Submitted:
15 June 2023
Posted:
15 June 2023
You are already at the latest version
Abstract
Activated platelets are involved in blood coagulation by exposing phosphatidylserine (PS), which serves as a substrate for assembling coagulation complexes. Platelets accelerate fibrin formation and thrombin generation, two final reactions of the coagulation cascade. We investigated the effects of antiplatelet drugs on platelet impact in these reactions and platelet ability to expose PS. Washed human platelets were incubated with acetylsalicylic acid (ASA), ticagrelor, ASA in combination with ticagrelor, ruciromab (glycoprotein IIb-IIIa antagonist) or prostaglandin E1 (PGE1). Platelets were not activated or activated by collagen, sedimented in multiwell plates and plasma was added after supernatant removal. Fibrin formation (clotting) was monitored in a recalcification assay by light absorbance and thrombin generation in a fluorogenic test. PS exposure was assessed by annexin V staining using flow cytometry. Ticagrelor (alone and in combination with ASA), ruciromab and PGE1, but not ASA prolonged the lag phase and decreased the maximum rate of plasma clotting, and decreased the peak and maximum rate of thrombin generation. Inhibition was observed when platelets were not treated with exogenous agonists (activation by endogenous thrombin) and pretreated with collagen. Ticagrelor (alone and in combination with ASA), ruciromab and PGE1, but not ASA decreased PS exposure on washed platelets activated by thrombin and by thrombin + collagen. PS exposure on activated platelets in whole blood was lower in patients with acute coronary syndrome receiving ticagrelor + ASA in comparison with donors free of medications. These results indicate that antiplatelet drugs are able to suppress platelet coagulation activity not only in vitro but also after administration to patients.
Keywords:
platelets
; blood coagulation
; fibrin
; thrombin generation
; antiplatelet drugs
; acetylsalicylic acid
; ticagrelor
; glycoprotein IIb-IIIa antagonists
Introduction
Platelets are involved in blood coagulation by activation-dependent exposure of negatively charged phosphatidylserine (PS) which serves as a substrate for assembling coagulating complexes. [1,2,3,4]. Platelets are able to accelerate thrombin generation and fibrin formation, two final reactions of coagulation cascade [1,2,3,4,5].
Antiplatelet drugs or antiaggregants are widely used for prevention and treatment of thrombotic diseases, including myocardial infarction and unstable angina (acute coronary syndrome, ACS), and ischemic stroke. Antiplatelet drugs are divided into several classes depending on the mechanism of their action: (1) acetylsalicylic acid (ASA), an inhibitor of thromboxane A2 (TXA2) synthesis [6], (2) antagonists of P2Y12 ADP receptors (clopidogrel, prasugrel and ticagrelor) [7], (3) antagonists (blockers) of platelet fibrinogen receptor, glycoprotein (GP) IIb-IIIa, (abciximab, eptifibatide, tirofiban and ruciromab (Monafram®, used in Russian Federation only) [8,9,10]. Efficacy of these drugs is routinely assessed by their ability to suppress platelet aggregation. ASA and P2Y12 antagonists effectively inhibit platelet aggregation induced by arachidonic acid (precursor of platelet TXA2) and by ADP respectively and produce milder effects on aggregation induced by strong agonists such as thrombin or TRAP (thrombin receptor activating peptide) and collagen [6,7]. GP IIb-IIIa antagonists block final reaction in platelet aggregation, formation of fibrinogen bridges between aggregating platelets, thus completely inhibiting all types of aggregation response [8,9,10]. Prostaglandin Е1 (PGE1) and prostacyclin (activators of adenylate cyclase) used primarily as vasodilating agents, also effectively inhibit platelet activation and aggregation [11].
The effects of all antiplatelet drugs on platelet aggregation has been systematically investigated in numerous experimental and clinical studies [6,7,8,9,10,11]. However, their effects on platelet coagulation activity are not thoroughly explored, and sometimes quite contradictory results are reported. It is still unclear how antiplatelet drugs influence platelet-dependent fibrin formation. There is evidence that antiplatelet drugs inhibit thrombin generation in the presence of platelets [12,13,14,15,16]. However for ASA and P2Y12 antagonists this effect was observed predominantly in the presence of specific agonists (arachidonic acid and ADP respectively) [12,13,14] and was depended on the presence and concentration of activated factor VII [13]. PS exposure on activated platelets was partially inhibited by P2Y12 receptor antagonists [17,18], while for different GP IIb-IIIa antagonists inhibitory effects [16,19,20,21], absence of the effects [22] and even paradox stimulatory [20,21,23] effects were demonstrated, while ASA has no effects in vitro [18] being slightly effective in patients [24].
In this study we explored the effects of all types of antiplatelet drugs (ASA, ticagrelor, GP II-IIIa antagonist and PGE1) on platelet-dependent thrombin generation, fibrin formation and on the ability of activated platelets to expose PS
Materials and Methods
Donors and Patients
Blood was collected from: (1) healthy volunteers free of any medication for at least two weeks before blood donation, and (2) patients with ACS (4-6 days after disease onset) receiving dual antiplatelet therapy, ASA (100 mg daily) + ticagrelor (90 mg x 2 daily). All patients were treated at Chazov National Medical Research Center of Cardiology. All participants signed informed concent and the study was approved by a local ethic committee of Chazov National Medical Research Center of Cardiology (protocol # 274, November 9 2021).
Blood Collection
For preparation of washed platelets blood was collected in acid citrate dextrose (ACD, 65 mM citric acid, 85 mM sodium citrate, 2% dextrose) at 6/1 blood/anticoagulant ratio. For the measurement of PS exposure in whole blood and platelet aggregation in platelet rich plasma (PRP) blood was collected into 3.8% sodium citrate at 9/1 blood/anticoagulant ratio.
Platelet Counting
Platelet count in whole blood, in PRP and in suspension of washed platelets (see below) was determined in an Abacus Junior B hematological analyzer (Diatron Ltd. Austria).
Washed Platelets
Washed platelets were prepared from ACD anticoagulated blood as described elsewhere [25] and resuspended in Tyrode/HEPES solution (137 mM NaCl, 2.7 mM KCl, 0.36 mM NaH2PO4, 0.1% dextrose, 2 mM CaCl2, 1 mM MgCl2, 0.35% BSA, 5 mM HEPES, pH 7.35) at 0.5 x 108/ml.
