
Submitted:
16 June 2023
Posted:
16 June 2023
You are already at the latest version
Abstract
Strong evidence indicates that insulin resistance contributes to the development of Alzheimer´s disease by promoting inflammation, oxidative stress, and blood vessel damage. Conversely, neuroinflammation induces insulin resistance by targeting insulin signaling molecules: the insulin receptor substrates 1 and 2 (IRS1/2). In addition, neuroinflammation is strongly linked to reduced levels of brain-derived neurotrophic factor (BDNF) expression. BDNF is a key factor for neuronal survival, and its expression is compromised in neurodegenerative disorders. In this study, we show that, in a triple-transgenic (3xTg) mice model of Alzheimer’s disease, the BDNF and IRS2 but not the IRS1 mRNA expression levels were reduced in the hippocampus, together with higher levels of TNFα, compared with wild-type mice. Subgroups of control and 3xTg mice received short and long treatments of Abscisic Acid (ABA), a phytohormone with anti-inflammatory and insulin-sensitizing capabilities. We found that the short ABA treatment can increase the IRS1 and IRS2 mRNA expression both in wild-type and 3xTg mice, concomitant with reducing pro-inflammatory cytokine TNFα in 3xTg mice. However, earlier, and thus longer treatments are required to rescue the BDNF mRNA levels. Our data strongly confirm that ABA administration is a potential treatment to prevent Alzheimer´s disease, via lowering neuroinflammation, potentially rescuing insulin signaling molecules and BDNF mRNA expression.
Keywords:
hippocampus
; Alzheimer’s disease
; neuroinflammation
; insulin signal
; phytohormones
; neurotrophic factor
; neuroprotector
1. Introduction
Alzheimer’s disease (AD) is the most common type of dementia, and it is characterized by progressive memory loss, cognitive impairment, and functional decline. The main histopathological hallmarks of AD are the accumulation of extracellular amyloid beta-peptide (Aβ) and hyperphosphorylated tau protein in intracellular neurofibrillary tangles that correlate with significant glucose hypometabolism. Scientific evidence has demonstrated that the onset of neurodegeneration occurs decades before the first clinical symptoms appear [1]. At these early stages, the neuroinflammatory status and insulin resistance are key players at the onset of the disease, as they can cause irreversible neuronal damage [2].
Late-onset AD has an idiopathic etiology; however, several risk factors have been identified, and metabolic syndrome leading to type II diabetes are considered major risks factors for AD [3]. One of the well-studied targets of insulin resistance is Tau phosphorylation, and the presence of hyperphosphorylated Tau in the patient blood serum is a significant predictive biomarker of AD [4]. Insulin signaling starts by the binding to the insulin receptor, and it is mediated by the insulin receptor substrates isoforms 1 and 2 (IRS1 and IRS2). We previously observed reduced levels of hippocampal IRS2 mRNA in an animal model of metabolic syndrome that also displayed cognitive impairment [5]. Furthermore, human studies have shown that IRS2 mRNA and protein expression is significantly decreased in the hippocampus and temporal [6] and prefrontal cortexes [7] of AD patients compared to control subjects. The administration of molecules with anti-inflammatory capability can rescue the IRS2 mRNA and protein levels, exemplified by ABA in rats with metabolic syndrome [5], and curcumin in a rat model of Alzheimer´s disease [8]. IRS2 rescuing, in all cases, is concomitant with cognitive function improvement.
Interestingly, insulin signaling is strongly linked with BDNF, a key neurotrophic factor for neuronal survival and plasticity [9]. In fact, diabetic patients display distinctly reduced BDNF serum levels compared to non-diabetic controls [10]. Low BDNF expression is strongly correlated with high brain inflammatory status [11]. Not only is the BDNF function reduced in Alzheimer´s disease, but it is also considered a fundamental target for disease management, given the fundamental role in neuroprotection [12].
In our previous studies, we evaluated two ABA treatments in a 3xTg model of AD: early ABA administration (duration of 5 months) and later (duration of 3 months). In this model, early treatment could effectively ameliorate memory impairment and prevent microglia switching to a reactive state [13]. According to the literature, and our previous findings, we hypothesized that the action of ABA would be mediated by enhancing the BDNF and IRS2 expression levels, thereby improving insulin signaling and preventing neurodegeneration.
2. Results
2.1. ABA Treatment Reduces Inflammatory Cytokine TNFα While Increasing BDNF mRNA Expression in Hippocampi of Triple-Transgenic Mice
We first evaluated the levels of cytokine tumor necrosis factor α (TNFα) to confirm the inflammatory status of the 3xTg mice and the effect of ABA (Figure 1A). We have previously shown that the microglia morphology in 3xTg mice is less ramified, corresponding to an M1/M2 status, compared to the age-matched controls. However, M1 and M2 differ in their cytokine profile, being M1 pro-inflammatory with higher levels of TNFα.
