vasomotor tone (impaired endothelium dependent vasodilation) and propensity to in situ thrombus formation. Morphometric analyses have shown that there are approximately 2200 capillaries per square millimeter in adult human hearts.23 It has been estimated that there are approximately 8 million capillaries in human heart. Coronary capillaries contain ~1/3 of myocardial blood (~45 ml) which moves with an average speed of 1 mm/sec (at resting state) under a hydrostatic pressure of ~30 mmHg.24, 64 In the setting of ischemia/reperfusion injury, coronary capillaries undergo constriction, obstruction, and compression, which leads to a marked reduction in the number of open vessels and diminished delivery of oxygen and nutrients to the surrounding tissue. Coronary capillaries are the main site of clogged microcirculation and MVO occurring as a response to ischemia and/or reperfusion. Coronary venules have a weak smooth muscle and consequently manifest weak muscular vascular responses (venular hydrostatic pressure is ~15 mmHg). However, coronary venules participate in the ischemia and/or reperfusion-related MVO by serving as a preferential site of leucocyte and platelet adhesion via expression of adhesion molecules and subsequent inflammation.64-65
Myocardial Ischemia
Total cessation or drastic reduction (>80%) of coronary blood flow results in severe myocardial ischemia in subtended myocardium. All cells and structures in the ischemic zone undergo various degrees of ischemic injury depending on the severity and duration of ischemia, metabolic demand at the time of blood flow cessation, preconditioning and presence of anti-ischemia agents. Previous studies that have investigated cardioprotective measures in ischemia/reperfusion models were potentially conceptually flawed in that they focused on the cardiomyocytes paying little attention to the microcirculation. This could be one reason of the failure of cardioprotective measures applied in (conceptually-flawed) ischemia/reperfusion studies to translate into clinical benefit.66 Microcirculation - the key component of MVO and CNR - is particularly vulnerable to ischemia and reperfusion injury. In the following material, we focused on the endothelial cells and other components of microcirculation whereas the impact of ischemia on cardiomyocytes was not addressed.
Endothelial cells are more resistant to ischemia than surrounding cardiomyocytes and may survive hypoxia for minutes to days following the installation of ischemia.10, 67 Endothelial cells are abundant in myocardium representing 3% to 5% of the myocardial volume or approximately 45% of total cells or 60% of nonmyocyte cells in the murine myocardium.68 Experimental studies using human umbilical vein endothelial cells showed that 75% of cells survived 24 hours of hypoxia69 and >50% of cells survived 48 hours of hypoxia.67 Myocardial concentration of high-energy phosphates (adenosine triphosphate [ATP] and creatine phosphate) is low in myocardium and can support contraction for only a few effective systoles. The oxygen present in capillaries (as oxyhemoglobin) and cardiomyocytes (oxymyoglobin) is exhausted after 8-10 seconds, which brings to almost total cessation of oxidative phosphorylation and effective myocardial contraction. At 15 to 20 seconds of ischemia (artery occlusion), anaerobic glycolysis supervenes as the only source of generation of ATP. At 60 seconds of ischemia anaerobic glycolysis slows markedly, primarily because of glyceraldehyde phosphate dehydrogenase inhibition by high NADH/NAD+ ratio and at 40 to 60 minutes of total ischemia anaerobic glycolysis essentially stops.70 The lack of aerobic metabolism and coronary blood flow lead to accumulation of various small-molecule catabolites in the cells and interstitial space of ischemic tissue including lactate, protons (tissue acidosis), ammonium, degraded nucleotide phosphates (adenosine diphosphate, adenosine), products of glycogen break-down (glucose-1-phosphate), glucose-6-phosphate and many other products of intermediary metabolism. A high concentration of these catabolites (particularly, protons and ammonia) is directly toxic to the cells (cardiomyocytes and endothelial cells) and contributes to the ischemia-related tissue injury. In addition, higher concentrations of these catabolites increase the osmotic load within the cells and interstitial space, which generates an osmotic gradient forcing the water to move inside the cells or from intra-capillary to interstitial space leading to cellular swelling and interstitial edema.71 Thus, endothelial cell swelling and interstitial edema are important mechanisms of MVO and CNR during myocardial ischemia.
Lack of high-energy phosphates (ATP) is associated with severe perturbations in ionic hemostasis in endothelial cells that contribute to ischemia-related endothelial dysfunction. Apart from further contributing to endothelial cell swelling and intra-capillary space obstruction as a response to ischemia, altered ionic hemostasis has other negative actions promoting endothelial cell dysfunction. Ischemic endothelial cells show increased concentration of calcium in cytoplasm, which activates endothelial contractile elements.72 Calcium-induced contraction of endothelial cell filaments reduces mechanical support for endothelial cell membrane promoting cytoplasmic budding or blebbing into the intra-capillary space.73 Actin filaments constitute 5–15% of the total protein in endothelial cells74 and actin cytoskeleton is critical for maintenance of endothelial barrier function.75 Blebs and their role in the intra-capillary obstruction have been described since inaugural structural studies of no-reflow2-3 and bled formation appears to be further favored by loss of antegrade pulsatile flow and increased shear stress.76 Increased intracytoplasmic calcium and calcium-induced filament contraction appear to change the cellular shape, which destabilizes cellular junctions and increase the inter-cellular permeability. There are other factors that destabilize endothelial inter-cellular junctions favoring increased permeability and interstitial edema during myocardial ischemia. Ischemia-induced expression of vascular endothelial growth factor (VEGF) - a major regulator of vascular permeability77 - and dissociation of VEGF receptor 2-vascular endothelial (VE)-cadherin (a cell adhesion protein of adherens junctions) complex lead to increased inter-endothelial cell permeability.78 VEGF activates Scr (a member of Src kinase family) via phosphorylation which leads to phosphorylation of tyrosine residues of VE-cadherin of the inter-endothelial cell junctions. This action promotes VE-cadherin internalization and reduces the amount of VE-cadherin in the inter-endothelial cell junctions.42, 79 The removal of VE-cadherin from the inter-cellular junctions further destabilizes intercellular connection and increase inter-cellular permeability. In experimental conditions VE-cadherin phosphorylation is also facilitated by increased shear stress.79 VEGF also activates endothelial nitric oxide synthase (eNOS) in the caveolae of endothelial cells80, further contributing to increased vascular permeability. Activated endothelial and circulating cells (platelets and neutrophils) show increased expression of adhesion molecules81, which may be further potentiated by subsequent reperfusion by thrombolysis or angioplasty.82 Exposed adhesion molecules mediate platelet and leukocyte endothelial interactions, which facilitates trapping of these cells in the ischemic microvascular space (discussed later under this subheading).
Glycocalyx is perhaps the earliest microcirculation components that is damaged in the course of ischemia/reperfusion. Glycocalyx is an important component of endothelial barrier. Glycocalyx represents a 0.5 µm thick carbohydrate-rich matrix that covers endothelium surface throughout the capillary system.83 The thickness of glycocalyx exceeds the length of extracellular domains of most endothelial adhesion molecules,84 which in normal conditions prevents the adhesion of circulating cells to endothelial cells. The highly hydrophilic nature of glycocalyx enables creation of a relatively fixed (albeit exchangeable) water layer on the surface of endothelial cells, which together with electrostatic interactions with circulating erythrocytes, reduces the capillary hematocrit compared with that found in the systemic circulation and facilitates the passage of blood through the capillaries.85 Of note, glycocalyx represents a dynamic fluid wall and a constituent of capillary barrier together with endothelial cells and basal membrane. Glycocalyx is degraded upon exposure to ischemia,86-87 reactive oxygen species (ROS),87-88 oxidized lipoproteins,89 acute hyperglycemia,90 tumor necrosis factor alpha (TNFα),91 matrix metalloproteinase (MMP) 2 and 9,92 inflammatory states and vigorous volume loading.93 Nitric oxide (NO) appears to be protective against glycocalyx shedding.94 Glycocalyx degradation (shedding) contributes to MVO and CNR by damaging the capillary barrier and increasing capillary permeability, which contributes to endothelial cell and interstitial edema95 and by enabling leukocyte96 and platelet97 adhesion to endothelial cells facilitating the entrapment of these cells in the intra-capillary space.
Various circulating cells, in particular platelets and neutrophils are recruited in the capillaries following myocardial ischemia and make a substantial contribution to ischemic injury, MVO and CNR.98 Platelets participate in ischemia and reperfusion-related capillary damage via a number of mechanisms. Following activation by ischemia, platelets expose their adhesion molecules and aggregate to endothelial cells (facilitated by glycocalyx shedding), neutrophils (platelet-neutrophil aggregates), erythrocytes (platelet-erythrocyte aggregates) and to each other (platelet-platelet aggregates) contributing to microcirculation plugging and obstruction. These cellular aggregates have been demonstrated in capillaries from the very first ultramicroscopic characterization of no-reflow.2-3 Moreover, a significant increase in the neutrophil-platelet aggregates and monocyte-platelet aggregates was shown in the coronary sinus blood samples of microsphere-induced CNR in Yorkshire pigs.99 Apart from mechanical blockage by aggregates and microthrombi, activated platelets release various biological active substances including nucleotides, proteases, platelet activating factor (PAF), ROS, adhesive proteins (fibronectin, von Willebrand factor, thrombospondin, P-selectin, glycoprotein IIb/IIIa, fibrinogen), vasoconstrictors such as thromboxane A2 and serotonin, growth factors, coagulation and complement system factors, various cytokines and chemokines, pro-angiogenic factors and microvesicles and exosomes.31, 63, 98, 100-102 These substances contribute to proteolytic destruction of endothelial cells and intercellular junctions (and increased permeability), intra-capillary blood coagulation, chemiotaxis and recruitment of leukocytes in microcirculation and promote inflammation, apoptosis and angiogenesis. Although in the early stage of ischemia/reperfusion, the release of these substances is detrimental and contribute to MVO and CNR, at longer term these mediators and cellular processes contribute to elimination of severely-damaged (or necrotic) tissue and pave the way for tissue healing and regeneration. However, it appears that the degree and nature of platelet contribution to ischemia and reperfusion-related injury depend on the state of platelet activation. Experimental studies of ischemia/reperfusion in rats and guinea pigs showed that ischemia/reperfusion damage was ameliorated and endothelial integrity was improved by platelet or platelet-derived perfusion.103-104 Platelet glycoprotein IIb/IIIa receptor blockage has reduced microvascular thrombosis in murine models of acute stroke105 and platelet depletion counteracts deleterious effects of acute hypercholesterolemia on infarct size and CNR in a ischemia/reperfusion model in rabbits.106 Clinical studies have also shown that glycoprotein IIb/IIIa receptor blockade with abciximab improved the recovery of microvascular perfusion and enhanced the recovery of contractile function in the area at risk in patients with AMI undergoing mechanical reperfusion with coronary stenting.107 These studies offer evidence on the participation of platelets in the damage of microcirculation during myocardial ischemia and reperfusion and cardioprotective effects of platelet inhibition.
Neutrophils are recruited early in the ischemic myocardium.98 Neutrophils transmigrate through endothelial cells by interaction with endothelial cell junction proteins due to the highly chemotactic milieu in the ischemic microcirculation.108 Activated neutrophils aggregate with other cells (endothelial cells, platelets, erythrocytes and with each other) and form neutrophil extracellular traps (NETs) clogging the microcirculation and impeding blood flow.109-110 Neutrophils are a major source of ROS111, myeloperoxidase112 and proteolytic enzymes (such as, elastase and metalloproteinase-9)113-114, which in turn, promote degradation of all components of capillary barrier (glycocalyx, endothelial cells and basal membrane) leading to vascular leakage, increased vascular permeability and excess edema. Following initial infiltration and activation, neutrophils and other inflammatory cells (monocyte/macrophages and lymphocytes) participate in the powerful local and systemic inflammatory response that develops in patients with AMI. The role of neutrophils and other circulating and resident cells in the pathophysiology of ischemia/reperfusion related MVO has been recently reviewed.98, 108
Growing evidence suggests that pericytes play an important role in the genesis of CNR. With a density of approximately 3.6x107 pericytes/cm3, pericyte is the second most frequent non-myogenic cell found in the heart in vitro.115 Pericytes express the contractile protein α-smooth muscle actin and under physiological (or pathological) conditions they contract, both in circumferential and longitudinal directions influencing the diameter and stiffness of capillaries. The close proximity of pericytes to sympathetic axons suggests that their tone may be under noradrenergic regulation.116 Pericytes has an established role in autoregulation of cerebral blood flow and contribute to vasoconstriction of cerebral capillaries and entrapment of erythrocytes and leukocytes in no-reflow zones following cerebral ischemia.117 In mouse models of cerebral ischemia, pericytes caused capillary constriction and obstructed erythrocyte passage despite reopening the middle cerebral artery.118 The role of pericytes in regulation of coronary blood flow is less clear; however, cardiac pericytes constrict coronary capillaries and reduce microvascular blood flow after ischemia, despite re-opening of the culprit artery.116 In rat models of ischemia and reperfusion, areas of capillary blockage co-localized with pericytes which showed a 37% diameter reduction. Notably, intravenous adenosine – a pericyte relaxant drug - increased the capillary diameter by 21% (at pericyte somata), decreased the capillary block by 25% and increased the perfusion volume by 57%.116 Recent evidence also strongly suggests that pericytes contract as a response to myocardial ischemia and they play an important role in CNR.119-120 Apart from adenosine116, there is also evidence that ischemic preconditioning inhibited the contraction of microvascular pericytes induced by cardiac ischemia/reperfusion injury suggesting that protective role of ischemic preconditioning may be at least partially mediated by its impact on pericyte function.121 Based on this evidence, it has been proposed that cardiac pericytes may represent a novel therapeutic target aiming at protection of coronary microcirculation and alleviation of MVO and CNR after AMI.31
Although there is a generally-held view that coronary circulation is maximally dilated during myocardial ischemia, this condition sets into operation a large number of systemic and local vasoconstrictor stimuli that impair the coronary vasodilator reserve and increase the vasoconstrictor tone of microcirculation, which may be alleviated by vasodilator drug therapy.122 Experimental studies have shown that arterioles undergoing ischemia/reperfusion fail to dilate under the effect of endothelium dependent vasoactive substances acetylcholine and bradykinin, which leads to reduced blood flow in the distal vascular bed showing that endothelium-dependent relaxation of coronary microvessels was markedly impaired during ischemia/reperfusion cycle.123-124 However, endothelium-independent relaxation to nitroglycerin or nitroprusside was not altered. Although, the underlying mechanisms of persistently increased vasoconstrictor tone in microcirculation undergoing ischemia/reperfusion remain unknown, excess alpha-adrenergic tone,125-126 angiotensin II,127-128 excessive production of ROS and cytokines like tumor necrosis factor alpha (TNFα),129-130 vasoconstrictor substances released from the culprit lesions including serotonin and thromboxane A2,130 endothelin131 or neuropeptide Y,132 most likely in combination, do play a role. A decrease in the availability of vasodilator mediators, particularly NO, as a consequence of ischemia/reperfusion contributes to heightened vascular tone in these conditions. Thus, inhibition133 or uncoupling of endothelial nitric oxide synthase)134, upregulation of arginase135-136 and increased production of ROS137 reduce NO availability affecting vascular tone (among other deleterious effects) during ischemia/reperfusion. Knowledge on the systemic or local mediators that enhance vasoconstrictor tone and contribute to impaired microvascular function during ischemia/reperfusion is important because pharmacological blockade of these mediators has been for decades the mainstay of therapy to prevent MVO and CNR in patients with STEMI. Although increased vasoconstrictor tone was considered as deleterious in that it contributes to obstructed microcirculation and MVO, we hypothesize that increased vasoconstrictor tone in the ischemic area may have a protective role as well. By obstructing the microcirculation, the increased vasoconstrictor tone confines ischemic products, necrotic debris and a large number of harmful catabolites and active substances to the ischemic area preventing them from spreading to surrounding viable myocardium or from entering the circulation. This may be at least one factor why vasodilator therapy fails to improve clinical outcome albeit it apparently may improve reperfusion. In addition, vasodilator therapy may preferentially dilate vessels surrounding ischemic region and shift the blood towards viable surrounding myocardium (microcirculation steal syndrome) worsening the reperfusion of ischemic region, apparently associated with improved reperfusion, at least by angiographic (TIMI flow grade) markers. However, these hypotheses need testing.
In aggregate, myocardial ischemia leads to various alterations affecting all components of microcirculation leading to various degrees of MVO and CNR depending on the duration and severity of coronary blood interruption, degree and availability of coronary collateral blood flow, metabolic demand (or anti-ischemic drugs) at the time of coronary occlusion and preconditioning due to pre-occlusion ischemic episodes. As stated above, endothelial cells – the principal cellular component of microcirculation – are relatively resistant to ischemia and may remain ultrastructurally intact up to 6 hours of ischemia in anesthetized cats.138 Similarly, in rat models of AMI, no clear damage to capillary endothelium occurred after 30 minutes of ischemia without reperfusion and no reduction of inter-endothelial cell junctions was observed after 90-minute occlusion of left anterior descending artery.139 Another study in mongrel dogs subjected to ligation of the circumflex branch showed mild swelling of endothelial cells but no totally occluded capillaries following prolonged periods of ischemia. This study suggested that ischemia-induced loss of vascular competence was unlikely to be due to intravascular thrombosis, endothelial cell swelling, or external compression by interstitial edema.140 Thus, shorter periods of ischemia (≤1 hour) are characterized by mild edema with almost no (or little) signs of cellular necrosis, inflammation or capillary injury. Longer periods of ischemia (≥2 hours) are characterized by uniform infarcted area (cellular death), marked infiltration by inflammatory cells (primarily neutrophils), and severely damaged capillaries and evident hemorrhage.92
Distal Embolization
Distal embolization or detachment of atherothrombotic fragments from the atherosclerotic plaque occurs spontaneously or during the primary PCI procedure as a result of guide-wire passage, lesion preparation and stent implantation. Angiographically visible distal embolization is documented in 11% to 17% of primary PCI procedures in patients with STEMI.141-144 However, the true incidence of lesser degrees of distal embolization appears to be much higher with one study showing that visible debris was retrieved in 73% of patients who received a distal embolization protection system.145 Histologically, the embolized material consists of a mixture of atheromatous debris, platelet aggregates, erythrocytes, fibrin, cholesterol crystals and inflammatory cells.146-149 Distal embolization is more frequent in atherosclerotic plaques with large volumes (particularly plaques with large necrotic core)150 and those with more intracoronary thrombus at the lesion site.151-152 Moreover, erythrocyte-rich thrombi, elevated glucose level on admission, larger culprit vessel, pre-balloon dilation and right coronary artery as culprit lesion have been identified as independently associated with a higher risk of distal embolization during the primary PCI procedures in patients with STEMI.144, 147 Although most studies have assessed distal embolization in patients with STEMI characterized by a large thrombus burden, it has been suggested that distal microembolization may occur during plaque erosion at the culprit lesion in patients presenting with non-STEMI.146 Since microthrombi preferentially end in well reperfused and viable myocardium (directed by blood stream), distal embolization kills potentially salvageable myocardium.153 One experimental study in dogs suggested that embolizing particles tend to flow away from the central infarcted area (forced by developing CNR) and accumulate in the infarct border contributing to infarct extension.154 The study suggested that embolizing particles are more important for infarct expansion than for CNR at least in the early phase of reperfusion. Distal embolization may contribute to CNR, increases biomarkers of myocardial necrosis, causes patchy microinfarcts that disproportionally impair the left ventricular function beyond the actual amount of damaged myocardium, increases infarct size and markedly reduces the efficacy of primary PCI and is associated with a poor clinical outcome.42, 143, 154-155 Distal embolization is not a sine qua non factor for the development of CNR and MVO and CNR may develop in patients with STEMI undergoing reperfusion even if distal embolization does not occur. However, distal embolization promotes MVO and CNR via a number of mechanisms (recently reviewed by Kleinbongard and Heusch146). First, distal embolization may cause physical (mechanical) obstruction at the arteriolar and/or microcirculation levels. Second, as shown by analysis of aspirates obtained in patients undergoing PCI, embolized material contains a number soluble vasoconstrictor substances that increase the vasoconstrictor tone at the arteriolar and microcirculation levels130-132 and contribute to MVO. Third, apart from mechanical obstruction, embolized material and active substances contained in it are prothrombotic and generate a highly prothrombotic milieu in microcirculation favoring platelet aggregation and in situ thrombosis.146, 156 Fourth, thrombotic and atheromatous debris (necrotic core content including foam cells, cell debris, lipids and crystalline cholesterol) are highly chemo-attractant and pro-inflammatory and induce a powerful local and systemic inflammatory response146, 157, which contribute to MVO and CNR. Experimental studies of microthrombi-induced CNR in Yorkshire pigs showed that distal embolization was associated with elevated levels of metalloproteinase-2 and a reduction in the activity of survival kinase (Akt) within the infarct zone 3 days after AMI, with both events helping to explain deleterious effects of distal embolization on infarct size.158 It has to be emphasized that the atheromatous debris fraction of the embolized material is extremely resistant to antithrombotic or thrombolytic agents used to treat patients with AMI and the only way to clear it is through inflammation or other processes set into operation by organism to clear necrotic tissue after AMI.
Reperfusion-Related Injury
While, endothelial cells and microcirculation in general are relatively resistant to ischemia (at least compared to cardiomyocytes), they are extremely sensitive to reperfusion-related injury –a condition first described by Jennings et al.159 in 1960 in canine hearts. There are at least 5 manifestations of reperfusion-related injury: reperfusion-induced arrhythmias, myocardial stunning, MVO, intra-myocardial hemorrhage and reperfusion-induced cell death (or lethal reperfusion injury).160-161 Although, myocardial ischemia and reperfusion appear to be opposite events in terms of blood interruption and restoration, they are similar in terms of molecular and cellular events that develop following both events and it appears that reperfusion-related injury completes the cellular damage initiated by ischemia. The underlying mechanisms for the explanation of the similarity between ischemia and reperfusion in terms cellular damage are unknown but both opposing events appear to produce the same set of mediators, ROS and activated metalloproteinases, that lead to cellular and tissue damage. The motif of reperfusion-related injury is unclear but it may represent an early scavenger mechanism to get rid of ischemia-induced irreversibly damaged cells.
Experimental studies in dogs by Kloner et al.10 involving clamping of proximal left circumflex coronary artery showed that fluorescent dye thioflavin S managed to penetrate ischemic myocardium after 40 minutes of ischemia followed by reperfusion. However, after 90 minutes of ischemia followed by reperfusion, thiofalvin S failed to penetrate the ischemic myocardium and perfusion defects were observed in subendocardium. Reperfusion failure was observed within seconds of clamp release and it was well established within the first few minutes. Notably perfusion defects were always found within the ischemic-necrotic zone but not in the surrounding viable myocardium not undergoing ischemia/reperfusion, strongly suggesting that CNR is due to microvascular damage within the zone of necrosis.10 Later studies showed that if proximal coronary arteries were occluded for a longer time (3 hours), then perfusion defects were more widespread and reached mid-myocardium and occasionally the outer layers of myocardium.162 These studies showed that the extent of CNR depends on the duration of ischemia, a finding that has been confirmed in the clinical studies as well.163 Histological ultrastructural studies showed a number of morphological alterations that underlie perfusion defects following reperfusion of ischemic myocardium. The most consistent finding was demonstration of areas of swollen endothelium and formation of intraluminal membrane-bound protrusions or blebs that obstruct the capillary lumen. Other (less frequently observed) markers of microcirculation damage included loss of pinocytotic vesicles, endothelial gaps, rupture of capillary walls with extravasation of red blood cells, deposits of fibrin tactoids in vicinity of endothelium gaps, platelet-leukocyte aggregates, and rouleaux structures of erythrocytes. The local edema involving endothelium and surrounding myocardium suggested an initial restoration of some blood flow which was later interrupted by reperfusion-induced MVO and swollen cardiomyocytes. Occasionally a capillary compressed (and consequently obstructed) by 2 swollen cardiomyocytes was seen. Reperfusion-induced hypercontracture of myocardium was also involved in the compression of microcirculation.164-165 Studies by Ambrosio et al.166 in open-chest dogs subjected to 90 minute occlusion of left circumflex coronary artery followed by reperfusion for 2 minutes or 3.5 hours showed that the extent of CNR area grows over the reperfusion time. Thus, the area of impaired reperfusion (absent thioflavin) was 9.5% of the initial area at risk in animals reperfused for 2 minutes and 25.9% of the area at risk in dogs reperfused for 3.5 hours. Importantly, serial measurements using microspheres showed that areas with adequate reperfusion at 30 minutes of reperfusion had a marked fall of perfusion at 3.5 hours of reperfusion. Another study in dogs undergoing 90-minute (balloon) occlusion of the left anterior descending coronary artery followed by reflow showed that the extent of MVO (assessed by hypo-enhanced regions on contrast-enhanced CMR) increased 3-fold over the 48 hours after reperfusion (3.2%, 6.7% and 9.9% of the left ventricular mass at 2, 6, and 48 hours, respectively).167 Similar findings were reported by Reffelmann and Kloner168 in a rabbit model of reperfusion. The area of CNR increased progressively from 12.2% after 2 minutes of reperfusion to 30.8% after 2 hours of reperfusion and to 34.9% of the initial area at risk after 8 hours of reperfusion. The expansion of CNR zone was fastest within the first 2 hours of reperfusion, finally encompassing ~80% of the infarct size. Moreover, regional myocardial blood flow was hyperemic at 2 minutes of reperfusion, decreased later and remained unchanged (plateau) between 2 and 8 hours of reperfusion. Notably, no hemorrhage was visible after 2 minutes of reperfusion but it reached a value of 37.3% of area at risk after 8 hours of reperfusion. One study that included patients with the first AMI showed that MCE-defined CNR present at 24 hours after reperfusion was sustained at one month in approximately 50% of the patients.28 These studies strongly suggested that CNR is primarily a reperfusion injury-related phenomenon.
Removal of mechanical obstacle in the proximal coronary arteries and blood flow restoration in ischemic myocardium is associated with a number of events within and outside the coronary microcirculation that further accentuate the ischemia-related injury. One of the earliest events that develop following blood flow restoration to ischemic myocardium is exacerbation of ischemia-initiated interstitial and cellular edema. An experimental study in dogs undergoing a 90-minute balloon occlusion of left circumflex artery followed by 60-minute reperfusion showed increased wall thickness in the reperfused myocardium due to tissue edema eventually leading to CNR because of mechanical compression.169 CMR imaging studies in pigs170 and humans171-172 have shown a bimodal pattern of myocardial edema following reperfusion. The early wave of edema appears to be due to exposure of a hyperosmotic interstitium (due to accumulation of catabolites produced during ischemia) to normo-osmotic blood at reperfusion, which creates an osmotic gradient forcing water to move from intravascular to interstitial space. The early wave of edema occurs immediately after reperfusion and markedly diminished at 24 hours as a result of catabolite washout from the interstitium.170 The second (late) wave of edema develops gradually following ischemia/reperfusion and is maximal around day 7 following reperfusion.170 The second wave of edema is explained by increased vascular permeability related to influx of inflammatory cells and healing process of the infarcted tissue.170 Interstitial and cardiomyocyte edema generate forces from outside microvasculature that tend to compress the capillary wall and increase the resistance to blood flow.
The loss of cellular competency to maintain ionic hemostasis in the setting of ischemia/reperfusion is associated with at least 2 consequences: cellular swelling and intracellular Ca2+ overload. Thus, ischemia-initiated endothelial cell edema is further accentuated by reperfusion worsening the ischemia-initiated MVO. Upon restoration of blood flow, the extracellular pH is rapidly restored which stimulates the Na+/H+ exchanger and Na+/HCO3- symporter leading to proton extrusion from the cells, rapid normalization of intracellular pH, massive Na+ influx, and intracellular Ca2+ overload.73, 160 The increased Ca2+ in endothelial cells leads to cellular retraction and intercellular gap formation and blebbing resulting in increased vascular permeability and obstruction of intra-capillary space. Occasionally, endothelial cell retraction is so severe that it may lead to a total detachment of endothelial cell from the basal membrane.92 In addition to cellular swelling, increased ATP availability upon restoration of blood flow, restored intracellular pH and abundant cytoplasmic Ca2+ favor the hypercontracture of cardiomyocytes and contraction band formation – a histological marker of reperfusion173 which further compresses the microvasculature. Reperfusion is associated with increased rates of cellular death. One of the most important deleterious effects of reperfusion is mitochondrial injury related to opening of mitochondrial permeability transition pore (MPTP) – a nonselective channel that enables movement of water, ions and low molecular weight solutes to cross the inner mitochondrial membrane. MPTP channel remained closed during ischemia under the inhibitory effect of acidosis and increased Ca2+ content.161, 174 Removal of the inhibitory effects of acidosis, Ca2+ overload and massive ROS production during reperfusion cause MPTP opening leading to loss mitochondrial inner membrane potential, oxidative phosphorylation/ATP production uncoupling, mitochondrial membrane rupture, release of apoptotic factors (cytochrome c) and cell death by necrosis.73, 160 Apart from necrosis other forms of cellular death particularly apoptosis participate in cellular loss following ischemia/reperfusion. Thus apoptosis is evidenced within minutes after initiation of ischemia173 and it persists over days to weeks into the postinfarction period in humans.175
Blood cells that are brought to ischemic microcirculation following reperfusion contribute further to microvasculature injury and MVO. Serial CMR imaging studies in pigs have shown that interstitial edema is maximal immediately after reperfusion, whereas the maximal content of neutrophils, macrophages, and collagen is observed at 24 hours, 4 days and 7 days after reperfusion.170 Neutrophils brought to ischemic microcirculation upon blood restoration are activated and tend to aggregate with other neutrophils, platelets or endothelial cells leading to microcirculation plugging and MVO. In addition, activated neutrophils produce inflammatory cytokines, ROS, elastase and metalloproteinases, which cause capillary destruction, vascular leakage and a strong inflammatory response. In addition, newly-brought platelets are activated in the highly prothrombotic milieu, as is microcirculation undergoing ischemia/reperfusion, and activated platelets aggregate causing further capillary plugging and release numerous active substances with vasopressor and prothrombotic effects further increasing vascular tone and microthrombi formation (see: Myocardial ischemia). In brief, newly arrived cells in microcirculation upon restoration of blood flow to previously ischemic microcirculation are activated and recruited in capillaries and further accentuate ischemia-induced MVO and CNR.
