Submitted:
07 July 2023
Posted:
10 July 2023
You are already at the latest version
Abstract
Huntington's disease (HD) is caused by an expansion of a CAG repeat in the gene that encodes the huntingtin protein (HTT). The exact function of HTT is still not fully understood, and previous studies have mainly focused on identifying proteins that interact with HTT to gain insights into its function. Numerous HTT-interacting proteins have been discovered, shedding light on the functions and structure of HTT. Most of these proteins interact with the N-terminal region of HTT. Among the various HTT-interacting proteins, huntingtin-associated protein 1 (HAP1) and HTT-interacting protein 1 (HIP1) have been extensively studied. Recent research has uncovered differences in the distribution of HAP1 in monkey and human brains compared to mice. This finding suggests that there may be species-specific variations in the regulation and function of HTT-interacting proteins. Understanding these differences could provide crucial insights into the development of HD. In this review, we will focus on the recent advancements in the study of HTT-interacting proteins, with particular attention to the differential distributions of HTT and HAP1 in larger animal models.
Keywords:
Huntingtin
; Protein interaction
; Huntington’s disease
; Polyglutamine
Introduction
Huntington's disease (HD) is an autosomal dominant inherited neurodegenerative disease. The primary cause of HD is the CAG expansion mutation of the huntingtin (HTT) gene, which is located on the fourth chromosome in humans, resulting in the clinical manifestation of HD [1]. In normal healthy individuals, the number of CAG repeats is typically less than 35, whereas individuals with HD have 36 or more CAG repeats. The range of CAG repeats in the majority of HD patients is usually between 40 and 50 and cause adult-onset symptoms. However, when the HTT gene has more than 60 CAG repeats, it typically leads to the onset of the disease during childhood or adolescence [1]. It is important to note that the earlier the disease onset, the longer the CAG repeat tends to be.
The CAG repeats in the HTT gene encode a polyglutamine (polyQ) tract such that expanded CAG repeats result in an expansion of a polyQ repeat in HTT. The pathological feature of HD is the preferential loss of medium spiny neurons (MSN) in the striatum, and neurodegeneration spreads to other regions of the brain as the disease progresses. The neurodegeneration causes dance-like involuntary movements, and as the disease advances, individuals gradually lose their ability to speak, perform daily routine tasks, and think. HD continues to progress over approximate 10 to 20 years, eventually leading to the death of the patient.
Protein-protein interactions are crucial for numerous cellular functions, and any malfunction or disruption in these interactions has been implicated in various pathological conditions [3]. HTT engages in interactions with a wide range of proteins. It serves to maintain an intracellular protein network and potentially plays vital roles in intracellular transportation or acts as a cytoskeleton scaffold [4,5]. Mutations in HTT can not only affect its own function but also impair the function of other proteins that interact with it. Therefore, studying the proteins that interact with HTT is of significant importance [4]. Investigating HTT-interacting proteins can enhance our understanding of the pathogenic mechanism of HD and may open up new avenues for the treatment of the disease.
In this review, we will provide an overview of the proteins that interact with HTT, which can be influenced by the polyQ numbers or its function may be important for HD development, and have been extensively investigated. Additionally, we will concentrate on the recent discoveries related to huntingtin-associated protein 1 (HAP1), with the objective of gaining new insights into the mechanisms underlying the pathogenesis of HD.
Structure of HTT and expression of mutant HTT
HTT, a soluble protein consisting of 3144 amino acids (348 kDa), is primarily expressed in the central nervous system and testis, particularly in the cerebral cortex of the brain [6]. The N-terminal 17 amino acids, also known as the N17 region, have been identified as a crucial region involved in HTT's localization, aggregation, and toxicity [7]. Immediately following the N17 region is the polyQ region, which contains up to 35 CAG repeats in individuals without HD. Subsequent to the polyQ stretch is a polyproline-rich region [8], known to interact with various proteins containing the SH3 domain [9]. After the polyproline-rich region, there are the HEAT repeat clusters (α-helix-loop-α-helix motif), which play a vital role in mediating interactions between HTT and other proteins.
Cytoplasmic retention signals and nuclear export signals have been identified at the N-terminus and C-terminus of HTT, respectively. The amino acid sequence of HTT contains significant post-translational modification sites, such as ubiquitination/sumoylation, phosphorylation, palmitoylation, acetylation, and proteolysis [10]. Many of these modification sites are located within or near four regions known as PEST domains, which consist of proline-rich, glutamic acid, serine, threonine, and predicted cleavage sites [11].
The expansion of CAG repeats in exon 1 of the HTT gene leads to the translation of a mutant HTT protein with an enlarged polyglutamine (PolyQ) tract in its N-terminal region. The major toxic form of HTT is believed to be the N-terminal fragments carrying the expanded polyQ, which can result from aberrant splicing [12] or proteolytic processes [13]. These mutant N-terminal fragments can enter the nucleus and become retained there through self-aggregation or oligomerization. Large aggregates form inclusion bodies, and HTT aggregation can disrupt the balance of interacting proteins and may be toxic. The toxic effects of mutant HTT include transcriptional dysregulation, synaptic dysfunction, mitochondrial toxicity, and impaired axonal transport [14].
By understanding the proteins that interact with HTT and contribute to its toxic effects, researchers can gain valuable insights into the molecular pathways involved in HD. The identification of HTT-interacting proteins involved in HD pathogenesis is crucial for defining the underlying mechanisms of the disease and finding potential targets for treatment.
HTT interacting proteins
The functions of HTT are still not fully understood. The interaction between HTT and other proteins influences the state, function, and other aspects of HTT and its interacting proteins, leading to related physiological changes [15]. Mutant HTT exists in at least two forms: diffuse (soluble) and aggregated (insoluble), both of which are cytotoxic [16]. The soluble form of mutant HTT can be cleared by the cellular metabolic system, such as the proteasome. However, the aggregated insoluble form of mutant HTT has reduced solubility, is highly compact, and is difficultly cleared by the proteasome. As a result, it accumulates and aggregates over time [17].
HTT-interacting proteins can bind to either normal or mutant HTT. The binding affinity of certain HTT-interacting proteins increases as the polyQ number (number of glutamine repeats) in mutant HTT increases, while other proteins show reduced binding affinity with increasing polyQ number (Table 1). We will select a few examples for further discussion.
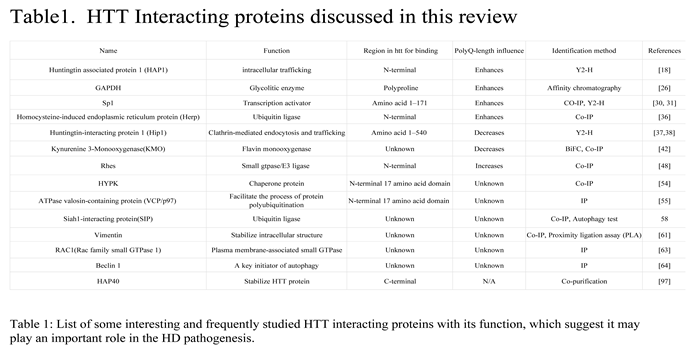
1.1. Proteins binding to HTT enhanced by Poly Q expansion
Huntingtin-associated protein 1 (HAP1)
HAP1, a neuronal protein, was initially discovered as the first Huntingtin interacting protein through the use of the yeast two-hybrid system (Y2H) [18]. Since its discovery, HAP1 has been extensively studied as one of the key proteins involved in HTT interactions. In rodents, HAP1 exists in two alternative spliced subtypes, which differ only at the very C-terminal end (amino acids 579-599 in HAP1A and 579-629 in HAP1B) [18,19]. Notably, HAP1 exhibits a stronger binding affinity to HTT with an expanded glutamine repeat compared to wild-type HTT [18,19]. HAP1 is predominantly expressed in the hypothalamus and has been found to interact with various proteins involved in intracellular trafficking, receptor function, transcription factors, cytoskeleton proteins, and even forms dimers and octamers through self-interaction [19,20]. Its role in intracellular transport/trafficking and receptor endocytosis has been well established [19,20]. Recent studies have also highlighted the protective role of HAP1 against mutant HTT (mHtt) accumulation, as knockdown of HAP1 in the presence of mHtt leads to neuronal cell death [21]. Both HAP1 and HTT are implicated in intracellular trafficking in neuronal cells, as they both interact with the microtubule machinery [17,20]. However, HAP1 is also involved in other cellular processes such as gene transcriptional regulation and signal transduction [22,23]. It is plausible that the interaction between HTT and HAP1 plays a crucial role in various cellular functions, and disruption of HTT mutation affects the function of both proteins.
Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH)
GAPDH, also known as glyceraldehyde-3-phosphate dehydrogenase, exhibits both glyceraldehyde-3-phosphate dehydrogenase and nitrosylase activities [24], making it involved in glycolysis and other cellular functions [25]. The reports on the interaction between mutant HTT (mHTT) and GAPDH are somehow inconsistent. Some studies have indicated that the polyQ repeats in the HTT protein specifically interact with GAPDH, and this interaction is enhanced with longer polyQ stretches [26,27]. On the other hand, other research has shown that mutant HTT binds to GAPDH through its interaction with Siah1, an E3 ligase, resulting in cell death [28]. Furthermore, mHtt with different N-terminal polymers disrupts GAPDH-mediated micro-mitophagy, leading to the accumulation of damaged mitochondria in cells. This impairment of micro-mitophagy may contribute to the pathological events observed in HD [28].
Transcription factor Sp1 (Sp1)
Sp1 is a transcription factor that can activate or repress transcription in response to physiological and pathological stimuli. It binds to GC-rich DNA motifs with high affinity and regulates the expression of numerous genes involved in various processes, including cell growth, apoptosis, cellular differentiation, and immune responses [29]. Previous studies have shown that HTT interacts with Sp1, and the expansion of polyglutamine enhances this interaction [30]. The binding of mHTT to Sp1 impairs its function, resulting in the inhibition of nuclear Sp1 binding to the promoter of the nerve growth factor receptor. This leads to reduced transcriptional activity in cultured cells or disrupts the coordinated transcriptional activity of Sp1 and TAFII130 in HD mouse models or patients with HD [31].
Another study investigating astroglial-specific mHtt expression demonstrated that mHtt reduces the expression of the glutamate transporter in astrocytes. This is achieved by increasing its binding to Sp1, thereby reducing the binding of Sp1 to the glutamate transporter promoter. Consequently, this causes glutamate excitotoxicity and neuronal death [32]. Other studies have found functional Sp1 responsive elements in the HTT gene promoter region. The expression of Sp1 enhances the transcription of HTT, while inhibiting Sp1-mediated transcription reduces the expression of mHtt [33]. Additionally, increased SP1 activity has been reported in cellular and transgenic HD mouse models [34]. The downstream effects of the interaction between Sp1 and mHtt are complex. The imbalance in Sp1-mediated HTT transcription, combined with the adverse effects of mHtt on downstream SP1-dependent gene expression, may contribute to the pathogenesis of HD at different disease stages.
Homocysteine-induced endoplasmic reticulum protein (Herp)
Herp, an E3 ubiquitin ligase, is the integral membrane protein regulated by the ER stress response pathway. It is involved in the ubiquitination and degradation of many mutant proteins [35]. It was found that Herp can bind to the overexpressed HTT N-terminus in HTT-transfected N2a cells, and this interaction is enhanced with the polyQ expanded HTT N-terminal fragments [36]. Mutant HTT transgenic mice show increased Herp expression in their brain regions, and Herp reduces the cytotoxicity of mHtt by inhibiting its aggregation and promoting its degradation [36].
Herp's N-terminal acts as a molecular chaperone, inhibiting protein aggregation, while its C-terminal acts as an E3 ubiquitin ligase, promoting the degradation of misfolded proteins through UPP [36]. Understanding the mechanisms by which Herp regulates HTT degradation and aggregation could have significant implications for the development of therapeutic strategies for protein misfolding diseases, such as HD. By targeting Herp or its associated pathways, it may be possible to enhance the clearance of misfolded proteins and alleviate the cytotoxic effects associated with their accumulation. Further research in this area is warranted to explore the full potential of Herp as a therapeutic target in HD.
2.2. Proteins binding to HTT inhibited by PolyQ expansion
Huntingtin-interacting protein 1 (Hip1)
Apart from HAP1, Hip1 is also one of the most extensively studied proteins that interact with HTT. The binding affinity between HTT and HIP1 decreases as the number of polyglutamine residues exceeds the pathogenic threshold [37,38]. HIP1 is a membrane-associated protein that plays a role in clathrin-mediated endocytosis and intracellular protein trafficking [39]. HIP1 exists in multiple variants, each with its own specific function, and they also work together to fulfill their roles.
In HD, the disruption of the normal HTT-HIP1 interaction may result in cytoskeletal defects and impaired membrane receptor trafficking in the brain, ultimately leading to neuronal cell death [39]. In addition to its interaction with HTT, HIP1 also interacts with other proteins and has been implicated in various medical conditions, including tumor genesis such as lung cancer and leukemia. HIP1 is also involved in the development of rheumatoid arthritis by modulating the PDGFR and Rac1 signaling pathways [40]. Furthermore, it has been reported that patients with epilepsy exhibit a deletion of the chromosome segment that contains HIP1 [41]. It is worth noting that HTT-interacting proteins have multiple other functions, which may be affected by mHTT.
Kynurenine 3-Monooxygenase (KMO)
Kynurenine 3-Monooxygenase (KMO) is another protein that interacts with normal HTT, but its interaction with mHTT is reduced [42]. Studies have demonstrated that KMO physically interacts with soluble HTT exon 1 protein fragments within living cells, primarily at the mitochondrial outer membrane [42]. Specifically, the expansion of the pathogenic polyglutamine tract in HTT leads to the formation of intracellular protein inclusions, which disrupts the interaction with KMO [42]. KMO is located in the mitochondrial outer membrane and is responsible for catalyzing the synthesis of 3-hydroxykynurenine from L-kynurenine [43]. It plays a crucial role in the degradation of tryptophan through the kynurenine pathway (KP), controlling the balance between the formation of kynurenic acid (KYNA) and the synthesis of downstream neurotoxic metabolites [44]. The neuroactive metabolites produced by the tryptophan-degrading enzyme kynurenine pathway (KP) are implicated in the pathophysiological processes of neurodegenerative diseases, including HD [44,45]. Furthermore, KMO has been reported to play important roles in renal disease and cancer progression [46], as well as cancer development [47]. Given its significance in cellular processes, KMO's interaction with HTT may regulate mitochondrial function. Further studies are required to delve into the intricacies of KMO-HTT interaction biology and comprehend their implications for HD pathogenesis.
Interacting protein that can enhance the mHTT toxicity
HTT-interacting proteins originate from various sources, some of which can reduce mHTT toxicity and protect neuronal cells. For instance, HAP1(21) has been shown to have these beneficial effects. On the other hand, there are proteins like Rhes [48] that enhance mHTT toxicity and lead to cell death. Rhes belongs to the Ras superfamily of small GTPases and functions as an E3 ligase, facilitating the attachment of small ubiquitin-like modifier (SUMO) to its targets. It has been found that Rhes initiates mHTT sumolation, resulting in the production of more soluble mHtt, which is believed to be more toxic than the aggregated form of mHtt [48].
Rhes is primarily found in the striatum, and knocking down Rhes has been shown to protect striatal neuronal cell death in an 3'-NP-induced HD mouse model [49]. Additionally, Rhes has been found to regulate autophagy activity in an HD knock-in mouse model [50,51]. Given that striatal neuronal death is selective in HD and Rhes is enriched in this brain region with a function that is crucial for protein aggregation-related diseases, Rhes emerges as a strong candidate for linking to HD pathogenesis. However, it is important to note that all the intriguing findings regarding the interaction between Rhes and mHtt have been generated using HD mouse models or cell models. It would be interesting to investigate whether these results can be replicated in large animal models of HD.
HTT interacting proteins that can alter mHtt aggregates
Many proteins that interact with HTT have been reported to be capable of altering mHtt aggregation. In this discussion, we will briefly explore some newly discovered HTT-interacting proteins. One such protein is HTT interacting protein K (HYPK), which belongs to the AAA ATPase family. HYPK binds to the first 17 amino acids of the N-terminus of HTT (HTT-N17), inhibiting the formation of mHtt aggregates and reducing neuronal cell death caused by mHtt [52,53]. HYPK's regulation of mHtt aggregation may be attributed to its chaperoning activity [54]. Another protein, VCP, acts as a decomposing enzyme that binds to mHtt and reduces its aggregation [55]. VCP's ability to break down protein aggregates stems from its depolymerizing enzyme function, which may guide the mHtt protein for degradation.
Additionally, Siah1-interacting protein (SIP) also plays a role in regulating mHtt aggregation. SIP is a multi-domain and multi-ligand protein that interacts with various proteins and plays important roles in the cell [56]. Siah1 is a central component of a multiprotein E3 ubiquitin ligase complex, and SIP binds to Siah1 within this complex [57]. Wild-type SIP increases mHtt ubiquitination, reduces mHtt protein levels, and decreases mHtt aggregation. However, an increase in SIP dimerization in HD MSN leads to a decrease in SIP's function in degrading mHtt through the ubiquitin-proteasome pathway, resulting in an increase in mHtt aggregation [58]. Protein interactions have an impact on the aggregation of mHtt, which, in turn, may affect the pathology of HD. Therefore, further research should be conducted to investigate the mechanisms of these protein interactions with mHtt, which could potentially lead to new directions in treating protein aggregation-related neurological disorders.
