
Submitted:
03 July 2023
Posted:
04 July 2023
You are already at the latest version
Abstract
Gliomas are aggressive, primary central nervous system tumours arising from glial cells. Glioblastomas are the most malignant. They are known for their poor prognosis or median overall survival. Current standard of care is overwhelmed by the heterogeneous, immunosuppressive tumour microenvironment promoting immune evasion and tumour proliferation. The advent of immunotherapy with its various modalities – immune checkpoint inhibitors, cancer vaccines, oncolytic viruses, chimeric antigen receptor T cells and NK cells have shown promise. Clinical trials incorporating combination therapies of the above have overcome the microenvironment resistance and yielded survival and prognostic benefit. Rolling these new therapies out in the real-world scenario in a low cost, high throughput manner is the unmet need of the hour. These will bring practice changing implications to the glioma treatment landscape. In this review article, we focus on describing the hallmarks of the glioma microenvironment and its interplay with the different emerging modalities of immunotherapy.
Keywords:
Gliomas
; Tumour Microenvironment
; Immunotherapy
; Immune Checkpoint Inhibitors
; Vaccines
Introduction
The mainstay of treatment of primary central nervous system tumours (PCNSTs) is surgical resection, followed by chemotherapy (ChT) and/or radiotherapy (RT) [1]. The degree of advancement is not as significant as other paediatric cancers, for example, molecularly targeted therapy for leukaemia. Unsurprisingly, PCNSTs account for most paediatric cancer related deaths [2]. One of the most prevalent types of PCNSTs are gliomas, notorious for their aggressive behaviour and poor prognosis i.e., median overall survival (mOS). Originating from various glial cell lines, they include astrocytoma, oligodendrocytoma, ependymoma and glioblastoma. These are classified as grade 1 to 4 according to the World Health Organization (WHO), which incorporates molecular and genomic features [3,4]. Glioblastoma (GB), formerly called glioblastoma multiforme (GBM), is the commonest malignant PCNST, representing 49% of them and having an incidence of 3.23 per 100,000 population [3]. With mOS of 14.6 months and a 5-year survival rate of 5% despite extensive surgical resections and adjuvant therapies, it is the centre of attention [1,3,4]. Resistance to standard treatments for gliomas stem principally from the heterogeneity of the tumour microenvironment (TME), which is immunosuppressive. It enables evasion of the immune system, which could partially explain the rapid disease progression [5]. Recently, novel treatment options, such as immunotherapy (IO) are being investigated upon. Understanding the operability of the TME in immune evasion would yield in potential efficacious IOs for high-grade gliomas. The aim of this review is to provide an outline of the immunobiological hallmarks of the TME of gliomas and the immunotherapeutic interplay to overcome immune evasion.
Hallmarks of the Tumour Microenvironment of Gliomas
- a)
- Cellular Armoury and the Blood Brain Barrier
The hallmarks of cancer describe a set of characteristics acquired by healthy cells as they transform into neoplastic entities. The interaction between the glioma cells and the TME is key for tumour proliferation and migration [6,7,8]. They secrete C–C motif chemokine ligand 2 (CCL2), which increases the formation of new blood vessels and attracts macrophages and microglia to the TME, resulting in enhanced tumour growth [9]. In addition, GB cells also release C-X-C motif ligand 8 (CXCL8), which modifies the extracellular matrix (ECM) through activating matrix metalloproteinases (MMPs) within in the TME [9,10,11]. Furthermore, through activation of tumour growth factor beta (TGF-β) and epidermal growth factor receptor (EGFR) signalling pathways, glioma cells can enhance their invasiveness [11].
A major part of the tumour bulk is comprised of immune cells such as tumour-associated myeloid cells (TAMCs) (11). TAMCs consists of tumour-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), dendritic cells (DCs), neutrophils, and microglia. These cells promote cancer growth directly by enhancing tumour cell proliferation and indirectly by generating an immunosuppressive microenvironment (Table 1) [11,12,13].
Microglia are present throughout the CNS and account for 10–20% of the non-neuronal cells. They are key in regulating the cerebral immunological homeostasis [14]. Along with resident CNS macrophages, they constitute TAMs [15]. TAMs can secrete either immunosuppressive factors such as IL-10 and TGF-β or produce antitumour-stimulating cytokines such as IL-12, TNF-α, according to the state of TME, whether ‘hot’ or ‘cold’ [16]. Activated TAMs can exist in two phenotypes, tumour-suppressive M1 or immune-suppressive M2 [14]. Increased accumulation of TAMS with the M2 phenotype were correlated with a higher tumour grade and lower mOS or poor outcomes in recurrent GB [17]. TAMs have a high degree of plasticity and therefore can be reprogrammed, thus providing opportunities of exploitation for treatment options.
DCs are ‘professional’ antigen-presenting cells (APCs) linking innate and adaptive immunity. They capture antigens and present them to T cells [13]. DCs are usually present in the meninges and choroid plexus but are not seen within the normal brain parenchyma [11]. On the contrary, in a glioma – infiltrated brain, they are harboured within the parenchyma [18]. Some animal studies have demonstrated that these are recruited to the TME in a similar way to NK cells via chemokines CCL5 and XCL1 [18]. DCs are also essential in the activation of antitumour immune responses and interact with other immune cells through integration of the various TME signals [13]. They can secrete cytokines such as IL-12, leading to increased recruitment of CD8+ T cells. However, they are still affected by TME immunosuppression, thus becoming regulatory DCs, which subsequently activate Treg [19]. This leads to downregulation of CD8+ T-cell recruitment [20]. Increased IL-10 secretion by macrophages leads to reduced IL-12 production and results in containing DC within the TME [18]. These mechanisms lead to inefficient DC differentiation and formation of impaired DCs in immature cellular states, causing immunosuppressive conditioning of the TME [20].
DC-based vaccines against GB are presently under construction and significant progress has been made over the past year [21,22].
The immune cells and the blood-brain barrier (BBB) are key to the TME’s adaptive alterations [8]. The BBB comprises a semipermeable membrane with endothelial cells, astrocyte foot processes and pericytes. This disconnects the brain from the peripheral immune system as evidenced by nil acute rejection of implanted grafts [23,24]. Naïve T cells cannot cross the BBB but activated T cells can [23]. The BBB thus tightly regulates substance entry into the brain parenchyma, due to which gliomas experience an overall decreased immune surveillance as compared to other tumours [14]. Furthermore, this tight regulation accounts for the poor therapeutic effectiveness of intravenous treatments. In gliomas, the tumour physically distorts the BBB and induces inflammation, which then causes the surrounding blood vessels to become leaky and compromised [23]. The inadequate blood flow creates hypoxic regions within the tumour due to insufficient oxygen delivery, and these areas then attract macrophages, which further enhances the immunosuppression of gliomas [19].
- b)
- The Lymphocytic Milieu
Naturally, the cytokine environment of the CNS is regulated towards helper T cell lymphocytes (Th2) to shield the brain against inflammatory destruction [19]. Gliomas exploit this response by enhancing tumour-infiltrating lymphocyte (TIL) production of Th2 cytokines [11,12].
Regulatory T cell (Treg) lymphocytes suppress the activity of effector T cells and DCs. Whilst no Treg are found in normal brain tissue, increased numbers of Treg cells are seen in a glioma-infiltrated brain. This offers the key ability of a glioma to evade the immune system as would be discussed in onward sections [11]. These cells are recruited to the TME by the secretion of chemokines such as CCL2 and CXCL12 by glioma cells. The number of Treg present is linked to the location and grade of the tumour [11,19]. They induce compromised APCs, which have decreased ability to activate tumour reactive T cells [19]. In addition, Treg secrete factors such as interleukin (IL-10) and transforming growth factor – beta (TGF-β), which inhibit the activity of other immune cells [13]. M2 phenotype macrophages and regulatory T cells (Treg) infiltrating the GB also leads to suppression of T-cell function [7]. In another context, Treg depletion was shown to improve OS rates in mice with glioma [19]. A study showed that this concept was successful in treating ovarian cancer [25].
Natural killer (NK) cells are CD3+, CD56+ and CD16+ innate lymphocytes that induce cytotoxic apoptosis in cells, therefore playing a vital role in the immune response [16]. NKs can recognise virally infected or malignant cells by their absent major histocompatibility complex (MHC) class I and cause apoptosis by exhibiting a combination of inhibitory as well as stimulatory receptors [11,12,13]. Studies have shown that NK-cell deficiencies were correlated with an increased incidence of certain cancers and GBs were one of them [26,27]. Furthermore, GB expresses human leukocyte antigen G (HLA-G), which further limits the action of NK cells, providing protection from NK-cell-mediated death [16]. NK-cell activity is also hindered by MDSCs by production of arginase and reactive oxygen species (ROS) [7].