Platelet Aggregation
All antiplatelet agents used in the study were tested for their ability to inhibit platelet aggregation. Platelet aggregation in PRP was recorded using standard turbidimetric method in a BIOLA aggregation analyzer (BIOLA, Russia ) as described previously [26]. PRP was left intact or supplemented with 0.2 mM ASA (Sigma-Aldrich, Saint Louis, MO), 1 µM ticagrelor (Sigma-Aldrich, Saint Louis, MO USA), 20 µg/ml ruciromab (F(ab)2 fragment of ant-GP IIb-IIIa monoclonal antibody FRaMon (CRC64)) (Mona Ltd, Moscow) or 1 µg/ml PGE1 (Sigma-Aldrich, Saint Louis, MO) and incubated for 5 min at 37oC. For ASA testing aggregation was induced by 1 mM arachidonic acid (Santa Cruz Biotechnology Heidelberg, Germany), for ticagrelor testing, by 20 µM ADP (AppliChem GmbH, Darmstadt, Germany) and for ruciromab and PGE1 testing, by 20 µM TRAP (sequence SFLLRN, provided by Dr. M.D. Ovchinnikov, Chazov National Medical Research Center of Cardiology). All test antiplatelet agents inhibited platelet aggregation induced by the corresponding agonists: ASA, PGE1 and ruciromab completely and ticagrelor by about 70% (Suppl. Fig. S1).
Plasma Recalcification Assay
Plasma recalcification assay in the presence of platelets was performed essentially as described previosly [4]. Washed platelets at 0.5 x 108/ml were incubated for 5 min at room temperature without any additions (control) or in the presence 0.2 mM ASA, 1 µM ticagrelor, 0.2 mM ASA + 1 µM ticagrelor, 20 µg/ml ruciromab or 1 µg/ml PGE1. After incubation platelets were left intact or activated by 10 µg/ml collagen (Revohem, Sysmex, UK) for 5 min at room temperature. Intact or activated platelets (100 µl, 0.5 x 107 per well) were added to 96-well cell culture flat bottom plates (Costar, Kennebunk, ME, Cat. # 3599) and sedimented for 5 min at 1500 g. After removal of the supernatant from sedimented platelets 50 µl CaCl2-free Tyrode/HEPES solution supplemented with 150 μg/ml corn trypsin inhibitor (provided by Dr. G.V. Shekhvatova, Institute of Protein, Pushchino, Russia) for partial inhibition of contact activation and 50 µl citrated plasma (pooled plasma from 3-4 donors depleted of endogenous microparticles by centrifugation for 90 min at 20000 g) was added into the wells. Control samples contained no platelets. Plasma was recalcified by adding 50 μl 25 mM CaCl2 (Diagnostica Stago, France). Fibrin formation (plasma clotting) was evaluated by changes in light absorbance at 450 nm (A450) for 60 min at 25°C in a Thermo Scientific Multiscan Go plate spectrophotometer (Thermo Fisher Scientific, Finland). The lag phase (time to reach 5% maximum A450 increase, min) and the maximum rate of fibrin formation (Vmax, maximum increase in A450 per min as percentage of total increase after 60 min (%A450/min) were determined.
Thrombin Generation Test
Thrombin generation test (TGT) in the presence of platelets was performed as described earlier [4]. Platelets were prepared and sedimented on the bottom of multiwell plates in the same way as for plasma recalcification assay (see above). All reagents used in TGT were from Diagnostica Stago (France). After removal of the supernatant from sedimented platelets 80 µl citrated plasma and 20 µl trigger PRP reagent (tissue factor (TF) and minimum amount of phospholipids) were added into the wells and incubated for 10 min, 37o С. Control samples contained no platelets. The reaction was started by 20 µl Fluo-Buffer (thrombin fluorogenic substrate and CaCl2). Final concentration of TF was 0.5 pM. In control samples thrombin generation was monitored with the use of “PPP reagent” that provided a final concentration of 5 pM for tissue factor and 4 µM for phospholipids. For calibration of fluorescent signal Thrombin Calibrator (750 nM Thrombin-α2-macroglobulin complex) was added instead of trigger reagent. Measurements were performed in a Fluoroscan Ascent plate fluorimeter (ThermoLab Systems, Finland). Thrombin generation curves were analyzed using Thrombinoscope software (Thrombinoscope BV, Netherlands). The following parameters were determined: lag phase, endogenous thrombin potential (ETP), peak, Vмакс.
Phosphatidylserine Exposure Washed Platelets
Phosphatidylserine (PS) exposure on the surface of activated washed platelets was evaluated by flow cytometry using staining with Annexin V-FITC essentially as described previously [4].
Washed platelets (0.5 x 108/ml) were left intact or activated for 15-min at 37oC without stirring by 10 U/ml human thrombin (Haematologic Technologies, Inc., Essex Junction, VT), or by thrombin in combination with 10 µg/ml collagen. Platelet suspension (50 µl) was then supplemented with 5 µl annexin V-FITC (BD Biosciences, San Jose, CA) and 3 µl CD42b-APC (BD Pharmingen, San Diego, CA) and incubated for 20 min at room temperature in the dark. Negative control contained no annexin V-FITC. Then Tyrode/HEPES solution (250 µl) was added and the samples were analyzed in a FACSCanto II flow cytometer using DivaTM Software (BD Biosciences, San Jose, CA). Platelets were gated according to their size and CD42b-positive staining. The percentage of annexin V-FITC (PS) positive platelets was calculated in comparison with negative control (platelet without annexin V-FITC).
Phosphatidylserine Exposure Whole Blood
Phosphatidylserine (PS) exposure on the surface of activated platelets was also measured in the whole blood. Blood from healthy volunteers and patients with ACS receiving dual antiplatelet therapy (ASA + ticagrelor) was collected into sodium citrate (see above). In order to prevent blood coagulation after the addition of Ca+ containing Tyrode/HEPES solution (calcium was required for effective PS exposure) blood was supplemented with thrombin inhibitor PPACK (D-Phe-Pro-Arg-clormetylketone) (Chinese peptide company, China) at a final concentration of 100 µM. Blood was diluted 20 fold, and platelets were not activated or activated by 20 µM TRAP and by 20 µM TRAP + 10 µg/ml collagen for 15-min at 37oC without stirring. Then, probes for flow cytometry were prepared and analyzed in the same way as for washed platelets.
Statistics
Statistical analysis was performed using Statistica 12 software (StatSoft. Inc. Tulsa, OK). Most of analyzed variables fit normal distribution (Shapiro-Wilk’s test). Data were expressed as means ± standard deviations (SD). The significance of differences was evaluated using paired t-test or t-test for means (as indicated).