Intramyocardial hemorrhage is one of the most severe manifestations of reperfusion-related injury that is closely linked with MVO and CNR.176-177 Although, ischemia may damage vascular barrier, increase vascular permeability and predispose to extravasation of erythrocytes, experimental studies in anesthetized dogs178 and autopsy studies in patients with AMI (recanalized with intracoronary streptokinase within 3.5 hours of ischemia)179 showed that intramyocardial hemorrhage is always observed following reperfusion but not in non-reperfused infarctions. Intramyocardial hemorrhage is always confined to the necrotic zone (not in the nonischemic tissue) predominantly in the central part of the necrosis and tends to diminish towards the border zone.179-180 The presence and extent of intramyocardial hemorrhage differs in the time period following reperfusion. Serial imaging studies in pigs have shown that the hemorrhage score (from 0 absent to 5 very severe) was 0 at 120 minutes, 2 at 24 hours, 4 at day 4 and 1 at day 7 after the reperfusion.170 CMR imaging and histological studies in dogs undergoing 4 hours of coronary occlusion followed by one hour of reperfusion showed that CMR-assessed hemorrhage size (decreased signal intensity zones) correlated closely with hemorrhage size defined by histology (correlation coefficient of 0.96).181 In dogs without reperfusion, no macroscopic or CMR-defined zones of hemorrhage were observed.181 The underlying factors of intramyocardial hemorrhage remain partially known. Marked increase in permeability gap formation (of sufficient diameter to allow passage of erythrocytes) in the vascular barrier after ischemia followed by reperfusion may lead to extravasation of red blood cells in the perivascular space. Platelet-activating factor (PAF) - a potent inflammatory mediator182 - and prolonged adhesion of neutrophils to endothelium may promote gap formation183 via basal membrane destruction and endothelial cell detachment via released active proteases183 and formation of neutrophil extracellular traps (NETs).110, 184 However, it is known that hemorrhage is more common after prolonged severe ischemia followed by reperfusion, which results in necrosis of endothelial cells, breakdown of basal membrane and destroyed microvessels.185-186 These studies support the notion that intramyocardial hemorrhage represents the most severe form of ischemia/reperfusion. Microvascular destruction and local consumption of coagulation factors due to coagulation cascade activation and intravascular microthrombi formation promoted by activated endothelium and inflammation has also been suggested as mechanisms for extravasation of erythrocytes and hemorrhage in the areas of MVO after AMI.187 Apart from being a manifestation of severity ischemia/reperfusion after AMI, intramyocardial hemorrhage per se aggravates MVO and CNR. First, intramyocardial hemorrhage aggravates extracellular compression exacerbating MVO. CMR imaging studies in swine undergoing circumflex coronary artery occlusion for 75 minutes by a balloon catheter and in patients with AMI showed an overlap and a close anatomic correlation between areas of intramyocardial hemorrhage and MVO (correlation coefficients of 0.85 and 0.87, respectively).187 In addition, CMR imaging studies showed that all patients with AMI and intramyocardial hemorrhage on T2* imaging had CMR-confirmed MVO188 or that 80% of patients with MVO had CMR evidence of intramyocardial hemorrhage.189 Second, intramyocardial hemorrhage is irreversible and induces a powerful and prolonged inflammatory response, which also contributes to MVO. Extravasated erythrocytes undergo destruction, which leads to iron release and deposition in the infarct zone. CMR imaging studies in canine models of ischemia/reperfusion showed that intramyocardial hemorrhage leads to iron deposition in the infract zone up to 2 months after the acute event and that newly recruited macrophages co-localize with iron deposits suggesting a prolonged inflammatory burden in the chronic phase of myocardial infarction.190 Intramyocardial hemorrhage appears to be more frequent after reperfusion by thrombolytic agents than primary angioplasty. In a series of 19 necropsies of patients undergoing thrombolytic therapy, 74% of infarcts treated with thrombolytic agents (but none of the infarcts undergoing balloon angioplasty alone) were hemorrhagic.191 Apart from thrombolytic agents, intramyocardial hemorrhage appears to be associated (or be more frequent) with larger infarct size, greater MVO, larger left ventricular dimensions, lower LVEF, anterior wall infarct location and glycoprotein IIb/IIIa inhibitor use. However, after adjustment, only anterior wall infarct location and the use of glycoprotein IIb/IIIa inhibitors were associated with higher odds of intramycardial hemorrhage.192 Intramyocardial hemorrhage is a determinant of infarct size, infarct expansion and reduced myocardial salvage after reperfusion.193 Intramyocardial hemorrhage is a strong correlate of adverse outcomes, which is stronger than infarct size188 and patients with MVO and intramyocardial hemorrhage have a worse prognosis than patients with MVO without intramyocardial hemorrhage.194
Ischemia/reperfusion is associated with a strong inflammatory response in the infarct zone. Although inflammatory response may remove necrotic tissue and promote scar formation and tissue healing, in acute phase it greatly contributes to MVO and CNR. Inflammatory response is predominantly mediated by neutrophils but also by monocytes, macrophages and lymphocytes. One experimental study in mongrel dogs in which a segment of a large epicardial coronary artery was deprived of blood flow for 3 hours followed by reperfusion, showed an influx of neutrophils within the media of ischemic/reperfused vessels but not in the nonischemic vessels. Moreover, electron microscopic analysis showed that neutrophils were often located between the endothelial cells and the elastic lamina in the ischemic/reperfused vessels.195 As stated earlier in this review neutrophils form aggregates with other cells and form NETs contributing to microcirculation plugging and impediment of blood flow and stick to endothelial cells via adhesion molecules (favored by endothelial cell activation and glycocalyx shedding) promoting endothelial damage and increased vascular permeability and leakage. The deleterious effects of neutrophils (and other immune cells) are predominantly mediated by release various active substances including inflammatory cytokines,196 MMPs (particularly MMP-9)113-114, ROS111, 134 and myeloperoxidase.112 The necrotic debris (among other stimuli) activates the NLRP3 inflammasome, which regulates caspase-1 activity, stimulates production (and release) of large amounts of cytokines (primarily IL-1β and IL-18) and promotes inflammatory cell death via pyroptosis.197 Inflammasome contributes to MVO and CNR by exacerbating endothelial cell damage and vascular leakage (promoting interstitial edema and microcirculation compression), heightening vasopressor tone and promoting cellular trapping and stasis and microthrombi formation in microcirculation.198-199 Patients with AMI who develop MVO and CNR have higher levels of several inflammatory cytokines in circulation, including C-reactive protein163, 200, interleukin 6 (IL-6)201 and interleukin 8 (IL-8)202 compared with patients who did not develop these phenomena. Patients developing CNR have significantly higher levels of myeloperoxidase at culprit lesions than patients without CNR.203-204 MPO produces a large number of highly reactive species which attack all known cellular components leading to reduced NO availability, endothelial dysfunction and impaired vasoreactivity.112 The role of inflammation in tissue healing is outside the scope of this review.
Of all mechanisms and mediators proposed to date to explain microcirculation damage in the setting of ischemia/reperfusion, ROS and MMPs, have received most attention for their role in microvascular damage and genesis of MVO. Ample evidence suggests that excess production of ROS and activation (or overexpression) of MMPs are underlying mechanisms of tissue damage (including microcirculation) during ischemia/reperfusion and their actions appear to be mutually dependent. In the setting of ischemia/reperfusion, ROS appear to have multiple cellular sources (endothelial cells, platelets, neutrophils or other immune cells and resident macrophages) and the main producers at molecular level are xanthine oxidase, NADPH oxidase, mitochondrial electron transport chain and uncoupled NO synthase.92, 134 Excess amounts of ROS interact with any biological structure in their vicinity rendering them dysfunctional. ROS are involved and play a critical role in almost all cellular and molecular events leading to MVO in the setting of ischemia/reperfusion. Likewise, activated MMPS appear to have multiple sources including endothelial cells, smooth muscle cells, inflammatory cells and resident macrophages.92 MMPs have a wide specificity and cleave a wide range of extracellular matrix components including glycocalyx, inter-endothelial cell junctions, basal membrane and extracellular substrates such as adhesion molecules, cytokines and chemokines.92 MMPs play a critical role in the MVO by participation in the increased vascular permeability and leakage, glycocalyx shedding, capillary destruction and intramyocardial hemorrhage. Detailed information on the biology of ROS and MMPs and their role in genesis of MVO during ischemia/reperfusion are provided in 2 excellent reviews by Granger and Kvietys.92, 134
Individual Susceptibility (Predisposing Factors) to CNR
The frequency of CNR after primary PCI differs widely. Aside from sensitivity of the method used to detect CNR and the timing of the assessment (already discussed), a number of other factors appear to predispose to CNR after primary PCI including, infarct size, pre-existing endothelial and microvascular dysfunction, cardiovascular risk factors, culprit lesion characteristics, invasiveness of coronary intervention and genetic predisposition to CNR.
Several experimental166, 205 and clinical163, 206 studies have shown that infarct size is a determinant of CNR, both in terms of frequency and extent. Expectedly, a higher frequency of CNR has been reported in clinical conditions that lead to larger infarct size such as culprit lesion location in the proximal left anterior descending artery206-207 and longer time-to-treatment interval.163, 208 Our group assessed the correlates of CNR in 1140 patients (108 with CNR) undergoing primary PCI. The study identified, advanced age, no smoking, previous myocardial infarction, Killip class, serum creatinine, C-reactive protein, time-to-treatment interval, LVEF, baseline TIMI flow and scintigraphic initial perfusion defect as correlates of CNR. However, after adjustment, only 4 variables – previous myocardial infarction, baseline TIMI flow, C-reactive protein and initial perfusion defect correlated independently with a higher risk of CNR after primary PCI.163 Since, a large necrosis is associated with more extensive local tissue destruction including vascular tissue, edema and mechanical compression, the association between infarct size and CNR is explainable. The incidence of CNR is markedly higher in patients with STEMI compared with patients with non-ST-segment elevation or patients undergoing elective PCI.209
Pre-existing endothelial and microvascular dysfunction are common in patients with AMI (related to cardiovascular risk factors or coronary atherosclerosis and/or myocardial diseases) and they likely contribute to MVO and CNR after primary PCI.210 Coronary artery segments distal to the atherosclerotic plaques undergo remodeling with hypertrophy of vascular wall and attenuation of vasomotor responses.211 Diseases associated with (or predisposing to) coronary microvascular dysfunction and the underlying mechanisms of this association have been reviewed.212 Indeed, pre-existing coronary endothelial and microvascular dysfunction increases the susceptibility of microcirculation to ischemia/reperfusion-related injury and facilitates the development of MVO and CNR.213-214
Culprit lesion morphology appears to impact on frequency of CNR after primary PCI. Atherothrombotic plaques responsible for ACS are larger and softer (contain more lipids, inflammation and thrombus) and are more prone to be fragmented (and embolized) during coronary interventions.153 One study showed that large lipid index (defined by optical coherence tomography and plaque burden (defined by and intravascular ultrasound) were associated with a higher risk of CNR after primary PCI.215 Furthermore, a long target lesion length, larger reference diameter, and high thrombus burden on angiography or large vessels with lipid pool–like image on ultrasound imaging are reported to be independent correlates of CNR.216-217 One angiographic study of patients with AMI identified the cutoff pattern of occlusion (an abrupt cutoff without taper before the occlusion) in the infarct-related artery, accumulated thrombus (> 5 mm) proximal to the occlusion, presence of floating thrombus, persistent contrast stasis distal to the obstruction, reference lumen diameter of infarct related artery ≥4 mm, and incomplete obstruction with presence of accumulated thrombus more than three times the reference lumen diameter of infarct-related artery as independent correlates of slow flow or CNR. Conversely, early reperfusion (<240 minutes), baseline TIMI flow ≥2 and taper pattern of occlusion in the infarct-related artery were independent correlates of freedom from slow flow or CNR after reperfusion.218 It has been reported that atherectomy and coronary stenting cause more frequently plaque fragmentation and embolization compared with balloon angioplasty.219 The SYNTAX score obtained in the diagnostic phase of primary PCI for STEMI can identify patients at risk for CNR with a cut-off of >21 identifying patients having double the risk for CNR compared to those with SINTAX score ≤21.220 The intervention in saphenous grafts, high pressure balloon inflation and debulking devises appear to increase the frequency of CNR, potentially due to more frequent distal embolization.221 A large study based on the National Cardiovascular Data Registry (NCDR) identified, longer lesion length, higher class C lesions, bifurcation lesions and impaired preprocedural TIMI flow as independent angiographic correlates of CNR.222 Apart from angiographic correlates, numerous biomarkers including blood cell-related markers (leukocyte and neutrophil count, mean platelet volume), thromboxane A2, markers of myocardial necrosis (creatine kinase and cardiac troponin), markers of inflammation (C-reactive protein and fibrinogen), Von Willebrand factor, tissue factor, natriuretic peptides and endothelin have been reported to be associated with CNR after primary PCI.223 The incidence of CNR after primary PCI appears to be higher in patients with elevated uric acid level224, impaired renal function225-226, higher systemic immune-inflammation index227, higher PRECISE-DAPT score228, lower vitamin D levels229, higher red blood-cell distribution width230 and higher soluble suppression of tumourigenicity 2.231
Cardiovascular risk factors predispose to MVO and CNR. Although diabetes mellitus is associated with generalized endothelial232 and microvascular233-234 dysfunction, and it greatly contributes to poor outcomes in patients with cardiovascular disease235, in patients with STEMI, pre-existing diabetes mellitus was not associated with more frequent (or extensive) MVO compared with patients without diabetes.236-237 However, several CMR-imaging studies have shown that acute hyperglycemia is associated with MVO in patients with STEMI.236, 238-239 One prospective study that excluded patients with diabetes showed that hyperglycemia on admission was an independent correlate of presence and size of MVO in patients with the first STEMI.240 It has been suggested that hyperglycemia predispose to MVO and CNR via a number of mechanisms including leukocyte plugging in capillaries, platelet activation (increased procoagulability), elevated catecholamine level (common in large infarcts), elevated free fatty acid levels and toxic metabolites impairing endothelial function, impaired endothelial-dependent vasodilatation, increased oxidative stress and inflammatory cytokines.238, 241 Arterial hypertension is commonly associated with endothelial dysfunction (impaired endothelium-dependent vasodilatation) mostly related to reduced availability of NO.242 However, arterial hypertension was not associated with invasive intracoronary parameters used to assess reperfusion injury in patients with AMI243 or CMR-defined MVO243-244 but it showed a questionable association (P=0.059) with intramyocardial hemorrhage.243 The association between hypercholesterolemia and CNR remains poorly investigated and controversial. In patients with normal coronary arteries and arterial hypertension, hypercholesterolemia was associated with depression of both basal and pharmacologically stimulated bioavailability of NO245 suggesting a role of these conditions in promoting endothelial dysfunction. Likewise an experimental study in rabbits fed by 2% cholesterol-enriched diets for 3 days showed that hypercholesterolemia was associated with larger infarct size and larger nonreperfused (no-reflow) zones suggesting that hypercholesterolemia increased infarct size by vascular obstruction.246 However, evidence that links hypercholesterolemia with MVO in patients with AMI undergoing reperfusion is weak. One CME-study found no difference in the incidence of CNR according to hypercholesterolemia in 293 patients with AMI undergoing successful primary PCI.247Another CMR imaging study showed modest association (adjusted odds ratio of 1.02) between elevated low-density lipoprotein (LDL)-cholesterol level and microvascular injury in 235 patients with STEMI undergoing primary PCI.248 A recent lipidomics study showed that phosphatidylcholine, alkylphosphatidylcholine, and sphingomyelin, were significantly elevated in plasma of patients with STEMI who developed CNR after primary PCI.249 Despite well-known deleterious effects of smoking on vasculature including endothelial dysfunction, vascular remodeling, increased thrombogenicity and proinflammatory action,250 recent CMR imaging studies in reperfused patients with STEMI did not show a significant difference in the presence or extent of MVO in smokers versus nonsmokers.251-253 However, smoking appears to favor intramyocardial hemorrhage.251, 253 A higher CHA2DS2-VASc Score is also associated with the increased risk of CNR after primary PCI.254-255 Pre-infarction angina appears to reduce the incidence of CNR potentially due to salutary effects of ischemic pre-conditioning.256-257
There appears to be a genetic predisposition to CNR. A 2007 case-control study showed that survivors of AMI who developed CNR after PCI had more compact fibrin network and resistance to lysis.258 The study suggested a genetic predisposition to CNR mediated by genetic factors that modulate fibrin clot properties. One study of patients with STEMI showed higher serum levels of SCUBE1 [signal peptide-CUB (complement C1r/C1 s)-EGF (epidermal growth factor)-like domain-containing protein 1] – a protein expressed in platelets and endothelial cells that could function as an adhesion molecule – in patients who developed CNR after primary PCI.259 The 1976 T/C polymorphism of the adenosine 2A receptor gene may increase susceptibility to microvascular injury and CNR.210 Single nucleotide polymorphisms in the VEGFA -vascular endothelial growth factor A - and CDKN2B-AS1 genes are associated with abnormal coronary flow reserve and microvascular dysfunction in patients without significant obstructive coronary artery disease referred for cardiac catheterization.260 Furthermore there appears to be sex-specific differences with genetic variation in alleles of MYH15 (Myosin Heavy Chain 15), VEGFA, and NT5E (5'-Nucleotidase Ecto) genes increasing the risk of coronary microvascular dysfunction in men.260 Patients with MVO after primary PCI show a sustained increase in the levels of platelet gp91phox (NOX2) - the catalytic subunit of NADPH oxidase – and 8-iso-PGF2α – a marker of lipid peroxidation - suggesting platelet-mediated ROS generation involvement in MVO.261 Despite this evidence, the role of genetic factors in pathophysiology of MVO and CNR remains to be better defined.
Impact of CNR on Clinical Outcome
Clinical presentation of CNR varies greatly. CNR can be clinically silent or manifest as angina, ST-segment (re)elevation, sudden hemodynamic deterioration while in the catheterization laboratory, malignant ventricular arrhythmias, early congestive heart failure (or cardiogenic shock) and cardiac death.262 At longer term, patients with AMI exhibiting CNR following reperfusion are prone to develop depressed left ventricular function and adverse left ventricular remodeling and have more frequent congestive heart failure or cardiogenic shock and reduced survival. In patients with the first AMI of anterior wall, Ito et al.263 showed that MCE-defined CNR was associated with progressive increase in left ventricular end-diastolic volume and early and more prolonged congestive heart failure in the postinfarction period with 3 of 47 patients with CNR dying of pump failure. The study strongly suggested that CNR is associated with subsequent adverse left ventricular remodeling. In the study by Resnic et al.264 that included 4264 patients with AMI undergoing primary PCI, CNR was highly predictive of postprocedural MI and in-hospital death. In the study by Brosh et al.265 that included 599 patients with STEMI undergoing primary PCI, CNR occurred more frequently after coronary stenting and required more often balloon pump counterpulsation. Patients with CNR had larger enzymatic infarct size, more frequent moderate-to-severe left ventricular dysfunction and higher 6-month mortality than patients without CNR. A large study based on NCRD data showed significantly larger infarct size (creatine kinase-MB: 133 vs. 76 ng/ml) and a higher incidence of in-hospital complications after PCI for AMI including mortality (12.6% vs. 3.8%), reinfraction (2.4% vs. 0.7%), cardiogenic shock (7.4% vs. 1.7%) and heart failure (5.2% vs. 2.1%) in patients with CNR compared with patients without CNR.222 The length of hospital stay was significantly longer in patients who developed CNR compared with patients who did not (mean: 5.9 days vs. 4.9 days).222 Of note, CNR was associated with worse outcomes in patients with STEMI and non-STEMI, but the incidence of all complications was higher in patients with STEMI compared with patients with non-STEMI.
Studies that have investigated the association between CNR and long-term outcome have further confirmed a worse long-term prognosis associated with CNR. Morishima et al.22 assessed the association between CNR and prognosis in 120 patients with the first AMI treated by PCI over a mean follow-up of 5.8 years. CNR was associated with a higher risk of cardiac death, all-cause death, malignant arrhythmias and congestive heart failure. After adjustment, CNR remained an independent correlate of cardiac death and adverse cardiac events. At follow-up, survivors with CNR had higher end-diastolic and end-systolic left ventricular volumes and plasma brain natriuretic peptide levels and lower LVEF strongly suggesting the role of CNR in adverse left ventricular remodeling. In the study by Bolognese et al.266 that included 124 patients with AMI, microvascular dysfunction assessed by intracoronary MCE, was the only independent correlate of cardiac death and combined adverse events (cardiac death, reinfarction, and heart failure) over a mean 46-month follow-up. Notably, starting from day one, left ventricular volumes increased progressively and were significantly higher at 6 months in patients with microvascular dysfunction compared with patients without microvascular dysfunction. Our group163 assessed the association of CNR with myocardial salvage (defined by repeat scintigraphy), left ventricular function at 6 months and mortality at one year in 1140 patients with STEMI undergoing primary PCI. In this study, CNR was associated with significantly larger initial perfusion defect and final infarct size and lower myocardial salvage by primary PCI (salvage index: 34% of the initial perfusion defect in patients with CNR versus 55% of the initial perfusion defect in patients without CNR). Notably, patients with CNR had a lower LVEF than patients without CNR (47.7±13.1% versus 54.2±13.9%) and less LVEF improvement at 6 months and higher mortality (16.7% vs. 5.5%) at one year. Our group49 also studied the association between CNR and 5-year mortality in 1406 patients with STEMI undergoing primary PCI. CNR was defined as a TIMI flow grade <3 or any TIMI flow grade combined with a TIMI perfusion grade of 0 to 1. CNR was diagnosed in 29% of the patients. Patients with CNR had significantly higher scintigraphic infarct size (15% of the left ventricle vs. 8% of the left ventricle) and higher 5-year mortality (18.2% vs. 9.5%). After adjustment, CNR remained significantly associated with the risk of 5-year mortality (adjusted hazard ratio of 1.66). Of note, CNR after primary PCI provided prognostic information that was independent of and beyond the information provided by the infarct size.
Several studies have assessed the association between transient CNR, defined as CNR during the primary PCI procedure in the absence of flow limiting obstructions at the culprit lesion that reversed to normal blood flow at the end of the procedure, with clinical outcome.267-270 In 1192 patients undergoing primary PCI in the setting of Primary Angioplasty in Myocardial Infarction (PAMI) trial, transient CNR occurred in 16 patients (1.3%).267 Patients with transient CNR had higher in-hospital (13% vs. 2%) and 6-month (31% vs. 3%) mortality. One study that compared transient CNR with persistent CNR reported a lower in-hospital mortality in patients with transient CNR. The study also identified a baseline TIMI flow grade of 3 and better renal function as correlates of transient CNR.268 A retrospective study of 414 patients with STEMI undergoing primary PCI showed a graded increase in the 6-month mortality in patients exhibiting slow flow (6.8%), transient CNR (14.1%) and persistent CNR (44.4%).269 Finally, in a large registry that included 4329 Korean patients with AMI (2668 patients with STEMI), the incidences of transient and persistent CNR were 5% and 1%, respectively. In patients with restored flow, transient and persistent CNR, the 3-year all-cause mortality was 18.1%, 24.4% and 46.7%, respectively; cardiac mortality was 7.9%, 13.1% and 40.0%, respectively.270 In this study, transient CNR was associated with higher long-term mortality compared with patients with restored flow and with a lower long-term mortality compared with patients with persistent CNR after primary PCI. The differences in mortality were mostly driven by differences in cardiac mortality. Transient CNR appears to correlate with the characteristics of culprit lesion. A study of patients with stable CAD, in whom the coronary plaques were assessed using the multidetector spiral computed tomography, showed that a low density plaque by computed tomography and reduced LVEF were identified as independent correlates of transient CNR.271 Likewise, in patients with AMI undergoing PCI, Iijima et al.272 showed that vessel area, plaque burden in the culprit lesion, ruptured plaque, lipid-like images and thrombus formation by intravascular ultrasound were significantly more frequent among patients with transient CNR than patients with reflow. However, after adjustment, only plaque burden and thrombus formation were independently associated with transient CNR. The underlying mechanisms of transient CNR remain unclear. However, coronary blood fluctuations during the primary PCI procedure may reflect vasoconstriction caused by release of vasoactive substances like thromboxane A2 and serotonin when thrombus at culprit lesion site is squeezed against the artery wall during balloon inflation.273 Furthermore, embolized fresh loose thrombotic material causing temporary vessel occlusion may be dissolved upon flow restoration facilitated by excess amounts of antithrombotic/anticoagulant drugs used during the primary PCI procedures. Finally, acute recruitment of vessels in the periphery of ischemic/necrotic region may restore TIMI flow grade by blood shifting towards myocardium surrounding the ischemic/necrotic region leading to a microcirculation steal syndrome. However, whether transient CNR at the end of primary PCI procedure heralds the development of persistent CNR remains to be explored.
Although it has been firmly established that both persistent and transient forms of CNR are associated with poor clinical outcome after primary PCI, the underlying mechanisms of this association remain unclear. Ample evidence suggests that infarct size – a well-known correlate of poor outcome after primary PCI for STEMI – is a strong correlate of CNR. In this regard, CNR is a manifestation of severe ischemic damage of the myocardium characterized by prolonged ischemia affecting extensive parts of the myocardium. In addition, factors that predispose to CNR (see material under subheading: Predisposing factors for CNR) predispose also to a poor clinical outcome, regardless of CNR. Although evidence supporting an association between infarct size and CNR is firm, whether CNR per se impacts on the infarct size remains controversial. Since, the zone of CNR is almost invariably located within the dead tissue (i.e., surrounded by dead tissue), it has been suggested that CNR does not cause ischemic cell death.165 However, clinical studies in patients with STEMI have shown that CNR is associated with larger final infarct size independent of initial infarct size and with reduced myocardial salvage by primary PCI.163 Apart from being a manifestation of severity of myocardial ischemia or worse cardiovascular risk profile, by impeding blood flow to necrotic tissue, CNR delays the removal of necrotic debris and impedes the arrival of cells and cytokines that are involved in the tissue healing.165 Experimental studies in rats showed that CNR persists up to one month after reperfusion and is associated with scar thinning and infarct expansion.274 Likewise, clinical studies have shown that MVO and CNR are most important correlates of left ventricular adverse remodeling after reperfusion.266, 275 Finally, intramyocardial hemorrhage is common in patients with CNR.189, 193 As already stated in this review, intramyocardial hemorrhage is associated with larger infarct size and infarct expansion after reperfusion193 and it is an independent correlate of major adverse cardiovascular events after reperfusion in patients with STEMI.276
Therapy of CNR
Over the years, many therapies have been tested to protect microcirculation and prevent or alleviate MVO and CNR in patients with STEMI undergoing reperfusion. Despite more than 50 years of research, little progress has been made in finding a treatment strategy of proven efficacy to be routinely used to prevent CNR in patients with STEMI. Notably, many therapies that showed benefits in reducing MVO and CNR in animals failed to result in similar benefits in clinical setting in patients with STEMI. Although some therapies have shown benefit in improving some markers of reperfusion, so far, no therapy applied to prevent or alleviate MVO or CNR has resulted in clinical benefit in terms of reduction of hard clinical endpoints such as mortality. Many therapies (mechanical strategies used to prevent distal embolization and various pharmacological agents) used before to treat CNR are no longer recommended to be used in patients with STEMI undergoing primary PCI due to futility reasons or worse clinical outcomes with them. Therapy against MVO and CNR includes nonpharmacological and pharmacological approaches. All pathophysiological mechanisms of CNR – distal embolization, myocardial ischemia and reperfusion-related injury – and predisposing factors (when possible) have been targeted by nonpharmacological or pharmacological preventive strategies as single or combined strategies. These therapies were applied before, during or after cardiac catheterization and primary PCI procedure to prevent or alleviate CNR (when it develops).
Therapy against Distal Embolization
Various nonpharmacological and pharmacological therapies have been tested to reduce the incidence of distal embolization in the setting of primary PCI procedures in patients with STEMI. Intuitively, direct stenting (stenting without pre-dilation) was expected to reduce MVO and CNR by reducing distal embolization. Early randomized studies277-278 with limited numbers of patients and meta-analyses279-280 suggested that direct stenting may reduce the incidence of CNR in patients with STEMI. In one meta-analysis, however, direct stenting was significantly better in reducing CNR in nonrandomized studies but not in randomized trials.279 A large recent study including patient-level data from 3 randomized trials showed no benefit of direct stenting in improving the markers of tissue reperfusion including ST-segment resolution or myocardial blush grade.281 The study also showed no direct stenting-by-thrombus aspiration interaction with respect to ST-segment resolution or myocardial blush grade. The fact that direct stenting did not produce a consistent action regarding reduction of the CNR may point to a modest impact of distal embolization in the pathophysiology of CNR.42
Distal protection devices (distal filters, distal occluders, proximal occluders or thrombus extraction devices) have been used to reduce distal embolization of atherothrombotic debris during the primary PCI. The EMERALD (Enhanced Myocardial Efficacy and Recovery by Aspiration of Liberated Debris) trial randomized 501 patients with STEMI to PCI plus balloon occlusion and aspiration distal microcirculatory protection system or PCI alone. Visible debris was retrieved from 73% of the patients. However, distal protection system failed to reduce CNR and had no significant impact on final TIMI flow, final corrected TIMI frame count, myocardial blush grade, ST-segment resolution >70%, infarct size or 6-month incidence of major adverse cardiovascular events (MACE).145 In the same vein, other randomized trials of distal filter protection showed that the routine use of distal protection devices during the primary PCI did not improve microvascular perfusion, limit infarct size or reduce the occurrence of MACE.282-283 Based on the results of these studies, distal protection devices are not recommended to be used as adjunct to primary PCI.
Mechanical or aspiration thrombectomy devices have also been used to reduce distal embolization in the setting of primary PCI. A 2013 meta-analysis summarized studies that have assessed the impact of aspiration (18 trials with 3936 patients) and mechanical thrombectomy (7 trials with 1598 patients) on tissue reperfusion and clinical outcome.284 In this meta-analysis, aspiration thrombectomy reduced the risk of MACE and all-cause mortality but not final infarct size or LVEF. With respect to markers of tissue reperfusion, aspiration thrombectomy improved ST-segment resolution and TIMI blush grade. On the other hand, mechanical thrombectomy did not improve TIMI blush grade, infarct size or the incidence of MACE; however, it improved ST-segment resolution at 60 minutes.284 The meta-analysis discouraged the use of mechanical thrombectomy but it offered some evidence that aspiration thrombectomy may be beneficial with respect to microcirculation protection and clinical outcome when used as adjunct to primary PCI. However, two large randomized trials showed no benefit in using aspiration thrombectomy in the setting or primary PCI.285-286 The Trial of Routine Aspiration Thrombectomy with PCI versus PCI alone in patients with STEMI (TOTAL) study even showed a significant increase in the incidence of stroke with aspiration thrombectomy.286 Notably, the TOTAL trial showed no difference in the incidence of angiographic CNR between aspiration thrombectomy and PCI alone groups (2.4% vs. 2.8%; P=0.28).286 These studies showed that aspiration thrombectomy does not protect microcirculation or reduce the incidence of CNR or improve clinical outcome when used as adjunct to primary PCI in patients with STEMI.
