
Submitted:
08 July 2023
Posted:
10 July 2023
You are already at the latest version
Abstract
Targeted protein degradation is an attractive technology for cancer treatment due to its ability to overcome the unpredictability of small molecule inhibitors that cause resistance mutations. In recent years, various targeted protein degradation strategies have been developed based on the ubiquitin-proteasome system in the cytoplasm or the autophagy-lysosomal system during endocytosis. In this review, we describe and compare technologies for targeted inhibition and targeted degradation of the epidermal growth factor receptor (EGFR), one of the major proteins responsible for the onset and progression of many types of cancer. In addition, we have developed an alternative strategy, called alloAUTO, based on the binding of new heterocyclic compounds to an allosteric site located in close proximity to the EGFR catalytic site. These compounds cause targeted degradation of the transmembrane receptor, simultaneously activating both systems of protein degradation in cells. Damage to EGFR signaling pathways promotes inactivation of Bim sensor protein phosphorylation, which leads to disintegration of the cytoskeleton, followed by detachment of cancer cells from the extracellular matrix and, ultimately, to cancer cell death. This hallmark of targeted cancer cell death suggests an advantage over other targeted protein degradation strategies, namely that the fewer cancer cells survive, the fewer chemotherapy-resistant mutants appear.
Keywords:
cancer chemotherapy
; transmembrane receptors
; EGFR
; targeted protein degradation
; furfuryl-quinolin-triazole-thiol chemicals
1. Introduction
Advances in the development of more effective drugs and therapeutic approaches [1,2] and prevention programs to reduce the harmful effects of tobacco on smokers [3] have determined success in reducing human cancer mortality over the past decade. Consistent with these data, the count of functional limitations among cancer survivors in the United States from 1999 to 2018 showed that their proportion increased from 57.0% to 70.1%, indicating a clear improvement in the health of cancer patients [4]. However, traditional cancer therapies have low specificity and cause serious side effects [5,6], and moreover, targeted protein inhibition cause drug resistance [7,8]. Consequently, cancer remains one of the leading causes of human death, and the treatment of various types of tumors is a huge challenge for humanity in the 21st century.
The identification and characterization of tumor-related hallmarks has contributed to the development of anti-target therapeutic strategies for more than 100 types of malignancies [9,10,11]. Currently, cancer treatment includes various approaches such as surgery, radiation therapy, hormonal therapy, immunotherapy, and chemotherapy. This review focuses on cancer chemotherapy, which uses two fundamentally different strategies: standard therapy and targeted therapy. Standard chemotherapeutic agents are cytotoxic because they kill cancer cells, while targeted chemotherapeutic agents are often cytostatic because they bind to tumor cells and block cell proliferation. Approved cytotoxic drugs have relatively low tumor specificity and high toxicity, while targeted therapy increases treatment specificity and appears to reduce secondary events in patients [12]. Accurate and reliable information about the key genes and proteins responsible for the respective cancer has become the cornerstone of targeted treatment.
Targeted protein inhibition accelerated the development of new chemotherapeutic drugs capable of inhibiting key proteins in tumor cells [13]. However, it became clear that the main disadvantage of inhibitors is the occurrence of drug-resistant mutations both in the gene encoding the target protein and in other associated DNA regions, which leads to the loss of the therapeutic effect [14]. The combined use of standard chemotherapy and targeted chemotherapy has been found to provide more personalized and effective treatment for cancer patients [15]. A striking example of modern therapy for aggressive cancer is intravenous treatment with platinum drugs in combination with targeted protein inhibition by chemotherapy, which improves the well-being of patients and prolongs their life [16]. However, inhibition of overexpressed and overactivated key proteins with small molecule inhibitors provides a limited duration of therapeutic effect on cancer.
Primary and secondary driver mutations resulting from targeted protein inhibition are often common causes of drug resistance in cancer treatment. In this review, we turn to data on the epidermal growth factor receptor (EGFR), which is one of the main tumor markers in many types of cancer. We highlight the shortcomings of the EGFR inhibition strategy in cancer treatment, leading to the need to develop an alternative strategy called targeted protein degradation (TPD). This promising strategy for the degradation of key proteins responsible for the pathological process offers new hopes for more successful cancer treatments in the future
2. Targeted inhibition of receptor tyrosine kinase EGFR
Transmembrane receptor tyrosine kinases control different signaling pathways that play an important role in many cellular processes [17]. ErbB1, ErbB2, ErbB3 and ErbB4 receptors belonging to the ErbB family are involved in cell proliferation, differentiation, invasion, angiogenesis and other functions. The canonical mechanism of EGFR activation uses the EGF ligand or other growth factors that bind to the extracellular region of the receptor and lead to structural rearrangements favorable for its dimerization, which is necessary for activation of the catalytic ATP-binding site in the cytoplasmic region (Figure 1A) [18]. Subsequent phosphorylation of tyrosine residues located in the tyrosine kinase domain triggers downstream signaling pathways required for normal cellular activity. However, overexpression and overactivation of EGFR in cells can provoke aberrant signaling leading to the development and progression of cancer [19]. It should be noted that EGFR can dimerize with other receptors of the ErbB family, as well as with other transmembrane receptors, and activate a wide range of cellular functions under the action of appropriate extracellular ligands [20].
The intracellular region of EGFR consists of a juxtamembrane portion, a tyrosine kinase domain, and a C-terminal tail that acts as a linker for interaction with other proteins after phosphorylation (Figure 1B) [21]. The kinase domain consists of five β-sheets and one α-helix containing the N-lobe and an α-helix containing the C-lobe as determined to 2.6 Å resolution. ATP-binding domain is located between the N-lobe and C-lobe of the tyrosine kinase domain. Phosphorylation of the substrate occurs by transfer of phosphorus from ATP to the tyrosine residue through the Asp813 residue of the catalytic loop.
The EGFR can be also activated by hydrogen peroxide (H2O2) generated during cognate ligand EGF binding to the receptor [22]. The binding of EGF to EGFR promotes the transformation of O2 to H2O2 through the membrane-located NADPH oxidase Nox2; then, this reactive oxygen species reacts with cysteine residue at the position 797 (Cys797) in the proximity to the ATP-binding site, leading to the transition of the thiolate anion (Cys-S) to sulfenic acid (Cys-SOH), which is required for activation of the receptor [23,24]. This cysteine is not directly required for catalysis, but can be used to irreversibly block the ATP-binding site by binding to a small chemical molecule. EGF-independent EGFR auto-phosphorylation has also been described in cells treated with small chemicals. In particular, the action of 4-nitrobenzoxadiazole derivatives rely on the formation of H2O2 by cytoplasmic superoxide dismutase, which is associated simultaneously with the inactivation of protein tyrosine phosphatase PTP-1B in cancer cells [25,26]. It was found that reactive H2O2 quickly binds to various proteins after the passage of lipophilic chemical agents through the cytoplasmic membrane [27]. However, hydrogen peroxide produced by these compounds both activates and inactivates the expression of a large number of targeted proteins that ensure the functionality of various cellular processes [26,28]. Therefore, such chemical agents serving as a source of hydrogen peroxide will provoke unpredictable effect on therapeutic efficacy and cannot be considered as a suitable candidate for cancer treatment [29].
Protein degradation in mammals depends on the initiation type of autophagy categorized as macro-autophagy, chaperone-mediated, and micro-autophagy [30]. Endocytosis of EGFR is a micro-autophagy process, which orchestrates cellular signaling networks, and can direct the fate of the receptor in cells. Ligand-bound EGFR undergoes endocytosis followed by recycling and/or degradation of the receptor by proteolytic enzymes in lysosomes fused to endosomes [31]. Low doses of EGF activate clathrin-dependent endocytosis, which promotes sustained EGFR signaling and is the main mechanism of EGFR endocytosis in tumors in vivo [32]. High doses of ligand additionally induce clathrin-independent endocytosis, which is the main lysosomal degradation pathway for reducing EGFR signaling [33]. Overall, endocytic degradation of EGFR likely influences the fate of the receptor through the mechanism of ubiquitination [34].
Small chemical molecules and neutralizing antibodies have been developed to inhibit proliferative phosphorylation in ErbB1 and ErbB2 mediated signaling pathways, which are considered important targets in cancer cells [35]. For more than two decades, targeting the ATP binding site in EGFR has become an attractive issue in medicinal chemistry. Inhibitors with reversible and irreversible mechanisms of action have been developed to inhibit the catalytic site and improve patient survival compared to platinum-based chemotherapy, the former standard of care [36].
Three generations of tyrosine kinase inhibitors (TKIs) for EGFR have been developed for the treatment of cancer patients, and the respective therapeutic molecules have been grouped according to their structure or target sites in the receptors [37]. Different competitive kinase inhibitors mimic ATP and interact with the hinge region at the kinase ATP site, but differ in the conformational state of the kinase they interact with [38].
According to FDA approved inhibitors, first-generation gefitinib [39] and erlotinib [40] are reversible inhibitors of the catalytic site in EGFR, which strongly inhibit the receptor when it is constitutively activated by in-frame deletion in exon 19 (del19) or L858R substitution of exon 21 [41,42]. These cancer-activating mutations, which occur in the tyrosine kinase domain of EGFR, have been classified as oncogenic drivers of non-small cell lung cancer (NSCLC), one of the most lethal cancers in the world [43]. Up to 70% of NSCLC patients with EGFR-mutant tumors develop brain metastases during the course of the disease and approximately 20% of patients receiving anti-EGFR TKIs [44,45]. First-generation TKIs, gefitinib and erlotinib, initially showed significant therapeutic response and prolonged survival in patients with non-small cell lung cancer (NSCLC). However, the secondary gatekeeper mutation T790M increased ATP binding affinity, caused relapse in most patients with NSCLC after 9-14 months of treatment, and led to resistance to gefitinib and erlotinib [46,47,48,49]. To overcome resistance, a number of new EGFR inhibitors have been developed targeting the receptor carrying the T790M activating mutation.
Second-generation afatinib and dacomitinib are irreversible inhibitors that interact with Cys797 in EGFR [50,51]. It is worth noting that cysteine is the most common covalent amino acid residue used to create irreversible bonds in drugs [52]. Afatinib and dacomitinib are active against exon 19 (del19) deletion and L858R mutation in exon 21, which are the most common activating EGFR mutations and oncogenes for many patients with NSCLC [53,54]. Unlike non-covalent inhibitors, covalent inhibitors block target proteins in two steps [55]. Covalent binding to target proteins first requires the formation of protein-target-inhibitor complexes via non-covalent binding, which is similar to the equilibrium process used by non-covalent inhibitors. In the second rate-determining step, the electrophilic head of the covalent inhibitor is appropriately positioned adjacent to the amino acid residues at the binding site.
Third-generation osimertinib and mobocertinib are irreversible inhibitors that can overcome the acquired T790M mutation in EGFR [56,57]. However, acquired resistance to these drugs has been identified due to C797X mutation in EGFR, mutations in the phosphoinositide 3-kinase (PI3K) and RAS/mitogen-activated protein kinase (MAPK) pathways, or due to ErbB2/Erb3 amplification and mesenchymal epithelial transition factor (MET) or due to alterations in cell cycle genes [58]. An important intrinsic mechanism of resistance to osimertinib is a mutation in exon 20 of EGFR, which was found to be sensitive to the inhibitor mobocertinib and the EGFR/MET bispecific antibody amivantamab. Thus, determining the mechanism of resistance to osimertinib therapy remains an important challenge in EGFR-induced cancer. Unfortunately, with regard to resistance to third-generation EGFR inhibitors, there was no major breakthroughs in cancer treatment [59]. However, clinical trials have shown that the combined use of EGFR TKIs may offer new hope for the treatment of cancer. A good example is the combination therapy of lung adenocarcinoma using first and third generation TKIs, erlotinib with osimertinib, in patients with C797S and T790M mutations [60]. These impressive results have stimulated the use of other combination therapies for resistant tumors, including platinum-based chemotherapy.
Targeting EGFR by allosteric binding to the ATP binding site has also been described as a novel therapeutic strategy to overcome drug resistance that emerge within this catalytic site [61]. Allosteric inhibitors were active against EGFRL858R/T790M/C797S mutants and these compounds were proposed as fourth generation drug candidates [61,62,63,64]. Non-allosteric molecules have also been developed with activity against osimertinib-resistant NSCLC, characterized by broad molecular heterogeneity, and EGFRL858R/T790M/C797S mutant cells [65]. However, allosteric compounds can still induce drug resistance mutations due to a similar strategy of inhibiting the EGFR adenosine triphosphate (ATP) binding site. Further clinical trials will allow selection of fourth-generation EGFR therapeutic agents.
It has been stated that TKIs of all generations often showed toxicity, as judged by the appearance of diarrhea, skin rashes, and other manifestations in patients. Patients undergoing cancer treatment complain of fatigue, cognitive impairment, depression, sleep disturbances, as well as diarrhea and skin rashes. These symptoms are often caused by the emergence of resistance mutations, making drugs ineffective in stopping cancer progression [66]. Regression to cancer is often the result of driver mutations in the EGFR gene itself, preventing drug binding to the ATP-binding pocket and reactivating EGFR signaling pathways suppressed by corresponding TKIs, which can lead to metastatic manifestations in the patient. However, according to the data of scaled DNA sequencing, a decrease in the therapeutic effect of an inhibitor is often associated with the appearance of various resistant mutations in the same tumor during the treatment of a patient [67]. This means that resistance mutations that lead to cancer progression occur both in the EGFR gene itself and in other genes providing associated functions that make aggressive tumors insensitive to further treatment.
Thus, the bottleneck of chemotherapy with EGFR inhibitors is the emergence of resistance mutations in the same tumor, which leads to a decrease in the effectiveness of cancer therapy. Therefore, anticancer chemotherapy clearly needed a more advanced strategy other than inhibition of a key protein in order to overcome the limitations of the inhibition mechanism.
