
Submitted:
26 June 2023
Posted:
26 June 2023
You are already at the latest version
Abstract
Fungal plant diseases are a major threat to food security worldwide. Current efforts to identify and list loci involved in different biological processes are more complicated than originally thought, even when complete genome assemblies are available. Despite numerous experimental and computational efforts to characterize gene functions in plants, about ~ 40% of protein-coding genes in the model plant A. thaliana are still not categorized in the Gene Ontology Biological Process (BP) annotation. In non-model organisms, such as sunflower (Helianthus annuus), the number of BP term annotation is far fewer, ~22%. In the current study we performed gene co-expression network analysis using eight terabytes of public transcriptome datasets and performed expression-based functional prediction to categorize and identify loci involved in the response to fungal pathogens. We were able to construct a reference gene network of healthy green tissue (GreenGCN) and a gene network of healthy and stressed root tissues (RootGCN). Both networks achieved robust high quality scores on the metrics of guilt-by-association and selective constraints versus gene connectivity. We were able to identify eight modules enriched in defense functions, of which, two out of the three modules in the RootGCN were also conserved in the GreenGCN, suggesting similar defense-related expression patterns. We identified 16 WRKY genes involved in defense related functions and 65 previously uncharacterized loci now linked to defense response. In addition, we identified and classified 122 loci previously identified within QTL or near candidate loci reported in GWAS studies of disease resistance in sunflower linked to defense response. In all, we have implemented a valuable strategy to better describe genes within specific biological processes.
Keywords:
transcriptomics
; co-expression networks
; fungal pathogens
; WRKY
; plant pathology
; meta-analysis
; multi-study analysis
; sunflower
; candidate genes
1. Introduction
Sunflower (Helianthus annuus L.) is one of the most important crops for the production of high-quality oil and seeds consumed by both humans and livestock. Last year’s FAO scientific review states that climate change represents an unprecedented challenge to food security, and due to its impact, plant diseases that ravage economically important crops are becoming more destructive and pose an increasing threat to food security and the environment [1]. A plant’s response to stress is complex, varies in time (e.g., early or delayed responses), and differs in space (e.g., whole plant, organs, tissues, or cell types). It is also governed by networks that are likely to involve numerous signaling pathways and genes [2]. Moreover, abiotic and biotic stresses can interact in complex ways. Over the past decades, many groups have conducted genetic, genomic and transcriptomic experiments at different stages and under different conditions to understand the regulatory and genetic basis for many stress related responses in sunflower [3,4,5,6,7]. Yet the identification of causal genes in these cases is very limited, mainly due to the lack of statistical power and poor experimental design. It is known that Helianthus species possess a rich spectrum of non-redundant defense mechanisms, many of which have been introduced into cultivated sunflowers from their wild relatives [8,9]. Moreover, fungal resistance in crops is typically governed by many interacting loci, which each offer small contributions [10]. Identifying those causal loci via strategies such as genome wide association studies (GWAS) and quantitative trait loci mapping (QTL mapping) is very challenging and cumbersome. The genomic regions surrounding GWAS markers and those encompassing QTL mapping intervals can be extensive, including hundreds of genes, only a few of which may be potentially causal. To make matters worse, the sunflower genome is only partially annotated. Only ~60% of the genes are annotated with at least one Gene Ontology (GO) term, and all annotations being in silico (inferred from phylogeny (IBA) or electronic (IEA) annotation). Moreover, only ~22% of gene products have at least one BP GO annotation, and only 864 (1.6%) out of the total gene products are annotated as “defense response”, whereas this figure rises to 8% in Arabidopsis thaliana [11,12].
To date, several novel automated function prediction methods have been developed to address this knowledge gap. Among them, top scoring methods incorporate algorithms utilizing gene co-expression data to infer biological functions through the Guilt-By-Association principle [13,14]. Historically, BLAST [15] and InterProScan [16] using sequence similarity and presence/absence of domains, respectively, have been the major annotation pipelines. The top scoring methods include machine learning techniques and novel ways to integrate genomic features, sequence intrinsic properties, protein-protein interactions, and other omics data and transfer these new annotations to unknown genes [17]. As expected, the assignment of molecular function (MF) GO terms significantly outperforms the assignment of BP GO terms in these machine learning methods [18]. Moreover, given the scarcity of data from different functional omics studies, such as protein interaction assays, in non-model organisms, such as sunflower, gene-expression based automated functional prediction remains of great importance, especially for the prediction of BP GO terms [13,19]. Therefore, re-analyzing transcriptome data as a whole enhances functional annotation by leveraging the collective knowledge gained from multiple experiments. To date, several authors have focused on multi-study co-expression networks to gain a deeper understanding of specific biological processes [20], for example in the model plant Arabidopsis thaliana [13,14]. The work of Depuydt et. al. (2021) [13] in Arabidopsis showed very promising results in annotating unknown Arabidopsis genes with multi-study co-expression networks, especially using the BP GO terms under the category “response to stimulus”. Taken together, these network analyses can provide new knowledge for the field under study.
In this work, we investigated the potential of gene co-expression networks generated from multi-study datasets from all public RNAseq bio-projects currently available for sunflower to identify and/or validate candidate loci for defense or fungal resistance. We focused on identifying and categorizing gene candidates for two major fungal diseases, Verticillium wilt and Sclerotinia head rot. Verticillium dahliae (Vd) is a soil-borne and seed-borne pathogen that is capable of infecting hundreds of plant species including sunflower. Infection by V. dahliae causes vascular discoloration and systemic wilting [21]. On the other hand, the necrotrophic fungus Sclerotinia sclerotiorum can behave as an air-borne and a soil-borne pathogen, and is the causal agent of three distinct diseases in sunflower (sclerotinia root rot, sclerotinia stem rot, and sclerotinia head rot) [22]. Altogether, these pathogens affect oil quality and under favourable conditions may lead to massive production loss [23]. Different studies have identified many candidate genes potentially involved in the resistance against these diseases [21,24,25,26] but as we mentioned, narrowing down the culprits is difficult.
Here we present the first large-scale co-expression network for sunflower and perform functional prediction to further characterize previous candidate loci for S. sclerotiorum and V. dahliae resistance and identify novel loci involved in defense response, specifically, to fungal pathogens. introduction should briefly place the study in a broad context and highlight why it is important. It should define the purpose of the work and its significance. The current state of the research field should be carefully reviewed and key publications cited. Please highlight controversial and diverging hypotheses when necessary. Finally, briefly mention the main aim of the work and highlight the principal conclusions. As far as possible, please keep the introduction comprehensible to scientists outside your particular field of research. References should be numbered in order of appearance and indicated by a numeral or numerals in square brackets—e.g., [1] or [2,3], or [4,5,6]. See the end of the document for further details on references.
2. Results
2.1. Construction of a Reference Weighted Gene Co-Expression Network from green healthy tissue
We generated a stress-free reference network in order to organize genes in biologically relevant clusters. We analyzed 733 sunflower samples of healthy photosynthetic tissues from 27 different studies (see File S1). Outlier samples were filtered out using a recursive process of read count normalization, batch effect correction and hierarchical clustering (Figure 1, see Material and Methods). As a result, a total of 15978 genes and 686 samples remained (Table 1). With this data, we calculated a co-expression network using WGCNA [27]. We selected a signed-hybrid formula with a power β = 4 as the soft-threshold to ensure a scale-free network (R2 = 0.97). The resulting network, from now on referred to as “GreenGCN”, presented 62 modules (named Green1 to Green62) grouping 13099 genes; the remaining 2879 genes (18%) were not included in any gene cluster and were listed in the module Green0. The network presented 198 “hub-genes”, i.e., genes with a scaled intramodular-connectivity (IMK) score of 0.9 or greater, of which 20 have no BP GO term annotation and 12 were described as “uncharacterized” or “unknown”. Module Green1 had the highest number of hub-genes, 26, well above the mean number of hub genes per module, 3.18 (SD± 3.37) (see File S2).
Figure 1.
Bioinformatic flowchart describing sample processing in our co-expression network analysis.
Figure 1.
Bioinformatic flowchart describing sample processing in our co-expression network analysis.