A strategy of deferred stenting – a two-step strategy of initial reperfusion by balloon angioplasty (or thrombus removal) followed by stent implantation hours or days thereafter - was used with the hope that it would reduce the rate of vessel dissection or distal embolization. In a study of 101 patients with STEMI with ≥1 factor for CNR, deferred stenting reduced the frequency of CNR (2% vs. 14%) and improved myocardial salvage assessed by CMR at 6 months.287 However the DANAMI-3-DEFER trial that included 510 patients with at least one CMR study showed that deferred stenting did not reduce the infarct size or the extent or MVO (43% vs. 42%) or increase myocardial salvage compared with conventional primary PCI.288 A 2018 meta-analysis of all randomized trials to that time with 1570 patients showed that a strategy of deferred stenting reduced the incidence of slow flow or CNR (8.8% vs. 16.6%) but not MVO assessed by CMR (48.4% vs, 52.6%; P=0.51).289 The treatment effect for slow flow or CNR and MVO correlated with a thrombus score grade >3 at the baseline angiography and with the total stent length implanted in the culprit coronary artery.
Mesh-covered stents have been developed to prevent distal embolization by trapping and excluding embolism-prone material at the level of culprit lesion in patients with STEMI. The MASTER (Safety and Efficacy Study of MGuard Stent After a Heart Attack) trial, which randomized 433 patients with STEMI to a mesh-covered stent (MGuard stent) or a bare-metal or drug-eluting stent, showed that mesh-covered stent improved complete ST-segment resolution ≥70% (57.8% vs. 44.7%; P=0.008) and postprocedural TIMI flow grade of 3 (91.7% vs. 82.9%; P=0.006) with no significant impact on infarct size assessed by CMR or 30-day incidence of MACE.290 Patients implanted with MGuard stent had a significantly higher one-year incidence of MACE (9.1% vs. 3.3%; P=0.02), driven by ischemia-driven target lesion revascularization.291 One study have suggested that MGuard stent may be beneficial in patients with high-thrombus burden.292 One observational study has shown that pressure controlled intermittent coronary sinus occlusion (PICSO) lowered the index of microcirculatory resistance in patients with STEMI and higher values of this metric before PCI (>40) and the CMR assessed infarct size at 6 months.293 This approach may reduce MVO by redistributing venous blood towards ischemic border which may enhance the wash-out of catabolites from the ischemic region.212
Glycoprotein 2b/3a receptor inhibitors were used to reduce thrombotic events including distal embolization and improve microcirculation due to their inhibitory effects on platelet aggregation. However clinical results with these agents have not been convincing. In the On-TIME-2 (Ongoing Tirofiban in Myocardial Infarction 2) trial, routine prehospital high-bolus dose of tirofiban improved ST-segment resolution before and one hour after PCI.294 The trial also showed that prehospital tirofiban reduced the incidence of MACE (death, recurrent myocardial infarction, urgent target vessel revascularization or thrombotic bail-out) at 30 days.294 A study of 162 patients developing angiographic CNR (defined as TIMI flow grade <3) after primary PCI randomized to receive intracoronary tirofiban (25 µg/kg) or placebo (0.9% isotonic saline solution) showed that intracoronary tirofiban improved TIMI flow grade and restored normal reperfusion in 26% of patients and in-hospital MACE were significantly lower in the tirofiban group (36% vs 19%; P=0.013).295 A recent study that included 226 patients with STEMI assigned to receive intravenous or intracoronary tirofiban showed that intracoronary tirofiban reduced the frequency and extent of MVO (36% vs. 56%; P=0.004), improved left ventricular end-diastolic volume at 6 months but had no effect on the MACE rate at one year compared with intravenous tirofiban.296 In the Intracoronary Abciximab and Aspiration Thrombectomy in Patients With Large Anterior Myocardial Infarction (INFUSE-AMI) trial, intracoronary abciximab but not aspiration thrombectomy, was associated with a significant reduction of infarct size by CMR at 30 days in patients with large anterior STEMI presenting early after symptom onset and undergoing primary PCI with bivalirudin anticoagulation. However, intracoronary abciximab was not associated with better tissue reperfusion as assessed by final TIMI flow grade, myocardial blush grade or complete (>70%) ST-segment resolution after PCI.297 The limited sample size did not allow a meaningful assessment of the impact of intracoronary abciximab on clinical outcomes. The AIDA STEMI (Abciximab Intracoronary versus intravenous Drug Application in STEMI) showed no difference in the 90-day incidence of MACE between patients assigned to intracoronary versus those assigned to intravenous abciximab; however the incidence of new congestive heart failure was lower with the intracoronary administration of the drug.298 The CMR substudy of the AIDA STEMI trial, showed no difference between the 2 strategies with respect to final infarct size, MVO, intramyocardial hemorrhage or LVEF at one-week CMR imaging.35 One recent study reported a strong association between the use of glycoprotein 2b/3a inhibitors and the risk of intramyocardial hemorrhage in patients with STEMI, which was associated with a poor outcome.192 Since a clear clinical benefit of glycoprotein 2b/3a inhibitors in reducing infarct size or MVO or improving clinical outcomes has never been proven, current guidelines gave a class IIa, level of evidence C, for the use of glycoprotein 2b/3a inhibitors for patients with STEMI and CNR.299
In summary, therapy against distal embolization has failed to produce beneficial clinical effects in a consistent manner. The reasons why this therapy failed to prevent or ameliorate CNR remains unclear. However, 2 putative mechanisms may be offered: first, therapy, in particular mechanical devices, may fail to prevent distal embolization or even may facilitate it at the time of device placement, and second, distal embolization may play a modest role in the pathophysiology of MVO and CNR. What is known from experimental and clinical studies is that MVO and CNR may develop in the setting of ischemia/reperfusion even in case of total absence of distal embolization.
Pharmacological Therapy
A large number of pharmacological agents or therapeutic strategies, alone or in combination, have been used to reduce infarct size, boost myocardial salvage and prevent or reduce reperfusion injury and MVO or CNR after primary PCI in patients with STEMI. Apart from antithrombotic (aspirin, clopidogrel, prasugrel and ticagrelor) and anticoagulant (unfractionated heparin and bivalirudin) drugs that are routinely used during the primary PCI, numerous drugs including, statins, angiotensin-converting enzyme inhibitors, calcium channel blockers (like verapamil), beta-blockers (like metoprolol), antioxidants (recombinant human superoxide dismutase, desferoxamine, edaravone and allopurinol), neutrophil and complement system inhibitors (like pexelizumab), antidiabetic drugs (like exenatide), glucose–insulin–potassium infusion, antiischemic agents (like trimetazidine), inhibitors of mitochondrial permeability transition pore opening (like cyclosporine), NO donors, Na+/H+ ion exchanger inhibitors (like cariporide), Na+/Ca2+ ion exchanger inhibitors (like caldaret), K+ATP channel agonists (like nicorandil), atrial natriuretic peptides, erythropoietin, adenosine, endothelin receptor inhibitors, protein kinase C inhibitors (like delcasertib) as well as therapeutic strategies of hypothermia, hyperoxemia and ischemic conditioning have been used alone or in combination to prevent MVO and CNR.300 Despite the fact that these therapies targeted almost all known pathophysiological mechanisms of reperfusion injury, MVO or CNR and improved some markers of reperfusion, none of them produced consistent clinical benefits in terms of improved clinical outcome. The most commonly used drugs to protect microcirculation, prevent MVO and CNR or reduce infarct size are shown below.
Adenosine – a purine nucleoside and a potent vasodilator of arterioles and microcirculation via binding to A2 receptors - has been tested as a means to prevent (or reverse) MVO and CNR after primary PCI. Experimental studies have suggested that adenosine ameliorates ischemia/reperfusion injury, limits infarct size and improves left ventricular function. However, most clinical research with this drug has been disappointed. Earlier studies showed that intravenous adenosine reduces infarct size when used as adjunct to thrombolytic drugs or primary PCI although with a neutral effect on clinical outcomes in patients with STEMI.301-302 The Intracoronary Nitroprusside Versus Adenosine in Acute Myocardial Infarction (REOPEN-AMI) trial showed that intracoronary high dose of adenosine, but not nitroprusside improved MVO assessed by ST-segment resolution. The angiographic MVO (TIMI flow grade ≤2 or 3 with a myocardial blush grade <2) or MACE (a composite of cardiac death, myocardial infarction, target lesion revascularization, and heart failure requiring hospitalization) at 30 days were not improved by adenosine.303 A randomized trial by Desmet et al.304 showed that selective high-dose intracoronary adenosine infused distal to the culprit lesion failed to promote myocardial salvage or reduce MVO in patients with STEMI. Similarly, the REperfusion Facilitated by LOcal adjunctive therapy in STEMI (REFLO-STEMI) trial found no significant difference in CMR-assessed infarct size or MVO in patients allocated to intracoronary high-dose adenosine, sodium nitroprusside or controls. On per-protocol analysis, however, the infarct size and the rate of MACE at 30 days and 6 months were increased and LVEF was reduced in the adenosine arm compared with control arm.305 A recent meta-analysis of 26 randomized controlled trials with 5843 patients with ACS undergoing revascularization showed that intracoronary or intravenous adenosine offered no clinical benefit in terms of reduction of MACE, all-cause mortality, non-fatal myocardial infarction or heart failure. In patients undergoing PCI, adenosine reduced myocardial blush grade 0–1 and TIMI flow grade 0–2 but had no effect on infarct size or LVEF. Of note, adenosine increased the risk of atrioventricular block and supraventricular and ventricular arrhythmias is studies with ischemia time >3 hours.306 Evidence from these studies do not support the use of adenosine as adjunctive to primary PCI.
Sodium nitrite – a NO donor - is a powerful arteriolar vasodilator with antiplatelet and inti-inflammatory properties. The Nitrates in Acute Myocardial Infarction (NIAMI) randomized 229 patients with STEMI to receive either an intravenous infusion of 70 μmol sodium nitrite or matching placebo. The drug failed to reduce infract size measured by CMR at 6-8 days. In addition there were no significant differences between nitrite and placebo with respect to area under the curve of troponin I and creatine kinase, left ventricular volumes and ejection fraction measured at 6-8 days and infarct size measured at 6 months.307 Along the same lines, in the REOPEN-AMI trial, a complete (≥70%) ST-segment resolution was observed in 71% of patients assigned to adenosine, 54% of patients assigned to nitroprusside and 51% in patients assigned to saline (P=0.009 for adenosine versus nitroprusside and P=0.75 for nitroprusside versus saline).303 Based on these data, there is no proven benefit of intravenous nitrite or nitroprusside as means to prevent or treat MVO and CNR after primary PCI.
Calcium channel blockers (verapamil, diltiazem and nicardipine) have been used to treat CNR almost immediately after the syndrome was recognized in humans, but the quality of studies has been poor.308 A 2015 meta-analysis of 8 randomized controlled trials with 494 patients showed that intracoronary verapamil/diltiazem injection significantly reduced the frequency of CNR.309 In a retrospective study of 72 patients with ACS, intracoronary nicardipine reversed CNR (defined as restoration of TIMI flow grade of 3) in 71 patients (98.6%) with no adverse hemodynamic or chronotropic effects.310 Intracoronary infusion of a cocktail consisting nicardipine and adenosine, appears to reduce the frequency of CNR during rotational atherectomy.311 Despite these encouraging results, the low quality of studies does not allow any solid recommendation for the use of calcium channel blockers for prevention of CNR in patients with STEMI.
The impact of beta-blocking agents on MVO and infarct size in patients with STEMI remains controversial. One study in Yorkshire pigs undergoing a 90-minute occlusion of left anterior descending coronary followed by reperfusion showed that intravenous metoprolol was associated with a 5-fold higher myocardial salvage and a significant improve in LVEF compared with placebo.312 This effect was at least partially explained by metoprolol-induced modulation of inflammatory response by inhibiting neutrophil migration and neutrophil-platelet aggregate formation.313 Pre-clinical studies have also shown that newer beta-blocking agents, carvedilol and nebivolol may protect microcirculation and reduce the frequency of CNR and infarct size. One ischemia/reperfusion study in swine showed that carvedilol reduced the area of CNR and infarct size. These effects were associated with reduced plasma and tissue endothelin-1 levels induced by carvedilol-induced activation of the KATP channels.314 Another study in mice with extensive AMI of anterior wall showed that nebivolol improved endothelium-dependent vasorelaxation, (potentially via NO-mediated improvement of endothelial function), inhibited cardiac NADPH oxidase activation after AMI, and improved left ventricular dysfunction at 4 weeks.315 The use of beta blockers in patients with STEMI have been less promising. The Effect of Metoprolol in Cardioprotection During an Acute Myocardial Infarction (METOCARD-CNIC) trial showed that early pre-reperfusion intravenous metoprolol (up to three 5-mg intravenous boluses of metoprolol tartrate 2 minutes apart) reduced infarct size estimated by CMR (21.2±11.5% vs. 25.1±13.9% of the left ventricle; P=0.029) and (slightly) improved LVEF in 270 patients with STEMI of anterior wall presenting within 6 hours form pain onset.316 However, the larger Early-Beta blocker Administration before reperfusion primary PCI in patients with ST-elevation Myocardial Infarction (EARLY-BAMI) trial randomized 683 patients with STEMI presenting within the first 12 hours in Killip class I to II without atrioventricular block to receive intravenous metoprolol (2x5 mg bolus) or placebo. In this trial, metoprolol did not reduce infarct size estimated by CMR at 30 days (15.3±11.0% vs 14.9±11.5% of the left ventricle) or improve LVEF (51.0±10.9% vs. 51.6±10.8%) compared with placebo.317 These data do not support the early intravenous beta-blockage to protect microcirculation, promote myocardial salvage or reduce infarct size in patients with STEMI undergoing primary PCI.
It was hoped that statins may improve microcirculation and have beneficial effects on the prevention or treatment of CNR through their pleiotropic effects including endothelium protection, microcirculation dilatation and antithrombotic and anti-inflammatory actions. The Efficacy of High-Dose AtorvaSTATIN Loading Before Primary Percutaneous Coronary Intervention in ST-Elevation Myocardial Infarction (STATIN STEMI) showed that high-dose (80 mg) atorvastatin pre-treatment improved angiographic MVO (corrected TIMI frame count and myocardial blush grade) and ST-segment resolution but not MACE at 30 days compared with low (10 mg) atorvastatin dose.318 A study by Hahn et al.319 randomized patients with STEMI undergoing primary PCI to receive atorvastatin 80 mg before PCI and for 5 days after PCI or 10 mg atorvastatin daily after PCI. The study showed no significant difference in the scintigraphic infarct size, myocardial blush grade 2/3 or complete ST-segment resolution at 60 minutes after PCI between the groups. The Statins Evaluation in Coronary Procedures and Revascularization (SECURE-PCI) trial randomized 4191 patients with ACS evaluated with coronary angiography to proceed with a PCI to receive 2 loading doses of 80 mg of atorvastatin (n = 2087) or matching placebo (n = 2104) before and 24 hours after a planned PCI. The trial showed that periprocedural use of loading doses of atorvastatin did not reduce the risk of MACE at 30 days. In the subgroup of patients with STEMI (n=1012), atorvastatin reduced the incidence of MACE but without treatment effect interaction.320 These studies dampened the enthusiasm for the periprocedural use of statins as means to protect microcirculation, reduce MVO or promote myocardial salvage in patients with STEMI undergoing primary PCI.
Evidence on the efficacy of intracoronary fibrinolysis with respect to MVO and CNR remains limited and inconsistent. The Trial of Low-dose Adjunctive alTeplase During prIMary PCI (T-TIME) assessed the impact of intracoronary fibrinolysis on MVO. Intracoronary alteplase at doses of 10 mg and 20 mg did not reduce MVO compared with placebo.321 However, the trial was stopped prematurely due to futility reasons. A recent meta-analysis of 6 randomized controlled trials with 890 patients showed that intracoronary fibrinolysis did not improve postprocedural TIMI flow 2 or 3, but it improved complete ST-segment resolution and was associated with a trend for fewer in-hospital MACE events with no difference in bleeding events compared with placebo.322 The Bivalirudin Infusion for Ventricular InfArction Limitation (BIVAL) study tested whether bivalirudin reduces infarct size compared with unfractionated heparin in 78 patients with large AMI undergoing primary PCI. Bivalirudin reduced the index of microcirculatory resistance but had no significant effect on infarct size, early CMR-assessed MVO or LVEF at 90 days compared with unfractionated heparin.323
Intracoronary epinephrine can mediate coronary vasodilatation at lower doses and consequently has been used mostly to reverse CNR. Navarese et al.324 tested intracoronary epinephrine in 30 consecutive patients with STEMI and established CNR (defined as TIMI flow grade 0-1 and myocardial blush grade 0-1). Intracoronary epinephrine restored a TIMI flow grade or 2 (64.3% vs. 12.5%) and 3 (28.6% vs. 18.8%) compared with patients who received conventional treatment. Intracoronary epinephrine significantly improved ST-segment resolution and LVEF and reduced the 30-day composite endpoint of death or heart failure. The Comparison of Intracoronary Epinephrine and Adenosine for No-Reflow in Normotensive Patients With Acute Coronary Syndrome (COAR) trial randomized 201 patients with ACS and CNR to intracoronary epinephrine or adenosine. Intracoronary epinephrine improved final TIMI flow grade of 3 and final corrected TIMI frame count but not final myocardial blush grade or in-hospital or short-term mortality or MACE compared with adenosine.325 Two observational studies showed that intracoronary epinephrine reversed CNR in 9 of 12 patients326 and in 74 of 81 patients327 with STEMI and persistent CNR after primary PCI. Although intracoronary epinephrine appears to successfully reverse CNR, it may increase the risk of malignant arrhythmias and the quality of studies does not allow a firm recommendation for its use to treat CNR.
Consistent with the role of platelets in the pathophysiology of MVO and CNR, suboptimal platelet response to dual antiplatelet therapy was associated with a greater extent of MVO.328 This has raised the interest on newer antiplatelet drugs which cause a deeper and more predictable platelet inhibition. However, whether the newer antiplatelet drugs protect microcirculation and improve reperfusion in patients with STEMI undergoing primary PCI remains controversial. Subgroup analyses from the Study of Platelet Inhibition and Patient Outcomes (PLATO)329 and Administration of Ticagrelor in the Cath Lab or in the Ambulance for New ST Elevation Myocardial Infarction to Open the Coronary Artery (ATLANTIC)330 trials showed no benefit of ticagrelor versus clopidogrel in improving reperfusion or reducing the frequency of CNR after primary PCI. In the same vein, the Reducing Micro Vascular Dysfunction in Acute Myocardial Infarction by Ticagrelor (REDUCE-MVI) trial that randomized 110 patients with STEMI to receive loading dose of ticagrelor or prasugrel showed no significant difference between the drugs in the index of microcirculatory resistance or its recovery over time. Intramyocardial hemorrhage was less frequent in patients who received ticagrelor (23% vs. 43%; P=0.04) but there was no significant difference in the infarct size on the one-month CMR imaging.331 A 2018 meta-analysis of 14 randomized trials and one observational study with 4162 patients showed that ticagrelor significantly reduced the frequency of CNR after primary PCI and reduced the risk of MACE up to 180 days compared with clopidogrel with no significant increase in the risk for bleeding.332 The efficacy of cangrelor to reduce MVO and infarct size in patients with STEMI is under investigation.333
Over the years, a plethora of drugs and strategies, alone or in combination, have been used to protect microcirculation and prevent MVO or CNR and reduce infarct size in patients with STEMI. Based on the central role of mitochondrial permeability transition pore opening in the reperfusion-related injury and the results of experimental studies, inhibitors of this pore (particularly cyclosporine A) were considered very promising drugs to protect microcirculation during the primary PCI procedures. After promising results with a small randomized trial of cyclosporine A in reducing infarct size,334 two randomized trials showed no benefit of cyclosporine A (administered intravenously at a dose of 2.5 mg per kg of weight before coronary recanalization) in reducing infarct size (estimated by CMR or high-sensitivity cardiac troponin), ST-segment resolution, LVEF improvement or prevention of adverse remodeling of left ventricle or incidence of MACE at 6 months and one year.335-336 Based on experimental evidence that hyperoxemia reduces formation lipid peroxide radicals, increases NO availability via expression of NO synthase and inhibits leukocyte adherence and plugging in microcirculation, intracoronary delivery of supersaturated oxygen was tested in the Acute Myocardial Infarction with Hyperoxemic Therapy (AMIHOT) trial. The trial showed that intracoronary supersaturated oxygen did not improve ST-segment resolution, regional wall motion by echocardiography or scintigraphic infarct size.337 In a subgroup analysis, patients with STEMI of anterior wall undergoing reperfusion within the first 6 hours showed improvement in regional wall motion and smaller infarct size. In the AMIHOT-2 trial that included patients with STEMI of anterior wall, intracoronary delivery of supersaturated oxygen reduced infarct size with no difference in 30-day MACE compared with placebo.338 However, a SWEDEHEART registry-based randomized trial showed no beneficial effects of routine supplemental oxygen in terms of one-year mortality in patients with suspected AMI and oxygen saturation ≥90%.339 In the IntraCoronary Hyper-oxemic Supersaturated Oxygen Therapy (IC-HOT) study, delivery of supersaturated oxygen in the left main coronary artery for 60 minutes after PCI in patients with anterior STEMI was safe.340 Although, hypothermia was hypothesized to reduce metabolic demand and inflammatory response, 3 randomized trials showed no beneficial effects of this therapeutic strategy in reducing the MVO or infarct size in patients with STEMI. Instead, an increase in the frequency of adverse events was observed.341-343 However, the strategy of elective intracoronary hypothermia during the primary PCI remains under investigation and the preliminary results indicate that this strategy is safe.344-345 Atrial natriuretic peptides suppress endothelin-1 production and activate reperfusion injury salvage kinase (RISK) cardioprotective pathway. Although an earlier study showed that atrial natriuretic peptide agonist carperitide reduced enzymatic infarct size and improved LVEF in patients with STEMI undergoing primary PCI,346 evidence on beneficial effects of these drugs on reducing MVO and CNR remains rather limited. Pre-clinical research suggested that glucagon-like peptide-1 agonist exenatide may reduce infarct size and improve ventricular function by reducing apoptosis and oxidative stress; however, human studies have given conflicting results. In one study with 172 patients with STEMI, exenatide (given intravenously 15 minutes before intervention and continued for 6 hours after the procedure) increased myocardial salvage and reduced CMR-assessed infarct size compared with placebo (saline).347 In another study of 58 patients with STEMI, patients who received exenatide showed a significant reduction of the absolute mass of delayed hyperenhancement on CMR compared with control patients.348 However in a more recent and larger trial of 191 patients with STEMI, intravenous exenatide (10 μg/h for 30 minutes before PCI and followed by 0.84 μg/h for 72 hours) failed to reduce infarct size after correction for the area at risk in the acute phase (37.1±18.8% vs. 39.3±20.1%; P=0.662) or final infarct size as a percentage of the left ventricle at 4 months (18.8±13.2 vs. 18.8±11.3%; P=0,965) compared with placebo.349 A 2009 meta-analysis by Iwakura et al.350 that included 17 studies showed that nicorandil - a hybrid of the KATP channel opener and nitrate - reduced the incidence of TIMI flow grade ≤2 by 37% (10 studies with 1337 patients) and improved LVEF by 3.7% with no impact on peak creatine kinase (11studies with 905 patients). The ITF-1697- a C-reactive protein-derived tetrapeptide – was tested in 402 patients with AMI in a multicenter, randomized, double blind, dose-finding placebo-controlled study. Post-procedural perfusion assessed by TIMI flow, corrected TIMI frame count, blush grade and ST-segment resolution, did not differ between the 0.1 and 1.0 μg/kg/min study arms or placebo.351 Consistent with the role of neutrophil extracellular traps (NETs) in reperfusion injury, a rat model showed that a combination DNase1 and recombinant tissue-type plasminogen activator reduced NET density, no-reflow area in the ischemic region and infarct size and ameliorated ischemia/reperfusion-induced adverse left ventricular remodeling.110 The impact of current anti-inflammatory therapies used in patients with ACS on MVO and CNR remains largely untested.212
Preclinical research suggested that ischemic postconditioning (transient episodes of deliberate inflation/deflation of an occluding balloon in the infarct-related artery) may protect microcirculation and reduce MVO and infarct size. Ischemic postconditioning has been tested as means to reduce the extent of MVO and CNR in patients with STEMI following favorable results obtained from preclinical research. Earlier studies with small numbers of patients suggested that ischemic postconditioning may reduce the infarct size estimated by CMR.352-353 However, a 2014 meta-analysis of 15 randomized trials with 1,545 patients showed no beneficial effects of ischemic postconditioning on ST-segment resolution, infarct size or any of the clinical outcomes (mortality, recurrent myocardial infarction, stent thrombosis or composite MACE) at 5 months after PCI.354 Along the same lines, the third Danish Study of Optimal Acute Treatment of Patients With ST Elevation Myocardial Infarction–Ischemic Postconditioning (DANAMI-3–iPOST) trial that randomized 1234 patients with STEMI presenting within 12 hours from symptom onset and a baseline TIMI flow grade of 0-1 to conventional primary PCI or postconditioning (4 cycles of 30-second balloon occlusions followed by 30 seconds of reperfusion immediately after opening of the infarct-related artery and before stent implantation) found no difference in CMR-estimated infarct size, extent of MVO, myocardial salvage index or LVEF at 3 months or MACE (death or hospitalization for heart failure) over a median of 38 months.355 Earlier studies also suggested that remote postconditioning (repetitive cycles of ischemia in a tissue remote from the heart) applied during the primary PCI may improve myocardial salvage and ST-segment resolution, reduce infarct size, and lower MACE and reduce infarct size in patients with STEMI.356-357 However, recent research did not support the benefits of remote postconditioning reported in earlier trials. The LIPSIA CONDITIONING trial that included 696 patients with STEMI showed that postconditioning alone did not improve myocardial salvage, infarct size or MVO358 or MACE at 3.6 years.359 However, combined ischemic postconditioning and remote postconditioning improved myocardial salvage358 and MACE oven a median of 3.6 years, mostly driven by reduced incidence of new congestive heart failure.359 However, the CONDI-2/ERIC-PPCI trial that randomized 5401 patients with STEMI undergoing primary PCI to remote postconditioning or control, did not show a benefit of remote postconditioning in reducing MACE at one year (death or hospitalization for heart failure: 8.6% vs. 9.4%; P=0.32).360
In summary, despite decades of intensive research and testing of numerous agents and therapeutic strategies targeting all known pathophysiological mechanisms of MVO and CNR, no satisfactory therapy has been found to prevent of reverse these phenomena and consistently improve the clinical outcome. Consequently, no MVO or CNR preventive therapies can be recommended to be routinely used in patients with STEMI undergoing primary PCI to prevent or reverse CNR. Of note, the reperfusion-related injury – the key mechanism of MVO and CNR - has proven to be very resistant to all tested agents and therapies. The promising results obtained from the preclinical research have largely failed to translate into benefit in clinical studies. Therapies that have resulted in myocardial salvage in the pre-PCI era failed to bring clinical benefit when used as adjunct to primary PCI. CMR imaging - the most sensitive and accurate diagnostic tool for estimating myocardial necrosis, MVO and intramyocardial hemorrhage in clinical setting – has proven to be highly conservative and restrictive in identifying therapies that could prevent or reverse MVO and CNR or improve clinical outcome. Although there is no evidence to support the routine use of any above-mentioned agents or therapies during the primary PCI, in patients with large thrombus burden at high risk for CNR, manual thrombus aspiration with direct stenting continue to be used to reduce distal embolization. Powerful antithrombotic drugs like glycoprotein 2b/3a may be used but they increase the risk of intramyocardial hemorrhage. Vasodilator therapy has been the mainstay of CNR therapy for almost 50 years in patients with AMI who develop persistent CNR and continue to be used in catheterization laboratories based on operator’s personal experience or intuition, albeit without evidence of a proven benefit. Even though vasodilator therapy may improve some markers of reperfusion (particularly angiographic markers), it has largely failed to reduce infarct size or improve clinical outcome.
Concluding Remarks
MVO and CNR are relatively frequent phenomena that develop following reperfusion therapy in patients with STEMI. CNR is highly dynamic in nature and develops gradually (over hours) and persists for days to weeks after primary PCI. The frequency of CNR or MVO after primary PCI differs widely depending on the sensitivity of the tools used for the diagnosis and timing of examination. Coronary angiography is readily available and most convenient to diagnose CNR but it is highly conservative due to suboptimal (too early) timing of examination (end of PCI procedure) and low sensitivity of angiographic criteria. CMR imaging is the most sensitive method to diagnose MVO and CMR that provides information on the presence, localization and extent of MVO. CMR imaging detects also intramyocardial hemorrhage and accurately estimates infarct size in clinical setting. Although myocardial ischemia and distal embolization (spontaneous or intervention-induced) contribute to the development of MVO and CNR, the reperfusion-related injury is the most important factor and key pathophysiological mechanism of these phenomena. MVO and CNR markedly negate the benefits of reperfusion therapy and contribute to poor clinical outcomes including adverse remodeling of left ventricle, worsening or new congestive heart failure and increased risk of mortality. Since MVO and CNR are important predictors of clinical outcome, patients with STEMI developing these phenomena are in need of careful risk stratification and tailored therapy. Despite of almost 50 years of research and targeting of all known factors (and mechanisms) involved in the pathophysiology of MVO and CNR, no therapy has been found that prevents or reverses these phenomena (when they develop) or provides consistent clinical benefit in patients with STEMI. Although many new therapies provided hope for the prevention of MVO and CNR, the results of recent research consisting of large and well-conducted studies were overwhelmingly disappointing. While no MVO or CNR preventive therapies can be recommended to be routinely used in patients with STEMI undergoing primary PCI, early reperfusion by primary PCI remains the only means to stop ongoing ischemia, reduce the infarct size and consequently reduce the occurrence and extent of MVO and CNR. For the time being, prevention or alleviation of MVO and CNR remain unmet goals in the therapy of STEMI that continue to be under intense research.
Funding
None.
Conflicts of interest
The authors have nothing to declare.
