
Submitted:
29 June 2023
Posted:
30 June 2023
You are already at the latest version
Abstract
The Cl- transporting proteins CFTR, SLC26A9, and anoctamin (ANO1; ANO6) appear to have more in common than anticipated initially. They all participate in the pathogenic process and clinical outcome of renal and airway diseases. In the present review we will therefore concentrate on recent findings concerning electrolyte transport in airways and kidney, and the role of CFTR, SLC26A9, and anoctamins (ANO1 and ANO6). A particular focus will be on the airway diseases cystic fibrosis and asthma, as well as renal alkalosis and polycystic kidney disease. In essence, latest findings are summarized that demonstrate CFTR as the only relevant secretory Cl- channel in airways under basal (non-stimulated) conditions and after stimulation by secretagogues. For proper CFTR function, expression of ANO1 and ANO6 appears to be a prerequisite. Evidence is summarized suggesting that the Cl- transporter SLC26A9 may have a reabsorptive rather than a Cl- secretory function in the airways. In renal collecting ducts bicarbonate secretion takes place due to synergistic tasks of CFTR and the Cl-/HCO3- transporter SLC26A4 (pendrin), which is likely to be supported by ANO1. Finally, in autosomal dominant polycystic kidney disease (ADPKD), the secretory function of CFTR in renal cyst formation might have been overestimated, while ANO1 as well as ANO6 turned out to be crucial in ADPKD, and therefore represent novel pharmacological targets for the treatment of polycystic kidney disease.
Keywords:
TMEM16A
; TMEM16F
; anoctamin
; SLC26A9
; CFTR
; pendrin
1. Introduction
A vast number of reports describe functional changes induced by either knockout or knockdown or by overexpression of the cystic fibrosis transmembrane conductance regulator (CFTR). Although CFTR is the essential Cl- channel for transepithelial Cl- secretion, many of the morphological and functional changes occurring during change of CFTR-expression are thought to be caused indirectly by up- or downregulation of intracellular signaling pathways or by metabolic changes, which affect the function of other, independent proteins. Along this line, physical protein-protein interactions are reported, resulting in functional coupling to partner proteins (e.g. the epithelial Na+ channel ENaC or the Cl-/HCO3- exchanger pendrin), thus controlling ion transport and other cellular and tissue functions. Meanwhile, large “interactomes” exist for CFTR, which lead to the somewhat provocative question, whether CFTR “acts with everything?” [1-6]. Indeed, CFTR is a true hub for kinases and crosstalk of cAMP and Ca2+ [7]. In this short review we will focus specifically on the interplay of CFTR with the Cl- transporter SLC26A9, the Ca2+ activated Cl- channel anoctamin 1 (ANO1) and the phospholipid scramblase anoctamin 6 (Ano6) in airways and kidney.
Airway secretion of bicarbonate (HCO3-) by CFTR and additional apical transporter
Early studies demonstrated that disruption of cftr in mice causes organ disease typical for cystic fibrosis (CF), such as meconium ileus, distal intestinal obstructions with mucus accumulation, blockage of pancreatic ducts and lacrimal gland dilatation, along with some developmental defects [8]. These initial studies were confirmed in a number of subsequent transgenic models for cystic fibrosis. However, a central aspect of CF pathology, namely the chronic inflammatory airway disease was hardly detectable in CF mice [9]. However, in a number of studies with a CF pig model, human CF pathology could be nicely reproduced [10,11]. In contrast to transgenic F508del-cftr mice, CF pigs demonstrated reduced airway surface pH, impaired bacterial killing and adhesive mucus that disrupts mucociliary transport [12-15]. It was concluded that dysfunctional CFTR leads to a lack of HCO3- secretion, thus causing acidification of the airway surface liquid (ASL), followed by mucus abnormalities, attenuation of airway defense, inflammation and a typical CF lung phenotype [14-17]. However, in another porcine CFTR-knockout model acidic ASL pH could not be detected [18,19]. In this study, micro pH-electrodes measurements were used to assess ASL-pH directly in small airways of lung sections from acutely sacrificed newborn piglets [18]. Pathological changes in these CFTR-/- lungs were not detected. Along this line, another study reported mucus accumulation preceding pulmonary infection in children with CF [20]. Moreover, using a novel luminescent technology integrated with fiberoptic probes, an acidic airway surface liquid pH could not be detected in children with cystic fibrosis [21]. This raises the question whether HCO3- may also use a secretory pathway that is different to CFTR.
SLC26A9 is expressed in the apical membrane of airways from CFTR-knockout piglets, but not in airways expressing CFTR-F508del
Recently, expression of the Cl-/SLC26A9 was found to be essentially absent in human F508del-CFTR/F508del-CFTR airways [22], which corresponds to the well-known inhibitory effect of F508del-CFTR on membrane expression of SLC26A9 [23,24]. In contrast, SLC26A9 is well expressed in the apical membrane of airway epithelial cells in non-CF lungs and in lungs from CFTR-knockout piglets [22]. Membrane expression of SLC26A9 in the absence of CFTR was also shown in cell culture, whereas coexpression with F508del-CFTR in cultured cells abrogates biosynthesis, trafficking and function of SLC26A9 [23-25]. We therefore speculate that normal ASL-pH measured in airways of CFTR-/- piglets is due to normal location and function of SLC26A9, which suggests that HCO3- can be secreted by SLC26A9 to the luminal side of airways.
Transport of HCO3- by SLC26A9 has been proposed in some studies [26-28], but was not found by other laboratories [29-32]. Additional tissue factors like epithelial polarization or coexpression with additional proteins like CFTR may affect SLC26A9 transport function. Along this line, the vast majority of SLC26A9 expressed in non-polarized cells remains in the cytosol, while it is nicely expressed in the apical membrane of polarized cells [33]. Coexpression with wtCFTR or complete absence of CFTR (CFTR knockout piglets) allows proper plasma membrane location of SLC26A9, in contrast to airways of CF-patients expressing a F508del-CFTR allele [22]. Like other SLC26A proteins (SLC26A3,4,6,8), also SLC26A9 may interact physically with CFTR via R (regulatory) and STAS (Sulphate Transporter and AntiSigma factor antagonist) domains, and probably through PDZ-domain interaction [29,34-38]. A recent study showed a contribution of SLC26A9 to airway bicarbonate secretion using the novel SLC26A9 inhibitor S9-A13. Online recordings of ASL pH in primary human nasal epithelial cells under thin film conditions indicated a sustained decrease in ASL-pH by S9-A13, while subsequent activation of CFTR was unable to re-alkalinize ASL-pH [33]. These initial results should now be confirmed by additional studies in vivo to confirm SLC26A9-dependent bicarbonate transport in airways. It will also be interesting to learn to what extend SLC26A9 contributes to airway HCO3- secretion, when compared to SLC26A4 (pendrin), which probably secretes most HCO3-, particularly during inflammation [39]. Along this line, the main task of SLC26A9 in airways and particularly in alveoli could be actually reabsorption of Cl- rather than Cl- secretion (reviewed in [40].
CFTR causes constitutive basal Cl- secretion in the airways
Airways demonstrate a basal Cl- secretion in the absence of secretagogues and cAMP or Ca2+-dependent stimulation. It should be noted that during patch clamp recordings a spontaneous basal CFTR activity was not observed in the absence of PKA- or PKC-dependent stimulation. Previous studies provided conflicting data as to the origin of basal Cl- secretion. While some studies suggested a spontaneous activity of CFTR causing basal Cl- secretion [18,41-43], others suggested SLC26A9 as the responsible transporter [23,25,29,44,45]. Previously separation of both conductances (CFTR and SLC26A) was difficult due to the lack of specific inhibitors for SLC26A9, off-target effects of CFTR [46] and because intracellular trafficking and activity of SLC26A9 depends on expression and function of CFTR [23,24,47-49].
In the study by Jo et al the SLC26A9-inhibitor S9-A13 exerted no inhibitory effect on airway Cl- transport in vitro or ex vivo, while C·FTRinh172 inhibited both basal and cAMP-induced Cl- secretion [33]. Interestingly, neither inhibition of adenosine receptors nor inhibition of adenylate cyclase blocked basal Cl- secretion, asking for additional mechanisms. It is possible that increase in protein kinase C (PKC) activity, e.g. through ATP-release and binding to purinergic receptors, may keep some CFTR active at basal cAMP levels and PKA-phosphorylation [50-53]. Moreover, the Hanrahan lab recently demonstrated expression of SLC26A4 (pendrin) in ciliated primary nasal and bronchial airway epithelial cells, where it enhances Cl- secretion by stimulated CFTR [39]. The molecular mechanism of STAS/R- domain interaction had been shown earlier for activation of CFTR by SLC26A6 [35]. As discussed in the next section, the Ca2+ activated Cl- channel ANO1 may contribute maintenance of a basal CFTR activity by a Ca2+ dependent mechanism.
Relationship between CFTR and anoctamins
Early studies showed cAMP/PKA and increase in intracellular Ca2+ as two independent second messenger pathways that lead to epithelial Cl- secretion [54]. The pharmacological tools to discriminate both Cl- conductances, however, were rather non-specific and thus our team could not clearly keep these conductances apart [55-57]. We and others also reported that CFTR seemingly “inhibits” endogenous Ca2+ activated Cl- currents (CaCC) in Xenopus oocytes, bovine pulmonary artery endothelium and isolated parotid acinar cells [58-62]. After molecular identification of the Ca2+ activated Cl- channel (CACC) as anoctamin 1 (ANO1), it was found that CFTR does not inhibit ANO1 but that ANO1 currents and CFTR currents are not additive, i.e. do not add up to the sum of both currents [63].
In fact, Ca2+- enhancing agonists such as purinergic or muscarinic ligands activate mostly CFTR-dependent secretion in airways [50,64,65], while ANO1 currents rapidly inactive due to mechanisms outlined in previous reports [66-69]. Therefore, after inhibition or in the absence of CFTR CACC is very short-lived and in the intestine there is not even an apical CACC (ANO1) [70-73]. Inactivation of ANO1 is reasonably well understood [66] [74] and therefore raises the question of whether direct pharmacological activation of ANO1 by ETX001 (ETD-002) can be successful in restoring Cl secretion in the airways of CF patients [75]. It should also mentioned that ANO1 is expressed at only very low levels in airways[76].
Activation of ANO1 in CF may be even counterproductive as ANO1 is a proinflammatory factor, enhances mucus production and mucus secretion, and supports pain sensation [77-81]. Moreover, during inflammatory airway diseases such as asthma and CF, ANO1 is upregulated in pulmonary arterial vessels where it supports airway constriction [76,82]. Finally, ANO1 supports the release of inflammatory cytokines such as IL-8 and accumulation of pulmonary CD-45 positive cells [77]. A phase 1 clinical trial with ETD-002 has been finished more than a year ago, but so far, no outcome has been reported.
Crosstalk between CFTR and ANO1
Studies reported attenuated expression of ANO1 in the apical membrane of airway epithelial cells, when coexpressed with F508del-CFTR [56,83]. We found evidence for an interaction of ANO1 and CFTR through PSD-95/Dlg/ZO-1 (PDZ) domain proteins, similar as described for SLC26A9 [23]. The functional interaction between ANO1 and CFTR is based on the crosstalk of intracellular Ca2+ and the intracellular cAMP signaling pathway. Crosstalk is facilitated by exchange protein directly activated by cAMP (EPAC1) and Ca2+ -sensitive adenylate cyclase type 1 (ADCY1). Assembly of such a local signalosome also depends on the presence of G-protein coupled receptors (GPCRs) [57,84].