Results
Effects of Antiplatelet Drugs on Platelet-Dependent Fibrin Formation
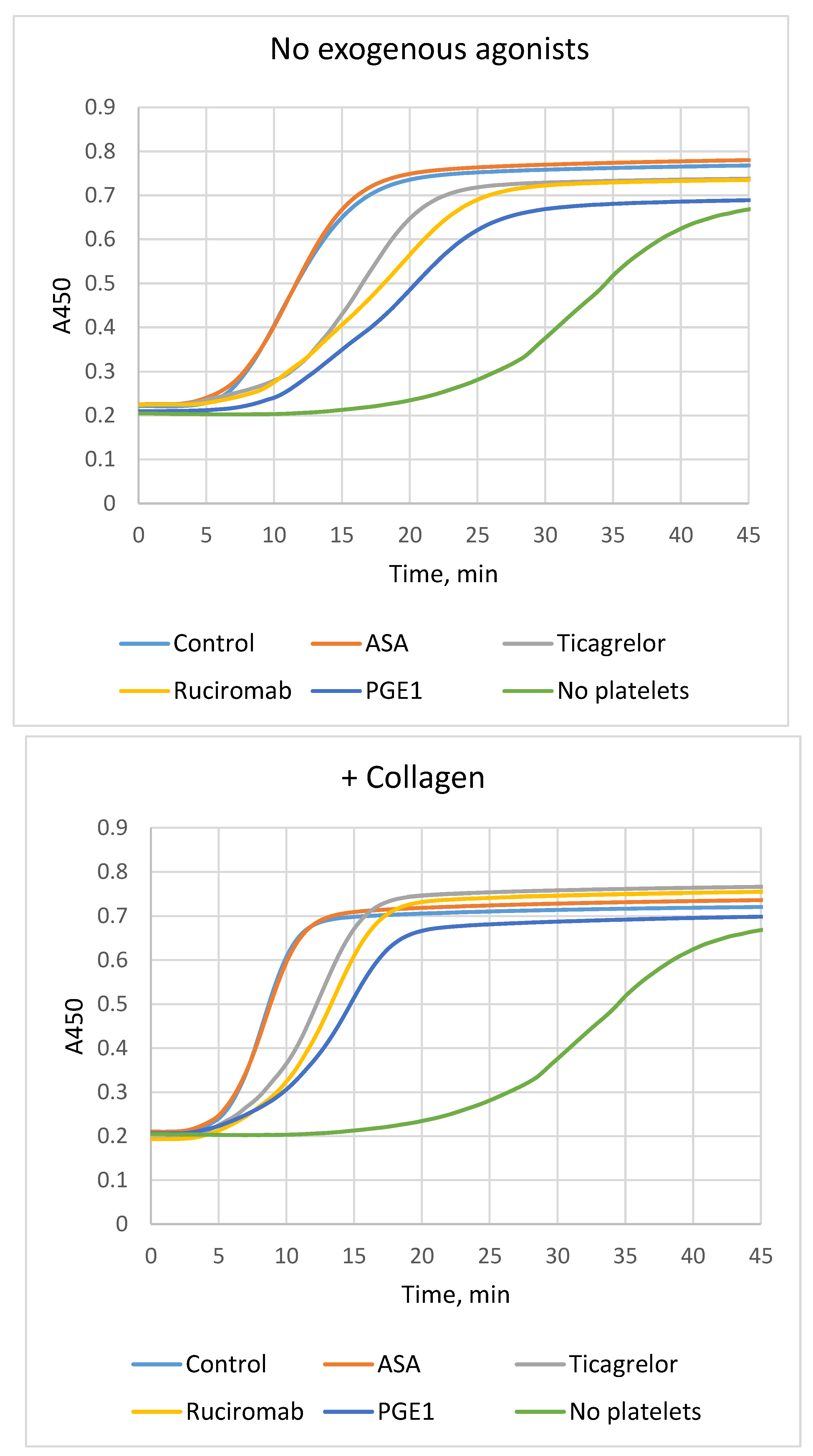
Fibrin formation in blood plasma (plasma clotting) was evaluated using modified recalcification assay [4]. Platelets accelerated fibrin formation shortening of the lag phase and increasing Vmax V maxin comparison with platelet-free samples. Platelet activation by collagen in addition to exogenous thrombin formed in plasma provided further acceleration (Fig. 1, Table 1). Ticagrelor, ruciromab and PGE1, partially reduced the rate of fibrin formation (prolongation of the lag phase and Vmax decrease). In contrast to other antiplatelet agents, ASA had no effect, both alone and in combination with ticagrelor. Similar results were obtained with platelets not treated with exogenous agonists (activation by endogenous thrombin) and with platelets pretreated with collagen. (Fig. 1, Table 1).
Figure 1.
Effects of antiplatelet drugs on fibrin formation (plasma clotting). Plasma recalcification assay was performed in the presence of platelets not treated with antiplatelet drugs (“Control”) or treated with 0.2 mM ASA (“ASA”), 1 µM ticagrelor (Ticagrelor), 20 µg/ml ruciromab, 1 µg/ml PGE1, or without platelets (“No platelets”). Platelets were not activated with exogenous agonists (upper panel, “No exogenous agonists”), or preactivated with 10 µg/ml collagen (low panel, “+ Collagen). Curves with ticagrelor combined with ASA were the same as with ticagrelor alone and are not shown. Representative results from 6-15 experiments. Statistical data are presented in Table 1.
Figure 1.
Effects of antiplatelet drugs on fibrin formation (plasma clotting). Plasma recalcification assay was performed in the presence of platelets not treated with antiplatelet drugs (“Control”) or treated with 0.2 mM ASA (“ASA”), 1 µM ticagrelor (Ticagrelor), 20 µg/ml ruciromab, 1 µg/ml PGE1, or without platelets (“No platelets”). Platelets were not activated with exogenous agonists (upper panel, “No exogenous agonists”), or preactivated with 10 µg/ml collagen (low panel, “+ Collagen). Curves with ticagrelor combined with ASA were the same as with ticagrelor alone and are not shown. Representative results from 6-15 experiments. Statistical data are presented in Table 1.