3. EGFR degradation by Protac technology
Targeted protein degradation (TPD) is a class of novel technologies based on chemically induced non-natural interactions between proteins of interest (POIs) and two major degradation systems, namely the ubiquitin-proteasome system (UPS) and the autophagy-lysosomal pathway. Targeted protein degradation by a bifunctional small molecule known as proteolysis-targeted chimeras (PROTACs) is one of the most exciting new technologies in medical chemistry and biotechnology. The advantage of this technology lies in its ability to attack and destroy druggable and non-druggable proteins in diseased cells and tissues, including cancer, immune disorders, viral infections, and neurodegenerative diseases [68].
TPD was first described as a heterobifunctional degrader PROTAC containing two chemical structures, one to recognize the protein of interest and the other to bind to the E3 ligase, resulting in the degradation of the targeted protein by UPS [69,70]. It was only after almost 15 years that the first catalytic small molecular degrader was developed [71], which has now become a breakthrough technology in chemical pharmacology to create a new generation of TPDs capable of eliminating proteins involved in the appearance and progression of cancer [72]. The chemical structure of PROTAC consists of three covalently linked moieties: (i) a structure to bind to the protein of interest, (ii) a structure to recognize the E3 ligase, and (iii) a linker to conjugate the two structures, allowing the formation of the E3-ligase-degrader-POI ternary complex [73]. The mechanism of action of a ternary complex, leading to polyubiquitination of the target protein and its subsequent degradation by the UPS, is shown in Figure 2.
UPS is a degradation mechanism of misfolded proteins in eukaryotic cells, to maintain intracellular protein homeostasis [74]. In this system, proteins to be degraded are covalently labeled with 76 amino acid ubiquitin (Ub), and the tagging process is catalyzed by three enzymes: ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzyme (E2), and ubiquitin ligase (E3) [75]. Free Ub is activated by E1 and then attached to the cysteine residue of E1; Ub-tagged E1 passes its Ub to cysteine of E2; E3 recruits the Ub-tagged E2 and E3 substrate for labeling ubiquitin at the lysine residue of the substrate. It is noteworthy that the human proteome contains more than 600 E3 ligases that provide specific recognition of substrates, two E1 proteins, and about forty E2 proteins. Repeated ubiquitination processes generate a poly-Ub chain (linked via Lys48 of Ub) on the target protein, which directs the substrate to the 26S proteasome for degradation [76].
A multiunit ATP-dependent protease called the 26S proteasome is involved in protein degradation by UPS, which is probably the most important protease in the cell, degrading proteins in the cytosol and nucleus [77]. In short, it removes the poly-Ub, unfolds the protein, and moves it into the inner chamber of the complexing protein. Synthesis of the 26S proteasome requires a significant amount of energy, and there are various mechanisms that regulate the adequate production of proteasome elements [78].
When designing the chemical structure in PROTAC, various strategies must be considered to select a small compound that will provide high affinity for the corresponding E3 ligase in recognition of the targeted protein (see Figure 2). The chemical synthesis and behavior of engineered protein degraders in diseased cells is detailed in recent reviews [79,80]. Below we summarize useful information on the evolution of protein degradation to eliminate wild-type and mutant EGFR in cancer cells.
The linker plays a key role as a molecular glue in the PROTAC structure. As far as the linker binds to two ligands, the given protein of interest and the E3 ligase, the binding sites in both proteins can influence not only binding selectivity but also other protein degradation parameters such as microsomal stability [81,82]. Moreover, the anchor point of the linker is important for degradation efficiency. Therefore, moving the anchor point from one site to another in the generated compound can have up to a ten-fold difference in effect on the target protein, as shown in threonine tyrosine kinase, when the value of the degradation coefficient (DC50) was shifted from 21.7 nM to 2.2 nM [83].
The binding strength between the warhead and the protein of interest is usually at a low nanomolar level. The structure of the ligand protein of interest can be slightly modified to provide suitable binding and optimize physicochemical properties, for example by replacing piperidine with an N,N-diethylamino group [84] or morpholine with piperazine [85]. The choice of the right target protein is important for the development of PROTAC since some proteins are highly expressed in normal tissues and their reduction can cause unpredictable toxicity. For example, ABT263-based VHLs recruiting PROTACs induce selective degradation of BCL-XL in tumor models of T-cell acute lymphoblastic leukemia while conserving platelets due to low VHL expression [86]. It is clear that such behavior of a potential drug is unattractive for cancer treatment.
To date, 60 proteins have been developed for PROTAC with four types of E3 ligases, including von Hippel Lindau ligases (VHL), Cereblon (CRBN), inhibitors of apoptosis proteins (IAP), and mouse double minute 2 homologue [87]. Currently, only E3 ligases, which are typically VHL, CRBN, and IAP, are tested for PROTAC activation. Expanding the E3 ligase toolbox is important not only to degrade a wider range of proteins, but also to potentially enable more selective recognition of tissue- and organ-specific proteins. Two E3 ligases, VHL and CRBN, are widely used in the development of targeted protein degraders due to their efficiency and good expression in cancer cells [88,89].
Linker length and chemical composition are probably the most important characteristic of the targeted protein degrader structure, as it can have a strong impact on permeability, degradation and specificity [90,91]. A shorter linker may prevent binding to the corresponding proteins due to steric hindrance. A longer linker may prevent the two ligands from being in close proximity, which is necessary for targeted ubiquitination. Different linkers based on polyethylene glycol, unsaturated alkane chains and heterocyclic rings, due to their polarity and flexibility, have been used as scaffold elements for PROTAC design [92].
Over the past seven years the PROTAC technology has been successfully applied to the degradation of numerous proteins in cancer [93,94,95]. With regard to targeting EGFR with PROTAC, it was important to elucidate whether this methodology could induce degradation of transmembrane proteins, given their limited cellular localization and questionable accessibility of membrane-bound receptors for ubiquitination via a cytosolic mechanism. In the first studies, different generations of EGFR inhibitors were used as the receptor-recognizing core to assess the effect of receptor degradation.
Two constructs based on gefitinib and afatinib were tested in cancer cells for degradation of EGFRdel19 by the and EGFRL858R/T790M mutants, respectively [90]. PROTACs have been shown to be able to induce degradation of active EGFR, ErbB2 and MET receptor tyrosine kinases, including both EGFR mutants, and at lower concentrations than comparable drugs that simply inhibit proteins. In control experiments using inactive diastereomeric compounds with identical physicochemical properties, degradation was shown to be enhanced compared to inhibition alone, highlighting the potential advantages of this pharmacological modality. These results strongly suggest that not only are RTKs substrates for post-translational degradation, but also that signaling inactivation and growth inhibition achieved by protein degraders are more potent and less susceptible to kinome rewiring than those achieved through RTK inhibition. Unlike EGFR inhibitors, the new degraders act on the entire targeted protein, resulting in the blocking of all receptor functions. This study has been instrumental in targeting the degradation of EGFR and other RTKs. Moreover, PROTAC has become something of a semolina for chemists and biologists interested in expanding the technological possibilities in the development of new therapeutics against various diseases, especially against cancer [96].
It was shown that small molecular degraders based on allosteric EGFR-TKI selectively inhibit the proliferation of cancer cells carrying EGFRL858R/T790M double mutant, and osimertinib-resistant triple mutants EGFRL858R/T790M/C797S and EGFRL858R/T790M/L718Q [64]. Unfortunately, these compounds were inactive at the wild-type receptor. However, given their high antiproliferative activity, allosteric compounds remain promising candidates for the development of suitable degraders of clinically relevant EGFR mutants.
Gefitinib-based PROTAC molecules have been shown to enhance degradation activity against mutant EGFRdel19 during cell starvation [97]. These compounds strongly induced the degradation of mutant but not wild-type EGFR in cancer cell lines and effectively suppressed the growth of lung cancer cells. Serum starvation has been found to enhance the degradation effect, which may lead to improved efficacy in tumors where serum deprivation is often observed. Overall proteomic analysis showed that the engineered compounds have high selectivity for EGFR. In pharmacokinetic studies in mice, one of the degraders, which recruits VHL E3 ligase, showed increased activity.
Osimertinib-based PROTAC compounds were synthesized and evaluated for cytotoxicity against NSCLC cell line. The selected compounds had high IC50 value of 0.413 μM in PC9 (EGFRdel19) and 0.657 μM in H1975 (EGFRL858R/T790M) cells [98]. The tested compounds arrested the growth of mutant cells in the G0/G1 phase by induction of apoptosis. Carnertinib-based covalent compounds have been also reported to cause high level degradation of EGFRdel19 mutant and antiproliferative activity against cancer cells, but moderate degradation of EGFRL858R/T790M and weak antiproliferative activity against this double mutant harboring cells [99]. In this study, the role of the autophagy-lysosomal pathway in EGFR degradation appeared to be more important than expected. A number of degraders have also been designed and developed by linking previously selected pyrido-[2, 3-d]pyrimidin-7-one based compound to the appropriate ligands and using linkers of various lengths to degrade the EGFRL858R/T790M double mutant in cancer cells [100]. One of the potent compounds selectively degraded the mutant receptor with a DC50 value of 5.9 nM, but had no apparent effect on wild-type EGFR. This study confirmed that the VHP ligase pathway provides the highest protein degradation among the ligases tested.
Another research group developed PROTACs inducing degradation of EGFRdel19 and EGFRL858R/T790M mutants in HCC827 and H1975 cells, respectively [101]. The use of starvation medium without fetal bovine serum enhanced EGFR degradation. An increased amount of the autophagy biomarker protein LC3α compared to its precursor LC3β [102] indicated autophagic degradation of the mutant EGFR, possibly via the autophagy-lysosomal system [103]. As shown later, the covalent binding strategy proved to be an effective approach in the development of degraders based on pyrimidine and purine structures [104]. However, compound containing a pyrimidine fragment exhibited moderate degradation of EGFR mutants, which prompted the authors to switch to the synthesis of purine-containing covalent degraders. One of purine-based agents significantly inhibited cell colony formation and growth of H1975 and HCC827 cell lines at concentrations as low as 30 nM and 1 nM, respectively, but was much weaker against cell harboring EGFR triple mutants. The compound remarkably reduced the phosphorylation level of EGFR, ERK, and AKT in H1975 and HCC827 cells at concentrations as low as 100 nM and 3 nM, respectively, while leaving phosphorylated B-RAF cells intact. Furthermore, the DC50 values of EGFRL858R/T790M and EGFRdel19 mutants were 1.56 nm and 0.49 nM, respectively. After complete consumption of new compounds, the content of mutant EGFRL858R/T790M began to recover rapidly.
It should be noted that acquired clinical resistance to EGFR inhibitors in cancer is usually the result of a tertiary mutation combining C797S mutation, MET amplification, and KRAS mutations [105], among which the C797S mutation in EGFR is dominant one [106]. New degraders were synthesized to eliminate the receptor upon expression of C797S mutation in triple EGFR mutant cells [107]. One of the allosteric compound-based degraders showed advantage in eliminating the triple mutant EGFRL858R/T790M/C797S from cancer cells while the degradation of another important triple mutant EGFRdel19/T790M/C797S was unsuccessful.
PROTACs provide cyclic ubiquitination of the targeted protein, resulting in a decrease in the actual number of active degraders capable of damaging the target protein in diseased cells. The technology is not tissue specific and readily distributes in non-target tissues after systemic administration, causing protein dysfunction in normal tissues [108]. Additionally, the side effects caused by off-target toxicity severely limit the continued use of targeted protein degradation [109,110]. Using antibody conjugates, degraders can be delivered specifically to cancer-associated target cells [111]. To overcome these limitations, pro-PROTACs were constructed by introducing endogenous and exogenous stimulus-responsive cellular motifs into key binding sites of hetero-bifunctional molecules [112,113]. A non-invasive exogenous stimulus was also applied to create photo-guided drug delivery using light and near-infrared activated PROTAC platforms [114,115,116].
A significant disadvantage of chemotherapy is associated with its negative effect on the immune system, while alternative immunotherapy with monoclonal antibodies (mAbs) has a positive effect on the immune system [117]. Various mAbs targeting the extracellular region of EGFR have been developed and approved by many laboratories to inhibit receptor activity in cancer. Among them, cetuximab, matuzumab, panitumumab, and necitumumab have been used to treat patients with NSCLC [118,119]. These anti-EGFR mAbs bind to the extracellular region of the receptor and block the ATP binding site, resulting in inhibition of EGFR activity [120].
To date, more than a dozen protein-degrading drugs have entered phase I clinical trials, and two therapeutic agents are in phase II trials for the treatment of ER+/HER2- breast cancer and castration-resistant prostate cancer [121,122,123]. However, the search for chemical and immunological inhibitors of EGFR in phase I and II clinical trials continues with the hope of discovering reliable therapeutic drugs [124].
4. Alternative strategies for EGFR degradation
Inspired by the attractiveness and success of small molecule disruptors, novel antibody-based degraders such as Lysosome-targeting chimera (LYTAC) [125], Autophagosome-binding compounds (ATTEC) [126], Autophagy-targeting chimera (AUTAC) [127], and AUTOphagy targeting chimera (AUTOTAC) [128] have recently been developed for cancer treatment. These approaches are based on the use of the autophagy-lysosomal degradation system, which is independent of the proteasomal degradation system [129] and ensures the degradation of misfolded proteins and damaged organelles through endocytosis, phagocytosis, and autophagy in cells [130,131,132,133]. The activity of such protein degraders in cancer cells is of great importance for the development of new drugs for the treatment of oncological, neurodegenerative, and other diseases.
A special place among these approaches is occupied by the LYTACs technology, which is designed to degrade extracellular proteins, including cell surface receptors, membrane proteins, and secreted proteins via the lysosomal pathway [125]. To implement this approach, the cation-independent mannose-6-phosphate receptor (CI-M6PR) was covalently conjugated with the corresponding monoclonal antibody to recognize the target protein located in the cytoplasmic membrane (Figure 3). The complex is then taken up by endocytosis and cleaved through the autophagic-lysosomal pathway. Cetuximab, an FDA-approved anti-EGFR antibody, resulted in 70% degradation of EGFR in HeLa cells. EGFR degradation by GalNAc-LYTAC attenuated EGFR signaling compared to antibody inhibition. LYTAC, containing a 3.4 kDa binding peptide coupled to a tri-GalNAc ligand, has been shown to degrade integrins and reduce cancer cell proliferation. The LYTAC methodology not only shows high degradation of target proteins, but also provides selectivity for cell types [130,134]. These impressive results under the leadership of Bertozzi C. R. (Nobel Prize in Chemistry 2022) in the field of developing a new technology for the targeted degradation of extracellular and membrane proteins are of great importance for expanding the range of targeted proteins in the treatment of cancer.