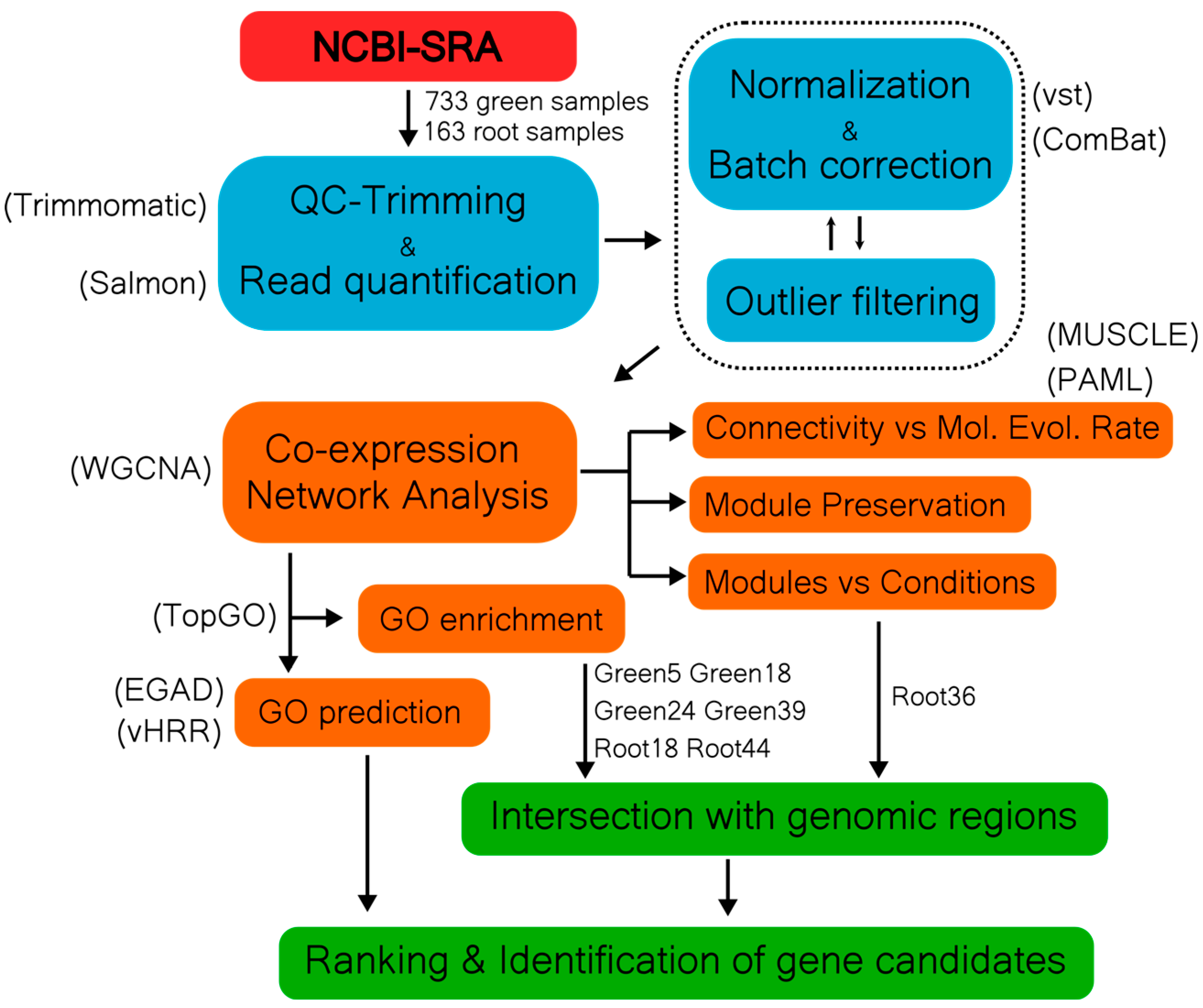

Table 1.
Number of samples used for the construction of the GreenGCN and the RootGCN.
| Samples condition | Initially | Post filtering | |
|---|---|---|---|
| GreenGCN | Healthy | 733 | 686 |
| RootGCN | Healthy | 134 | 127 |
| V. dahliae infection | 14 | 14 | |
| R. irregulare infection | 6 | 6 | |
| O. cumana infection | 9 | 3 |
In order to relate and integrate the modules within a biological context, we performed Gene Ontology (GO) term enrichment tests on all 62 modules. We observed that all modules were enriched significantly with one or more GO terms (adjusted p-value <0.05), with the largest module (Green1) enriched in GO terms such as “growth”, “cell division”, and “plant-type cell wall biogenesis”, among others. The complete GO enrichment data for the 62 modules can be found in File S3. To test the network’s performance, we evaluated its capacity to connect genes with shared GO terms. Using the genes adjacencies matrix, we obtained a mean Guilt-By-Association-AUROC (GBA-AUC) of 0.82. Moreover, using the simple grouping by modules as a network, we still measured a robust mean GBA-AUC of 0.70. Furthermore, as a supplementary quality measure, we found a negative correlation between gene connectivity and dN/dS ratio (Supplementary materials Figure S1). To further characterize the network and check its consistency, we investigated the two modules with the tightest correlation between their genes (highest “density”) after correction by module size, Green20 and Green35 (Supplementary materials Figure S2). Both modules were highly enriched for “photosynthesis”, as well as enriched for cellular compartments such as “thylakoid”, “photosynthetic membrane”, and “chloroplast”. These results are in line with the prime biological functions of plant green tissues, thus attesting to the reliability of GreenGCN as a reference co-expression network to explore relationships among genes.
2.2. Construction of a weighted gene co-expression network from stressed and control root tissue
In order to study the root tissue under control and stressed conditions, we generated a new network. We collected a total of 163 samples, of which 134 were “control” conditions and 29 corresponded to “biological disturbances”, namely infections with fungal pathogen V. dahliae (14), arbuscular mycorrhizal fungi Rhizoglomus irregulare (6), and the parasitic plant Orobanche cumana (9) (see File S1). The initial cohort of 163 samples from 11 different studies was filtered down to 150 samples (Table 1), containing 20850 genes. We selected a soft-threshold β=6 resulting in a scale-free network fit R2=0.92. The resulting network, from now on referred to as “RootGCN”, had 77 modules (named Root1 to Root77), with the largest module (Root1) grouping 8.36% of genes, and only 2.68% of genes remained unconnected in module Root0. We observed 293 “hub-genes” with IMK≥0.9, of which 82 were genes with no BP GO term annotation, and 23 were described as “uncharacterized” or “unknown function”. The mean number of “hub genes” per module was 3.77 (SD ± 3.0). In this network, modules Root3 and Root24 had the highest number of hub-genes, 16 and 14 respectively (see File S2). In addition to identifying modules enriched in biotic defense functions, we anticipated discovering co-expression modules that display correlations specific to the comparison between control versus infected samples. Gene Ontology (GO) term enrichment tests showed that all modules were enriched significantly with one or more GO terms (p-value <0.05), (File S3). Interestingly, we found module Root5 highly enriched for root development and root hair elongation, whereas, as expected, we observed no enrichment for chlorophyll biosynthesis in any module. Applying the same Guilt-by-Association prediction method as before, we measured a GBA-AUC of 0.79 using the adjacency matrix. Moreover, we again observed an inverse correlation between gene connectivity and dN/dS ratios, as expected (Figure S1). To further check consistency in this network, we again analyzed the highest density module, Root37, which is highly enriched in cell wall biosynthesis processes, an important process in root development [28] (Supplementary materials Figure S2).
2.3. Identification of modules related to fungal resistance
Since we were interested in defense responses against pathogens, particularly against phytopathogenic fungi, we selected modules enriched in immune functions, and as reinforcement, we also searched for enrichment in WRKY genes, given they have been frequently linked to defense responses [29]. We first searched for modules significantly enriched in the GO term “response to fungus” (GO:0009620) and its direct descendants. We identified four modules: Green16, Green18, Green24 and Root18, specifically enriched in the term “defense response to fungus” among others. Expanding our scope, we searched the term “defense response” (GO:0006952) and all its child terms, also detecting modules Green5, Green39 and Root44. Modules Green5, Green18, Green24, Green39, Root18, Root36 and Root44 were all significantly enriched for WRKY-like genes, and furthermore, these modules’ eigengenes were all positively correlated among each other (within each GCN) (Figure 2). Interestingly, we observed that module Green16 was inversely correlated to Green5, Green18 and Green39, while harboring no WRKY-like proteins.
Furthermore, in the RootGCN it was possible to test traits-to-modules correlation, using the sample’s condition (control or infected) as trait. Seven modules were significantly correlated (adjusted pval<0.01), in descending order: Root0, Root36, Root21, Root1, Root14 and Root35. None of these modules were significantly enriched for defense-related functions, but it caught our attention how the module Root36 was enriched in WRKY-like genes (Table 2) and how its eigengene was inversely correlated to modules Root18 and Root44 (Figure 2).