Reduced plasma membrane expression of CFTR in the absence of ANO1
Cell-specific knockout of ANO1 in ciliated airway epithelial cells abolished Ca2+ activated Cl- currents and largely reduced Ca2+-dependent Cl- secretion in mouse airways. Moreover, Ca2+ dependent Cl- transport was abolished in intestinal epithelial cells from epithelial-specific ANO1-knockout mice [56]. However, we also reported the surprising finding, that in parallel to the loss of ANO1-dependent transport, also CFTR-dependent Cl- transport was lost in these ANO1-knockout animals [56] (Figure 1). In both airways and intestine, we found that expression of CFTR in the apical membrane was largely attenuated, if not abolished. It should be noted that expression of ANO1 in mouse airways is very low, while clear expression of ANO1 is detected in colonic epithelial cells, which however, is mainly located in the basolateral membrane [73,85].
How are these findings be explained? From earlier studies we know that ANO1 tethers the endoplasmic reticulum (ER) near the plasma membrane (PM), via binding to the inositol trisphosphate receptor (IP3R). Due to this, IP3 - mediated Ca2+-release from the ER and store-operated Ca2+ influx are strongly improved in airway sub-apical or colonic sub-basolateral membrane compartment [86-88]. Along this line it is of note that extended synaptotagmin-1 (ESYT1), another ER-PM tether was found to further enhance PM-expression of ANO1 and Ca2+ signaling [89]. PM-expression of CFTR requires exocytosis due to high local Ca2+ levels facilitated by ANO1. Exocytosis is further supported by ANO6. Using knockout mice for ANO1 and ANO6 and a number of human cell lines we and others showed that both ANO1 and ANO6 are important for PM-insertion and activation of CFTR [76,77,90-93]. In this context, Ca2+-dependent activation of PKC could play a role [50,94]. Enhanced sub-membranous Ca2+ may further support CFTR activity by via Ca2+-activated adenylate cyclases and EPAC. Both anoctamins are equally important for mucus secretion by goblet cells and release of lysozyme and other antimicrobial factors by Paneth cells [95]. We may speculate that ANO1 serves local Ca2+ signaling and not or at least not primarily Cl- secretion.
Patients carrying a loss of function mutation in ANO1 lack of CFTR currents
The first two patients expressing the ANO1-variant c.897+3_897+6delAAGT were reported recently. These patients express a dysfunctional ANO1 and lack of Ca2+ activated Cl- currents [96]. The two reported siblings presented in early infancy with reduced intestinal peristalsis and recurrent episodes of hemorrhagic diarrhea. Analysis of isolated primary airway epithelial cells obtained from one of the patients reproduced the results obtained earlier in tissue specific ANO1-knockout mice [56]: Apart from the absence of Ca2+ activated Cl- transport, CFTR Cl- currents were also completely absent possibly due to a lack of expression of CFTR in the apical membrane. Moreover, analysis of cells obtained from a heterozygous sibling showed reduced Cl- secretion [96]. Rather surprising, the patients did not show a CF-like lung phenotype, although sweat tests were positive, indicating defective CFTR Cl- conductance. This is even more surprising given the fact that both Ca2+ - activated ANO1 and cAMP-activated CFTR Cl- conductances were absent. Cytokine levels measured in sputum samples obtained from one of the ANO1-patients were largely reduced when compared to cytokine levels measured in samples from two CF patients (Figure 2). While this may provide further evidence for the pro-inflammatory role of ANO1 [81], it also raises questions regarding the true contribution of apical Cl- conductance for CF pathology [78].
CFTR and ANO1/ANO6 in cell death
Within the pathogenic relationships of CFTR with other proteins, the phospholipid scramblase ANO6 was found to have a role in CFTR-dependent cell death [98]. CFTR had been proposed to release glutathione (GSH) from airway epithelial cells to be enriched in the apical airway surface liquid, which will neutralize reactive oxygen species (ROS) [99-101]. Apparently GSH-efflux does not change cytosolic GSH content [102] and we were unable to detect different ROS levels depending on expression of CFTR [103]. However, we observed an enhanced activity of ANO6 in the presence of wtCFTR. As in most other cell types, ANO6 is also expressed in airway cells epithelial cells where it can scramble plasma membrane phospholipids, which leads to cell death [104]. The importance of ANO6 for regulated cell death is also demonstrated in ANO6 knockout mice. In these animals the number of apoptotic cells within the intestinal epithelium was strongly reduced [103].
In vivo inoculation with P. aeruginosa or Staphylococcus aureus induced lipid peroxidation in lungs of CFTR-knockout mice and wild type animals. Exposure of human airway epithelial cells to P. aeruginosa induced an increase in reactive oxygen species (ROS) and caused lipid peroxidation and cell death. P. aeruginosa induced cell death, was independent of expression of wt-CFTR or F508del-CFTR [105]. In contrast, knockout of ANO1 clearly reduced cell death, probably because ANO1 supports Ca2+ - dependent activation of ANO6 and phospholipid scrambling [106].
Bicarbonate is secreted in renal collecting ducts, which requires CFTR, Pendrin and possibly ANO1
CFTR is expressed in tubular epithelial cells of human kidneys, where it affects different transport functions [107]. Early studies suggested a role of CFTR for renal bicarbonate (HCO3-) transport [108,109], which was later confirmed for many other epithelial organs [17]. HCO3- excretion was found to be largely reduced in people with CF, particularly when patients were challenged with the hormone secretin, which binds to its receptor and increases intracellular cAMP. A defect in renal bicarbonate excretion can lead to metabolic alkalosis occasionally observed in CF patients. Detailed studies in mice lacking expression of CFTR or the HCO3- transporter SLC26A4 (pendrin) finally uncovered the molecular mechanism [110,111]. Physical interaction of CFTR with pendrin and/or Cl- recycling via CFTR drives tubular release of HCO3- through apical pendrin and urinary excretion. This process takes place in ß-intercalated cells of the renal collecting duct, which coexpress all CFTR, pendrin and receptors for secretin [110]. ANO1 is colocalized together with pendrin (and CFTR) in the apical membrane of renal ß-intercalated cells and may support the activity of CFTR [110] (Figure 3).
CFTR and ANO1 in polycystic kidney disease: which one counts?
Autosomal Dominant Polycystic Kidney Disease (ADPKD) is the most common monogenic kidney disease affecting approximately 1:1000 individuals often resulting in end-stage renal disease [112]. Mutations in either the PKD1 (~78%) or PKD2 (~15%) gene [113] cause formation of multiple renal cysts which originate from renal tubule epithelial cells, predominantly principal cells of the collecting duct [114,115]. The cysts grow continuously over years and cause compression of the adjacent intact nephrons, which results in a decline of renal function [116]. Two key features are identified for cyst growth: change from an absorptive towards a secretory epithelium and abnormal proliferation of cyst epithelial cells [117]. It is assumed that the major secretory force for cyst fluid secretion is apical cAMP-dependent Cl- secretion and several studies suggested CFTR as the essential Cl- channel [118-120].
However, recently Cabrita et al. demonstrated that cyst growth in ADPKD is prevented by pharmacological and genetic inhibition of the calcium activated chloride channel ANO1 [121]. Loss of PKD1 increased expression of ANO1 and CFTR and induced Cl− secretion in murine kidneys. Importantly, upregulated ANO1 enhanced intracellular Ca2+ signaling and proliferation of PKD1-deficient renal epithelial cells. In contrast, increase in Ca2+ signaling, cell proliferation and CFTR expression was not observed in PKD1/ANO1 double knockout mice. In a sophisticated renal collecting duct M1 cell organoid model and in primary renal epithelial cells, cell proliferation and Cl- secretion was also dependent on enhanced expression of ANO1 [122,123]. Knockdown of PKD1 or PKD2 increased basal intracellular Ca2+ levels and enhanced purinergic Ca2+ release from endoplasmic reticulum. Ca2+ signals, proliferation, and Cl- secretion were largely reduced by knockdown or blockade of ANO1. ANO1 is therefore central to enhanced Ca2+ release from IP3-sensitive ER Ca2+ stores, and is a central player in ADPKD caused by mutations in PKD1 and PKD2. The data strongly suggest pharmacological inhibition of ANO1 to slow down progression of ADPKD.
Concerning disease progression, male gender is a major risk factor [124,125]. Talbi et al. found that kidneys from PKD1 knockout mice had a more pronounced phenotype in males compared to females. Proliferation of cells from the cyst epithelium was enhanced in male when compared to female kidneys. This was paralleled by higher basal intracellular Ca2+ concentrations in cells isolated from PKD1 knockout males. These results again suggest enhanced intracellular Ca2+ levels contributing to enhanced proliferation and cyst development in male kidneys. Notably, incubation of renal cells with dihydrotestosterone enhanced basal Ca2+ levels and ATP-stimulated ANO1 currents [126]. Similar results were obtained in a mouse model for autosomal recessive polycystic kidney disease (ARPKD) [127]. Finally, polycystic kidneys are under constant oxidative stress, which causes lipid peroxidation [128]. Lipid peroxidation has been shown to activate ANO1 and to drives cyst growth [129].
Targeting ANO1 or CFTR in ADPKD?
Inhibitors of Cl- currents such as duiphenylamine-2-carboxylate and knockdown of CFTR by antisense oligo-nucleotides inhibited cAMP-activated Cl- currents in cyst cells [130]; 8807590). CFTRinh-172 or Ph-GlyH-101 reduced cyst growth of renal MDCK cells, in a metanephric mouse kidney model and a rapidly progressive neonatal Pkd1 knockout mouse model [119,120]. In three CF patients with concomitant ADPKD, diseases progress was delayed when compared to their siblings without CF [131,132]. However, the ADPKD-protective effect by CF was not confirmed in a subsequent report [133] and CFTR expression in isolated ADPKD cyst cells was shown to be very heterogeneous [118,130,134]. It is therefore not entirely conclusive to inhibit CFTR to slow down cyst progression. Moreover, both CFTRinh-172 or Ph-GlyH-101 have pronounced off-target effects and affect intracellular Ca2+ signals which actually inhibits ANO1 [46].
Initial studies showed that Ca2+-activated ANO1 Cl- currents contribute to cyst growth [135,136]. ATP is released by cyst cells, accumulates in the cyst lumen and activates ANO1 via stimulation of purinergic receptors [115,135,137]. By contrast, scavenging of ATP by apyrase, the P2Y2 receptor antagonist suramin, and knockdown of P2Y2 inhibited cyst growth [115,138]. These studies and the subsequent work outlined above [121,122] suggested ANO1 as the relevant pharmacological target to inhibit in ADPKD.
Taken together, in mouse studies ANO1 is a dominant driver of secretion-dependent cyst enlargement, while we also found that knockout of CFTR had no significant impact on cyst growth [139] (Figure 4). Nevertheless, it is important to keep in mind that the physiological contribution of ANO1 in the mouse is probably greater while the contribution of CFTR is lower than in humans. In mouse, CFTR shows lower activity in the airways but has a more pronounced contribution to intestinal transport [140]. In the kidneys of healthy mice, CFTR is only clearly expressed in ß-intercalated cells, where it controls HCO3- secretion [110]. Interestingly, a recent report shows that application of VX-809 (Lumacaftor) in a Pkd1 knockout and the Pkd1RC/RC mouse model reduced cyst growth [141,142]. These findings were explained by a cellular translocalization of CFTR and the Na+/H+ exchanger 3. A clinical phase 2, placebo-controlled, randomized trial investigates the efficacy and safety of the CFTR corrector GLPG2737 in ADPKD patients (NCT04578548) [143]. More studies are required to analyse the contribution of ANO1 to cyst formation in human tissue. Central aspects of ANO1 are its obvious pro-proliferative and de-differentiating properties [98], which after all may have a larger impact on cyst progression than fluid secretion.