Here we show that, as expected, at the age of 8 months, untreated 3xTg mice have significantly higher levels of TNFα than wild-type animals (*** p = 0.0004; t = 4.34; df = 17). Confirming our previous effect on inflammatory microglia, ABA administration significantly prevented the increase in the TNFα mRNA levels in the 3xTg mice hippocampi, both at 3 months (* p = 0.042; t = 2.298; df = 11) and 5 months (* p = 0.036; t = 2.33; df = 13) of treatment.
Next, we measured the brain neurotrophic factor (BDNF) (Figure 1B), given its relevance in neuron survival and activity, and the evidence showing that BDNF activity is compromised in Alzheimer´s models [14]. We confirmed the reduced BDNF mRNA levels in the hippocampi of the untreated 3xTg mice compared to the wild type (** p = 0.009; t = 2.268; df = 16). Next, we evaluated the potential benefit of ABA treatment and found that 3 months of ABA exposure did not increase the BDNF mRNA levels; however, with the earlier and longer ABA treatment, the hippocampal BDNF levels were significantly higher than in the untreated 3xTg mice (**** p < 0.0001; t = 8.865; df = 12). Interestingly, we found that 5 months of ABA exposure also increased the hippocampal BDNF in the wild-type mice (** p = 0.002; t = 3.79; df = 13)
2.2. ABA Treatment Increases mRNA Levels of Insulin Substrate Receptors 1 and 2 (IRS1/2)
Next, we determined the levels of IRS1 and IRS2 at 3 and 5 months of ABA treatment (Figure 2). We observed that, at 8 months of age, the IRS2 levels (Figure 2A) were significantly lower in the hippocampi of the 3xTg mice compared to the wild type (## p = 0.0075; t = 3.34; df = 10). Early ABA treatment (5 months duration) rescued the levels of hippocampal IRS2 mRNA expression in the 3xTg mice compared to untreated mice (* p = 0.043; t = 2.35; df = 9). The later ABA treatment (3 months) increased the expression but did not reach statistical significance. In the wild-type mice, the short ABA treatment induced a significant increase in the IRS2 mRNA expression (*** p = 0.0003; t = 5.372; df = 8)). The increases in IRS2 mRNA obtained with the long ABA treatment (5 months) did not reach statistical significance. On the contrary, the IRS1 levels were not different in the untreated 3xTg mice compared to the wild-type mice at 8 months of age. In the wild-type mice, ABA treatment significantly increased the IRS1 mRNA levels, both the short (* p = 0.0215; t = 2.642; df = 12) and long (* p = 0.0311; t = 2.44; df = 12) treatments, while the increase observed in the 3xTg mice with 3 months of ABA treatment was not significant, but the long treatment induced significant growth (* p = 0.0115; t = 2.980; df = 12).
3. Discussion
In this study, using the 3xTg mice model of AD, we confirmed that the 8-month-old mice displayed higher levels of TNFα and lower levels of BDNF mRNA, as expected. Interestingly, the short ABA treatment (3 months) that started when the mice where 5 months old was sufficient to significantly decrease the TNFα mRNA levels in the 3xTg mice to control levels. However, the earlier ABA treatment started at 3 months old; thus, longer ABA exposure was required to observe an increase in the BDNF mRNA levels, suggesting that, in this model, the BDNF levels are regulated by inflammation. The relationship between BDNF and inflammation is bidirectional, and other studies have shown that reductions in inflammatory markers are a consequence of BDNF increase [15]. Because our previous studies [13] have shown that memory improvement by ABA in this model requires longer treatment, the present data are in line with the fact that lowering inflammation is not sufficient, but it is likely a mandatory step to rescue BDNF and cognitive function.
The mechanism by which ABA exerts its effects has not been completely elucidated, and some studies indicate that ABA may be a PPAR-γ agonist (brain tissue [16]; in T-cells [17]), or indirectly activate PPAR-γ expression via LANC-2 (microglia cells [18]). However, other studies indicate that ABA’s function may be PPAR-γ-independent (macrophages) [19]. This discrepancy may be due to the different cell types analyzed, and it requires further research to fully elucidate the ABA mechanism of action. Our data would be in accordance with ABA as a PPAR-γ agonist, as PPAR-γ can decrease the TNFα mRNA levels through NFkB activation and, in parallel, activate the BDNF promoter [20]. Moreover, this action is associated with improving brain injury, as demonstrated in a neuropsychiatric systemic lupus erythematosus [21], and with alleviating memory impairment in different rat models of neuroinflammation, either by LPS administration [22] or metabolic syndrome [23].
We also analyzed the IRS1 and IRS2 mRNA levels, given the importance of insulin resistance in AD. We found that the IRS2 (but not IRS1) mRNA levels were reduced in the 8-month-old 3xTg mice compared to the wild-type controls. This is concomitant with the reduction in spatial memory that we previously observed in this model [13]. These data agree with the hypothesis stating that IRS2 deficiency could be the link between type II diabetes [24] and AD (for review, see [25,26]). Moreover, these findings are in accordance with our previous studies in which we found that the IRS2 but not IRS1 gene expression was reduced in the brain of a rat metabolic syndrome model [23]. Interestingly, we also found that physiological aging is accompanied by a reduction in both the IRS1 and IRS2 levels [27], which agrees with the notion that AD accelerates aging pathology.