Among the total 119 WRKY-like genes in the annotation of the sunflower genome, 37 were present in GreenGCN and 66 in RootGCN; of which 22 and 12 were included in the previously mentioned modules (Green5, Green18, Green24, Green39, Root18, Root44 and Root36). WRKY-like genes were not only enriched in these modules but some also had central roles in them, evidenced by their high IMK (Table 2). For example, module Green24, was the most enriched in WRKYs, harboring 8 out of 185 total genes, with four of them having IMK>0.6. Coherently, most of the sunflower homologs to Arabidopsis WRKYs in the modules of interest were described to be involved in defense response against pathogens (Table 2). As another layer of evidence, only 9 of the hub genes (IMK>0.8) inside these modules were previously annotated as related to defense functions; yet, we identified 16 hub genes, so far not annotated as defense-related, which had homologs in A. thaliana linked to such processes (Table 3, also see File S4).
Figure 2.
Eigengenes correlations between modules of interest in GreenGCN and RootGCN networks. Asterisks indicate p-value < 1e-10.
Figure 2.
Eigengenes correlations between modules of interest in GreenGCN and RootGCN networks. Asterisks indicate p-value < 1e-10.
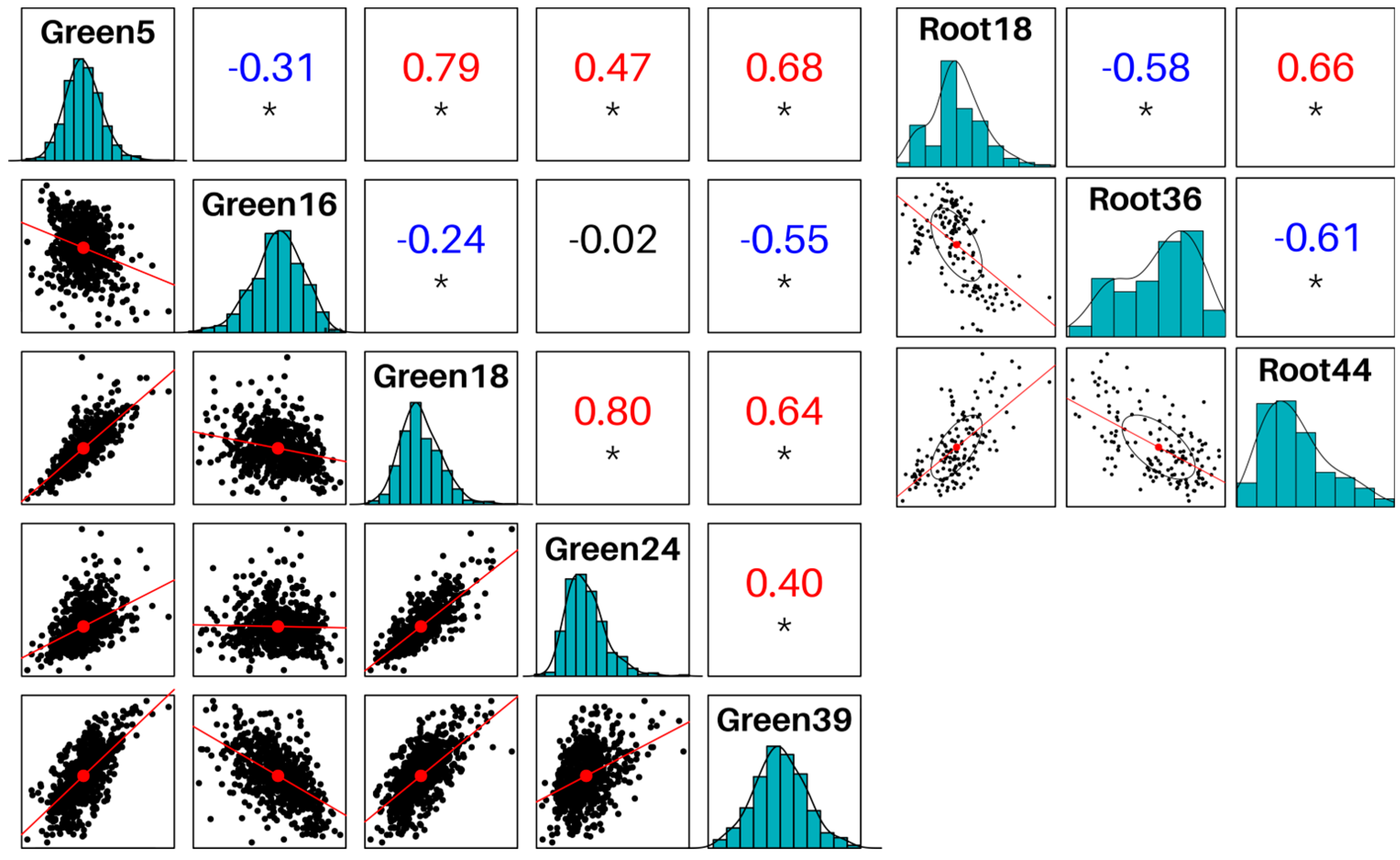
Table 2.
Putative WRKY genes present in modules of interest and their corresponding closest homologs in A. thaliana. Asterisked genes of A . thaliana are specifically associated with defense response against pathogens according to the TAIR database.
Table 2.
Putative WRKY genes present in modules of interest and their corresponding closest homologs in A. thaliana. Asterisked genes of A . thaliana are specifically associated with defense response against pathogens according to the TAIR database.
| Sunflower gene | Closest homolog in A. thaliana | |||||
|---|---|---|---|---|---|---|
| Gene name | Module | IMK | Homolog | Identity % | Q-cov % | E-value |
| HanXRQChr10g0294651 | Green5 | 0.3 | AtWRKY70 * | 43.31 | 19 | 2E-23 |
| HanXRQChr16g0506841 | Green5 | 0.28 | AtWRKY6 | 54.02 | 81 | 8E-137 |
| HanXRQChr16g0508961 | Green5 | 0.26 | AtWRKY7 * | 49.72 | 95 | 1E-95 |
| HanXRQChr08g0211091 | Green5 | 0.21 | AtWRKY4 * | 53.71 | 97 | 2E-144 |
| HanXRQChr15g0480431 | Green5 / Root18 | 0.13 / 0.18 | AtWRKY28 * | 40.48 | 100 | 3E-61 |
| HanXRQChr11g0329641 | Green5 | 0.08 | AtWRKY41 * | 46.55 | 39 | 1E-29 |
| HanXRQChr10g0306731 | Green5 | 0.07 | AtWRKY1 * | 43.62 | 65 | 6E-68 |
| HanXRQChr10g0281391 | Green5 | 0.05 | AtWRKY21 | 48.11 | 100 | 2E-106 |
| HanXRQChr03g0071411 | Green18 | 0.71 | AtWRKY51 * | 53.78 | 80 | 8E-39 |
| HanXRQChr14g0460611 | Green18 / Root44 | 0.41 / 0.78 | AtWRKY70 * | 42.94 | 54 | 2E-37 |
| HanXRQChr04g0113641 | Green18 | 0.09 | AtWRKY21 | 49.62 | 100 | 5E-113 |
| HanXRQChr16g0509771 | Green24 | 0.89 | AtWRKY33 * | 46.56 | 88 | 2E-117 |
| HanXRQChr09g0274431 | Green24 / Root18 | 0.69 / 0.97 | AtWRKY40 * | 53.17 | 99 | 8E-99 |
| HanXRQChr06g0166901 | Green24 | 0.67 | AtWRKY41 * | 37.25 | 100 | 3E-46 |
| HanXRQChr16g0499381 | Green24 / Root18 | 0.64 / 0.85 | AtWRKY40 * | 51.49 | 99 | 1E-93 |
| HanXRQChr03g0084521 | Green24 | 0.37 | AtWRKY11 * | 42.01 | 91 | 2E-46 |
| HanXRQChr03g0088861 | Green24 | 0.37 | AtWRKY6 | 47.67 | 93 | 7E-132 |
| HanXRQChr05g0142161 | Green24 | 0.28 | AtWRKY11 * | 37.70 | 95 | 2E-44 |
| HanXRQChr08g0216831 | Green24 / Root44 | 0.18 / 0,24 | AtWRKY70 * | 40.19 | 63 | 4E-37 |
| HanXRQChr08g0228641 | Green39 / Root18 | 0.79 / 0.38 | AtWRKY33 * | 46.65 | 91 | 3E-113 |
| HanXRQChr09g0264011 | Green39 / Root36 | 0.31 / 0.01 | AtWRKY4 * | 43.05 | 97 | 3E-107 |
| HanXRQChr16g0505941 | Green39 / Root18 | 0.29 / 0.05 | AtWRKY7 * | 47.15 | 94 | 1E-81 |
| HanXRQChr11g0348481 | Root18 | 0.17 | AtWRKY53 * | 40.96 | 98 | 1E-74 |
| HanXRQChr17g0544771 | Root36 | 0.71 | AtWRKY72 * | 38.29 | 81 | 6E-60 |
| HanXRQChr11g0336511 | Root36 | 0.42 | AtWRKY6 | 36.49 | 94 | 1E-72 |
| HanXRQChr08g0209791 | Root44 | 0.32 | AtWRKY75 * | 92.13 | 35 | 2E-55 |
Table 3.