Inhibitors of ANO1
Given the promising results obtained by genetic and pharmacological inhibition of ANO1, ANO1 has qualified as a potential target for the treatment of ADPKD. ANO1 function can be addressed by drugs that have already been approved for other indications like niclosamide or benzbromarone [121]. Niclosamide is an essential oral anthelminthic drug used for decades to treat parasitic infections, but it is meant for short-term use [144]. Benzbromarone is a uricosuric drug that has been used in the treatment of gout over the last 30 years. Although withdrawn by Sanofi for safety reasons after reports of hepatotoxicity, it is still marketed in several countries by other drug companies (drugs.com/international/benzbromarone.html). Hepatotoxicity is rare and occurs in 1:17000 patients [145]. For comparison, the only drug approved for treatment of ADPKD, the vasopressin-2-receptor antagonist tolvaptan, has a hepatotoxic risk of 1:3000. Therefore, many experts in the field have questioned withdrawal of benzbromarone [145].
Conclusion
The present review summarizes recent findings for CFTR, SLC26A9 and the anoctamins 1 and 6 in airways and kidney. It becomes clear that these ion channels and transports cannot only be examined individually but should be analyzed in the context of their molecular and functional interaction. This is particularly important in pharmacotherapy and the choice of the choice of the right pharmacological target.
Authors contributions: Conceptualization, K.K., J.O., A.K., R.S., B.B.; methodology., J.O., A.K., J.H.P., T.M., R.S., B.B.; formal analysis, J.O., A.K., B.B.; writing—original draft preparation, K.K., R.S., B.B.; writing—review and editing, ., J.O., A.K., J.H.P., T.M., R.S., B.B. All authors have read and agreed to the published version of the manuscript.
Funding
Supported by DFG Transregio-SFB, Project-ID 509149993, TRR 374 (Project A3).
Institutional Review Board Statement
All animal experiments complied with the general guidelines for animal research, in accordance with the United Kingdom Animals Act, 1986, and associated guidelines, and EU Directive 2010/ 63/EU for animal experiments. All animal experiments were approved by the local Ethics Committee of the Government of Unterfranken/Wurzburg/Germany (AZ: 55.2-2532-2-677) and were conducted according to the guidelines of the American Physiologic Society and German Law for the Welfare of Animals.
Data Availability Statement
MDPI Research Data Policies at https://www.mdpi.com/ethics.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kunzelmann, K. CFTR: Interacting with everything? News Physiol Sciences 2001, 17, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Venable, J.; LaPointe, P.; Hutt, D.M.; Koulov, A.V.; Coppinger, J.; Gurkan, C.; Kellner, W.; Matteson, J.; Plutner, H. , et al. Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell 2006, 127, 803–815. [Google Scholar] [CrossRef] [PubMed]
- Pankow, S.; Bamberger, C.; Calzolari, D.; Martínez-Bartolomé, S.; Lavallée-Adam, M.; Balch, W.E.; Yates, J.R. , 3rd. ∆F508 CFTR interactome remodelling promotes rescue of cystic fibrosis. Nature 2015, 528, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.H.; Snider, J.; Birimberg-Schwartz, L.; Ip, W.; Serralha, J.C.; Botelho, H.M.; Lopes-Pacheco, M.; Pinto, M.C.; Moutaoufik, M.T.; Zilocchi, M. , et al. CFTR interactome mapping using the mammalian membrane two-hybrid high-throughput screening system. Molecular systems biology 2022, 18, e10629. [Google Scholar] [CrossRef]
- Pinto, M.C.; Botelho, H.M.; Silva, I.A.L.; Railean, V.; Neumann, B.; Pepperkok, R.; Schreiber, R.; Kunzelmann, K.; Amaral, M.D. Systems Approaches to Unravel Molecular Function: High-content siRNA Screen Identifies TMEM16A Traffic Regulators as Potential Drug Targets for Cystic Fibrosis. J Mol Biol 2022, 167436. [Google Scholar] [CrossRef]
- Pereira, C.; Mazein, A.; Farinha, C.M.; Gray, M.A.; Kunzelmann, K.; Ostaszewski, M.; Balaur, I.; Amaral, M.D.; Falcao, A.O. CyFi-MAP: an interactive pathway-based resource for cystic fibrosis. Sci Rep 2021, 11, 22223. [Google Scholar] [CrossRef]
- Kunzelmann, K.; Mehta, A. CFTR: a hub for kinases and cross-talk of cAMP and Ca. FEBS J 2013, 280, 4417–4429. [Google Scholar] [CrossRef]
- Ratcliff, R.; Evans, M.J.; Cuthbert, A.W.; MacVinish, L.J.; Foster, D.; Anderson, J.R.; Colledge, W.H. Production of a severe cystic fibrosis mutation in mice by gene targeting. Nat. Genet 1993, 4, 35–41. [Google Scholar] [CrossRef]
- Wilke, M.; Buijs-Offerman, R.M.; Aarbiou, J.; Colledge, W.H.; Sheppard, D.N.; Touqui, L.; Bot, A.; Jorna, H.; De Jonge, H.R.; Scholte, B.J. Mouse models of cystic fibrosis: phenotypic analysis and research applications. J Cyst. Fibros 2011, 10 Suppl 2, 9. [Google Scholar] [CrossRef]
- Chen, J.H.; Stoltz, D.A.; Karp, P.H.; Ernst, S.E.; Pezzulo, A.A.; Moninger, T.O.; Rector, M.V.; Reznikov, L.R.; Launspach, J.L.; Chaloner, K. , et al. Loss of anion transport without increased sodium absorption characterizes newborn porcine cystic fibrosis airway epithelia. Cell 2010, 143, 911–923. [Google Scholar] [CrossRef]
- Ostedgaard, L.S.; Meyerholz, D.K.; Chen, J.H.; Pezzulo, A.A.; Karp, P.H.; Rokhlina, T.; Ernst, S.E.; Hanfland, R.A.; Reznikov, L.R.; Ludwig, P.S. , et al. The {Delta}F508 Mutation Causes CFTR Misprocessing and Cystic Fibrosis-Like Disease in Pigs. Sci. Transl. Med 2011, 3, 74ra24. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, K.A.; Schroder, R.L.; Skaaning-Jensen, B.; Strobaek, D.; Olesen, S.P.; Christophersen, P. Activation of the human intermediate-conductance Ca(2+)-activated K(+) channel by 1-ethyl-2-benzimidazolinone is strongly Ca(2+)-dependent. Biochim. Biophys. Acta 1999, 1420, 231–240. [Google Scholar] [CrossRef]
- Stoltz, D.A.; Meyerholz, D.K.; Pezzulo, A.A.; Ramachandran, S.; Rogan, M.P.; Davis, G.J.; Hanfland, R.A.; Wohlford-Lenane, C.; Dohrn, C.L.; Bartlett, J.A. , et al. Cystic fibrosis pigs develop lung disease and exhibit defective bacterial eradication at birth. Sci. Transl. Med 2010, 2, 29ra31. [Google Scholar] [CrossRef] [PubMed]
- Pezzulo, A.A.; Tang, X.X.; Hoegger, M.J.; Alaiwa, M.H.; Ramachandran, S.; Moninger, T.O.; Karp, P.H.; Wohlford-Lenane, C.L.; Haagsman, H.P.; van Eijk, M. , et al. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature 2012, 487, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Hoegger, M.J.; Fischer, A.J.; McMenimen, J.D.; Ostedgaard, L.S.; Tucker, A.J.; Awadalla, M.A.; Moninger, T.O.; Michalski, A.S.; Hoffman, E.A.; Zabner, J. , et al. Impaired mucus detachment disrupts mucociliary transport in a piglet model of cystic fibrosis. Science 2014, 345, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Shah, V.S.; Ernst, S.; Tang, X.X.; Karp, P.H.; Parker, C.P.; Ostedgaard, L.S.; Welsh, M.J. Relationships among CFTR expression, HCO3- secretion, and host defense may inform gene- and cell-based cystic fibrosis therapies. Proceedings of the National Academy of Sciences of the United States of America 2016, 113, 5382–5387. [Google Scholar] [CrossRef]
- Kunzelmann, K.; Schreiber, R.; Hadorn, H.B. Bicarbonate in cystic fibrosis. Journal of Cystic Fibrosis 2017, 16, 653–662. [Google Scholar] [CrossRef]
- Benedetto, R.; Centeio, R.; Ousingsawat, J.; Schreiber, R.; Janda, M.; Kunzelmann, K. Transport properties in CFTR-/- knockout piglets suggest normal airway surface liquid pH and enhanced amiloride-sensitive Na(+) absorption. Pflugers Arch 2020, 472, 1507–1519. [Google Scholar] [CrossRef]
- Klymiuk, N.; Mundhenk, L.; Kraehe, K.; Wuensch, A.; Plog, S.; Emrich, D.; Langenmayer, M.C.; Stehr, M.; Holzinger, A.; Kroner, C. , et al. Sequential targeting of CFTR by BAC vectors generates a novel pig model of cystic fibrosis. Journal of molecular medicine (Berlin, Germany) 2012, 90, 597–608. [Google Scholar] [CrossRef]
- Esther, C.R., Jr.; Muhlebach, M.S.; Ehre, C.; Hill, D.B.; Wolfgang, M.C.; Kesimer, M.; Ramsey, K.A.; Markovetz, M.R.; Garbarine, I.C.; Forest, M.G. , et al. Mucus accumulation in the lungs precedes structural changes and infection in children with cystic fibrosis. Science translational medicine 2019, 11. [Google Scholar] [CrossRef]
- Schultz, A.; Puvvadi, R.; Borisov, S.M.; Shaw, N.C.; Klimant, I.; Berry, L.J.; Montgomery, S.T.; Nguyen, T.; Kreda, S.M.; Kicic, A. , et al. Airway surface liquid pH is not acidic in children with cystic fibrosis. Nature communications 2017, 8, 1409. [Google Scholar] [CrossRef] [PubMed]
- Ousingsawat, J.; Centeio, R.; Schreiber, R.; Kunzelmann, K. Expression of SLC26A9 in Airways and Its Potential Role in Asthma. International journal of molecular sciences 2022, 23, 2998. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, C.A.; Mitra, S.; Mishra, S.K.; Wang, X.; Zhao, Y.; Pilewski, J.M.; Madden, D.R.; Frizzell, R.A. The CFTR trafficking mutation F508del inhibits the constitutive activity of SLC26A9. Am J Physiol Lung Cell Mol Physiol 2017, 312, L912–l925. [Google Scholar] [CrossRef] [PubMed]
- Needham, P.G.; Goeckeler-Fried, J.L.; Zhang, C.; Sun, Z.; Wetzel, A.R.; Bertrand, C.A.; Brodsky, J.L. SLC26A9 is selected for endoplasmic reticulum associated degradation (ERAD) via Hsp70-dependent targeting of the soluble STAS domain. Biochem J 2021, 478, 4203–4220. [Google Scholar] [CrossRef]
- Ousingsawat, J.; Schreiber, R.; Kunzelmann, K. Differential contribution of SLC26A9 to Cl(-) conductance in polarized and non-polarized epithelial cells. J Cell Physiol 2011, 227, 2323–2329. [Google Scholar] [CrossRef]
- Chang, M.H.; Plata, C.; Zandi-Nejad, K.; Sindic, A.; Sussman, C.R.; Mercado, A.; Broumand, V.; Raghuram, V.; Mount, D.B.; Romero, M.F. Slc26a9-Anion Exchanger, Channel and Na(+) Transporter. J Membr. Biol 2009, 228, 125–140. [Google Scholar] [CrossRef]
- Xu, J.; Henriksnäs, J.; Barone, S.; Witte, D.; Shull, G.E.; Forte, J.G.; Holm, L.; Soleimani, M. SLC26A9 is expressed in gastric surface epithelial cells, mediates Cl-/HCO3- exchange, and is inhibited by NH4+. Am J Physiol Cell Physiol 2005, 289, C493–C505. [Google Scholar] [CrossRef]
- Demitrack, E.S.; Soleimani, M.; Montrose, M.H. Damage to the gastric epithelium activates cellular bicarbonate secretion via SLC26A9 Cl(-)/HCO(3)(-). American journal of physiology. Gastrointestinal and liver physiology 2010, 299, G255–G264. [Google Scholar] [CrossRef]