Effects of Antiplatelet Drugs on Platelet-Dependent Thrombin Generation
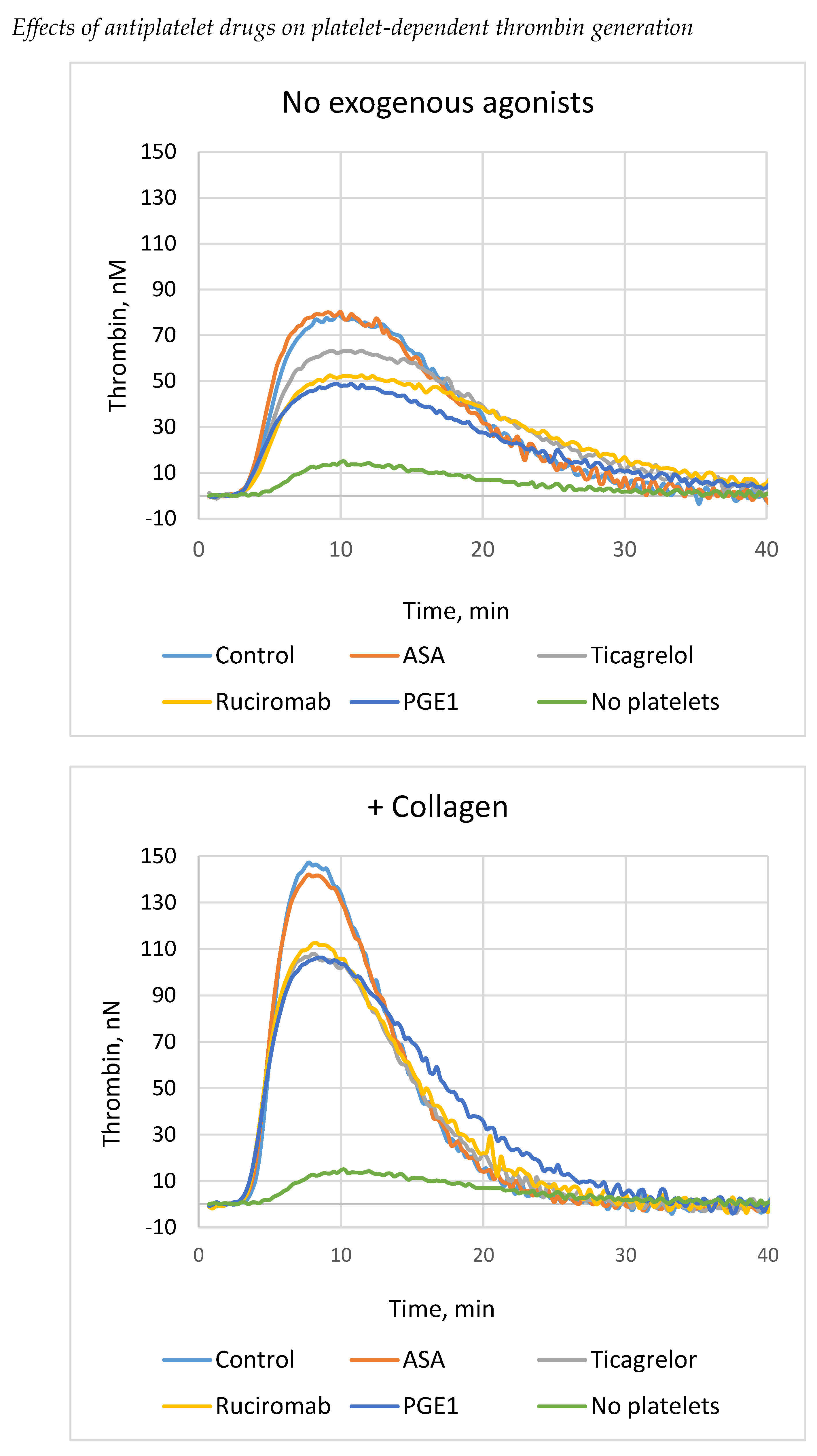
Thrombin generation in blood plasma was measured using TGT protocol adapted to assess platelet contribution (low TF, and minimum phospholipids). Platelets were prepared in the same way as for recalcification assay. Platelets considerably increased thrombin generation in comparison with platelet-free samples, the stimulatory effect being enhanced after their activation with collagen (Fig. 2, Table 2). Ticagrelor, ruciromab and PGE1 significantly decreased the peak and Vmax of thrombin generation, and produced negligible effect on the lag phase and the ETP (only PGE1 slightly shortened the lag phase and decreased the ETP in samples without collagen). Thus, these antiplatelet drugs inhibited primarily platelet-dependent acceleration of thrombin generation but do not affect duration of the initiation phase (presumably depended on TF) and the total amount of generated thrombin. Again, as in a recalcification assay, ASA did not change any parameters of thrombin generation and failed to potentiate effects of ticagrelor. Similar results were obtained with platelets not treated with exogenous agonists (activation by endogenous thrombin) and with platelets pretreated with collagen. (Fig. 2, Table 2.).
Figure 2.
Effects of antiplatelet drugs on thrombin generation.. TGT test was performed in the presence of platelets not treated with antiplatelet drugs (“Control”) or treated with 0.2 mM ASA (“ASA”), 1 µM ticagrelor (Ticagrelor), 20 µg/ml ruciromab, 1 µg/ml PGE1, or without platelets (“No platelets”). Platelets were not activated with exogenous agonists (upper panel, “No exogenous agonists”), or preactivated with 10 µg/ml collagen (low panel, “+ Collagen). Curves with ticagrelor combined with ASA were the same as with ticagrelor alone and are not shown. Representative results from 5-9 experiments. Statistical data are presented in Table 2.
Figure 2.
Effects of antiplatelet drugs on thrombin generation.. TGT test was performed in the presence of platelets not treated with antiplatelet drugs (“Control”) or treated with 0.2 mM ASA (“ASA”), 1 µM ticagrelor (Ticagrelor), 20 µg/ml ruciromab, 1 µg/ml PGE1, or without platelets (“No platelets”). Platelets were not activated with exogenous agonists (upper panel, “No exogenous agonists”), or preactivated with 10 µg/ml collagen (low panel, “+ Collagen). Curves with ticagrelor combined with ASA were the same as with ticagrelor alone and are not shown. Representative results from 5-9 experiments. Statistical data are presented in Table 2.
Effects of Antiplatelet Drugs on PS Exposure
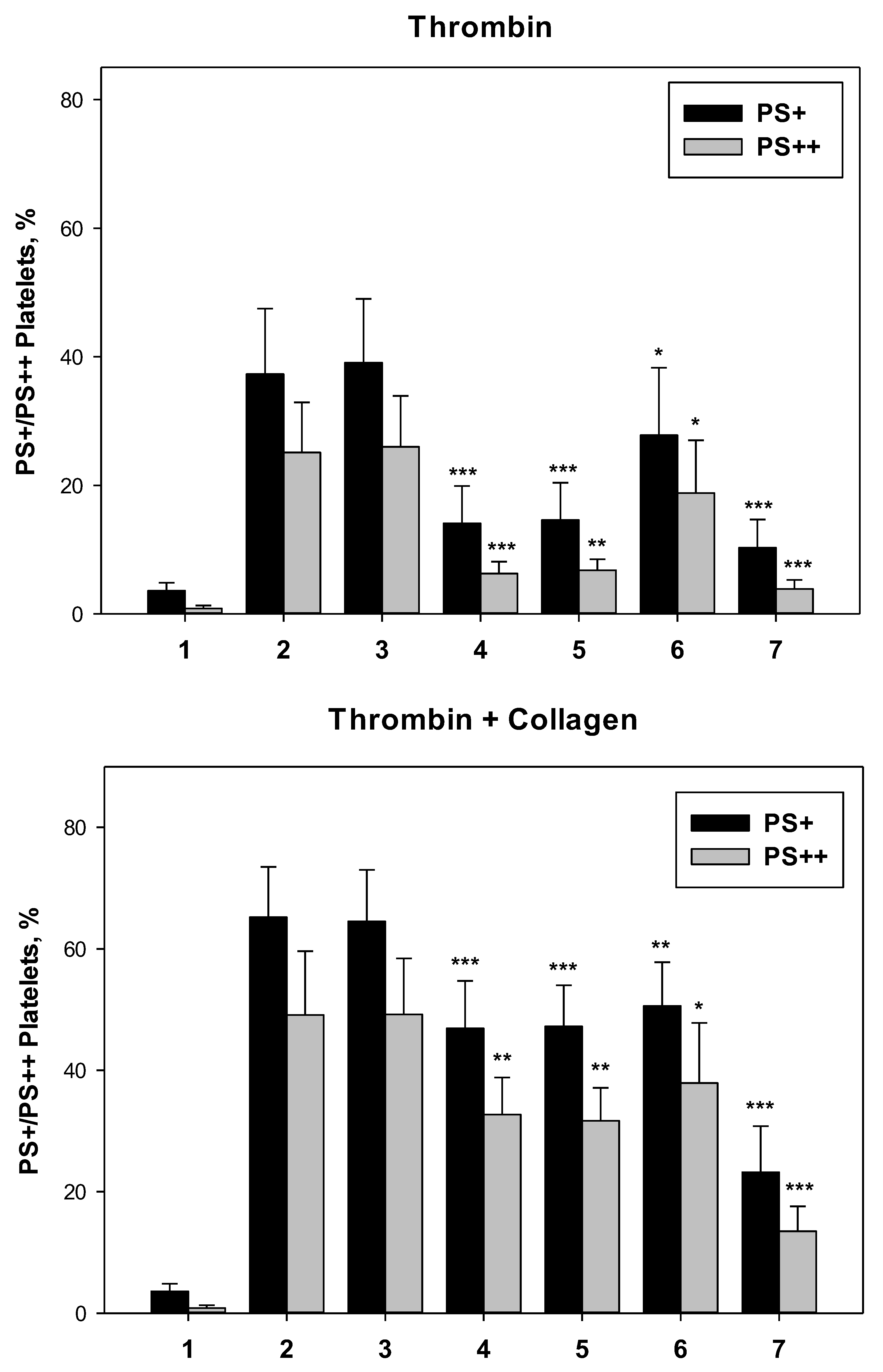