Hub genes (IMK>0.8) from modules of interest and their closest homologs in A. thaliana related to defense functions.
Table 3.
Hub genes (IMK>0.8) from modules of interest and their closest homologs in A. thaliana related to defense functions.
| Sunflower gene | Closest homolog in A. thaliana | |||||
|---|---|---|---|---|---|---|
| Name | Module | IMK | Homolog | Identity % | Q-cov % | E-value |
| HanXRQChr01g0014161 | Green5 | 1.0 | AT5G48380 | 60.54 | 90 | 0 |
| HanXRQChr04g0107431 | Green5/Root44 | 0.96 / 0.9 | AT1G34420 | 48.14 | 99 | 0 |
| HanXRQChr02g0045301 | Green5 | 0.82 | AT3G48090 | 38.99 | 98 | 1E-147 |
| HanXRQChr09g0248321 | Green24 / Root18 | 0.94 / 0.60 | AT5G12010 | 67.32 | 89 | 0 |
| HanXRQChr02g0040711 | Green24 | 0.89 | AT1G18740 | 60.21 | 98 | 5E-163 |
| HanXRQChr15g0496321 | Green24 | 0.89 | AT2G40140 | 51.39 | 94 | 1E-178 |
| HanXRQChr03g0086901 | Green24 | 0.88 | AT3G56880 | 34.54 | 99 | 2E-18 |
| HanXRQChr16g0504131 | Green24 | 0.86 | AT2G40140 | 51.25 | 97 | 0 |
| HanXRQChr13g0399921 | Green39 | 1 | AT3G09830 | 66.92 | 88 | 0 |
| HanXRQChr12g0366961 | Green39 | 0.81 | AT1G30755 | 49.69 | 100 | 0 |
| HanXRQChr10g0300021 | Root36 | 0.96 | AT5G01050 | 53.74 | 98 | 0 |
| HanXRQChr05g0161441 | Root36 | 0.94 | AT1G22400 | 52.50 | 98 | 0 |
| HanXRQChr07g0205991 | Root44 / Green18 | 1.0 / 0.39 | AT1G08450 | 70.12 | 95 | 0 |
| HanXRQChr17g0553831 | Root44 | 0.89 | AT5G42510 | 42.47 | 64 | 1E-38 |
| HanXRQChr01g0001251 | Root44 | 0.84 | AT3G54040 | 54.02 | 70 | 5E-58 |
| HanXRQChr04g0123531 | Root44 | 0.83 | AT3G60450 | 55.42 | 93 | 5E-93 |
2.4. Module preservation among GreenGCN and RootGCN
We noticed how modules of interest from the RootGCN were enriched in genes also present in modules of interest from the GreenGCN (Table 4), this again suggested the possibility of modules being conserved between the two tissues. To test this hypothesis we analyzed the topology preservation between the Green and Root GCNs.
Table 4.
Shared genes between modules of interest. Asterisks indicate significant enrichment (p-value < 0.01) by Fisher test.
Table 4.
Shared genes between modules of interest. Asterisks indicate significant enrichment (p-value < 0.01) by Fisher test.
| Modules | Root18 | Root36 | Root44 |
|---|---|---|---|
| Green5 | 20* | 6 | 19* |
| Green16 | 1 | 0 | 0 |
| Green18 | 34* | 1 | 12* |
| Green24 | 24* | 0 | 1 |
| Green39 | 5 | 3 | 1 |
There were 14481 genes present in both networks, with overall similar expression and connectivity ranks between networks (see Figure S3), with significant Pearson correlation coefficients of 0.68 and 0.5 respectively. Using GreenGCN modules as the reference, 31 out of 63 seem markedly preserved (Zsummary score > 10 and medianRank < 40) and two appear markedly not preserved (Zsummary score < 2). On one hand, module Green14 (involved in cellular metabolic compound salvage in mitochondria) is well preserved (Zsummary score of 69.4 and medianRank of 3) as may be expected. On the other hand, Green47 with a Zsummary score 1.2 and 40 genes, is involved in cellular biosynthetic processes in the chloroplast envelope and it is markedly not preserved. Moreover, another module involved in photosynthesis, Green20 has a Zsummary score of 4.3 with 94 genes and is not preserved as we would expect.
Among the modules of interest we identified, the Green24 and Root44 modules were particullarly preserved having Zsummary scores >10 and relatively low medianRank scores (Table 5). On the contrary, the modules Green5, Green18 and Root18 didn’t pass the Zsummary threshold despite having larger module sizes, also reflected by their medianRank. Modules Green16, Green39 and Root36 showed marked non-preservation. This suggests that some possible defense-related processes influenced by WRKY genes (modules Green24 and Root44) have similar co-expression patterns in both studied tissues.
Table 5.
Preservation of modules of interest between GreenGCN and RootGCN. Modules Green24 and Root44 showed signs of consistent preservation among the two GCNs.
Table 5.
Preservation of modules of interest between GreenGCN and RootGCN. Modules Green24 and Root44 showed signs of consistent preservation among the two GCNs.
| Module | Number of shared genes | Zsummary | MedianRank |
|---|---|---|---|
| Green5 | 530 | 8.81 | 58 |
| Green16 | 186 | 3.12 | 61 |
| Green18 | 247 | 9.24 | 51 |
| Green24 | 181 | 12.67 | 39 |
| Green39 | 90 | 2.06 | 56 |
| Root18 | 209 | 8.58 | 61 |
| Root36 | 62 | 1.95 | 72 |
| Root44 | 42 | 12.65 | 10 |
2.5. Functional prediction of “unknown/uncharacterized” genes in defense modules
The selected eight defense-related modules (Green5, Green16, Green18, Green24, Green39, Root18, Root36 and Root44) included a total of 431 genes with no GO term annotation and 162 genes coding for proteins described as of “unknown” or “uncharacterized” function (HeliaGene Sunflower annotation). Interestingly, by simply searching the closest homologs in model species, we found that 125 of these unannotated genes had homologs associated with defense functions (File S4). Moreover, 30 genes from this subgroup had IMK > 0.6, meaning they may play important roles in these gene clusters. These results reaffirmed our interest in the selected modules and show the value of constructing co-expression networks to characterize gene functions, particularly for under-annotated biological processes such as “defense response”.
Therefore we used our co-expression data to perform functional inference to further characterize loci of interest. We used a strategy that combined two co-expression-based functional annotation algorithms (EGAD and vHRR; see Material and Methods). This approach rendered 881 genes predicted by both methods with defense-related GO terms, of which 482 were included in at least one module of interest (File S5). More interestingly, 65 of the 881 newly annotated genes were previously described as of “unknown function”, with 10 of them being central genes (IMK > 0,7). From the 119 WRKY-like genes annotated in the sunflower genome, 68 were included in at least one of our GCNs; of which 16 were predicted by both methods to be related to defense responses. Of these proteins, 15 were present in modules of interest (see File S5).
2.6. Functional prediction of candidate genes associated with resistance to V. dahliae and S. sclerotiorum
After completion and analysis of our networks, we compiled lists of candidate genes previously associated with resistance to sunflower fungal pathogens V. dahliae and S. sclerotiorum to assess the performance of our networks in predicting gene functions. We collected three groups of genes: Group A, a list of 26 candidate genes from Filippi et al. (2020) [25]; Group B, genes that were close (±1 megabase) to markers from four QTL mapping and genome-wide association studies: Zubrzycki et al. (2017), Filippi et al. (2020), Filippi et al. (2022) and unpublished data from Montecchia et al. [25,30,31]; and Group C, genes that were in between markers flanking QTL regions from one study [32]. It is noteworthy that group B and group C employed different germplasm in their respective studies which allows for a broader range of genetic characteristics to be captured. The full list of genes totaled 12,485 unique candidate loci, of which 5,101 genes were present in at least one of the studied GCNs, with 439 genes being included in our predefined modules of interest. Finally, of this intersection, we focused only on the genes that were also predicted as related to defense functions by EGAD and vHRR methods. The resulting list included 122 unique genes, of which 1 was from group A (candidate genes from Filippi 2020), 40 were from the close vicinity of 43 associated markers of group B, and the remaining 81 genes were included in 13 QTL regions from group C (see File S5). Interestingly, 7 of the 122 genes are linked to markers from different studies, giving further support to their biological significance. Furthermore, of the 122 total loci, 43 had no previous BP GO term annotation, 3 were WRKY transcription factors, and 8 were described as “unknown” or “uncharacterized” proteins.