- Bertrand, C.A.; Zhang, R.; Pilewski, J.M.; Frizzell, R.A. SLC26A9 is a constitutively active, CFTR-regulated anion conductance in human bronchial epithelia. J Gen. Physiol 2009, 133, 421–438. [Google Scholar] [CrossRef]
- Dorwart, M.R.; Shcheynikov, N.; Wang, Y.; Stippec, S.; Muallem, S. SLC26A9 is a Cl(-) channel regulated by the WNK kinases. J Physiol 2007, 584, 333–345. [Google Scholar] [CrossRef]
- Loriol, C.; Dulong, S.; Avella, M.; Gabillat, N.; Boulukos, K.; Borgese, F.; Ehrenfeld, J. Characterization of SLC26A9, facilitation of Cl(-) transport by bicarbonate. Cell Physiol Biochem 2008, 22, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Walter, J.D.; Sawicka, M.; Dutzler, R. Cryo-EM structures and functional characterization of murine Slc26a9 reveal mechanism of uncoupled chloride transport. Elife 2019, 8, e46986. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Centeio, R.; Park, J.; Ousingsawat, J.; Jeon, D.K.; Talbi, K.; Schreiber, R.; Ryu, K.; Kahlenberg, K.; Somoza, V. , et al. The SLC26A9 inhibitor S9-A13 provides no evidence for a role of SLC26A9 in airway chloride secretion but suggests a contribution to regulation of ASL pH and gastric proton secretion. Faseb j 2022, 36, e22534. [Google Scholar] [CrossRef] [PubMed]
- Ko, S.B.; Shcheynikov, N.; Choi, J.Y.; Luo, X.; Ishibashi, K.; Thomas, P.J.; Kim, J.Y.; Kim, K.H.; Lee, M.G.; Naruse, S. , et al. A molecular mechanism for aberrant CFTR-dependent HCO(3)(-) transport in cystic fibrosis. EMBO J 2002 Nov. 1. ;21. (21. ):5662. -72 2002, 21, 5662–5672. [Google Scholar]
- Ko, S.B.; Zeng, W.; Dorwart, M.R.; Luo, X.; Kim, K.H.; Millen, L.; Goto, H.; Naruse, S.; Soyombo, A.; Thomas, P.J. , et al. Gating of CFTR by the STAS domain of SLC26 transporters. Nat. Cell Biol 2004, 6, 343–350. [Google Scholar] [CrossRef]
- Rakonczay, Z., Jr.; Hegyi, P.; Hasegawa, M.; Inoue, M.; You, J.; Iida, A.; Ignáth, I.; Alton, E.W.; Griesenbach, U.; Ovári, G. , et al. CFTR gene transfer to human cystic fibrosis pancreatic duct cells using a Sendai virus vector. J Cell Physiol 2008, 214, 442–455. [Google Scholar] [CrossRef]
- Rode, B.; Dirami, T.; Bakouh, N.; Rizk-Rabin, M.; Norez, C.; Lhuillier, P.; Lorès, P.; Jollivet, M.; Melin, P.; Zvetkova, I. , et al. The testis anion transporter TAT1 (SLC26A8) physically and functionally interacts with the cystic fibrosis transmembrane conductance regulator channel: a potential role during sperm capacitation. Hum Mol Genet 2012, 21, 1287–1298. [Google Scholar] [CrossRef]
- El Khouri, E.; Toure, A. Functional interaction of the cystic fibrosis transmembrane conductance regulator with members of the SLC26 family of anion transporters (SLC26A8 and SLC26A9): physiological and pathophysiological relevance. The international journal of biochemistry & cell biology 2014, 52, 58–67. [Google Scholar] [CrossRef]
- Kim, D.; Huang, J.; Billet, A.; Abu-Arish, A.; Goepp, J.; Matthes, E.; Tewfik, M.A.; Frenkiel, S.; Hanrahan, J.W. Pendrin Mediates Bicarbonate Secretion and Enhances Cystic Fibrosis Transmembrane Conductance Regulator Function in Airway Surface Epithelia. American journal of respiratory cell and molecular biology 2019, 60, 705–716. [Google Scholar] [CrossRef]
- Kunzelmann, K.; Centeio, R.; Ousingsawat, J.; Talbi, K.; Seidler, U.; Schreiber, R. SLC26A9 in airways and intestine: secretion or absorption? Channels (Austin) 2023, 17, 2186434. [Google Scholar] [CrossRef]
- Ishibashi, K.; Okamura, K.; Yamazaki, J. Involvement of apical P2Y2 receptor-regulated CFTR activity in muscarinic stimulation of Cl(-) reabsorption in rat submandibular gland. Am J Physiol Regul. Integr. Comp Physiol 2008, 294, R1729–R1736. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lytle, C.; Quinton, P.M. Predominant constitutive CFTR conductance in small airways. Respir. Res 2005, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Salinas, D.; Nielson, D.W.; Verkman, A.S. Hyperacidity of secreted fluid from submucosal glands in early cystic fibrosis. Am J Physiol Cell Physiol 2006, 290, C741–C749. [Google Scholar] [CrossRef]
- Salomon, J.J.; Spahn, S.; Wang, X.; Fullekrug, J.; Bertrand, C.A.; Mall, M.A. Generation and functional characterization of epithelial cells with stable expression of SLC26A9 Cl- channels. Am J Physiol Lung Cell Mol Physiol 2016, 310, L593–L602. [Google Scholar] [CrossRef]
- Larsen, M.B.; Choi, J.J.; Wang, X.; Myerburg, M.M.; Frizzell, R.A.; Bertrand, C.A. Separating the contributions of SLC26A9 and CFTR to anion secretion in primary human bronchial epithelia (HBE). Am J Physiol Lung Cell Mol Physiol 2021, 321, L1147–L1160. [Google Scholar] [CrossRef]
- Lin, J.; Gettings, S.M.; Talbi, K.; Schreiber, R.; Taggart, M.J.; Preller, M.; Kunzelmann, K.; Althaus, M.; Gray, M.A. Pharmacological inhibitors of the cystic fibrosis transmembrane conductance regulator exert off-target effects on epithelial cation channels. Pflugers Arch 2023, 475, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Thomas, D.Y.; Hanrahan, J.W. The anion transporter SLC26A9 localizes to tight junctions and is degraded by the proteasome when co-expressed with F508del-CFTR. J Biol Chem 2019, 294, 18269–18284. [Google Scholar] [CrossRef]
- Pinto, M.C.; Quaresma, M.C.; Silva, I.A.L.; Railean, V.; Ramalho, S.S.; Amaral, M.D. Synergy in Cystic Fibrosis Therapies: Targeting SLC26A9. International journal of molecular sciences 2021, 22, 13064. [Google Scholar] [CrossRef]
- Avella, M.; Loriol, C.; Boulukos, K.; Borgese, F.; Ehrenfeld, J. SLC26A9 stimulates CFTR expression and function in human bronchial cell lines. J Cell Physiol 2011, 226, 212–223. [Google Scholar] [CrossRef]
- Palmer, M.L.; Lee, S.Y.; Carlson, D.; Fahrenkrug, S.; O'Grady, S.M. Stable knockdown of CFTR establishes a role for the channel in P2Y receptor-stimulated anion secretion. J Cell Physiol 2006, 206, 759–770. [Google Scholar] [CrossRef]
- Chappe, V.; Hinkson, D.A.; Howell, L.D.; Evagelidis, A.; Liao, J.; Chang, X.B.; Riordan, J.R.; Hanrahan, J.W. Stimulatory and inhibitory protein kinase C consensus sequences regulate the cystic fibrosis transmembrane conductance regulator. Proc. Natl. Acad. Sci. U. S. A 2004, 101, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Mathews, C.J.; Hanrahan, J.W. Phosphorylation by protein kinase C is required for acute activation of cystic fibrosis transmembrane conductance regulator by protein kinase A. J. Biol. Chem 1997, 272, 4978–4984. [Google Scholar] [CrossRef] [PubMed]
- Winpenny, J.P.; McAlroy, H.L.; Gray, M.A.; Argent, B.E. Protein kinase C regulates the magnitude and stability of CFTR currents in pancreatic duct cells. Am. J. Physiol 1995, 268, C823–C828. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.P.; Welsh, M.J. Calcium and cAMP activate different chloride channels in the apical membrane of normal and cystic fibrosis epithelia. Proc. Natl. Acad. Sci. USA 1991, 88, 6003–6007. [Google Scholar] [CrossRef] [PubMed]
- Kunzelmann, K.; Kubitz, R.; Grolik, M.; Warth, R.; Greger, R. Small conductance Cl- channels in HT29 cells: activation by Ca2+, hypotonic cell swelling and 8-Br-cGMP. Pflügers Arch 1992, 421, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Benedetto, R.; Ousingsawat, J.; Wanitchakool, P.; Zhang, Y.; Holtzman, M.J.; Amaral, M.; Rock, J.R.; Schreiber, R.; Kunzelmann, K. Epithelial Chloride Transport by CFTR Requires TMEM16A. Scientific Reports 2017, 7, 12397. [Google Scholar] [CrossRef]
- Lerias, J.; Pinto, M.; Benedetto, R.; Schreiber, R.; Amaral, M.; Aureli, M.; Kunzelmann, K. Compartmentalized crosstalk of CFTR and TMEM16A (ANO1) through EPAC1 and ADCY1. Cell Signal 2018, 44, 10–19. [Google Scholar] [CrossRef]
- Kunzelmann, K.; Mall, M.; Briel, M.; Hipper, A.; Nitschke, R.; Ricken, S.; Greger, R. The cystic fibrosis transmembrane conductance regulator attenuates the endogenous Ca2+ activated Cl- conductance in Xenopus ooyctes. Pflügers Arch 1997, 434, 178–181. [Google Scholar] [CrossRef]
- Wei, L.; Vankeerberghen, A.; Cuppens, H.; Eggermont, J.; Cassiman, J.J.; Droogmans, G.; Nilius, B. Interaction between calcium-activated chloride channels and the cystic fibrosis transmembrane conductance regulator. Pflugers Arch 1999, 438, 635–641. [Google Scholar] [CrossRef]
- Wei, L.; Vankeerberghen, A.; Cuppens, H.; Cassiman, J.J.; Droogmans, G.; Nilius, B. The C-terminal part of the R-domain, but not the PDZ binding motif, of CFTR is involved in interaction with Ca2+ -activated Cl- channels. Pflügers Arch 2001, 442, 280–285. [Google Scholar] [CrossRef]
- Perez-Cornejo, P.; Arreola, J. Regulation of Ca(2+)-activated chloride channels by cAMP and CFTR in parotid acinar cells. Biochem. Biophys. Res. Commun 2004, 316, 612–617. [Google Scholar] [CrossRef]
- Ousingsawat, J.; Kongsuphol, P.; Schreiber, R.; Kunzelmann, K. CFTR and TMEM16A are Separate but Functionally Related Cl Channels. Cell Physiol Biochem 2011, 28, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Kunzelmann, K.; Tian, Y.; Martins, J.R.; Faria, D.; Kongsuphol, P.; Ousingsawat, J.; Wolf, L.; Schreiber, R. Cells in focus: Airway epithelial cells-Functional links between CFTR and anoctamin dependent Cl(-) secretion. Int. J. Biochem. Cell Biol 2012, 44, 1897–1900. [Google Scholar] [CrossRef] [PubMed]
- Namkung, W.; Finkbeiner, W.E.; Verkman, A.S. CFTR-Adenylyl Cyclase I Association Is Responsible for UTP Activation of CFTR in Well-Differentiated Primary Human Bronchial Cell Cultures. Mol. Biol. Cell 2010, 21, 2639–2648. [Google Scholar] [CrossRef] [PubMed]
- Billet, A.; Hanrahan, J.W. The secret life of CFTR as a calcium-activated chloride channel. J Physiol 2013, 591, 5273–5278. [Google Scholar] [CrossRef] [PubMed]
- Le, S.C.; Jia, Z.; Chen, J.; Yang, H. Molecular basis of PIP2-dependent regulation of the Ca(2+)-activated chloride channel TMEM16A. Nature communications 2019, 10, 3769. [Google Scholar] [CrossRef]
- Ko, W.; Jung, S.R.; Kim, K.W.; Yeon, J.H.; Park, C.G.; Nam, J.H.; Hille, B.; Suh, B.C. Allosteric modulation of alternatively spliced Ca(2+)-activated Cl(-) channels TMEM16A by PI(4,5)P(2) and CaMKII. Proceedings of the National Academy of Sciences of the United States of America 2020. [Google Scholar] [CrossRef]