References
- Majno, G.; Ames, A.; Chiang, J.; Wright, R. L. No Reflow after Cerebral Ischaemia. Lancet 1967, 2(7515), 569–570. [Google Scholar] [CrossRef]
- Ames, A., 3rd; Wright, R. L.; Kowada, M.; Thurston, J. M.; Majno, G. Cerebral ischemia. II. The no-reflow phenomenon. Am J Pathol 1968, 52(2), 437–53. [Google Scholar] [PubMed]
- Chiang, J.; Kowada, M.; Ames, A., 3rd; Wright, R. L.; Majno, G. Cerebral ischemia. III. Vascular changes. Am J Pathol 1968, 52(2), 455–76. [Google Scholar] [PubMed]
- Harman, J. W. The Significance of Local Vascular Phenomena in the Production of Ischemic Necrosis in Skeletal Muscle. American Journal of Pathology 1948, 24(3), 625–641. [Google Scholar] [PubMed]
- Sheehan, H. L.; Davis, J. C. Renal Ischaemia with Failed Reflow. J Pathol Bacteriol 1959, 78(1), 105–&. [Google Scholar] [CrossRef]
- Flores, J.; DiBona, D. R.; Beck, C. H.; Leaf, A. The role of cell swelling in ischemic renal damage and the protective effect of hypertonic solute. J Clin Invest 1972, 51(1), 118–26. [Google Scholar] [CrossRef]
- Kovacs, K.; Carroll, R.; Tapp, E. Temporary Ischaemia of Rat Adrenal Gland. J Pathol Bacteriol 1966, 91(1), 235–&. [Google Scholar] [CrossRef] [PubMed]
- Krug, A.; Du Mesnil de, R.; Korb, G. Blood supply of the myocardium after temporary coronary occlusion. Circ Res 1966, 19(1), 57–62. [Google Scholar] [CrossRef] [PubMed]
- Willms-Kretschmer, K.; Majno, G. Ischemia of the skin. Electron microscopic study of vascular injury. Am J Pathol 1969, 54(3), 327–53. [Google Scholar]
- Kloner, R. A.; Ganote, C. E.; Jennings, R. B. The "no-reflow" phenomenon after temporary coronary occlusion in the dog. J Clin Invest 1974, 54(6), 1496–508. [Google Scholar] [CrossRef]
- Kloner, R. A.; Ganote, C. E.; Whalen, D. A., Jr.; Jennings, R. B. Effect of a transient period of ischemia on myocardial cells. II. Fine structure during the first few minutes of reflow. Am J Pathol 1974, 74(3), 399–422. [Google Scholar]
- Jefferson, G. "Arterial Embolectomy". Br Med J 1934, 2(3858), 1090–4. [Google Scholar] [CrossRef]
- Griffiths, D. L. Volkmann's ischaemic contracture. Br. J. Surg. 1940, 28, 239–260. [Google Scholar] [CrossRef]
- Schofer, J.; Montz, R.; Mathey, D. G. Scintigraphic evidence of the "no reflow" phenomenon in human beings after coronary thrombolysis. J Am Coll Cardiol 1985, 5(3), 593–8. [Google Scholar] [CrossRef]
- Bates, E. R.; Krell, M. J.; Dean, E. N.; O'Neill, W. W.; Vogel, R. A. Demonstration of the "no-reflow" phenomenon by digital coronary arteriography. Am J Cardiol 1986, 57(1), 177–8. [Google Scholar] [CrossRef] [PubMed]
- Pomerantz, R. M.; Kuntz, R. E.; Diver, D. J.; Safian, R. D.; Baim, D. S. Intracoronary verapamil for the treatment of distal microvascular coronary artery spasm following PTCA. Cathet Cardiovasc Diagn 1991, 24(4), 283–5. [Google Scholar] [CrossRef]
- Feld, H.; Lichstein, E.; Schachter, J.; Shani, J. Early and late angiographic findings of the "no-reflow" phenomenon following direct angioplasty as primary treatment for acute myocardial infarction. Am Heart J 1992, 123(3), 782–4. [Google Scholar] [CrossRef]
- Wilson, R. F.; Laxson, D. D.; Lesser, J. R.; White, C. W. Intense microvascular constriction after angioplasty of acute thrombotic coronary arterial lesions. Lancet 1989, 1(8642), 807–11. [Google Scholar] [CrossRef]
- Ito, H.; Tomooka, T.; Sakai, N.; Yu, H.; Higashino, Y.; Fujii, K.; Masuyama, T.; Kitabatake, A.; Minamino, T. Lack of myocardial perfusion immediately after successful thrombolysis. A predictor of poor recovery of left ventricular function in anterior myocardial infarction. Circulation 1992, 85(5), 1699–705. [Google Scholar] [CrossRef] [PubMed]
- Piana, R. N.; Paik, G. Y.; Moscucci, M.; Cohen, D. J.; Gibson, C. M.; Kugelmass, A. D.; Carrozza, J. P., Jr.; Kuntz, R. E.; Baim, D. S. Incidence and treatment of 'no-reflow' after percutaneous coronary intervention. Circulation 1994, 89(6), 2514–8. [Google Scholar] [CrossRef] [PubMed]
- Morishima, I.; Sone, T.; Mokuno, S.; Taga, S.; Shimauchi, A.; Oki, Y.; Kondo, J.; Tsuboi, H.; Sassa, H. Clinical significance of no-reflow phenomenon observed on angiography after successful treatment of acute myocardial infarction with percutaneous transluminal coronary angioplasty. Am Heart J 1995, 130(2), 239–43. [Google Scholar] [CrossRef] [PubMed]
- Morishima, I.; Sone, T.; Okumura, K.; Tsuboi, H.; Kondo, J.; Mukawa, H.; Matsui, H.; Toki, Y.; Ito, T.; Hayakawa, T. Angiographic no-reflow phenomenon as a predictor of adverse long-term outcome in patients treated with percutaneous transluminal coronary angioplasty for first acute myocardial infarction. J Am Coll Cardiol 2000, 36(4), 1202–9. [Google Scholar] [CrossRef] [PubMed]
- Rakusan, K.; Flanagan, M. F.; Geva, T.; Southern, J.; Van Praagh, R. Morphometry of human coronary capillaries during normal growth and the effect of age in left ventricular pressure-overload hypertrophy. Circulation 1992, 86(1), 38–46. [Google Scholar] [CrossRef]
- Kaul, S.; Jayaweera, A. R. Coronary and myocardial blood volumes: noninvasive tools to assess the coronary microcirculation? Circulation 1997, 96(3), 719–24. [Google Scholar]
- Kaul, S. Evaluating the 'no reflow' phenomenon with myocardial contrast echocardiography. Basic Res Cardiol 2006, 101(5), 391–9. [Google Scholar] [CrossRef] [PubMed]
- Lanzer, P.; Leigh, R.; Berry, C.; van de Hoef, T.; Heiss, W. D.; Senior, R.; Schwartz, A.; Rischpler, C.; Liebeskind, D. Salutary reperfusion is the ultimate target of ST-Segment Elevation Myocardial Infarction (STEMI) and Acute Ischemic Stroke (AIS) interventions and should be routinely assessed in standard clinical and research protocols. Card Res Med 2017, 1, 20–34. [Google Scholar]
- Heusch, G. Coronary microvascular obstruction: the new frontier in cardioprotection. Basic Res Cardiol 2019, 114(6), 45. [Google Scholar] [CrossRef]
- Galiuto, L.; Lombardo, A.; Maseri, A.; Santoro, L.; Porto, I.; Cianflone, D.; Rebuzzi, A. G.; Crea, F. Temporal evolution and functional outcome of no reflow: sustained and spontaneously reversible patterns following successful coronary recanalisation. Heart 2003, 89(7), 731–7. [Google Scholar] [CrossRef]
- Wei, K.; Jayaweera, A. R.; Firoozan, S.; Linka, A.; Skyba, D. M.; Kaul, S. Basis for detection of stenosis using venous administration of microbubbles during myocardial contrast echocardiography: bolus or continuous infusion? J Am Coll Cardiol 1998, 32(1), 252–60. [Google Scholar] [CrossRef]
- Vogel, R.; Indermuhle, A.; Reinhardt, J.; Meier, P.; Siegrist, P. T.; Namdar, M.; Kaufmann, P. A.; Seiler, C. The quantification of absolute myocardial perfusion in humans by contrast echocardiography: algorithm and validation. J Am Coll Cardiol 2005, 45(5), 754–62. [Google Scholar] [CrossRef]
- Hausenloy, D. J.; Chilian, W.; Crea, F.; Davidson, S. M.; Ferdinandy, P.; Garcia-Dorado, D.; van Royen, N.; Schulz, R.; Heusch, G.; Action, E.-C. C. The coronary circulation in acute myocardial ischaemia/reperfusion injury: a target for cardioprotection. Cardiovasc Res 2019, 115(7), 1143–1155. [Google Scholar] [CrossRef]
- Shah, B. N.; Chahal, N. S.; Bhattacharyya, S.; Li, W.; Roussin, I.; Khattar, R. S.; Senior, R. The feasibility and clinical utility of myocardial contrast echocardiography in clinical practice: results from the incorporation of myocardial perfusion assessment into clinical testing with stress echocardiography study. J Am Soc Echocardiogr 2014, 27(5), 520–30. [Google Scholar] [CrossRef]
- Ndrepepa, G.; Mehilli, J.; Schulz, S.; Iijima, R.; Keta, D.; Byrne, R. A.; Pache, J.; Seyfarth, M.; Schomig, A.; Kastrati, A. Prognostic significance of epicardial blood flow before and after percutaneous coronary intervention in patients with acute coronary syndromes. J Am Coll Cardiol 2008, 52(7), 512–7. [Google Scholar] [CrossRef]
- Henriques, J. P.; Zijlstra, F.; van 't Hof, A. W.; de Boer, M. J.; Dambrink, J. H.; Gosselink, M.; Hoorntje, J. C.; Suryapranata, H. Angiographic assessment of reperfusion in acute myocardial infarction by myocardial blush grade. Circulation 2003, 107(16), 2115–9. [Google Scholar] [CrossRef]
- Eitel, I.; Wohrle, J.; Suenkel, H.; Meissner, J.; Kerber, S.; Lauer, B.; Pauschinger, M.; Birkemeyer, R.; Axthelm, C.; Zimmermann, R.; Neuhaus, P.; Brosteanu, O.; de Waha, S.; Desch, S.; Gutberlet, M.; Schuler, G.; Thiele, H. Intracoronary compared with intravenous bolus abciximab application during primary percutaneous coronary intervention in ST-segment elevation myocardial infarction: cardiac magnetic resonance substudy of the AIDA STEMI trial. J Am Coll Cardiol 2013, 61(13), 1447–54. [Google Scholar] [CrossRef]
- Ndrepepa, G.; Alger, P.; Fusaro, M.; Kufner, S.; Seyfarth, M.; Keta, D.; Mehilli, J.; Schomig, A.; Kastrati, A. Impact of perfusion restoration at epicardial and tissue levels on markers of myocardial necrosis and clinical outcome of patients with acute myocardial infarction. EuroIntervention 2011, 7(1), 128–35. [Google Scholar] [CrossRef]
- Gibson, C. M.; Cannon, C. P.; Daley, W. L.; Dodge, J. T., Jr.; Alexander, B., Jr.; Marble, S. J.; McCabe, C. H.; Raymond, L.; Fortin, T.; Poole, W. K.; Braunwald, E. TIMI frame count: a quantitative method of assessing coronary artery flow. Circulation 1996, 93(5), 879–88. [Google Scholar] [CrossRef] [PubMed]
- Gibson, C. M.; Murphy, S. A.; Rizzo, M. J.; Ryan, K. A.; Marble, S. J.; McCabe, C. H.; Cannon, C. P.; Van de Werf, F.; Braunwald, E. Relationship between TIMI frame count and clinical outcomes after thrombolytic administration. Thrombolysis In Myocardial Infarction (TIMI) Study Group. Circulation 1999, 99(15), 1945–50. [Google Scholar] [CrossRef]
- Hamada, S.; Nishiue, T.; Nakamura, S.; Sugiura, T.; Kamihata, H.; Miyoshi, H.; Imuro, Y.; Iwasaka, T. TIMI frame count immediately after primary coronary angioplasty as a predictor of functional recovery in patients with TIMI 3 reperfused acute myocardial infarction. J Am Coll Cardiol 2001, 38(3), 666–71. [Google Scholar] [CrossRef] [PubMed]
- Ohara, Y.; Hiasa, Y.; Takahashi, T.; Yamaguchi, K.; Ogura, R.; Ogata, T.; Yuba, K.; Kusunoki, K.; Hosokawa, S.; Kishi, K.; Ohtani, R. Relation between the TIMI frame count and the degree of microvascular injury after primary coronary angioplasty in patients with acute anterior myocardial infarction. Heart 2005, 91(1), 64–7. [Google Scholar] [CrossRef] [PubMed]
- van 't Hof, A. W.; Liem, A.; Suryapranata, H.; Hoorntje, J. C.; de Boer, M. J.; Zijlstra, F. Angiographic assessment of myocardial reperfusion in patients treated with primary angioplasty for acute myocardial infarction: myocardial blush grade. Zwolle Myocardial Infarction Study Group. Circulation 1998, 97(23), 2302–6. [Google Scholar] [CrossRef] [PubMed]
- Bouleti, C.; Mewton, N.; Germain, S. The no-reflow phenomenon: State of the art. Arch Cardiovasc Dis 2015, 108(12), 661–74. [Google Scholar] [CrossRef]
- Allencherril, J.; Jneid, H.; Atar, D.; Alam, M.; Levine, G.; Kloner, R. A.; Birnbaum, Y. Pathophysiology, Diagnosis, and Management of the No-Reflow Phenomenon. Cardiovasc Drugs Ther 2019, 33(5), 589–597. [Google Scholar] [CrossRef]
- Vicente, J.; Mewton, N.; Croisille, P.; Staat, P.; Bonnefoy-Cudraz, E.; Ovize, M.; Revel, D. Comparison of the angiographic myocardial blush grade with delayed-enhanced cardiac magnetic resonance for the assessment of microvascular obstruction in acute myocardial infarctions. Catheter Cardiovasc Interv 2009, 74(7), 1000–7. [Google Scholar] [CrossRef] [PubMed]
- Nijveldt, R.; van der Vleuten, P. A.; Hirsch, A.; Beek, A. M.; Tio, R. A.; Tijssen, J. G.; Piek, J. J.; van Rossum, A. C.; Zijlstra, F. Early electrocardiographic findings and MR imaging-verified microvascular injury and myocardial infarct size. JACC Cardiovasc Imaging 2009, 2(10), 1187–94. [Google Scholar] [CrossRef]
- Kampinga, M. A.; Nijsten, M. W.; Gu, Y. L.; Dijk, W. A.; de Smet, B. J.; van den Heuvel, A. F.; Tan, E. S.; Zijlstra, F. Is the myocardial blush grade scored by the operator during primary percutaneous coronary intervention of prognostic value in patients with ST-elevation myocardial infarction in routine clinical practice? Circ Cardiovasc Interv 2010, 3(3), 216–23. [Google Scholar] [CrossRef] [PubMed]
- Sorajja, P.; Gersh, B. J.; Costantini, C.; McLaughlin, M. G.; Zimetbaum, P.; Cox, D. A.; Garcia, E.; Tcheng, J. E.; Mehran, R.; Lansky, A. J.; Kandzari, D. E.; Grines, C. L.; Stone, G. W. Combined prognostic utility of ST-segment recovery and myocardial blush after primary percutaneous coronary intervention in acute myocardial infarction. Eur Heart J 2005, 26(7), 667–74. [Google Scholar] [CrossRef]
- Nijveldt, R.; Beek, A. M.; Hirsch, A.; Stoel, M. G.; Hofman, M. B.; Umans, V. A.; Algra, P. R.; Twisk, J. W.; van Rossum, A. C. Functional recovery after acute myocardial infarction: comparison between angiography, electrocardiography, and cardiovascular magnetic resonance measures of microvascular injury. J Am Coll Cardiol 2008, 52(3), 181–9. [Google Scholar] [CrossRef]
- Ndrepepa, G.; Tiroch, K.; Fusaro, M.; Keta, D.; Seyfarth, M.; Byrne, R. A.; Pache, J.; Alger, P.; Mehilli, J.; Schomig, A.; Kastrati, A. 5-year prognostic value of no-reflow phenomenon after percutaneous coronary intervention in patients with acute myocardial infarction. J Am Coll Cardiol 2010, 55(21), 2383–9. [Google Scholar] [CrossRef]
- Wong, D. T.; Leung, M. C.; Richardson, J. D.; Puri, R.; Bertaso, A. G.; Williams, K.; Meredith, I. T.; Teo, K. S.; Worthley, M. I.; Worthley, S. G. Cardiac magnetic resonance derived late microvascular obstruction assessment post ST-segment elevation myocardial infarction is the best predictor of left ventricular function: a comparison of angiographic and cardiac magnetic resonance derived measurements. Int J Cardiovasc Imaging 2012, 28(8), 1971–81. [Google Scholar] [PubMed]
- Wong, D. T.; Leung, M. C.; Das, R.; Liew, G. Y.; Teo, K. S.; Chew, D. P.; Meredith, I. T.; Worthley, M. I.; Worthley, S. G. Intracoronary ECG during primary percutaneous coronary intervention for ST-segment elevation myocardial infarction predicts microvascular obstruction and infarct size. Int J Cardiol 2013, 165(1), 61–6. [Google Scholar] [CrossRef]
- Appelbaum, E.; Abraham, J. M.; Pride, Y. B.; Harrigan, C. J.; Peters, D. C.; Biller, L. H.; Manning, W. J.; Gibson, C. M. Association of Thrombolysis in Myocardial Infarction Myocardial Perfusion Grade with cardiovascular magnetic resonance measures of infarct architecture after primary percutaneous coronary intervention for ST-segment elevation myocardial infarction. Am Heart J 2009, 158(1), 84–91. [Google Scholar] [CrossRef] [PubMed]
- Gibson, C. M.; Kirtane, A. J.; Morrow, D. A.; Palabrica, T. M.; Murphy, S. A.; Stone, P. H.; Scirica, B. M.; Jennings, L. K.; Herrmann, H. C.; Cohen, D. J.; McCabe, C. H.; Braunwald, E.; Group, T. S. Association between thrombolysis in myocardial infarction myocardial perfusion grade, biomarkers, and clinical outcomes among patients with moderate- to high-risk acute coronary syndromes: observations from the randomized trial to evaluate the relative PROTECTion against post-PCI microvascular dysfunction and post-PCI ischemia among antiplatelet and antithrombotic agents-Thrombolysis In Myocardial Infarction 30 (PROTECT-TIMI 30). Am Heart J 2006, 152(4), 756–61. [Google Scholar]
- Bulluck, H.; Dharmakumar, R.; Arai, A. E.; Berry, C.; Hausenloy, D. J. Cardiovascular Magnetic Resonance in Acute ST-Segment-Elevation Myocardial Infarction: Recent Advances, Controversies, and Future Directions. Circulation 2018, 137(18), 1949–1964. [Google Scholar] [CrossRef] [PubMed]
- Mather, A. N.; Lockie, T.; Nagel, E.; Marber, M.; Perera, D.; Redwood, S.; Radjenovic, A.; Saha, A.; Greenwood, J. P.; Plein, S. Appearance of microvascular obstruction on high resolution first-pass perfusion, early and late gadolinium enhancement CMR in patients with acute myocardial infarction. J Cardiovasc Magn Reson 2009, 11, 33. [Google Scholar] [CrossRef]
- Nijveldt, R.; Hofman, M. B.; Hirsch, A.; Beek, A. M.; Umans, V. A.; Algra, P. R.; Piek, J. J.; van Rossum, A. C. Assessment of microvascular obstruction and prediction of short-term remodeling after acute myocardial infarction: cardiac MR imaging study. Radiology 2009, 250(2), 363–70. [Google Scholar] [CrossRef] [PubMed]
- Weir, R. A.; Murphy, C. A.; Petrie, C. J.; Martin, T. N.; Balmain, S.; Clements, S.; Steedman, T.; Wagner, G. S.; Dargie, H. J.; McMurray, J. J. Microvascular obstruction remains a portent of adverse remodeling in optimally treated patients with left ventricular systolic dysfunction after acute myocardial infarction. Circ Cardiovasc Imaging 2010, 3(4), 360–7. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A. J.; Al-Saadi, N.; Abdel-Aty, H.; Schulz-Menger, J.; Messroghli, D. R.; Friedrich, M. G. Detection of acutely impaired microvascular reperfusion after infarct angioplasty with magnetic resonance imaging. Circulation 2004, 109(17), 2080–5. [Google Scholar] [CrossRef]
- de Waha, S.; Patel, M. R.; Granger, C. B.; Ohman, E. M.; Maehara, A.; Eitel, I.; Ben-Yehuda, O.; Jenkins, P.; Thiele, H.; Stone, G. W. Relationship between microvascular obstruction and adverse events following primary percutaneous coronary intervention for ST-segment elevation myocardial infarction: an individual patient data pooled analysis from seven randomized trials. Eur Heart J 2017, 38(47), 3502–3510. [Google Scholar] [CrossRef] [PubMed]
- Guerra, E.; Hadamitzky, M.; Ndrepepa, G.; Bauer, C.; Ibrahim, T.; Ott, I.; Laugwitz, K. L.; Schunkert, H.; Kastrati, A. Microvascular obstruction in patients with non-ST-elevation myocardial infarction: a contrast-enhanced cardiac magnetic resonance study. Int J Cardiovasc Imaging 2014, 30(6), 1087–95. [Google Scholar] [CrossRef]
- Wehrens, X. H.; Doevendans, P. A.; Ophuis, T. J.; Wellens, H. J. A comparison of electrocardiographic changes during reperfusion of acute myocardial infarction by thrombolysis or percutaneous transluminal coronary angioplasty. Am Heart J 2000, 139(3), 430–6. [Google Scholar] [CrossRef] [PubMed]
- Santoro, G. M.; Valenti, R.; Buonamici, P.; Bolognese, L.; Cerisano, G.; Moschi, G.; Trapani, M.; Antoniucci, D.; Fazzini, P. F. Relation between ST-segment changes and myocardial perfusion evaluated by myocardial contrast echocardiography in patients with acute myocardial infarction treated with direct angioplasty. Am J Cardiol 1998, 82(8), 932–7. [Google Scholar] [CrossRef] [PubMed]
- Konijnenberg, L. S. F.; Damman, P.; Duncker, D. J.; Kloner, R. A.; Nijveldt, R.; van Geuns, R. M.; Berry, C.; Riksen, N. P.; Escaned, J.; van Royen, N. Pathophysiology and diagnosis of coronary microvascular dysfunction in ST-elevation myocardial infarction. Cardiovasc Res 2020, 116(4), 787–805. [Google Scholar] [CrossRef]
- Kaul, S.; Ito, H. Microvasculature in acute myocardial ischemia: part I: evolving concepts in pathophysiology, diagnosis, and treatment. Circulation 2004, 109(2), 146–9. [Google Scholar] [CrossRef] [PubMed]
- Kurose, I.; Anderson, D. C.; Miyasaka, M.; Tamatani, T.; Paulson, J. C.; Todd, R. F.; Rusche, J. R.; Granger, D. N. Molecular determinants of reperfusion-induced leukocyte adhesion and vascular protein leakage. Circ Res 1994, 74(2), 336–43. [Google Scholar] [CrossRef] [PubMed]
- Heusch, G. The Coronary Circulation as a Target of Cardioprotection. Circ Res 2016, 118(10), 1643–58. [Google Scholar] [CrossRef]
- Stempien-Otero, A.; Karsan, A.; Cornejo, C. J.; Xiang, H.; Eunson, T.; Morrison, R. S.; Kay, M.; Winn, R.; Harlan, J. Mechanisms of hypoxia-induced endothelial cell death. Role of p53 in apoptosis. J Biol Chem 1999, 274(12), 8039–45. [Google Scholar] [CrossRef]
- Kalogeris, T.; Baines, C. P.; Krenz, M.; Korthuis, R. J. Ischemia/Reperfusion. Compr Physiol 2016, 7(1), 113–170. [Google Scholar]
- Baldea, I.; Teacoe, I.; Olteanu, D. E.; Vaida-Voievod, C.; Clichici, A.; Sirbu, A.; Filip, G. A.; Clichici, S. Effects of different hypoxia degrees on endothelial cell cultures-Time course study. Mech Ageing Dev 2018, 172, 45–50. [Google Scholar] [CrossRef]
- Jennings, R. B.; Reimer, K. A. The cell biology of acute myocardial ischemia. Annu Rev Med 1991, 42, 225–46. [Google Scholar] [CrossRef]
- Steenbergen, C.; Hill, M. L.; Jennings, R. B. Volume regulation and plasma membrane injury in aerobic, anaerobic, and ischemic myocardium in vitro. Effects of osmotic cell swelling on plasma membrane integrity. Circ Res 1985, 57(6), 864–75. [Google Scholar] [CrossRef]
- Kasseckert, S. A.; Schafer, C.; Kluger, A.; Gligorievski, D.; Tillmann, J.; Schluter, K. D.; Noll, T.; Sauer, H.; Piper, H. M.; Abdallah, Y. Stimulation of cGMP signalling protects coronary endothelium against reperfusion-induced intercellular gap formation. Cardiovasc Res 2009, 83(2), 381–7. [Google Scholar] [CrossRef] [PubMed]
- Ndrepepa, G.; Colleran, R.; Kastrati, A. Reperfusion injury in ST-segment elevation myocardial infarction: the final frontier. Coron Artery Dis 2017, 28(3), 253–262. [Google Scholar] [CrossRef]
- Patterson, C. E.; Lum, H. Update on pulmonary edema: the role and regulation of endothelial barrier function. Endothelium 2001, 8(2), 75–105. [Google Scholar] [CrossRef]
- Waschke, J.; Curry, F. E.; Adamson, R. H.; Drenckhahn, D. Regulation of actin dynamics is critical for endothelial barrier functions. Am J Physiol Heart Circ Physiol 2005, 288(3), H1296–305. [Google Scholar] [CrossRef]
- Maxwell, L.; Gavin, J. B. The role of post-ischaemic reperfusion in the development of microvascular incompetence and ultrastructural damage in the myocardium. Basic Res Cardiol 1991, 86(6), 544–53. [Google Scholar] [CrossRef]
- Dvorak, H. F. Discovery of vascular permeability factor (VPF). Exp Cell Res 2006, 312(5), 522–6. [Google Scholar] [CrossRef]
- Weis, S. M.; Cheresh, D. A. Pathophysiological consequences of VEGF-induced vascular permeability. Nature 2005, 437(7058), 497–504. [Google Scholar] [CrossRef] [PubMed]
- Orsenigo, F.; Giampietro, C.; Ferrari, A.; Corada, M.; Galaup, A.; Sigismund, S.; Ristagno, G.; Maddaluno, L.; Koh, G. Y.; Franco, D.; Kurtcuoglu, V.; Poulikakos, D.; Baluk, P.; McDonald, D.; Grazia Lampugnani, M.; Dejana, E. Phosphorylation of VE-cadherin is modulated by haemodynamic forces and contributes to the regulation of vascular permeability in vivo. Nat Commun 2012, 3, 1208. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Venema, V. J.; Venema, R. C.; Tsai, N.; Behzadian, M. A.; Caldwell, R. B. VEGF-induced permeability increase is mediated by caveolae. Invest Ophthalmol Vis Sci 1999, 40(1), 157–67. [Google Scholar]
- Scotland, R. S.; Cohen, M.; Foster, P.; Lovell, M.; Mathur, A.; Ahluwalia, A.; Hobbs, A. J. C-type natriuretic peptide inhibits leukocyte recruitment and platelet-leukocyte interactions via suppression of P-selectin expression. Proc Natl Acad Sci U S A 2005, 102(40), 14452–7. [Google Scholar] [CrossRef] [PubMed]
- Kerner, T.; Ahlers, O.; Reschreiter, H.; Buhrer, C.; Mockel, M.; Gerlach, H. Adhesion molecules in different treatments of acute myocardial infarction. Crit Care 2001, 5(3), 145–50. [Google Scholar] [CrossRef]
- Vink, H.; Duling, B. R. Identification of distinct luminal domains for macromolecules, erythrocytes, and leukocytes within mammalian capillaries. Circulation Research 1996, 79(3), 581–589. [Google Scholar] [CrossRef]
- Kolarova, H.; Ambruzova, B.; Sindlerova, L. S.; Klinke, A.; Kubala, L. Modulation of Endothelial Glycocalyx Structure under Inflammatory Conditions. Mediat Inflamm 2014, 2014. [Google Scholar]
- Ostergaard, L.; Kristiansen, S. B.; Angleys, H.; Frokiaer, J.; Hasenkam, J. M.; Jespersen, S. N.; Botker, H. E. The role of capillary transit time heterogeneity in myocardial oxygenation and ischemic heart disease. Basic Research in Cardiology 2014, 109(3). [Google Scholar] [CrossRef] [PubMed]
- Ishiharajima, S.; Aida, T.; Nakagawa, R.; Kameyama, K.; Sugano, K.; Oguro, T.; Asano, G. Early membrane damage during ischemia in rat heart. Exp Mol Pathol 1986, 44(1), 1–6. [Google Scholar] [CrossRef] [PubMed]