3. Discussion
To our knowledge, this is the first large scale multi study co-expression network reported for sunflower. The objective was to arrange genes into biologically significant clusters while investigating their connections, classify previously reported candidate loci and deduce their possible function, and to discover new potential candidates for fungal disease resistance. For this, we constructed two weighted gene co-expression networks (GCNs) from different tissues of sunflower plants: green healthy tissue and stressed/healthy root tissue. It has been shown that merging samples from different tissues and studies generates networks with less gene connectivity and fewer modules[33]. Hence, in our work, we decided to separate samples according to tissue origin and construct different networks to compare them later. Moreover, it is known that in both simulated and real datasets, approximately 100 samples or more are required to capture transcriptional genome-wide regulations in a consistent and robust manner [32]. Therefore, we did not undertake the analysis of other relevant tissues such as the stem of fully grown plants or capitula since there were not enough samples to construct robust networks.
Co-expression network analyses have become a popular tool to find associations between complex expression data in plants [34,35,36,37,38,39]. The use of public transcriptomic data, while increasing the heterogeneity, also increases sensibly the sample size, enhancing the possibility to establish genuine associations and provide valuable insights into gene co-expression patterns and their potential roles in defense responses in sunflower. In our work, the inclusion of 733 samples in the GreenGCN made it possible to organize 25% of the genes in the sunflower’s genome in 62 modules, while the inclusion of 163 samples in the RootGCN grouped 40% of the genes in the sunflower’s genome in 77 modules. Both networks exhibited a scale-free topology, indicating a robust organization of gene co-expression [40]. The identification of hub-genes with high intramodular connectivity scores suggests their importance in coordinating gene expression within the modules. To further establish a biological context for the modules, a Gene Ontology (GO) term enrichment analysis was performed. All modules of both networks showed significant enrichment for one or more GO terms, further supporting the functional relevance of the identified gene clusters. Notably, module Green1 stood out with the highest number of hub genes, indicating its potential significance in the biological processes of growth, cell division, and plant-type cell wall biogenesis. Moreover, the GO term analysis of the RootGCN modules revealed significant enrichment for defense responses, consistent with the control versus infected condition of the samples. Additionally, the networks’ performances were evaluated by measuring the Guilt-By-Association-AUROC (GBA-AUC), which quantifies each network’s ability to connect genes with shared GO terms. The obtained GBA-AUC values indicated a high level of functional coherence within each network. The analysis revealed a negative correlation between gene connectivity and dN/dS ratios, consistent with Masalia et al (2017) [21] where genes with less overall connectivity appeared to be under more intense selective pressure. In summary, the networks serve as a valuable tool for investigating potential connections between genes and advancing towards the field of systems biology in sunflower.
The identification of modules related to fungal disease resistance was a key focus of the study. We identified eight defense-related modules, significantly enriched in GO terms associated with defense responses and/or WRKY genes: Green5, Green16, Green18, Green24, Green39, Root18, Root36, and Root44. Within them, the intramodular connectivity scores pointed out the main genetic drivers of each as hub genes. Among the 40 hub genes with predicted function, nine were directly associated to defense responses through their GO-Term annotation, while other 16 hub genes were linked to them through their A. thaliana homologs. Additionally, the positive correlation between the eigengenes of these modules within each GCN suggested a coordinated regulation in defense processes. Notably, module Green16 exhibited an inverse correlation with other defense-related modules, indicating potential functional distinctions. Supportively, no WRKY genes were found in this module. The WRKY family is considered as one of the largest transcription factor (TF) families in higher plants [23,33], with 119 loci in sunflower [27,34]. Members of the WRKY-family are involved in several stress related processes, including defense response and biotic or abiotic stress responses [23]. However, only eight sunflower WRKY genes are annotated with the GO term “defense response” or its child terms in the HanXRQv1 genome. In sunflower, previous findings by Giacomelli et al. (2010) [24], Liu et al. (2020) [27], Li et al. (2020) [35] and Filippi et al (2020) [18] implicated WRKY genes in fungal defense responses. In this study, we also identified homologs of sunflower WRKY genes in our networks that were previously associated with defense responses in other plant species (see Table 2). The presence of these sunflower gene homologs within the “defense modules”, along with their high intramodular connectivity, provides evidence for their function in a defense regulatory role. In support of the predictive ability of the networks, the WRKY hub of the Green24 module, HanXRQChr16g0509771 (IMK=0.89), without prior GO defense-related annotation, was found by Peluffo (2010) and Giacomelli et al. (2010) (under the name HaWRKY7) to be differentially expressed upon infection with S. sclerotiorum in two independent studies[41,42]. Furthermore, using this candidate gene as a marker in an association mapping study, Filippi et al. (2015) found it to be associated with low severity of Sclerotinia head rot [43]. Furthermore, another five WRKY genes (four present in Green24 and one in Green39) were described by Giacomelli et al. (2010)[41] to have differential expression under S. sclerotiorum infection: HanXRQChr16g0499381, HanXRQChr03g0088861, HanXRQChr03g0084521, HanXRQChr08g0216831 and HanXRQChr16g0505941 (IMKs = 0.64, 0.37, 0.37 and 0.18 respectively) (Supp. Table S1). Lastly, the WRKY-like genes HanXRQChr14g0460611 and HanXRQChr15g0480431 from modules Green18 (IMK=0,41) and Green5 (IMK=0,13), respectively, have been shown to be upregulated in roots under R. irregulare infection [44]. It is worth mentioning that among the total of 26 WRKY genes identified in the studied modules, 8 genes are found in both the Green and Root modules of interest. This finding suggests a potential link or association between these gene clusters in different tissues. Our comprehensive analysis, based on public data, presents compelling evidence supporting the involvement of multiple sunflower WRKY genes in defense responses.
In addition to the WRKY family, a large number of other genes have been included in our defense-related modules with potential functional connections. To illustrate the functional potential of defense-related networks, we selected the gene AT3G55800, a homolog of the hub gene HanXRQChr14g0459921 in the Green16 module, which is described as a sedoheptulose biphosphatase. According to the Protein-Protein Interaction Networks of STRING Database (© STRING CONSORTIUM 2023), this protein has been reported to be coexpressed with several fructose-bisphosphate aldolases and glyceraldehyde-3-phosphate dehydrogenases, among others. In addition to its role in the process of carbohydrate biosynthesis, this protein, along with three other metabolic enzymes involved in glycolysis and the pentose phosphate pathway, decreased during the response to Pseudomonas syringae pv tomato DC3000 in A. thaliana leaves [45]. At the same time, the ATP synthase subunit CF1δ, which is encoded by the gene AT4G09650 and plays a central role in oxidative or photosynthetic phosphorylation, decreased in abundance. Remarkably, the Green16 module is highly enriched in predicted enzymes of carbohydrate metabolism, including several fructose biphosphatases, glyceraldehyde-3-phosphate dehydrogenases, sedoheptulose biphosphatases, and malate dehydrogenases, many of which play a central role in this module. Green16 also contains a homolog of AT4G09650, HanXRQChr01g0001181. Moreover, this gene was down-regulated in a tolerant inbred line at early stages of infection with the fungus S. sclerotiorum in sunflower, suggesting a common functional role in pathogen-associated molecular pattern (PAMP)-triggered immunity [46]. Several authors have identified modulation of primary metabolic pathways during the defense response, although the specific players in each species remain to be elucidated [47,48,49].
Previous studies identified various loci involved in the quantitative resistance to V. dahliae and S. sclerotiorum, mostly through comprehensive analysis in biparental mapping, association mapping, or transcriptome differential expression studies [25,26,30,31,32,46] (citas). In an attempt to provide a biological context to the candidate genes derived either by co-localizing with a molecular marker in GWAS studies or by being located in between markers of QTL studies, we focused on the candidates predicted as related to defense functions in our modules of interest. This strategy allowed to select and annotate by association the candidates immersed in a defense-related biological context. The resulting list included 122 unique genes, of which 1 was from group A, 40 were from the close vicinity of 44 associated markers of group B, and the remaining 81 genes were included in 13 QTL regions from group C (see File S5). Interestingly, 7 of the 122 genes had been repeatedly identified by different studies. Furthermore, of the 122 total loci, 43 have no previous BP GO term annotation, 3 are WRKY transcription factors, and 8 are described as “unknown” or “uncharacterized” proteins.