Proteins with impaired function when interacting with mHtt
Proteins with known functions are valuable targets for studying the mechanisms of HD when their functions are altered by interacting with mHTT. When a protein interacts with mHtt, its own function can also be affected. For example, Xu et al. identified Vimentin as the preferential interaction factor of mHtt [59]. Vimentin normally forms a cage-like structure around aggregates in neural stem cells (NSCs) to maintain cellular proteostasis [60]. However, in HD NSCs, mHtt impairs the ability of Vimentin to form a cage around the aggregates in response to proteotoxic stress, resulting in a decline in the proliferation and neurogenesis of the NSCs [59].
Another example of mHtt affecting the function of an interacting protein is illustrated by its ability to retard the reactivity of Rac1 to BDNF-stimulated growth activity in human NSCs and a mouse HD model [61]. Rac1, a member of the Rho family of GTPases, is an intracellular transducer known to regulate multiple signaling pathways that control cytoskeleton organization, transcription, and cell proliferation [62]. Rac1 is a downstream target of growth factor receptor/PI3 kinase activity and is critical for actin-dependent membrane remodeling in response to growth signaling [62]. HTT normally regulates Rac1 activity as part of a coordinated response to growth factor signaling, but this function is altered by mHtt in the early stages of HD [63].
Beclin 1, another example of impaired function due to interaction with mHtt, is a key component of autophagy [64,65]. Beclin 1 is sequestered by mHtt, and this interaction affects the degradation of long-lived protein aggregates through autophagy. HTT has been reported to potentially serve as a scaffold for selective autophagy [7], but mHtt may lose this function and aberrantly interact with different components of the autophagy system, resulting in impaired autophagy function in various HD models [4,67,68].
New insights in HAP1
HAP1, the first HTT-associated protein identified through yeast two-hybrid screening, has been extensively studied by numerous researchers across various model systems. Notably, several differences have been observed between rodents and large animal models [60,70,71]. As a result, the distribution of HAP1 in rodents and primates has been investigated, along with its potential function in primate brains [72]. In this paragraph, we aim to provide a comprehensive overview of the latest findings for the distribution of HAP1 and HTT. These findings could contribute to a better understanding of HAP1's role in HD and its potential implications for therapeutic interventions.
(1) HAP1 in the primate brains
Human HAP1 (hHAP1) was discovered a few years later through similarity PCR cloning, following the discovery of rat HAP1. It shares 62% identity with rat HAP1 across its entire sequence and 82% amino acid identity in the putative HTT-binding region [73]. Human HAP1 also binds to HTT, and this binding is enhanced by increased polyQ numbers. Interestingly, a previous study found that the expression level of hHAP1 is low in the human brain striatum and is significantly decreased in the HD brain striatum [73]. Some researchers have questioned its role in HD pathogenesis, as rodent Hap1 is primarily enriched in the hypothalamus, which is not the most affected brain region in HD [4,18]. However, since Hap1 has shown a protective effect in HD KI mouse striatum [74], it is worth further investigating whether HAP1 is distributed differently in primate brains compared to rodent brains and if it indeed participates in HD pathogenesis. Mutation of HTT has been found to be fully responsible for causing HD [75], and HTT mRNA and protein were reported to be ubiquitously distributed throughout the body in rodents and humans [76]. Therefore, selective neurodegeneration cannot be fully explained solely by the mutation in HTT. The selective neurodegeneration in HD raises the possibility of mHtt affecting neuronal protein function through abnormal protein-protein interactions and causing specific neuronal cell death [77,78]. In HD, the medium spiny neurons (MSN) in the striatum are the most vulnerable neuronal cells, and brains from patients with HD show severe loss of MSN in the striatum at the late stages of HD [79,80].
In the phenomenon of selective neurodegeneration, when a mutant protein is expressed ubiquitously in all cells, there are two possible explanations for the pathogenesis of the disease. The first is that the specific mutant protein is enriched in the brain region that is most affected. The second is that a protein or proteins with an important function in this brain region are being affected by the mutant protein. In the case of HD, the mutant protein is found to be distributed in all cells, with the highest expression observed in the brain and testis [76,77,78,79].
Following the discovery that the mutant huntingtin (HTT) protein causes HD and is ubiquitously distributed in the body, many researchers began searching for HTT-interacting proteins. One such protein is HAP1, which is enriched in the brain and was the first reported HTT-interacting protein [18]. Subsequently, many other HTT-interacting proteins have been identified, some of which are neuronal-enriched while others are not. Additionally, some interactions are enhanced by polyQ expansion, while others do not show this enhancement [80,81,82]. Many of these studies have been conducted using rodent models, human cell lines, and drosophila models for confirmation. Only a few interactions have been discovered through human tissue studies [80,81,82].
Given that large animal models have physiological functions, anatomical structures, metabolic pathways, and behavioral phenotypes more similar to humans than rodents [83,84], it is important to consider investigation of the disease pathogenesis, particularly in relation to brain diseases, using large animal models.
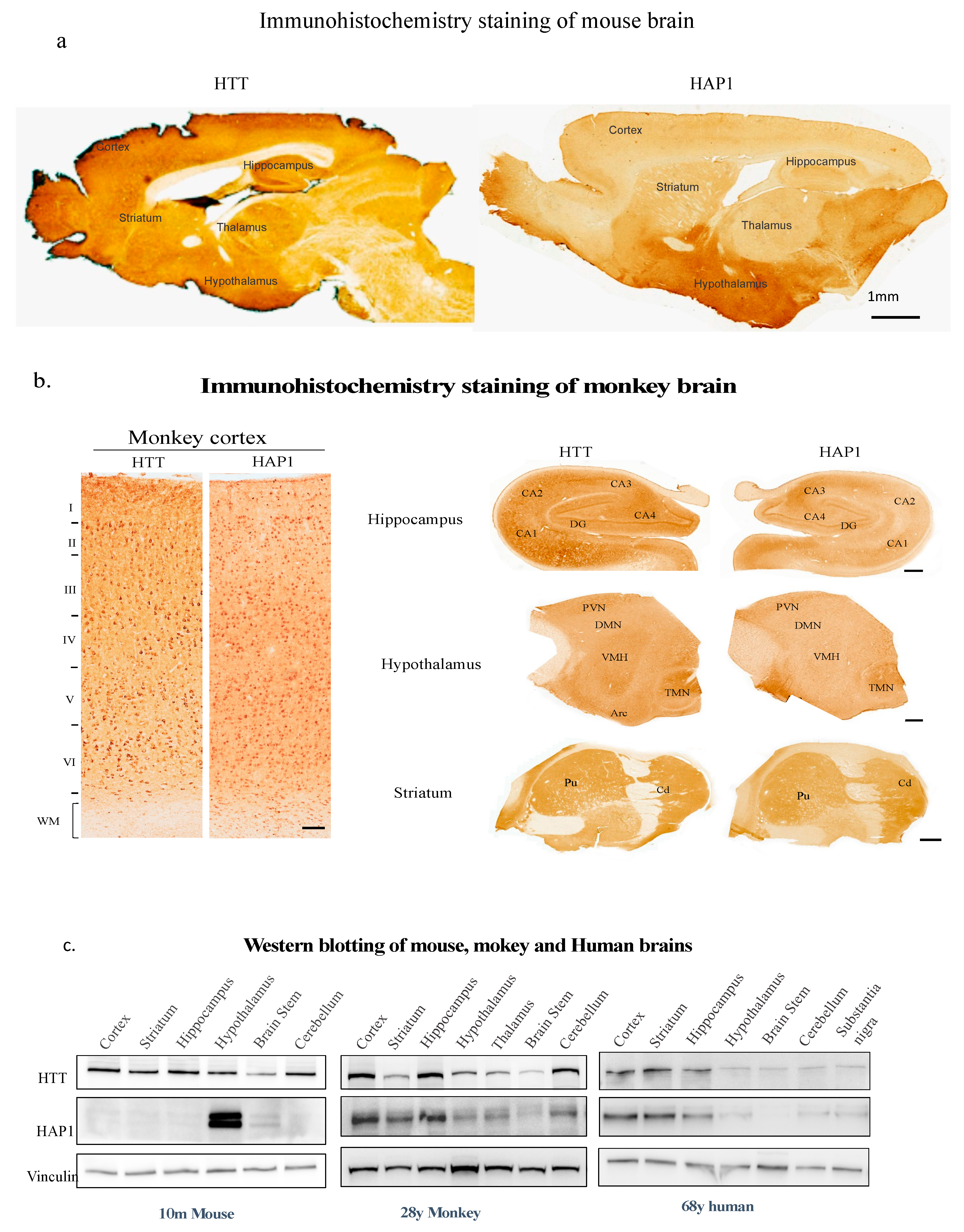
Our recent study compared the expression patterns of primate HAP1 and HTT in monkey brains. The results showed that HAP1 and HTT are both expressed in similar brain areas, including the striatum, cortex, hippocampus, and hypothalamus (Figure 1b) [72]. In contrast to the highly enriched expression of Hap1 in the rodent hypothalamus (Figure 1a and 1c) [18,85], the levels of HAP1 and HTT expression in different regions of the monkey brain are relatively similar, with higher abundance observed in the striatum, cortex, and hippocampus (Figure 1b). Immunofluorescent staining of the monkey brain striatum and cortex revealed that both Hap1 and HTT are expressed in the cytoplasm of the same cells [72]. Furthermore, immunoprecipitation of HAP1 with rabbit anti-hHAP1 from the monkey brain cortex resulted in the significant co-precipitation of HTT. In contrast, there was negligible Hap1 in the mouse cortex that could be precipitated by the HAP1 antibody. These findings indicate that there is a higher presence of HAP1 and HTT in the monkey cortex, where they interact with each other in vivo [72].