Phosphatidylserine (PS) exposure on the surface of activated platelets was measured by flow cytometry using annexin V as a specific marker. Washed platelets were activated by thrombin alone or in combination with collagen. On histograms we identified three regions: PS-, platelets not exposing PS, PS+, all platelets exposing PS, and PS++, platelets with high level of PS exposure (Fig. 3). Ticagrelor, ruciromab and PGE1 decreased the number of PS+ and PS++ platelets after their activation with thrombin and thrombin + collagen, while ASA was ineffective both alone and in combination with ticagrelor (Fig. 3, Fig. 4).
Platelets in the whole blood obtained from healthy volunteers free of any medications and patients with ACS receiving dual antiplatelet therapy (ASA + ticagrelor) were activated by TRAP or TRAP + collagen. TRAP was used instead of thrombin in order to avoid blood coagulation. In whole blood TRAP less effectively stimulated PS than thrombin added to washed platelets (compare data in Fig. 4 and Table 3) which agrees with our previous results obtained for both agonists in washed platelets [4]. PS exposure was significantly lower in patients than in healthy volunteers after platelet activation with TRAP alone and with TRAP + collagen (Table 3) indicating that antiplatelet drugs (ticagrelor in particular) suppress this reaction not only in vitro but also in vivo after their administration to patients.
Figure 3.
Effects of antiplatelet drugs on PS exposure. Washed platelets. Flow cytometry. Annexin V-FITC is used as PS marker. Vertical lines – borders between PS- (95% of negative control without annexin V-FITC, not shown), PS+ and PS++ regions. Platelets were not treated with antiplatelet drugs (A1, A2 and B1, B2) or treated with 0.2 mM ASA (C1, C2), 1 µM ticagrelor (D1, D2), 20 µg/ml ruciromab (E1, E2), 1 µg/ml PGE1 (F1, F2). Platelets were not activated (A1, A2), or were activated with 10 U/ml thrombin (left panel, B2-F2) or with 10 U/ml thrombin + 10 µg/ml collagen (right panel, (B2-F2). Histograms with ticagrelor combined with ASA were the same as with ticagrelor alone and are not shown. Representative histograms from 6 experiments. Statistical data are presented in Fig. 4.
Figure 3.
Effects of antiplatelet drugs on PS exposure. Washed platelets. Flow cytometry. Annexin V-FITC is used as PS marker. Vertical lines – borders between PS- (95% of negative control without annexin V-FITC, not shown), PS+ and PS++ regions. Platelets were not treated with antiplatelet drugs (A1, A2 and B1, B2) or treated with 0.2 mM ASA (C1, C2), 1 µM ticagrelor (D1, D2), 20 µg/ml ruciromab (E1, E2), 1 µg/ml PGE1 (F1, F2). Platelets were not activated (A1, A2), or were activated with 10 U/ml thrombin (left panel, B2-F2) or with 10 U/ml thrombin + 10 µg/ml collagen (right panel, (B2-F2). Histograms with ticagrelor combined with ASA were the same as with ticagrelor alone and are not shown. Representative histograms from 6 experiments. Statistical data are presented in Fig. 4.
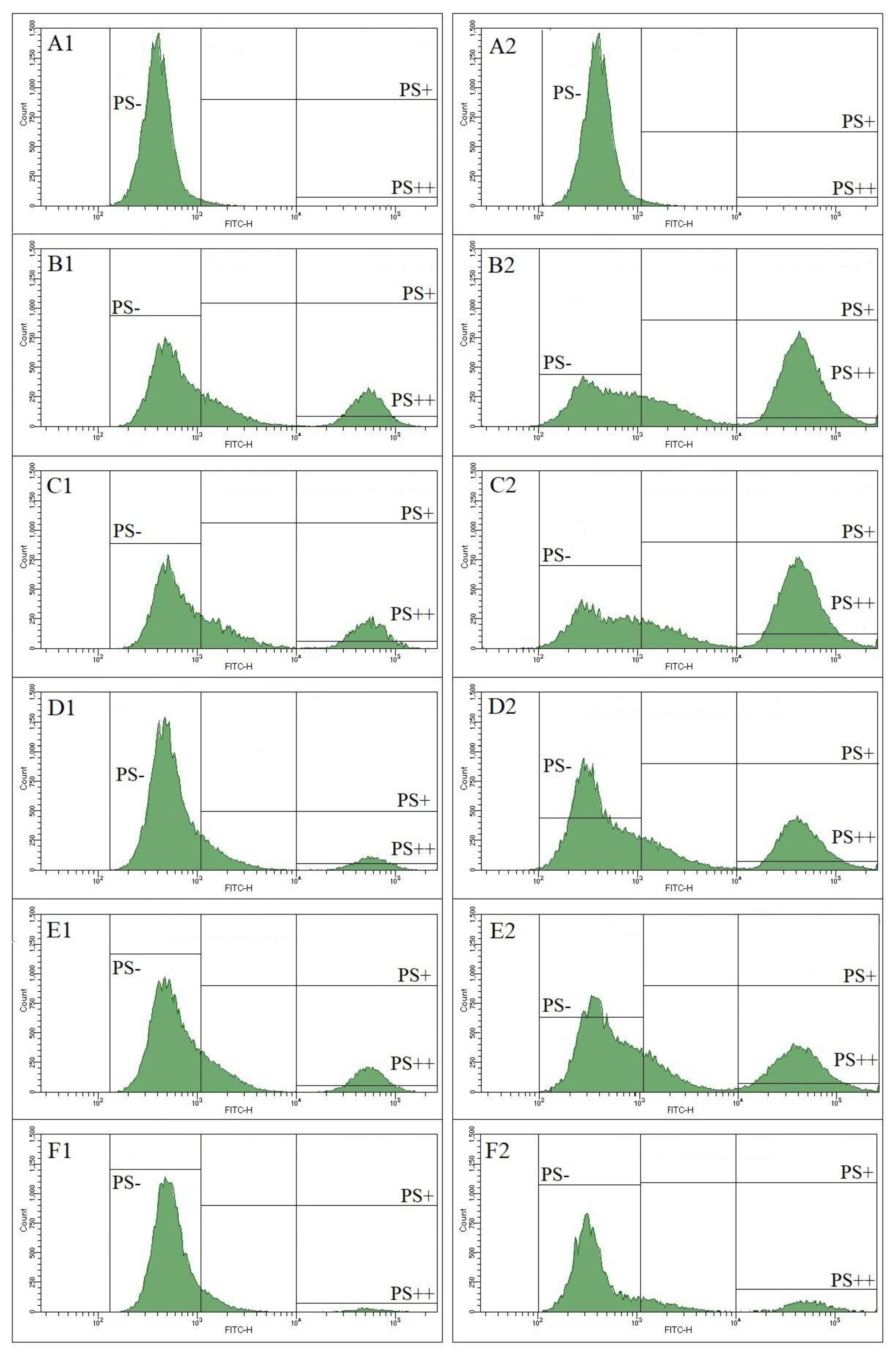
Figure 4.