Overall, the aforementioned findings present a novel approach to exploiting the great amount of data generated in sunflower to date. Although only a moderate proportion of the sunflower genome could be included in our networks, they highlight the presence of genetic drivers and group co-expressed elements, expanding our understanding of the associations established during different biological processes taking place. The insights into disease resistance in sunflowers are of particular interest for breeding programs. The identification of WRKY genes with a recurrent and central role in defense-related modules presents a compelling example of promising breeding targets. The dominant role of these transcription factors can potentially achieve more comprehensive and substantial effects on multiple loci associated with defense responses by modulating the expression of various downstream genes. This could also apply to other hub-genes, allowing for fine-tuning the breeding strategies and leading to the development of sunflower varieties with enhanced defense capabilities against fungal pathogens. The defense-related annotation resource generated in this study is available in File S5, where it can be explored by the sunflower and plant community to facilitate future research, as these novel annotations might provide the missing link in defense-related pathways.
4. Material and methods
4.1. Data acquisition
We explored all the publicly available sunflower RNA-seq samples from the Sequence Read Archive (SRA) (NCBI) and downloaded samples originating from either: 1) photosynthetic tissues under control conditions, and 2) radical tissues under either control conditions or infected by pathogens. The total 898 samples comprised 31 SRA studies and 391 different genotypes, detailed in File S1. SSR markers from Zubrzycki et al. (2017) [31] were mapped to the sunflower reference genome HanXRQv1.2 [7] to identify their genomic position.
4.2. Quality Control and Mapping of Data
The sequencing quality of each sample was evaluated with FastQC [50] and any adaptors and low quality reads were removed with Trimmomatic [51]. We quantified the expression of our reference transcriptome via Salmon v1.5.2 [52], using the genome HanXRQ version r1.2 as our reference to produce 1 unique transcript per gene (coding and non-coding), totaling 56917 distinct sequences [7]. The approach used by Salmon estimates abundance uncertainty due to random sampling and the ambiguity introduced by multi-mapping reads, a key feature when mapping to a transcriptome reference. The mapping rates ranged from 1% to 97%, with a median of 75%. Overall, samples from wild-cultivar species had lower mapping percentages, as well as samples from infected tissues probably due to low efficiency in RNA extraction from sunflowers. Samples with less than 3 million read counts were discarded.
4.3. Co-expression network analysis
For each group of samples, we applied a recursive process of: filtering genes with less than 2 counts per million (CPM) in ¾ of samples; normalizing read counts via variance stabilizing transformation from the “DESeq2” R package [53]; adjusting batch effects arising from sample origin via an empirical Bayesian method described by Johnson et al. [54] implemented in the R package “sva” [55]; and excluding visual outliers in hierarchical clustering of Pearson distances. With these matrices, we calculated weighted gene co-expression networks using the R package “WGCNA” [27]. We used bicor correlation, signed-hybrid weighting, and signed Topological Overlap Matrices. Appropriate β soft thresholds were picked by maximizing the networks’ fit to a free scale topology. Merge cut height, and deep split parameters were 0.25 and 2 respectively, and minimal module size was set to 30.
4.4. Guilt-by-Association network performance evaluation
We used the EGAD R package [56] to evaluate the network’s capacity to connect genes with shared Gene Ontology (GO) terms as a measure of quality. Based on the Guilt-by-Association principle, networks can be evaluated by hiding a subset of genes GO terms and test whether the hidden GO terms could be predicted from the remaining annotated genes given their expression correlation. GO data for the HaXRQ 1.2 sunflower genome was downloaded from https://www.heliagene.org/. For evaluation, only GO terms with 20 to 300 annotated genes present in the evaluated network were used, amounting between 556 and 624 GO terms depending on the network. The predictions’ performances were measured by AUROC values in 5-fold cross-validation.
4.5. Estimation of dN/dS ratios
We estimated molecular evolution rates of each sunflower gene using PAML’s yn00 model [57] with Arabidopsis thaliana as reference. We downloaded A. thaliana CDS from Phytozome v12 (https://phytozome-next.jgi.doe.gov/), translated the sequences, and identified putative sunflower orthologs by using reciprocal best BLASTP hits with an e-value threshold of 1E-10. Then, the resulting 10,425 orthologs pairs were aligned using MUSCLE v.5.0.1428 [58]; we used pal2nal [59] to transform the protein alignments into nucleotide alignments before feeding them to the PAML algorithm. Rates of non-synonymous substitutions per non-synonymous site (dN), synonymous substitutions per synonymous site (dS), and estimates of adaptive evolution (ω = dN/dS) were compared via linear regression against estimates of gene connectivity in each network.
4.6. GO enrichment analysis
GO enrichment of modules was analyzed via the R/Bioconductor package TopGO [60] using Fisher’s exact test as statistic (considering the complete list of 56917 unique genes as background references) and two methods of dealing with the GO graph structure: “classic” and “weight”. P-values below 0.05 were considered significant enrichments, p-values calculated with the “weight” method do not require multiple testing correction, as per authors’ indications [61].
4.7. Module-condition relationship
To identify modules that were significantly associated with biological conditions, correlations between module eigengenes (MEs) (i.e., the first principal component of the module, which represents the overall expression level of the module) [62] and conditions were computed using binomial generalized linear models implemented in R in the base package stats.
4.8. Module Preservation Analysis
To assess the preservation of modules from one co-expression network to the other, we used the WGCNA package function modulePreservation to calculate the composite preservation statistics “Zsummary” and “medianRank”. Higher values of Zsummary indicate stronger preservation of the module being compared between the networks. Zsummary scores higher than 10 indicate consistent preservation of the module, while scores lower than 2 indicate consistent disruption. However, Zsummary tends to be dependent of module size, meaning bigger modules will have inflated Zsummary scores, since it is more significant to observe preserved connectivity patterns among hundreds of genes than to observe the same among only a few [63]. On the other hand, medianRank is size-independent and lower values means more preserved modules, however medianRank measure is dependent of the total number of modules. As reference, modules with medianRanks higher than ⅔ of the total module number are considered non-preserved [64].
4.9. Gene function prediction
We used the mentioned EGAD package along with the methods vHRR [13] to predict biological functions on network genes.
For the EGAD method, we used the same adjacency matrices calculated by WGCNA. For each network, the optimum threshold for each GO term was picked according to the maximum F1 score, calculated using the known annotations. For the vHRR pipeline, we used the filtered and normalized data also used as input for WGCNA. As input annotations we used the aforementioned GO data. The performances of the prediction methods were evaluated by F1 scores over the known annotations.
Overall performance comparison of these methods is shown in Figure S4. In order to prioritize genes with possible defense functions, we chose to consider both methods as complementary. We selected a list of BP GO terms linked to defense response and related terms to predict new annotations. This list is detailed in File S5, page 2, GO terms not present in any of the genes from the GCNs are omitted. Our strategy was to focus on genes that were annotated by both methods with any defense-related GO term. That is, for example, we would select a gene predicted as “defense to fungus” by EGAD using the GreenGCN data, and “response to stress” by vHRR using RootGCN data.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org., Figure S1: Gene connectivities versus molecular evolution rates; Figure S2: Module densities versus size; Figure S3: Comparison of expression and connectivity ranks between GCNs; Figure S4: Comparison of prediction performance of EGAD and vHRR; Table S1: title. File S1: Samples metadata. File S2: Genes characteristics and modules. File S3: GO enrichment analysis. File S4: BlastP hits of uncharacterized proteins from defense modules. File S5: Gene functional prediction.
Author Contributions
AR: VL, and MR planned and designed the research. AR performed the experiments. AR, MF, SG, and MR analyzed the data. AR, MF, VL, NP, and MR wrote the manuscript. NP supplied the resources. NP, VL and MR reviewed and validated the manuscript with the contribution of all authors. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Agency for the Promotion of Research, Technological Development and Innovation, Ministry of Science, Technology and Innovation PICT 2018-03810; PICT 2019-1698; PICT 2017-1021.
Data Availability Statement
All data is in the public domain.
Acknowledgments
This work used computational resources from BIOCAD, IABIMo-UDD INTA-CONICET, Programa de Sustentabilidad y Competitividad Forestal/BID 2853/Oc-Ar, Consorcio Argentino de Tecnología Genómica, MinCyT PPL 2011 004.