Despite significant progress in technological diversity, targeted protein chemotherapy does not lead to the death of cancer cells. It can be assumed that TPD is rather useful in slowing down the progression of cancer, but not in completely stopping the development and progression of the tumor. Therefore, the search for other therapeutic strategies for the degradation and elimination of key cancer proteins is an important therapeutic challenge.
5. New allosteric chemicals bind to EGFR and lead to cancer cell death
In 2019, we described a targeted protein degradation based on the use of allosteric small molecules that can degrade a transmembrane receptor and lead to the death of cancer cells [135]. The elucidation of the molecular processes involved allowed us to explain the mechanism of directed EGFR degradation and postulate an alternative vision of cancer chemotherapy.
We designed and synthesized furfuryl derivatives of 4-allyl-5-[2-(4'-alkoxyphenyl)-quinolin-4-yl]-4H-1,2,4-triazole-3-thiol (briefly FQTT), by combining three scaffolds into one molecule (Figure 4A). Alkyl ester substituents of various lengths were attached to the benzene ring to obtain modified compounds capable of increasing the sensitivity of the target protein to the action of proteases. Western blotting showed a significant reduction in EGFR and tyrosine phosphorylation bands in breast cancer, prostate cancer, and lung cancer cells treated with FQTT. In addition, a decrease in the amount of some proteins not associated with EGFR signaling pathways was observed, suggesting the involvement of protein degradation [134,135]. MDA MB-468 triple negative breast cancer cells lack expression of the estrogen receptor, progesterone receptor, and ErbB2, but overexpress EGFR and tend to metastasize to major organs [136]. MDA MB-468 cells were used to investigate the reason for the decrease in EGFR levels caused by new chemicals. Notably, the cytotoxicity of the most active compounds VM26 and VM25 at IC50 of 12.6 μM and 14.4 μM, respectively, is similar to that of gefitinib at IC50 of 15.2 μM, a well-known anti-EGFR drug in cancer therapy.
FQTT compounds bind to an allosteric pocket located in close proximity to the EGFR catalytic site and cause only a short-term and very weak inhibition of EGFR tyrosine phosphorylation in cells in the presence of EGF ligand. Molecular dynamics modeling has shown that short-chain like methyl or ethyl in the compounds is not bulky enough to fill the hydrophobic allosteric pocket, while longer chain like butyl or pentyl almost completely occupies this space (Figure 1C). This can lead to the interaction of long-chain alkyl ethers with the Met766 residue located in the αCβC loop near the ATP-binding site and cause possible degradation of EGFR.
A distinct increase in the amount of the autophagy biomarker LC3β was revealed due to the transition of LC3α to LC3β in cancer cells treated with FQTT derivatives [135]. The transition of LC3α to LC3β indicates an increase in the activity of autophagosomes responsible for the endocytic degradation of EGFR, previously proven by other authors [137]. Therefore, the significant degradation of the transmembrane receptor by small allosteric chemicals observed in our study (Figure 4B), initiated by lysosomes during endocytosis, was called allosteric autophagy (alloAUTO).
Heat shock chaperones (Hsp) bind to more than 700 misfolded proteins and protect them from ubiquitination and subsequent degradation by the 26S proteasome during proliferation, invasion, metastasis, and death of cancer cells [138]. The Hsp90a chaperone is highly expressed in cancer cells, and suppression of the defense mechanism leads to the degradation of misfolded client proteins by cellular proteasomes [139]. Interestingly, Hsp90 recognizes the αCβ4 loop in the intracellular domain of EGFR tyrosine kinase and likely binds to it, resulting in blocking of the catalytic site [140,141]. We demonstrated that EGFR degradation by FQTT derivatives is associated with a decrease in the amount of a Hsp90α detected by immunoprecipitation with anti-EGFR mAb [142]. This means that a decrease in EGFR folding should promote greater degradation of the receptor protein in addition to the degradation of EGFR caused by the autophagy-lysosome system in endosomes.
Incubation of cancer cells for more than two hours with FQTT derivatives leads to significant degradation of EGFR, which is accompanied by a decrease in the content of cytoskeletal proteins β-actin and α-tubulin, commonly used as load controls in Western blotting. This unexpected effect was caused by the detachment of cells from the surface of immunological wells. The action of each compound on cell detachment was quantified by counting the amount of different proteins in the cell culture supernatant collected over adherent cells after treatment with the respective compound.
Traces of EGFR were detected in cells attached to the extracellular matrix (ECM), while EGFR almost completely disappeared from detached cells, especially in serum-free media compared to serum-supplemented media after exposure to FQTT derivatives. It was suggested that, in addition to the degradation of EGFR, these chemical compounds cause a strong detachment of cancer cells from the extracellular matrix, especially under conditions of cell starvation for EGF or glutamine.
Nutrient deficiency is known to disrupt the EGFR-driven signaling cascade, leading to cell detachment from the ECM and ultimately programmed cell death known as anoikis (a Greek word meaning “loss of home”) [143]. The metastatic progression of inflammatory tumors suggests that cancer cells are more resistant to anoikis compared to healthy cells. The cytoskeleton consists of actin polymers and microtubules formed by tubulin polymers, which, together with other proteins, allow integrins to attach to ECM [144]. The phosphorylation status of EGFR in downstream signaling pathways determines the functional state of integrins, which are transmembrane receptors that mediate cell adhesion to the ECM. Importantly, EGFR regulates normal cytoskeletal function via the MAPK/ERK pathway by phosphorylation of the proapoptotic Bim, a sensor protein important for interaction with microtubules [145]. Interruption of this signaling pathway by blocking Bim phosphorylation leads to sequestration of the cytoskeleton and detachment of healthy cells.
Defective regulation of apoptosis is considered one of the hallmarks of cancer [10]. The apoptosis pathway is controlled by the Bcl-2 protein family, which contains both pro-apoptotic and pro-survival members that balance the decision between cell life and death. The Bcl-2 family of proteins plays a critical role in apoptosis initiated by internal signaling by regulating the integrity of the mitochondrial outer membrane (MOM) [146]. The study of the dynamic interaction between proteins of the BCL-2 family and how they control the apoptotic death of healthy and diseased cells remains an important task in the treatment of malignant tumors. Three isoforms of Bim result from alternative mRNA splicing and give rise to BimEL, BimL and BimS proteins [147]. All three Bim isoforms contain a BH3 domain required for binding other Bcl-2 family proteins and a C-terminal sequence for binding to the MOM protein [148]. Overexpression of the pro-apoptotic Bim protein has been found in the cytoskeleton and mitochondria, and this protein is constitutively overexpressed in prostate and breast cancer cells, as well as in primary tumor cells [149]. Recent studies on the dynamic interactions between BCL-2 family proteins and how they control the apoptotic death of healthy and diseased cells have opened up new avenues for therapeutic intervention [150]. How Bim is activated in different types of cancer cells is still unknown.
To understand whether the destabilization of the cytoskeletal mechanism is related to the status of the Bim sensor protein, protein expression was assessed using immunofluorescence imaging and Western blotting. Kinetic analyzes showed a transient and significant increase in BimEL expression after one hour of exposure of cells to FQTT compounds in serum-deprived medium, followed by a decrease in the level of this protein after three hours of exposure (Figure 4C). Notably, high level of BimEL expression was associated with a decrease in EGFR expression after one hour. The amount of lysosomal protease LAMP-2 and cytoskeletal protein β-actin decreased later compared to EGFR. This two-rate decrease in the abundance of functionally unrelated proteins (Figure 5A) appears to reflect two processes: early and rapid degradation of EGFR by endocytosis, followed by slower degradation and disintegration of the cytoskeleton due to Bim sequestration.
To find out which major nutritional factors are involved in Bim-induced sequestration, protein profiles were compared in serum-deprived cultures after addition of EGF or glutamine or both for 6 hours. The addition of glutamine or a mixture of glutamine with EGF, and to a lesser extent, the addition of EGF alone, increased BimEL expression compared to vehicle (Figure 5B). In addition, a significant increase in the rate of BimEL Ser69 phosphorylation was found, which is likely due to an earlier increase in tyrosine phosphorylation in EGFR at Tyr1068 leading to the activation of signaling pathways. Thus, replenishment of the medium with a fresh portion of glutamine improved the functional state of Bim protein in cancer cells not exposed to FQTT derivatives.
Serum-deprived cells exposed to VM26 after addition of EGF, glutamine, or both showed slightly increased levels of expression of EGFR, LAMP-2, β-actin, and probably cleaved caspase 3, compared with low expression of these proteins in cells exposed to VM26 only [135]. BimEL expression was slightly reduced after EGF, glutamine, or both were added to VM26-treated culture, while this protein was more expressed in untreated culture after nutrient addition. Meanwhile, nutrient supplementation increased BimEL phosphorylation at Ser69 in culture not exposed to VM26, but strongly suppressed Bim phosphorylation, regardless of addition of EGF, glutamine, or both to culture exposed to VM26. It becomes clear that suppression of BimEL phosphorylation is associated with blocking of EGFR phosphorylation required for Bim activation. These data demonstrate the ability of allosteric degraders of EGFR to influence metabolic and energy balance in glutamine-deprived cancer cells.
Why does glutamine deficiency increase the ability of FQTT compounds to kill cancer cells? First, cancer cells grow rapidly and require more energy for protein synthesis than normal cells. Second, glutamine is converted to α-ketoglutarate, which feeds the tricarboxylic acid cycle with more ATP. Third, EGFR uses ATP to activate the catalytic site and to phosphorylate more than thirty amino acids, including seven tyrosine residues, when the receptor undergoes endocytosis [151]. Fourthly, glutamine is not stable at 37°C and the addition of glutamine to the culture medium is always necessary. Fifth, the ATP-dependent activity of a large amount of the Hsp90α chaperone is required to correct many misfolded proteins and protect them from degradation of the 26S proteasome [139]. Therefore, the dependence of EGFR activation and associated signaling processes on the content of glutamine in cells can be formulated simply as "no glutamine, no EGFR signaling".
6. New vision on cancer chemotherapy
Cell death is a fundamental biological function that completes various processes in human life, such as embryonic development or the removal of damaged cells in diseases. Based on the molecular mechanism of action of factors that stimulate death, biological death is represented by two main categories: programmed cell death and unprogrammed cell death [152,153]. According to this classification, unprogrammed cell death includes one type of cell suicide, namely necrosis, while twelve other types of cell death are recognized as programmed non-apoptotic cell death [154]. Combining different scientific views on the relationship between oncogenesis and programmed cell death is an important issue for the development of effective therapeutic agents against cancer. In this regard, anoikis is a type of programmed apoptotic cell death that behaves differently than programmed non-apoptotic cell death. Signaling pathways triggered by EGFR protect normal and cancer cells from anoikis [155,156].
FQTT compounds bind to a hydrophobic allosteric pocket located in the immediate vicinity of the ATP binding site in EGFR (see Figure 1C). An important role in this binding seems to be played by reorientation of chemicals from Arg803 to Arg841, which is consistent with the participation of Arg841 in the dynamic changes preceding the sulfenylation of Cys797 [135,157]. This probably leads to the interaction of longer alkyl ether chains of compounds with Met766 in the aCb4 loop located near the ATP-binding site. This rearrangement may accelerate and/or enhance the endocytic degradation of EGFR. Induced EGFR depletion leads to sequestration of Bim, which provokes the breakdown of the cytoskeleton. Two different authentic pathways, endocytic and cytoplasmic degradation, promote cell detachment. This course of logically connected events reflects the functional interplay that precedes the death of cancer cells.
The effect of EGFR degradation by FQTT compounds has fundamentally different consequences for cells than the inactivation of tyrosine kinase activity by inhibitors [158,159]. Targeting EGFR degradation has an advantage over EGFR inhibition because it promotes a more specific interruption of Bim phosphorylation leading to the death of cancer cells. Unlike cytostatic TKIs against EGFR, allosteric degraders of EGFR affect cell survival rather than growth and induce cancer cell death like cytotoxic molecules. This unexpected biological scenario is reminiscent of the return of «immortal» cancer cells to programmed cell death, anoikis. This means that EGFR allosteric inhibitors are not "cancer cell killers", but rather molecules that restore the lost ability of cancer cells to die like normal cells after a limited number of generations. Notably, cancer cell death resulting in tumor size reduction by allosteric FQTT compounds has been confirmed in vivo in a mouse model of sarcoma [160].
The proposed mechanism of targeted protein degradation suggests that allosteric degraders of EGFR are promising agents for chemotherapy of human metastatic tumors (Figure 6). Shutting down phosphorylation pathways by potent TKIs in proliferating cancer cells creates selective conditions for the emergence of different mutants through alternative mechanisms, such as H2O2 release, in the branched EGFR interactome in the tumor microenvironment. Conversely, protein degradation due to EGFR depletion results in cancer cell death, leaving fewer cells to proliferate, reducing the chance of new resistance mutations emerging. Thus, we believe that this study opens up opportunities to attenuate metastatic progression and reduce drug resistance in malignant tumors associated with aberrant behavior of transmembrane receptors in cancer cells.
7. Conclusions
Targeted protein degradation by chemotherapeutic agents has a number of drawbacks, including a rapid return to protein synthesis with full utilization of the therapeutic molecule in cancer cells. The treatment of tumors caused by transmembrane receptors requires a better understanding of the molecular mechanisms to improve the technological reliability of the degradation of target proteins. Our data on cancer cell death during the degradation of EGFR by chemical compounds are also of interest for the development of other transmembrane proteins degradation. In particular, the evaluation of compounds derived from FQTT in preventing the progression of tumors caused by an EGFR-paired receptor can be important for further researches. Cancer cell death by alloAUTO strategy look likes pro-apoptotic anoikis, which does not rule out other pathways leading to cancer cell death under the action of other agents. Elucidating this question could lead to new, more attractive strategies for the simultaneous degradation of proteins and the destruction of cancer cells in order to prevent the emergence of resistant mutants. Therefore, the clear advantages of technologies aimed at the degradation of key transmembrane proteins in tumor cells hold out hope for improved cancer treatment.