The expression patterns of HAP1 in monkey brains differ significantly from those in mice. Additionally, the parallel expression of HAP1 and HTT in primate brains emphasizes the importance of studying these proteins in primate brains. The expression pattern of HAP1 suggests its involvement in the pathogenesis of HD, and the interaction between mHtt and HAP1 may impact the function of these proteins, thereby affecting the pathology of HD. This is especially evident when both proteins are expressed in the same cells in the most vulnerable brain regions [86,87]. It would be interesting to conduct a comprehensive study on HAP1 and HTT using a relevant HD model, such as the HD knock-in pig model [88], or a more precise monkey HD model that can replicate the selective neuropathology observed in human patients.
(1) HAP1 is neuroprotective in monkey tissue
Mutation of a protein can result in either the loss of its own function or the gain of a toxic function through binding to other proteins, which could be its regular interactor or a protein that normally does not interact with the mutant protein. It has been found that both HAP1 and HTT participate in intracellular transport [15,19]. The consequence of this interaction could be that the mutant protein binds more tightly to the interactor and causes its loss of function. Indeed, research has reported impaired intracellular transport in the presence of mHtt [5,89,90].
Proteins that interact with HTT may have a protective role in the intracellular environment, and their deletion may be detrimental to the cells. For instance, in the presence of mutant HTT, deletion of Hap1 caused striatal cell death in the HD KI 140Q mouse model [21], even though the expression level of Hap1 in the mouse brain is not the highest in the striatum [18,85]. This result suggests that Hap1 is protective to mouse striatal neuronal cells, which agrees with previous RNA expression study results [91].
In a large animal model that more closely resembles humans in terms of physiology, anatomy, metabolism, certain proteins might distribute differently and have functions different from those in rodents. Our study showed that HAP1 distributes differently in the monkey brain compared to rodent brains. When we knocked out HAP1 using CRISPR/Cas9 in the monkey brain slice culture with the expression of mHtt, it also caused a significant amount of cell death [72]. This result further confirms that HAP1 indeed participates in the HD pathogenesis in the monkey brain.
(1) Other functions of HAP1 in human
Up to this point, seven isoforms of human HAP1 (hHAP1) transcripts have been documented in the NCBI gene database. These isoforms share a conserved N-terminal region but diverge from the middle of the transcript, suggesting potential alternative splicing events. As a result, these isoforms give rise to seven distinct HAP1 proteins. It has been observed that hHAP1 transcripts are predominantly expressed in the brain and stomach, with higher levels detected in the stomach compared to the brain. Researchers have demonstrated that hHAP1 is localized in the human digestive tract [92].
HAP1 has been implicated in various types of cancer, including gastric cancer [93], acute lymphoblastic leukemia (ALL) [23], breast cancer [94,95], and lung cancer [96]. These findings suggest a potential role for hHAP1 in regulating cell growth and cell death pathways. However, due to the existence of seven isoforms of hHAP1, the distribution of these isoforms within the human body and their specific functions in different systems remain unclear. It is important to note that although rodent HAP1 has been extensively studied, it only presents with two isoforms. Therefore, the distribution and function of rodent HAP1 cannot be directly extrapolated to explain the expression and function of hHAP1.
Conclusion and future remarks
HD is caused by a poly CAG expansion mutation in the HTT gene. HTT was discovered 30 years ago through positional cloning [75]. However, the exact function of HTT is still not fully understood. Knocking out HTT in mice resulted in embryonic death, while humans with large CAG repeat mutations can still be born alive. To understand the selective neuronal cell death in HD, many researchers have conducted studies to identify HTT-interacting proteins that may explain the vulnerability of striatal neurons. Over the past 30 years, the majority of HTT-interacting proteins were found to bind to the N-terminus of the HTT protein, while only a few were found to bind to the very C-terminal, such as HAP40 [97]. HAP40 was recently fund to be complex with wild type HTT to stabilize HTT protein in a HTT cryo-electron microscopy structure study [98]. Interestingly, recent several studies revealed that HAP40 not only bind to HTT C-terminal but also participate in HD pathogenesis [99,100]. It will be interesting to find out how this HTT C-terminal binding protein modulates HD development. The findings from mouse studies and the identification of interacting proteins have greatly contributed to our understanding of the biological function of HTT. Some of these proteins are specific to neurons, such as HAP1 [18], while others are not. Only a small number of proteins were found to be highly expressed in the striatum, such as Rhes [48,49,50,51].
Most of the HTT interacting proteins were found from studying mouse or cell models of HD. So far, none of the HTT-interacting proteins has been shown clearly to participate in the HD pathogenesis or is the only modulator that is directly involved in the HD manifestation. Only a few studies from human HD samples have shown that HTT-interacting proteins can modify the age of onset of HD [101,102]. This could be due to the fact that mHtt affects the functions of many interacting proteins, and HD is a result of the abnormal function of all the affected proteins, which aligns with the theory of a gain of toxic function mutation. Alternatively, HD could result from mHtt actually losing its own function, as suggested by a few studies that have shown mHtt affecting animal development at an early stage [102,103,104]. However, most adult patients with HD do not manifest clinical symptoms until middle age, suggesting that the loss of normal function of HTT may not be the major player in the disease development. A third possibility could be that the interacting proteins behave differently in different animal species due to the significant differences between humans and rodents [69,70]. For example, both HAP1 and HTT are important for the proliferation and differentiation of mouse neuro-stem cells (mNSC), but they function differently in human NSC (hNSC) [72]. The loss of HAP1 and HTT will impact the cell proliferation and neural development of mNSC, and it will also affect the expression of many gene families related to neuronal development in mice [72]. However, in hNSC, the loss of HAP1 does not affect neurogenesis and does not lead to significant changes in gene transcription [72], indicating that HAP1 in primate neurons may not be important for the early development of neurons but may be crucial for maintaining the function of mature neurons. On the other hand, HTT deficiency can affect hNSC neuronal differentiation and development and have a widespread impact on gene transcription [72].
Although HAP1 and HTT function differently in primate neurons for neuronal development, both HAP1 and HTT are expressed in the monkey brain in a very similar pattern and associate with each other. Knocking down HAP1 in monkey brain slice culture resulted in significant neuronal loss in the presence of mHtt. This result suggests that dysfunction of HAP1 may promote mHtt-mediated neurotoxicity in the primate brain, which supports the view that HAP1 dysfunction is associated with age-dependent neurodegeneration in the presence of mHTT in HD [72].
The distinct brain distribution of rodent HAP1 and primate HAP1, as well as the findings from the comparative study on HTT and HAP1 in mouse and primate brains, highlight an important point. Despite the high degree of similarity between rodent and primate genomes, individual proteins can exhibit different distribution patterns and functions in their respective brains. This discrepancy may underlie the significant differences observed between large animal and small animal models when studying human brain disorders [69]. Therefore, it is crucial to consider the disparities between primate and rodent animal models when selecting an appropriate model for studying brain diseases or analyzing the results of human disease studies in animal models. The primate brain HAP1 study serves as just one example of the differences between humans and rodents. Conducting a more detailed comparison of HAP1 or other promising HTT-interacting proteins' distribution between human and primate brains would further validate the current findings.