Effects of antiplatelet drugs on PS exposure. Washed platelets. Platelets were not treated with antiplatelet drugs (1 and 2) or treated with 0.2 mM ASA (3), 1 µM ticagrelor (4), 0.2 mM ASA + 1 µM ticagrelor (5), 20 µg/ml ruciromab (6), 1 µg/ml PGE1 (7). Platelets were not activated (1) or were activated with 10 U/ml thrombin (upper panel, “Thrombin”), or with 10 U/ml thrombin + 10 µg/ml collagen (low panel, “Thrombin + Collagen). PS exposure was evaluated using flow cytometry (see. Fig. 3). Means ± SD for the percentage of PS+ (black columns) and PS++ (grey columns) platelets are presented (n = 6). *p <0.05, **p <0.01, ***p <0.001 – Significance of differences from probes without antiplatelet drugs (paired t-test).
Figure 4.
Effects of antiplatelet drugs on PS exposure. Washed platelets. Platelets were not treated with antiplatelet drugs (1 and 2) or treated with 0.2 mM ASA (3), 1 µM ticagrelor (4), 0.2 mM ASA + 1 µM ticagrelor (5), 20 µg/ml ruciromab (6), 1 µg/ml PGE1 (7). Platelets were not activated (1) or were activated with 10 U/ml thrombin (upper panel, “Thrombin”), or with 10 U/ml thrombin + 10 µg/ml collagen (low panel, “Thrombin + Collagen). PS exposure was evaluated using flow cytometry (see. Fig. 3). Means ± SD for the percentage of PS+ (black columns) and PS++ (grey columns) platelets are presented (n = 6). *p <0.05, **p <0.01, ***p <0.001 – Significance of differences from probes without antiplatelet drugs (paired t-test).
Discussion
We studied effects of antiplatelet drugs on platelet-dependent fibrin formation (in plasma recalcification assay) and thrombin generation (in TGT), and on PS exposure on the surface of activated platelets. Several antiplatelet drugs with different mechanisms of action have been tested: ASA, inhibitor of TXA2 synthesis, ticagrelor, antagonist of P2Y12 ADP receptor, ruciromab, GP IIb-IIIa blocker, and PGE1, adenylate cyclase activator. The combination of ASA with ticagrelor was also tested, since this type of dual antiplatelet therapy it commonly used in the cardiovascular patients particularly in ACS patients.
In plasma recalcification assay and in TGT and we used platelets sedimented on the bottom of plastic multiwells. It was shown in our previous study [4] that platelets under applied conditions (see “Materials and methods” for details) formed a monolayer on plastic surface. Testing of sedimented platelets, but not platelets in suspension, prevented the effect of platelet aggregation on their coagulation activity and removal of the supernatant allowed us to avoid the effects procoagulant microparticles formed by activated platelets. It was reported that both platelet aggregation and formation of microparticles affect platelet-dependent thrombin generation [27]
Thrombin and collagen are the most powerful platelet agonists. At the sites of vascular injury, thrombin is formed from activated prothrombin due to TF-dependent initiation of the coagulation cascade reactions, and subendothelial collagen from the vessel wall is exposed to the blood due to endothelial cell injury. In recalcification assay and TGT platelets were activated either by endogenous thrombin formed in blood plasma or by endogenous thrombin and exogenous collagen. PS exposure on platelet surface was also induced by thrombin (in this case exogenous) or TRAP (in the whole blood) alone or in combination with collagen.
We tested for the first time the effects of different antiplatelet drugs on platelet accelerated fibrin formation (plasma clotting) using modified plasma recalcification assay. Ticagrelor (both alone and in combination with ASA), ruciromab and PGE1, but not ASA were able to prolong the lag phase and to decrease the maximum rate of fibrin formation. Activation of platelets by exogenous thrombin or thrombin + collagen stimulated TXA2 synthesis in platelets and release of ADP from their dense granules. Efficacy of ticagrelor but not ASA suggested that platelet-derived ADP but not TXA2 is involved in stimulation of platelet-dependent fibrin formation (at least under applied conditions). Arima et al [28] tested blood obtained from patients with ischemic heart disease receiving ASA or ASA + clopidogrel and from control subjects free of antiplatelet drugs in a flow system where fibrin thrombus formation was evaluated in recalcified blood stimulated by surface immobilized TF and collagen. They found no differences between these groups; however, the inefficacy of both drugs can be explained by a high impact of TF in coagulation in their system (diminishing platelet contribution). Inhibitory effects of ruciromab (fragment of anti-IIb-IIIa blocking antibody) on platelet-dependent fibrin formation suggested the involvement of GP IIb-IIIa in this reaction. These results are generally in agreement with the early findings by Goto et al [20], and Ramström et al. [21] who detected the inhibitory effect of another antibody derived GP IIb-IIIa blocker abciximab (but not of low molecular weight antagonists) on spontaneous blood clotting and coalin induced PRP clotting respectively with additional platelet activation in both cases.