- c)
- Immunosuppressive factors and immune evasion
The glioma microenvironment secretes a variety of immunosuppressive factors, such as transforming growth factor – beta 2 (TGF-β2), prostaglandin E2 (PGE2), interleukins (IL-1, IL-10) and fibrinogen-like protein 2 (FGL2). These factors collectively further suppress effector T cell activity [11]. In addition, Treg cells and MDSCs further prevent the normal NK-cell- and CTL-mediated cytotoxic reactions [13,14]. TGF-β1 and IL-10 skew TAMCs toward the immunosuppressive M2 phenotype, which then along with Treg secrete further TGF-β1 and IL-10, hence suppressing the immune system [12]. This immunosuppressive phenotype enables aggressive tumour proliferation and invasion, while inhibiting the normal antitumour immune responses [13].
Gliomas also express programmed death-ligand 1 (PD-L1), which is the primary ligand of programmed cell death protein 1 (PD-1), resulting in T-cell exhaustion and anergy [19]. T-cell anergy is a common tolerance mechanism in which T cells are functionally inactivated, thus unable to coordinate a response after encountering an antigen, but remain in a prolonged, hyporesponsive state. Both types of anergies i.e., clonal/in vitro and adaptive/in vivo, are seen in GB [28]. In clonal anergy, ineffective Ras/mitogen-activated protein kinase (Ras/MAPK) pathway activation and defective co-stimulation leads to impaired T-cell activation. Adaptive anergy, on the other hand, has persistent low-level antigen stimulation causing T-cell desensitisation which leads to defective nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), decreased IL-2 release and impaired T-cell amplification [12,28].
Immune evasion depends on the anatomical site of the tumour within the CNS and the intrinsic cell-to-cell interactions among the tumour and the immune cells [11,12,13]. Gliomas cause the evasion by reducing the overall recruitment of immune cells, while increasing that of microglial cells [14]. These microglia appear like immature APCs, lacking the ability to provide T-cell-mediated immunity. As mentioned above, gliomas release immunosuppressant cytokines such as transforming TGF-β, IL-10 and cyclooxygenase 2 (COX-2), while simultaneously inhibiting signal transducer and activator of transcription 3 (STAT-3), thus enhancing the immunosuppressive microenvironment [19]. Hypoxia within TME due to impaired blood vessels and greater usage of oxygen by tumour cells results in activation of the immunosuppressive STAT3 pathway. This STAT3 pathway leads to creation of hypoxia inducible factor-1 alpha (HIF-1α), stimulation of Treg cells and synthesis of vascular endothelial growth factor (VEGF). VEGF then further alters the vasculature, inhibits DC development, antigen presentation and T-cell infiltration into tumours [20].
Antigen recognition following presentation is essential for T-cell-mediated immunity (CMI), and this relies on the expression of major histocompatibility complex (MHC) molecules [6]. Invading gliomas downregulate the expression of MHC proteins and costimulatory molecules such as CD80 and CD86 on their surface, leading to reduced immune recognition and activation of cytotoxic T cells (CTLs) [7,12]. As mentioned above, the IL-10 and TGF-β enriched immunosuppressive TME of gliomas leads to loss of MHC expression on microglia [19]. Furthermore, reduced expression of MHC class I proteins was also present on glioma stem cells, in turn adding to T-CMI resistance and leading to increased tumour proliferation [14].
Blockage of chemotactic agents with antibodies or therapeutic drugs have supressed the recruitment of suppressor cells. TGF-β is key in the development of Treg cells and is upregulated in gliomas [12,19]. Antisense phosphorothioate oligodeoxynucleotide trabedersen (AP 12009) has been shown to successfully inhibit TGF-β expression in vitro and in animal models the inhibition of TGF beta pathways among gliomas helped to re-establish immune surveillance [29]. Inhibiting the cytokine production of glioma cells thus decreases their ability to proliferate, thus reducing its capacity to recruit immunosuppressive cells [30].
Immunotherapy and the Interplay
- a)
- Immunotherapy Landscape in Glioma
The standard of care (SOC) for GB is surgical resection in conjunction with RT and chemotherapy, mainly with temozolomide (TMZ), as per the Stupp protocol [1]. High-dose steroid, most commonly dexamethasone, is also administered to reduce vasogenic cerebral oedema. All these treatments in context further suppress the immune system. For example, pancytopenia and TMZ-induced lymphopenia are common side effects. Even a reduced dose of dexamethasone can lead to fewer immune cells in the TME, posing a challenge for clinical oncologists to weigh the benefit of reducing vasogenic oedema against the immunosuppressive side effects of steroids and consider using the lowest dose possible [19].
The immune system can be exploited in several ways. Active immunotherapeutic agents include cancer vaccines whereas passive counterparts comprise monoclonal antibodies (MABs) or adoptive cell transfer (ACT). Immunotherapies can be used as monotherapy, combination therapy or as an adjunct alongside alongside surgery, ChT or RT to enhance efficacy. Although they exist for other cancer primaries, they are presently under development in the GB treatment landscape [31]. The heterogeneous nature of the glioma TME and its adaptability allows enhanced immunosuppression and increased proliferation of tumour. SOC alone is unlikely to improve prognosis or survival. IO has emerged as a promising avenue for the treatment of gliomas, possibly in combination with SOC [32,33].
Understanding the mechanisms of immune evasion will facilitate the advancement of effective IO. The focus of glioma IO research has centred on four approaches: immune checkpoint inhibitors (ICIs), cancer vaccines, oncolytic viruses (OVs), chimeric antigen receptor (CAR) T and NK cell therapy (Table 2).
- b)
- Immune Checkpoint Inhibitors
T-cell activity is mediated through integrating both stimulatory and inhibitory signals, collectively termed immune checkpoints. These prevent the immune system from attacking one’s own cells. However, some cancer cells can manipulate these checkpoints within the TME to evade the immune system, allowing proliferation. ICIs are a ground-breaking class of humanised immunoglobulin G (IgG) monoclonal antibodies (mAbs) that have revolutionised cancer treatment in the last decade by enabling the immune system to recognise and attack cancer cells effectively [34].
There are three principal types of ICIs. Nivolumab, pembrolizumab and cemiplimab are anti-PD-1 IgG4 mAbs that target the inhibitor receptor PD-1 on activated T cells, NK cells, B cells, macrophages, and several subsets of DCs, thus activating immune cells by interfering with the CD28-costimulatory signalling pathway. Atezolizumab, durvalumab and avelumab are anti-PD-L1 IgG1 mAbs that target PD-L1, the main ligand of PD-1, along with PD-L2, which is constitutively expressed on APCs within the TME as well as a wide range of tumours, such as lung, breast, and melanoma, thereby disinhibiting the migration and activation of T cells to seek and destroy PD-L1-expressing cancer cells [35,36]. Ipilimumab is an anti-CTLA-4 IgG1 mAb that targets CTLA-4, which normally governs the amplitude of T-cell activation, thereby blocking the normally immunosuppressive effect of the CD28-costimulatory signalling pathway of T cells and increasing their activation and proliferation (Figure 1) [34].
Ipilimumab was first approved to treat melanoma, but when combined with nivolumab it can also be used to treat advanced renal cell carcinoma (RCC), microsatellite instability/deficient mismatch repair (MSI-H/dMMR) metastatic colorectal carcinoma (mCRC), malignant pleural mesothelioma (MPM), non-small-cell lung carcinoma (NSCLC) and hepatocellular carcinoma (HCC) [34,37]. In the recurrent GB setting, monotherapy with PD-1 blockade yielded a mOS comparable with that of bevacizumab [38], an anti-IgG1 mAb targeted against VEGF-A known to prolong median progression-free survival (mPFS) [39]. In mice with GB, combining stereotactic RT (SRT) with PD-1 blockade resulted in 75% complete pathologic response (CPR) by activating macrophages, highlighting a novel immunologic mechanism underlying the interaction between RT and ICIs [40]. Although an international phase 3 trial demonstrated longer mOS from TMZ with RT than nivolumab with RT, leaving the SOC for glioblastoma unchanged as of now [41].
Gliomas manipulate pathways to inactivate T cells within the TME. As described, PD-1 is an inhibitory membrane protein present on activated T cells to dampen the immune response. It is activated by ligands PD-L1 and PD-L2 found on tumour cells and infiltrating immune cells [42]. An increased presence of PD-L1 was associated with a higher GB grade and poorer prognosis in glioma patients [32]. Nivolumab alone did not demonstrate any prognostic benefit for relapsed GB. However, it is presently being explored as adjunct with RT and/or TMZ in newly diagnosed GB [43]. Two recent studies demonstrated that anti-PD-1 mAbs in combination with surgical resection leads to significantly improved mOS in GB as compared to adjuvant therapy alone [43,44]. Other studies using different mAbs have also found similar results. However, larger-scale RCTs are required to robustly prove the efficacy of the neoadjuvant approach. Using combinations of different IOs might be a potential management approach to overcome the highly heterogeneous nature of gliomas.