Author Contributions
V.S; writing - original draft preparation, writing - review and editing, visualization, project administration. N.I; data curation, R.S; formal analysis.
Funding
Previously published experimental research was initially funded by the National Academy of Sciences of the Republic of Armenia and the Agence Nationale de la Recherche (grant ANR-07-RIB-012, France) and was subsequently supported by CNRS and ProtNeteomix. The review article was prepared and written without financial support.
Acknowledgements
This review is dedicated to the former colleague of the authors, Professor Melkon Iradyan (1942-2018), who developed and synthesized the described anticancer drugs in this study at the Institute of Fine Organic Chemistry named after A. L. Mndzhoyan (Yerevan, Armenia).
Conflict of Interest and other Ethics Statements
The authors have no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Soneji S., Beltrán-Sánchez H., Sox H.C. Assessing progress in reducing the burden of cancer mortality, 1985–2005. J. Clin. Oncol., 2014, 32, 444-448. [CrossRef]
- Miller K.D., Nogueira L., Devasia T., Mariotto A.B., Yabroff K.R., Jemal A., Kramer J., Siegel R.L. Cancer treatment and survivorship statistics, 2022. CA Cancer J. Clin., 2022, 72(5):409-436. [CrossRef]
- Willemsen M.C., Mons U., Fernández E. Tobacco control in Europe: progress and key challenges. Tob Control 2022, 31:160–163. [CrossRef]
- Patel V.R., Qasim Hussaini S.M., Blaes A.H., Morgans A.K., Haynes A.B., Adamson A.S., Gupta A. Trends in the prevalence of functional limitations among US cancer survivors, 1999–2018. JAMA Oncol., 11 May 2023. [CrossRef]
- Jotte R.M., Spigel D.R. Advances in molecular-based personalized non-small-cell lung cancer therapy: targeting epidermal growth factor receptor and mechanisms of resistance. Cancer Med., 2015, 4(11), 1621-1632. [CrossRef]
- Castañeda A.M., Meléndez C.M., Uribe D., Pedroza-Díaz J. Synergistic effects of natural compounds and conventional chemotherapeutic agents: recent insights for the development of cancer treatment strategies. Heliyon, 2022, 8(6), e09519. [CrossRef]
- Seshacharyulu P., Ponnusamy M.P., Haridas D., Jain M., Ganti A.K., Batra S.K. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin. Ther. Targets. 2012, 16(1), 15-31. [CrossRef]
- Patnaik S.K., Chandrasekar M.J.N., Nagarjuna P., Ramamurthi D., Swaroop A.K. Targeting of ErbB1, ErbB2, and their dual targeting using small molecules and natural peptides: blocking EGFR cell signaling pathways in cancer: A mini-review. Mini Rev. Med. Chem., 2022, 21, 22(22), 2831-2846. [CrossRef]
- Hanahan D., Weinberg R.A. The hallmarks of cancer. Cell, 2000, 100(1), 57-70. [CrossRef]
- Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell, 2011, 144(5), 646-674. [CrossRef]
- Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discov. 2022 Jan;12(1):31-46. [CrossRef]
- Falzone L., Salomone S., Libra M. Evolution of cancer pharmacological treatments at the turn of the third millennium. Front. Pharmacol., 2018, 9:1300. [CrossRef]
- Du Z., Lovly C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol Cancer., 2018, 17(1), 58. [CrossRef]
- Levantini E., Maroni G., Del Re M., Tenen D.G. EGFR signaling pathway as therapeutic target in human cancers. Semin. Cancer Biol., 2022, 85, 253-275. [CrossRef]
- Pucci, C., Martinelli C., Ciofani G. Innovative approaches for cancer treatment: current perspectives and new challenges. Ecancermedicalscience, 2019, 13:961. [CrossRef]
- Zhang C., Xu C., Gao X., Yao Q. Platinum-based drugs for cancer therapy and anti-tumor strategies. Theranostics, 2022, 12(5), 2115-2132. [CrossRef]
- Schlessinger J. Receptor tyrosine kinases: legacy of the first two decades. Cold Spring Harb. Perspect. Biol., 2014, 6(3), a008912. [CrossRef]
- Sigismund S., Avanzato D., Lanzetti L. Emerging functions of the EGFR in cancer. Mol Oncol. 2018, 12(1), 3-20. [CrossRef]
- Uribe M.L., Marrocco I., Yarden Y. EGFR in Cancer: Signaling Mechanisms, Drugs, and Acquired Resistance. Cancers (Basel). 2021, 13(11), 2748. [CrossRef]
- Dawson J.P., Berger M.B., Lin C.C., Schlessinger J., Lemmon M.A., Ferguson K.M. Epidermal growth factor receptor dimerization and activation require ligand-induced conformational changes in the dimer interface. Mol. Cell. Biol. 2005 Sep, 25(17), 7734-42. [CrossRef]
- Stamos J., Sliwkowski M.X., Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 2002 Nov 29, 277(48), 46265-46272. [CrossRef]
- Bae Y.S., Kang S.W., Seo M.S., Baines I.C., Tekle E., Chock P.B., Rhee S.G. Epidermal growth factor (EGF)-induced generation of hydrogen peroxide. Role in EGF receptor-mediated tyrosine phosphorylation. J. Biol. Chem. 1997, 272(1), 217-221. [CrossRef]
- Paulsen C.E., Truong T.H., Garcia F.J., Homann A., Gupta V., Leonard S.E., Carroll K.S. Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nat. Chem. Biol. 2011 Dec 11, 8(1), 57-64. [CrossRef]
- Pan J., Carroll K. S. Chemical biology approaches to study protein cysteine sulfenylation. Biopolymers. 2014 Feb, 101(2), 165-172. [CrossRef]
- Sakanyan V., Angelini M., Le Béchec M., Lecocq M.F., Benaiteau F., Rousseau B., Gyulkhandanyan A., Gyulkhandanyan L., Logé C., Reiter E., Roussakis C., Fleury F. Screening and discovery of nitro-benzoxadiazole compounds activating epidermal growth factor receptor (EGFR) in cancer cells. Sci. Rep. 2014 Feb 5, 4:3977. [CrossRef]
- Sakanyan V., Hulin P., Alves de Sousa R., Silva V.A., Hambardzumyan A., Nedellec S., Tomasoni C., Logé C., Pineau C., Roussakis C., Fleury F., Artaud I. Activation of EGFR by small compounds through coupling the generation of hydrogen peroxide to stable dimerization of Cu/Zn SOD1. Sci. Rep. 2016, 6, 21088. [CrossRef]
- Sakanyan V., Benaiteau F., Alves de Sousa R., Pineau C., Artaud, I. Straightforward detection of reactive compound binding to multiple proteins in cancer cells: Towards a better understanding of electrophilic stress. Ann. Clin. Exp. Metabol. 2016, 1, 1006.
- Silva V.A., Lafont F., Benhelli-Mokrani H., Breton M.L., Hulin P., Chabot T., Paris F., Sakanyan V., Fleury F. Rapid diminution in the level and activity of DNA-dependent protein kinase in cancer cells by a reactive nitro-benzoxadiazole compound. Int. J. Mol. Sci. 2016 May 11, 17(5), 703. [CrossRef]
- Sakanyan V. Reactive Chemicals and Electrophilic Stress in Cancer: A Minireview. High Throughput. 2018 Apr 27, 7(2), 12. [CrossRef]
- Parzych K.R., Klionsky D.J. An overview of autophagy: morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014 Jan 20, 20(3), 460-473. [CrossRef]
- von Zastrow M., Sorkin A. Mechanisms for Regulating and Organizing Receptor Signaling by Endocytosis. Annu. Rev. Biochem. 2021 Jun 20, 90, 709-737. [CrossRef]
- Tomas A., Futter C.E., Eden E.R. EGF receptor trafficking: consequences for signaling and cancer. Trends Cell Biol. 2014 Jan, 24(1), 26-34. [CrossRef]
- Caldieri G., Malabarba M.G., Di Fiore P.P., Sigismund S. EGFR Trafficking in Physiology and Cancer. Prog. Mol. Subcell. Biol. 2018, 57, 235-272. [CrossRef]
- Zhou Y., Sakurai H. New trend in ligand-induced EGFR trafficking: A dual-mode clathrin-mediated endocytosis model. J. Proteomics. 2022 Mar 20, 255, 104503. [CrossRef]
- Mendelsohn J., Baselga J. Epidermal growth factor receptor targeting in cancer. Semin Oncol., 2006, 33(4):369-385. [CrossRef]
- Cohen P., Cross D., Jänne P.A. Kinase drug discovery 20 years after imatinib. Nat. Rev. Drug Discov., 2021, 20, 551−569. [CrossRef]
- Red Brewer M., Yun C.H., Lai D., Lemmon M.A., Eck M.J., Pao W. Mechanism for activation of mutated epidermal growth factor receptors in lung cancer. Proc. Natl. Acad. Sci. USA. 2013, 110(38), E3595-604. Erratum in: Proc. Natl. Acad. Sci. USA, 2013, 110(50), 20344. [CrossRef]
- Arter C., Trask L., Ward S., Yeoh S., Bayliss R. Structural features of the protein kinase domain and targeted binding by small-molecule inhibitors. J. Biol. Chem. 2022 Aug, 298(8), 102247. [CrossRef]
- Herbst R.S., Fukuoka M., Baselga J. Gefitinib - a novel targeted approach to treating cancer. Nat. Rev. Cancer, 2004, 4, 956−965. [CrossRef]
- Cappuzzo F., Ciuleanu T., Stelmakh L., Cicenas S., Szczésna A., Juhász E., Esteban E., Molinier O., Brugger W., Melezínek I., Klingelschmitt G., Klughammer B., Giaccone G. SATURN investigators. Erlotinib as maintenance treatment in advanced non-small-cell lung cancer: a multicentre, randomised, placebo-controlled phase 3 study. Lancet Oncol. 2010 Jun, 11(6), 521-529. [CrossRef]
- Jänne P.A. Challenges of detecting EGFR T790M in gefitinib/erlotinib-resistant tumours. Lung Cancer. 2008 Jun, 60 Suppl 2, S3-9. [CrossRef]
- Shah R., Lester J.F. Tyrosine kinase inhibitors for the yreatment of EGFR mutation-positive Non-Small-Cell Lung Cancer: A clash of the generations. Clin. Lung Cancer. 2020 May, 21(3), e216-e228. [CrossRef]
- Hirsch F. R., Scagliotti G. V., Mulshine J. L., Kwon R., Curran W. J., Wu Y.-L., Paz-Ares L. Lung cancer: current therapies and new targeted treatments. Lancet 2017, 389, 299−311. [CrossRef]
- Li L., Luo S., Lin H., Yang H., Chen H., Liao Z., Lin W., Zheng W., Xie X. Correlation between EGFR mutation status and the incidence of brain metastases in patients with non-small cell lung cancer. J. Thorac. Dis. 2017, 9, 2510–2520. [CrossRef]
- Russo A., Franchina T., Ricciardi G.R.R., Picone A., Ferraro G., Zanghì M., Toscano G., Giordano A., Adamo V. A decade of EGFR inhibition in EGFR-mutated non-small-cell lung cancer (NSCLC): Old successes and future perspectives. Oncotarget 2015, 6, 26814–26825. [CrossRef]
- Kobayashi S., Boggon T. J., Dayaram T., Janne P. A., Kocher O., Meyerson M., Johnson B. E., Eck M. J., Tenen D. G., Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 786−792. [CrossRef]
- Pao W., Miller V. A., Politi K. A., Riely G. J., Somwar R., Zakowski M. F., Kris M. G., Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005 Mar, 2(3):e73. [CrossRef]
- Godin-Heymann N., Ulkus L., Brannigan B. W., McDermott U., Lamb J., Maheswaran S., Settleman J., Haber D. A. The T790M “gatekeeper” mutation in EGFR mediates resistance to low concentrations of an irreversible EGFR inhibitor. Mol. Cancer Ther. 2008 Apr, 7(4), 874-879. [CrossRef]
- Yun C. H., Mengwasser K. E., Toms A. V., Woo M. S., Greulich H., Wong K. K., Meyerson M., Eck M. J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 2070−2075. [CrossRef]
- Yu H.A., Riely G.J. Second-generation epidermal growth factor receptor tyrosine kinase inhibitors in lung cancers. J. Natl. Compr. Canc. Netw. 2013 Feb 1, 11(2), 161-169. [CrossRef]
- Meng Y., Yu B., Huang H., Peng Y., Li E., Yao Y., Song C., Yu W., Zhu K., Wang K., Yi D., Du J., Chang J. Discovery of dosimertinib, a highly potent, selective, and orally efficacious deuterated EGFR targeting clinical candidate for the treatment of Non-Small-Cell Lung Cancer. J. Med. Chem., 2021, 64, 925−937.
- Liu Q., Sabnis Y., Zhao Z., Zhang T., Buhrlage S.J., Jones L.H., Gray N.S. Developing irreversible inhibitors of the protein kinase cysteinome. Chem. Biol. 2013 Feb 21, 20(2), 146-159. [CrossRef]
- da Cunha Santos G., Shepherd F. A., Tsao M. S. EGFR mutations and lung cancer. Annu. Rev. Pathol., 2011, 6, 49−69.