In TGT we used the protocol recommended for the evaluation of platelet effects (low amount of TF and minimum phospholipids). Our results obtained in TGT correlated with the results obtained in plasma recalcification assay. Ticagrelor (both alone and in combination with ASA), ruciromab and PGE1, but not ASA, decreased the peak and the maximum rate of thrombin generation. Inhibitory effects on thrombin generation were previously demonstrated for different types of antiplatelet agents [12,13,14,15,16]. However for ASA and P2Y12 antagonists these effects were detected primarily in the presence of arachidonic acid, precursor of TXA2, and ADP, respectively [12,13,14]. We do not add these agonists, nevertheless, TXA2 synthesis and ADP release from dense granules can be stimulated in platelets activated by endogenous thrombin and thrombin + collagen. A decrease of thrombin generation by ticagrelor and inefficacy of ASA confirmed the results obtained in recalcification assay suggesting that platelet-derived ADP but not TXA2 participates in realization of platelet coagulation activity. Berezovskaya et al [29] have shown that thrombin generation in PRP in patients with coronary artery disease on dual antiplatelet therapy, ASA + clopidogrel, was lower than in healthy donors. These data indicated that combination of ASA with P2Y12 antagonist (in this case clopidogrel) could suppress thrombin generation not only in vitro but also in in patients receiving this treatment. Our results with the GP IIb-IIIa blocker ruciromab in TGT confirm the ability of this type of antiplatelet drugs to inhibit thrombin generation which was earlier demonstrated for another antibody derived preparation abciximab [15,16] and for low molecular weight preparations (peptide eptifibatide and peptidomimetic tirofiban) [16].
Phosphatidylserine (PS) exposure on the surface of activated platelets is responsible for platelet-dependent acceleration of coagulation reactions [1,2,3,4]. Testing the effects of antiplatelet agents on PS exposure we expectedly found that similarly to recalcification assay and TGT ticagrelor, ruciromab and PGE1, but not ASA were able to decrease the level of PS exposure on activated platelets. Kotova et al [18] also found that in vitro ASA produce no effect on formation of so called “coated” platelets positive for PS (and for some other markers). However Prodan et al [24] detected significant effects of ASA on the level of “coated” platelets when the drug was taken by patients. This discrepancy between ASA effects in vitro and in vivo has no reasonable explanation and need to be further investigated. Inhibition of PS exposure by ticagrelor is generally consistent with previous findings [17,18] although with other P2Y12 receptor antagonists. Contradictory evidence was obtained for the effects of GP IIb-IIIa blockers on PS exposure: suppression of PS exposure [16,19,20,21], no effects [22] and even paradox stimulatory effects on this reaction [20,21,23]. However unexpected stimulation of PS exposure was detected only for low molecular weight agents [20,21,23] and not in all papers [16], while results with antibody-derived preparation abciximab demonstrating its inhibitory action [19,20,21] are consistent with our data obtained with antibody derived preparation ruciromab.
In order to demonstrate that antiplatelet drugs can decrease platelet procoagulant activity not only in in vitro experiments but also upon their administration to patients we performed a pilot study which demonstrated that PS exposure on activated platelets from ACS patients receiving standard dual antiplatelet therapy – ASA + ticagrelol was lower than in healthy subjects free of antiplatelet drugs. However, further and wider investigations in patients and/or volunteers are required for studying (1) the effects of aspirin separate administration on PS exposure, and (2) the action of different antiplatelet therapy directly on platelet-dependent coagulation reactions (fibrin formation and thrombin generation).
Conclusions
Complex investigation of the effects of different antiplatelet drugs on platelet procoagulant activities, including their ability to accelerate fibrin formation (plasma clotting) and thrombin generation, and to expose PS on their surface has shown in in vitro all these reactions were suppressed by the antagonist of P2Y12 ADP receptor ticagrelor, the GP IIb-IIIa (fibrinogen receptor) blocker rucirimab, and the adenylate cyclase activator PGE1, but not by ASA, an inhibitor of TXA2 synthesis. Limited clinical study demonstrated that the combination of ASA + ticagrelor reduced PS exposure on activated platelets upon administration to ACS patients.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Fig. S1. Inhibition of platelet aggregation in PRP by antiplatelet drugs.
Author Contributions
I.A.M. – investigation, data analysis, writing; A.B.D. – conception, design, investigation, data analysis, writing; O.A.A. – investigation; data analysis; S.G.K. – investigation, data analysis; A.K.A. – patients’ selection and characteristics; D.V.P – clinical conception, design; A.V.M. - conception, design, data analysis, writing.
Funding
This research was funded by the Russian Science Foundation grant number 22-15-00005.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Ethics Committee of the Chazov National Medical Research Center of Cardiology (protocol # 274 of 29 November 2021).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Platelets in Thrombotic and Non-Thrombotic Disorders; Springer Science and Business Media LLC: Dordrecht, GX, Netherlands, 2017; pp. 447–462. ISBN 9783319474601.
- Swieringa, F.; Spronk, H.M.; Heemskerk, J.W.; van der Meijden, P.E. Integrating platelet and coagulation activation in fibrin clot formation. Res. Pr. Thromb. Haemost. 2018, 2, 450–460. [Google Scholar] [CrossRef]
- Wielders, S.J.H.; Ungethüm, L.; Reutelingsperger, C.P.M.; Bevers, E.M.; Lindhout, T. Factor Xa-driven thrombin generation in plasma: Dependency on the aminophospholipid density of membranes and inhibition by phospholipid-binding proteins. Arthritis Res. Ther. 2007, 98, 1056–1062. [Google Scholar] [CrossRef]
- Muravlev, I.A.; Dobrovolsky, A.B.; Antonova, O.A.; Khaspekova, S.G.; Mazurov, A.V. Effects of platelets activated by different agonists on fibrin formation and thrombin generation. Platelets 2023, 34, 2139365. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Konings, J.; de Laat, B.; Hackeng, T.M.; Roest, M. Added Value of Blood Cells in Thrombin Generation Testing. Arthritis Res. Ther. 2021, 121, 1574–1587. [Google Scholar] [CrossRef] [PubMed]
- Patrono, C. Aspirin. In: Platelets. 4th edition. (Eds. Michelson A.D., Cattaneo M., Frelinger III A.L., Newman P.J.) Elsevier. Academic Press. Amsterdam, Netherlands, 2019. pp. 921–936.
- Cattaneo, M. P2Y12 Antagonists. In: Platelets. 4th Edition.( Eds. Michelson A.D., Cattaneo M., Frelinger III A.L., Newman P.J.) Elsevier. Academic Press. Amsterdam, Netherlands. 2019. pp. 937–956.
- Mahtta, D.; Bavry, A.A. αIIbβ3 (GPIIb-IIIa) Antagonists. In: Platelets. 4th edition (Eds. Michelson A.D., Cattaneo M., Frelinger III A.L., Newman P.J.) Elsevier. Academic Press. Amsterdam, Netherlands. 2019. pp. 957–971.