Although some investigations suggest that a reduction in IRS2 [28], and specifically in IGF1R/IRS2 signaling [29,30], can ameliorate AD, our current data are in line with other studies showing that IRS2 decline correlates with impaired spatial memory and emotional responses [31]. Furthermore, our findings agree with the pivotal role of IRS2 in NMDA-receptor-dependent synaptic transmission [32]. Moreover, the beneficial effects observed with ABA administration effects are supported by those reported for exendin (an anti-diabetic molecule) administration in a mice model of Alzheimer´s disease [33]. Furthermore, thiazolidinediones (TZD), which are known PPAR-γ agonists, also increase IRS2 [34] and IRS1 [35] expression, which supports that the action mechanism of ABA may be via PPAR-γ. However, there is no direct evidence to suggest that PPAR-γ directly activates the IRS2 promoter, but it indirectly interacts with transcription factors that bind and regulate the IRS2 promoter (for example, transcription factor Sp1) [36]. Likewise, the IRS1 promoter can be activated by Sp1 [37], and we indeed observed that ABA also increased the IRS1 gene expression, both in the wild-type and 3xTg mice, which could agree with the common activating mechanisms. Because 3xTg mice do not show brain IRS1 expression alteration compared to the wild type, it is unclear whether the ABA effect on IRS1 contributes to the improvement in the 3xTg mice memory function.
Interestingly, the short ABA treatment elevated the IRS1 and IRS2 mRNA levels in the wild-type mice, whereas 5 months of treatment was required to influence the 3xTg mice, suggesting a slower promoter activation in these mice.
Further studies are required to understand whether the key factor is the early intervention or the long exposure time. In conclusion, our data support that early ABA administration is a promising intervention to reduce inflammation-induced brain damage in Alzheimer’s models, effectively targeting the gene expression regulation of key molecules (BDNF, IRS1/2) for neuronal survival and optimal function.
4. Materials and Methods
4.1. Mice and Treatment
Mice were treated as in [13]. The procedures followed directive 86/609/EEC of the European Community on the protection of animals used for experimental and other scientific purposes. The experiments were approved by the Ethics Committee of the University Jaume I (approval number 2014/VSC/PEA00209). Briefly, control and transgenic mice were divided randomly into three experimental groups: ABA 3M and ABA 5M animals were supplemented with ABA (Fernandez-Rapado, Spain) in their drinking water (20 mg/L) for 3 or 5 months, respectively. Control groups were administered vehicle for 3 or 5 months (and pooled together for the analysis). Mice were fed ad libitum for 8 months. After the behavioral tests, mice were anesthetized with pentobarbital (120 mg/kg Eutanax, Barcelona, Spain) and sacrificed by quick decapitation. Brains were quickly removed, snap-frozen, and kept at −80 °C until RNA extraction.
4.2. RNA Extraction and Real-Time Quantitative Polymerase Chain Reaction
Total RNA was extracted from the mouse hippocampus (n = 5–9) using the RNeasy LipidTissue Mini Kit (Qiagen), according to the manufacturer’s protocol. We disrupted and homogenized fatty tissue in 1 mL QIAzol Lysis Reagent using the TissueRuptor. Then, the homogenate was incubated at room temperature (15–25 °C) for 5 min, and after this, 200 μL of chloroform was added. After incubating the sample for 2–3 min and centrifuging it (12,000× g for 15 min at 4 °C), the upper (aqueous phase) was transferred to a new tube with 1 volume of 70% of ethanol. An amount of 500 μL of the sample was transferred to an RNeasy Mini spin column in a 2 mL collection tube, and after centrifugation (15 s at 8000× g), the flow-through was discarded. This step was performed twice. An amount of 700 μL of Buffer RW1 was added to the RNeasy column and was centrifuged at 8000× g, and the flow-through was discarded. After, we added 500 μL Buffer RPE to the RNeasy column twice to dry membrane, and the flow-through was discarded. Finally, the RNA samples were resuspended in 40 μL of nuclease-free water.
RNA concentration and quantification of total RNA was performed using Thermo Scientific Nanodrop 2000c, with the OD260/OD280. Genomic DNA was removed using DNase I, RNase-free (Life Technologies, Carlsbad, CA, USA), for 30 min at 37 °C. The reaction was stopped by the addition of 1 μL of EDTA for 10 min at 65 °C. The first strand of cDNA was synthesized using the Prime- Script™ RT Reagent Kit (Perfect Real Time) (Takara Bio Inc., Shiga, Japan). For each reaction, 1 μg of RNA was used for reverse transcription, in a mixture of 4 μL of PrimeScript Buffer, 1 μL of PrimeScript RT, 1 μL of Oligo dT Primer (50 μM), and 1 μL of random primer (100 μM). Enzyme mix was adjusted to a final volume of 20 μL at room temperature. The mixture was incubated at 37 °C for 15 min and heated at 85 °C for 5 min to terminate the reaction. DNA concentration and quantification of total DNA was performed using Thermo Scientific Nanodrop 2000c, with the OD260/OD280, and the cDNA was subsequently stored at −20 °C.