- Herbst R. S., Morgensztern D., Boshoff C. The biology and management of non-small cell lung cancer. Nature, 2018, 553, 446− 454. [CrossRef]
- Huang F., Han X., Xiao X., Zhou J. Covalent warheads targeting cysteine residue: the promising approach in drug development. Molecules. 2022 Nov 10, 27(22), 7728. [CrossRef]
- , Ercan D., Chen L.., Yun C. H., Li D., Capelletti M., Cortot A. B., Chirieac L., Iacob R. E., Padera R., Engen J. R., Wong,K. K., Eck M. J., Gray N. S., Janne, P. A. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature, 2009, 462, 1070−1074. [CrossRef]
- Finlay M. R., Anderton M., Ashton S., Ballard P., Bethel P. A., Box M. R., Bradbury R. H., Brown S. J., Butterworth S., Campbell A., Chorley C., Colclough N., Cross D. A., Currie G. S., Grist M., Hassall L., Hill G. B., James D., James M., Kemmitt P., Klinowska T., Lamont G., Lamont S. G., Martin N., McFarland H. L., Mellor M. J., Orme J. P., Perkins D., Perkins P., Richmond G., Smith P., Ward R. A., Waring M. J., Whittaker D., Wells S., Wrigley G. L. Discovery of a potent and selective EGFR inhibitor (AZD9291) of both sensitizing and T790M resistance mutations that spares the wild type form of the receptor. J. Med. Chem., 2014, 57, 8249−8267. [CrossRef]
- Shah M.P., Neal J.W. Targeting acquired and intrinsic resistance mechanisms in Epidermal Growth Factor Receptor mutant Non-Small-Cell Lung Cancer. Drugs. 2022 Apr, 82(6), 649-662. [CrossRef]
- He J., Zhou Z., Sun X., Yang Z., Zheng P., Xu S., Zhu W. The new opportunities in medicinal chemistry of fourth-generation EGFR inhibitors to overcome C797S mutation. Eur. J. Med. Chem. 2021 Jan 15, 210, 112995. [CrossRef]
- Wang Z., Yang J.J., Huang J., Ye J.Y., Zhang X.C., Tu H.Y., Han-Zhang H., Wu Y.L. Lung adenocarcinoma harboring EGFR T790M and in trans C797S responds to combination therapy of first- and third-generation EGFR TKIs and shifts allelic configuration at resistance. J. Thorac. Oncol. 2017 Nov,12(11), 1723-1727. [CrossRef]
- Jia Y., Yun C.H., Park E., Ercan D., Manuia M., Juarez J., Xu C., Rhee K., Chen T, Zhang H, Palakurthi S, Jang J, Lelais G, DiDonato M, Bursulaya B, Michellys PY, Epple R., Marsilje T.H., McNeill M., Lu W., Harris J., Bender S., Wong K.K., Jänne P.A., Eck M.J. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature, 2016, 534(7605), 129-132. [CrossRef]
- Wang S., Song Y., Liu D. EAI045: The fourth-generation EGFR inhibitor overcoming T790M and C797S resistance. Cancer Lett., 2017, 385:51-54. [CrossRef]
- To C., Jang J., Chen T., Park E., Mushajiang M., De Clercq D.J.H., Xu M., Wang S, Cameron MD, Heppner DE, Shin BH, Gero TW, Yang A, Dahlberg SE, Wong KK, Eck M.J., Gray N.S., Jänne P.A. Single and dual targeting of mutant EGFR with an allosteric inhibitor. Cancer Discov., 2019, 9(7), 926-943. [CrossRef]
- Jang J., To C., De Clercq D.J.H., Park E., Ponthier C.M., Shin B.H., Mushajiang M., Nowak R.P., Fischer E.S., Eck M.J., Jänne P.A., Gray N.S. Mutant-selective allosteric EGFR degraders are effective against a broad range of drug-resistant mutations. Angew. Chem. Int. Ed. Engl., 2020, 59(34), 14481-14489. [CrossRef]
- Dong H., Ye X., Zhu Y., Shen H., Shen H, Chen W., Ji M., Zheng M., Wang K., Cai Z., Sun H., Xiao Y., Yang P. Discovery of potent and wild-type-sparing fourth-generation EGFR Inhibitors for treatment of osimertinib-resistance NSCLC. J. Med. Chem., 2023, May 4. [CrossRef]
- Marasco M., Misale S. Resistance is futile with fourth-generation EGFR inhibitors. Nat. Cancer, 2022, 3(4):381-383. [CrossRef]
- Xu L., Xu B., Wang J., Gao Y., He X., Xie T., Ye X.Y. Recent advances of novel fourth generation EGFR inhibitors in overcoming C797S mutation of lung cancer therapy. Eur. J. Med. Chem., 2023, 245(Pt 1):114900. [CrossRef]
- Chirnomas D., Hornberger K.R., Crews C.M. Protein degraders enter the clinic - a new approach to cancer therapy. Nat. Rev. Clin. Oncol. 2023 Apr, 20(4), 265-278. [CrossRef]
- Sakamoto K.M., Kim K.B., Kumagai A., Mercurio F., Crews C.M., Deshaies R.J. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA, 2001, 98(15), 8554-8559. [CrossRef]
- Sakamoto K.M., Kim K.B., Verma R., Ransick A., Stein B., Crews C.M., Deshaies R.J. Development of Protacs to target cancer-promoting proteins for ubiquitination and degradation. Mol. Cell. Proteomics. 2003 Dec; 2(12), 1350-8. [CrossRef]
- Bondeson D., Mares A., Smith I., Ko E., Campos S., Miah A.H., Mulholland K.E., Routly N., Buckley D.L., Gustafson J.L., Zinn N., Grandi P., Shimamura S., Bergamini G., Faelth-Savitski M., Bantscheff M., Cox C., Gordon D.A., Willard R.R., Flanagan J.J., Casillas L.N., Votta B.J., den Besten W., Famm K., Kruidenier L., Carter P.S., Harling J.D., Churcher I., Crews C.M. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015, 11, 611–617. [CrossRef]
- Dale B., Cheng M., Park K.S., Kaniskan H.Ü., Xiong Y., Jin J. Advancing targeted protein degradation for cancer therapy. Nat. Rev. Cancer, 2021, 21(10), 638-654. [CrossRef]
- Békés M., Langley D.R., Crews CM. PROTAC targeted protein degraders: the past is prologue. Nat. Rev. Drug Discov. 2022 Mar, 21(3), 181-200. [CrossRef]
- Hipp M.S., Kasturi P., Hartl F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019 Jul, 20(7), 421-435. [CrossRef]
- Li X., Pu W., Zheng Q., Ai M., Chen S., Peng Y. Proteolysis-targeting chimeras (PROTACs) in cancer therapy. Mol Cancer. 2022 Apr 11, 21(1), 99. [CrossRef]
- Yau R., Rape M. The increasing complexity of the ubiquitin code. Nat. Cell Biol. 2016 May 27, 18(6), 579-586. [CrossRef]
- Bard J.A.M., Goodall E.A., Greene E.R., Jonsson E., Dong K.C., Martin A. Structure and function of the 26S proteasome. Annu. Rev. Biochem. 2018 Jun 20, 87, 697-724. [CrossRef]
- Wolska-Washer A., Smolewski, P. Targeting protein degradation pathways in tumors: Focusing on their role in hematological malignancies. Cancers (Basel). 2022 Aug 3, 14(15), 3778. [CrossRef]
- Ishida T., Ciulli A. E3 ligase ligands for PROTACs: How they were found and How to discover new ones. SLAS Discovery 2021, 26, 484−502. [CrossRef]
- Li M., Zhi Y., Liu B., Yao Q. Advancing strategies for proteolysis-targeting chimera design. J. Med. Chem. 2023 Feb 23; 66(4), 2308-2329. [CrossRef]
- Smith B. E., Wang S. L., Jaime-Figueroa S., Harbin A., Wang J., Hamman B. D., Crews C. M. Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nat. Commun. 2019 Jan 10, 10(1), 131. [CrossRef]
- Kofink C., Trainor N., Mair B., Wohrle S., Wurm M., Mischerikow N., Roy M.J., Bader G., Greb P., Garavel G., Diers E., McLennan R., Whitworth C., Vetma V., Rumpel K., Scharnweber M., Fuchs J.E., Gerstberger T., Cui Y., Gremel G., Chetta P., Hopf S., Budano N., Rinnenthal J., Gmaschitz G., Mayer M., Koegl M., Ciulli A., Weinstabl H., Farnaby W. A selective and orally bioavailable VHL-recruiting PROTAC achieves SMARCA2 degradation in vivo. Nat. Commun. 2022 Oct 10, 13(1), 5969. [CrossRef]
- Lu J., Huang, Y., Huang J., He R., Huang M., Lu X., Xu Y., Zhou F., Zhang Z., Ding K. Discovery of the first examples of threonine tyrosine kinase PROTAC degraders. J. Med. Chem. 2022 Feb 10, 65(3), 2313-2328. [CrossRef]
- Hu J., Hu B., Wang M., Xu F., Miao B., Yang C.-Y., Wang M., Liu Z. Hayes D.F., Chinnaswamy K., Delproposto J., Stuckey J., Wang S. Discovery of ERD-308 as a highly potent proteolysis targeting chimera (PROTAC) degrader of estrogen receptor (ER). J. Med. Chem. 2019, 62, 1420−1442.
- Yokoo H., Shibata N., Naganuma M., Murakami Y., Fujii K., Ito T., Aritake K., Naito M., Demizu Y. Development of a hematopoietic prostaglandin D synthase-degradation inducer. ACS Med. Chem. Lett. 2021, 12, 236−241. [CrossRef]
- Khan S., Zhang X., Lv D., Zhang Q., He Y., Zhang P., Liu X., Thummuri D., Yuan Y., Wiegand J.S., Pei J., Zhang W., Sharma A., McCurdy C.R., Kuruvilla V.M., Baran N., Ferrando A.A., Kim Y., Rogojina A., Houghton P.J., Huang G., Hromas R., Konopleva M., Zheng G., Zhou D. A selective BCL-XL PROTAC degrader achieves safe and potent antitumor activity. Nat. Med. 2019, 25, 1938−1947. [CrossRef]
- Li X., Song Y. Proteolysis-targeting chimera (PROTAC) for targeted protein degradation and cancer therapy. J. Hematol. Oncol. 2020, 13, 50.
- Zhao Q., Lan T., Su S., Rao Y. Induction of apoptosis in MDAMB-231 breast cancer cells by a PARP1-targeting PROTAC small molecule. Chem. Commun. (Camb). 2019 Jan 2, 55(3), 369-372. [CrossRef]
- Yan W., Pan B., Shao J., Lin H., Li H. Feasible column chromatography-free, multi-gram scale synthetic process of VH032 amine, which could enable rapid PROTAC library construction. ACS Omega. 2022 Jul 19, 7(30), 26015-26020. [CrossRef]
- Burslem G. M., Smith B. E., Lai A. C., Jaime-Figueroa S., McQuaid D. C., Bondeson D. P., Toure M., Dong H., Qian Y., Wang J., Crew A. P., Hines J., Crews C. M. The advantages of targeted protein degradation over inhibition: An RTK case study. Cell Chem. Biol. 2018, 25, 67−77.e3. [CrossRef]
- Jones L.H., Mitchell C.A., Loberg L., Pavkovic M., Rao M., Roberts R., Stamp K., Volak L., Wittwer M.B., Pettit S. Targeted protein degraders: a call for collective action to advance safety assessment. Nat. Rev. Drug Discovery. 2022, 21, 401−402. [CrossRef]
- Zeng S., Huang W., Zheng X., Cheng L., Zhang Z., Wang, J., Shen Z. Proteolysis targeting chimera (PROTAC) in drug discovery paradigm: Recent progress and future challenges. Eur. J. Med. Chem. 2021, 210, 112981. [CrossRef]
- Ottis, P.; Crews, C. M. Proteolysis-targeting chimeras: induced protein degradation as a therapeutic strategy. ACS Chem. Biol. 2017 Apr 21, 12(4), 892-898. [CrossRef]
- Bondeson D.P., Smith B.E., Burslem G.M., Buhimschi A.D., Hines J., Jaime-Figueroa S., Wang J., Hamman B. D., Ishchenko A., Crews C. M. Lessons in PROTAC design from selective degradation with a promiscuous warhead. Cell Chem. Biol. 2018 Jan 18, 25(1), 78-87.e5. [CrossRef]
- Cromm P. M., Samarasinghe K.T.G., Hines J., Crews C. M. Addressing kinase-independent functions of Fak via PROTAC mediated degradation. J. Am. Chem. Soc. 2018 Dec 12, 140(49), 17019-17026. [CrossRef]
- Burslem G.M., Crews C.M. Proteolysis-targeting chimeras as therapeutics and tools for biological discovery. Cell. 2020 Apr 2,181(1), 102-114. [CrossRef]
- Cheng M., Yu X., Lu K., Xie L., Wang L., Meng F., Han X., Chen X., Liu J., Xiong Y., Jin J. Discovery of potent and selective epidermal growth factor receptor (EGFR) bifunctional small-molecule degraders. J. Med. Chem. 2020, 63, 1216−1232. [CrossRef]
- He K., Zhang Z., Wang W., Zheng X., Wang X., Zhang X. Discovery and biological evaluation of proteolysis targeting chimeras (PROTACs) as EGFR degraders based on osimertinib and lenalidomide. Bioorg. Med. Chem. Lett. 2020 Jun 15, 30(12), 127167. [CrossRef]
- Qu X., Liu H., Song X., Sun N., Zhong H., Qiu X., Yang X., Jiang B. Effective degradation of EGFRL858R+T790M mutant proteins by CRBN-based PROTACs through both proteosome and autophagy/lysosome degradation systems. Eur. J. Med. Chem. 2021 Jun 5, 218, 113328. [CrossRef]
- Zhang X., Xu F., Tong L., Zhang T., Xie H., Lu X., Ren X., Ding K. Design and synthesis of selective degraders of EGFRL858R/T790M mutant. Eur. J. Med. Chem. 2020 Apr 15, 192, 112199. [CrossRef]
- Zhao H.-Y., Yang X.-Y., Lei H., Xi X.-X., Lu S.-M., Zhang J.-J., Xin M., Zhang S.-Q. Discovery of potent small molecule PROTACs targeting mutant EGFR. Eur. J. Med. Chem. 2020 Dec 15, 208, 112781. [CrossRef]
- Kabeya Y., Mizushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000 Nov 1, 19(21), 5720-5728. [CrossRef]
- Zhang H., Zhao H.-Y., Xi X.-X., Liu Y.-J., Xin M., Mao S., Zhang J.-J., Lu A. X., Zhang S.-Q. Discovery of potent epidermal growth factor receptor (EGFR) degraders by proteolysis targeting chimera (PROTAC). Eur. J. Med. Chem. 2020 Mar 1, 189, 112061. [CrossRef]
- Zhao H.-Y., Wang H.P., Mao Y.Z., Zhang H., Xin M., Xi X-X., Lei H., Mao S., Li D.H, Zhang S-Q. Discovery of potent PROTACs targeting EGFR mutants through the optimization of covalent EGFR ligands. J. Med. Chem. 2022 Mar 24; 65(6), 4709-4726. [CrossRef]
- Ricordel C., Friboulet L., Facchinetti F., Soria J. C. Molecular mechanisms of acquired resistance to third-generation EGFR-TKIs in EGFR T790M-mutant lung cancer. Ann. Oncol. 2018 Jan 1, 29(suppl_1), 128-137. [CrossRef]
- Wang S. H., Tsui S. T., Liu C., Song Y. P., Liu D. L. EGFR C797S mutation mediates resistance to third-generation inhibitors in T790M-positive non-small cell lung cancer. J. Hematol. Oncol. 2016 Jul 22, 9(1), 59. [CrossRef]
- Zhang H., Xie R., AI-Furas H., Li Y., Wu Q., Li J., Xu F., Xu T. Design, synthesis, and biological evaluation of novel EGFR PROTACs targeting del19/T790M/C797S mutation. ACS Med. Chem. Lett. 2022 Jan 14, 13(2), 278-283. [CrossRef]
- Chen Y., Tandon I., Heelan W., Wang Y.X., Tang W.P., Hu Q.Y. Proteolysis-targeting chimera (PROTAC) delivery system: advancing protein degraders towards clinical translation. Chem. Soc. Rev. 2022, 51, 5330−5350.