- Schreiber, R.; Ousingsawat, J.; Wanitchakool, P.; Sirianant, L.; Benedetto, R.; Reiss, K.; Kunzelmann, K. Regulation of TMEM16A/ANO1 and TMEM16F/ANO6 ion currents and phospholipid scrambling by Ca2+ and plasma membrane lipid. J Physiology (London) 2018, 596, 217–229. [Google Scholar] [CrossRef]
- Talbi, K.; Ousingsawat, J.; Centeio, R.; Schreiber, R.; Kunzelmann, K. Calmodulin-Dependent Regulation of Overexpressed but Not Endogenous TMEM16A Expressed in Airway Epithelial Cells. Membranes 2021, 11, 723. [Google Scholar] [CrossRef]
- Mall, M.; Gonska, T.; Thomas, J.; Schreiber, R.; Seydewitz, H.H.; Kuehr, J.; Brandis, M.; Kunzelmann, K. Modulation of Ca2+ activated Cl- secretion by basolateral K+ channels in human normal and cystic fibrosis airway epithelia. Pediatric Research 2003, 53, 608–618. [Google Scholar] [CrossRef]
- Mall, M.; Bleich, M.; Greger, R.; Schürlein, M.; Kühr, J.; Seydewitz, H.H.; Brandis, M.; Kunzelmann, K. Cholinergic ion secretion in human colon requires co-activation by cAMP. Am J Physiol 1998, 275, G1274–G1281. [Google Scholar] [PubMed]
- Faria, D.; Schreiber, R.; Kunzelmann, K. CFTR is activated through stimulation of purinergic P2Y2 receptors. Pflügers Arch 2009, 457, 1373–1380. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.; Faria, D.; Skryabin, B.V.; Rock, J.R.; Kunzelmann, K. Anoctamins support calcium-dependent chloride secretion by facilitating calcium signaling in adult mouse intestine. Pflügers Arch 2015, 467, 1203–1213. [Google Scholar] [CrossRef] [PubMed]
- Tembo, M.; Wozniak, K.L.; Bainbridge, R.E.; Carlson, A.E. Phosphatidylinositol 4,5-bisphosphate (PIP2) and Ca(2+) are both required to open the Cl(-) channel TMEM16A. J Biol Chem 2019. [Google Scholar] [CrossRef]
- Danahay, H.L.; Lilley, S.; Fox, R.; Charlton, H.; Sabater, J.; Button, B.; McCarthy, C.; Collingwood, S.P.; Gosling, M. TMEM16A Potentiation: A Novel Therapeutic Approach for the Treatment of Cystic Fibrosis. American journal of respiratory and critical care medicine 2020. [Google Scholar] [CrossRef]
- Centeio, R.; Ousingsawat, J.; Cabrita, I.; Schreiber, R.; Talbi, K.; Benedetto, R.; Doušová, T.; Verbeken, E.K.; De Boeck, K.; Cohen, I. , et al. Mucus Release and Airway Constriction by TMEM16A May Worsen Pathology in Inflammatory Lung Disease. International journal of molecular sciences 2021, 22. [Google Scholar] [CrossRef]
- Benedetto, R.; Cabrita, I.; Schreiber, R.; Kunzelmann, K. TMEM16A is indispensable for basal mucus secretion in airways and intestine. FASEB J 2019, 33, 4502–4512. [Google Scholar] [CrossRef]
- Kunzelmann, K.; Ousingsawat, J.; Cabrita, I.; Doušová, T.; Bähr, A.; Janda, M.; Schreiber, R.; Benedetto, R. TMEM16A in Cystic Fibrosis: Activating or Inhibiting? Frontiers in pharmacology 2019, 29, 10:13. [Google Scholar] [CrossRef]
- Bai, W.; Liu, M.; Xiao, Q. The diverse roles of TMEM16A Ca(2+)-activated Cl(-) channels in inflammation. Journal of advanced research 2021, 33, 53–68. [Google Scholar] [CrossRef]
- Liu, B.; Linley, J.E.; Du, X.; Zhang, X.; Ooi, L.; Zhang, H.; Gamper, N. The acute nociceptive signals induced by bradykinin in rat sensory neurons are mediated by inhibition of M-type K+ channels and activation of Ca2+-activated Cl- channels. J. Clin. Invest 2010, 120, 1240–1252. [Google Scholar] [CrossRef]
- Lee, B.; Cho, H.; Jung, J.; Yang, Y.D.; Yang, D.J.; Oh, U. Anoctamin 1 contributes to inflammatory and nerve-injury induced hypersensitivity. Mol. Pain 2014. [Google Scholar] [CrossRef] [PubMed]
- Miner, K.; Labitzke, K.; Liu, B.; Elliot, R.; Wang, P.; Henckels, K.; Gaida, K.; Elliot, R.; Chen, J.J.; Liu, L. , et al. Drug Repurposing: The Anthelmintics Niclosamide and Nitazoxanide Are Potent TMEM16A Antagonists That Fully Bronchodilate Airways. Frontiers in pharmacology 2019, 14, 10:51. [Google Scholar] [CrossRef]
- Ruffin, M.; Voland, M.; Marie, S.; Bonora, M.; Blanchard, E.; Blouquit-Laye, S.; Naline, E.; Puyo, P.; Le Rouzic, P.; Guillot, L. , et al. Anoctamin 1 Dysregulation Alters Bronchial Epithelial Repair in Cystic Fibrosis. Biochim. Biophys. Acta 2013, 1832, 2340–2351. [Google Scholar] [CrossRef] [PubMed]
- Lobo, M.J.; Amaral, M.D.; Zaccolo, M.; Farinha, C.M. EPAC1 activation by cAMP stabilizes CFTR at the membrane by promoting its interaction with NHERF1. J Cell Sci 2016. [Google Scholar] [CrossRef]
- He, Q.; Halm, S.T.; Zhang, J.; Halm, D.R. Activation of the basolateral membrane Cl conductance essential for electrogenic K secretion suppresses electrogenic Cl secretion. Exp. Physiol 2011, 96, 305–316. [Google Scholar] [CrossRef]
- Cabrita, I.; Benedetto, R.; Fonseca, A.; Wanitchakool, P.; Sirianant, L.; Skryabin, B.V.; Schenk, L.K.; Pavenstadt, H.; Schreiber, R.; Kunzelmann, K. Differential effects of anoctamins on intracellular calcium signals. Faseb j 2017, 31, 2123–2134. [Google Scholar] [CrossRef]
- Kunzelmann, K.; Cabrita, I.; Wanitchakool, P.; Ousingsawat, J.; Sirianant, L.; Benedetto, R.; Schreiber, R. Modulating Ca2+signals: a common theme for TMEM16, Ist2, and TMC. Pflügers Arch 2016, 468, 475–490. [Google Scholar] [CrossRef]
- Jin, X.; Shah, S.; Du, X.; Zhang, H.; Gamper, N. Activation of Ca2+-activated Cl- channel ANO1 by localized Ca2+ signals. J Physiol 2016, 594, 19–30. [Google Scholar] [CrossRef]
- Lerias, J.R.; Pinto, M.C.; Botelho, H.M.; Awatade, N.T.; Quaresma, M.C.; Silva, I.A.L.; Wanitchakool, P.; Schreiber, R.; Pepperkok, R.; Kunzelmann, K. , et al. A novel microscopy-based assay identifies extended synaptotagmin-1 (ESYT1) as a positive regulator of anoctamin 1 traffic. Biochim Biophys Acta 2018, 1865, 421–431. [Google Scholar] [CrossRef]
- Benedetto, R.; Ousingsawat, J.; Cabrita, I.; Pinto, M.; Lerias, J.; Wanitchakool, P.; Schreiber, R.; Kunzelmann, K. Plasma membrane localized TMEM16 Proteins are Indispensable for expression of CFTR. J Mol Med 2019, 97, 711–722. [Google Scholar] [CrossRef]
- Cabrita, I.; Benedetto, R.; Schreiber, R.; Kunzelmann, K. Niclosamide repurposed for the treatment of inflammatory airway disease. JCI insight 2019, 8, 128414. [Google Scholar] [CrossRef] [PubMed]
- Yimnual, C.; Satitsri, S.; Ningsih, B.N.S.; Rukachaisirikul, V.; Muanprasat, C. A fungus-derived purpactin A as an inhibitor of TMEM16A chloride channels and mucin secretion in airway epithelial cells. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie 2021, 139, 111583. [Google Scholar] [CrossRef]
- Centeio, R.; Ousingsawat, J.; schreiber, R.; Kunzelmann, K. CLCA1 Regulates Airway Mucus Production and Ion Secretion Through TMEM16A International journal of molecular sciences 2021, 22, 5133. 22. [CrossRef]
- Chappe, V.; Hinkson, D.A.; Zhu, T.; Chang, X.B.; Riordan, J.R.; Hanrahan, J.W. Phosphorylation of protein kinase C sites in NBD1 and the R domain control CFTR channel activation by PKA. J Physiol 2003, 548, 39–52. [Google Scholar] [CrossRef]
- Schreiber, R.; Cabrita, I.; Kunzelmann, K. Paneth cell secretion in vivo requires expression of Tmem16a and Tmem16f. Gastro Hep Advances 2022, 1, 1088–1098. [Google Scholar] [CrossRef]
- Park, J.H.; Ousingsawat, J.; Cabrita, I.; Bettels, R.E.; Große-Onnebrink, J.; Schmalstieg, C.; Biskup, S.; Reunert, J.; Rust, S.; Schreiber, R. , et al. TMEM16A deficiency: a potentially fatal neonatal disease resulting from impaired chloride currents. J Med Genet 2020, 58, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Sagel, S.D.; Chmiel, J.F.; Konstan, M.W. Sputum biomarkers of inflammation in cystic fibrosis lung disease. Proceedings of the American Thoracic Society 2007, 4, 406–417. [Google Scholar] [CrossRef]
- Kunzelmann, K.; Ousingsawat, J.; Benedetto, R.; Cabrita, I.; Schreiber, R. Contribution of Anoctamins to Cell Survival and Cell Death. Cancers 2019, 19, E382. [Google Scholar] [CrossRef]
- Cantin, A.M.; North, S.L.; Hubbard, R.C.; Crystal, R.G. Normal alveolar epithelial lining fluid contains high levels of glutathione. Journal of applied physiology (Bethesda, Md. : 1985) 1987, 63, 152–157. [Google Scholar] [CrossRef]
- Rahman, I.; MacNee, W. Oxidative stress and regulation of glutathione in lung inflammation. The European respiratory journal 2000, 16, 534–554. [Google Scholar] [CrossRef]
- Kogan, I.; Ramjeesingh, M.; Li, C.; Kidd, J.F.; Wang, Y.; Leslie, E.M.; Cole, S.P.; Bear, C.E. CFTR directly mediates nucleotide-regulated glutathione flux. EMBO J 2003, 22, 1981–1989. [Google Scholar] [CrossRef]
- Gao, L.; Kim, K.J.; Yankaskas, J.R.; Forman, H.J. Abnormal glutathione transport in cystic fibrosis airway epithelia. Am J Physiol 1999, 277, L113–L118. [Google Scholar] [CrossRef]
- Simoes, F.; Ousingsawat, J.; Wanitchakool, P.; Fonseca, A.; Cabrita, I.; Benedetto, R.; Schreiber, R.; Kunzelmann, K. CFTR supports cell death through ROS-dependent activation of TMEM16F (anoctamin 6). Pflugers Arch 2018, 470, 305–314. [Google Scholar] [CrossRef]
- Ousingsawat, J.; Wanitchakool, P.; Kmit, A.; Romao, A.M.; Jantarajit, W.; Schreiber, S.; Kunzelmann, K. Anoctamin 6 mediates effects essential for innate immunity downstream of P2X7-receptors in macrophages. Nat. Commun 2015, 6, 6245. [Google Scholar] [CrossRef]
- Ousingsawat, J.; Schreiber, R.; Gulbins, E.; Kamler, M.; Kunzelmann, K. P. aeruginosa Induced Lipid Peroxidation Causes Ferroptotic Cell Death in Airways. Cell Physiol Biochem 2021, 55, 590–604. [Google Scholar] [CrossRef]
- Ousingsawat, J.; Schreiber, R.; Kunzelmann, K. TMEM16F/Anoctamin 6 in Ferroptotic Cell Death. Cancers 2019, 11, pii–E625. [Google Scholar] [CrossRef] [PubMed]
- Hegyi, P.; Seidler, U.; Kunzelmann, K. CFTR-beyond the airways: Recent findings on the role of the CFTR channel in the pancreas, the intestine and the kidneys. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society 2023. [Google Scholar] [CrossRef] [PubMed]
- Bretscher, D.; Schneider, A.; Hagmann, R.; Hadorn, B.; Howald, B.; Lüthy, C.; Oetliker, O. Response of renal handling of sodium and bicarbonate to secretin in normals and in patients with cystic fibrosis. Pediatric Research 1974, 8, 899. [Google Scholar] [CrossRef]
- Windstetter, D.; Schaefer, F.; Scharer, K.; Reiter, K.; Eife, R.; Harms, H.K.; Bertele-Harms, R.; Fiedler, F.; Tsui, L.C.; Reitmeir, P. , et al. Renal function and renotropic effects of secretin in cystic fibrosis. Eur. J Med. Res 1997, 2, 431–436. [Google Scholar] [PubMed]