- Czarnowska, E.; Karwatowskaprokopczuk, E. Ultrastructural Demonstration of Endothelial Glycocalyx Disruption in the Reperfused Rat-Heart - Involvement of Oxygen-Free Radicals. Basic Research in Cardiology 1995, 90(5), 357–364. [Google Scholar] [CrossRef]
- Rubio-Gayosso, I.; Platts, S. H.; Duling, B. R. Reactive oxygen species mediate modification of glycocalyx during ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol 2006, 290(6), H2247–56. [Google Scholar] [CrossRef]
- Vink, H.; Constantinescu, A. A.; Spaan, J. A. Oxidized lipoproteins degrade the endothelial surface layer : implications for platelet-endothelial cell adhesion. Circulation 2000, 101(13), 1500–2. [Google Scholar] [CrossRef]
- Nieuwdorp, M.; Mooij, H. L.; Kroon, J.; Atasever, B.; Spaan, J. A.; Ince, C.; Holleman, F.; Diamant, M.; Heine, R. J.; Hoekstra, J. B.; Kastelein, J. J.; Stroes, E. S.; Vink, H. Endothelial glycocalyx damage coincides with microalbuminuria in type 1 diabetes. Diabetes 2006, 55(4), 1127–32. [Google Scholar] [CrossRef]
- Chappell, D.; Hofmann-Kiefer, K.; Jacob, M.; Rehm, M.; Briegel, J.; Welsch, U.; Conzen, P.; Becker, B. F. TNF-alpha induced shedding of the endothelial glycocalyx is prevented by hydrocortisone and antithrombin. Basic Res Cardiol 2009, 104(1), 78–89. [Google Scholar] [CrossRef] [PubMed]
- Granger, D. N.; Kvietys, P. R. Reperfusion therapy-What's with the obstructed, leaky and broken capillaries? Pathophysiology 2017, 24(4), 213–228. [Google Scholar] [CrossRef]
- Hahn, R. G.; Patel, V.; Dull, R. O. Human glycocalyx shedding: Systematic review and critical appraisal. Acta Anaesthesiol Scand 2021, 65(5), 590–606. [Google Scholar] [CrossRef]
- Bruegger, D.; Rehm, M.; Jacob, M.; Chappell, D.; Stoeckelhuber, M.; Welsch, U.; Conzen, P.; Becker, B. F. Exogenous nitric oxide requires an endothelial glycocalyx to prevent postischemic coronary vascular leak in guinea pig hearts. Crit Care 2008, 12(3), R73. [Google Scholar] [CrossRef]
- van den Berg, B. M.; Vink, H.; Spaan, J. A. The endothelial glycocalyx protects against myocardial edema. Circ Res 2003, 92(6), 592–4. [Google Scholar] [CrossRef]
- Chappell, D.; Dorfler, N.; Jacob, M.; Rehm, M.; Welsch, U.; Conzen, P.; Becker, B. F. Glycocalyx protection reduces leukocyte adhesion after ischemia/reperfusion. Shock 2010, 34(2), 133–9. [Google Scholar] [CrossRef] [PubMed]
- Chappell, D.; Brettner, F.; Doerfler, N.; Jacob, M.; Rehm, M.; Bruegger, D.; Conzen, P.; Jacob, B.; Becker, B. F. Protection of glycocalyx decreases platelet adhesion after ischaemia/reperfusion: an animal study. Eur J Anaesthesiol 2014, 31(9), 474–81. [Google Scholar] [CrossRef]
- Bonaventura, A.; Montecucco, F.; Dallegri, F. Cellular recruitment in myocardial ischaemia/reperfusion injury. Eur J Clin Invest 2016, 46(6), 590–601. [Google Scholar] [CrossRef]
- Charron, T.; Jaffe, R.; Segev, A.; Bang, K. W.; Qiang, B.; Sparkes, J. D.; Butany, J.; Dick, A. J.; Freedman, J.; Strauss, B. H. Effects of distal embolization on the timing of platelet and inflammatory cell activation in interventional coronary no-reflow. Thromb Res 2010, 126(1), 50–5. [Google Scholar] [CrossRef]
- Battinelli, E. M.; Markens, B. A.; Italiano, J. E., Jr. Release of angiogenesis regulatory proteins from platelet alpha granules: modulation of physiologic and pathologic angiogenesis. Blood 2011, 118(5), 1359–69. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, M.; Wang, X.; Peter, K. Platelets in cardiac ischaemia/reperfusion injury: a promising therapeutic target. Cardiovasc Res 2019, 115(7), 1178–1188. [Google Scholar] [CrossRef]
- Schanze, N.; Bode, C.; Duerschmied, D. Platelet Contributions to Myocardial Ischemia/Reperfusion Injury. Front Immunol 2019, 10, 1260. [Google Scholar] [CrossRef]
- Yang, B. C.; Virmani, R.; Nichols, W. W.; Mehta, J. L. Platelets protect against myocardial dysfunction and injury induced by ischemia and reperfusion in isolated rat hearts. Circ Res 1993, 72(6), 1181–90. [Google Scholar] [CrossRef]
- Heindl, B.; Zahler, S.; Welsch, U.; Becker, B. F. Disparate effects of adhesion and degranulation of platelets on myocardial and coronary function in postischaemic hearts. Cardiovasc Res 1998, 38(2), 383–94. [Google Scholar] [CrossRef] [PubMed]
- Choudhri, T. F.; Hoh, B. L.; Zerwes, H. G.; Prestigiacomo, C. J.; Kim, S. C.; Connolly, E. S., Jr.; Kottirsch, G.; Pinsky, D. J. Reduced microvascular thrombosis and improved outcome in acute murine stroke by inhibiting GP IIb/IIIa receptor-mediated platelet aggregation. J Clin Invest 1998, 102(7), 1301–10. [Google Scholar] [CrossRef]
- Golino, P.; Maroko, P. R.; Carew, T. E. Efficacy of platelet depletion in counteracting the detrimental effect of acute hypercholesterolemia on infarct size and the no-reflow phenomenon in rabbits undergoing coronary artery occlusion-reperfusion. Circulation 1987, 76(1), 173–80. [Google Scholar] [CrossRef] [PubMed]
- Neumann, F. J.; Blasini, R.; Schmitt, C.; Alt, E.; Dirschinger, J.; Gawaz, M.; Kastrati, A.; Schomig, A. Effect of glycoprotein IIb/IIIa receptor blockade on recovery of coronary flow and left ventricular function after the placement of coronary-artery stents in acute myocardial infarction. Circulation 1998, 98(24), 2695–701. [Google Scholar] [CrossRef]
- Ong, S. B.; Hernandez-Resendiz, S.; Crespo-Avilan, G. E.; Mukhametshina, R. T.; Kwek, X. Y.; Cabrera-Fuentes, H. A.; Hausenloy, D. J. Inflammation following acute myocardial infarction: Multiple players, dynamic roles, and novel therapeutic opportunities. Pharmacol Ther 2018, 186, 73–87. [Google Scholar] [CrossRef]
- Gotz, A. K.; Zahler, S.; Stumpf, P.; Welsch, U.; Becker, B. F. Intracoronary formation and retention of micro aggregates of leukocytes and platelets contribute to postischemic myocardial dysfunction. Basic Res Cardiol 2005, 100(5), 413–21. [Google Scholar] [CrossRef] [PubMed]
- Ge, L.; Zhou, X.; Ji, W. J.; Lu, R. Y.; Zhang, Y.; Zhang, Y. D.; Ma, Y. Q.; Zhao, J. H.; Li, Y. M. Neutrophil extracellular traps in ischemia-reperfusion injury-induced myocardial no-reflow: therapeutic potential of DNase-based reperfusion strategy. Am J Physiol Heart Circ Physiol 2015, 308(5), H500–9. [Google Scholar] [CrossRef]
- Jaeschke, H.; Smith, C. W. Mechanisms of neutrophil-induced parenchymal cell injury. J Leukoc Biol 1997, 61(6), 647–53. [Google Scholar] [CrossRef] [PubMed]
- Ndrepepa, G. Myeloperoxidase - A bridge linking inflammation and oxidative stress with cardiovascular disease. Clin Chim Acta 2019, 493, 36–51. [Google Scholar] [CrossRef] [PubMed]
- Romanic, A. M.; Harrison, S. M.; Bao, W.; Burns-Kurtis, C. L.; Pickering, S.; Gu, J.; Grau, E.; Mao, J.; Sathe, G. M.; Ohlstein, E. H.; Yue, T. L. Myocardial protection from ischemia/reperfusion injury by targeted deletion of matrix metalloproteinase-9. Cardiovasc Res 2002, 54(3), 549–58. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, M.; Wedin, K.; Brown, M. D.; Keller, C.; Evans, A. J.; Smolen, J.; Burns, A. R.; Rossen, R. D.; Michael, L.; Entman, M. Matrix-dependent mechanism of neutrophil-mediated release and activation of matrix metalloproteinase 9 in myocardial ischemia/reperfusion. Circulation 2001, 103(17), 2181–7. [Google Scholar] [CrossRef]
- Nees, S.; Weiss, D. R.; Senftl, A.; Knott, M.; Forch, S.; Schnurr, M.; Weyrich, P.; Juchem, G. Isolation, bulk cultivation, and characterization of coronary microvascular pericytes: the second most frequent myocardial cell type in vitro. Am J Physiol Heart Circ Physiol 2012, 302(1), H69–84. [Google Scholar] [CrossRef] [PubMed]
- O'Farrell, F. M.; Mastitskaya, S.; Hammond-Haley, M.; Freitas, F.; Wah, W. R.; Attwell, D. Capillary pericytes mediate coronary no-reflow after myocardial ischaemia. Elife 2017, 6. [Google Scholar] [CrossRef]
- Hall, C. N.; Reynell, C.; Gesslein, B.; Hamilton, N. B.; Mishra, A.; Sutherland, B. A.; O'Farrell, F. M.; Buchan, A. M.; Lauritzen, M.; Attwell, D. Capillary pericytes regulate cerebral blood flow in health and disease. Nature 2014, 508(7494), 55–60. [Google Scholar] [CrossRef]
- Yemisci, M.; Gursoy-Ozdemir, Y.; Vural, A.; Can, A.; Topalkara, K.; Dalkara, T. Pericyte contraction induced by oxidative-nitrative stress impairs capillary reflow despite successful opening of an occluded cerebral artery. Nat Med 2009, 15(9), 1031–7. [Google Scholar] [CrossRef]
- Costa, M. A.; Paiva, A. E.; Andreotti, J. P.; Cardoso, M. V.; Cardoso, C. D.; Mintz, A.; Birbrair, A. Pericytes constrict blood vessels after myocardial ischemia. J Mol Cell Cardiol 2018, 116, 1–4. [Google Scholar] [CrossRef]
- Methner, C.; Cao, Z.; Mishra, A.; Kaul, S. Mechanism and potential treatment of the "no reflow" phenomenon after acute myocardial infarction: role of pericytes and GPR39. Am J Physiol Heart Circ Physiol 2021, 321(6), H1030–H1041. [Google Scholar] [CrossRef]
- Li, Q.; Guo, Z.; Wu, C.; Tu, Y.; Wu, Y.; Xie, E.; Yu, C.; Sun, W.; Li, X.; Zheng, J.; Gao, Y. Ischemia preconditioning alleviates ischemia/reperfusion injury-induced coronary no-reflow and contraction of microvascular pericytes in rats. Microvasc Res 2022, 142, 104349. [Google Scholar] [CrossRef] [PubMed]
- Canty, J. M., Jr.; Klocke, F. J. Reduced regional myocardial perfusion in the presence of pharmacologic vasodilator reserve. Circulation 1985, 71(2), 370–7. [Google Scholar] [CrossRef] [PubMed]
- VanBenthuysen, K. M.; McMurtry, I. F.; Horwitz, L. D. Reperfusion after acute coronary occlusion in dogs impairs endothelium-dependent relaxation to acetylcholine and augments contractile reactivity in vitro. J Clin Invest 1987, 79(1), 265–74. [Google Scholar] [CrossRef]
- Quillen, J. E.; Sellke, F. W.; Brooks, L. A.; Harrison, D. G. Ischemia-reperfusion impairs endothelium-dependent relaxation of coronary microvessels but does not affect large arteries. Circulation 1990, 82(2), 586–94. [Google Scholar] [CrossRef] [PubMed]
- Malliani, A.; Schwartz, P. J.; Zanchetti, A. A sympathetic reflex elicited by experimental coronary occlusion. Am J Physiol 1969, 217(3), 703–9. [Google Scholar] [CrossRef]
- Gregorini, L.; Marco, J.; Kozakova, M.; Palombo, C.; Anguissola, G. B.; Marco, I.; Bernies, M.; Cassagneau, B.; Distante, A.; Bossi, I. M.; Fajadet, J.; Heusch, G. Alpha-adrenergic blockade improves recovery of myocardial perfusion and function after coronary stenting in patients with acute myocardial infarction. Circulation 1999, 99(4), 482–90. [Google Scholar] [CrossRef]
- Shimizu, M.; Wang, Q. D.; Sjoquist, P. O.; Ryden, L. The angiotensin II AT1-receptor antagonist candesartan improves functional recovery and reduces the no-reflow area in reperfused ischemic rat hearts. J Cardiovasc Pharmacol 1999, 34(1), 78–81. [Google Scholar] [CrossRef]
- Ryckwaert, F.; Colson, P.; Guillon, G.; Foex, P. Cumulative effects of AT1 and AT2 receptor blockade on ischaemia-reperfusion recovery in rat hearts. Pharmacol Res 2005, 51(6), 497–502. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, H.; Belmadani, S.; Wu, J.; Xu, X.; Elford, H.; Potter, B. J.; Zhang, C. Role of TNF-alpha-induced reactive oxygen species in endothelial dysfunction during reperfusion injury. Am J Physiol Heart Circ Physiol 2008, 295(6), H2242–9. [Google Scholar] [CrossRef]
- Kleinbongard, P.; Bose, D.; Baars, T.; Mohlenkamp, S.; Konorza, T.; Schoner, S.; Elter-Schulz, M.; Eggebrecht, H.; Degen, H.; Haude, M.; Levkau, B.; Schulz, R.; Erbel, R.; Heusch, G. Vasoconstrictor potential of coronary aspirate from patients undergoing stenting of saphenous vein aortocoronary bypass grafts and its pharmacological attenuation. Circ Res 2011, 108(3), 344–52. [Google Scholar] [CrossRef]
- Kleinbongard, P.; Baars, T.; Mohlenkamp, S.; Kahlert, P.; Erbel, R.; Heusch, G. Aspirate from human stented native coronary arteries vs. saphenous vein grafts: more endothelin but less particulate debris. Am J Physiol Heart Circ Physiol 2013, 305(8), H1222-9. [Google Scholar] [CrossRef] [PubMed]
- Herring, N.; Tapoulal, N.; Kalla, M.; Ye, X.; Borysova, L.; Lee, R.; Dall'Armellina, E.; Stanley, C.; Ascione, R.; Lu, C. J.; Banning, A. P.; Choudhury, R. P.; Neubauer, S.; Dora, K.; Kharbanda, R. K.; Channon, K. M.; Oxford Acute Myocardial Infarction, S. Neuropeptide-Y causes coronary microvascular constriction and is associated with reduced ejection fraction following ST-elevation myocardial infarction. Eur Heart J 2019, 40(24), 1920–1929. [Google Scholar] [CrossRef]
- Xue, H. M.; He, G. W.; Huang, J. H.; Yang, Q. New strategy of endothelial protection in cardiac surgery: use of enhancer of endothelial nitric oxide synthase. World J Surg 2010, 34(7), 1461–9. [Google Scholar] [CrossRef]
- Granger, D. N.; Kvietys, P. R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol 2015, 6, 524–551. [Google Scholar] [CrossRef] [PubMed]
- Hein, T. W.; Zhang, C.; Wang, W.; Chang, C. I.; Thengchaisri, N.; Kuo, L. Ischemia-reperfusion selectively impairs nitric oxide-mediated dilation in coronary arterioles: counteracting role of arginase. FASEB J 2003, 17(15), 2328–30. [Google Scholar] [CrossRef]
- Schreckenberg, R.; Weber, P.; Cabrera-Fuentes, H. A.; Steinert, I.; Preissner, K. T.; Bencsik, P.; Sarkozy, M.; Csonka, C.; Ferdinandy, P.; Schulz, R.; Schluter, K. D. Mechanism and consequences of the shift in cardiac arginine metabolism following ischaemia and reperfusion in rats. Thromb Haemost 2015, 113(3), 482–93. [Google Scholar]
- Zhou, T.; Chuang, C. C.; Zuo, L. Molecular Characterization of Reactive Oxygen Species in Myocardial Ischemia-Reperfusion Injury. Biomed Res Int 2015, 2015, 864946. [Google Scholar] [CrossRef] [PubMed]
- Viehman, G. E.; Ma, X. L.; Lefer, D. J.; Lefer, A. M. Time course of endothelial dysfunction and myocardial injury during coronary arterial occlusion. Am J Physiol 1991, 261 (3 Pt 2), H874–81. [Google Scholar] [CrossRef]
- Hollander, M. R.; de Waard, G. A.; Konijnenberg, L. S.; Meijer-van Putten, R. M.; van den Brom, C. E.; Paauw, N.; de Vries, H. E.; van de Ven, P. M.; Aman, J.; Van Nieuw-Amerongen, G. P.; Hordijk, P. L.; Niessen, H. W.; Horrevoets, A. J.; Van Royen, N. Dissecting the Effects of Ischemia and Reperfusion on the Coronary Microcirculation in a Rat Model of Acute Myocardial Infarction. PLoS One 2016, 11(7), e0157233. [Google Scholar]
- Gavin, J. B.; Seelye, R. N.; Nevalainen, T. J.; Armiger, L. C. The effect of ischaemia on the function and fine structure of the microvasculature of myocardium. Pathology 1978, 10(2), 103–11. [Google Scholar] [CrossRef]
- Lonborg, J.; Kelbaek, H.; Helqvist, S.; Holmvang, L.; Jorgensen, E.; Saunamaki, K.; Klovgaard, L.; Kaltoft, A.; Botker, H. E.; Lassen, J. F.; Thuesen, L.; Terkelsen, C. J.; Kofoed, K. F.; Clemmensen, P.; Kober, L.; Engstrom, T. The impact of distal embolization and distal protection on long-term outcome in patients with ST elevation myocardial infarction randomized to primary percutaneous coronary intervention--results from a randomized study. Eur Heart J Acute Cardiovasc Care 2015, 4(2), 180–8. [Google Scholar] [CrossRef] [PubMed]
- Napodano, M.; Peluso, D.; Marra, M. P.; Frigo, A. C.; Tarantini, G.; Buja, P.; Gasparetto, V.; Fraccaro, C.; Isabella, G.; Razzolini, R.; Iliceto, S. Time-dependent detrimental effects of distal embolization on myocardium and microvasculature during primary percutaneous coronary intervention. JACC Cardiovasc Interv 2012, 5(11), 1170–7. [Google Scholar] [CrossRef] [PubMed]
- Henriques, J. P.; Zijlstra, F.; Ottervanger, J. P.; de Boer, M. J.; van 't Hof, A. W.; Hoorntje, J. C.; Suryapranata, H. Incidence and clinical significance of distal embolization during primary angioplasty for acute myocardial infarction. Eur Heart J 2002, 23(14), 1112–7. [Google Scholar] [CrossRef] [PubMed]
- Yameogo, N. V.; Guenancia, C.; Porot, G.; Stamboul, K.; Richard, C.; Gudjoncik, A.; Hamblin, J.; Buffet, P.; Lorgis, L.; Cottin, Y. Predictors of angiographically visible distal embolization in STEMI. Herz 2020, 45(3), 288–292. [Google Scholar] [CrossRef]
- Stone, G. W.; Webb, J.; Cox, D. A.; Brodie, B. R.; Qureshi, M.; Kalynych, A.; Turco, M.; Schultheiss, H. P.; Dulas, D.; Rutherford, B. D.; Antoniucci, D.; Krucoff, M. W.; Gibbons, R. J.; Jones, D.; Lansky, A. J.; Mehran, R.; Enhanced Myocardial, E.; Recovery by Aspiration of Liberated Debris, I. Distal microcirculatory protection during percutaneous coronary intervention in acute ST-segment elevation myocardial infarction: a randomized controlled trial. JAMA 2005, 293(9), 1063–72. [Google Scholar] [CrossRef]
- Kleinbongard, P.; Heusch, G. A fresh look at coronary microembolization. Nat Rev Cardiol 2022, 19(4), 265–280. [Google Scholar] [CrossRef]
- Yunoki, K.; Naruko, T.; Inoue, T.; Sugioka, K.; Inaba, M.; Iwasa, Y.; Komatsu, R.; Itoh, A.; Haze, K.; Yoshiyama, M.; Becker, A. E.; Ueda, M. Relationship of thrombus characteristics to the incidence of angiographically visible distal embolization in patients with ST-segment elevation myocardial infarction treated with thrombus aspiration. JACC Cardiovasc Interv 2013, 6(4), 377–85. [Google Scholar] [CrossRef]
- Abela, G. S.; Kalavakunta, J. K.; Janoudi, A.; Leffler, D.; Dhar, G.; Salehi, N.; Cohn, J.; Shah, I.; Karve, M.; Kotaru, V. P. K.; Gupta, V.; David, S.; Narisetty, K. K.; Rich, M.; Vanderberg, A.; Pathak, D. R.; Shamoun, F. E. Frequency of Cholesterol Crystals in Culprit Coronary Artery Aspirate During Acute Myocardial Infarction and Their Relation to Inflammation and Myocardial Injury. Am J Cardiol 2017, 120(10), 1699–1707. [Google Scholar] [CrossRef]
- Katayama, Y.; Taruya, A.; Kashiwagi, M.; Ozaki, Y.; Shiono, Y.; Tanimoto, T.; Yoshikawa, T.; Kondo, T.; Tanaka, A. No-reflow phenomenon and in vivo cholesterol crystals combined with lipid core in acute myocardial infarction. Int J Cardiol Heart Vasc 2022, 38, 100953. [Google Scholar] [CrossRef]
- Kawaguchi, R.; Oshima, S.; Jingu, M.; Tsurugaya, H.; Toyama, T.; Hoshizaki, H.; Taniguchi, K. Usefulness of virtual histology intravascular ultrasound to predict distal embolization for ST-segment elevation myocardial infarction. J Am Coll Cardiol 2007, 50(17), 1641–6. [Google Scholar] [CrossRef]
- Napodano, M.; Ramondo, A.; Tarantini, G.; Peluso, D.; Compagno, S.; Fraccaro, C.; Frigo, A. C.; Razzolini, R.; Iliceto, S. Predictors and time-related impact of distal embolization during primary angioplasty. Eur Heart J 2009, 30(3), 305–13. [Google Scholar] [CrossRef]
- Fokkema, M. L.; Vlaar, P. J.; Svilaas, T.; Vogelzang, M.; Amo, D.; Diercks, G. F.; Suurmeijer, A. J.; Zijlstra, F. Incidence and clinical consequences of distal embolization on the coronary angiogram after percutaneous coronary intervention for ST-elevation myocardial infarction. Eur Heart J 2009, 30(8), 908–15. [Google Scholar] [CrossRef]
- Falk, E.; Thuesen, L. Pathology of coronary microembolisation and no reflow. Heart 2003, 89(9), 983–5. [Google Scholar] [CrossRef] [PubMed]
- Skyschally, A.; Walter, B.; Heusch, G. Coronary microembolization during early reperfusion: infarct extension, but protection by ischaemic postconditioning. Eur Heart J 2013, 34(42), 3314–21. [Google Scholar] [CrossRef]
- Carlsson, M.; Martin, A. J.; Ursell, P. C.; Saloner, D.; Saeed, M. Magnetic resonance imaging quantification of left ventricular dysfunction following coronary microembolization. Magn Reson Med 2009, 61(3), 595–602. [Google Scholar] [CrossRef]
- Tucker, B.; Vaidya, K.; Cochran, B. J.; Patel, S. Inflammation during Percutaneous Coronary Intervention-Prognostic Value, Mechanisms and Therapeutic Targets. Cells 2021, 10(6). [Google Scholar] [CrossRef]
- Quinones, A.; Saric, M. The cholesterol emboli syndrome in atherosclerosis. Curr Atheroscler Rep 2013, 15(4), 315. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R. M.; Lim, S. Y.; Qiang, B.; Osherov, A. B.; Ghugre, N. R.; Noyan, H.; Qi, X.; Wolff, R.; Ladouceur-Wodzak, M.; Berk, T. A.; Butany, J.; Husain, M.; Wright, G. A.; Strauss, B. H. Distal coronary embolization following acute myocardial infarction increases early infarct size and late left ventricular wall thinning in a porcine model. J Cardiovasc Magn Reson 2015, 17, 106. [Google Scholar] [CrossRef]
- Jennings, R. B.; Sommers, H. M.; Smyth, G. A.; Flack, H. A.; Linn, H. Myocardial necrosis induced by temporary occlusion of a coronary artery in the dog. Arch Pathol 1960, 70, 68–78. [Google Scholar] [PubMed]
- Hausenloy, D. J.; Yellon, D. M. Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J Clin Invest 2013, 123(1), 92–100. [Google Scholar] [CrossRef]
- Frohlich, G. M.; Meier, P.; White, S. K.; Yellon, D. M.; Hausenloy, D. J. Myocardial reperfusion injury: looking beyond primary PCI. Eur Heart J 2013, 34(23), 1714–22. [Google Scholar] [CrossRef]
- Kloner, R. A.; Alker, K. J. The Effect of Streptokinase on Intramyocardial Hemorrhage, Infarct Size, and the No-Reflow Phenomenon during Coronary Reperfusion. Circulation 1984, 70(3), 513–521. [Google Scholar] [CrossRef] [PubMed]
- Ndrepepa, G.; Tiroch, K.; Keta, D.; Fusaro, M.; Seyfarth, M.; Pache, J.; Mehilli, J.; Schoemig, A.; Kastrati, A. Predictive Factors and Impact of No Reflow After Primary Percutaneous Coronary Intervention in Patients With Acute Myocardial Infarction. Circ-Cardiovasc Inte 2010, 3(1), 27–33. [Google Scholar] [CrossRef] [PubMed]
- Kloner, R. A.; Rude, R. E.; Carlson, N.; Maroko, P. R.; DeBoer, L. W.; Braunwald, E. Ultrastructural evidence of microvascular damage and myocardial cell injury after coronary artery occlusion: which comes first? Circulation 1980, 62(5), 945–52. [Google Scholar] [CrossRef]
- Kloner, R. A.; King, K. S.; Harrington, M. G. No-reflow phenomenon in the heart and brain. Am J Physiol Heart Circ Physiol 2018, 315(3), H550–H562. [Google Scholar] [CrossRef]
- Ambrosio, G.; Weisman, H. F.; Mannisi, J. A.; Becker, L. C. Progressive impairment of regional myocardial perfusion after initial restoration of postischemic blood flow. Circulation 1989, 80(6), 1846–61. [Google Scholar] [CrossRef]
- Rochitte, C. E.; Lima, J. A.; Bluemke, D. A.; Reeder, S. B.; McVeigh, E. R.; Furuta, T.; Becker, L. C.; Melin, J. A. Magnitude and time course of microvascular obstruction and tissue injury after acute myocardial infarction. Circulation 1998, 98(10), 1006–14. [Google Scholar] [CrossRef]
- Reffelmann, T.; Kloner, R. A. Microvascular reperfusion injury: rapid expansion of anatomic no reflow during reperfusion in the rabbit. Am J Physiol Heart Circ Physiol 2002, 283(3), H1099–107. [Google Scholar] [CrossRef]
- Turschner, O.; D'Hooge, J.; Dommke, C.; Claus, P.; Verbeken, E.; De Scheerder, I.; Bijnens, B.; Sutherland, G. R. The sequential changes in myocardial thickness and thickening which occur during acute transmural infarction, infarct reperfusion and the resultant expression of reperfusion injury. Eur Heart J 2004, 25(9), 794–803. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Jimenez, R.; Sanchez-Gonzalez, J.; Aguero, J.; Garcia-Prieto, J.; Lopez-Martin, G. J.; Garcia-Ruiz, J. M.; Molina-Iracheta, A.; Rossello, X.; Fernandez-Friera, L.; Pizarro, G.; Garcia-Alvarez, A.; Dall'Armellina, E.; Macaya, C.; Choudhury, R. P.; Fuster, V.; Ibanez, B. Myocardial edema after ischemia/reperfusion is not stable and follows a bimodal pattern: imaging and histological tissue characterization. J Am Coll Cardiol 2015, 65(4), 315–323. [Google Scholar] [CrossRef]
- Alkhalil, M.; Borlotti, A.; De Maria, G. L.; Gaughran, L.; Langrish, J.; Lucking, A.; Ferreira, V.; Kharbanda, R. K.; Banning, A. P.; Channon, K. M.; Dall'Armellina, E.; Choudhury, R. P. Dynamic changes in injured myocardium, very early after acute myocardial infarction, quantified using T1 mapping cardiovascular magnetic resonance. J Cardiovasc Magn Reson 2018, 20(1), 82. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Jimenez, R.; Barreiro-Perez, M.; Martin-Garcia, A.; Sanchez-Gonzalez, J.; Aguero, J.; Galan-Arriola, C.; Garcia-Prieto, J.; Diaz-Pelaez, E.; Vara, P.; Martinez, I.; Zamarro, I.; Garde, B.; Sanz, J.; Fuster, V.; Sanchez, P. L.; Ibanez, B. Dynamic Edematous Response of the Human Heart to Myocardial Infarction: Implications for Assessing Myocardial Area at Risk and Salvage. Circulation 2017, 136(14), 1288–1300. [Google Scholar] [CrossRef]