- Raina K., Lu J., Qian Y., Altieri M., Gordon D., Rossi A.M.K., Wang J., Chen X., Dong H.Q., Siu K., Winkler J.D., Crew A.P., Crews C.M., Coleman K.G. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 7124−7129.
- Moreau K., Coen M., Zhang A.X., Pachl F., Castaldi M.P., Dahl G., Boyd H., Scott C., Newham P. Proteolysis-targeting chimeras in drug development: A safety perspective. Brit. J. Pharmacol. 2020, 177, 1709−1718. [CrossRef]
- Hong K.B., An H. Degrader-antibody conjugates: Emerging new modality. J. Med. Chem. 2023 Jan 12, 66(1), 140-148. [CrossRef]
- Li K., Crews C.M. PROTACs: past, present and future. Chem. Soc. Rev. 2022 Jun 20, 51(12), 5214-5236. [CrossRef]
- Dragovich P.S. Degrader-antibody conjugates. Chem. Soc. Rev. 2022 May 23, 51(10), 3886-3897. [CrossRef]
- Yu N., Huang L., Zhou Y., Xue T., Chen Z., Han G. Near-infrared-light activatable nanoparticles for deep-tissue-penetrating wireless optogenetics. Adv. Health. Mater. 2019 Mar, 8(6), e1801132. [CrossRef]
- Zhang Y., Zhang Y., Song G., He Y., Zhang X., Liu Y., Ju H. A DNA-azobenzene nanopump fueled by upconversion luminescence for controllable intracellular drug release. Angew. Chem. Int. Ed. Engl. 2019 Dec 9, 58(50), 18207-18211. [CrossRef]
- He Q., Zhou L., Yu D., Zhu R., Chen Y., Song M., Liu X., Liao Y., Ding T., Fan W., Yu W. Near-infrared-activatable PROTAC nanocages for controllable target protein degradation and on-demand antitumor therapy. J. Med. Chem. 2023 Jun 6. [CrossRef]
- Schirrmacher V. From chemotherapy to biological therapy: A review of novel concepts to reduce the side effects of systemic cancer treatment (Review). Int. J. Oncol. 2019 Feb, 54(2), 407-419. [CrossRef]
- Pirker, R. EGFR-directed monoclonal antibodies in non-small cell lung cancer. Target. Oncol. 2013 Mar, 8(1), 47-53. [CrossRef]
- Tabasinezhad M., Omidinia E., Talebkhan Y., Omrani M.D., Mahboudi F., Ghaedi H., Wenzel W. The effects of somatic mutations on EGFR interaction with anti-EGFR monoclonal antibodies: Implication for acquired resistance. Proteins, 2020 Jan, 88(1), 3-14. [CrossRef]
- Capdevila J., Elez E., Macarulla T., Ramos F.J., Ruiz-Echarri M., Tabernero J. Anti-epidermal growth factor receptor monoclonal antibodies in cancer treatment. Cancer Treat. Rev. 2009 Jun, 35(4), 354-363. [CrossRef]
- Ermondi G., Garcia-Jimenez D., Caron G. PROTACs and building blocks: The 2D chemical space in very early drug discovery. Molecules. 2021 Jan 28, 26(3), 672. [CrossRef]
- Weng G., Cai X., Cao D., Du H., Shen C., Deng Y., He Q., Yang B., Li D., Hou T. PROTAC-DB 2.0: an updated database of PROTACs. Nucleic Acids Res. 2023 Jan 6, 51(D1), D1367-D1372. [CrossRef]
- O'Brien Laramy M.N., Luthra S., Brown M.F., Bartlett D.W. Delivering on the promise of protein degraders. Nat Rev Drug Discov. 2023 May, 22(5), 410-427. [CrossRef]
- Li S., de Camargo Correia G.S., Wang J., Manochakian R., Zhao Y., Lou Y. Emerging targeted therapies in advanced Non-Small-Cell Lung Cancer. Cancers (Basel). 2023 May 24, 15(11), 2899. [CrossRef]
- Banik S.M., Pedram K., Wisnovsky S., Ahn G., Riley N.M., Bertozzi C.R. Lysosome- targeting chimaeras for degradation of extracellular proteins. Nature, 584 (2020) 291–297. [CrossRef]
- Li Z., Zhu C., Ding Y., Fei Y., Lu B. ATTEC: a potential new approach to target proteinopathies. Autophagy, 2020 Jan, 16(1), 185-187. [CrossRef]
- Takahashi D., Arimoto H. Targeting selective autophagy by AUTAC degraders. Autophagy, 2020 Apr, 16(4), 765-766. [CrossRef]
- Ji C.H., Lee M.J., Kim H.Y., Heo A. J., Park D.Y., Kim Y.K., Kim B.Y., Kwon Y.T. Targeted protein degradation via the autophagy-lysosome system: AUTOTAC (AUTOphagy-TArgeting Chimera). Autophagy, 2022, 18, 2259−2262.
- Ballabio A., Bonifacino J.S. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 101−118. [CrossRef]
- Ahn G., Banik S.M., Bertozzi C.R. Degradation from the outside in: Targeting extracellular and membrane proteins for degradation through the endolysosomal pathway. Cell Chem. Biol. 2021 Jul 15, 28(7), 1072-1080. [CrossRef]
- Hong D, Zhou B, Zhang B, Ren H, Zhu L, Zheng G, Ge M, Ge J. Recent advances in the development of EGFR degraders: PROTACs and LYTACs. Eur. J. Med. Chem. 2022 Sep 5, 239, 114533. [CrossRef]
- Maity P., Chatterjee J., Patil K.T., Arora S., Katiyar M.K., Kumar M., Samarbakhsh A., Joshi G., Bhutani P., Chugh M., Gavande N.S., Kumar R. Targeting the epidermal growth factor receptor with molecular degraders: state-of-the-art and future opportunities. J. Med. Chem. 2023 Mar 9, 66(5), 3135-3172. [CrossRef]
- Li X., Liu Q., Xie X., Peng C., Pang Q., Liu B., Han B. Application of novel degraders employing autophagy for expediting medicinal research. J. Med. Chem. 2023 Feb 9, 66(3), 1700-1711. [CrossRef]
- Ahn G., Banik S.M., Miller C.L., Riley N.M., Cochran J.R., Bertozzi C.R. LYTACs that engage the asialoglycoprotein receptor for targeted protein degradation. Nat. Chem. Biol. 2021, 17, 937-946. [CrossRef]
- Iradyan M., Iradyan N., Hulin P., Hambardzumyan A., Gyulkhandanyan A., Alves de Sousa R., Hessani A., Roussakis C., Bollot G., Bauvais C., Sakanyan V. Targeting degradation of EGFR through the allosteric site leads to cancer cell detachment-promoted death. Cancers (Basel). 2019 Aug 1, 11(8), 1094. [CrossRef]
- Foulkes W.D., Smith I.E., Reis-Filho J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010 Nov 11, 363(20),1938-1948. [CrossRef]
- Tanida I., Ueno T., Kominami E. LC3 conjugation system in mammalian autophagy. Int. J. Biochem. Cell. Biol. 2004 Dec, 36(12), 2503-2518. [CrossRef]
- Taipale M., Jarosz D.F., Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat. Rev. Mol. Cell. Biol. 2010 Jul, 11(7), 515-528. [CrossRef]
- Miyata Y., Nakamoto H., Neckers L. The therapeutic target Hsp90 and cancer hallmarks. Curr. Pharm. Des. 2013, 19(3), 347-365. [CrossRef]
- Citri A., Harari D., Shohat G., Ramakrishnan P., Gan J., Lavi S., Eisenstein M., Kimchi A., Wallach D., Pietrokovski S., Yarden Y. Hsp90 recognizes a common surface on client kinases. J. Biol. Chem. 2006 May 19, 281(20), 14361-14369. [CrossRef]
- Pick E., Kluger Y., Giltnane J.M., Moeder C., Camp R.L., Rimm D.L., Kluger H.M. High HSP90 expression is associated with decreased survival in breast cancer. Cancer Res. 2007 Apr 1, 67(7), 2932-2937. [CrossRef]
- Sakanyan V.A., Iradyan M.A., Iradyan N.S. Development of targeted EGFR degradation for cancer treatment. Nat. Academy Sci. Armenia Reports. 2022, 122(3), 218-227. [CrossRef]
- Paoli P., Giannoni E., Chiarugi P. Anoikis molecular pathways and its role in cancer progression. Biochim. Biophys. Acta. 2013 Dec,1833(12), 3481-3498. [CrossRef]
- V lahakis A., Debnath J. The Interconnections between autophagy and integrin-mediated cell adhesion. J. Mol. Biol. 2017 Feb 17, 429(4), 515-530. [CrossRef]
- Reginato M.J., Mills K.R., Paulus J.K., Lynch D.K., Sgroi D.C., Debnath J., Muthuswamy S.K., Brugge J.S. Integrins and EGFR coordinately regulate the pro-apoptotic protein Bim to prevent anoikis. Nat. Cell. Biol. 2003 Aug, 5(8), 733-740. [CrossRef]
- Chi X., Nguyen D., Pemberton J.M., Osterlund E.J., Liu Q., Brahmbhatt H., Zhang Z., Lin J., Leber B., Andrews D.W. The carboxyl-terminal sequence of bim enables bax activation and killing of unprimed cells. eLife. 2020, 9:e44525. [CrossRef]
- O’Connor L., Strasser A., O’Reilly L.A., Hausmann G., Adams J.M., Cory S., Huang D.C. Bim: a novel member of the Bcl-2 family that promotes apoptosis. EMBO J. 1998, 17, 384–395. [CrossRef]
- Wilfling F., Weber A., Potthoff S., Vögtle F.N., Meisinger C., Paschen S.A., Häcker G. BH3-only proteins are tail-anchored in the outer mitochondrial membrane and can initiate the activation of bax. Cell Death Differentiation 2012, 19, 1328–1336. [CrossRef]
- Gogada R., Yadav N., Liu J., Tang S., Zhang D., Schneider A., Seshadri A., Sun L., Aldaz C.M., Tang D.G., Chandra D. Bim, a proapoptotic protein, up-regulated via transcription factor E2F1-dependent mechanism, functions as a prosurvival molecule in cancer. J. Biol. Chem. 2013 Jan 4, 288(1), 368-381. [CrossRef]
- Singh R., Letai A., Sarosiek, K. Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 20, 175–193 (2019). [CrossRef]
- Tong J., Taylor P., Moran M.F. Proteomic analysis of the epidermal growth factor receptor (EGFR) interactome and post-translational modifications associated with receptor endocytosis in response to EGF and stress. Mol. Cell. Proteomics. 2014 Jul, 13(7), 1644-1658. [CrossRef]
- Galluzzi L., Vitale I., Aaronson S.A., Abrams J.M., Adam D., Agostinis P., Alnemri E.S., Altucci L., Amelio I., Andrews D.W. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ., 2018, 25(3), 486-541. [CrossRef]
- Tang D., Kang R., Berghe T.V., Vandenabeele P., Kroemer G. The molecular machinery of regulated cell death. Cell Res., 2019, 29(5), 347-364. [CrossRef]
- Yan G., Elbadawi M., Efferth T. Multiple cell death modalities and their key features (Review). World Acad. Sci. J., 2020, 2, 39-48. [CrossRef]
- Le Gall M., Chambard J.C., Breittmayer J.P., Grall D., Pouyssegur J., Obberghen-Schilling E. The p42/p44 MAP kinase pathway prevents apoptosis induced by anchorage and serum removal. Mol. Biol. Cell, 2000, 11, 1103–1112. [CrossRef]
- Quadros M.R., Peruzzi F., Kari C., Rodeck U. Complex regulation of signal transducers and activators of transcription 3 activation in normal and malignant keratinocytes. Cancer Res., 2004, 64, 3934–3939. [CrossRef]
- Chamberlain P.P., Hamann L.G. Development of targeted protein degradation therapeutics. Nat. Chem. Biol. 2019 Oct, 15(10), 937-944. [CrossRef]
- Iradyan M.A., Iradyan N.S., Hambardzumyn A.A., Panosyan H.A., Tamazyan R.A., Ayvazyan A.G., Hovhannisyan G.S., Alves de sousa R., Sakanyan V.A. Selective N-, S-alkylation of 4-allyl-3-[2-(4-alkoxyphenyl)-quinolin-4-yl]-4,5-dihydro-1h-1,2,4-triazole-5-thiones with substituted benzylchlorides. synthesis, docking analysis and cytotoxic action. Chem. J. Arm. 2018, 71, 389–406.