- c)
- Therapeutic cancer vaccines
Cancer vaccines can be preventive or therapeutic (Figure 1). Preventive ones such as those targeting human papillomavirus (HPV) and hepatitis B virus (HBV), have been successful in reducing the risk of cervical and hepatocellular cancer respectively [45,46]. In contrast, therapeutic counterparts aim to stimulate the immune system to recognise and attack existing cancer cells [47]. These are an example of active IO as they work predominantly through activation of CTLs via presentation of tumour-associated antigens (TAAs) by APCs such as DCs. DC-based vaccines involve extracting DCs and exposing them to TAAs before being reintroduced into the patient's body; whereas tumour cell-based vaccines utilise whole tumour cells or specific antigens from the cancer cells to stimulate the immune system. They can be administered in numerous ways. The first method involves the administration of TAAs, which will then be presented to T cells by APCs to invoke an immune response. The second way is priming autologous DCs ex vivo with the patient’s TAAs and then re-administering these cells intradermally to the patient, a technique termed DC vaccination [22].
Bacillus Calmette-Guérin (BCG) in 1990 became the first ever immunotherapy and therapeutic cancer vaccine which was licensed for use in superficial early-stage bladder cancer [48]. Sipuleucel-T in 2010 became the first DC-based vaccine, with approval for asymptomatic hormone-refractory prostate cancer [49]. Finally in 2022, among both the newly diagnosed and recurrent GB setting, a study found that adding autologous tumour lysate-loaded DC vaccine (DCVax-L) to SOC resulted in significant mOS benefit. An even greater relative survival benefit was noted amongst those who would have fared worse with SOC [22]. DCVax-L is not yet approved by the Food and Drug Administration (FDA) in the US or the Medicines and Healthcare products Regulatory Agency (MHRA) in the UK. However, in the UK setting, it has recently been made available for private use [50], and the National Institutes of Health and Care Excellence (NICE) are conducting a technology appraisal of the clinical and cost effectiveness of DCVax-L for newly diagnosed GB [51].
However, there are several challenges in developing effective treatments, namely the need for better identification of TAAs, strategies to overcome immune evasion, and optimisation of vaccine delivery and adjuvant use. Additionally, the development of combination therapies synergistic with cancer vaccines i.e., ICIs or targeted therapies, may lead to more durable responses. As research in these areas continues, cancer vaccines may become an essential tool in the fight against cancer [47].
- d)
- Chimeric Antigen Receptor T and NK cells
This adoptive approach involves the genetic modification of patient-derived T cells to express CARs to recognise specific TAAs. These engineered T-cells are then infused back into the patient, where they can target and kill cancer cells. CAR-T cell therapy has shown success in the haematological malignancy landscape, specifically diffuse large B-cell lymphoma (DLBCL) [52] and B-cell acute lymphoblastic leukaemia (B-ALL) [53].
Clinical trials of CAR-T for gliomas have primarily focused on targeting TAAs such as IL-13 receptor alpha 2 (IL-13Rα2) [54], EGFR variant III (EGFRvIII) [55,56] and human EGFR 2 (HER2) [57]. EGFRvIII, for instance, is a tumour-specific mutant of EGFR found in a subset of GB and has been associated with poor prognosis [58]. However, a phase 1 trial of EGFRvIII targeted CAR-T cells demonstrated only transient reductions in tumour size and EGFRvIII expression in select patients (Figure 2) [56].
Translating to the glioma setting is challenging due to TAA heterogeneity, the immunosuppressive microenvironment, and the BBB [59,60]. The heterogenous expression of TAAs can result in the escape of antigen-negative tumour cells, leading to relapse [61]. Strategies to target multiple antigens simultaneously using dual or multi-antigen targeting of CAR T cells, which could avoid antigen escape within the TME are in the pipeline [57]. The immunosuppressive glioma TME consisting of Treg cells, MDSCs and TAMs, as well as inhibitory molecules like PD-L1 can impair the function and persistence of CAR-T [61]. Incorporating cell-intrinsic PD-1 checkpoint blockade within CAR T cells by engineering the expression of PD-1 dominant negative receptor (DNR), a decoy receptor that binds PD-L1 on tumour cells, thus disrupting the inhibitory action of this TME element and maintaining T-cell activation is a promising strategy [62]. Another strategy is combining CAR T-cell therapy with cell-extrinsic PD-1 blockade with ICIs such as nivolumab [60].
The BBB can physically limit the trafficking of systemically infused CAR T cells into the brain and the tumour site [59]. Strategies to improve CAR-T infiltration across this anatomical barrier into the CNS include direct intracranial administration, such as intratumoural or intraventricular infusion [63,64]. Crossing the physiologic BBB is then dependent on appropriate matched expression of adhesion molecules and chemokine receptors, namely CXCR3 and CCR5, to facilitate endothelial adhesion and translocation. However, these tumour-bound ligands are typically expressed in very low quantities. So, another strategy being explored is the engineering of CAR T cells that express better-matched chemokine receptors [65]. Once CAR-T enters the brain parenchyma, they encounter the immunosuppressive glioma TME, which induce T-cell exhaustion and apoptosis as previously described. To recruit Treg, gliomas overproduce factors like indoleamine 2,3-dioxygenase 1 (IDO-1) and glioma stem cell (GSC)-derived pericytes secrete CCL5; whereas cerebral stromal cells produce immunosuppressive cytokines (TGF-β, IL-10) [66]. Following the failure of ICI monotherapy, attention is now on combining therapies to simultaneously block multiple drivers of T cell exhaustion, such as with bispecific antibodies targeting TGF-β, PD-L1 and CD27, or with existing elements of SOC like RT and TMZ or targeting CCR4 to reduce Treg migration and disrupting immunosuppressive stromal components of the TME [66].
CAR NK-cell therapy is another potential therapeutic avenue for GB. Unlike T cells, NK cells as mentioned before are part of the innate immune system [67]. They directly recognise and eliminate cancer cells without prior antigen experience via an MHC-independent mechanism [68]. Activated NK cells release various cytotoxic molecules like IFN-γ which induce tumour apoptosis. Another mechanism is FcγRIIIA/CD16a mediated antibody-dependent cellular cytotoxicity (ADCC) [69]. Moreover, NK cells also regulate and activate the adaptive immune response through molecular crosstalk with DCs, enhancing tumour antigen presentation to modulate T-CMI antitumour responses. By switching from conventional CAR-T cell to NK signalling domains, CAR NK cells exhibit improved tumour-killing function. The targets being explored for CAR NK-cells in GB are like those of CAR-T therapies [67].
Initial trials of NK-cell therapy for GB have focused on autologous approaches, utilising ex-vivo-expanded activated NK cells derived from the patients' peripheral blood mononuclear cells (PBMC). These autologous adoptive therapies have demonstrated safety and shown durable responses recurrent GB [70]. To note is the limited cytotoxicity of autologous NK cells against GB. In contrast, allogeneic NK cells sourced from healthy donors are highly cytotoxic have minimal risk of graft-versus-host disease (GvHD) [71]. Therefore, allogeneic therapy holds promise for generating off-the-shelf cellular therapy products, bypassing inhibitory signals, and simplifying manufacturing processes. Current studies have demonstrated their safety and efficacy in haematological malignancies, along with some success in the solid tumour landscape [72].
Whilst preclinical models have demonstrated the efficacy of CAR-NK in orthotopic mouse xenograft models, several barriers persist [69]. GBs restrict NK-cell infiltration and downregulate target antigens. As previously described, the TME releases inhibitory cytokines and chemokines such as TGF-β to evade NK-cell-mediated oncolysis. Combining NK cells with TGF-β inhibitors or other agents like cationic supramolecular inhibitors and ICIs, shows potential in overcoming these obstacles [73]. However, technical challenges in CAR-NK development, large-scale manufacturing, and need to create bespoke molecules remain major limiting factors for all types of CAR therapies. This warrants the optimisation of gene modification and expansion methods for successful clinical trials of CAR NK-cell and T-cell therapies for GB [67].
- e)
- Oncolytic virotherapy
OVs or oncolytic virotherapy (OVT) represents a novel treatment strategy in cancer immunotherapy due to its dual mechanisms of action i.e., directly lysing cancer cells and modulating the TME to stimulate anti-tumour responses. OVs selectively replicate within cancer cells leading to their apoptotic destruction, known as oncolysis [74]. As OV-infected cancer cells die, they release tumour antigens which get taken up by APCs and presented to T cells, educating them to identify and kill the cancer cells, thus promoting an adaptive immune response [75]. Oncolysis leads to the release of damage-associated molecular patterns (DAMPs) and pro-inflammatory cytokines. These further stimulate the immune system, converting the ‘cold’ immunosuppressive TME, like that of GB, into a ‘hot’ immunostimulatory one, like that of melanoma, lending OVT, facilitating synergism with other IOs like ICIs and CAR-T [76]. OVs can also be genetically engineered to express immunomodulatory molecules boosting the immune response i.e., promoting drug activation or directly inhibiting tumour growth. Currently seven OV platforms are under investigation in neuro-oncology. DNA viruses include herpes simplex virus 1 (HSV-1), adenovirus (AdV), vaccinia virus and parvovirus whereas RNA viruses include poliovirus (PV), reovirus and measles virus. Each platform has its pros and cons and different modes of delivery [75].