Primers were designed using the Primer3 software tool (http://primer3.ut.ee/ (accessed on November 2018) (Table 1). RT-PCR was performed in a volume of 10 μL with 5 μL of Maxima SYBR Green/ROX qPCR Master Mix (2X) (Applied Biosystems Life Technologies, Carlsbad, CA, USA), 1 μL of primer, and 1 μL of cDNA. All PCR reactions were performed under the following conditions: initial cycle at 98 °C for 10 min, followed by 40 cycles at 98 °C for 10 s, 60 °C for 10 s, and 72 °C for 20 s. Gene expression in the hippocampus was quantified using a StepOnePlus Real-Time PCR system (Applied Biosystems Life Technologies). Each sample was tested in triplicate, and data were analyzed using the comparative critical threshold method, with the amount of target gene normalized to the housekeeping gene GAPDH. Relative gene expression was calculated using 2−ΔΔCt relative to control.
4.3. Statistics
Data were expressed as mean ± SEM using Graph Pad Prism version 8. Data were subjected to an unpaired Student t-test, two-tail, with a confidence of 95%, for the identification of the statistical differences between means. Gaussian distribution was monitored with the Shapiro–Wilk normality test.
Author Contributions
Conceptualization, A.M.S.-P.; methodology, V.E.-F.; software, L.A.B. and A.M.S.-P.; formal analysis, A.M.S.-P.; investigation, L.A.B., A.C.R., and V.E.-F.; data curation, V.E.-F., L.A.B., and A.M.S.-P.; writing—original draft preparation, A.M.S.-P., writing—review and editing, L.A.B. and A.C.R.; visualization, L.A.B.; supervision, A.M.S.-P.; project administration, A.M.S.-P.; funding acquisition, A.M.S.-P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by university grants, codes: UJI-B2018-01 and UJI-B2021-21 to A.M.S.-P.
Institutional Review Board Statement
The procedures followed directive 86/609/EEC of the European Community on the protection of animals used for experimental and other scientific purposes. The experiments were approved by the Ethics Committee of the University Jaume I (approval number 2014/VSC/PEA00209).
Informed Consent Statement
Not applicable.
Data Availability Statement
in process.
Acknowledgments
The authors want to thank the generous donations from the Association of Alzheimer Families, AFA, Castello.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Beason-Held, L.L.; Goh, J.O.; An, Y.; Kraut, M.A.; O’Brien, R.J.; Ferrucci, L.; Resnick, S.M. Changes in Brain Function Occur Years before the Onset of Cognitive Impairment. J. Neurosci. 2013, 33, 18008–18014. [Google Scholar] [CrossRef] [PubMed]
- Vinuesa, A.; Pomilio, C.; Gregosa, A.; Bentivegna, M.; Presa, J.; Bellotto, M.; Saravia, F.; Beauquis, J. Inflammation and Insulin Resistance as Risk Factors and Potential Therapeutic Targets for Alzheimer’s Disease. Front. Neurosci. 2021, 15, 653651. [Google Scholar] [CrossRef] [PubMed]
- Berlanga-Acosta, J.; Guillén-Nieto, G.; Rodríguez-Rodríguez, N.; Bringas-Vega, M.L.; García-del-Barco-Herrera, D.; Berlanga-Saez, J.O.; García-Ojalvo, A.; Valdés-Sosa, M.J.; Valdés-Sosa, P.A. Insulin Resistance at the Crossroad of Alzheimer Disease Pathology: A Review. Front. Endocrinol. 2020, 11, 560375. [Google Scholar] [CrossRef]
- Qin, W.; Li, F.; Jia, L.; Wang, Q.; Li, Y.; Wei, Y.; Li, Y.; Jin, H.; Jia, J. Phosphorylated Tau 181 Serum Levels Predict Alzheimer’s Disease in the Preclinical Stage. Front. Aging Neurosci. 2022, 14, 900773. [Google Scholar] [CrossRef]