- Veinot, J. P.; Gattinger, D. A.; Fliss, H. Early apoptosis in human myocardial infarcts. Hum Pathol 1997, 28(4), 485–92. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D. J.; Candilio, L.; Evans, R.; Ariti, C.; Jenkins, D. P.; Kolvekar, S.; Knight, R.; Kunst, G.; Laing, C.; Nicholas, J.; Pepper, J.; Robertson, S.; Xenou, M.; Clayton, T.; Yellon, D. M.; Investigators, E. T. Remote Ischemic Preconditioning and Outcomes of Cardiac Surgery. N Engl J Med 2015, 373(15), 1408–17. [Google Scholar] [CrossRef] [PubMed]
- Abbate, A.; Bussani, R.; Biondi-Zoccai, G. G.; Rossiello, R.; Silvestri, F.; Baldi, F.; Biasucci, L. M.; Baldi, A. Persistent infarct-related artery occlusion is associated with an increased myocardial apoptosis at postmortem examination in humans late after an acute myocardial infarction. Circulation 2002, 106(9), 1051–4. [Google Scholar] [CrossRef] [PubMed]
- Kidambi, A.; Mather, A. N.; Motwani, M.; Swoboda, P.; Uddin, A.; Greenwood, J. P.; Plein, S. The effect of microvascular obstruction and intramyocardial hemorrhage on contractile recovery in reperfused myocardial infarction: insights from cardiovascular magnetic resonance. J Cardiovasc Magn Reson 2013, 15(1), 58. [Google Scholar] [CrossRef]
- Kandler, D.; Lucke, C.; Grothoff, M.; Andres, C.; Lehmkuhl, L.; Nitzsche, S.; Riese, F.; Mende, M.; de Waha, S.; Desch, S.; Lurz, P.; Eitel, I.; Gutberlet, M. The relation between hypointense core, microvascular obstruction and intramyocardial haemorrhage in acute reperfused myocardial infarction assessed by cardiac magnetic resonance imaging. Eur Radiol 2014, 24(12), 3277–88. [Google Scholar] [CrossRef]
- Higginson, L. A.; White, F.; Heggtveit, H. A.; Sanders, T. M.; Bloor, C. M.; Covell, J. W. Determinants of myocardial hemorrhage after coronary reperfusion in the anesthetized dog. Circulation 1982, 65(1), 62–9. [Google Scholar] [CrossRef] [PubMed]
- Mathey, D. G.; Schofer, J.; Kuck, K. H.; Beil, U.; Kloppel, G. Transmural, haemorrhagic myocardial infarction after intracoronary streptokinase. Clinical, angiographic, and necropsy findings. Br Heart J 1982, 48(6), 546–51. [Google Scholar] [CrossRef] [PubMed]
- McNamara, J. J.; Lacro, R. V.; Yee, M.; Smith, G. T. Hemorrhagic infarction and coronary reperfusion. J Thorac Cardiovasc Surg 1981, 81(4), 498–501. [Google Scholar] [CrossRef]
- Lotan, C. S.; Bouchard, A.; Cranney, G. B.; Bishop, S. P.; Pohost, G. M. Assessment of postreperfusion myocardial hemorrhage using proton NMR imaging at 1.5 T. Circulation 1992, 86(3), 1018–25. [Google Scholar] [CrossRef]
- Adamson, R. H.; Zeng, M.; Adamson, G. N.; Lenz, J. F.; Curry, F. E. PAF- and bradykinin-induced hyperpermeability of rat venules is independent of actin-myosin contraction. Am J Physiol Heart Circ Physiol 2003, 285(1), H406–17. [Google Scholar] [CrossRef]
- Inauen, W.; Granger, D. N.; Meininger, C. J.; Schelling, M. E.; Granger, H. J.; Kvietys, P. R. Anoxia-reoxygenation-induced, neutrophil-mediated endothelial cell injury: role of elastase. Am J Physiol 1990, 259 (3 Pt 2), H925–31. [Google Scholar] [CrossRef]
- Gupta, A. K.; Joshi, M. B.; Philippova, M.; Erne, P.; Hasler, P.; Hahn, S.; Resink, T. J. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Lett 2010, 584(14), 3193–7. [Google Scholar] [CrossRef] [PubMed]
- Tarikuz Zaman, A. K.; Spees, J. L.; Sobel, B. E. Attenuation of cardiac vascular rhexis: a promising therapeutic target. Coron Artery Dis 2013, 24(3), 245–52. [Google Scholar] [CrossRef] [PubMed]
- Zeman, A. K. M. T.; French, C. J.; Spees, J. L.; Binbrek, A. S.; Sobel, B. E. Vascular rhexis in mice subjected to non-sustained myocardial ischemia and its therapeutic implications. Exp Biol Med 2011, 236(5), 598–603. [Google Scholar] [CrossRef] [PubMed]
- Robbers, L. F.; Eerenberg, E. S.; Teunissen, P. F.; Jansen, M. F.; Hollander, M. R.; Horrevoets, A. J.; Knaapen, P.; Nijveldt, R.; Heymans, M. W.; Levi, M. M.; van Rossum, A. C.; Niessen, H. W.; Marcu, C. B.; Beek, A. M.; van Royen, N. Magnetic resonance imaging-defined areas of microvascular obstruction after acute myocardial infarction represent microvascular destruction and haemorrhage. Eur Heart J 2013, 34(30), 2346–53. [Google Scholar] [CrossRef]
- Carrick, D.; Haig, C.; Ahmed, N.; McEntegart, M.; Petrie, M. C.; Eteiba, H.; Hood, S.; Watkins, S.; Lindsay, M. M.; Davie, A.; Mahrous, A.; Mordi, I.; Rauhalammi, S.; Sattar, N.; Welsh, P.; Radjenovic, A.; Ford, I.; Oldroyd, K. G.; Berry, C. Myocardial Hemorrhage After Acute Reperfused ST-Segment-Elevation Myocardial Infarction: Relation to Microvascular Obstruction and Prognostic Significance. Circ Cardiovasc Imaging 2016, 9(1), e004148. [Google Scholar] [CrossRef]
- Bulluck, H.; Rosmini, S.; Abdel-Gadir, A.; White, S. K.; Bhuva, A. N.; Treibel, T. A.; Fontana, M.; Ramlall, M.; Hamarneh, A.; Sirker, A.; Herrey, A. S.; Manisty, C.; Yellon, D. M.; Kellman, P.; Moon, J. C.; Hausenloy, D. J. Residual Myocardial Iron Following Intramyocardial Hemorrhage During the Convalescent Phase of Reperfused ST-Segment-Elevation Myocardial Infarction and Adverse Left Ventricular Remodeling. Circ Cardiovasc Imaging 2016, 9(10), e004940. [Google Scholar] [CrossRef]
- Kali, A.; Kumar, A.; Cokic, I.; Tang, R. L.; Tsaftaris, S. A.; Friedrich, M. G.; Dharmakumar, R. Chronic manifestation of postreperfusion intramyocardial hemorrhage as regional iron deposition: a cardiovascular magnetic resonance study with ex vivo validation. Circ Cardiovasc Imaging 2013, 6(2), 218–28. [Google Scholar] [CrossRef]
- Waller, B. F.; Rothbaum, D. A.; Pinkerton, C. A.; Cowley, M. J.; Linnemeier, T. J.; Orr, C.; Irons, M.; Helmuth, R. A.; Wills, E. R.; Aust, C. Status of the myocardium and infarct-related coronary artery in 19 necropsy patients with acute recanalization using pharmacologic (streptokinase, r-tissue plasminogen activator), mechanical (percutaneous transluminal coronary angioplasty) or combined types of reperfusion therapy. J Am Coll Cardiol 1987, 9(4), 785–801. [Google Scholar]
- Amier, R. P.; Tijssen, R. Y. G.; Teunissen, P. F. A.; Fernandez-Jimenez, R.; Pizarro, G.; Garcia-Lunar, I.; Bastante, T.; van de Ven, P. M.; Beek, A. M.; Smulders, M. W.; Bekkers, S.; van Royen, N.; Ibanez, B.; Nijveldt, R. Predictors of Intramyocardial Hemorrhage After Reperfused ST-Segment Elevation Myocardial Infarction. J Am Heart Assoc 2017, 6(8). [Google Scholar] [CrossRef]
- Liu, T.; Howarth, A. G.; Chen, Y.; Nair, A. R.; Yang, H. J.; Ren, D.; Tang, R.; Sykes, J.; Kovacs, M. S.; Dey, D.; Slomka, P.; Wood, J. C.; Finney, R.; Zeng, M.; Prato, F. S.; Francis, J.; Berman, D. S.; Shah, P. K.; Kumar, A.; Dharmakumar, R. Intramyocardial Hemorrhage and the "Wave Front" of Reperfusion Injury Compromising Myocardial Salvage. J Am Coll Cardiol 2022, 79(1), 35–48. [Google Scholar] [CrossRef] [PubMed]
- Wu, K. C.; Zerhouni, E. A.; Judd, R. M.; Lugo-Olivieri, C. H.; Barouch, L. A.; Schulman, S. P.; Blumenthal, R. S.; Lima, J. A. Prognostic significance of microvascular obstruction by magnetic resonance imaging in patients with acute myocardial infarction. Circulation 1998, 97(8), 765–72. [Google Scholar] [CrossRef] [PubMed]
- Kloner, R. A.; Giacomelli, F.; Alker, K. J.; Hale, S. L.; Matthews, R.; Bellows, S. Influx of neutrophils into the walls of large epicardial coronary arteries in response to ischemia/reperfusion. Circulation 1991, 84(4), 1758–72. [Google Scholar] [CrossRef] [PubMed]
- Hansen, P. R. Role of neutrophils in myocardial ischemia and reperfusion. Circulation 1995, 91(6), 1872–85. [Google Scholar] [CrossRef]
- Toldo, S.; Abbate, A. The NLRP3 inflammasome in acute myocardial infarction. Nat Rev Cardiol 2018, 15(4), 203–214. [Google Scholar] [CrossRef]
- Montone, R. A.; La Vecchia, G. Interplay between inflammation and microvascular obstruction in ST-segment elevation myocardial infarction: The importance of velocity. Int J Cardiol 2021, 339, 7–9. [Google Scholar] [CrossRef]
- Mayr, A.; Klug, G.; Schocke, M.; Trieb, T.; Mair, J.; Pedarnig, K.; Pachinger, O.; Jaschke, W.; Metzler, B. Late microvascular obstruction after acute myocardial infarction: relation with cardiac and inflammatory markers. Int J Cardiol 2012, 157(3), 391–6. [Google Scholar] [CrossRef]
- Reindl, M.; Reinstadler, S. J.; Feistritzer, H. J.; Klug, G.; Tiller, C.; Mair, J.; Mayr, A.; Jaschke, W.; Metzler, B. Relation of inflammatory markers with myocardial and microvascular injury in patients with reperfused ST-elevation myocardial infarction. Eur Heart J Acute Cardiovasc Care 2017, 6(7), 640–649. [Google Scholar] [CrossRef]
- Guo, F.; Dong, M.; Ren, F.; Zhang, C.; Li, J.; Tao, Z.; Yang, J.; Li, G. Association between local interleukin-6 levels and slow flow/microvascular dysfunction. J Thromb Thrombolysis 2014, 37(4), 475–82. [Google Scholar] [CrossRef]
- Shetelig, C.; Limalanathan, S.; Hoffmann, P.; Seljeflot, I.; Gran, J. M.; Eritsland, J.; Andersen, G. O. Association of IL-8 With Infarct Size and Clinical Outcomes in Patients With STEMI. J Am Coll Cardiol 2018, 72(2), 187–198. [Google Scholar] [CrossRef]
- Funayama, H.; Ishikawa, S. E.; Sugawara, Y.; Kubo, N.; Momomura, S.; Kawakami, M. Myeloperoxidase may contribute to the no-reflow phenomenon in patients with acute myocardial infarction. Int J Cardiol 2010, 139(2), 187–92. [Google Scholar] [CrossRef] [PubMed]
- Stamboul, K.; Zeller, M.; Rochette, L.; Cottin, Y.; Cochet, A.; Leclercq, T.; Porot, G.; Guenancia, C.; Fichot, M.; Maillot, N.; Vergely, C.; Lorgis, L. Relation between high levels of myeloperoxidase in the culprit artery and microvascular obstruction, infarct size and reverse remodeling in ST-elevation myocardial infarction. Plos One 2017, 12(7). [Google Scholar] [CrossRef]
- Reffelmann, T.; Hale, S. L.; Li, G.; Kloner, R. A. Relationship between no reflow and infarct size as influenced by the duration of ischemia and reperfusion. Am J Physiol Heart Circ Physiol 2002, 282(2), H766–72. [Google Scholar] [CrossRef] [PubMed]
- Bouleti, C.; Mathivet, T.; Serfaty, J. M.; Vignolles, N.; Berland, E.; Monnot, C.; Cluzel, P.; Steg, P. G.; Montalescot, G.; Germain, S. Angiopoietin-like 4 serum levels on admission for acute myocardial infarction are associated with no-reflow. International Journal of Cardiology 2015, 187, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Niccoli, G.; Lanza, G. A.; Shaw, S.; Romagnoli, E.; Gioia, D.; Burzotta, F.; Trani, C.; Mazzari, M. A.; Mongiardo, R.; De Vita, M.; Rebuzzi, A. G.; Luscher, T. F.; Crea, F. Endothelin-1 and acute myocardial infarction: a no-reflow mediator after successful percutaneous myocardial revascularization. Eur Heart J 2006, 27(15), 1793–8. [Google Scholar] [CrossRef]
- Nallamothu, B. K.; Bradley, E. H.; Krumholz, H. M. Time to treatment in primary percutaneous coronary intervention. N Engl J Med 2007, 357(16), 1631–8. [Google Scholar] [CrossRef]
- Durante, A.; Camici, P. G. Novel insights into an "old" phenomenon: the no reflow. Int J Cardiol 2015, 187, 273–80. [Google Scholar] [CrossRef]
- Niccoli, G.; Scalone, G.; Lerman, A.; Crea, F. Coronary microvascular obstruction in acute myocardial infarction. Eur Heart J 2016, 37(13), 1024–33. [Google Scholar] [CrossRef]
- Sorop, O.; Merkus, D.; de Beer, V. J.; Houweling, B.; Pistea, A.; McFalls, E. O.; Boomsma, F.; van Beusekom, H. M.; van der Giessen, W. J.; VanBavel, E.; Duncker, D. J. Functional and structural adaptations of coronary microvessels distal to a chronic coronary artery stenosis. Circ Res 2008, 102(7), 795–803. [Google Scholar] [CrossRef]
- Vaidya, K.; Tucker, B.; Patel, S.; Ng, M. K. C. Acute Coronary Syndromes (ACS)-Unravelling Biology to Identify New Therapies-The Microcirculation as a Frontier for New Therapies in ACS. Cells 2021, 10(9). [Google Scholar] [CrossRef]
- Lerman, A.; Holmes, D. R.; Herrmann, J.; Gersh, B. J. Microcirculatory dysfunction in ST-elevation myocardial infarction: cause, consequence, or both? Eur Heart J 2007, 28(7), 788–97. [Google Scholar] [CrossRef] [PubMed]
- Ng, M. K.; Yong, A. S.; Ho, M.; Shah, M. G.; Chawantanpipat, C.; O'Connell, R.; Keech, A.; Kritharides, L.; Fearon, W. F. The index of microcirculatory resistance predicts myocardial infarction related to percutaneous coronary intervention. Circ Cardiovasc Interv 2012, 5(4), 515–22. [Google Scholar] [CrossRef]
- Soeda, T.; Higuma, T.; Abe, N.; Yamada, M.; Yokoyama, H.; Shibutani, S.; Ong, D. S.; Vergallo, R.; Minami, Y.; Lee, H.; Okumura, K.; Jang, I. K. Morphological predictors for no reflow phenomenon after primary percutaneous coronary intervention in patients with ST-segment elevation myocardial infarction caused by plaque rupture. Eur Heart J Cardiovasc Imaging 2017, 18(1), 103–110. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, P.; Rekwal, L.; Sinha, S. K.; Nath, R. K.; Khanra, D.; Singh, A. P. Predictors of no-reflow phenomenon following percutaneous coronary intervention for ST-segment elevation myocardial infarction. Ann Cardiol Angeiol (Paris) 2021, 70(3), 136–142. [Google Scholar] [CrossRef]
- Tanaka, A.; Kawarabayashi, T.; Nishibori, Y.; Sano, T.; Nishida, Y.; Fukuda, D.; Shimada, K.; Yoshikawa, J. No-reflow phenomenon and lesion morphology in patients with acute myocardial infarction. Circulation 2002, 105(18), 2148–52. [Google Scholar] [CrossRef]
- Yip, H. K.; Chen, M. C.; Chang, H. W.; Hang, C. L.; Hsieh, Y. K.; Fang, C. Y.; Wu, C. J. Angiographic morphologic features of infarct-related arteries and timely reperfusion in acute myocardial infarction: predictors of slow-flow and no-reflow phenomenon. Chest 2002, 122(4), 1322–32. [Google Scholar] [CrossRef]
- Mehran, R.; Dangas, G.; Mintz, G. S.; Lansky, A. J.; Pichard, A. D.; Satler, L. F.; Kent, K. M.; Stone, G. W.; Leon, M. B. Atherosclerotic plaque burden and CK-MB enzyme elevation after coronary interventions : intravascular ultrasound study of 2256 patients. Circulation 2000, 101(6), 604–10. [Google Scholar] [CrossRef] [PubMed]
- Magro, M.; Nauta, S. T.; Simsek, C.; Boersma, E.; van der Heide, E.; Regar, E.; van Domburg, R. T.; Zijlstra, F.; Serruys, P. W.; van Geuns, R. J. Usefulness of the SYNTAX score to predict "no reflow" in patients treated with primary percutaneous coronary intervention for ST-segment elevation myocardial infarction. Am J Cardiol 2012, 109(5), 601–6. [Google Scholar] [CrossRef]
- Kaur, G.; Baghdasaryan, P.; Natarajan, B.; Sethi, P.; Mukherjee, A.; Varadarajan, P.; Pai, R. G. Pathophysiology, Diagnosis, and Management of Coronary No-Reflow Phenomenon. Int J Angiol 2021, 30(1), 15–21. [Google Scholar] [PubMed]
- Harrison, R. W.; Aggarwal, A.; Ou, F. S.; Klein, L. W.; Rumsfeld, J. S.; Roe, M. T.; Wang, T. Y. ; American College of Cardiology National Cardiovascular Data, R., Incidence and outcomes of no-reflow phenomenon during percutaneous coronary intervention among patients with acute myocardial infarction. Am J Cardiol 2013, 111(2), 178–84. [Google Scholar] [PubMed]
- Galiuto, L.; Paraggio, L.; Liuzzo, G.; de Caterina, A. R.; Crea, F. Predicting the no-reflow phenomenon following successful percutaneous coronary intervention. Biomark Med 2010, 4(3), 403–20. [Google Scholar] [PubMed]
- Hu, X.; Yang, X.; Li, X.; Li, G.; Zhou, Y.; Dong, H. Elevated uric acid is related to the no-/slow-reflow phenomenon in STEMI undergoing primary PCI. Eur J Clin Invest 2022, 52(4), e13719. [Google Scholar] [PubMed]
- Kai, T.; Oka, S.; Hoshino, K.; Watanabe, K.; Nakamura, J.; Abe, M.; Watanabe, A. Renal Dysfunction as a Predictor of Slow-Flow/No-Reflow Phenomenon and Impaired ST Segment Resolution After Percutaneous Coronary Intervention in ST-Elevation Myocardial Infarction With Initial Thrombolysis in Myocardial Infarction Grade 0. Circ J 2021, 85(10), 1770–1778. [Google Scholar]
- Savic, L.; Mrdovic, I.; Asanin, M.; Stankovic, S.; Lasica, R.; Krljanac, G.; Rajic, D.; Simic, D. The Impact of Kidney Function on the Slow-Flow/No-Reflow Phenomenon in Patients Treated with Primary Percutaneous Coronary Intervention: Registry Analysis. J Interv Cardiol 2022, 2022. [Google Scholar]
- Esenboga, K.; Kurtul, A.; Yamanturk, Y. Y.; Tan, T. S.; Tutar, D. E. Systemic immune-inflammation index predicts no-reflow phenomenon after primary percutaneous coronary intervention. Acta Cardiol 2022, 77(1), 59–65. [Google Scholar]
- Selcuk, M.; Cinar, T.; Saylik, F.; Demiroz, O.; Yildirim, E. The Association of a PRECISE-DAPT Score With No-Reflow in Patients With ST-Segment Elevation Myocardial Infarction. Angiology 2022, 73(1), 68–72. [Google Scholar]
- Sen, O.; Sen, S. B.; Topuz, A. N.; Topuz, M. Vitamin D level predicts angiographic no-reflow phenomenon after percutaneous coronary intervention in patients with ST segment elevation myocardial infarction. Biomark Med 2021, 15(15), 1357–1366. [Google Scholar]
- Sun, Y.; Ren, J.; Li, L.; Wang, C. S.; Yao, H. C. RDW as A Predictor for No-Reflow Phenomenon in DM Patients with ST-Segment Elevation Myocardial Infarction Undergoing Primary Percutaneous Coronary Intervention. J Clin Med, 2023; 12, 3. [Google Scholar]
- Sondergaard, F. T.; Beske, R. P.; Frydland, M.; Moller, J. E.; Helgestad, O. K. L.; Jensen, L. O.; Holmvang, L.; Goetze, J. P.; Engstrom, T.; Hassager, C. Soluble ST2 in plasma is associated with post-procedural no-or-slow reflow after primary percutaneous coronary intervention in ST-elevation myocardial infarction. Eur Heart J Acute Cardiovasc Care 2023, 12(1), 48–52. [Google Scholar]
- Hadi, H. A.; Suwaidi, J. A. Endothelial dysfunction in diabetes mellitus. Vasc Health Risk Manag 2007, 3(6), 853–76. [Google Scholar] [PubMed]
- Murthy, V. L.; Naya, M.; Foster, C. R.; Gaber, M.; Hainer, J.; Klein, J.; Dorbala, S.; Blankstein, R.; Di Carli, M. F. Association between coronary vascular dysfunction and cardiac mortality in patients with and without diabetes mellitus. Circulation 2012, 126(15), 1858–68. [Google Scholar]
- Di Carli, M. F.; Janisse, J.; Grunberger, G.; Ager, J. Role of chronic hyperglycemia in the pathogenesis of coronary microvascular dysfunction in diabetes. J Am Coll Cardiol 2003, 41(8), 1387–93. [Google Scholar]
- Rawshani, A.; Rawshani, A.; Gudbjornsdottir, S. Mortality and Cardiovascular Disease in Type 1 and Type 2 Diabetes. N Engl J Med 2017, 377(3), 300–301. [Google Scholar] [PubMed]
- Eitel, I.; Hintze, S.; de Waha, S.; Fuernau, G.; Lurz, P.; Desch, S.; Schuler, G.; Thiele, H. Prognostic Impact of Hyperglycemia in Nondiabetic and Diabetic Patients With ST-Elevation Myocardial Infarction Insights From Contrast-Enhanced Magnetic Resonance Imaging. Circ-Cardiovasc Imag 2012, 5(6), 708–718. [Google Scholar]
- Reinstadler, S. J.; Stiermaier, T.; Eitel, C.; Metzler, B.; de Waha, S.; Fuernau, G.; Desch, S.; Thiele, H.; Eitel, I. Relationship between diabetes and ischaemic injury among patients with revascularized ST-elevation myocardial infarction. Diabetes Obes Metab 2017, 19(12), 1706–1713. [Google Scholar]
- Iwakura, K.; Ito, H.; Ikushima, M.; Kawano, S.; Okamura, A.; Asano, K.; Kuroda, T.; Tanaka, K.; Masuyama, T.; Hori, M.; Fujii, K. Association between hyperglycemia and the no-reflow phenomenon in patients with acute myocardial infarction. J Am Coll Cardiol 2003, 41(1), 1–7. [Google Scholar]
- Ota, S.; Tanimoto, T.; Orii, M.; Hirata, K.; Shiono, Y.; Shimamura, K.; Matsuo, Y.; Yamano, T.; Ino, Y.; Kitabata, H.; Yamaguchi, T.; Kubo, T.; Tanaka, A.; Imanishi, T.; Akasaka, T. Association between hyperglycemia at admission and microvascular obstruction in patients with ST-segment elevation myocardial infarction. J Cardiol 2015, 65(3-4), 272–277. [Google Scholar] [PubMed]
- Jensen, C. J.; Eberle, H. C.; Nassenstein, K.; Schlosser, T.; Farazandeh, M.; Naber, C. K.; Sabin, G. V.; Bruder, O. Impact of hyperglycemia at admission in patients with acute ST-segment elevation myocardial infarction as assessed by contrast-enhanced MRI. Clin Res Cardiol 2011, 100(8), 649–659. [Google Scholar]
- Barrett, E. J.; Liu, Z. Q.; Khamaisi, M.; King, G. L.; Klein, R.; Klein, B. E. K.; Hughes, T. M.; Craft, S.; Freedman, B. I.; Bowden, D. W.; Vinik, A. I.; Casellini, C. M. Diabetic Microvascular Disease: An Endocrine Society Scientific Statement. J Clin Endocr Metab 2017, 102(12), 4343–4410. [Google Scholar]
- Panza, J. A.; Casino, P. R.; Kilcoyne, C. M.; Quyyumi, A. A. Role of Endothelium-Derived Nitric-Oxide in the Abnormal Endothelium-Dependent Vascular Relaxation of Patients with Essential-Hypertension. Circulation 1993, 87(5), 1468–1474. [Google Scholar]
- Carrick, D.; Haig, C.; Maznyczka, A. M.; Carberry, J.; Mangion, K.; Ahmed, N.; May, V. T. Y.; McEntegart, M.; Petrie, M. C.; Eteiba, H.; Lindsay, M.; Hood, S.; Watkins, S.; Davie, A.; Mahrous, A.; Mordi, I.; Ford, I.; Radjenovic, A.; Welsh, P.; Sattar, N.; Wetherall, K.; Oldroyd, K. G.; Berry, C. Hypertension, Microvascular Pathology, and Prognosis After an Acute Myocardial Infarction. Hypertension 2018, 72(3), 720–730. [Google Scholar] [PubMed]
- Reinstadler, S. J.; Stiermaier, T.; Eitel, C.; Saad, M.; Metzler, B.; de Waha, S.; Fuernau, G.; Desch, S.; Thiele, H.; Eitel, I. Antecedent hypertension and myocardial injury in patients with reperfused ST-elevation myocardial infarction. J Cardiovasc Magn R 2016, 18. [Google Scholar]
- Quyyumi, A. A.; Mulcahy, D.; Andrews, N. P.; Husain, S.; Panza, J. A.; Cannon, R. O. 3rd, Coronary vascular nitric oxide activity in hypertension and hypercholesterolemia. Comparison of acetylcholine and substance P. Circulation 1997, 95(1), 104–10. [Google Scholar] [PubMed]
- Golino, P.; Maroko, P. R.; Carew, T. E. The effect of acute hypercholesterolemia on myocardial infarct size and the no-reflow phenomenon during coronary occlusion-reperfusion. Circulation 1987, 75(1), 292–8. [Google Scholar]
- Iwakura, K.; Ito, H.; Kawano, S.; Okamura, A.; Kurotobi, T.; Date, M.; Inoue, K.; Fujii, K. Chronic pre-treatment of statins is associated with the reduction of the no-reflow phenomenon in the patients with reperfused acute myocardial infarction. Eur Heart J 2006, 27(5), 534–9. [Google Scholar] [PubMed]
- Reindl, M.; Reinstadler, S. J.; Feistritzer, H. J.; Theurl, M.; Basic, D.; Eigler, C.; Holzknecht, M.; Mair, J.; Mayr, A.; Klug, G.; Metzler, B. Relation of Low-Density Lipoprotein Cholesterol With Microvascular Injury and Clinical Outcome in Revascularized ST-Elevation Myocardial Infarction. J Am Heart Assoc 2017, 6(10). [Google Scholar]
- Surendran, A.; Ismail, U.; Atefi, N.; Bagchi, A. K.; Singal, P. K.; Shah, A.; Aliani, M.; Ravandi, A. Lipidomic Predictors of Coronary No-Reflow. Metabolites 2023, 13(1). [Google Scholar]
- Messner, B.; Bernhard, D. Smoking and cardiovascular disease: mechanisms of endothelial dysfunction and early atherogenesis. Arterioscler Thromb Vasc Biol 2014, 34(3), 509–15. [Google Scholar]
- Symons, R.; Masci, P. G.; Francone, M.; Claus, P.; Barison, A.; Carbone, I.; Agati, L.; Galea, N.; Janssens, S.; Bogaert, J. Impact of active smoking on myocardial infarction severity in reperfused ST-segment elevation myocardial infarction patients: the smoker's paradox revisited. Eur Heart J 2016, 37(36), 2756–2764. [Google Scholar]
- Reinstadler, S. J.; Eitel, C.; Fuernau, G.; de Waha, S.; Desch, S.; Mende, M.; Metzler, B.; Schuler, G.; Thiele, H.; Eitel, I. Association of smoking with myocardial injury and clinical outcome in patients undergoing mechanical reperfusion for ST-elevation myocardial infarction. Eur Heart J Cardiovasc Imaging 2017, 18(1), 39–45. [Google Scholar] [CrossRef]
- Haig, C.; Carrick, D.; Carberry, J.; Mangion, K.; Maznyczka, A.; Wetherall, K.; McEntegart, M.; Petrie, M. C.; Eteiba, H.; Lindsay, M.; Hood, S.; Watkins, S.; Davie, A.; Mahrous, A.; Mordi, I.; Ahmed, N.; Teng Yue May, V.; Ford, I.; Radjenovic, A.; Welsh, P.; Sattar, N.; Oldroyd, K. G.; Berry, C. Current Smoking and Prognosis After Acute ST-Segment Elevation Myocardial Infarction: New Pathophysiological Insights. JACC Cardiovasc Imaging 2019, 12(6), 993–1003. [Google Scholar] [CrossRef] [PubMed]