- Berg, P.; Svendsen, S.L.; Sorensen, M.V.; Larsen, C.K.; Andersen, J.F.; Jensen-Fangel, S.; Jeppesen, M.; Schreiber, R.; Cabrita, I.; Kunzelmann, K. , et al. Impaired Renal HCO(3) (-) Excretion in Cystic Fibrosis. J Am Soc Nephrol 2020, 31, 1711–1727. [Google Scholar] [CrossRef] [PubMed]
- Berg, P.; Svendsen, S.L.; Sorensen, M.V.; Schreiber, R.; Kunzelmann, K.; Leipziger, J. The molecular mechanism of CFTR- and secretin-dependent renal bicarbonate excretion. J Physiol. 2021, 599, 3003–3011. [Google Scholar] [CrossRef] [PubMed]
- Torres, V.E.; Harris, P.C.; Pirson, Y. Autosomal dominant polycystic kidney disease. Lancet 2007, 369, 1287–1301. [Google Scholar] [CrossRef] [PubMed]
- Senum, S.R.; Li, Y.S.M.; Benson, K.A.; Joli, G.; Olinger, E.; Lavu, S.; Madsen, C.D.; Gregory, A.V.; Neatu, R.; Kline, T.L. , et al. Monoallelic IFT140 pathogenic variants are an important cause of the autosomal dominant polycystic kidney-spectrum phenotype. Am J Hum Genet 2022, 109, 136–156. [Google Scholar] [CrossRef] [PubMed]
- Raphael, K.L.; Strait, K.A.; Stricklett, P.K.; Miller, R.L.; Nelson, R.D.; Piontek, K.B.; Germino, G.G.; Kohan, D.E. Inactivation of Pkd1 in principal cells causes a more severe cystic kidney disease than in intercalated cells. Kidney Int 2009, 75, 626–633. [Google Scholar] [CrossRef]
- Buchholz, B.; Teschemacher, B.; Schley, G.; Schillers, H.; Eckardt, K.U. Formation of cysts by principal-like MDCK cells depends on the synergy of cAMP- and ATP-mediated fluid secretion. J Mol. Med 2011, 89, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Grantham, J.J.; Mulamalla, S.; Swenson-Fields, K.I. Why kidneys fail in autosomal dominant polycystic kidney disease. Nature reviews. Nephrology 2011, 7, 556–566. [Google Scholar] [CrossRef]
- Terryn, S.; Ho, A.; Beauwens, R.; Devuyst, O. Fluid transport and cystogenesis in autosomal dominant polycystic kidney disease. Biochim. Biophys. Acta 2011. [Google Scholar] [CrossRef]
- Davidow, C.J.; Maser, R.L.; Rome, L.A.; Calvet, J.P.; Grantham, J.J. The cystic fibrosis transmembrane conductance regulator mediates transepithelial fluid secretion by human autosomal dominant polycystic kidney disease epithelium in vitro. Kidney Int 1996, 50, 208–218. [Google Scholar] [CrossRef]
- Magenheimer, B.S.; St John, P.L.; Isom, K.S.; Abrahamson, D.R.; De Lisle, R.C.; Wallace, D.P.; Maser, R.L.; Grantham, J.J.; Calvet, J.P. Early embryonic renal tubules of wild-type and polycystic kidney disease kidneys respond to cAMP stimulation with cystic fibrosis transmembrane conductance regulator/Na(+),K(+),2Cl(-) Co-transporter-dependent cystic dilation. J Am Soc Nephrol 2006, 17, 3424–3437. [Google Scholar] [CrossRef]
- Yang, B.; Sonawane, N.D.; Zhao, D.; Somlo, S.; Verkman, A.S. Small-Molecule CFTR Inhibitors Slow Cyst Growth in Polycystic Kidney Disease. J Am Soc. Nephrol 2008, 19, 1300–1310. [Google Scholar] [CrossRef]
- Cabrita, I.; Kraus, A.; Scholz, J.K.; Skoczynski, K.; Schreiber, R.; Kunzelmann, K.; Buchholz, B. Cyst growth in ADPKD is prevented by pharmacological and genetic inhibition of TMEM16A in vivo. Nature communications 2020, 11, 4320. [Google Scholar] [CrossRef]
- Cabrita, I.; Buchholz, B.; Schreiber, R.; Kunzelmann, K. TMEM16A drives renal cyst growth by augmenting Ca(2+) signaling in M1 cells. Journal of molecular medicine (Berlin, Germany) 2020, 98, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Cabrita, I.; Talbi, K.; Kunzelmann, K.; Schreiber, R. Loss of PKD1 and PKD2 share common effects on intracellular Ca2+ signaling. Cell Calcium 2021, 97, 102413. [Google Scholar] [CrossRef] [PubMed]
- Gabow, P.A.; Johnson, A.M.; Kaehny, W.D.; Kimberling, W.J.; Lezotte, D.C.; Duley, I.T.; Jones, R.H. Factors affecting the progression of renal disease in autosomal-dominant polycystic kidney disease. Kidney Int 1992, 41, 1311–1319. [Google Scholar] [CrossRef]
- Stewart, J.H. End-stage renal failure appears earlier in men than in women with polycystic kidney disease. Am J Kidney Dis 1994, 24, 181–183. [Google Scholar] [CrossRef] [PubMed]
- Talbi, K.; Cabrita, I.; Schreiber, R.; Kunzelmann, K. Gender-Dependent Phenotype in Polycystic Kidney Disease Is Determined by Differential Intracellular Ca2+ Signals. International journal of molecular sciences 2021, 22, 6019. [Google Scholar] [CrossRef]
- Nakanishi, K.; Sweeney, W.E., Jr.; Macrae, D.K.; Cotton, C.U.; Avner, E.D. Role of CFTR in autosomal recessive polycystic kidney disease. J Am Soc Nephrol 2001, 12, 719–725. [Google Scholar] [CrossRef]
- Menon, V.; Rudym, D.; Chandra, P.; Miskulin, D.; Perrone, R.; Sarnak, M. Inflammation, oxidative stress, and insulin resistance in polycystic kidney disease. Clinical journal of the American Society of Nephrology : CJASN 2011, 6, 7–13. [Google Scholar] [CrossRef]
- Schreiber, R.; Buchholz, B.; Kraus, A.; Schley, G.; Scholz, J.; Ousingsawat, J.; Kunzelmann, K. Lipid peroxidation drives renal cyst growth in vitro through activation of TMEM16A. J Am Soc Nephrol 2019, 30, 228–242. [Google Scholar] [CrossRef]
- Hanaoka, K.; Devuyst, O.; Schwiebert, E.M.; Wilson, P.D.; Guggino, W.B. A role for CFTR in human autosomal dominant polycystic kidney disease. Am J Physiol 1996, 270, C389–C399. [Google Scholar] [CrossRef]
- O'Sullivan, D.A.; Torres, V.E.; Gabow, P.A.; Thibodeau, S.N.; King, B.F.; Bergstralh, E.J. Cystic fibrosis and the phenotypic expression of autosomal dominant polycystic kidney disease. Am J Kidney Dis 1998, 32, 976–983. [Google Scholar] [CrossRef]
- Xu, N.; Glockner, J.F.; Rossetti, S.; Babovich-Vuksanovic, D.; Harris, P.C.; Torres, V.E. Autosomal dominant polycystic kidney disease coexisting with cystic fibrosis. J Nephrol 2006, 19, 529–534. [Google Scholar] [PubMed]
- Persu, A.; Devuyst, O.; Lannoy, N.; Materne, R.; Brosnahan, G.; Gabow, P.A.; Pirson, Y.; Verellen-Dumoulin, C. CF gene and cystic fibrosis transmembrane conductance regulator expression in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2000, 11, 2285–2296. [Google Scholar] [CrossRef] [PubMed]
- Brill, S.R.; Ross, K.E.; Davidow, C.J.; Ye, M.; Grantham, J.J.; Caplan, M.J. Immunolocalization of ion transport proteins in human autosomal dominant polycystic kidney epithelial cells. Proc. Natl. Acad. Sci. U. S. A 1996, 93, 10206–10211. [Google Scholar] [CrossRef]
- Buchholz, B.; Faria, D.; Schley, G.; Schreiber, R.; Eckardt, K.U.; Kunzelmann, K. Anoctamin 1 induces calcium-activated chloride secretion and tissue proliferation in polycystic kidney disease. Kidney Int 2014, 85, 1058–1067. [Google Scholar] [CrossRef]
- Buchholz, B.; Schley, G.; Faria, D.; Kroening, S.; Willam, C.; Schreiber, R.; Klanke, B.; Burzlaff, N.; Kunzelmann, K.; Eckardt, K.U. Hypoxia-Inducible Factor-1a Causes Renal Cyst Expansion through Calcium-Activated Chloride Secretion. J Am Soc Nephrol 2014, 25, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Schwiebert, E.M.; Wallace, D.P.; Braunstein, G.M.; King, S.R.; Peti-Peterdi, J.; Hanaoka, K.; Guggino, W.B.; Guay-Woodford, L.M.; Bell, P.D.; Sullivan, L.P. , et al. Autocrine extracellular purinergic signaling in epithelial cells derived from polycystic kidneys. Am. J Physiol Renal Physiol 2002, 282, F763–F775. [Google Scholar] [CrossRef]
- Kraus, A.; Grampp, S.; Goppelt-Struebe, M.; Schreiber, R.; Kunzelmann, K.; Peters, D.J.; Leipziger, J.; Schley, G.; Schodel, J.; Eckardt, K.U. , et al. P2Y2R is a direct target of HIF-1alpha and mediates secretion-dependent cyst growth of renal cyst-forming epithelial cells. Purinergic signalling 2016, 12, 687–695. [Google Scholar] [CrossRef]
- Talbi, K.; Cabrita, I.; Kraus, A.; Hofmann, S.; Skoczynski, K.; Kunzelmann, K.; Buchholz, B.; Schreiber, R. The chloride channel CFTR is not required for cyst growth in an ADPKD mouse model. Faseb j 2021, 35, e21897. [Google Scholar] [CrossRef]
- Guilbault, C.; Saeed, Z.; Downey, G.P.; Radzioch, D. Cystic fibrosis mouse models. American journal of respiratory cell and molecular biology 2007, 36, 1–7. [Google Scholar] [CrossRef]
- Yanda, M.K.; Cha, B.; Cebotaru, C.V.; Cebotaru, L. Pharmacological reversal of renal cysts from secretion to absorption suggests a potential therapeutic strategy for managing autosomal dominant polycystic kidney disease. J Biol Chem 2019, 294, 17090–17104. [Google Scholar] [CrossRef]
- Yanda, M.K.; Cebotaru, L. VX-809 mitigates disease in a mouse model of autosomal dominant polycystic kidney disease bearing the R3277C human mutation. Faseb j 2021, 35, e21987. [Google Scholar] [CrossRef] [PubMed]
- van Koningsbruggen-Rietschel, S.; Conrath, K.; Fischer, R.; Sutharsan, S.; Kempa, A.; Gleiber, W.; Schwarz, C.; Hector, A.; Van Osselaer, N.; Pano, A. , et al. GLPG2737 in lumacaftor/ivacaftor-treated CF subjects homozygous for the F508del mutation: A randomized phase 2A trial (PELICAN). Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society 2020, 19, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Mook, R.A., Jr.; Premont, R.T.; Wang, J. Niclosamide: Beyond an antihelminthic drug. Cell Signal 2018, 41, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Graham, G.G.; Williams, K.M.; Day, R.O. A benefit-risk assessment of benzbromarone in the treatment of gout. Was its withdrawal from the market in the best interest of patients? Drug safety 2008, 31, 643–665. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Attenuated CFTR-dependent Cl- secretion in in mice with intestinal epithelial knockout of ANO1.A) Ussing chamber recordings obtained under open circuit conditions, as described in [56]. Stimulation of colonic epithelia with IBMX (100 µM) and forskolin (2 µM) induced a pronounced voltage deflection, which was strongly attenuated in a colonic tissue obtained from a mouse lacking expression of ANO1. B) Calculated equivalent short circuit currents indicate strongly attenuated CFTR-dependent (I/F-stimulated) and Ca2+ - dependent (ATP (100 µM) -stimulated) Cl- secretion. Mean ± SEM (number of experiments). §significantly reduced when compared to basal Isc in +/+ tissues (ANOVA). #significantly reduced when compared to stimulated Isc in +/+ tissues (ANOVA). For methods see [56].