References
- Tabrizi, S.J.; Flower, M.D.; Ross, C.A.; Wild, E.J. Huntington disease: new insights into molecular pathogenesis and therapeutic opportunities. Nat. Rev. Neurol. 2020, 16, 529–546. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Tabrizi, S.J. Clinical Features of Huntington’s Disease. In Polyglutamine Disorders. Advances in Experimental Medicine and Biology; Nóbrega, C., Pereira de Almeida, L., Eds.; Springer: Cham, Switzerland, 2018; Volume 1049, pp. 1–28. [Google Scholar] [CrossRef]
- Martino, E.; Chiarugi, S.; Margheriti, F.; Garau, G. Mapping, Structure and Modulation of PPI. Front. Chem. 2021, 9, 718405. [Google Scholar] [CrossRef] [PubMed]
- Wanker, E.E.; Ast, A.; Schindler, F.; Trepte, P.; Schnoegl, S. The pathobiology of perturbed mutant huntingtin protein–protein interactions in Huntington's disease. J. Neurochem. 2019, 151, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Saudou, F.; Humbert, S. The Biology of Huntingtin. Neuron 2016, 89, 910–926. [Google Scholar] [CrossRef] [PubMed]
- Parsons, M.P.; Raymond, L.A. Huntington Disease. Neurobiol. Brain Disord. 2015, 303–320. [Google Scholar] [CrossRef]
- Ochaba, J.; Lukacsovich, T.; Csikos, G.; Zheng, S.; Margulis, J.; Salazar, L.; Mao, K.; Lau, A.L.; Yeung, S.Y.; Humbert, S.; et al. Potential function for the Huntingtin protein as a scaffold for selective autophagy. Proc. Natl. Acad. Sci. 2014, 111, 16889–16894. [Google Scholar] [CrossRef]
- Pandey, N.K.; Isas, J.M.; Rawat, A.; Lee, R.V.; Langen, J.; Pandey, P.; Langen, R. The 17-residue-long N terminus in huntingtin controls stepwise aggregation in solution and on membranes via different mechanisms. J. Biol. Chem. 2018, 293, 2597–2605. [Google Scholar] [CrossRef]
- Pigazzini, M.L.; Lawrenz, M.; Margineanu, A.; Schierle, G.S.K.; Kirstein, J. An Expanded Polyproline Domain Maintains Mutant Huntingtin Soluble in vivo and During Aging. Front. Mol. Neurosci. 2021, 14, 721749. [Google Scholar] [CrossRef]
- Gao, Y.-G.; Yan, X.-Z.; Song, A.-X.; Chang, Y.-G.; Gao, X.-C.; Jiang, N.; Zhang, Q.; Hu, H.-Y. Structural Insights into the Specific Binding of Huntingtin Proline-Rich Region with the SH3 and WW Domains. Structure 2006, 14, 1755–1765. [Google Scholar] [CrossRef]
- Arbez, N.; Ratovitski, T.; Roby, E.; Chighladze, E.; Stewart, J.C.; Ren, M.; Wang, X.; Lavery, D.J.; Ross, C.A. Post-translational modifications clustering within proteolytic domains decrease mutant huntingtin toxicity. J. Biol. Chem. 2017, 292, 19238–19249. [Google Scholar] [CrossRef]
- Neueder, A.; Landles, C.; Ghosh, R.; Howland, D.; Myers, R.H.; Faull, R.L.M.; Tabrizi, S.J.; Bates, G.P. The pathogenic exon 1 HTT protein is produced by incomplete splicing in Huntington’s disease patients. Sci. Rep. 2017, 7, 1307. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Yang, S.; Jing, L.; Huang, L.; Chen, L.; Zhao, X.; Yang, W.; Pan, Y.; Yin, P.; Qin, Z.S.; et al. Truncation of mutant huntingtin in knock-in mice demonstrates exon1 huntingtin is a key pathogenic form. Nat. Commun. 2020, 11, 2582. [Google Scholar] [CrossRef] [PubMed]
- Sathasivam, K.; Neueder, A.; Gipson, T.A.; Landles, C.; Benjamin, A.C.; Bondulich, M.K.; Smith, D.L.; Faull, R.L.M.; Roos, R.A.C.; Howland, D.; et al. Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc. Natl. Acad. Sci. 2013, 110, 2366–2370. [Google Scholar] [CrossRef] [PubMed]
- Schulte, J.; Littleton, J.T. The biological function of the Huntingtin protein and its relevance to Huntington’s Disease pathology. Curr. Trends Neurol. 2011, 5, 65–78. [Google Scholar] [PubMed]
- Li, S.-H.; Li, X.-J. Huntington and its Role in Neuronal Degeneration. Neurosci. 2004, 10, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Caron, N.S.; Banos, R.; Yanick, C.; Aly, A.E.; Byrne, L.M.; Smith, E.D.; Xie, Y.; Smith, S.E.; Potluri, N.; Black, H.F.; et al. Mutant Huntingtin Is Cleared from the Brain via Active Mechanisms in Huntington Disease. J. Neurosci. 2021, 41, 780–796. [Google Scholar] [CrossRef]
- Li, X.-J.; Li, S.-H.; Sharp, A.H.; Nucifora, F.C.; Schilling, G.; Lanahan, A.; Worley, P.; Snyder, S.H.; Ross, C.A. A huntingtin-associated protein enriched in brain with implications for pathology. Nature 1995, 378, 398–402. [Google Scholar] [CrossRef]
- Wu, L.L.-Y.; Zhou, X.-F. Huntingtin associated protein 1 and its functions. Cell Adhes. Migr. 2009, 3, 71–76. [Google Scholar] [CrossRef]
- Li, X.-J.; Li, S.-H. HAP1 and intracellular trafficking. Trends Pharmacol. Sci. 2005, 26, 1–3. [Google Scholar] [CrossRef]
- Liu, Q.; Cheng, S.; Yang, H.; Zhu, L.; Pan, Y.; Jing, L.; Tang, B.; Li, S.; Li, X.-J. Loss of Hap1 selectively promotes striatal degeneration in Huntington disease mice. Proc. Natl. Acad. Sci. 2020, 117, 20265–20273. [Google Scholar] [CrossRef]
- Zhao, X.; Chen, A.; Wang, Z.; Xu, X.-H.; Tao, Y. Biological functions and potential therapeutic applications of huntingtin-associated protein 1: progress and prospects. Clin. Transl. Oncol. 2022, 24, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Kang, S.; Wang, X.; Rosales, J.L.; Gao, X.; Byun, H.-G.; Jin, Y.; Fu, S.; Wang, J.; Lee, K.-Y. HAP1 loss confers l-asparaginase resistance in ALL by downregulating the calpain-1-Bid-caspase-3/12 pathway. Blood 2019, 133, 2222–2232. [Google Scholar] [CrossRef] [PubMed]
- Tisdale, E.J. Glyceraldehyde-3-phosphate Dehydrogenase Is Phosphorylated by Protein Kinase Cι/λ and Plays a Role in Microtubule Dynamics in the Early Secretory Pathway. J. Biol. Chem. 2002, 277, 3334–3341. [Google Scholar] [CrossRef] [PubMed]
- Tristan, C.; Shahani, N.; Sedlak, T.W.; Sawa, A. The diverse functions of GAPDH: Views from different subcellular compartments. Cell. Signal. 2011, 23, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Burke, J.R.; Enghild, J.J.; Martin, M.E.; Jou, Y.-S.; Myers, R.M.; Roses, A.D.; Vance, J.M.; Strittmatter, W.J. Huntingtin and DRPLA proteins selectively interact with the enzyme GAPDH. Nat. Med. 1996, 2, 347–350. [Google Scholar] [CrossRef] [PubMed]
- Bae, B.-I.; Hara, M.R.; Cascio, M.B.; Wellington, C.L.; Hayden, M.R.; Ross, C.A.; Ha, H.C.; Li, X.-J.; Snyder, S.H.; Sawa, A. Mutant Huntingtin: Nuclear translocation and cytotoxicity mediated by GAPDH. Proc. Natl. Acad. Sci. 2006, 103, 3405–3409. [Google Scholar] [CrossRef]
- Hwang, S.; Disatnik, M.-H.; Mochly-Rosen, D. Impaired GAPDH -induced mitophagy contributes to the pathology of Huntington's disease. EMBO Mol. Med. 2015, 7, 1307–1326. [Google Scholar] [CrossRef]
- Vizcaíno, C.; Mansilla, S.; Portugal, J. Sp1 transcription factor: A long-standing target in cancer chemotherapy. Pharmacol. Ther. 2015, 152, 111–124. [Google Scholar] [CrossRef]