- Sánchez-Sarasúa, S.; Moustafa, S.; García-Avilés, Á.; López-Climent, M.F.; Gómez-Cadenas, A.; Olucha-Bordonau, F.E.; Sánchez-Pérez, A.M. The Effect of Abscisic Acid Chronic Treatment on Neuroinflammatory Markers and Memory in a Rat Model of High-Fat Diet Induced Neuroinflammation. Nutr. Metab. 2016, 13, 73. [Google Scholar] [CrossRef]
- Moloney, A.M.; Griffin, R.J.; Timmons, S.; O’Connor, R.; Ravid, R.; O’Neill, C. Defects in IGF-1 Receptor, Insulin Receptor and IRS-1/2 in Alzheimer’s Disease Indicate Possible Resistance to IGF-1 and Insulin Signalling. Neurobiol. Aging 2010, 31, 224–243. [Google Scholar] [CrossRef]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; De La Monte, S.M. Impaired Insulin and Insulin-like Growth Factor Expression and Signaling Mechanisms in Alzheimer’s Disease—Is This Type 3 Diabetes? J. Alzheimer’s Dis. 2005, 7, 63–80. [Google Scholar] [CrossRef]
- Das, T.K.; Chakrabarti, S.K.; Zulkipli, I.N.; Hamid, M.R.W.A. Curcumin Ameliorates the Impaired Insulin Signaling Involved in the Pathogenesis of Alzheimer’s Disease in Rats. J. Alzheimers. Dis. Rep. 2019, 3, 59–70. [Google Scholar] [CrossRef]
- Edelmann, E.; Leßmann, V.; Brigadski, T. Pre- and Postsynaptic Twists in BDNF Secretion and Action in Synaptic Plasticity. Neuropharmacology 2014, 76 (Pt. C), 610–627. [Google Scholar] [CrossRef]
- Moosaie, F.; Mohammadi, S.; Saghazadeh, A.; Dehghani, F.; Id, N.R. Brain-Derived Neurotrophic Factor in Diabetes Mellitus : A Systematic Review and Meta- Analysis. PLoS ONE 2023, 18, e0268816. [Google Scholar] [CrossRef]
- Porter, G.A.; O’Connor, J.C. Brain-Derived Neurotrophic Factor and Inflammation in Depression: Pathogenic Partners in Crime? World J. Psychiatry 2022, 12, 77–97. [Google Scholar] [CrossRef]
- Gao, L.; Zhang, Y.; Sterling, K.; Song, W. Brain-Derived Neurotrophic Factor in Alzheimer’s Disease and Its Pharmaceutical Potential. Transl. Neurodegener. 2022, 11, 1–34. [Google Scholar] [CrossRef]
- Espinosa-Fernández, V.; Mañas-Ojeda, A.; Pacheco-Herrero, M.; Castro-Salazar, E.; Ros-Bernal, F.; Sánchez-Pérez, A.M. Early Intervention with ABA Prevents Neuroinflammation and Memory Impairment in a Triple Transgenic Mice Model of Alzheimer’s Disease. Behav. Brain Res. 2019, 374, 112106. [Google Scholar] [CrossRef]
- Choi, S.H.; Bylykbashi, E.; Chatila, Z.K.; Lee, S.W.; Pulli, B.; Clemenson, G.D.; Kim, E.; Rompala, A.; Oram, M.K.; Aronson, J.; et al. Exercise on Cognition in an Alzheimer’s Mouse Model. Science 2018, 361, eaan8821. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Zhu, X.; Si, J. Angelica Polysaccharide Ameliorates Memory Impairment in Alzheimer’s Disease Rat through Activating BDNF/TrkB/CREB Pathway. Exp. Biol. Med. 2020, 245, 1–10. [Google Scholar] [CrossRef]
- Kooshki, R.; Anaeigoudari, A.; Abbasnejad, M.; Askari-Zahabi, K.; Esmaeili-Mahani, S. Abscisic Acid Interplays with PPARγ Receptors and Ameliorates Diabetes-Induced Cognitive Deficits in Rats. Avicenna J. Phytomed. 2021, 11, 247–257. [Google Scholar] [CrossRef]
- Guri, A.J.; Evans, N.P.; Hontecillas, R.; Bassaganya-Riera, J. T Cell PPAR γ Is Required for the Anti-Inflammatory Efficacy of Abscisic Acid against Experimental IBD. J. Nutr. Biochem. 2011, 22, 812–819. [Google Scholar] [CrossRef] [PubMed]