Figure 1.
Attenuated CFTR-dependent Cl- secretion in in mice with intestinal epithelial knockout of ANO1.A) Ussing chamber recordings obtained under open circuit conditions, as described in [56]. Stimulation of colonic epithelia with IBMX (100 µM) and forskolin (2 µM) induced a pronounced voltage deflection, which was strongly attenuated in a colonic tissue obtained from a mouse lacking expression of ANO1. B) Calculated equivalent short circuit currents indicate strongly attenuated CFTR-dependent (I/F-stimulated) and Ca2+ - dependent (ATP (100 µM) -stimulated) Cl- secretion. Mean ± SEM (number of experiments). §significantly reduced when compared to basal Isc in +/+ tissues (ANOVA). #significantly reduced when compared to stimulated Isc in +/+ tissues (ANOVA). For methods see [56].
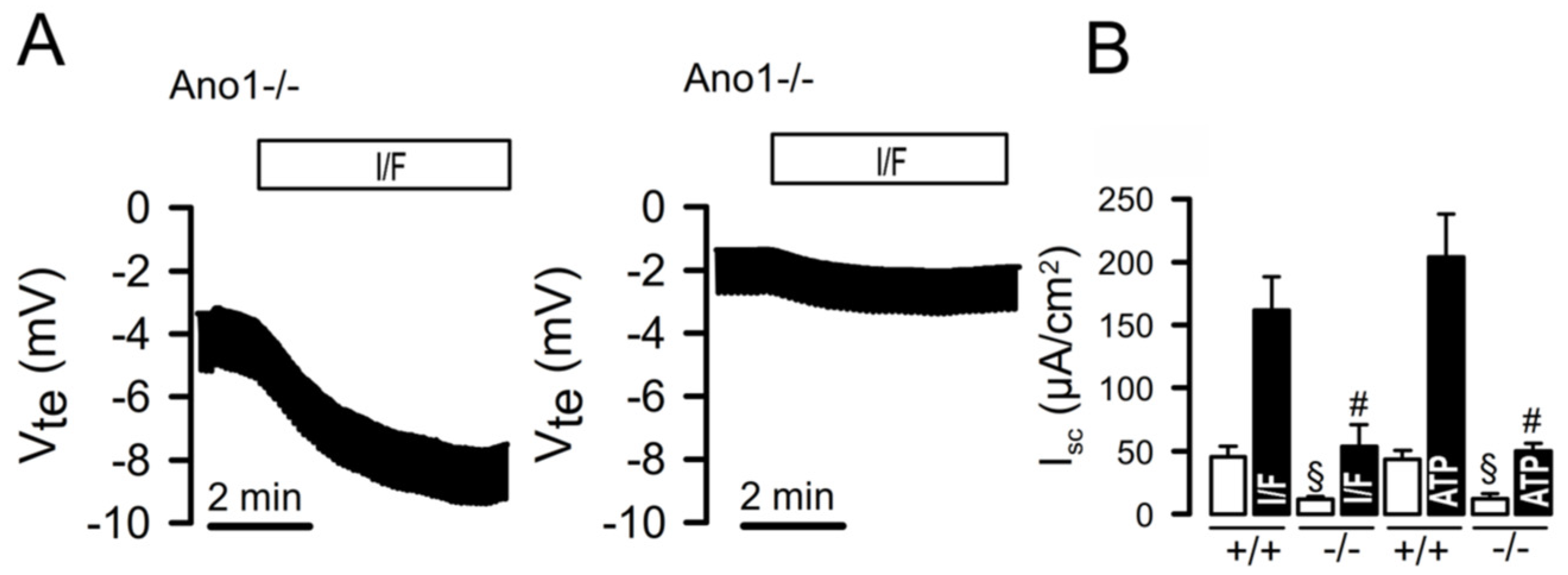
Figure 2.
Cytokines in a sputum sample from a patient carrying the ANO1 los-of-function variant c.897+3_897+6delAAGT are strongly reduced when compared to samples from CF patients. Sputum samples were obtained from a healthy volunteer, the patient carrying the ANO1 variant c.897+3_897+6delAAGT, and from two CF patients and the concentration of the cytokines IL-8 and IL-1ß were determined. Although the ANO1-patient lacks of CFTR function in addition to the defect in ANO1 Ca2+ -dependent Cl- secretion, cytokines were strongly reduced when compared to CF patients carrying known CFTR mutations. Mean ± SEM (number of measurements). #significantly enhanced when compared to the healthy volunteer (ANOVA). $significantly enhanced when compared to the ANO1 patients (ANOVA). For methods see [97].
Figure 2.
Cytokines in a sputum sample from a patient carrying the ANO1 los-of-function variant c.897+3_897+6delAAGT are strongly reduced when compared to samples from CF patients. Sputum samples were obtained from a healthy volunteer, the patient carrying the ANO1 variant c.897+3_897+6delAAGT, and from two CF patients and the concentration of the cytokines IL-8 and IL-1ß were determined. Although the ANO1-patient lacks of CFTR function in addition to the defect in ANO1 Ca2+ -dependent Cl- secretion, cytokines were strongly reduced when compared to CF patients carrying known CFTR mutations. Mean ± SEM (number of measurements). #significantly enhanced when compared to the healthy volunteer (ANOVA). $significantly enhanced when compared to the ANO1 patients (ANOVA). For methods see [97].
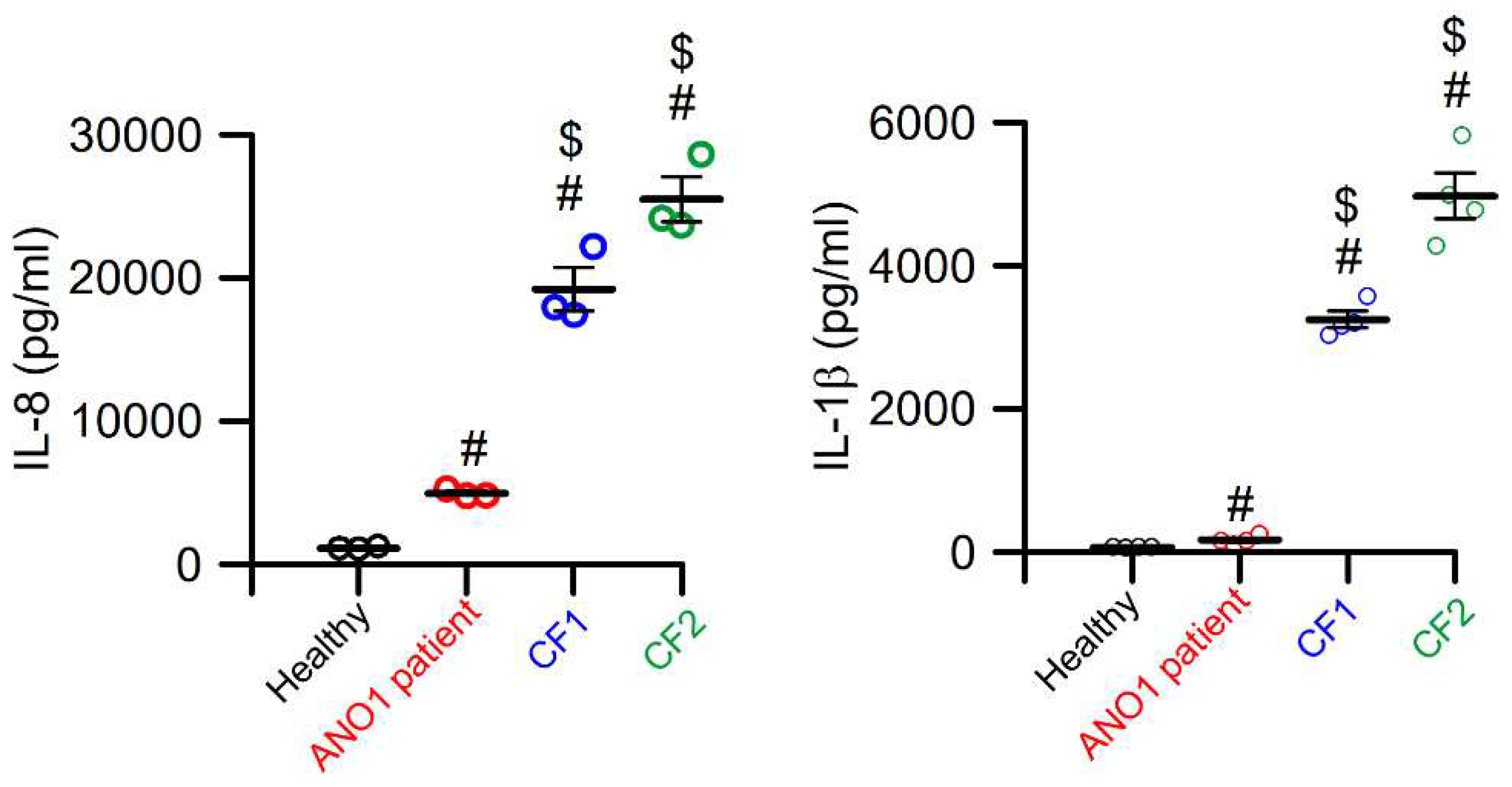
Figure 3.