In 2022, teserpaturev became the world’s first OVT approved for glioma based on the landmark Japanese single-arm phase 2 trial. A third-generation oncolytic HSV-1 called G47Δ was delivered intratumorally via a stereotactic neurosurgical procedure to 19 patients with either residual or recurrent GB. The primary endpoint of 1-year survival rate after G47Δ initiation was 84.2%, which is a substantial improvement from 30%. The mOS was 20.2 months after G47Δ initiation and 28.8 months from the initial surgery, which is significantly longer than standard mOS of under a year with existing therapies. The best overall response in 2 years was a partial response in 1 patient and stable disease in 18 patients. On MRI, oncolysis was suggested by characteristic enlargement and contrast clearing within the target lesion after each repeated G47Δ administration. Tumour biopsies showed increasing numbers of tumour infiltrating CD4+ and CD8+ lymphocytes, indicating an immune response, as well as persistently low numbers of FOXP3+ Treg, indicating decreased immune suppression within the TME (Figure 3) [77].
However, several challenges that need to be addressed for OVT to be adopted as a real world modality. These include the immune potential to neutralise OVs prior to tumour infection, ability of OVs to infect and kill all types of cancer cells, and ensuring the safety of using live viruses. Ongoing strategies include combination of OVT with standard therapies [75,76]. RT can enhance OV replication in tumour cells by altering gene expression. For instance, by upregulating human transcription factor Y-box binding protein 1 (YB-1) in the GB cell nuclei to upregulate replication of oncolytic AdV dl520 [78]. Another recent phase 1 trial of AdV-tk, an oncolytic AdV engineered to express HSV thymidine kinase (HSV-tk), demonstrated a safe RT and OVT combination in paediatric high-grade gliomas [79]. OVT is also showing promise for overcoming TMZ resistance, i.e., the oncolytic paramyxovirus Newcastle disease virus (NDV) inhibits the Akt signalling pathway and enhances the antitumour effect of TMZ [80]. Another example is the combination of oncolytic AdV DNX-2401 with TMZ which greatly enhances the CD8+ recognition of GB cells [81].
The combination of OVT with other IO modalities is particularly attractive as it offers direct glioma TME immunomodulation, which is the principal limiting factor. Looking at ICIs, monotherapies yielded lacklustre results, and combination therapies resulted in severe adverse reactions, especially with anti-PD1 and anti-CTLA-4 mAbs together [76]. However, OVs can increase the effectiveness of other IO modalities in GB by essentially reprogramming the TME to enhance the antitumour properties of the other immunotherapies and allow synergism [75,82]. OVs were shown to induce the upregulation of PD-1 on T cells and PD-L1 on tumour cells, thereby increasing the sensitivity of gliomas to ICIs [83]. Also, a phase 2 trial of oncolytic AdV DNX-2401 with anti-PD1 pembrolizumab achieved a median OS of 12.5 months [84].
The combination of OVs with CAR-T and CAR-NK have also shown promising results in the face of poor penetration when used alone and the highly immunosuppressive glioma TME. For example, loading a CAR-T cell with tumour-specific mAbs can help overcome the on-target/off-tumour cross-reactivity of some CAR-T cells with both glioma and normal cells. As in Lp2 CAR-T cells loaded with LpMab-2 to target podoplanin (PDPN)-expressing glioma cells whilst sparing PDPN-expressing normal cells, when used with G47Δ [85]. Oncolytic HSV-1 (oHSV-1) enhanced the therapeutic efficacy of CD70-targeted CAR-T by increasing intratumoural T and NK-cell infiltration and IFN-γ release within the GB TME [86]. When used in combination with B7-H3 CAR-T, an oncolytic AdV loaded with CXCL11, called oAds-CXCL11, led to increased infiltration of CD8+, NKs and M1-polarised macrophages, as well as decreased levels of MDSCs, Treg and M2-polarised marophages, when compared to B7-H3 CAR-T alone in mice [82]. The combination of OV-IL15C, an oncolytic HSV-1 that expresses IL15/IL15Rα fusion protein, and off-the-shelf EGFR-CAR-NK showed a synergism in inhibiting tumour growth and improving survival in mice compared to using either as monotherapy. This was associated with higher levels of NK and CD8+ infiltration and activation within the brain, as well as increased persistence of CAR-NK. These findings were noted in an immunocompetent model [87]. These combinations represent a significant frontier in the development of IOs targeting gliomas [75].
Conclusions
Gliomas including glioblastomas are notorious for poor prognosis. Existing standard of care regimens are neither highly effective nor offer a lucrative survival benefit. The tumour microenvironment has a challenging heterogenous, immunosuppressive milieu facilitating immune evasion and tumour proliferation. Immunotherapy modalities including ICIs, cancer vaccines, OVT, CAR-T and CAR-NK are emerging game changers. Combination therapies using these are increasingly being translated onto the glioma setting as TME shortcomings are being overcome. The clinical trials in the pipeline over the last decade have shown promising results in efficacy and survival outcomes. Rolling out these multimodal, immunomodulatory cocktail therapies in the real-world scenario is an unmet need of the hour. If executed in a low cost, high throughput manner, landscape changes in the mainstay of glioma therapy are expected.
Author Contributions
Conceptualization, A.V., and S.B.; methodology, C-A.L.; software, A.G., S-D.S.; validation, S.A., E.S. and C.C.; formal analysis, M.S.; investigation, E.R.; resources, E.S.; data curation, M.S., C.C., and E.R.; writing—original draft preparation, A.V.; writing—review and editing, C-A.L., A.V., A.G., S.A., C.C., E.R., and S.B.; visualization, S-D.S.; supervision, S.B.; project administration, E.S.; funding acquisition, M.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. European Organisation for Research and Treatment of Cancer Brain Tumour and Radiation Oncology Groups; National Cancer Institute of Canada Clinical Trials Group. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet. Oncol. 2009, 10, 459–466.
- Pollack, I.F.; Agnihotri, S.; Broniscer, A. Childhood brain tumors: current management, biological insights, and future directions. J. Neurosurg. Pediatr. 2019, 23, 261–273. [Google Scholar] [PubMed]
- Ostrom, Q.T.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2014-2018. Neuro. Oncol. 2021, 23, iii–iii105. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro. Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Soeda, A.; Hara, A.; Kunisada, T.; Yoshimura, S.; Iwama, T.; Park, D.M. The evidence of glioblastoma heterogeneity. Sci. Rep. 2015, 5, 7979. [Google Scholar] [CrossRef]
- Rolle, C.E.; Sengupta, S.; Lesniak, M.S. Mechanisms of immune evasion by gliomas. Adv. Exp. Med. Biol. 2012, 746, 53–76. [Google Scholar] [PubMed]
- Magaña-Maldonado, R.; Chávez-Cortez, E.G.; Olascoaga-Arellano, N.K.; López-Mejía, M.; Maldonado-Leal, F.M.; Sotelo, J.; Pineda, B. Immunological Evasion in Glioblastoma. Biomed. Res. Int. 2016, 2016, 7487313. [Google Scholar] [CrossRef]
- Gieryng, A.; Pszczolkowska, D.; Walentynowicz, K.A.; Rajan, W.D.; Kaminska, B. Immune microenvironment of gliomas. Lab. Invest. 2017, 97, 498–518. [Google Scholar] [CrossRef]
- Groblewska, M.; Litman-Zawadzka, A.; Mroczko, B. The Role of Selected Chemokines and Their Receptors in the Development of Gliomas. Int. J. Mol. Sci. 2020, 21, 3704. [Google Scholar] [CrossRef]
- Chen, Z.; Mou, L.; Pan, Y.; Feng, C.; Zhang, J.; Li, J. CXCL8 Promotes Glioma Progression By Activating The JAK/STAT1/HIF-1α/Snail Signaling Axis. Onco. Targets. Ther. 2019, 12, 8125–8138. [Google Scholar] [CrossRef] [PubMed]
- Dapash, M.; Hou, D.; Castro, B.; Lee-Chang, C.; Lesniak, MS. The Interplay between Glioblastoma and Its Microenvironment. Cells. 2021, 10, 2257. [Google Scholar] [CrossRef] [PubMed]
- DeCordova, S.; Shastri, A.; Tsolaki, A.G.; Yasmin, H.; Klein, L.; Singh, S.K.; Kishore, U. Molecular Heterogeneity and Immunosuppressive Microenvironment in Glioblastoma. Front. Immunol. 2020, 11, 1402. [Google Scholar] [CrossRef]
- Chen, H.; Li, M.; Guo, Y.; Zhong, Y.; He, Z.; Xu, Y.; Zou, J. Immune response in glioma's microenvironment. Innov. Surg. Sci. 2021, 5, 20190001. [Google Scholar] [CrossRef]
- Brown, N.F.; Carter, T.J.; Ottaviani, D.; Mulholland, P. Harnessing the immune system in glioblastoma. Br. J. Cancer. 2018, 119, 1171–1181. [Google Scholar] [CrossRef]