- Maixner, D.W.; Christy, D.; Kong, L.; Viatchenko-Karpinski, V.; Horner, K.A.; Hooks, S.B.; Weng, H.R. Phytohormone Abscisic Acid Ameliorates Neuropathic Pain via Regulating LANCL2 Protein Abundance and Glial Activation at the Spinal Cord. Mol. Pain 2022, 18, 1–18. [Google Scholar] [CrossRef]
- Bassaganya-Riera, J.; Guri, A.J.; Lu, P.; Climent, M.; Carbo, A.; Sobral, B.W.; Horne, W.T.; Lewis, S.N.; Bevan, D.R.; Hontecillas, R. Abscisic Acid Regulates Inflammation via Ligand-Binding Domain-Independent Activation of Peroxisome Proliferator-Activated Receptor γ. J. Biol. Chem. 2011, 286, 2504–2516. [Google Scholar] [CrossRef]
- Kariharan, T.; Nanayakkara, G.; Parameshwaran, K.; Bagasrawala, I.; Ahuja, M.; Abdel-Rahman, E.; Amin, A.T.; Dhanasekaran, M.; Suppiramaniam, V.; Amin, R.H. Central Activation of PPAR-Gamma Ameliorates Diabetes Induced Cognitive Dysfunction and Improves BDNF Expression. Neurobiol. Aging 2015, 36, 1451–1461. [Google Scholar] [CrossRef]
- Li, X.; Xu, S.; Liu, J.; Zhao, Y.; Han, H.; Li, X.; Wang, Y. Treatment with 1,25-Dihydroxyvitamin D3 Delays Choroid Plexus Infiltration and BCSFB Injury in MRL/Lpr Mice Coinciding with Activation of the PPARγ/NF-ΚB/TNF-α Pathway and Suppression of TGF-β/Smad Signaling. Inflammation 2022, 46, 556–572. [Google Scholar] [CrossRef] [PubMed]
- Beheshti, F.; Hosseini, M.; Hashemzehi, M.; Soukhtanloo, M.; Khazaei, M.; Naser Shafei, M. The Effects of PPAR-γ Agonist Pioglitazone on Hippocampal Cytokines, Brain-Derived Neurotrophic Factor, Memory Impairment, and Oxidative Stress Status in Lipopolysaccharidetreated Rats. Iran J. Basic Med. Sci. 2019, 22, 940–948. [Google Scholar] [CrossRef] [PubMed]
- Ribes-Navarro, A.; Atef, M.; Sánchez-Sarasúa, S.; Beltrán-Bretones, M.T.; Olucha-Bordonau, F.; Sánchez-Pérez, A.M. Abscisic Acid Supplementation Rescues High Fat Diet-Induced Alterations in Hippocampal Inflammation and IRSs Expression. Mol. Neurobiol. 2019, 56, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Brady, M.J. IRS2 Takes Center Stage in the Development of Type 2 Diabetes. J. Clin. Investig. 2004, 114, 886–888. [Google Scholar] [CrossRef]
- Athanasaki, A.; Melanis, K.; Tsantzali, I.; Stefanou, M.I.; Ntymenou, S.; Paraskevas, S.G.; Kalamatianos, T.; Boutati, E.; Lambadiari, V.; Voumvourakis, K.I.; et al. Type 2 Diabetes Mellitus as a Risk Factor for Alzheimer’s Disease: Review and Meta-Analysis. Biomedicines 2022, 10, 778. [Google Scholar] [CrossRef] [PubMed]
- Rojas, M.; Chávez-Castillo, M.; Bautista, J.; Ortega, Á.; Nava, M.; Salazar, J.; Díaz-Camargo, E.; Medina, O.; Rojas-Quintero, J.; Bermúdez, V. Alzheimer’s Disease and Type 2 Diabetes Mellitus: Pathophysiologic and Pharmacotherapeutics Links. World J. Diabetes 2021, 12, 745–766. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Sarasúa, S.; Meseguer-Beltrán, M.; García-Díaz, C.; Beltrán-Bretones, M.T.; ElMlili, N.; Sánchez-Pérez, A.M. IRS1 Expression in Hippocampus Is Age-Dependent and Is Required for Mature Spine Maintenance and Neuritogenesis. Mol. Cell. Neurosci. 2022, 118, 103693. [Google Scholar] [CrossRef]
- Killick, R.; Scales, G.; Leroy, K.; Causevic, M.; Hooper, C.; Irvine, E.E.; Choudhury, A.I.; Drinkwater, L.; Kerr, F.; Al-Qassab, H.; et al. Deletion of Irs2 Reduces Amyloid Deposition and Rescues Behavioural Deficits in APP Transgenic Mice. Biochem. Biophys. Res. Commun. 2009, 386, 257–262. [Google Scholar] [CrossRef]