- Ipek, G.; Onuk, T.; Karatas, M. B.; Gungor, B.; Osken, A.; Keskin, M.; Oz, A.; Tanik, O.; Hayiroglu, M. I.; Yaka, H. Y.; Ozturk, R.; Bolca, O. CHA2DS2-VASc Score is a Predictor of No-Reflow in Patients With ST-Segment Elevation Myocardial Infarction Who Underwent Primary Percutaneous Intervention. Angiology 2016, 67(9), 840–5. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Hu, M.; Ma, S.; Niu, T. New R(2)-CHA(2)DS(2)-VASc score predicts no-reflow phenomenon and long-term prognosis in patients with ST-segment elevation myocardial infarction after primary percutaneous coronary intervention. Front Cardiovasc Med 2022, 9, 899739. [Google Scholar] [CrossRef] [PubMed]
- Karila-Cohen, D.; Czitrom, D.; Brochet, E.; Faraggi, M.; Seknadji, P.; Himbert, D.; Juliard, J. M.; Assayag, P.; Steg, P. G. Decreased no-reflow in patients with anterior myocardial infarction and pre-infarction angina. Eur Heart J 1999, 20(23), 1724–30. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Anzai, T.; Yoshikawa, T.; Asakura, Y.; Ishikawa, S.; Mitamura, H.; Ogawa, S. Absence of preinfarction angina is associated with a risk of no-reflow phenomenon after primary coronary angioplasty for a first anterior wall acute myocardial infarction. Int J Cardiol 2000, 75(2-3), 253–60. [Google Scholar] [CrossRef]
- Zalewski, J.; Undas, A.; Godlewski, J.; Stepien, E.; Zmudka, K. No-reflow phenomenon after acute myocardial infarction is associated with reduced clot permeability and susceptibility to lysis. Arterioscler Thromb Vasc Biol 2007, 27(10), 2258–65. [Google Scholar] [CrossRef] [PubMed]
- Bolayir, H. A.; Gunes, H.; Kivrak, T.; Sahin, O.; Akaslan, D.; Kurt, R.; Bolayir, A.; Imadoglu, O. The role of SCUBE1 in the pathogenesis of no-reflow phenomenon presenting with ST segment elevation myocardial infarction. Anatol J Cardiol 2017, 18(2), 122–127. [Google Scholar] [CrossRef]
- Yoshino, S.; Cilluffo, R.; Best, P. J.; Atkinson, E. J.; Aoki, T.; Cunningham, J. M.; de Andrade, M.; Choi, B. J.; Lerman, L. O.; Lerman, A. Single nucleotide polymorphisms associated with abnormal coronary microvascular function. Coron Artery Dis 2014, 25(4), 281–9. [Google Scholar] [CrossRef]
- Niccoli, G.; Celestini, A.; Calvieri, C.; Cosentino, N.; Falcioni, E.; Carnevale, R.; Nocella, C.; Fracassi, F.; Roberto, M.; Antonazzo, R. P.; Pignatelli, P.; Crea, F.; Violi, F. Patients with microvascular obstruction after primary percutaneous coronary intervention show a gp91phox (NOX2) mediated persistent oxidative stress after reperfusion. Eur Heart J Acute Cardiovasc Care 2013, 2(4), 379–88. [Google Scholar] [CrossRef]
- Galasso, G.; Schiekofer, S.; D'Anna, C.; Gioia, G. D.; Piccolo, R.; Niglio, T.; Rosa, R. D.; Strisciuglio, T.; Cirillo, P.; Piscione, F.; Trimarco, B. No-reflow phenomenon: pathophysiology, diagnosis, prevention, and treatment. A review of the current literature and future perspectives. Angiology 2014, 65(3), 180–9. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Maruyama, A.; Iwakura, K.; Takiuchi, S.; Masuyama, T.; Hori, M.; Higashino, Y.; Fujii, K.; Minamino, T. Clinical implications of the 'no reflow' phenomenon. A predictor of complications and left ventricular remodeling in reperfused anterior wall myocardial infarction. Circulation 1996, 93(2), 223–8. [Google Scholar] [CrossRef] [PubMed]
- Resnic, F. S.; Wainstein, M.; Lee, M. K.; Behrendt, D.; Wainstein, R. V.; Ohno-Machado, L.; Kirshenbaum, J. M.; Rogers, C. D.; Popma, J. J.; Piana, R. No-reflow is an independent predictor of death and myocardial infarction after percutaneous coronary intervention. Am Heart J 2003, 145(1), 42–6. [Google Scholar] [CrossRef]
- Brosh, D.; Assali, A. R.; Mager, A.; Porter, A.; Hasdai, D.; Teplitsky, I.; Rechavia, E.; Fuchs, S.; Battler, A.; Kornowski, R. Effect of no-reflow during primary percutaneous coronary intervention for acute myocardial infarction on six-month mortality. Am J Cardiol 2007, 99(4), 442–5. [Google Scholar] [CrossRef]
- Bolognese, L.; Carrabba, N.; Parodi, G.; Santoro, G. M.; Buonamici, P.; Cerisano, G.; Antoniucci, D. Impact of microvascular dysfunction on left ventricular remodeling and long-term clinical outcome after primary coronary angioplasty for acute myocardial infarction. Circulation 2004, 109(9), 1121–6. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R. H.; Harjai, K. J.; Boura, J.; Cox, D.; Stone, G. W.; O'Neill, W.; Grines, C. L.; Primary Angioplasty in Myocardial Infarction, I. Prognostic significance of transient no-reflow during primary percutaneous coronary intervention for ST-elevation acute myocardial infarction. Am J Cardiol 2003, 92(12), 1445–7. [Google Scholar] [CrossRef]
- Jinnouchi, H.; Sakakura, K.; Wada, H.; Arao, K.; Kubo, N.; Sugawara, Y.; Funayama, H.; Momomura, S.; Ako, J. Transient no reflow following primary percutaneous coronary intervention. Heart Vessels 2014, 29(4), 429–36. [Google Scholar] [CrossRef]
- Fujii, T.; Masuda, N.; Nakano, M.; Nakazawa, G.; Shinozaki, N.; Matsukage, T.; Ogata, N.; Yoshimachi, F.; Ikari, Y. Impact of transient or persistent slow flow and adjunctive distal protection on mortality in ST-segment elevation myocardial infarction. Cardiovasc Interv Ther 2015, 30(2), 121–30. [Google Scholar] [CrossRef]
- Kim, M. C.; Cho, J. Y.; Jeong, H. C.; Lee, K. H.; Park, K. H.; Sim, D. S.; Yoon, N. S.; Youn, H. J.; Kim, K. H.; Hong, Y. J.; Park, H. W.; Kim, J. H.; Jeong, M. H.; Cho, J. G.; Park, J. C.; Seung, K. B.; Chang, K.; Ahn, Y. Long-Term Clinical Outcomes of Transient and Persistent No Reflow Phenomena following Percutaneous Coronary Intervention in Patients with Acute Myocardial Infarction. Korean Circ J 2016, 46(4), 490–8. [Google Scholar] [CrossRef]
- Nakazawa, G.; Tanabe, K.; Onuma, Y.; Yachi, S.; Aoki, J.; Yamamoto, H.; Higashikuni, Y.; Yagishita, A.; Nakajima, H.; Hara, K. Efficacy of culprit plaque assessment by 64-slice multidetector computed tomography to predict transient no-reflow phenomenon during percutaneous coronary intervention. Am Heart J 2008, 155(6), 1150–7. [Google Scholar] [CrossRef]
- Iijima, R.; Shinji, H.; Ikeda, N.; Itaya, H.; Makino, K.; Funatsu, A.; Yokouchi, I.; Komatsu, H.; Ito, N.; Nuruki, H.; Nakajima, R.; Nakamura, M. Comparison of coronary arterial finding by intravascular ultrasound in patients with "transient no-reflow" versus "reflow" during percutaneous coronary intervention in acute coronary syndrome. Am J Cardiol 2006, 97(1), 29–33. [Google Scholar] [CrossRef]
- Ndrepepa, G.; Colleran, R.; Kastrati, A. No-reflow after percutaneous coronary intervention: a correlate of poor outcome in both persistent and transient forms. EuroIntervention 2018, 14(2), 139–141. [Google Scholar] [CrossRef]
- Reffelmann, T.; Hale, S. L.; Dow, J. S.; Kloner, R. A. No-reflow phenomenon persists long-term after ischemia/reperfusion in the rat and predicts infarct expansion. Circulation 2003, 108(23), 2911–7. [Google Scholar] [CrossRef] [PubMed]
- Galiuto, L.; Garramone, B.; Scara, A.; Rebuzzi, A. G.; Crea, F.; La Torre, G.; Funaro, S.; Madonna, M.; Fedele, F.; Agati, L.; Investigators, A. The extent of microvascular damage during myocardial contrast echocardiography is superior to other known indexes of post-infarct reperfusion in predicting left ventricular remodeling: results of the multicenter AMICI study. J Am Coll Cardiol 2008, 51(5), 552–9. [Google Scholar] [CrossRef]
- Husser, O.; Monmeneu, J. V.; Sanchis, J.; Nunez, J.; Lopez-Lereu, M. P.; Bonanad, C.; Chaustre, F.; Gomez, C.; Bosch, M. J.; Hinarejos, R.; Chorro, F. J.; Riegger, G. A.; Llacer, A.; Bodi, V. Cardiovascular magnetic resonance-derived intramyocardial hemorrhage after STEMI: Influence on long-term prognosis, adverse left ventricular remodeling and relationship with microvascular obstruction. Int J Cardiol 2013, 167(5), 2047–54. [Google Scholar] [CrossRef] [PubMed]
- Loubeyre, C.; Morice, M. C.; Lefevre, T.; Piechaud, J. F.; Louvard, Y.; Dumas, P. A randomized comparison of direct stenting with conventional stent implantation in selected patients with acute myocardial infarction. J Am Coll Cardiol 2002, 39(1), 15–21. [Google Scholar] [CrossRef] [PubMed]
- Gasior, M.; Gierlotka, M.; Lekston, A.; Wilczek, K.; Zebik, T.; Hawranek, M.; Wojnar, R.; Szkodzinski, J.; Piegza, J.; Dyrbus, K.; Kalarus, Z.; Zembala, M.; Polonski, L. Comparison of outcomes of direct stenting versus stenting after balloon predilation in patients with acute myocardial infarction (DIRAMI). Am J Cardiol 2007, 100(5), 798–805. [Google Scholar] [CrossRef]
- Azzalini, L.; Millan, X.; Ly, H. Q.; L'Allier, P. L.; Jolicoeur, E. M. Direct stenting versus pre-dilation in ST-elevation myocardial infarction: a systematic review and meta-analysis. J Interv Cardiol 2015, 28(2), 119–31. [Google Scholar] [CrossRef]
- Li, C.; Zhang, B.; Li, M.; Liu, J.; Wang, L.; Liu, Y.; Wang, Z.; Wen, S. Comparing Direct Stenting With Conventional Stenting in Patients With Acute Coronary Syndromes: A Meta-Analysis of 12 Clinical Trials. Angiology 2016, 67(4), 317–25. [Google Scholar] [CrossRef]
- Mahmoud, K. D.; Jolly, S. S.; James, S.; Dzavik, V.; Cairns, J. A.; Olivecrona, G. K.; Renlund, H.; Gao, P.; Lagerqvist, B.; Alazzoni, A.; Kedev, S.; Stankovic, G.; Meeks, B.; Frobert, O.; Zijlstra, F. Clinical impact of direct stenting and interaction with thrombus aspiration in patients with ST-segment elevation myocardial infarction undergoing percutaneous coronary intervention: Thrombectomy Trialists Collaboration. Eur Heart J 2018, 39(26), 2472–2479. [Google Scholar] [CrossRef]
- Gick, M.; Jander, N.; Bestehorn, H. P.; Kienzle, R. P.; Ferenc, M.; Werner, K.; Comberg, T.; Peitz, K.; Zohlnhofer, D.; Bassignana, V.; Buettner, H. J.; Neumann, F. J. Randomized evaluation of the effects of filter-based distal protection on myocardial perfusion and infarct size after primary percutaneous catheter intervention in myocardial infarction with and without ST-segment elevation. Circulation 2005, 112(10), 1462–9. [Google Scholar] [CrossRef] [PubMed]
- Kelbaek, H.; Terkelsen, C. J.; Helqvist, S.; Lassen, J. F.; Clemmensen, P.; Klovgaard, L.; Kaltoft, A.; Engstrom, T.; Botker, H. E.; Saunamaki, K.; Krusell, L. R.; Jorgensen, E.; Hansen, H. H.; Christiansen, E. H.; Ravkilde, J.; Kober, L.; Kofoed, K. F.; Thuesen, L. Randomized comparison of distal protection versus conventional treatment in primary percutaneous coronary intervention: the drug elution and distal protection in ST-elevation myocardial infarction (DEDICATION) trial. J Am Coll Cardiol 2008, 51(9), 899–905. [Google Scholar] [CrossRef] [PubMed]
- Kumbhani, D. J.; Bavry, A. A.; Desai, M. Y.; Bangalore, S.; Bhatt, D. L. Role of aspiration and mechanical thrombectomy in patients with acute myocardial infarction undergoing primary angioplasty: an updated meta-analysis of randomized trials. J Am Coll Cardiol 2013, 62(16), 1409–18. [Google Scholar] [CrossRef]
- Frobert, O.; Lagerqvist, B.; Olivecrona, G. K.; Omerovic, E.; Gudnason, T.; Maeng, M.; Aasa, M.; Angeras, O.; Calais, F.; Danielewicz, M.; Erlinge, D.; Hellsten, L.; Jensen, U.; Johansson, A. C.; Karegren, A.; Nilsson, J.; Robertson, L.; Sandhall, L.; Sjogren, I.; Ostlund, O.; Harnek, J.; James, S. K. Thrombus Aspiration during ST-Segment Elevation Myocardial Infarction. New Engl J Med 2013, 369(17), 1587–1597. [Google Scholar] [CrossRef]
- Jolly, S. S.; Cairns, J. A.; Yusuf, S.; Meeks, B.; Pogue, J.; Rokoss, M. J.; Kedev, S.; Thabane, L.; Stankovic, G.; Moreno, R.; Gershlick, A.; Chowdhary, S.; Lavi, S.; Niemela, K.; Steg, P. G.; Bernat, I.; Xu, Y.; Cantor, W. J.; Overgaard, C. B.; Naber, C. K.; Cheema, A. N.; Welsh, R. C.; Bertrand, O. F.; Avezum, A.; Bhindi, R.; Pancholy, S.; Rao, S. V.; Natarajan, M. K.; ten Berg, J. M.; Shestakovska, O.; Gao, P.; Widimsky, P.; Dzavik, V.; Investigators, T. Randomized trial of primary PCI with or without routine manual thrombectomy. N Engl J Med 2015, 372(15), 1389–98. [Google Scholar] [CrossRef] [PubMed]
- Carrick, D.; Oldroyd, K. G.; McEntegart, M.; Haig, C.; Petrie, M. C.; Eteiba, H.; Hood, S.; Owens, C.; Watkins, S.; Layland, J.; Lindsay, M.; Peat, E.; Rae, A.; Behan, M.; Sood, A.; Hillis, W. S.; Mordi, I.; Mahrous, A.; Ahmed, N.; Wilson, R.; Lasalle, L.; Genereux, P.; Ford, I.; Berry, C. A randomized trial of deferred stenting versus immediate stenting to prevent no- or slow-reflow in acute ST-segment elevation myocardial infarction (DEFER-STEMI). J Am Coll Cardiol 2014, 63(20), 2088–2098. [Google Scholar] [CrossRef]
- Lonborg, J.; Engstrom, T.; Ahtarovski, K. A.; Nepper-Christensen, L.; Helqvist, S.; Vejlstrup, N.; Kyhl, K.; Schoos, M. M.; Ghotbi, A.; Goransson, C.; Bertelsen, L.; Holmvang, L.; Pedersen, F.; Jorgensen, E.; Saunamaki, K.; Clemmensen, P.; De Backer, O.; Klovgaard, L.; Hofsten, D. E.; Kober, L.; Kelbaek, H.; Investigators, D.-. . Myocardial Damage in Patients With Deferred Stenting After STEMI: A DANAMI-3-DEFER Substudy. J Am Coll Cardiol 2017, 69(23), 2794–2804. [Google Scholar] [CrossRef]
- Cassese, S.; Belle, L.; Ndrepepa, G.; Bosson, J. L.; Fusaro, M.; Lonborg, J.; Ahtarovski, K. A.; Kelbaek, H.; Fusaro, M. Deferred vs Immediate Stenting in Primary Percutaneous Coronary Intervention: A Collaborative Meta-analysis of Randomized Trials With Cardiac Magnetic Resonance Imaging Data. Can J Cardiol 2018, 34(12), 1573–1580. [Google Scholar] [CrossRef]
- Stone, G. W.; Abizaid, A.; Silber, S.; Dizon, J. M.; Merkely, B.; Costa, R. A.; Kornowski, R.; Abizaid, A.; Wojdyla, R.; Maehara, A.; Dressler, O.; Brener, S. J.; Bar, E.; Dudek, D. Prospective, Randomized, Multicenter Evaluation of a Polyethylene Terephthalate Micronet Mesh-Covered Stent (MGuard) in ST-Segment Elevation Myocardial Infarction The MASTER Trial. Journal of the American College of Cardiology 2012, 60(19), 1975–1984. [Google Scholar] [CrossRef]
- Dudek, D.; Dziewierz, A.; Brener, S. J.; Abizaid, A.; Merkely, B.; Costa, R. A.; Bar, E.; Rakowski, T.; Kornowski, R.; Dressler, O.; Abizaid, A.; Silber, S.; Stone, G. W. Mesh-Covered Embolic Protection Stent Implantation in ST-Segment-Elevation Myocardial Infarction Final 1-Year Clinical and Angiographic Results From the MGUARD for Acute ST Elevation Reperfusion Trial. Circ-Cardiovasc Inte 2015, 8(2). [Google Scholar]
- Romaguera, R.; Gomez-Hospital, J. A.; Sanchez-Elvira, G.; Gomez-Lara, J.; Ferreiro, J. L.; Roura, G.; Gracida, M.; Homs, S.; Teruel, L.; Cequier, A. MGuard Mesh-Covered Stent for Treatment of ST-Segment Elevation Myocardial Infarction with High Thrombus Burden Despite Manual Aspiration. J Interv Cardiol 2013, 26(1), 1–7. [Google Scholar] [CrossRef]
- De Maria, G. L.; Alkhalil, M.; Borlotti, A.; Wolfrum, M.; Gaughran, L.; Dall'Armellina, E.; Langrish, J. P.; Lucking, A. J.; Choudhury, R. P.; Kharbanda, R. K.; Channon, K. M.; Banning, A. P. Index of microcirculatory resistance-guided therapy with pressure-controlled intermittent coronary sinus occlusion improves coronary microvascular function and reduces infarct size in patients with ST-elevation myocardial infarction: the Oxford Acute Myocardial Infarction - Pressure-controlled Intermittent Coronary Sinus Occlusion study (OxAMI-PICSO study). EuroIntervention 2018, 14(3), e352–e359. [Google Scholar]
- Van't Hof, A. W.; Ten Berg, J.; Heestermans, T.; Dill, T.; Funck, R. C.; van Werkum, W.; Dambrink, J. H.; Suryapranata, H.; van Houwelingen, G.; Ottervanger, J. P.; Stella, P.; Giannitsis, E.; Hamm, C. ; Ongoing Tirofiban In Myocardial infarction Evaluation 2 study, g., Prehospital initiation of tirofiban in patients with ST-elevation myocardial infarction undergoing primary angioplasty (On-TIME 2): a multicentre, double-blind, randomised controlled trial. Lancet 2008, 372(9638), 537–46. [Google Scholar] [PubMed]
- Akpek, M.; Sahin, O.; Sarli, B.; Baktir, A. O.; Saglam, H.; Urkmez, S.; Ergin, A.; Oguzhan, A.; Arinc, H.; Kaya, M. G. Acute Effects of Intracoronary Tirofiban on No-Reflow Phenomena in Patients With ST-Segment Elevated Myocardial Infarction Undergoing Primary Percutaneous Coronary Intervention. Angiology 2015, 66(6), 560–7. [Google Scholar] [CrossRef]
- Ma, Q. M.; Ma, Y.; Wang, X. N.; Li, S. S.; Yu, T. T.; Duan, W. L.; Wu, J. K.; Wen, Z. Y.; Jiao, Y. D.; Sun, Z. Q.; Hou, Y. Intracoronary compared with intravenous bolus tirofiban on the microvascular obstruction in patients with STEMI undergoing PCI: a cardiac MR study. Int J Cardiovas Imag 2020, 36(6), 1121–1132. [Google Scholar] [CrossRef] [PubMed]
- Stone, G. W.; Maehara, A.; Witzenbichler, B.; Godlewski, J.; Parise, H.; Dambrink, J. H.; Ochala, A.; Carlton, T. W.; Cristea, E.; Wolff, S. D.; Brener, S. J.; Chowdhary, S.; El-Omar, M.; Neunteufl, T.; Metzger, D. C.; Karwoski, T.; Dizon, J. M.; Mehran, R.; Gibson, C. M.; Investigators, I.-A. Intracoronary abciximab and aspiration thrombectomy in patients with large anterior myocardial infarction: the INFUSE-AMI randomized trial. JAMA 2012, 307(17), 1817–26. [Google Scholar] [CrossRef]
- Thiele, H.; Wohrle, J.; Hambrecht, R.; Rittger, H.; Birkemeyer, R.; Lauer, B.; Neuhaus, P.; Brosteanu, O.; Sick, P.; Wiemer, M.; Kerber, S.; Kleinertz, K.; Eitel, I.; Desch, S.; Schuler, G. Intracoronary versus intravenous bolus abciximab during primary percutaneous coronary intervention in patients with acute ST-elevation myocardial infarction: a randomised trial. Lancet 2012, 379(9819), 923–931. [Google Scholar] [CrossRef]
- Ibanez, B.; James, S.; Agewall, S.; Antunes, M. J.; Bucciarelli-Ducci, C.; Bueno, H.; Caforio, A. L. P.; Crea, F.; Goudevenos, J. A.; Halvorsen, S.; Hindricks, G.; Kastrati, A.; Lenzen, M. J.; Prescott, E.; Roffi, M.; Valgimigli, M.; Varenhorst, C.; Vranckx, P.; Widimsky, P.; Group, E. S. C. S. D. 2017 ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation: The Task Force for the management of acute myocardial infarction in patients presenting with ST-segment elevation of the European Society of Cardiology (ESC). Eur Heart J 2018, 39(2), 119–177. [Google Scholar]
- Ndrepepa, G. Improving myocardial injury, infarct size, and myocardial salvage in the era of primary PCI for STEMI. Coron Artery Dis 2015, 26(4), 341–55. [Google Scholar] [CrossRef]
- Mahaffey, K. W.; Puma, J. A.; Barbagelata, N. A.; DiCarli, M. F.; Leesar, M. A.; Browne, K. F.; Eisenberg, P. R.; Bolli, R.; Casas, A. C.; Molina-Viamonte, V.; Orlandi, C.; Blevins, R.; Gibbons, R. J.; Califf, R. M.; Granger, C. B. Adenosine as an adjunct to thrombolytic therapy for acute myocardial infarction: results of a multicenter, randomized, placebo-controlled trial: the Acute Myocardial Infarction STudy of ADenosine (AMISTAD) trial. J Am Coll Cardiol 1999, 34(6), 1711–20. [Google Scholar] [CrossRef]
- Ross, A. M.; Gibbons, R. J.; Stone, G. W.; Kloner, R. A.; Alexander, R. W.; Investigators, A.-I. A randomized, double-blinded, placebo-controlled multicenter trial of adenosine as an adjunct to reperfusion in the treatment of acute myocardial infarction (AMISTAD-II). J Am Coll Cardiol 2005, 45(11), 1775–80. [Google Scholar] [CrossRef]
- Niccoli, G.; Rigattieri, S.; De Vita, M. R.; Valgimigli, M.; Corvo, P.; Fabbiocchi, F.; Romagnoli, E.; De Caterina, A. R.; La Torre, G.; Lo Schiavo, P.; Tarantino, F.; Ferrari, R.; Tomai, F.; Olivares, P.; Cosentino, N.; D'Amario, D.; Leone, A. M.; Porto, I.; Burzotta, F.; Trani, C.; Crea, F. Open-label, randomized, placebo-controlled evaluation of intracoronary adenosine or nitroprusside after thrombus aspiration during primary percutaneous coronary intervention for the prevention of microvascular obstruction in acute myocardial infarction: the REOPEN-AMI study (Intracoronary Nitroprusside Versus Adenosine in Acute Myocardial Infarction). JACC Cardiovasc Interv 2013, 6(6), 580–9. [Google Scholar] [PubMed]
- Desmet, W.; Bogaert, J.; Dubois, C.; Sinnaeve, P.; Adriaenssens, T.; Pappas, C.; Ganame, J.; Dymarkowski, S.; Janssens, S.; Belmans, A.; Van de Werf, F. High-dose intracoronary adenosine for myocardial salvage in patients with acute ST-segment elevation myocardial infarction. Eur Heart J 2011, 32(7), 867–77. [Google Scholar] [CrossRef]
- Nazir, S. A.; McCann, G. P.; Greenwood, J. P.; Kunadian, V.; Khan, J. N.; Mahmoud, I. Z.; Blackman, D. J.; Been, M.; Abrams, K. R.; Shipley, L.; Wilcox, R.; Adgey, A. A.; Gershlick, A. H. Strategies to attenuate micro-vascular obstruction during P-PCI: the randomized reperfusion facilitated by local adjunctive therapy in ST-elevation myocardial infarction trial. Eur Heart J 2016, 37(24), 1910–9. [Google Scholar] [CrossRef]
- Laborante, R.; Bianchini, E.; Restivo, A.; Ciliberti, G.; Galli, M.; Vergallo, R.; Rodolico, D.; Zito, A.; Princi, G.; Leone, A. M.; Aurigemma, C.; Romagnoli, E.; Montone, R. A.; Burzotta, F.; Trani, C.; Crea, F.; D'Amario, D. Adenosine as adjunctive therapy in acute coronary syndrome: a meta-analysis of randomized controlled trials. Eur Heart J Cardiovasc Pharmacother 2023, 9(2), 173–182. [Google Scholar] [CrossRef]
- Siddiqi, N.; Neil, C.; Bruce, M.; MacLennan, G.; Cotton, S.; Papadopoulou, S.; Feelisch, M.; Bunce, N.; Lim, P. O.; Hildick-Smith, D.; Horowitz, J.; Madhani, M.; Boon, N.; Dawson, D.; Kaski, J. C.; Frenneaux, M.; investigators, N. Intravenous sodium nitrite in acute ST-elevation myocardial infarction: a randomized controlled trial (NIAMI). Eur Heart J 2014, 35(19), 1255–62. [Google Scholar] [CrossRef]
- Annibali, G.; Scrocca, I.; Aranzulla, T. C.; Meliga, E.; Maiellaro, F.; Musumeci, G. "No-Reflow" Phenomenon: A Contemporary Review. J Clin Med 2022, 11(8). [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Cheng, Z.; Gu, Y.; Peng, D. Short-Term Effects of Verapamil and Diltiazem in the Treatment of No Reflow Phenomenon: A Meta-Analysis of Randomized Controlled Trials. Biomed Res Int 2015, 2015, 382086. [Google Scholar] [CrossRef]
- Huang, R. I.; Patel, P.; Walinsky, P.; Fischman, D. L.; Ogilby, J. D.; Awar, M.; Frankil, C.; Savage, M. P. Efficacy of intracoronary nicardipine in the treatment of no-reflow during percutaneous coronary intervention. Catheter Cardiovasc Interv 2006, 68(5), 671–6. [Google Scholar] [CrossRef] [PubMed]
- Fischell, T. A.; Haller, S.; Pulukurthy, S.; Virk, I. S. Nicardipine and adenosine "flush cocktail" to prevent no-reflow during rotational atherectomy. Cardiovasc Revasc Med 2008, 9(4), 224–8. [Google Scholar] [CrossRef]
- Ibanez, B.; Prat-Gonzalez, S.; Speidl, W. S.; Vilahur, G.; Pinero, A.; Cimmino, G.; Garcia, M. J.; Fuster, V.; Sanz, J.; Badimon, J. J. Early metoprolol administration before coronary reperfusion results in increased myocardial salvage analysis of ischemic myocardium at risk using cardiac magnetic resonance. Circulation 2007, 115(23), 2909–2916. [Google Scholar] [CrossRef]
- Garcia-Prieto, J.; Villena-Gutierrez, R.; Gomez, M.; Bernardo, E.; Pun-Garcia, A.; Garcia-Lunar, I.; Crainiciuc, G.; Fernandez-Jimenez, R.; Sreeramkumar, V.; Bourio-Martinez, R.; Garcia-Ruiz, J. M.; Del Valle, A. S.; Sanz-Rosa, D.; Pizarro, G.; Fernandez-Ortiz, A.; Hidalgo, A.; Fuster, V.; Ibanez, B. Neutrophil stunning by metoprolol reduces infarct size. Nat Commun 2017, 8, 14780. [Google Scholar] [CrossRef]
- Zhao, J. L.; Yang, Y. J.; Pei, W. D.; Sun, Y. H.; Zhai, M.; Liu, Y. X.; Gao, R. L. Carvedilol reduces myocardial no-reflow by decreasing endothelin-1 via activation of the ATP-sensitive K+ channel. Perfusion 2008, 23(2), 111–5. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, S. A.; Doerries, C.; Manes, C.; Speer, T.; Dessy, C.; Lobysheva, I.; Mohmand, W.; Akbar, R.; Bahlmann, F.; Besler, C.; Schaefer, A.; Hilfiker-Kleiner, D.; Luscher, T. F.; Balligand, J. L.; Drexler, H.; Landmesser, U. Nebivolol exerts beneficial effects on endothelial function, early endothelial progenitor cells, myocardial neovascularization, and left ventricular dysfunction early after myocardial infarction beyond conventional beta1-blockade. J Am Coll Cardiol 2011, 57(5), 601–11. [Google Scholar] [CrossRef]
- Ibanez, B.; Macaya, C.; Sanchez-Brunete, V.; Pizarro, G.; Fernandez-Friera, L.; Mateos, A.; Fernandez-Ortiz, A.; Garcia-Ruiz, J. M.; Garcia-Alvarez, A.; Iniguez, A.; Jimenez-Borreguero, J.; Lopez-Romero, P.; Fernandez-Jimenez, R.; Goicolea, J.; Ruiz-Mateos, B.; Bastante, T.; Arias, M.; Iglesias-Vazquez, J. A.; Rodriguez, M. D.; Escalera, N.; Acebal, C.; Cabrera, J. A.; Valenciano, J.; Perez de Prado, A.; Fernandez-Campos, M. J.; Casado, I.; Garcia-Rubira, J. C.; Garcia-Prieto, J.; Sanz-Rosa, D.; Cuellas, C.; Hernandez-Antolin, R.; Albarran, A.; Fernandez-Vazquez, F.; de la Torre-Hernandez, J. M.; Pocock, S.; Sanz, G.; Fuster, V. Effect of early metoprolol on infarct size in ST-segment-elevation myocardial infarction patients undergoing primary percutaneous coronary intervention: the Effect of Metoprolol in Cardioprotection During an Acute Myocardial Infarction (METOCARD-CNIC) trial. Circulation 2013, 128(14), 1495–503. [Google Scholar] [PubMed]