- Iradyan M.A., Iradyan N.S., Hambardzumyan A.A., Minasyan N.S., Roussakis C., Sakanyan V.A. Synthesis of furfuryl derivatives of 4-allyl-1-(4-hydroxy-3-nitrobenzyl)-3-[2- (4-alkoxyphenyl)-quinolin-4-yl]-4,5-dihydro-1H-1,2,4-triazole-5-thions and their toxicity in cancer cells. Chem. J. Arm. 2018, 71, 559–570.
- Iradyan M.A., Iradyan N.S., Hambardzumyan A.A., Nersesyan L.E., Aharonyan A.S., Danielyan I.S., Muradyan R.E., Paronikyan R.V., Stepanyan G.M. Docking analysis and some biological properties of furfuryl derivatives of 4-allyl-5-[2-(4-alkoxyphenyl) quinolin-4-yl]-4H-1,2,4-triazol-3-thiol. Biol. J. Arm. 2018, 70, 100–107.
Figure 1.
EGFR dimerization (A), structural features of the dimeric form of EGFR (B), and binding of FQTT compounds VM3 (orange), VM25 (pink), VM26 (green), and gefitinib (yellow) to EGFR (C).
Figure 1.
EGFR dimerization (A), structural features of the dimeric form of EGFR (B), and binding of FQTT compounds VM3 (orange), VM25 (pink), VM26 (green), and gefitinib (yellow) to EGFR (C).
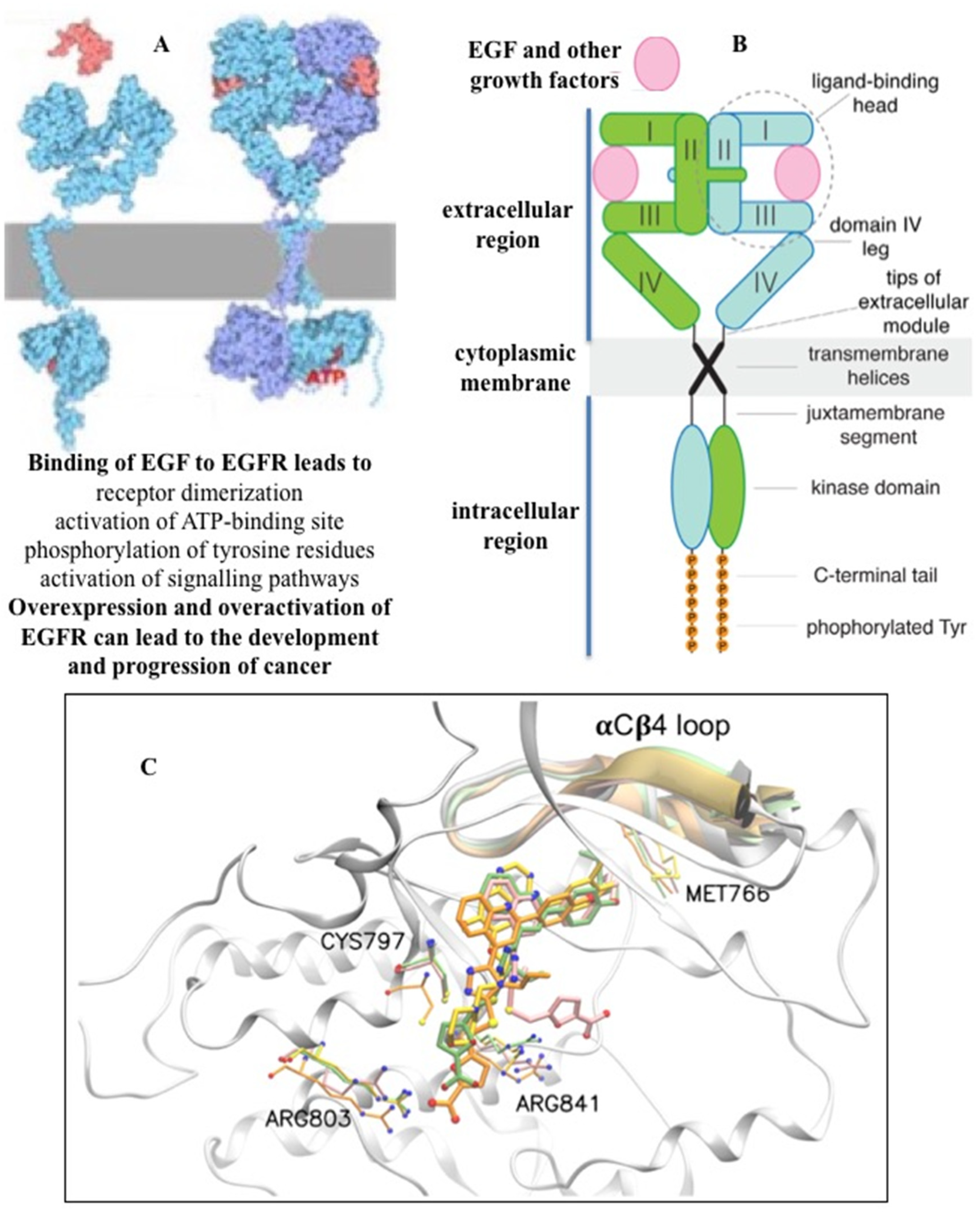
Figure 2.
Development process for a heterobifunctional PROTAC compound designed to degrade a target protein. Molecular image of a small degrader (circled) showing its binding to ubiquitin ligase (blue) and targeted protein (green) [73].
Figure 2.
Development process for a heterobifunctional PROTAC compound designed to degrade a target protein. Molecular image of a small degrader (circled) showing its binding to ubiquitin ligase (blue) and targeted protein (green) [73].
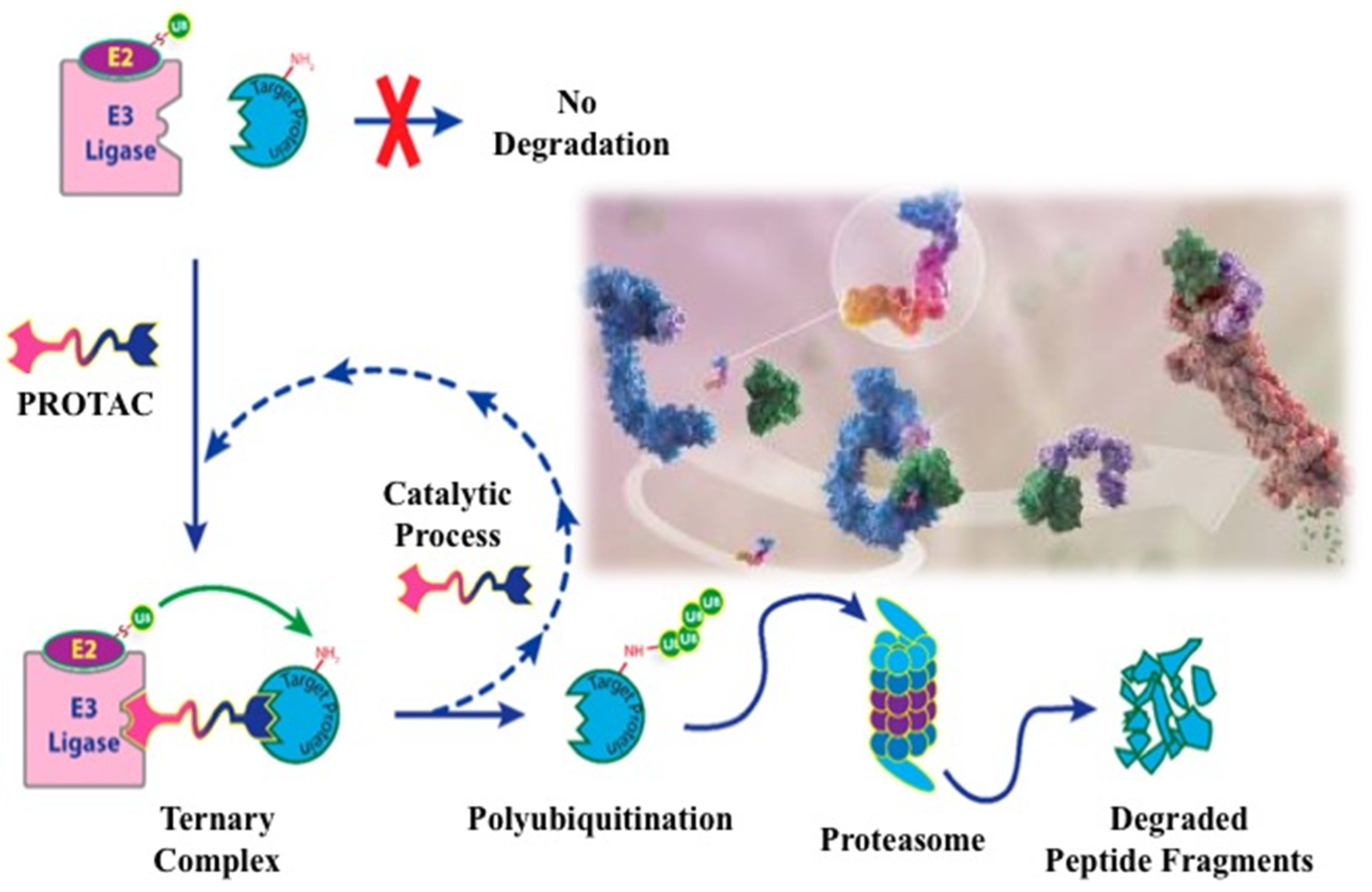
Figure 3.
Functionality of LYTAC for targeted and cell-specific degradation of proteins [130]. A - First-generation LYTACs use the cation-independent mannose-6-phosphate receptor (CI-M6PR) to degrade EGFR: B - GalNAc-LYTAC captures the liver-specific asialoglycoprotein receptor (ASPGR) to specifically target hepatocytes; C - Tri-GalNAc-DBCO ligand structure for ASGPR targeting.
Figure 3.
Functionality of LYTAC for targeted and cell-specific degradation of proteins [130]. A - First-generation LYTACs use the cation-independent mannose-6-phosphate receptor (CI-M6PR) to degrade EGFR: B - GalNAc-LYTAC captures the liver-specific asialoglycoprotein receptor (ASPGR) to specifically target hepatocytes; C - Tri-GalNAc-DBCO ligand structure for ASGPR targeting.
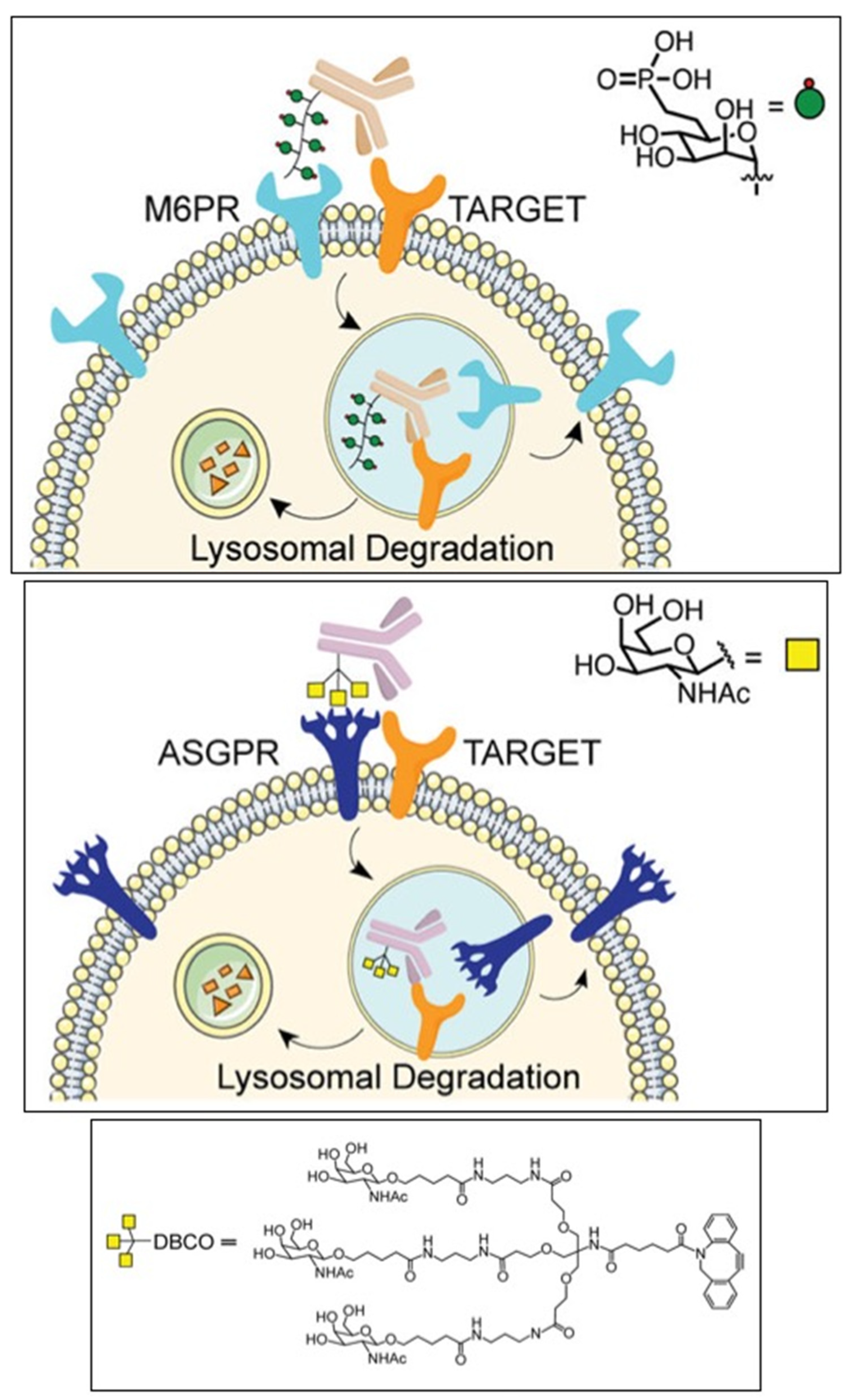
Figure 4.