- Tanokashira, D.; Fukuokaya, W.; Taguchi, A. Involvement of Insulin Receptor Substrates in Cognitive Impairment and Alzheimer’s Disease. Neural Regen. Res. 2019, 14, 1330–1334. [Google Scholar] [CrossRef]
- Freude, S.; Hettich, M.M.; Schumann, C.; Stöhr, O.; Koch, L.; Köhler, C.; Udelhoven, M.; Leeser, U.; Müller, M.; Kubota, N.; et al. Neuronal IGF-1 Resistance Reduces Aβ Accumulation and Protects against Premature Death in a Model of Alzheimer’s Disease. FASEB J. 2009, 23, 3315–3324. [Google Scholar] [CrossRef] [PubMed]
- Tanokashira, D.; Wang, W.; Maruyama, M.; Kuroiwa, C.; White, M.F.; Taguchi, A. Irs2 Deficiency Alters Hippocampus-Associated Behaviors during Young Adulthood. Biochem. Biophys. Res. Commun. 2021, 559, 148–154. [Google Scholar] [CrossRef]
- Costello, D.A.; Claret, M.; Al-Qassab, H.; Plattner, F.; Irvine, E.E.; Choudhury, A.I.; Giese, K.P.; Withers, D.J.; Pedarzani, P. Brain Deletion of Insulin Receptor Substrate 2 Disrupts Hippocampal Synaptic Plasticity and Metaplasticity. PLoS ONE 2012, 7, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Bomfim, T.R.; Forny-germano, L.; Sathler, L.B.; Brito-moreira, J.; Houzel, J.; Decker, H.; Silverman, M.A.; Kazi, H.; Melo, H.M.; Mcclean, P.L.; et al. An Anti-Diabetes Agent Protects the Mouse Brain from Defective Insulin Signaling Caused by Alzheimer’s Disease– Associated Aβ Oligomers. J. Clin. Investig. 2012, 122, 1339–1353. [Google Scholar] [CrossRef] [PubMed]
- Smith, U.; Gogg, S.; Johansson, A.; Olausson, T.; Rotter, V.; Svalstedt, B. Thiazolidinediones (PPARγ Agonists) but Not PPAR α Agonists Increase IRS-2 Gene Expression in 3T3-L1 and Human Adipocytes 1. FASEB J. 2001, 15, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Hammarstedt, A.; Smith, U. Thiazolidinediones (PPARγ Ligands) Increase IRS-1, UCP-2 and C/EBPα Expression, but Not Transdifferentiation, in L6 Muscle Cells. Diabetologia 2003, 46, 48–52. [Google Scholar] [CrossRef]
- Singh, A.K.; Battu, A.; Mohareer, K.; Hasnain, S.E.; Ehtesham, N.Z. Transcription of Human Resistin Gene Involves an Interaction of Sp1 with Peroxisome Proliferator- Activating Receptor Gamma (PPARγ). PLoS ONE 2010, 5, e9912. [Google Scholar] [CrossRef]
- Panno, M.L.; Mauro, L.; Marsico, S.; Bellizzi, D.; Rizza, P.; Morelli, C.; Salerno, M.; Giordano, F.; Ando, S. Evidence That the Mouse Insulin Receptor Substrate-1 Belongs to the Gene Family on Which the Promoter Is Activated by Estrogen Receptor α through Its Interaction with Sp1. J. Mol. Endocrinol. 2006, 36, 91–105. [Google Scholar] [CrossRef]
Figure 1.
ABA treatment (A) reduces hippocampal TNF and (B) increases BDNF expression as measured by qPCR. Data are represented as the mean ± of at least 5–9 independent subjects in triplicate. Data were analyzed by Student t-test (* p < 0.05, ** p < 0.01, and **** p < 0.0001 for ABA effect; ## p < 0.01 and ### p < 0.001 for genotype effect).
Figure 1.
ABA treatment (A) reduces hippocampal TNF and (B) increases BDNF expression as measured by qPCR. Data are represented as the mean ± of at least 5–9 independent subjects in triplicate. Data were analyzed by Student t-test (* p < 0.05, ** p < 0.01, and **** p < 0.0001 for ABA effect; ## p < 0.01 and ### p < 0.001 for genotype effect).
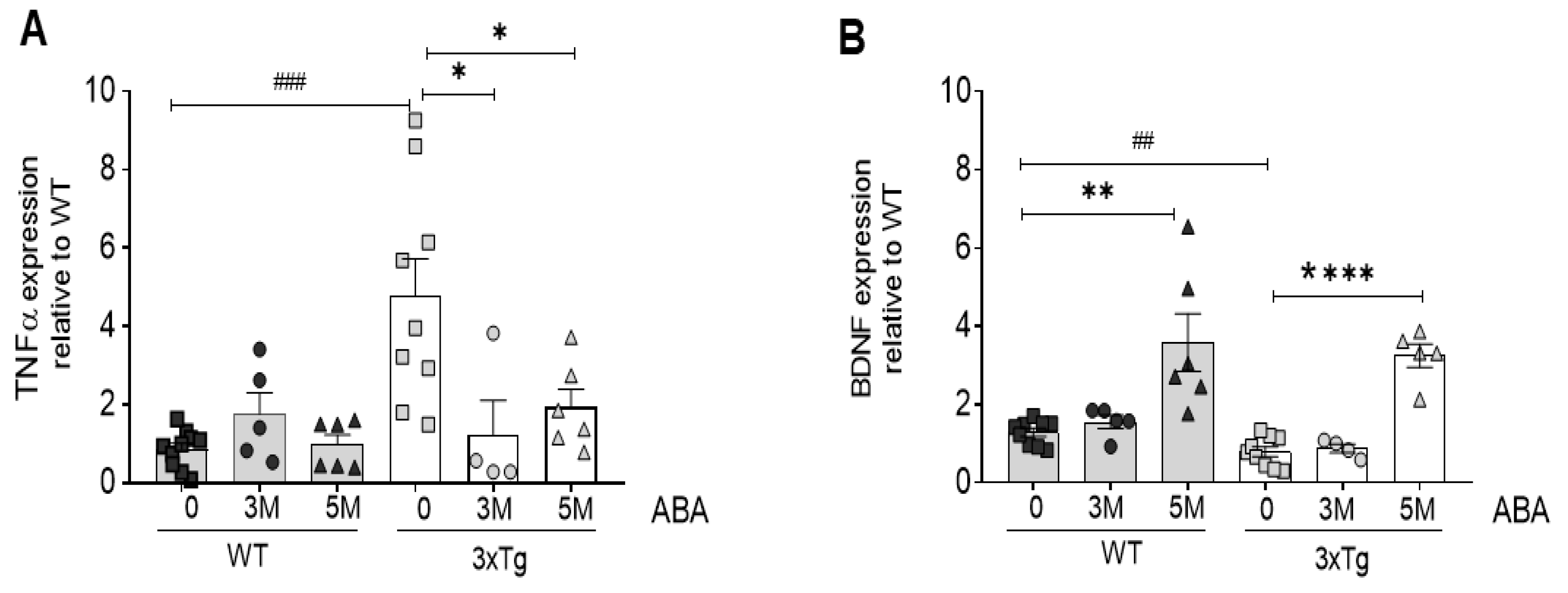
Figure 2.