- Pombo Antunes, A.R.; Scheyltjens, I.; Duerinck, J.; Neyns, B.; Movahedi, K.; Van Ginderachter, J.A. Understanding the glioblastoma immune microenvironment as basis for the development of new immunotherapeutic strategies. Elife. 2020, 9, e52176. [Google Scholar] [CrossRef] [PubMed]
- Price, G.; Bouras, A.; Hambardzumyan, D.; Hadjipanayis, C.G. Current knowledge on the immune microenvironment and emerging immunotherapies in diffuse midline glioma. EBioMedicine. 2021, 69, 103453. [Google Scholar] [CrossRef] [PubMed]
- Lu-Emerson, C.; Snuderl, M.; Kirkpatrick, N.D.; Goveia, J.; Davidson, C.; Huang, Y.; Riedemann, L.; Taylor, J.; Ivy, P.; Duda, D.G.; et al. Increase in tumor-associated macrophages after antiangiogenic therapy is associated with poor survival among patients with recurrent glioblastoma. Neuro. Oncol. 2013, 15, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Böttcher, J.P.; Reis e Sousa, C. The Role of Type 1 Conventional Dendritic Cells in Cancer Immunity. Trends. Cancer. 2018, 4, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, M.M.; Sankey, E.W.; Ryan, K.J.; Chongsathidkiet, P.; Lorrey, S.J.; Wilkinson, D.S.; Fecci, P.E. Immune suppression in gliomas. J. Neurooncol. 2021, 151, 3–12. [Google Scholar] [CrossRef]
- Hetze, S.; Sure, U.; Schedlowski, M.; Hadamitzky, M.; Barthel, L. Rodent Models to Analyze the Glioma Microenvironment. ASN. Neuro. 2021, 13, 17590914211005074. [Google Scholar] [CrossRef]
- Lieberman, N.A.P.; DeGolier, K.; Kovar, H.M.; Davis, A.; Hoglund, V.; Stevens, J.; Winter, C.; Deutsch, G.; Furlan, S.N.; Vitanza, N.A.; et al. Characterization of the immune microenvironment of diffuse intrinsic pontine glioma: implications for development of immunotherapy. Neuro. Oncol. 2019, 21, 83–94. [Google Scholar] [CrossRef]
- Liau, L.M.; Ashkan, K.; Brem, S.; Campian, J.L.; Trusheim, J.E.; Iwamoto, F.M.; Tran, D.D.; Ansstas, G.; Cobbs, C.S.; Heth, J.A.; et al. Association of Autologous Tumor Lysate-Loaded Dendritic Cell Vaccination With Extension of Survival Among Patients With Newly Diagnosed and Recurrent Glioblastoma: A Phase 3 Prospective Externally Controlled Cohort Trial. JAMA. Oncol. 2023, 9, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Dubois, L.G.; Campanati, L.; Righy, C.; D'Andrea-Meira, I.; Spohr, T.C.; Porto-Carreiro, I.; Pereira, C.M.; Balça-Silva, J.; Kahn, S.A.; DosSantos, M.F.; et al. Gliomas and the vascular fragility of the blood brain barrier. Front. Cell. Neurosci. 2014, 8, 418. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold. Spring. Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef]
- Fecci, P.E.; Mitchell, D.A.; Whitesides, J.F.; Xie, W.; Friedman, A.H.; Archer, G.E.; Herndon, J.E. 2nd.; Bigner, D.D.; Dranoff, G.; Sampson, J.H. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer. Res. 2006, 66, 3294–3302.
- Fares, J.; Fares, M.Y.; Fares, Y. Natural killer cells in the brain tumor microenvironment: Defining a new era in neuro-oncology. Surg. Neurol. Int. 2019, 10, 43. [Google Scholar] [CrossRef]
- Zhang, L.; Yu, H.; Xue, Y.; Liu, Y. Decreased natural killer cells in diffuse intrinsic pontine glioma patients. Childs. Nerv. Syst. 2020, 36, 1345–1346. [Google Scholar] [CrossRef] [PubMed]
- Zarek, P.E.; Huang, C.T.; Lutz, E.R.; Kowalski, J.; Horton, M.R.; Linden, J.; Drake, C.G.; Powell, J.D. A2A receptor signaling promotes peripheral tolerance by inducing T-cell anergy and the generation of adaptive regulatory T cells. Blood. 2008, 111, 251–259. [Google Scholar] [CrossRef]
- Jaschinski, F.; Rothhammer, T.; Jachimczak, P.; Seitz, C.; Schneider, A.; Schlingensiepen, K.H. The antisense oligonucleotide trabedersen (AP 12009) for the targeted inhibition of TGF-β2. Curr. Pharm. Biotechnol. 2011, 12, 2203–2213. [Google Scholar] [CrossRef]
- Christofides, A.; Kosmopoulos, M.; Piperi, C. Pathophysiological mechanisms regulated by cytokines in gliomas. Cytokine. 2015, 71, 377–384. [Google Scholar] [CrossRef]
- Shah, P.V.; Arrieta, V.A.; Lee-Chang, C.; Sonabend, A.M. Cancer Immunoediting in Gliomas: Recent Advances and Implications for Immunotherapy. J. Cell. Immunol. 2020, 2, 352–358. [Google Scholar]
- Al-Kharboosh, R.; ReFaey, K.; Lara-Velazquez, M.; Grewal, S.S.; Imitola, J.; Quiñones-Hinojosa, A. Inflammatory Mediators in Glioma Microenvironment Play a Dual Role in Gliomagenesis and Mesenchymal Stem Cell Homing: Implication for Cellular Therapy. Mayo. Clin. Proc. Innov. Qual. Outcomes. 2020, 4, 443–459. [Google Scholar] [CrossRef]
- Najem, H.; Khasraw, M.; Heimberger, A.B. Immune Microenvironment Landscape in CNS Tumors and Role in Responses to Immunotherapy. Cells. 2021, 10, 2032. [Google Scholar] [CrossRef] [PubMed]
- Shiravand, Y.; Khodadadi, F.; Kashani, S.M.A.; Hosseini-Fard, S.R.; Hosseini, S.; Sadeghirad, H.; Ladwa, R.; O'Byrne, K.; Kulasinghe, A. Immune Checkpoint Inhibitors in Cancer Therapy. Curr. Oncol. 2022, 29, 3044–3060. [Google Scholar] [CrossRef]
- Revythis, A.; Shah, S.; Kutka, M.; Moschetta, M.; Ozturk, M.A.; Pappas-Gogos, G.; Ioannidou, E.; Sheriff, M.; Rassy, E.; Boussios, S. Unraveling the Wide Spectrum of Melanoma Biomarkers. Diagnostics. (Basel). 2021, 11, 1341. [Google Scholar] [CrossRef]
- Boussios, S.; Rassy, E.; Samartzis, E.; Moschetta, M.; Sheriff, M.; Pérez-Fidalgo, J.A.; Pavlidis, N. Melanoma of unknown primary: New perspectives for an old story. Crit. Rev. Oncol. Hematol. 2021, 158, 103208. [Google Scholar] [CrossRef]
- Adeleke, S.; Haslam, A.; Choy, A.; Diaz-Cano, S.; Galante, J.R.; Mikropoulos, C.; Boussios, S. Microsatellite instability testing in colorectal patients with Lynch syndrome: lessons learned from a case report and how to avoid such pitfalls. Per. Med. 2022, 19, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P. ; Bähr, O, et al. Effect of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA. Oncol. 2020, 6, 1003–1010.
- Ameratunga, M.; Pavlakis, N.; Wheeler, H.; Grant, R.; Simes, J.; Khasraw, M. Anti-angiogenic therapy for high-grade glioma. Cochrane. Database. Syst. Rev. 2018, 11, CD008218. [Google Scholar] [CrossRef]
- Stessin, A.M.; Clausi, M.G.; Zhao, Z.; Lin, H.; Hou, W.; Jiang, Z.; Duong, T.Q.; Tsirka, S.E.; Ryu, S. Repolarized macrophages, induced by intermediate stereotactic dose radiotherapy and immune checkpoint blockade, contribute to long-term survival in glioma-bearing mice. J. Neurooncol. 2020, 147, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Omuro, A.; Brandes, A.A.; Carpentier, A.F.; Idbaih, A.; Reardon, D.A.; Cloughesy, T.; Sumrall, A.; Baehring, J.; van den Bent, M.; Bähr, O.; et al. Radiotherapy combined with nivolumab or temozolomide for newly diagnosed glioblastoma with unmethylated MGMT promoter: An international randomized phase III trial. Neuro. Oncol. 2023, 25, 123–134. [Google Scholar] [CrossRef]
- Scheffel, T.B.; Grave, N.; Vargas, P.; Diz, F.M.; Rockenbach, L.; Morrone, F.B. Immunosuppression in Gliomas via PD-1/PD-L1 Axis and Adenosine Pathway. Front. Oncol. 2021, 10, 617385. [Google Scholar] [CrossRef]
- Schalper, K.A.; Rodriguez-Ruiz, M.E.; Diez-Valle, R.; López-Janeiro, A.; Porciuncula, A.; Idoate, M.A.; Inogés, S.; de Andrea, C.; López-Diaz de Cerio, A.; Tejada, S.; et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat. Med. 2019, 25, 470–476. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Ismail, A.; Pappas-Gogos, G.; Boussios, S. HPV and Cervical Cancer: A Review of Epidemiology and Screening Uptake in the UK. Pathogens. 2023, 12, 298. [Google Scholar] [CrossRef] [PubMed]
- Flores, J.E.; Thompson, A.J.; Ryan, M.; Howell, J. The Global Impact of Hepatitis B Vaccination on Hepatocellular Carcinoma. Vaccines. (Basel). 2022, 10, 793. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.J.; Svensson-Arvelund, J.; Lubitz, G.S.; Marabelle, A.; Melero, I.; Brown, B.D.; Brody, J.D. Cancer vaccines: the next immunotherapy frontier. Nat. Cancer. 2022, 3, 911–926. [Google Scholar] [CrossRef]
- Morales, A. Treatment of carcinoma in situ of the bladder with BCG: a phase II trial. Cancer. Immunol. Immunother. 1980, 9, 69–72. [Google Scholar] [CrossRef]
- Small, E.J.; Schellhammer, P.F.; Higano, C.S.; Redfern, C.H.; Nemunaitis, J.J.; Valone, F.H.; Verjee, S.S.; Jones, L.A.; Hershberg, R.M. Placebo-controlled phase III trial of immunologic therapy with sipuleucel-T (APC8015) in patients with metastatic, asymptomatic hormone refractory prostate cancer. J. Clin. Oncol. 2006, 24, 3089–3094. [Google Scholar] [CrossRef]
- Northwest Biotherapeutics. New Patient Inquiry. Available at https://nwbio.com/patients-information-form/ Accessed April 1, 2023.