Targeted protein degradation in cancer cells with heterocyclic compounds [135]. A - Furfuryl derivatives of 4-allyl-5-[2-(4'-alkoxyphenyl)quinolin-4-yl]-4H-1,2,4-triazole-3-thiol (FQTTs); B - compounds VM26 and VM25 dose-dependent degradation of EGFR and other proteins and reduced phosphorylation of EGFR; C - Bim response to the compound VM26 detected by immunofluorescence imaging.
Figure 4.
Targeted protein degradation in cancer cells with heterocyclic compounds [135]. A - Furfuryl derivatives of 4-allyl-5-[2-(4'-alkoxyphenyl)quinolin-4-yl]-4H-1,2,4-triazole-3-thiol (FQTTs); B - compounds VM26 and VM25 dose-dependent degradation of EGFR and other proteins and reduced phosphorylation of EGFR; C - Bim response to the compound VM26 detected by immunofluorescence imaging.
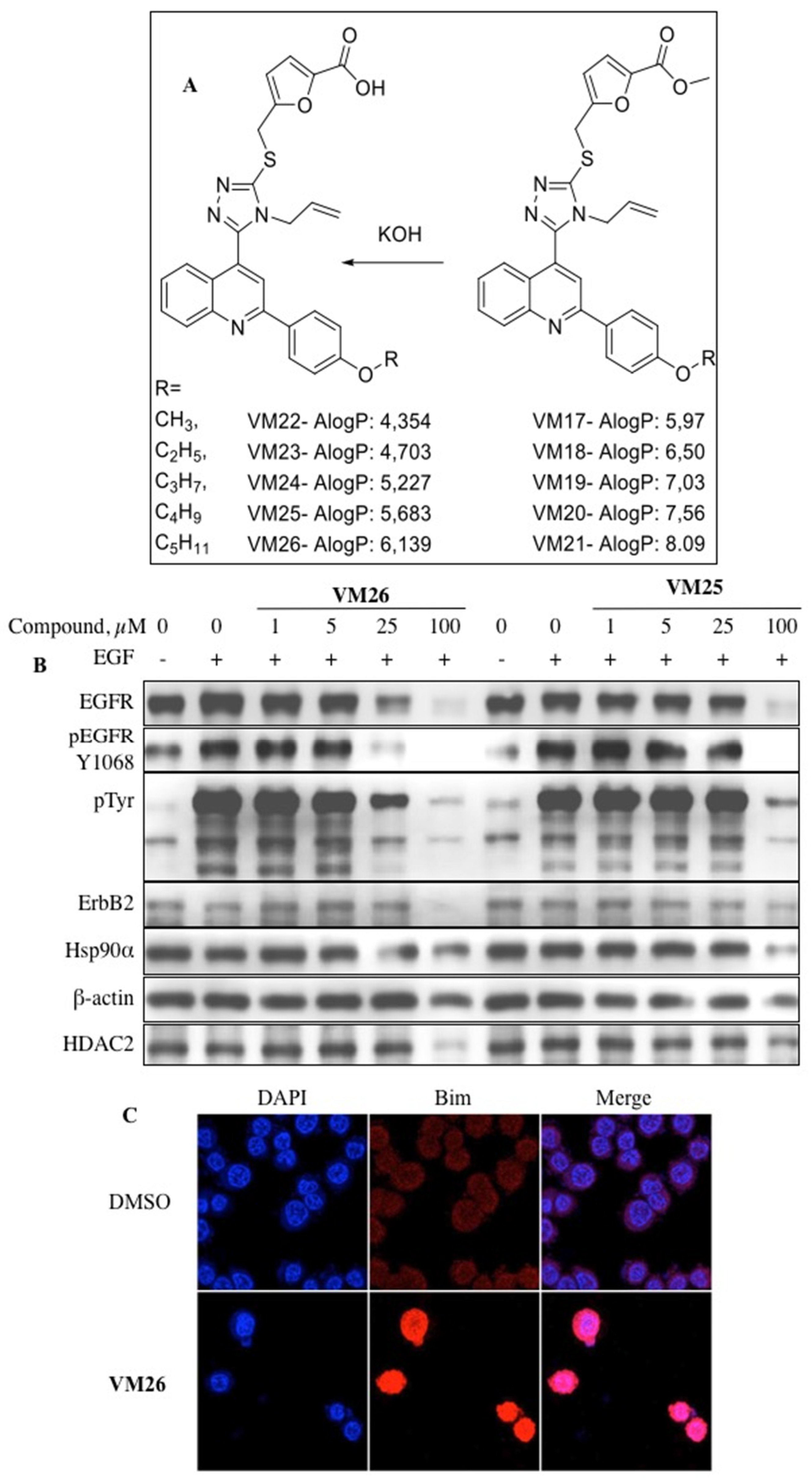
Figure 5.
Sequestration of Bim in cancer cells by allosteric degraders of EGFR [135]. A - Two-step protein degradation in starved cells vs non-starved cells considering 100% of each protein in non-starved cells; B - impact of EGF and glutamine on protein expression and phosphorylation in untreated cells and treated with VM26 in serum-deprived medium.
Figure 5.
Sequestration of Bim in cancer cells by allosteric degraders of EGFR [135]. A - Two-step protein degradation in starved cells vs non-starved cells considering 100% of each protein in non-starved cells; B - impact of EGF and glutamine on protein expression and phosphorylation in untreated cells and treated with VM26 in serum-deprived medium.
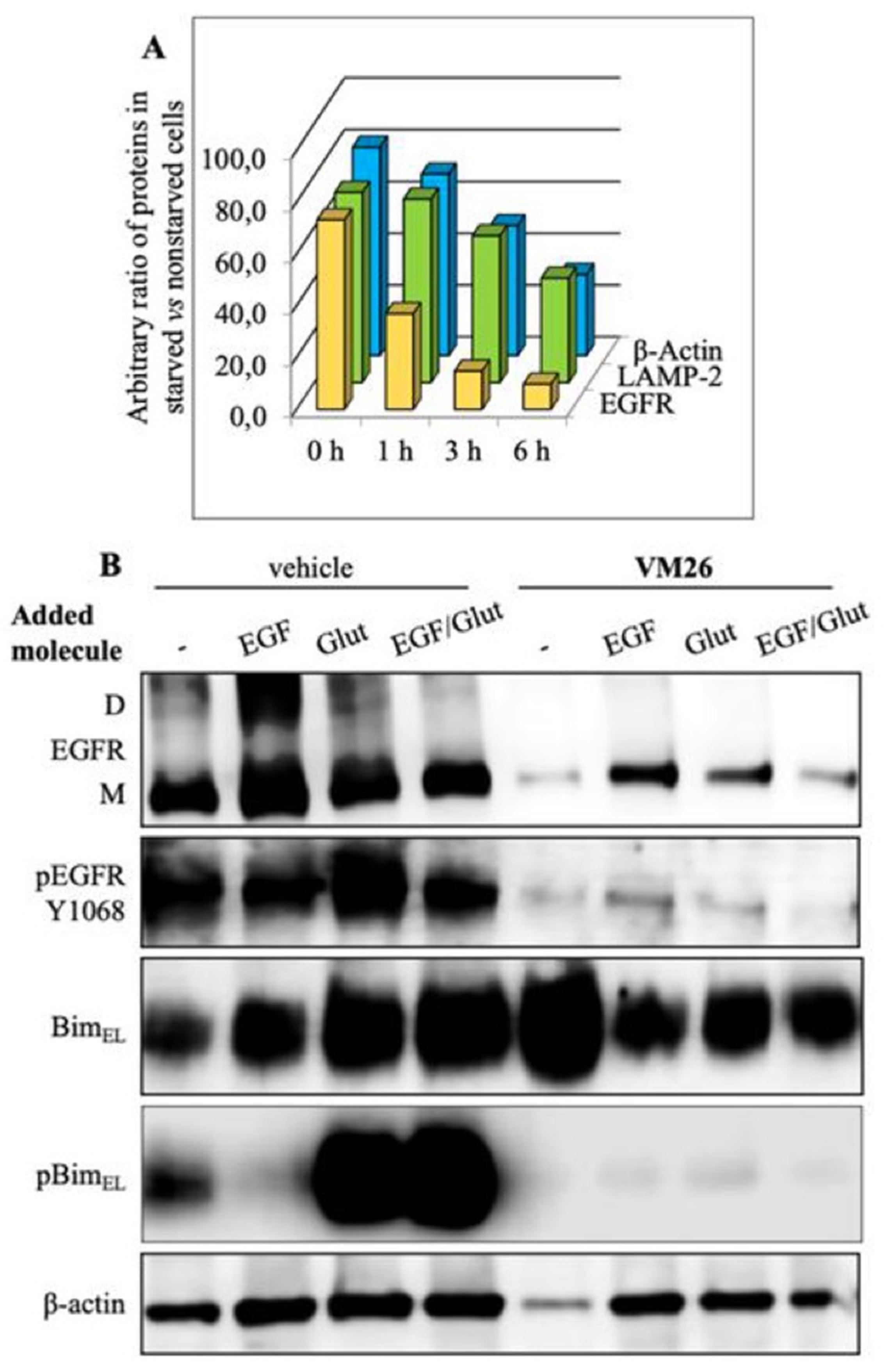
Figure 6.
Mechanism of action of EGFR-specific allosteric degraders on cancer cells. A - Role of Bim protein in connection of cells to the extracellular matrix [144). B - FQTT compounds bind to a allosteric site in EGFR, inducing degradation of the receptor in endosomes. Depletion of EGFR leads to sequestration of Bim, followed by disintegration of the cytoskeleton and detachment of cancer cells from the extracellular matrix. Glutamine starvation causes a deficiency of α-ketoglutarate and an inability of cells to replenish the tricarboxylic acid (TCA) cycle and produce ATP. Double starvation of EGF and glutamine reinforces the cytoskeleton disintegration leading to cancer cell detachment-promoted death [135,142].
Figure 6.
Mechanism of action of EGFR-specific allosteric degraders on cancer cells. A - Role of Bim protein in connection of cells to the extracellular matrix [144). B - FQTT compounds bind to a allosteric site in EGFR, inducing degradation of the receptor in endosomes. Depletion of EGFR leads to sequestration of Bim, followed by disintegration of the cytoskeleton and detachment of cancer cells from the extracellular matrix. Glutamine starvation causes a deficiency of α-ketoglutarate and an inability of cells to replenish the tricarboxylic acid (TCA) cycle and produce ATP. Double starvation of EGF and glutamine reinforces the cytoskeleton disintegration leading to cancer cell detachment-promoted death [135,142].
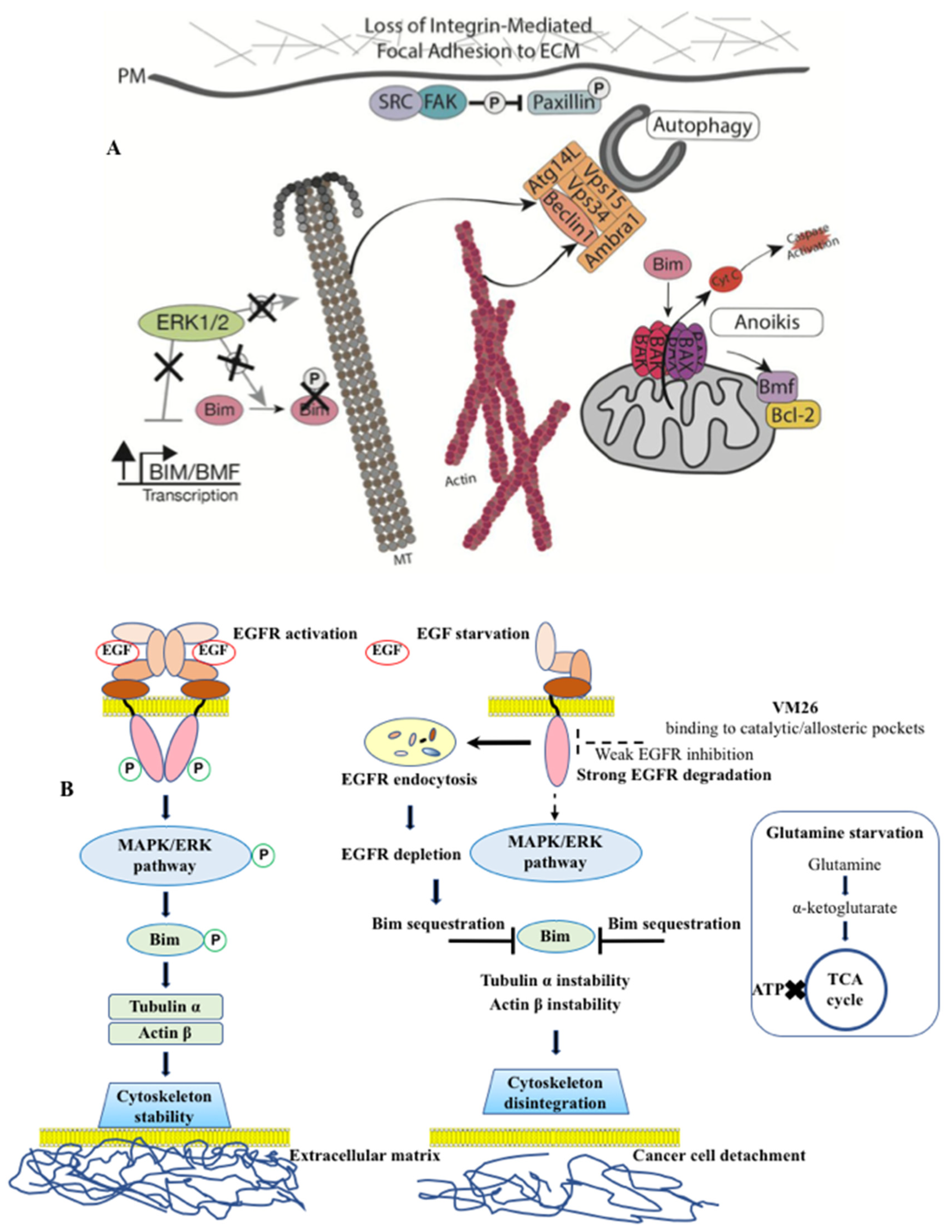
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