- Li, S.-H.; Cheng, A.L.; Zhou, H.; Lam, S.; Rao, M.; Li, H.; Li, X.-J. Interaction of Huntington Disease Protein with Transcriptional Activator Sp1. Mol. Cell. Biol. 2002, 22, 1277–1287. [Google Scholar] [CrossRef]
- Dunah, A.W.; Jeong, H.; Griffin, A.; Kim, Y.-M.; Standaert, D.G.; Hersch, S.M.; Mouradian, M.M.; Young, A.B.; Tanese, N.; Krainc, D. Sp1 and TAFII130 Transcriptional Activity Disrupted in Early Huntington's Disease. Science 2002, 296, 2238–2243. [Google Scholar] [CrossRef]
- Bradford, J.; Shin, J.-Y.; Roberts, M.; Wang, C.-E.; Li, X.-J.; Li, S. Expression of mutant huntingtin in mouse brain astrocytes causes age-dependent neurological symptoms. Proc. Natl. Acad. Sci. 2009, 106, 22480–22485. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Luo, Y.; Ly, P.T.; Cai, F.; Zhou, W.; Zou, H.; Song, W. Sp1 regulates human huntingtin gene expression. J Mol Neurosci. 2012, 47, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Norflus, F.; Singh, B.; Swindell, M.K.; Buzescu, R.; Bejarano, M.; Chopra, R.; Zucker, B.; Benn, C.L.; DiRocco, D.P.; et al. Sp1 Is Up-regulated in Cellular and Transgenic Models of Huntington Disease, and Its Reduction Is Neuroprotective. J. Biol. Chem. 2006, 281, 16672–16680. [Google Scholar] [CrossRef]
- Kokame, K.; Agarwala, K.L.; Kato, H.; Miyata, T. Herp, a New Ubiquitin-like Membrane Protein Induced by Endoplasmic Reticulum Stress. J. Biol. Chem. 2000, 275, 32846–32853. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Cao, L.; Liang, X.; Du, A.; Peng, T.; Li, H. Herp Promotes Degradation of Mutant Huntingtin: Involvement of the Proteasome and Molecular Chaperones. Mol. Neurobiol. 2018, 55, 7652–7668. [Google Scholar] [CrossRef]
- Kalchman, M.A.; Koide, H.B.; McCutcheon, K.; Graham, R.K.; Nichol, K.; Nishiyama, K.; Kazemi-Esfarjani, P.; Lynn, F.C.; Wellington, C.; Metzler, M.; et al. HIP1, a human homologue of S. cerevisiae Sla2p, interacts with membrane-associated huntingtin in the brain. Nat. Genet. 1997, 16, 44–53. [Google Scholar] [CrossRef]
- Wanker, E.E.; Rovira, C.; Scherzinger, E.; Hasenbank, R.; Wälter, S.; Tait, D.; Colicelli, J.; Lehrach, H. HIP-I: A huntingtin interacting protein isolated by the yeast two-hybrid system. Hum. Mol. Genet. 1997, 6, 487–495. [Google Scholar] [CrossRef]
- Waelter, S.; Scherzinger, E.; Hasenbank, R.; Nordhoff, E.; Lurz, R.; Goehler, H.; Gauss, C.; Sathasivam, K.; Bates, G.P.; Lehrach, H.; et al. The huntingtin interacting protein HIP1 is a clathrin and alpha-adaptin-binding protein involved in receptor-mediated endocytosis. Hum. Mol. Genet. 2001, 10, 1807–1817. [Google Scholar] [CrossRef]
- Laragione, T.; Brenner, M.; Lahiri, A.; Gao, E.; Harris, C.; Gulko, P.S. Huntingtin-interacting protein 1 (HIP1) regulates arthritis severity and synovial fibroblast invasiveness by altering PDGFR and Rac1 signalling. Ann. Rheum. Dis. 2018, 77, 1627–1635. [Google Scholar] [CrossRef]
- Nicita, F.; Garone, G.; Spalice, A.; Savasta, S.; Striano, P.; Pantaleoni, C.; Spartà, M.V.; Kluger, G.; Capovilla, G.; Pruna, D.; et al. Epilepsy is a possible feature in Williams-Beuren syndrome patients harboring typical deletions of the 7q11.23 critical region. Am. J. Med Genet. Part A 2016, 170, 148–155. [Google Scholar] [CrossRef]
- Swaih, A.M.; Breda, C.; Sathyasaikumar, K.V.; Allcock, N.; Collier, M.E.W.; Mason, R.P.; Feasby, A.; Herrera, F.; Outeiro, T.F.; Schwarcz, R.; et al. Kynurenine 3-Monooxygenase Interacts with Huntingtin at the Outer Mitochondrial Membrane. Biomedicines 2022, 10, 2294. [Google Scholar] [CrossRef] [PubMed]
- Sathyasaikumar, K.V.; de la Cruz, V.P.; Pineda, B.; Cervantes, G.I.V.; Ortega, D.R.; Donley, D.W.; Severson, P.L.; West, B.L.; Giorgini, F.; Fox, J.H.; et al. Cellular Localization of Kynurenine 3-Monooxygenase in the Brain: Challenging the Dogma. Antioxidants 2022, 11, 315. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Shao, M.; Wu, T. Kynurenine-3-monooxygenase: A new direction for the treatment in different diseases. Food Sci. Nutr. 2020, 8, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Zwilling, D.; Huang, S.-Y.; Sathyasaikumar, K.V.; Notarangelo, F.M.; Guidetti, P.; Wu, H.-Q.; Lee, J.; Truong, J.; Andrews-Zwilling, Y.; Hsieh, E.W.; et al. Kynurenine 3-Monooxygenase Inhibition in Blood Ameliorates Neurodegeneration. Cell 2011, 145, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Zhang, A.; Binnie, M.; McGuire, K.; Webster, S.P.; Hughes, J.; Howie, S.E.M.; Mole, D.J. Kynurenine 3-monooxygenase is a critical regulator of renal ischemia–reperfusion injury. Exp. Mol. Med. 2019, 51, 1–14. [Google Scholar] [CrossRef]
- Park, S.-Y.; Nam, J.-S. Kynurenine pathway enzyme KMO in cancer progression: A tip of the Iceberg. EBioMedicine 2020, 55, 102762. [Google Scholar] [CrossRef]
- Subramaniam, S.; Sixt, K.M.; Barrow, R.; Snyder, S.H. Rhes, a Striatal Specific Protein, Mediates Mutant-Huntingtin Cytotoxicity. Science 2009, 324, 1327–1330. [Google Scholar] [CrossRef]
- Mealer, R.G.; Subramaniam, S.; Snyder, S.H. Rhes Deletion Is Neuroprotective in the 3-Nitropropionic Acid Model of Huntington's Disease. J. Neurosci. 2013, 33, 4206–4210. [Google Scholar] [CrossRef]
- Ramirrez-Jarquin, U.N.; Manish Sharmaa, M.; Zhou, W.; Shahani, N.; Subramaniam, S. Deletion of SUMO1 attenuates behavioral andanatomical deficits by regulating autophagic activities in Huntington disease. Proc. Natl. Acad. Sci. USA 2022, 119, e2107187119. [Google Scholar] [CrossRef]
- Mealer, R.G.; Murray, A.J.; Shahani, N.; Subramaniam, S.; Snyder, S.H. Rhes, a Striatal-selective Protein Implicated in Huntington Disease, Binds Beclin-1 and Activates Autophagy. J. Biol. Chem. 2014, 289, 3547–3554. [Google Scholar] [CrossRef]
- DiGiovanni, L.F.; Mocle, A.J.; Xia, J.; Truant, R. Huntingtin N17 domain is a reactive oxygen species sensor regulating huntingtin phosphorylation and localization. Hum. Mol. Genet. 2016, 25, 3937–3945. [Google Scholar] [CrossRef] [PubMed]
- Raychaudhuri, S.; Sinha, M.; Mukhopadhyay, D.; Bhattacharyya, N.P. HYPK, a Huntingtin interacting protein, reduces aggregates and apoptosis induced by N-terminal Huntingtin with 40 glutamines in Neuro2a cells and exhibits chaperone-like activity. Hum. Mol. Genet. 2008, 17, 240–255. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, K.R.; Bhattacharyya, N.P. Chaperone protein HYPK interacts with the first 17 amino acid region of Huntingtin and modulates mutant HTT-mediated aggregation and cytotoxicity. Biochem. Biophys. Res. Commun 2015, 456, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.K.; Roy, A.; Ranjan, A. The ATP ase VCP /p97 functions as a disaggregase against toxic Huntingtin-exon1 aggregates. FEBS Lett. 2018, 592, 2680–2692. [Google Scholar] [CrossRef] [PubMed]