- National Institute for Health and Care Excellence. DCVax-L for treating newly diagnosed glioblastoma multiforme [ID836]. Available at https://www.nice.org.uk/guidance/indevelopment/gid-ta10143 Accessed April 1, 2023.
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. ACT IV trial investigators. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet. Oncol. 2017, 18, 1373–1385.
- O'Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef]
- Bielamowicz, K.; Fousek, K.; Byrd, T.T.; Samaha, H.; Mukherjee, M.; Aware, N.; Wu, M.F.; Orange, J.S.; Sumazin, P.; Man, T.K.; et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro. Oncol. 2018, 20, 506–518. [Google Scholar] [CrossRef] [PubMed]
- Del Vecchio, C.A.; Giacomini, C.P.; Vogel, H.; Jensen, K.C.; Florio, T.; Merlo, A.; Pollack, J.R.; Wong, A.J. EGFRvIII gene rearrangement is an early event in glioblastoma tumorigenesis and expression defines a hierarchy modulated by epigenetic mechanisms. Oncogene. 2013, 32, 2670–2681. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.J.; Mashouf, L.A.; Lim, M. CAR T Cell Therapy in Primary Brain Tumors: Current Investigations and the Future. Front. Immunol. 2022, 13, 817296. [Google Scholar] [CrossRef] [PubMed]
- Marofi, F.; Achmad, H.; Bokov, D.; Abdelbasset, W.K.; Alsadoon, Z.; Chupradit, S.; Suksatan, W.; Shariatzadeh, S.; Hasanpoor, Z.; Yazdanifar, M.; et al. Hurdles to breakthrough in CAR T cell therapy of solid tumors. Stem. Cell. Res. Ther. 2022, 13, 140. [Google Scholar] [CrossRef]
- Jackson, S.; Meeks, C.; Vézina, A.; Robey, R.W.; Tanner, K.; Gottesman, M.M. Model systems for studying the blood-brain barrier: Applications and challenges. Biomaterials. 2019, 214, 119217. [Google Scholar] [CrossRef]
- Cherkassky, L.; Morello, A.; Villena-Vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, D.R.; Sadelain, M.; Adusumilli, P.S. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Invest. 2016, 126, 3130–3144. [Google Scholar] [CrossRef]
- Priceman, S.J.; Tilakawardane, D.; Jeang, B.; Aguilar, B.; Murad, J.P.; Park, A.K.; Chang, W.C.; Ostberg, J.R.; Neman, J.; Jandial, R.; et al. Regional Delivery of Chimeric Antigen Receptor-Engineered T Cells Effectively Targets HER2+ Breast Cancer Metastasis to the Brain. Clin. Cancer. Res. 2018, 24, 95–105. [Google Scholar] [CrossRef]
- Mulazzani, M.; Fräßle, S.P.; von Mücke-Heim, I.; Langer, S.; Zhou, X.; Ishikawa-Ankerhold, H.; Leube, J.; Zhang, W.; Dötsch, S.; Svec, M.; et al. Long-term in vivo microscopy of CAR T cell dynamics during eradication of CNS lymphoma in mice. Proc. Natl. Acad. Sci. U S A. 2019, 116, 24275–24284. [Google Scholar]
- Newick, K.; O'Brien, S.; Moon, E.; Albelda, S.M. CAR T Cell Therapy for Solid Tumors. Annu. Rev. Med. 2017, 68, 139–152. [Google Scholar] [CrossRef]
- Singh, K.; Hotchkiss, K.M.; Patel, K.K.; Wilkinson, D.S.; Mohan, A.A.; Cook, S.L.; Sampson, J.H. Enhancing T Cell Chemotaxis and Infiltration in Glioblastoma. Cancers. (Basel). 2021, 13, 5367. [Google Scholar] [CrossRef]
- Wang, G.; Wang, W. Advanced Cell Therapies for Glioblastoma. Front. Immunol. 2022, 13, 904133. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Burger, M.C.; Zhang, C.; Harter, P.N.; Romanski, A.; Strassheimer, F.; Senft, C.; Tonn, T.; Steinbach, J.P.; Wels, W.S. CAR-Engineered NK Cells for the Treatment of Glioblastoma: Turning Innate Effectors Into Precision Tools for Cancer Immunotherapy. Front. Immunol. 2019, 10, 2683. [Google Scholar] [CrossRef]
- Lim, J.; Park, Y.; Ahn, J.W.; Sim, J.; Kang, S.J.; Hwang, S.; Chun, J.; Choi, H.; Kim, S.H.; Chun, D.H.; et al. Autologous adoptive immune-cell therapy elicited a durable response with enhanced immune reaction signatures in patients with recurrent glioblastoma: An open label, phase I/IIa trial. PloS. One. 2021, 16, e0247293. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Ramanathan, A.; Lorimer, I.A.J. Engineered cells as glioblastoma therapeutics. Cancer. Gene. Ther. 2022, 29, 156–166. [Google Scholar] [CrossRef]
- Beier, C.P.; Kumar, P.; Meyer, K.; Leukel, P.; Bruttel, V.; Aschenbrenner, I.; Riemenschneider, M.J.; Fragoulis, A.; Rümmele, P.; Lamszus, K.; et al. The cancer stem cell subtype determines immune infiltration of glioblastoma. Stem. Cells. Dev. 2012, 21, 2753–2761. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: a new class of immunotherapy drugs. Nat. Rev. Drug. Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef]
- Fudaba, H.; Wakimoto, H. Oncolytic virus therapy for malignant gliomas: entering the new era. Expert. Opin. Biol. Ther. 2023, 23, 269–282. [Google Scholar] [CrossRef]
- Qi, Z.; Long, X.; Liu, J.; Cheng, P. Glioblastoma microenvironment and its reprogramming by oncolytic virotherapy. Front. Cell. Neurosci. 2022, 16, 819363. [Google Scholar] [CrossRef]
- Todo, T.; Ito, H.; Ino, Y.; Ohtsu, H.; Ota, Y.; Shibahara, J.; Tanaka, M. Intratumoral oncolytic herpes virus G47∆ for residual or recurrent glioblastoma: a phase 2 trial. Nat. Med. 2022, 28, 1630–1639. [Google Scholar] [CrossRef] [PubMed]
- Bieler, A.; Mantwill, K.; Holzmüller, R.; Jürchott, K.; Kaszubiak, A.; Stärk, S.; Glockzin, G.; Lage, H.; Grosu, A.L.; Gansbacher, B.; et al. Impact of radiation therapy on the oncolytic adenovirus dl520: implications on the treatment of glioblastoma. Radiother. Oncol. 2008, 86, 419–427. [Google Scholar] [CrossRef]
- Kieran, M.W.; Goumnerova, L.; Manley, P.; Chi, S.N.; Marcus, K.J.; Manzanera, A.G.; Polanco, M.L.S.; Guzik, B.W.; Aguilar-Cordova, E.; Diaz-Montero, C.M.; et al. Phase I study of gene-mediated cytotoxic immunotherapy with AdV-tk as adjuvant to surgery and radiation for pediatric malignant glioma and recurrent ependymoma. Neuro. Oncol. 2019, 21, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Chen, Y.; Hong, X.; Liu, X.; Su, X.; Li, S.; Dong, X.; Zhao, G.; Li, Y. Newcastle disease virus enhances the growth-inhibiting and proapoptotic effects of temozolomide on glioblastoma cells in vitro and in vivo. Sci. Rep. 2018, 8, 11470. [Google Scholar] [CrossRef]
- Kleijn, A.; van den Bossche, W.; Haefner, E.S.; Belcaid, Z.; Burghoorn-Maas, C.; Kloezeman, J.J.; Pas, S.D.; Leenstra, S.; Debets, R.; de Vrij, J.; et al. The Sequence of Delta24-RGD and TMZ Administration in Malignant Glioma Affects the Role of CD8+T Cell Anti-tumor Activity. Mol. Ther. Oncolytics. 2017, 5, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhang, Z.; Zhong, K.; Wang, Z.; Yang, N.; Tang, X.; Li, H.; Lu, Q.; Wu, Z.; Yuan, B.; et al. CXCL11-armed oncolytic adenoviruses enhance CAR-T cell therapeutic efficacy and reprogram tumor microenvironment in glioblastoma. Mol. Ther. 2023, 31, 134–153. [Google Scholar] [CrossRef]
- Samson, A.; Scott, K.J.; Taggart, D.; West, E.J.; Wilson, E.; Nuovo, G.J.; Thomson, S.; Corns, R.; Mathew, R.K.; Fuller, M.J.; et al. Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci. Transl. Med. 2018, 10, eaam7577. [Google Scholar] [CrossRef]
- Zadeh, G.; Daras, M.; Cloughesy, T.F.; Colman, H.; Kumthekar, P.U.; Chen, C.C.; Aiken, R.; Groves, M.D.; Ong, S.; Ramakrishna, R.; et al. LTBK-04. Phase 2 Multicenter Study of the Oncolytic Adenovirus DNX-2401 (tasadenoturev) in Combination with Pembrolizumab for Recurrent Glioblastoma; Captive Study (KEYNOTE-192). Neuro. Oncol. 2020, 22, ii237.