Conflicts of Interest
The authors declare no conflict of interest.
References
- IPPC Secretariat Scientific review of the impact of climate change on plant pests; FAO on behalf of the IPPC Secretariat, 2021; ISBN 978-92-5-134435-4.
- Maron, L.G.; Piñeros, M.A.; Kochian, L.V.; McCouch, S.R. Redefining “stress resistance genes”, and why it matters. J. Exp. Bot. 2016, 67, 5588–5591. [Google Scholar] [CrossRef] [PubMed]
- Dimitrijevic, A.; Horn, R. Sunflower hybrid breeding: from markers to genomic selection. Front. Plant Sci. 2017, 8, 2238. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Shi, H.; Yu, H.; Ma, Y.; Hu, H.; Han, Z.; Zhang, Y.; Zhen, Z.; Yi, L.; Hou, J. Combined GWAS and transcriptome analyses provide new insights into the response mechanisms of sunflower against drought stress. Front. Plant Sci. 2022, 13, 847435. [Google Scholar] [CrossRef]
- Guo, S.; Zuo, Y.; Zhang, Y.; Wu, C.; Su, W.; Jin, W.; Yu, H.; An, Y.; Li, Q. Large-scale transcriptome comparison of sunflower genes responsive to Verticillium dahliae. BMC Genomics 2017, 18, 42. [Google Scholar] [CrossRef]
- Ramu, V.S.; Paramanantham, A.; Ramegowda, V.; Mohan-Raju, B.; Udayakumar, M.; Senthil-Kumar, M. Transcriptome Analysis of Sunflower Genotypes with Contrasting Oxidative Stress Tolerance Reveals Individual- and Combined- Biotic and Abiotic Stress Tolerance Mechanisms. PLoS ONE 2016, 11, e0157522. [Google Scholar] [CrossRef]
- Badouin, H.; Gouzy, J.; Grassa, C.J.; Murat, F.; Staton, S.E.; Cottret, L.; Lelandais-Brière, C.; Owens, G.L.; Carrère, S.; Mayjonade, B.; Legrand, L.; Gill, N.; Kane, N.C.; Bowers, J.E.; Hubner, S.; Bellec, A.; Bérard, A.; Bergès, H.; Blanchet, N.; Boniface, M.-C.; Langlade, N.B. The sunflower genome provides insights into oil metabolism, flowering and Asterid evolution. Nature 2017, 546, 148–152. [Google Scholar] [CrossRef]
- Seiler, G.J.; Qi, L.L.; Marek, L.F. Utilization of sunflower crop wild relatives for cultivated sunflower improvement. Crop Sci. 2017, 57, 1083–1101. [Google Scholar] [CrossRef]
- Mason, C.M.; Bowsher, A.W.; Crowell, B.L.; Celoy, R.M.; Tsai, C.-J.; Donovan, L.A. Macroevolution of leaf defenses and secondary metabolites across the genus Helianthus. New Phytol. 2016, 209, 1720–1733. [Google Scholar] [CrossRef]
- Cowger, C.; Brown, J.K.M. Durability of quantitative resistance in crops: greater than we know? Annu. Rev. Phytopathol. 2019, 57, 253–277. [Google Scholar] [CrossRef]
- Carbon, S.; Ireland, A.; Mungall, C.J.; Shu, S.; Marshall, B.; Lewis, S. ; AmiGO Hub; Web Presence Working Group AmiGO: online access to ontology and annotation data. Bioinformatics 2009, 25, 288–289. [Google Scholar] [CrossRef]
- Berardini, T.Z.; Mundodi, S.; Reiser, L.; Huala, E.; Garcia-Hernandez, M.; Zhang, P.; Mueller, L.A.; Yoon, J.; Doyle, A.; Lander, G.; Moseyko, N.; Yoo, D.; Xu, I.; Zoeckler, B.; Montoya, M.; Miller, N.; Weems, D.; Rhee, S.Y. Functional annotation of the Arabidopsis genome using controlled vocabularies. Plant Physiol. 2004, 135, 745–755. [Google Scholar] [CrossRef]
- Depuydt, T.; Vandepoele, K. Multi-omics network-based functional annotation of unknown Arabidopsis genes. Plant J. 2021, 108, 1193–1212. [Google Scholar] [CrossRef]
- Di Persia, L.; Lopez, T.; Arce, A.; Milone, D.H.; Stegmayer, G. exp2GO: improving prediction of functions in the Gene Ontology with expression data. IEEE/ACM Trans Comput Biol Bioinform. [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Zdobnov, E.M.; Apweiler, R. InterProScan--an integration platform for the signature-recognition methods in InterPro. Bioinformatics 2001, 17, 847–848. [Google Scholar] [CrossRef]
- Mahood, E.H.; Kruse, L.H.; Moghe, G.D. Machine learning: A powerful tool for gene function prediction in plants. Appl. Plant Sci. 2020, 8, e11376. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.; Jiang, Y.; Bergquist, T.R.; Lee, A.J.; Kacsoh, B.Z.; Crocker, A.W.; Lewis, K.A.; Georghiou, G.; Nguyen, H.N.; Hamid, M.N.; Davis, L.; Dogan, T.; Atalay, V.; Rifaioglu, A.S.; Dalkıran, A.; Cetin Atalay, R.; Zhang, C.; Hurto, R.L.; Freddolino, P.L.; Zhang, Y.; Friedberg, I. The CAFA challenge reports improved protein function prediction and new functional annotations for hundreds of genes through experimental screens. Genome Biol. 2019, 20, 244. [Google Scholar] [CrossRef]
- Mutwil, M.; Usadel, B.; Schütte, M.; Loraine, A.; Ebenhöh, O.; Persson, S. Assembly of an interactive correlation network for the Arabidopsis genome using a novel heuristic clustering algorithm. Plant Physiol. 2010, 152, 29–43. [Google Scholar] [CrossRef]
- Rao, X.; Dixon, R.A. Co-expression networks for plant biology: why and how. Acta Biochim Biophys Sin (Shanghai) 2019, 51, 981–988. [Google Scholar] [CrossRef]
- Montecchia, J.F.; Fass, M.I.; Cerrudo, I.; Quiroz, F.J.; Nicosia, S.; Maringolo, C.A.; Di Rienzo, J.; Troglia, C.; Hopp, H.E.; Escande, A.; González, J.; Álvarez, D.; Heinz, R.A.; Lia, V.V.; Paniego, N.B. On-field phenotypic evaluation of sunflower populations for broad-spectrum resistance to Verticillium leaf mottle and wilt. Sci. Rep. 2021, 11, 11644. [Google Scholar] [CrossRef]
- Mathew, F.; Harveson, R.; Block, C.; Gulya, T.; Ryley, M.; Thompson, S.; Markell, S. Sclerotinia diseases of sunflower. PHI 2020. [CrossRef]
- Gulya, T.; Rashid, K.Y.; Marisevic, S.M. ; Gulya K Y Rashid, S N Masirevic, T.J. Sunflower diseases. In Sunflower Technology and Production; (ed.), I. A. A. S., Ed.; ASA, CSSA, and SSSA, Madison, WI., 1997; Vol. 35, pp. 263–379, ISBN 9780891182276.
- Zubryzcki, J.; Fusari, C.; Maringolo, C.; Dirienzo, J.; Cervigni, G.; Nishinakamasu, V.; Filippi, C.; Troglia, C.; Quiroz, F.; Alvarez, D.; Escande, A.; Hopp, H.E.; Heinz, R.; Lia, V.; Paniego, N. Biparental QTL and Association Mapping for Sclerotinia head rot resistance in cultivated sunflower. In Asagir Mar Del Plata {&} Balcarce 2012; Argentina; Mar del Plata, Argentina, 2012; Vol. II.