- Topolska-Woś, A.M.; Chazin, W.J.; Filipek, A. CacyBP/SIP—Structure and variety of functions. Biochimica et Biophysica Acta (BBA)-General Subjects 2016, 1860, 79–85. [Google Scholar] [CrossRef]
- Santelli, E.; Leone, M.; Li, C.; Fukushima, T.; Preece, N.E.; Olson, A.J.; Ely, K.R.; Reed, J.C.; Pellecchia, M.; Liddington, R.C.; et al. Structural Analysis of Siah1-Siah-interacting Protein Interactions and Insights into the Assembly of an E3 Ligase Multiprotein Complex. J. Biol. Chem. 2005, 280, 34278–34287. [Google Scholar] [CrossRef]
- Latoszek, E.; Wiweger, M.; Ludwiczak, J.; Dunin-Horkawicz, S.; Kuznicki, J.; Czeredys, M. Siah-1-interacting protein regulates mutated huntingtin protein aggregation in Huntington’s disease models. Cell Biosci. 2022, 12. [Google Scholar] [CrossRef]
- Xu, H.; Bensalel, J.; Raju, S.; Capobianco, E.; Lu, M.L.; Wei, J. Characterization of huntingtin interactomes and their dynamic responses in living cells by proximity proteomics. J. Neurochem. 2023, 164, 512–528. [Google Scholar] [CrossRef]
- Morrow, C.S.; Porter, T.J.; Xu, N.; Arndt, Z.P.; Ako-Asare, K.; Heo, H.J.; Thompson, E.A.; Moore, D.L. Vimentin Coordinates Protein Turnover at the Aggresome during Neural Stem Cell Quiescence Exit. Cell Stem Cell 2020, 26, 558–568. [Google Scholar] [CrossRef]
- Bauer, P.; Hudec, R.; Goswami, A.; Kurosawa, M.; Matsumoto, G.; Mikoshiba, K.; Nukina, N. ROCK-phosphorylated vimentin modifies mutant huntingtin aggregation via sequestration of IRBIT. Mol. Neurodegener. 2012, 7, 43–43. [Google Scholar] [CrossRef]
- Chi, X.; Wang, S.; Huang, Y.; Stamnes, M.; Chen, J.-L. Roles of Rho GTPases in Intracellular Transport and Cellular Transformation. Int. J. Mol. Sci. 2013, 14, 7089–7108. [Google Scholar] [CrossRef] [PubMed]
- Tousley, A.; Iuliano, M.; Weisman, E.; Sapp, E.; Zhang, N.; Vodicka, P.; Alexander, J.; Aviolat, H.; Gatune, L.; Reeves, P.; et al. Rac1 Activity Is Modulated by Huntingtin and Dysregulated in Models of Huntington’s Disease. J. Huntington's Dis. 2019, 8, 53–69. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Bento, C.F.; Ricketts, T.; Vicinanza, M.; Siddiqi, F.; Pavel, M.; Squitieri, F.; Hardenberg, M.C.; Imarisio, S.; Menzies, F.M.; et al. Polyglutamine tracts regulate beclin 1-dependent autophagy. Nature 2017, 545, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Shibata, M.; Lu, T.; Furuya, T.; Degterev, A.; Mizushima, N.; Yoshimori, T.; MacDonald, M.; Yankner, B.; Yuan, J. Regulation of Intracellular Accumulation of Mutant Huntingtin by Beclin 1. Perspect. Surg. 2006, 281, 14474–14485. [Google Scholar] [CrossRef]
- Pircs, K.; Drouin-Ouellet, J.; Horváth, V.; Gil, J.; Rezeli, M.; Garza, R.; A Grassi, D.; Sharma, Y.; St-Amour, I.; Harris, K.; et al. Distinct subcellular autophagy impairments in induced neurons from patients with Huntington's disease. Brain 2022, 145, 3035–3057. [Google Scholar] [CrossRef]
- Croce, K.R.; Yamamoto, A. A role for autophagy in Huntington's disease. Neurobio. Dis. 2019, 122, 16–22. [Google Scholar] [CrossRef]
- Valionyte, E.; Yang, Y.; Roberts, S.L.; Kelly, J.; Lu, B.; Luo, S. Lowering Mutant Huntingtin Levels and Toxicity: Autophagy-Endolysosome Pathways in Huntington's Disease. J. Mol. Bio. 2020, 432, 2673–2691. [Google Scholar] [CrossRef]
- Yang, W.; Chen, X.; Li, S.; Li, X.-J. Genetically modified large animal models for investigating neurodegenerative diseases. Cell Biosci. 2021, 11. [Google Scholar] [CrossRef]
- Yin, P.; Li, S.; Li, X.-J.; Yang, W. New pathogenic insights from large animal models of neurodegenerative diseases. Protein Cell 2022, 13, 707–720. [Google Scholar] [CrossRef]
- Li, C.; Li, J.; Lai, L.; Li, S.; Yan, S. Genetically engineered pig models of neurological diseases. Ageing Neurodegener. Dis. 2022, 2, 13. [Google Scholar] [CrossRef]
- Chen, X.; Sun, Y.; Chen, L.; Chen, X.-S.; Pan, M.; Zhang, Y.; Wang, Q.; Yang, W.; Yin, P.; He, D.; et al. Differential expression and roles of Huntingtin and Huntingtin-associated protein 1 in the mouse and primate brains. Cell. Mol. Life Sci. 2022, 79, 554. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-H.; Hosseini, S.H.; Gutekunst, C.-A.; Hersch, S.M.; Ferrante, R.J.; Li, X.-J. A Human HAP1 Homologue. J. Biol. Chem. 1998, 273, 19220–19227. [Google Scholar] [CrossRef] [PubMed]
- Bertaux, F.; Sharp, A.H.; A Ross, C.; Lehrach, H.; Bates, G.P.; Wanker, E. HAP1-huntingtin interactions do not contribute to the molecular pathology in Huntington's disease transgenic mice. FEBS Lett. 1998, 426, 229–232. [Google Scholar] [CrossRef] [PubMed]
- A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell 1993, 72, 971–983. [CrossRef] [PubMed]
- Li, S.H.; Schilling, G.; Young, W.S.; Li, X.; Margolis, R.L.; Stine, O.C.; Wagster, M.V.; Abbott, M.H.; Franz, M.L.; Ranen, N.G.; et al. Huntington's disease gene (IT15) is widely expressed in human and rat tissues. Neuron 1993, 11, 985–993. [Google Scholar] [CrossRef] [PubMed]
- Strong, T.V.; Tagle, D.A.; Valdes, J.M.; Elmer, L.W.; Boehm, K.; Swaroop, M.; Kaatz, K.W.; Collins, F.S.; Albin, R.L. Widespread expression of the human and rat Huntington's disease gene in brain and nonneural tissues. Nat. Genet. 1993, 5, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Sharp, A.H.; Loev, S.J.; Schilling, G.; Li, S.-H.; Li, X.-J.; Bao, J.; Wagster, M.V.; Kotzuk, J.A.; Steiner, J.P.; Lo, A.; et al. Widespread expression of Huntington's disease gene (IT15) protein product. Neuron 1995, 14, 1065–1074. [Google Scholar] [CrossRef]
- Landwehrmeyer, G.B.; McNeil, S.M.; Dure, L.S.; Ge, P.; Aizawa, H.; Huang, Q.; Ambrose, C.M.; Duyao, M.P.; Bird, E.D.; Bonilla, E.; et al. Huntington's disease gene: Regional and cellular expression in brain of normal and affected individuals. Ann. Neurol. 1995, 37, 218–230. [Google Scholar] [CrossRef]
- Harjes, P.; Wanker, E. The hunt for huntingtin function: interaction partners tell many different stories. Trends Biochem. 2003, 28, 425–433. [Google Scholar] [CrossRef]
- Kaltenbach, L.S.; Romero, E.; Becklin, R.R.; Chettier, R.; Bell, R.; Phansalkar, A.; Strand, A.; Torcassi, C.; Savage, J.; Hurlburt, A.; et al. Huntingtin Interacting Proteins Are Genetic Modifiers of Neurodegeneration. PLOS Genet. 2007, 3, e82. [Google Scholar] [CrossRef]
- Miller, J and Hughes, R.E. Protein Interactions and Target Discovery in Huntington’s Disease. Neurobiology of Huntington Disease (Applications to Drug Discovery) Frontiers in Neuroscience 2011, ISBN: 978-0-8493-9000.