ANO1 is colocalized with pendrin in the apical membrane of ß-intercalated cells.A) Immunocytochemistry demonstrating colocalization of ANO1 and pendrin in the apical membrane of ß-intercalated cells of mouse collecting duct. For methods see [110]. B) Model showing the molecular mechanisms for HCO3- excretion by collecting duct ß-intercalated cells. Blood HCO3- is taken up into ß-intercalated cells and is transported by pendrin into the collecting duct lumen in exchange with Cl- which recycles via colocalized CFTR. In addition CFTR may directly interact with CFTR (?). Colocalized ANO1 tethers the endoplasmic reticulum (ER) to the apical membrane and facilitates efficient Ca2+ signaling in the apical compartment which supports insertion of CFTR into the apical membrane and its activation. Increase in blood secretin leads to activation of basolateral secretin receptors (SCTR), which further activates CFTR and HCO3- excretion. For methods see [110].
Figure 3.
ANO1 is colocalized with pendrin in the apical membrane of ß-intercalated cells.A) Immunocytochemistry demonstrating colocalization of ANO1 and pendrin in the apical membrane of ß-intercalated cells of mouse collecting duct. For methods see [110]. B) Model showing the molecular mechanisms for HCO3- excretion by collecting duct ß-intercalated cells. Blood HCO3- is taken up into ß-intercalated cells and is transported by pendrin into the collecting duct lumen in exchange with Cl- which recycles via colocalized CFTR. In addition CFTR may directly interact with CFTR (?). Colocalized ANO1 tethers the endoplasmic reticulum (ER) to the apical membrane and facilitates efficient Ca2+ signaling in the apical compartment which supports insertion of CFTR into the apical membrane and its activation. Increase in blood secretin leads to activation of basolateral secretin receptors (SCTR), which further activates CFTR and HCO3- excretion. For methods see [110].
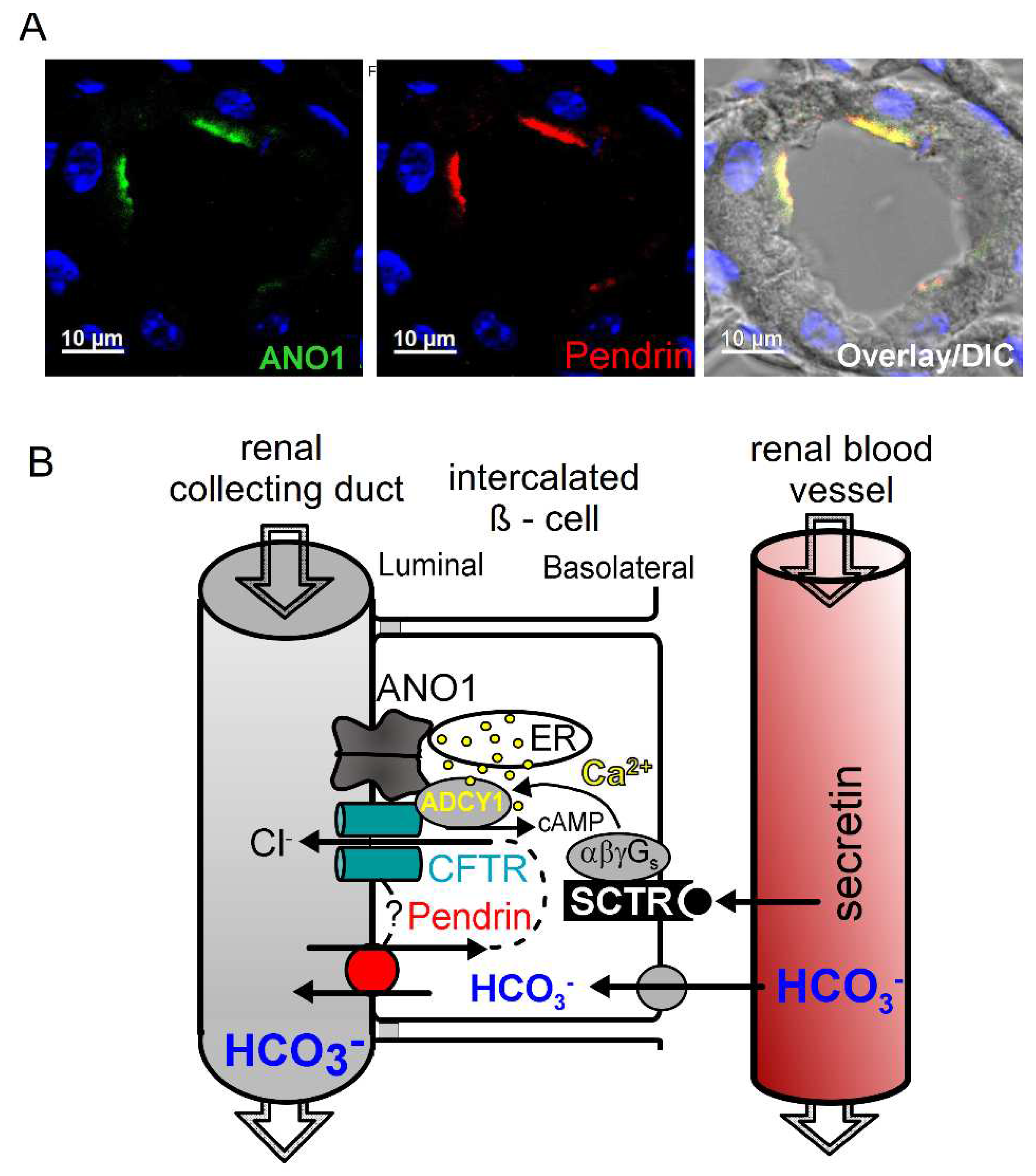
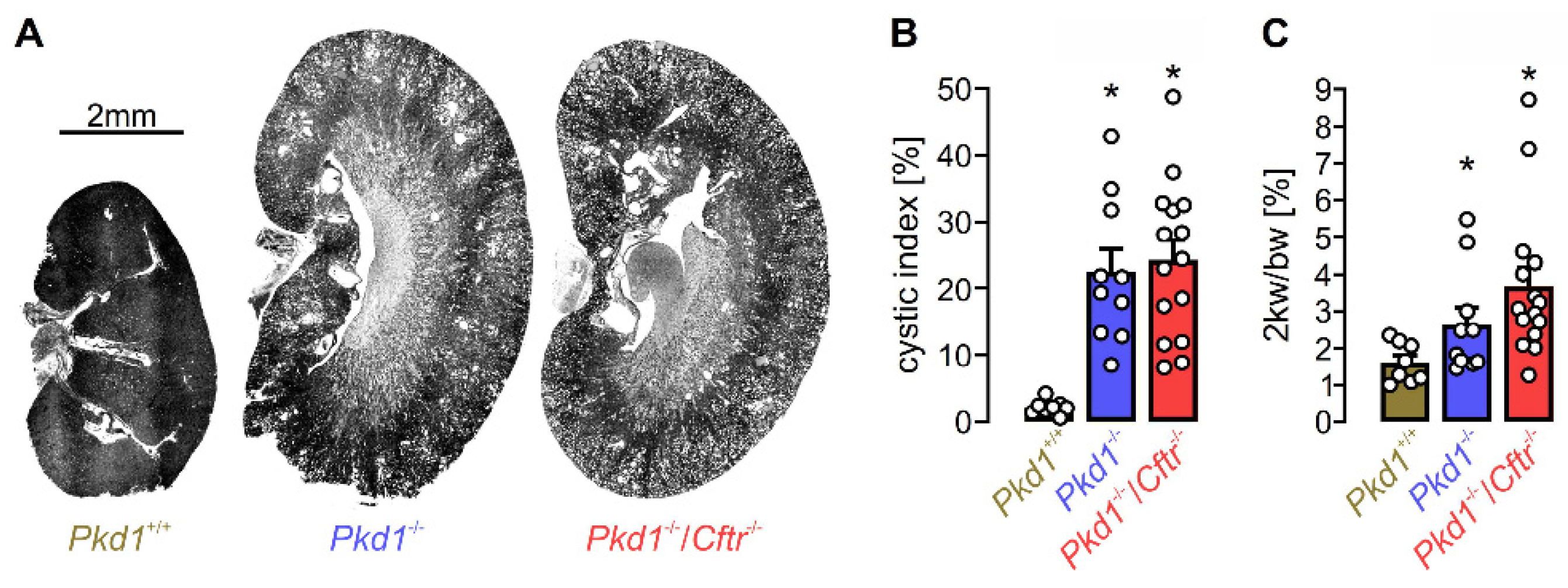
Figure 4.
Knockout of Cftr does not affect cyst growth in an ADPKD mouse model. KspCreERT2; Pkd1lox;lox mice (Pkd1-/-; n=10) and KspCreERT2; Pkd1lox;lox/Cftrlox;lox mice (Pkd1-/-/Cftr-/-; n=15) received daily intraperitoneal injection of tamoxifen (2 mg/kg body weight dissolved in 5% ethanol and 95% neutral oil at postnatal days 20-22) to induce tubule-specific deletion of Pkd1 or co-deletion of Pkd1 and Cftr. Non-induced KspCreERT2; Pkd1lox;lox mice (Pkd1+/+; n=8) served as controls. Analyses were performed 10 weeks after induction with tamoxifen. A) Representative kidney sections at the end of the experiment. For methods see [121]. B) Analysis of the cystic indices defined as the ratio of cortical cystic area divided by the whole cortex area. C) Two-kidney weight per body weight ratio. No significant effect of CFTR-knockout was found [139]. Bars show means ± SEM, dots indicate individual values. *significant increase compared to Pkd1+/+ (p<0.05; one-way ANOVA, Posthoc test: Tukey); n.s. means no statistical difference (one-way ANOVA, Posthoc test: Tukey and unpaired t-test). For methods see [139].
Figure 4.
Knockout of Cftr does not affect cyst growth in an ADPKD mouse model. KspCreERT2; Pkd1lox;lox mice (Pkd1-/-; n=10) and KspCreERT2; Pkd1lox;lox/Cftrlox;lox mice (Pkd1-/-/Cftr-/-; n=15) received daily intraperitoneal injection of tamoxifen (2 mg/kg body weight dissolved in 5% ethanol and 95% neutral oil at postnatal days 20-22) to induce tubule-specific deletion of Pkd1 or co-deletion of Pkd1 and Cftr. Non-induced KspCreERT2; Pkd1lox;lox mice (Pkd1+/+; n=8) served as controls. Analyses were performed 10 weeks after induction with tamoxifen. A) Representative kidney sections at the end of the experiment. For methods see [121]. B) Analysis of the cystic indices defined as the ratio of cortical cystic area divided by the whole cortex area. C) Two-kidney weight per body weight ratio. No significant effect of CFTR-knockout was found [139]. Bars show means ± SEM, dots indicate individual values. *significant increase compared to Pkd1+/+ (p<0.05; one-way ANOVA, Posthoc test: Tukey); n.s. means no statistical difference (one-way ANOVA, Posthoc test: Tukey and unpaired t-test). For methods see [139].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