- Chalise, L.; Kato, A.; Ohno, M.; Maeda, S.; Yamamichi, A.; Kuramitsu, S.; Shiina, S.; Takahashi, H.; Ozone, S.; Yamaguchi, J.; et al. Efficacy of cancer-specific anti-podoplanin CAR-T cells and oncolytic herpes virus G47Δ combination therapy against glioblastoma. Mol. Ther. Oncolytics. 2022, 26, 265–274. [Google Scholar] [CrossRef]
- Zhu, G.; Zhang, J.; Zhang, Q.; Jin, G.; Su, X.; Liu, S.; Liu, F. Enhancement of CD70-specific CAR T treatment by IFN-γ released from oHSV-1-infected glioblastoma. Cancer. Immunol. Immunother. 2022, 71, 2433–2448. [Google Scholar] [CrossRef]
- Ma, R.; Lu, T.; Li, Z.; Teng, K.Y.; Mansour, A.G.; Yu, M.; Tian, L.; Xu, B.; Ma, S.; Zhang, J.; et al. An Oncolytic Virus Expressing IL15/IL15Rα Combined with Off-the-Shelf EGFR-CAR NK Cells Targets Glioblastoma. Cancer. Res. 2021, 81, 3635–3648. [Google Scholar] [CrossRef]
Figure 1.
Mechanism of action of Immune checkpoint inhibitors and cancer vaccines and combinatorial therapies for glioblastoma. Dendritic cell (DC), T-cell receptor (TCR), Cytotoxic T lymphocytes (CTLs), major histocompatibility complex (MHC).
Figure 1.
Mechanism of action of Immune checkpoint inhibitors and cancer vaccines and combinatorial therapies for glioblastoma. Dendritic cell (DC), T-cell receptor (TCR), Cytotoxic T lymphocytes (CTLs), major histocompatibility complex (MHC).
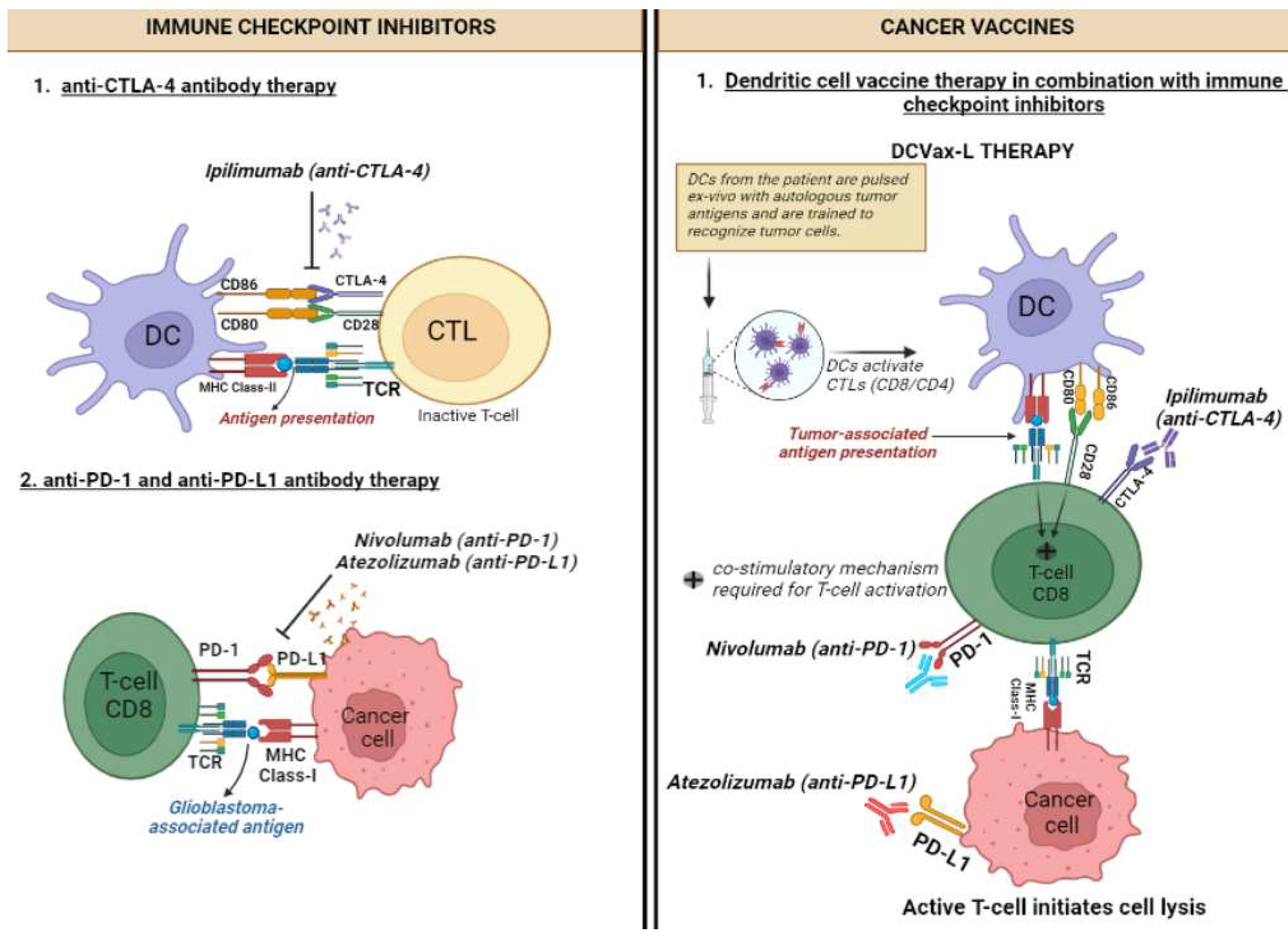
Figure 2.
(A) Structure of anti-EGFRvIII specific CAR-T cells: It consists of a single-chain fragment variable (scFv) for anti-EGFRvIII monoclonal antibody along with CD3ζ (signaling domain for TCR). The intracellular domain consists of costimulatory domains (CD28, 4-1BB, OX40) which are necessary for T-cell activation. (B) Mechanism of action: anti-EGFRvIII specific CAR-T cells recognize EGFRvIII antigens present in the glioblastoma cells and this attachment leads to the release of perforin leading to cytotoxic degranulation.
Figure 2.
(A) Structure of anti-EGFRvIII specific CAR-T cells: It consists of a single-chain fragment variable (scFv) for anti-EGFRvIII monoclonal antibody along with CD3ζ (signaling domain for TCR). The intracellular domain consists of costimulatory domains (CD28, 4-1BB, OX40) which are necessary for T-cell activation. (B) Mechanism of action: anti-EGFRvIII specific CAR-T cells recognize EGFRvIII antigens present in the glioblastoma cells and this attachment leads to the release of perforin leading to cytotoxic degranulation.