- Li, X.-J.; Li, S. Influence of Species Differences on the Neuropathology of Transgenic Huntington's Disease Animal Models. J. Genet. Genom. 2012, 39, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Eaton, S.L.; Wishart, T.M. Bridging the gap: large animal models in neurodegenerative research. Mamm. Genome 2017, 28, 324–337. [Google Scholar] [CrossRef] [PubMed]
- Sheng, G.; Xu, X.; Lin, Y.-F.; Wang, C.-E.; Rong, J.; Cheng, D.; Peng, J.; Jiang, X.; Li, S.-H.; Li, X.-J. Huntingtin-associated protein 1 interacts with Ahi1 to regulate cerebellar and brainstem development in mice. J. Clin. Investig. 2008, 118, 2785–2795. [Google Scholar] [CrossRef] [PubMed]
- Vonsattel, J.-P.; Myers, R.H.; Stevens, T.J.; Ferrante, R.J.; Bird, E.D.; Richardson, E.P., Jr. Neuropathological Classification of Huntington's Disease. J. Neuropathol. Exp. Neurol. 1985, 44, 559–577. [Google Scholar] [CrossRef]
- Vonsattel, J.P.G.; Keller, C.; Pilar Amaya, M.d. Neuropathology of Huntington's Disease. Handbook of clinical neurology; Elsevier: 2008; Volume 89, pp. 599-618.
- Yan, S.; Tu, Z.C.; Liu, Z.M.; Fan, N.N.; Yang, H.M.; Yang, S.; Yang, W.L.; Zhao, Y.; Ouyang, Z.; Lai, C.D.; et al. A Huntingtin Knockin Pig Model Recapitulates Features of Selective Neurodegeneration in Huntington’s Disease. Cell 2018, 173, 989–1002. [Google Scholar] [CrossRef] [PubMed]
- Caviston, J.P.; Holzbaur, E.L. Huntingtin as an essential integrator of intracellular vesicular trafficking. Trends Cell Biol. 2009, 19, 147–155. [Google Scholar] [CrossRef]
- Millecamps, S.; Julien, J.-P. Axonal transport deficits and neurodegenerative diseases. Nat. Rev. Neurosci. 2013, 14, 161–176. [Google Scholar] [CrossRef]
- Page, K.J.; Potter, L.; Aronni, S.; Everitt, B.J.; Dunnett, S.B. The expression of Huntingtin-associated protein (HAP1) mRNA in developing, adult and ageing rat CNS: implications for Huntington's disease neuropathology. Eur. J. Neurosci. 1998, 10, 1835–1845. [Google Scholar] [CrossRef]
- Li, T.; Li, S.; Gao, X.; Cai, Q.; Li, X.-J. Expression and Localization of Huntingtin-Associated Protein 1 (HAP1) in the Human Digestive System. Dig. Dis. Sci. 2019, 64. [Google Scholar] [CrossRef]
- Qu, Y.-M.; Chen, A.; Zhao, X.; Wang, Z.; Guo, D.; Shao, S.-L.; Tao, Y.-Y.; Li, Q.-J.; Wang, M.-Y.; Ma, W.-S. Huntingtin-associated protein 1 is a potential tumor suppressor for gastric cancer. Mol. Biol. Rep. 2023, 50, 1517–1531. [Google Scholar] [CrossRef]
- Zhu, L.; Song, X.; Tang, J.; Wu, J.; Ma, R.; Cao, H.; Ji, M.; Jing, C.; Wang, Z. Huntingtin-associated protein 1: A potential biomarker of breast cancer. Oncol. Rep. 2013, 29, 1881–1887. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, J.-y.; Yin, L.; Wu, J.-z.; Guo, W.-j.; Wu, J.-f.; Chen, M.; Xia, Y.-y.; Tang, J.-h.; Ma, Y.-c.; et al. HAP1 gene expression is associated with radiosensitivity in breast cancer cells. Biochem. Biophys. Res. 2015, 456, 162–166. [Google Scholar] [CrossRef]
- Wang, W.; Yan, H.; Zhang, Q.; Song, W.; Li, H.; Xu, J. Evaluating the association of polymorphisms in the HAP1 gene with lung cancer risk: a meta-analysis. Tumor Biol. 2014, 35, 10825–10831. [Google Scholar] [CrossRef]
- Peters, M.F.; Ross, C.A. Isolation of a 40-kDa Huntingtin-associated Protein. J. Biol. Chem. 2001, 276, 3188–3194. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Huang, B.; Cheng, J.; Seefelder, M.; Engler, T.; Pfeifer, G.; Oeckl, P.; Otto, M.; Moser, F.; Maurer, M.; et al. The cryo-electron microscopy structure of huntingtin. Nature 2018, 555, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Seefelder, M.; Buck, E.; Engler, T.; Lindenberg, K.S.; Klein, F.; Landwehrmeyer, G.B.; Kochanek, S. HAP40 protein levels are huntingtin-dependent and decrease in Huntington disease. Neurobiol. Dis. 2021, 158, 105476. [Google Scholar] [CrossRef] [PubMed]
- Harding, R.J.; Deme, J.C.; Hevler, J.F.; Tamara, S.; Lemak, A.; Cantle, J.P.; Szewczyk, M.M.; Begeja, N.; Goss, S.; Zuo, X.; et al. Huntingtin structure is orchestrated by HAP40 and shows a polyglutamine expansion-specific interaction with exon 1. Commun. Biol. 2021, 4, 1374. [Google Scholar] [CrossRef]
- Metzger, S.; Rong, J.; Nguyen, H.-P.; Cape, A.; Tomiuk, J.; Soehn, A.S.; Propping, P.; Freudenberg-Hua, Y.; Freudenberg, J.; Tong, L.; et al. Huntingtin-associated protein-1 is a modifier of the age-at-onset of Huntington's disease. Hum. Mol. Genet. 2008, 17, 1137–1146. [Google Scholar] [CrossRef]
- Lee, J.-M.; Huang, Y.; Orth, M.; Gillis, T.; Siciliano, J.; Hong, E.; Mysore, J.S.; Lucente, D.; Wheeler, V.C.; Seong, I.S.; et al. Genetic modifiers of Huntington disease differentially influence motor and cognitive domains. Am. J. Hum. Genet. 2022, 109, 885–899. [Google Scholar] [CrossRef]
- Bhide, P.G.; Day, M.; Sapp, E.; Schwarz, C.; Sheth, A.; Kim, J.; Young, A.B.; Penney, J.; Golden, J.; Aronin, N.; et al. Expression of Normal and Mutant Huntingtin in the Developing Brain. J. Neurosci. 1996, 16, 5523–5535. [Google Scholar] [CrossRef]
- Barnat, M.; Capizzi, M.; Aparicio, E.; Boluda, S.; Wennagel, D.; Kacher, R.; Kassem, R.; Lenoir, S.; Agasse, F.; Braz, B.Y.; et al. Huntington’s disease alters human neurodevelopment. Science 2020, 369, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Molero, A.E.; Arteaga-Bracho, E.E.; Chen, C.H.; Gulinello, M.; Winchester, M.L.; Pichamoorthy, N.; Gokhan, S.; Khodakhah, K.; Mehler, M.F. Selective expression of mutant huntingtin during development recapitulates characteristic features of Huntington’s disease. Proc. Natl. Acad. Sci. 2016, 113, 5736–5741. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
HAP1 and HTT distribution in mouse and Monkey brains (adapted from Chen et al., 2022). a). Immunohistochemistry staining of mouse brain sagittal section with antibodies to HAP1 and HTT. mouse HAP1 is enriched in the hypothalamic and middle brain region, while HTT is distributed more broadly in the mouse brain. b). Immunohistochemistry staining of monkey brain with anti-HAP1 or anti-HTT specific antibodies. The HAP1 and HTT showed a very similar brain distribution pattern in the monkey brain. c). Western blotting of proteins extracted from brains of 10 months old mice or 28year old monkey. The western blotting further support the immunohistochemistry result that Hap1 is enriched in mouse hypothalamus while distribute evenly as the HTT in monkey brain. note the distribution of monkey HAP1 is very similar to HAP1 in human brain.
Figure 1.
HAP1 and HTT distribution in mouse and Monkey brains (adapted from Chen et al., 2022). a). Immunohistochemistry staining of mouse brain sagittal section with antibodies to HAP1 and HTT. mouse HAP1 is enriched in the hypothalamic and middle brain region, while HTT is distributed more broadly in the mouse brain. b). Immunohistochemistry staining of monkey brain with anti-HAP1 or anti-HTT specific antibodies. The HAP1 and HTT showed a very similar brain distribution pattern in the monkey brain. c). Western blotting of proteins extracted from brains of 10 months old mice or 28year old monkey. The western blotting further support the immunohistochemistry result that Hap1 is enriched in mouse hypothalamus while distribute evenly as the HTT in monkey brain. note the distribution of monkey HAP1 is very similar to HAP1 in human brain.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