ABA administration increases IRS mRNA expression in hippocampi as measured by qPCR. (A) IRS1 expression increases in WT mice as soon as 3 months of ABA treatment, but in 3xTg mice, only at 5 months. (B) IRS2 increases in WT animals at 3 months of treatment in WT animals. Data are represented as the mean ± of at least 5–9 independent subjects in triplicate. Data were analyzed by Student t-test (* p < 0.05 and ** p < 0.01 for ABA effect, and ## p < 0.01 for genotype effect).
Figure 2.
ABA administration increases IRS mRNA expression in hippocampi as measured by qPCR. (A) IRS1 expression increases in WT mice as soon as 3 months of ABA treatment, but in 3xTg mice, only at 5 months. (B) IRS2 increases in WT animals at 3 months of treatment in WT animals. Data are represented as the mean ± of at least 5–9 independent subjects in triplicate. Data were analyzed by Student t-test (* p < 0.05 and ** p < 0.01 for ABA effect, and ## p < 0.01 for genotype effect).
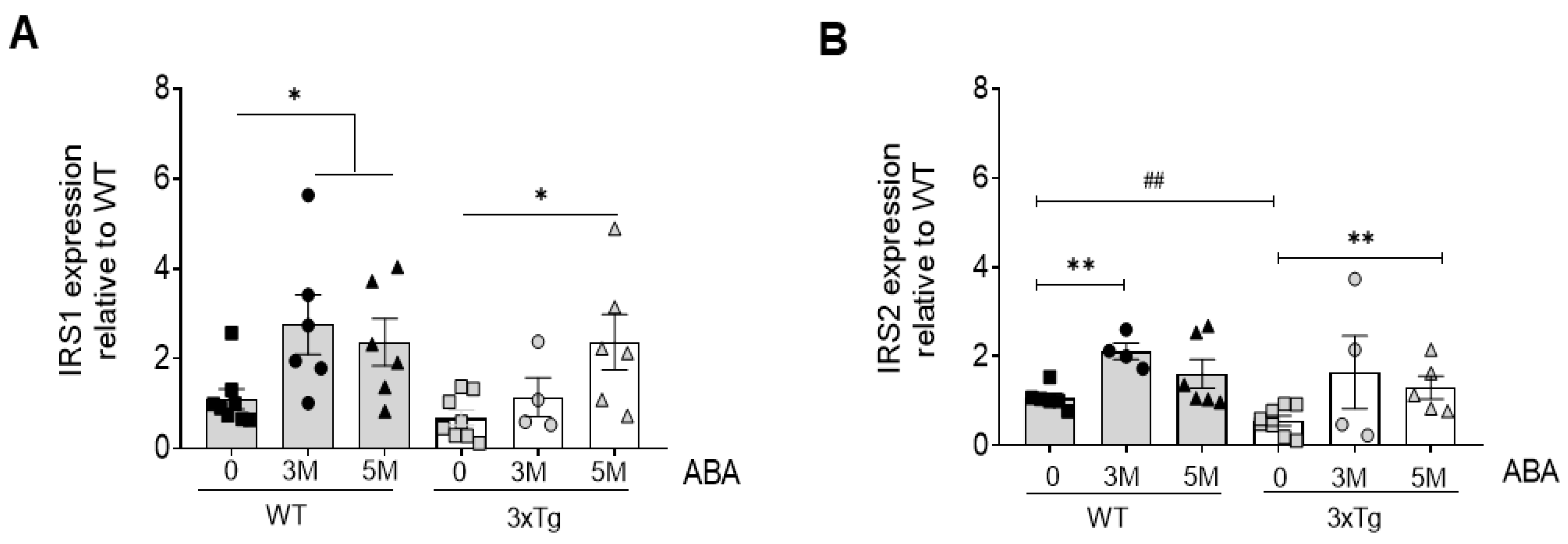

Table 1.
Primer sequences [23].
Table 1.
Primer sequences [23].
| Gene | Forward Primer (5′-3′) | Reverse Primer (5′-3′) |
|---|---|---|
| IRS1 | CCTGACATTGGAGGTGGGTC | TGGGGATCTTCTGGGCCATA |
| IRS2 | GCAGCCAGGAGACAAGAACT | AGCGCTTCACTCTTTCACGA |
| BDNF | TACCTGGATGCCGCAAACAT | AGTTGGCCTTTGGATACCGG |
| TNFα | GACCCTCACACTCAGATCA | TGCTACGACGTGGGCTACG |
| GAPDH | TGCCCCCATGTTTGTGATG | TGGTGGTGCAGGATGCATT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