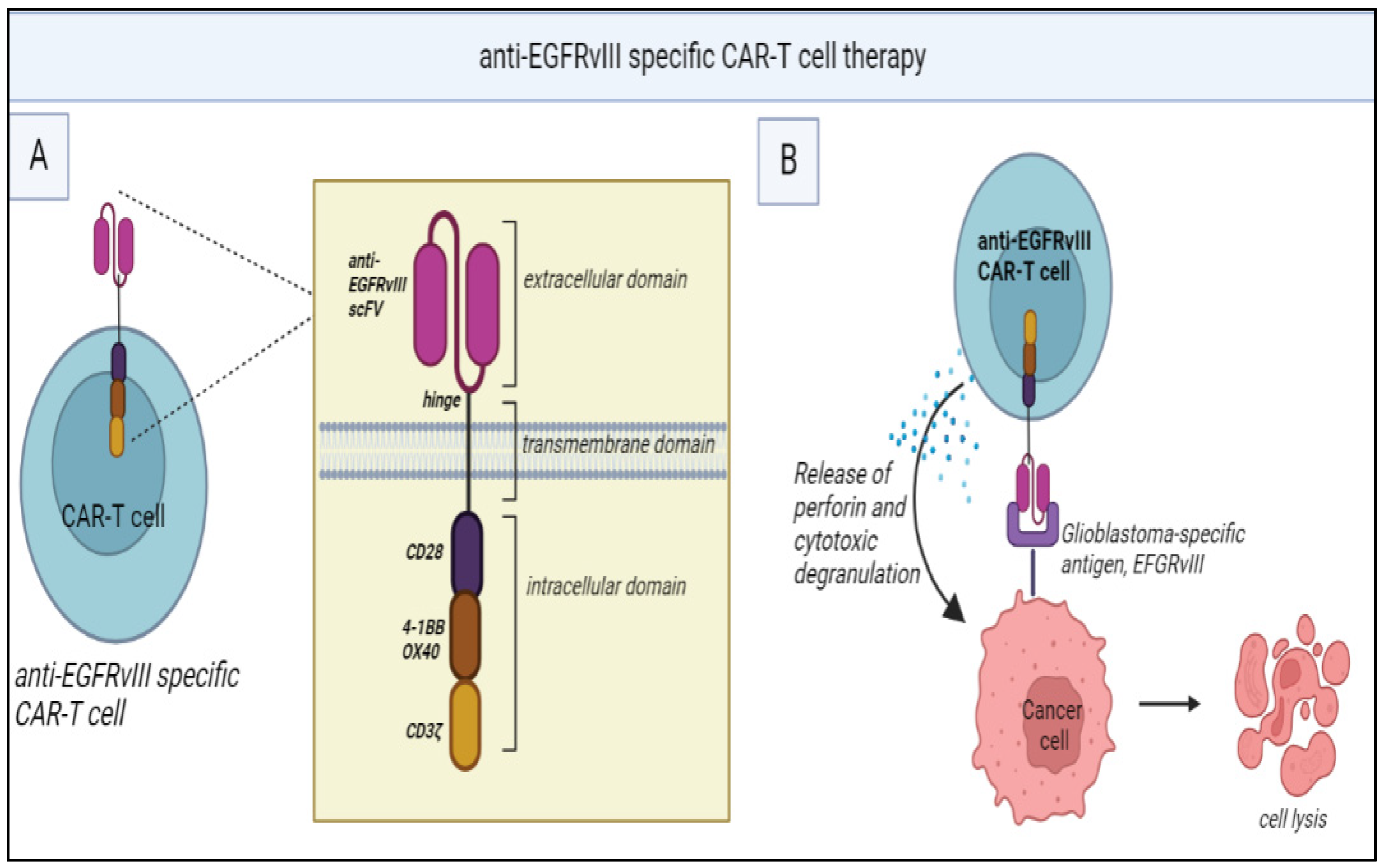
Figure 3.
Oncolytic viral therapy with G47Δ: G47Δ enters the tumor cell through receptor-mediated endocytosis. Once inside the cell, it undergoes viral replication leading to the release of virus progeny. Apoptosis takes place and leads to the release of cytokines such as interferons. IFNs activate the antigen-presenting cells (APCs) like DCs which further mature the cytotoxic T-lymphocytes such as CD8+ T-cells leading to immune stimulation.
Figure 3.
Oncolytic viral therapy with G47Δ: G47Δ enters the tumor cell through receptor-mediated endocytosis. Once inside the cell, it undergoes viral replication leading to the release of virus progeny. Apoptosis takes place and leads to the release of cytokines such as interferons. IFNs activate the antigen-presenting cells (APCs) like DCs which further mature the cytotoxic T-lymphocytes such as CD8+ T-cells leading to immune stimulation.
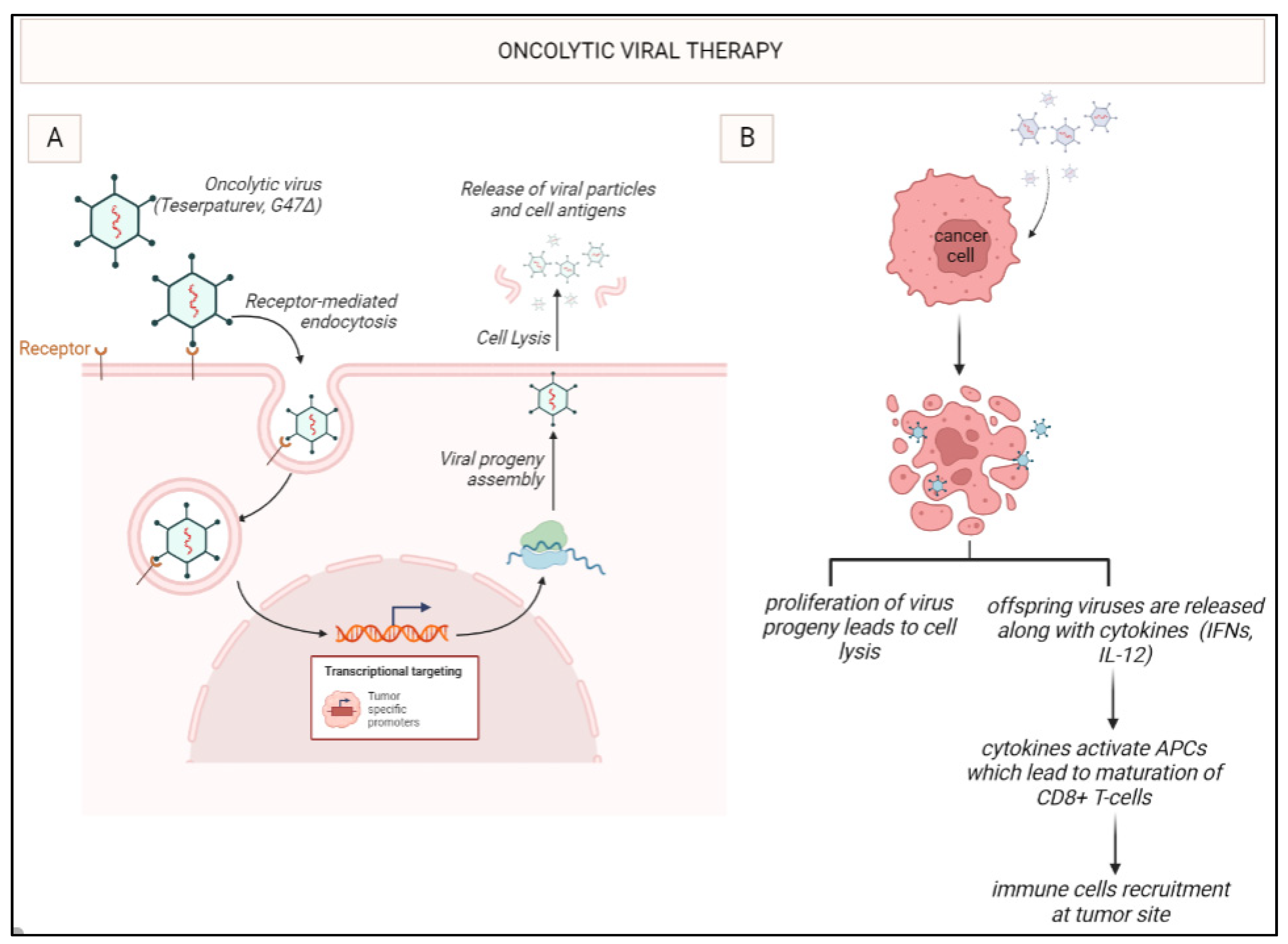

Table 1.
Principal cells of the tumour microenvironment of gliomas.
| Cell type | Function within the tumour microenvironment (TME) |
|---|---|
| Glioma cells |
|
| Tumour-associated macrophages and microglia (TAMs) |
|
| Regulatory T (Treg) cells |
|
| Natural kills (NK) cells |
|
| Dendritic cells (DCs) |
|
| Myeloid-derived suppressor cells (MDSCs) |
|
Table 2.
Recently developed immunotherapies for glioblastoma.
| Immunotherapy | Description |
|---|---|
| Immune checkpoint inhibitors (ICIs) | Monoclonal antibodies that block either the programmed cell death protein 1 (PD-1) or cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) pathways, resulting in the activation of T cells to target cancer cells |
| Cancer vaccines | Immunogenic agents designed to stimulate antigen presentation and immune activation against cancer cells |
| T-cell therapies | T cells are genetically engineered to express chimeric antigen receptors (CARs) that can recognize specific tumour antigens |
| Oncolytic virotherapy (OVT) | Engineered viruses selectively infect and kill cancer cells, inducing an immune response against tumour antigens |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