- Filippi, C.V.; Zubrzycki, J.E.; Di Rienzo, J.A.; Quiroz, F.J.; Puebla, A.F.; Alvarez, D.; Maringolo, C.A.; Escande, A.R.; Hopp, H.E.; Heinz, R.A.; Paniego, N.B.; Lia, V.V. Unveiling the genetic basis of Sclerotinia head rot resistance in sunflower. BMC Plant Biol. 2020, 20, 322. [Google Scholar] [CrossRef]
- Talukder, Z.I.; Hulke, B.S.; Qi, L.; Scheffler, B.E.; Pegadaraju, V.; McPhee, K.; Gulya, T.J. Candidate gene association mapping of Sclerotinia stalk rot resistance in sunflower (Helianthus annuus L.) uncovers the importance of COI1 homologs. Theoretical and Applied Genetics 2014. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 2008, 9, 559. [Google Scholar] [CrossRef]
- Somssich, M.; Khan, G.A.; Persson, S. Cell wall heterogeneity in root development of arabidopsis. Front. Plant Sci. 2016, 7, 1242. [Google Scholar] [CrossRef]
- Wani, S.H.; Anand, S.; Singh, B.; Bohra, A.; Joshi, R. WRKY transcription factors and plant defense responses: latest discoveries and future prospects. Plant Cell Rep. 2021, 40, 1071–1085. [Google Scholar] [CrossRef] [PubMed]
- Filippi, C.V.; Corro Molas, A.; Dominguez, M.; Colombo, D.; Heinz, N.; Troglia, C.; Maringolo, C.; Quiroz, F.; Alvarez, D.; Lia, V.; Paniego, N. Genome-Wide Association Studies in Sunflower: Towards Sclerotinia sclerotiorum and Diaporthe/Phomopsis Resistance Breeding. Genes (Basel) 2022, 13. [Google Scholar] [CrossRef]
- Zubrzycki, J.E.; Maringolo, C.A.; Filippi, C.V.; Quiróz, F.J.; Nishinakamasu, V.; Puebla, A.F.; Di Rienzo, J.A.; Escande, A.; Lia, V.V.; Heinz, R.A.; Hopp, H.E.; Cervigni, G.D.L.; Paniego, N.B. Main and epistatic QTL analyses for Sclerotinia Head Rot resistance in sunflower. PLoS ONE 2017, 12, e0189859. [Google Scholar] [CrossRef]
- Talukder, Z.I.; Underwood, W.; Misar, C.G.; Seiler, G.J.; Cai, X.; Li, X.; Qi, L. Genomic Insights Into Sclerotinia Basal Stalk Rot Resistance Introgressed From Wild Helianthus praecox Into Cultivated Sunflower (Helianthus annuus L.). Front. Plant Sci. 2022, 13, 840954. [Google Scholar] [CrossRef] [PubMed]
- Liesecke, F.; De Craene, J.-O.; Besseau, S.; Courdavault, V.; Clastre, M.; Vergès, V.; Papon, N.; Giglioli-Guivarc’h, N.; Glévarec, G.; Pichon, O.; Dugé de Bernonville, T. Improved gene co-expression network quality through expression dataset down-sampling and network aggregation. Sci. Rep. 2019, 9, 14431. [Google Scholar] [CrossRef] [PubMed]
- You, Q.; Zhang, L.; Yi, X.; Zhang, K.; Yao, D.; Zhang, X.; Wang, Q.; Zhao, X.; Ling, Y.; Xu, W.; Li, F.; Su, Z. Co-expression network analyses identify functional modules associated with development and stress response in Gossypium arboreum. Sci. Rep. 2016, 6, 38436. [Google Scholar] [CrossRef]
- Gupta, C.; Pereira, A. Recent advances in gene function prediction using context-specific coexpression networks in plants. [version 1; peer review: 2 approved]. F1000Res. 2019, 8. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Liu, X.; Zhou, J.; Deng, H.; Zhang, G.; Xiao, Y.; Tang, W. WGCNA Analysis Identifies the Hub Genes Related to Heat Stress in Seedling of Rice (Oryza sativa L.). Genes (Basel) 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, Y.; Shi, H.; Hu, H.; Yi, L.; Hou, J. Time-course transcriptome and WGCNA analysis revealed the drought response mechanism of two sunflower inbred lines. PLoS ONE 2022, 17, e0265447. [Google Scholar] [CrossRef]
- Du, J.; Wang, S.; He, C.; Zhou, B.; Ruan, Y.-L.; Shou, H. Identification of regulatory networks and hub genes controlling soybean seed set and size using RNA sequencing analysis. J. Exp. Bot. 2017, 68, 1955–1972. [Google Scholar] [CrossRef]
- Zainal-Abidin, R.-A.; Harun, S.; Vengatharajuloo, V.; Tamizi, A.-A.; Samsulrizal, N.H. Gene Co-Expression Network Tools and Databases for Crop Improvement. Plants 2022, 11. [Google Scholar] [CrossRef]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, Article17. [Google Scholar] [CrossRef]
- Giacomelli, J.I.; Ribichich, K.F.; Dezar, C.A.; Chan, R.L. Expression analyses indicate the involvement of sunflower WRKY transcription factors in stress responses, and phylogenetic reconstructions reveal the existence of a novel clade in the Asteraceae. Plant Sci. 2010, 178, 398–410. [Google Scholar] [CrossRef]
- Peluffo, L. Caracterización de los mecanismos de defensa a Sclerotinia sclerotiorum, agente causal de la Podredúmbre Húmeda de Girasol, a través del estudio de perfiles metabólicos y transcripcionales. Doctoral dissertation, Instituto de Biotecnología, CICVyA, INTA-Castelar, 2010.
- Filippi, C.V. Diversidad genómica y mapeo por asociación para la resistencia a la podredumbre húmeda del capítulo causada por Sclerotinia sclerotiorum en girasol. Doctoral dissertation, Instituto Nacional de Tecnología Agropecuaria (INTA) Castelar. Instituto de Biotecnología. Centro de Investigación en Ciencias Veterinarias y Agronómicas (CICVyA), 2015.
- Liu, A.; Liu, C.; Lei, H.; Wang, Z.; Zhang, M.; Yan, X.; Yang, G.; Ren, J. Phylogenetic analysis and transcriptional profiling of WRKY genes in sunflower (Helianthus annuus L.): Genetic diversity and their responses to different biotic and abiotic stresses. Industrial Crops and Products 2020, 148, 112268. [Google Scholar] [CrossRef]
- Jones, A.M.E.; Thomas, V.; Bennett, M.H.; Mansfield, J.; Grant, M. Modifications to the Arabidopsis defense proteome occur prior to significant transcriptional change in response to inoculation with Pseudomonas syringae. Plant Physiol. 2006, 142, 1603–1620. [Google Scholar] [CrossRef]
- Fass, M.I.; Rivarola, M.; Ehrenbolger, G.F.; Maringolo, C.A.; Montecchia, J.F.; Quiroz, F.; García-García, F.; Blázquez, J.D.; Hopp, H.E.; Heinz, R.A.; Paniego, N.B.; Lia, V.V. Exploring sunflower responses to Sclerotinia head rot at early stages of infection using RNA-seq analysis. Sci. Rep. 2020, 10, 13347. [Google Scholar] [CrossRef] [PubMed]
- Rojas, C.M.; Senthil-Kumar, M.; Tzin, V.; Mysore, K.S. Regulation of primary plant metabolism during plant-pathogen interactions and its contribution to plant defense. Front. Plant Sci. 2014, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Jiang, Y.; Ritchie, E.S.; Macho, A.P.; Yu, F.; Wu, D. Manipulation of plant metabolism by pathogen effectors: more than just food. FEMS Microbiol. Rev. 2023, 47. [Google Scholar] [CrossRef] [PubMed]
- Morkunas, I.; Ratajczak, L. The role of sugar signaling in plant defense responses against fungal pathogens. Acta Physiol. Plant. 2014, 36, 1607–1619. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A quality control tool for high throughput sequence data.
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. BioRxiv 2014. [CrossRef]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef]
- Ballouz, S.; Weber, M.; Pavlidis, P.; Gillis, J. EGAD: ultra-fast functional analysis of gene networks. Bioinformatics 2017, 33, 612–614. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Suyama, M.; Torrents, D.; Bork, P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006, 34, W609–12. [Google Scholar] [CrossRef] [PubMed]
- Alexa, A.; Rahnenfuhrer, J. topGO: Enrichment Analysis for Gene Ontology. 2019.
- Alexa, A.; Rahnenführer, J. Gene set enrichment analysis with topGO. 2023.
- Langfelder, P.; Horvath, S. Eigengene networks for studying the relationships between co-expression modules. BMC Syst. Biol. 2007, 1, 54. [Google Scholar] [CrossRef] [PubMed]
- Bakhtiarizadeh, M.R.; Hosseinpour, B.; Shahhoseini, M.; Korte, A.; Gifani, P. Weighted Gene Co-expression Network Analysis of Endometriosis and Identification of Functional Modules Associated With Its Main Hallmarks. Front. Genet. 2018, 9, 453. [Google Scholar] [CrossRef]
- Langfelder, P.; Luo, R.; Oldham, M.C.; Horvath, S. Is my network module preserved and reproducible? PLoS Comput. Biol. 2011, 7, e1001057. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