- Roolvink, V.; Ibanez, B.; Ottervanger, J. P.; Pizarro, G.; van Royen, N.; Mateos, A.; Dambrink, J. E.; Escalera, N.; Lipsic, E.; Albarran, A.; Fernandez-Ortiz, A.; Fernandez-Aviles, F.; Goicolea, J.; Botas, J.; Remkes, W.; Hernandez-Jaras, V.; Kedhi, E.; Zamorano, J. L.; Navarro, F.; Alfonso, F.; Garcia-Lledo, A.; Alonso, J.; van Leeuwen, M.; Nijveldt, R.; Postma, S.; Kolkman, E.; Gosselink, M.; de Smet, B.; Rasoul, S.; Piek, J. J.; Fuster, V.; van 't Hof, A. W. J.; Investigators, E.-B. Early Intravenous Beta-Blockers in Patients With ST-Segment Elevation Myocardial Infarction Before Primary Percutaneous Coronary Intervention. J Am Coll Cardiol 2016, 67(23), 2705–2715. [Google Scholar] [CrossRef]
- Kim, J. S.; Kim, J.; Choi, D.; Lee, C. J.; Lee, S. H.; Ko, Y. G.; Hong, M. K.; Kim, B. K.; Oh, S. J.; Jeon, D. W.; Yang, J. Y.; Cho, J. R.; Lee, N. H.; Cho, Y. H.; Cho, D. K.; Jang, Y. Efficacy of High-Dose Atorvastatin Loading Before Primary Percutaneous Coronary Intervention in ST-Segment Elevation Myocardial Infarction The STATIN STEMI Trial. Jacc-Cardiovasc Inte 2010, 3(3), 332–339. [Google Scholar] [CrossRef]
- Hahn, J. Y.; Kim, H. J.; Choi, Y. J.; Jo, S. H.; Kim, H. J.; Lee, S.; Ahn, K. J.; Song, Y. B.; Choi, J. H.; Choi, S. H.; Choi, Y. J.; Lee, K. H.; Lee, S. H.; Gwon, H. C. Effects of atorvastatin pretreatment on infarct size in patients with ST-segment elevation myocardial infarction undergoing primary percutaneous coronary intervention. Am Heart J 2011, 162(6), 1026–33. [Google Scholar] [CrossRef] [PubMed]
- Berwanger, O.; Santucci, E. V.; de Barros, E. S. P. G. M.; Jesuino, I. A.; Damiani, L. P.; Barbosa, L. M.; Santos, R. H. N.; Laranjeira, L. N.; Egydio, F. M.; Borges de Oliveira, J. A.; Dall Orto, F. T. C.; Beraldo de Andrade, P.; Bienert, I. R. C.; Bosso, C. E.; Mangione, J. A.; Polanczyk, C. A.; Sousa, A.; Kalil, R. A. K.; Santos, L. M.; Sposito, A. C.; Rech, R. L.; Sousa, A. C. S.; Baldissera, F.; Nascimento, B. R.; Giraldez, R.; Cavalcanti, A. B.; Pereira, S. B.; Mattos, L. A.; Armaganijan, L. V.; Guimaraes, H. P.; Sousa, J.; Alexander, J. H.; Granger, C. B.; Lopes, R. D.; Investigators, S.-P. Effect of Loading Dose of Atorvastatin Prior to Planned Percutaneous Coronary Intervention on Major Adverse Cardiovascular Events in Acute Coronary Syndrome: The SECURE-PCI Randomized Clinical Trial. JAMA 2018, 319(13), 1331–1340. [Google Scholar] [CrossRef]
- McCartney, P. J.; Eteiba, H.; Maznyczka, A. M.; McEntegart, M.; Greenwood, J. P.; Muir, D. F.; Chowdhary, S.; Gershlick, A. H.; Appleby, C.; Cotton, J. M.; Wragg, A.; Curzen, N.; Oldroyd, K. G.; Lindsay, M.; Rocchiccioli, J. P.; Shaukat, A.; Good, R.; Watkins, S.; Robertson, K.; Malkin, C.; Martin, L.; Gillespie, L.; Ford, T. J.; Petrie, M. C.; Macfarlane, P. W.; Tait, R. C.; Welsh, P.; Sattar, N.; Weir, R. A.; Fox, K. A.; Ford, I.; McConnachie, A.; Berry, C.; CorcoAran, D.; Davie, A.; Hood, S.; Kelly, J.; Krysztofiak, T.; McNulty, V.; Orchard, V.; Wales, J.; Aggarwal, S.; Ali, T.; Andrews, F.; Browning, N.; Fairbairn, T. A.; Hardwick, R.; Hasleton, J.; Kunadian, B.; Magapu, P.; Palmer, N.; Stables, R.; Taylerson, C.; Tong, H.; Twiss, S.; Velavan, P.; Banks, K.; Corbett, S.; Mahmoudi, M.; Nicholas, Z.; Wilkinson, J.; Allan, M.; Anderson, M.; Blackman, D. J.; Blaxill, J.; Cunnington, M.; Kodoth, V.; Kuria, J.; Lindsay, S.; McLenachan, J.; Somers, K.; Veerasamy, M.; Abraham, R.; Adlam, D.; Coleman, S.; Hudson, I.; Ladwiniec, A.; McCann, G. P.; Parker, E.; Roberts, E.; Wormleighton, J.; Abdelaal, E.; Contractor, H.; Duran, B.; Egdell, R.; Ferguson, S.; Giannopoulou, S.; Sastry, S.; Sarma, J.; Schmitt, M.; Quinn, S.; Archbold, A.; Andiapen, M.; Evwierhoma, C.; Jain, A.; Jones, D.; Kennon, S.; Moon, J.; Mathur, A.; Richards, A.; Sirker, A.; Austin, D.; de Belder, M.; Carter, J.; Cunningham, N.; Dale, R.; Hall, J.; Mack, S.; Manders, T.; Maredia, N.; McLeod, K.; Settimo, L.; Sutton, A.; Swanson, N.; Williams, P.; Wright, R.; Radford, E.; Wrigley, B.; Das, R.; Jones, S.; Wealleans, V.; Cruden, N.; Flint, L.; Buchan, R.; Bucciarelli-Ducci, C.; Johnson, T.; Johny, V.; McCullagh, L.; Murphy, A.; Petrie, C. J.; Dinnett, E.; Ford, G. A.; Lang, C. C.; Norrie, J.; Kharbanda, R.; Labinjoh, C.; Baird, G.; Pairman, S.; Jamieson, D.; Weeden, S.; Van-Melckebeke, J.; Douglas, E.; Surtees, P.; Ross, B.; Brindley, G.; McGowan, S.; Gault, E. J.; Stuart, D.; Travers, M.; Jones, M.; Kean, S.; Watson, C.; Miller, E.; Bolger, E.; Thomson, G.; Butler, E.; Cooney, J.; Kennedy, J.; Leishman, E.; Carberry, J.; Radjenovic, A.; Grp, T.-T. Effect of Low-Dose Intracoronary Alteplase During Primary Percutaneous Coronary Intervention on Microvascular Obstruction in Patients With Acute Myocardial Infarction A Randomized Clinical Trial. Jama-J Am Med Assoc 2019, 321(1), 56–68. [Google Scholar] [CrossRef]
- Alyamani, M.; Campbell, S.; Navarese, E.; Welsh, R. C.; Bainey, K. R. Safety and Efficacy of Intracoronary Thrombolysis as Adjunctive Therapy to Primary PCI in STEMI: A Systematic Review and Meta-analysis. Canadian Journal of Cardiology 2021, 37(2), 339–346. [Google Scholar] [CrossRef]
- van Geuns, R. J.; Sideris, G.; Van Royen, N.; El Mahmoud, R.; Diletti, R.; Bal Dit Sollier, C.; Garot, J.; Van Der Hoeven, N. W.; Cortese, B.; Ding, L.; Lechthaler, I.; Deliargyris, E. N.; Anthopoulos, P.; Drouet, L. Bivalirudin infusion to reduce ventricular infarction: the open-label, randomised Bivalirudin Infusion for Ventricular InfArction Limitation (BIVAL) study. EuroIntervention 2017, 13(5), e540–e548. [Google Scholar] [CrossRef]
- Navarese, E. P.; Frediani, L.; Kandzari, D. E.; Caiazzo, G.; Cenname, A. M.; Cortese, B.; Piva, T.; Mucaj, A.; Tumscitz, C.; Paparoni, F.; Larosa, C.; Bisceglia, T.; Menozzi, M.; Gurbel, P. A.; Kubica, J. Efficacy and safety of intracoronary epinephrine versus conventional treatments alone in STEMI patients with refractory coronary no-reflow during primary PCI: The RESTORE observational study. Catheter Cardiovasc Interv 2021, 97(4), 602–611. [Google Scholar] [CrossRef]
- Khan, K. A.; Qamar, N.; Saghir, T.; Sial, J. A.; Kumar, D.; Kumar, R.; Qayyum, D.; Yasin, U.; Jalbani, J.; Karim, M. Comparison of Intracoronary Epinephrine and Adenosine for No-Reflow in Normotensive Patients With Acute Coronary Syndrome (COAR Trial). Circ Cardiovasc Interv 2022, 15(2), e011408. [Google Scholar] [CrossRef] [PubMed]
- Aksu, T.; Guler, T. E.; Colak, A.; Baysal, E.; Durukan, M.; Sen, T.; Guray, U. Intracoronary epinephrine in the treatment of refractory no-reflow after primary percutaneous coronary intervention: a retrospective study. BMC Cardiovasc Disord 2015, 15, 10. [Google Scholar] [CrossRef] [PubMed]
- Darwish, A.; Frere, A. F.; Abdelsamie, M.; Awady, W. E.; Gouda, M. Intracoronary epinephrine versus adenosine in the management of refractory no-reflow phenomenon: a single-center retrospective cohort study. Ann Saudi Med 2022, 42(2), 75–82. [Google Scholar] [CrossRef]
- Massalha, E.; Oren, D.; Goitein, O.; Brodov, Y.; Fardman, A.; Younis, A.; Berkovitch, A.; Raibman-Spector, S.; Konen, E.; Maor, E.; Fefer, P.; Segev, A.; Beigel, R.; Matetzky, S. Post-ST-Segment-Elevation Myocardial Infarction Platelet Reactivity Is Associated With the Extent of Microvascular Obstruction and Infarct Size as Determined by Cardiac Magnetic Resonance Imaging. J Am Heart Assoc 2022, 11(3), e020973. [Google Scholar] [CrossRef] [PubMed]
- Steg, P. G.; James, S.; Harrington, R. A.; Ardissino, D.; Becker, R. C.; Cannon, C. P.; Emanuelsson, H.; Finkelstein, A.; Husted, S.; Katus, H.; Kilhamn, J.; Olofsson, S.; Storey, R. F.; Weaver, W. D.; Wallentin, L.; Group, P. S. Ticagrelor versus clopidogrel in patients with ST-elevation acute coronary syndromes intended for reperfusion with primary percutaneous coronary intervention: A Platelet Inhibition and Patient Outcomes (PLATO) trial subgroup analysis. Circulation 2010, 122(21), 2131–41. [Google Scholar] [CrossRef]
- Montalescot, G.; van 't Hof, A. W.; Lapostolle, F.; Silvain, J.; Lassen, J. F.; Bolognese, L.; Cantor, W. J.; Cequier, A.; Chettibi, M.; Goodman, S. G.; Hammett, C. J.; Huber, K.; Janzon, M.; Merkely, B.; Storey, R. F.; Zeymer, U.; Stibbe, O.; Ecollan, P.; Heutz, W. M.; Swahn, E.; Collet, J. P.; Willems, F. F.; Baradat, C.; Licour, M.; Tsatsaris, A.; Vicaut, E.; Hamm, C. W.; Investigators, A. Prehospital ticagrelor in ST-segment elevation myocardial infarction. N Engl J Med 2014, 371(11), 1016–27. [Google Scholar] [CrossRef]
- van Leeuwen, M. A. H.; van der Hoeven, N. W.; Janssens, G. N.; Everaars, H.; Nap, A.; Lemkes, J. S.; de Waard, G. A.; van de Ven, P. M.; van Rossum, A. C.; Ten Cate, T. J. F.; Piek, J. J.; von Birgelen, C.; Escaned, J.; Valgimigli, M.; Diletti, R.; Riksen, N. P.; van Mieghem, N. M.; Nijveldt, R.; van Royen, N. Evaluation of Microvascular Injury in Revascularized Patients With ST-Segment-Elevation Myocardial Infarction Treated With Ticagrelor Versus Prasugrel. Circulation 2019, 139(5), 636–646. [Google Scholar] [CrossRef]
- Dai, W.; Ye, Z.; Li, L.; Su, Q. Effect of preoperative loading dose ticagrelor and clopidogrel on no-reflow phenomenon during intervention in patients with ST-segment elevation myocardial infarction undergoing primary percutaneous coronary intervention: a systematic review and meta-analysis. Drug Des Devel Ther 2018, 12, 2039–2049. [Google Scholar]
- Bulluck, H.; Chan, M. H. H.; Bryant, J. A.; Chai, P.; Chawla, A.; Chua, T. S.; Chung, Y. C.; Fei, G.; Ho, H. H.; Ho, A. F. W.; Hoe, A. J.; Imran, S. S.; Lee, C. H.; Lim, S. H.; Liew, B. W.; Yun, P. L. Z.; Hock, M. O. E.; Paradies, V.; Roe, M. T.; Teo, L.; Wong, A. S.; Wong, E.; Wong, P. E.; Watson, T.; Chan, M. Y.; Tan, J. W.; Hausenloy, D. J. Platelet inhibition to target reperfusion injury trial: Rationale and study design. Clin Cardiol 2019, 42(1), 5–12. [Google Scholar] [CrossRef] [PubMed]
- Piot, C.; Croisille, P.; Staat, P.; Thibault, H.; Rioufol, G.; Mewton, N.; Elbelghiti, R.; Cung, T. T.; Bonnefoy, E.; Angoulvant, D.; Macia, C.; Raczka, F.; Sportouch, C.; Gahide, G.; Finet, G.; Andre-Fouet, X.; Revel, D.; Kirkorian, G.; Monassier, J. P.; Derumeaux, G.; Ovize, M. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N Engl J Med 2008, 359(5), 473–81. [Google Scholar] [CrossRef]
- Cung, T. T.; Morel, O.; Cayla, G.; Rioufol, G.; Garcia-Dorado, D.; Angoulvant, D.; Bonnefoy-Cudraz, E.; Guerin, P.; Elbaz, M.; Delarche, N.; Coste, P.; Vanzetto, G.; Metge, M.; Aupetit, J. F.; Jouve, B.; Motreff, P.; Tron, C.; Labeque, J. N.; Steg, P. G.; Cottin, Y.; Range, G.; Clerc, J.; Claeys, M. J.; Coussement, P.; Prunier, F.; Moulin, F.; Roth, O.; Belle, L.; Dubois, P.; Barragan, P.; Gilard, M.; Piot, C.; Colin, P.; De Poli, F.; Morice, M. C.; Ider, O.; Dubois-Rande, J. L.; Unterseeh, T.; Le Breton, H.; Beard, T.; Blanchard, D.; Grollier, G.; Malquarti, V.; Staat, P.; Sudre, A.; Elmer, E.; Hansson, M. J.; Bergerot, C.; Boussaha, I.; Jossan, C.; Derumeaux, G.; Mewton, N.; Ovize, M. Cyclosporine before PCI in Patients with Acute Myocardial Infarction. N Engl J Med 2015, 373(11), 1021–31. [Google Scholar] [CrossRef] [PubMed]
- Ottani, F.; Latini, R.; Staszewsky, L.; La Vecchia, L.; Locuratolo, N.; Sicuro, M.; Masson, S.; Barlera, S.; Milani, V.; Lombardi, M.; Costalunga, A.; Mollichelli, N.; Santarelli, A.; De Cesare, N.; Sganzerla, P.; Boi, A.; Maggioni, A. P.; Limbruno, U.; Investigators, C. Cyclosporine A in Reperfused Myocardial Infarction: The Multicenter, Controlled, Open-Label CYCLE Trial. J Am Coll Cardiol 2016, 67(4), 365–374. [Google Scholar] [CrossRef]
- O'Neill, W. W.; Martin, J. L.; Dixon, S. R.; Bartorelli, A. L.; Trabattoni, D.; Oemrawsingh, P. V.; Atsma, D. E.; Chang, M.; Marquardt, W.; Oh, J. K.; Krucoff, M. W.; Gibbons, R. J.; Spears, J. R.; Investigators, A. Acute Myocardial Infarction with Hyperoxemic Therapy (AMIHOT): a prospective, randomized trial of intracoronary hyperoxemic reperfusion after percutaneous coronary intervention. J Am Coll Cardiol 2007, 50(5), 397–405. [Google Scholar] [CrossRef]
- Stone, G. W.; Martin, J. L.; de Boer, M. J.; Margheri, M.; Bramucci, E.; Blankenship, J. C.; Metzger, D. C.; Gibbons, R. J.; Lindsay, B. S.; Weiner, B. H.; Lansky, A. J.; Krucoff, M. W.; Fahy, M.; Boscardin, W. J.; Investigators, A.-I. T. Effect of supersaturated oxygen delivery on infarct size after percutaneous coronary intervention in acute myocardial infarction. Circ Cardiovasc Interv 2009, 2(5), 366–75. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, R.; James, S. K.; Jernberg, T.; Lindahl, B.; Erlinge, D.; Witt, N.; Arefalk, G.; Frick, M.; Alfredsson, J.; Nilsson, L.; Ravn-Fischer, A.; Omerovic, E.; Kellerth, T.; Sparv, D.; Ekelund, U.; Linder, R.; Ekstrom, M.; Lauermann, J.; Haaga, U.; Pernow, J.; Ostlund, O.; Herlitz, J.; Svensson, L.; Investigators, D. X. S. Oxygen Therapy in Suspected Acute Myocardial Infarction. N Engl J Med 2017, 377(13), 1240–1249. [Google Scholar] [CrossRef]
- David, S. W.; Khan, Z. A.; Patel, N. C.; Metzger, D. C.; Wood, F. O.; Wasserman, H. S.; Lotfi, A. S.; Hanson, I. D.; Dixon, S. R.; LaLonde, T. A.; Genereux, P.; Ozan, M. O.; Maehara, A.; Stone, G. W. Evaluation of intracoronary hyperoxemic oxygen therapy in acute anterior myocardial infarction: The IC-HOT study. Catheter Cardiovasc Interv 2019, 93(5), 882–890. [Google Scholar] [CrossRef]
- Erlinge, D.; Gotberg, M.; Lang, I.; Holzer, M.; Noc, M.; Clemmensen, P.; Jensen, U.; Metzler, B.; James, S.; Botker, H. E.; Omerovic, E.; Engblom, H.; Carlsson, M.; Arheden, H.; Ostlund, O.; Wallentin, L.; Harnek, J.; Olivecrona, G. K. Rapid endovascular catheter core cooling combined with cold saline as an adjunct to percutaneous coronary intervention for the treatment of acute myocardial infarction. The CHILL-MI trial: a randomized controlled study of the use of central venous catheter core cooling combined with cold saline as an adjunct to percutaneous coronary intervention for the treatment of acute myocardial infarction. J Am Coll Cardiol 2014, 63(18), 1857–65. [Google Scholar] [PubMed]
- Nichol, G.; Strickland, W.; Shavelle, D.; Maehara, A.; Ben-Yehuda, O.; Genereux, P.; Dressler, O.; Parvataneni, R.; Nichols, M.; McPherson, J.; Barbeau, G.; Laddu, A.; Elrod, J. A.; Tully, G. W.; Ivanhoe, R.; Stone, G. W.; Investigators, V. Prospective, multicenter, randomized, controlled pilot trial of peritoneal hypothermia in patients with ST-segment- elevation myocardial infarction. Circ Cardiovasc Interv 2015, 8(3), e001965. [Google Scholar] [CrossRef] [PubMed]
- Keeble, T. R.; Karamasis, G. V.; Noc, M.; Sredniawa, B.; Aradi, D.; Neskovic, A. N.; Arheden, H.; Erlinge, D.; Holzer, M. Effect of Intravascular Cooling on Microvascular Obstruction (MVO) in Conscious Patients with ST-Elevation Myocardial Infarction Undergoing Primary PCI: Results from the COOL AMI EU Pilot Study. Cardiovasc Revasc Med 2019, 20(9), 799–804. [Google Scholar] [CrossRef] [PubMed]
- El Farissi, M.; Keulards, D. C. J.; van 't Veer, M.; Zelis, J. M.; Berry, C.; De Bruyne, B.; Engstrom, T.; Frobert, O.; Piroth, Z.; Oldroyd, K. G.; Tonino, P. A. L.; Pijls, N. H. J.; Otterspoor, L. C. Selective intracoronary hypothermia in patients with ST-elevation myocardial infarction. Rationale and design of the EURO-ICE trial. EuroIntervention 2021, 16(17), 1444–1446. [Google Scholar] [CrossRef] [PubMed]
- El Farissi, M.; Good, R.; Engstrom, T.; Oldroyd, K. G.; Karamasis, G. V.; Vlaar, P. J.; Lonborg, J. T.; Teeuwen, K.; Keeble, T. R.; Mangion, K.; De Bruyne, B.; Frobert, O.; De Vos, A.; Zwart, B.; Snijder, R. J. R.; Brueren, G. R. G.; Palmers, P. J.; Wijnbergen, I. F.; Berry, C.; Tonino, P. A. L.; Otterspoor, L. C.; Pijls, N. H. J. Safety of Selective Intracoronary Hypothermia During Primary Percutaneous Coronary Intervention in Patients With Anterior STEMI. JACC Cardiovasc Interv 2021, 14(18), 2047–2055. [Google Scholar] [CrossRef]
- Kitakaze, M.; Asakura, M.; Kim, J.; Shintani, Y.; Asanuma, H.; Hamasaki, T.; Seguchi, O.; Myoishi, M.; Minamino, T.; Ohara, T.; Nagai, Y.; Nanto, S.; Watanabe, K.; Fukuzawa, S.; Hirayama, A.; Nakamura, N.; Kimura, K.; Fujii, K.; Ishihara, M.; Saito, Y.; Tomoike, H.; Kitamura, S.; investigators, J. W. Human atrial natriuretic peptide and nicorandil as adjuncts to reperfusion treatment for acute myocardial infarction (J-WIND): two randomised trials. Lancet 2007, 370(9597), 1483–93. [Google Scholar] [CrossRef]
- Lonborg, J.; Vejlstrup, N.; Kelbaek, H.; Botker, H. E.; Kim, W. Y.; Mathiasen, A. B.; Jorgensen, E.; Helqvist, S.; Saunamaki, K.; Clemmensen, P.; Holmvang, L.; Thuesen, L.; Krusell, L. R.; Jensen, J. S.; Kober, L.; Treiman, M.; Holst, J. J.; Engstrom, T. Exenatide reduces reperfusion injury in patients with ST-segment elevation myocardial infarction. European Heart Journal 2012, 33(12), 1491–1499. [Google Scholar] [CrossRef]
- Woo, J. S.; Kim, W.; Ha, S. J.; Kim, J. B.; Kim, S. J.; Kim, W. S.; Seon, H. J.; Kim, K. S. Cardioprotective Effects of Exenatide in Patients With ST-Segment-Elevation Myocardial Infarction Undergoing Primary Percutaneous Coronary Intervention Results of Exenatide Myocardial Protection in Revascularization Study. Arterioscl Throm Vas 2013, 33(9), 2252–2260. [Google Scholar] [CrossRef]
- Roos, S. T.; Timmers, L.; Biesbroek, P. S.; Nijveldt, R.; Kamp, O.; van Rossum, A. C.; van Hout, G. P. J.; Stella, P. R.; Doevendans, P. A.; Knaapen, P.; Velthuis, B. K.; van Royen, N.; Voskuil, M.; Nap, A.; Appelman, Y. No benefit of additional treatment with exenatide in patients with an acute myocardial infarction. International Journal of Cardiology 2016, 220, 809–814. [Google Scholar] [CrossRef]
- Iwakura, K.; Ito, H.; Okamura, A.; Koyama, Y.; Date, M.; Higuchi, Y.; Inoue, K.; Kimura, R.; Nagai, H.; Imai, M.; Toyoshima, Y.; Ozawa, M.; Ito, N.; Okazaki, Y.; Shibuya, M.; Suenaga, H.; Kubota, A.; Fujii, K. Nicorandil Treatment in Patients With Acute Myocardial Infarction - A Meta-Analysis. Circ J 2009, 73(5), 925–931. [Google Scholar] [CrossRef]
- Dirksen, M. T.; Laarman, G.; van 't Hof, A. W. J.; Guagliumi, G.; Tonino, W. A. L.; Tavazzi, L.; Duncker, D. J. G. M.; Simoons, M. L.; Investigators, P.-M. The effect of ITF-1697 on reperfusion in patients undergoing primary angioplasty - Safety and efficacy of a novel tetrapeptide, ITF-1697. European Heart Journal 2004, 25(5), 392–400. [Google Scholar] [CrossRef]
- Lonborg, J.; Holmvang, L.; Kelbaek, H.; Vejlstrup, N.; Jorgensen, E.; Helqvist, S.; Saunamaki, K.; Clemmensen, P.; Treiman, M.; Jensen, J. S.; Engstrom, T. ST-Segment resolution and clinical outcome with ischemic postconditioning and comparison to magnetic resonance. Am Heart J 2010, 160(6), 1085–91. [Google Scholar] [CrossRef]
- Thuny, F.; Lairez, O.; Roubille, F.; Mewton, N.; Rioufol, G.; Sportouch, C.; Sanchez, I.; Bergerot, C.; Thibault, H.; Cung, T. T.; Finet, G.; Argaud, L.; Revel, D.; Derumeaux, G.; Bonnefoy-Cudraz, E.; Elbaz, M.; Piot, C.; Ovize, M.; Croisille, P. Post-conditioning reduces infarct size and edema in patients with ST-segment elevation myocardial infarction. J Am Coll Cardiol 2012, 59(24), 2175–81. [Google Scholar] [CrossRef]
- Khalili, H.; Patel, V. G.; Mayo, H. G.; de Lemos, J. A.; Brilakis, E. S.; Banerjee, S.; Bavry, A. A.; Bhatt, D. L.; Kumbhani, D. J. Surrogate and Clinical Outcomes Following Ischemic Postconditioning During Primary Percutaneous Coronary Intervention of ST-Segment Elevation Myocardial Infarction: A Meta-Analysis of 15 Randomized Trials. Catheter Cardio Inte 2014, 84(6), 978–986. [Google Scholar] [CrossRef] [PubMed]
- Engstrom, T.; Kelbaek, H.; Helqvist, S.; Hofsten, D. E.; Klovgaard, L.; Clemmensen, P.; Holmvang, L.; Jorgensen, E.; Pedersen, F.; Saunamaki, K.; Ravkilde, J.; Tilsted, H. H.; Villadsen, A.; Aaroe, J.; Jensen, S. E.; Raungaard, B.; Botker, H. E.; Terkelsen, C. J.; Maeng, M.; Kaltoft, A.; Krusell, L. R.; Jensen, L. O.; Veien, K. T.; Kofoed, K. F.; Torp-Pedersen, C.; Kyhl, K.; Nepper-Christensen, L.; Treiman, M.; Vejlstrup, N.; Ahtarovski, K.; Lonborg, J.; Kober, L.; T, T. D. S. O. A. Effect of Ischemic Postconditioning During Primary Percutaneous Coronary Intervention for Patients With ST-Segment Elevation Myocardial Infarction A Randomized Clinical Trial. Jama Cardiol 2017, 2(5), 490–497. [Google Scholar] [CrossRef]
- McLeod, S. L.; Iansavichene, A.; Cheskes, S. Remote Ischemic Perconditioning to Reduce Reperfusion Injury During Acute ST-Segment-Elevation Myocardial Infarction: A Systematic Review and Meta-Analysis. Journal of the American Heart Association 2017, 6(5). [Google Scholar] [CrossRef]
- Elbadawi, A.; Ha, L. D.; Abuzaid, A. S.; Crimi, G.; Azzouz, M. S. Meta-Analysis of Randomized Trials on Remote Ischemic Conditioning During Primary Percutaneous Coronary Intervention in Patients With ST-Segment Elevation Myocardial Infarction. American Journal of Cardiology 2017, 119(6), 832–838. [Google Scholar] [CrossRef] [PubMed]
- Eitel, I.; Stiermaier, T.; Rommel, K. P.; Fuernau, G.; Sandri, M.; Mangner, N.; Linke, A.; Erbs, S.; Lurz, P.; Boudriot, E.; Mende, M.; Desch, S.; Schuler, G.; Thiele, H. Cardioprotection by combined intrahospital remote ischaemic perconditioning and postconditioning in ST-elevation myocardial infarction: the randomized LIPSIA CONDITIONING trial. European Heart Journal 2015, 36(44), 3049–3057. [Google Scholar] [CrossRef]
- Stiermaier, T.; Jensen, J. O.; Rommel, K. P.; de Waha-Thiele, S.; Fuernau, G.; Desch, S.; Thiele, H.; Eitel, I. Combined Intrahospital Remote Ischemic Perconditioning and Postconditioning Improves Clinical Outcome in ST-Elevation Myocardial Infarction. Circ Res 2019, 124(10), 1482–1491. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D. J.; Kharbanda, R. K.; Moller, U. K.; Ramlall, M.; Aaroe, J.; Butler, R.; Bulluck, H.; Clayton, T.; Dana, A.; Dodd, M.; Engstrom, T.; Evans, R.; Lassen, J. F.; Christensen, E. F.; Garcia-Ruiz, J. M.; Gorog, D. A.; Hjort, J.; Houghton, R. F.; Ibanez, B.; Knight, R.; Lippert, F. K.; Lonborg, J. T.; Maeng, M.; Milasinovic, D.; More, R.; Nicholas, J. M.; Jensen, L. O.; Perkins, A.; Radovanovic, N.; Rakhit, R. D.; Ravkilde, J.; Ryding, A. D.; Schmidt, M. R.; Riddervold, I. S.; Sorensen, H. T.; Stankovic, G.; Varma, M.; Webb, I.; Terkelsen, C. J.; Greenwood, J. P.; Yellon, D. M.; Botker, H. E.; Investigators, C.-E.-P. Effect of remote ischaemic conditioning on clinical outcomes in patients with acute myocardial infarction (CONDI-2/ERIC-PPCI): a single-blind randomised controlled trial. Lancet 2019, 394(10207), 1415–1424. [Google Scholar] [CrossRef]
Figure 1.
Pathophysiological mechanisms of microvascular obstruction and coronary no-reflow. Pathophysiological events that develop at epicardial coronary artery and microcirculation (capillary) levels are shown. Black arrows denote forces generated outside the capillaries that compress microcirculation. Numbers show the following components of microvascular obstruction: 1, atherothrombotic debris; 2, blebs; 3, neutrophil aggregates; 4, fibrin; 5, platelet aggregates; 6, neutrophil extracellular traps (NETs); 7, monocytes; 8; erythrocyte aggregates; 9, inflammasome.
Figure 1.
Pathophysiological mechanisms of microvascular obstruction and coronary no-reflow. Pathophysiological events that develop at epicardial coronary artery and microcirculation (capillary) levels are shown. Black arrows denote forces generated outside the capillaries that compress microcirculation. Numbers show the following components of microvascular obstruction: 1, atherothrombotic debris; 2, blebs; 3, neutrophil aggregates; 4, fibrin; 5, platelet aggregates; 6, neutrophil extracellular traps (NETs); 7, monocytes; 8; erythrocyte aggregates; 9, inflammasome.
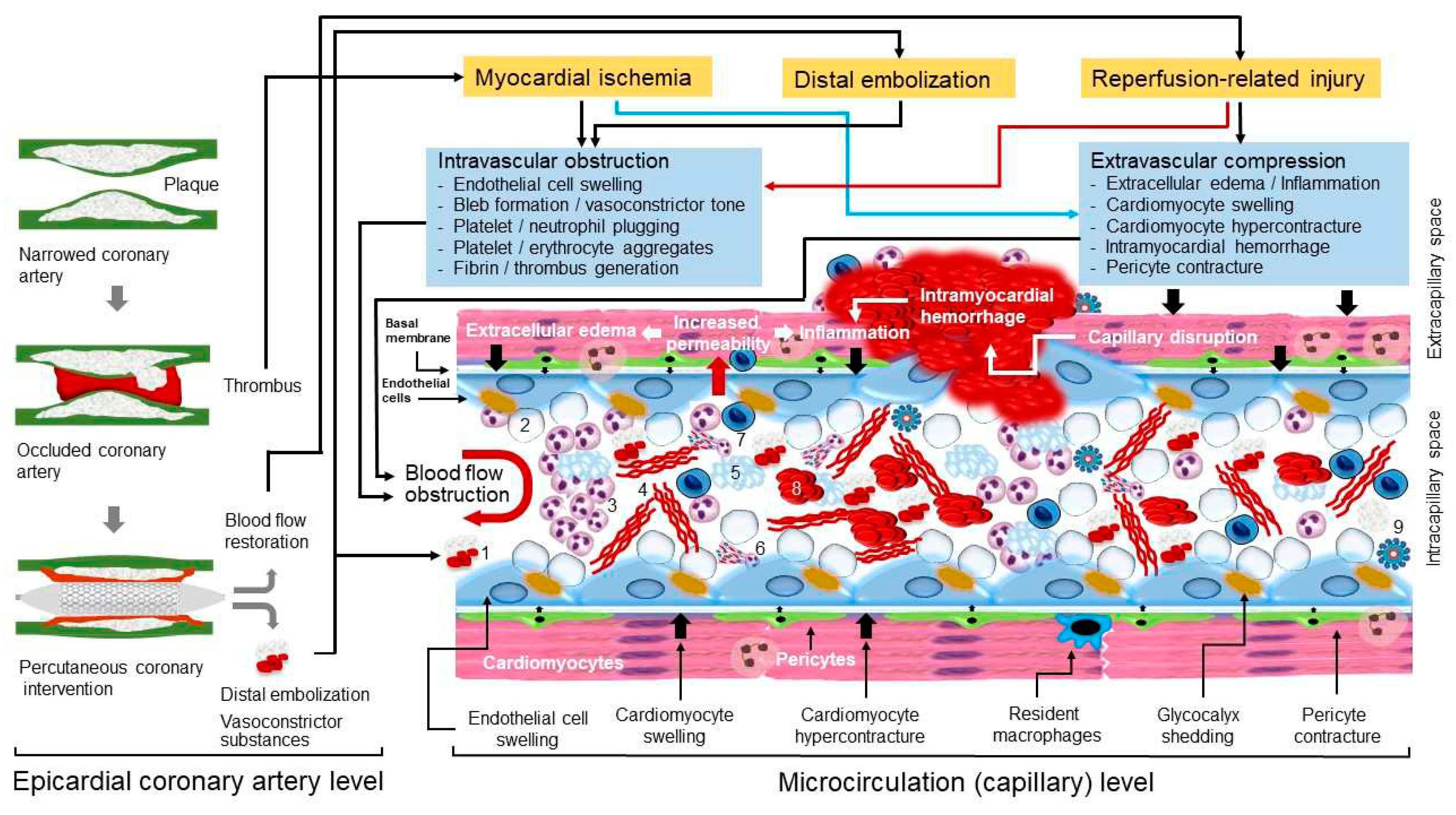
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