- Mazurov, A.; Pevzner, D.; Antonova, O.; Byzova, T.; Khaspekova, S.; Semenov, A.; Vlasik, T.; Samko, A.; Staroverov, I.; Ruda, M. Safety, inhibition of platelet aggregation and pharmacokinetics of F(ab′) 2 fragments of the anti-glycoprotein IIb-IIIa monoclonal antibody FRaMon in high-risk coronary angioplasty. Platelets 2002, 13, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Mazurov, A.V.; Pevzner, D.V.; Vlasik, T.N.; Ruda, M.I. Antiplatelet effects of glycoproteins IIb-IIIa antagonist monafram. Ross. Fiziol. Zh. Im. I. M. Sechenova. 2004, 90, 586–599. (in Russian). [Google Scholar] [PubMed]
- Braune, S.; Küpper, J.-H.; Jung, F. Effect of Prostanoids on Human Platelet Function: An Overview. Int. J. Mol. Sci. 2020, 21, 9020. [Google Scholar] [CrossRef]
- Scazziota, A.; Rouvier, J.; Gonzalez, C.; Altman, R. Effect of Sodium Arachidonate on Thrombin Generation through Platelet Activation – Inhibitory Effect of Aspirin. Arthritis Res. Ther. 2000, 84, 1109–1112. [Google Scholar] [CrossRef]
- Altman, R.; Scazziota, A.; Herrera, M.D.L.; Gonzalez, C. Recombinant factor VIIa reverses the inhibitory effect of aspirin or aspirin plus clopidogrel on in vitro thrombin generation. J. Thromb. Haemost. 2006, 4, 2022–2027. [Google Scholar] [CrossRef] [PubMed]
- van der Meijden, P.E.J.; Schoenwaelder, S.M.; Feijge, M.A.H.; Cosemans, J.M.E.M.; Munnix, I.C.A.; Wetzker, R.; Heller, R.; Jackson, S.P.; Heemskerk, J.W.M. Dual P2Y12 receptor signaling in thrombin-stimulated platelets – involvement of phosphoinositide 3-kinase β but not γ isoform in Ca2+ mobilization and procoagulant activity. FEBS J. 2008, 275, 371–385. [Google Scholar] [CrossRef]
- Reverter, J.C.; Béguin, S.; Kessels, H.; Kumar, R.; Hemker, H.C.; Coller, B.S. Inhibition of platelet-mediated, tissue factor-induced thrombin generation by the mouse/human chimeric 7E3 antibody. Potential implications for the effect of c7E3 Fab treatment on acute thrombosis and "clinical restenosis". J. Clin. Investig. 1996, 98, 863–874. [Google Scholar] [CrossRef] [PubMed]
- van der Meijden, P.E.J.; Feijge, M.A.H.; Swieringa, F.; Gilio, K.; Nergiz-Unal, R.; Hamulyák, K.; Heemskerk, J.W.M. Key role of integrin αIIbβ3 signaling to Syk kinase in tissue factor-induced thrombin generation. Cell. Mol. Life Sci. 2012, 69, 3481–3492. [Google Scholar] [CrossRef] [PubMed]
- Léon, C.; Ravanat, C.; Freund, M.; Cazenave, J.-P.; Gachet, C.; M, K.; L, C.; H, C.; J, F.; P, G.; et al. Differential Involvement of the P2Y 1 and P2Y 12 Receptors in Platelet Procoagulant Activity. Arter. Thromb. Vasc. Biol. 2003, 23, 1941–1947. [Google Scholar] [CrossRef] [PubMed]
- Kotova, Y.N.; Ataullakhanov, F.I.; Panteleev, M.A. Formation of coated platelets is regulated by the dense granule secretion of adenosine 5′diphosphate acting via the P2Y12 receptor. J. Thromb. Haemost. 2008, 6, 1603–1605. [Google Scholar] [CrossRef] [PubMed]
- Pedicord, D.L.; Thomas, B.E.; Mousa, S.A.; Dicker, I.B. Glycoprotein IIb/IIIa Receptor Antagonists Inhibit the Development of Platelet Procoagulant Activity. Thromb. Res. 1998, 90, 247–258. [Google Scholar] [CrossRef]
- Goto, S.; Tamura, N.; Li, M.; Handa, M.; Ikeda, Y.; Handa, S.; Ruggeri, Z.M. Different effects of various anti-GPIIb-IIIa agents on shear-induced platelet activation and expression of procoagulant activity. J. Thromb. Haemost. 2003, 1, 2022–2030. [Google Scholar] [CrossRef]
- Ramström, S.; Rånby, M.; Lindahl, T.L. Platelet phosphatidylserine exposure and procoagulant activity in clotting whole blood – different effects of collagen, TRAP and calcium ionophore A23187. Thromb. Haemost. 2003, 89, 132–141. [Google Scholar]
- Topalov, N.N.; Kotova, Y.N.; Vasil’ev, S.A.; Panteleev, M.A. Identification of signal transduction pathways involved in the formation of platelet subpopulations upon activation. Br. J. Haematol. 2012, 157, 105–115. [Google Scholar] [CrossRef]
- Jones, M.L.; Harper, M.T.; Aitken, E.W.; Williams, C.M.; Poole, A.W. RGD-ligand mimetic antagonists of integrin αIIbβ3 paradoxically enhance GPVI-induced human platelet activation. J. Thromb. Haemost. 2010, 8, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Prodan, C.I.; Joseph, P.M.; Vincent, A.S.; Dale, G.L. Coated-platelet levels are influenced by smoking, aspirin, and selective serotonin reuptake inhibitors. J. Thromb. Haemost. 2007, 5, 2149–2151. [Google Scholar] [CrossRef] [PubMed]
- Mazurov, A.V.; Vinogradov, D.V.; Kabaeva, N.V.; Antonova, G.N.; A Romanov, Y.; Viasik, T.N.; Antonov, A.S.; Smirnov, V.N. A Monoclonal Antibody, VM64, Reacts with a 130 kDa Glycoprotein Common to Platelets and Endothelial Cells: Heterogeneity in Antibody Binding to Human Aortic Endothelial Cells. Arthritis Res. Ther. 1991, 66, 494–499. [Google Scholar] [CrossRef]
- Khaspekova, S.G.; Zyuryaev, I.T.; Yakushkin, V.V.; Naimushin, Y.A.; Sirotkina, O.V.; Zaytseva, N.O.; Ruda, M.Y.; Mazurov, A.V. Mean platelet volume: Interrelation with platelet aggregation activity and glycoprotein IIb-IIIa and Ib expression levels. Biochem. (Moscow) Suppl. Ser. B: Biomed. Chem. 2014, 8, 134–142. [Google Scholar] [CrossRef]
- Didelot, M.; Docq, C.; Wahl, D.; Lacolley, P.; Regnault, V.; Lagrange, J. Platelet aggregation impacts thrombin generation assessed by calibrated automated thrombography. Platelets 2018, 29, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Arima, Y.; Kaikita, K.; Ishii, M.; Ito, M.; Sueta, D.; Oimatsu, Y.; Sakamoto, K.; Tsujita, K.; Kojima, S.; Nakagawa, K.; et al. Assessment of platelet-derived thrombogenicity with the total thrombus-formation analysis system in coronary artery disease patients receiving antiplatelet therapy. J. Thromb. Haemost. 2016, 14, 850–859. [Google Scholar] [CrossRef]
- Berezovskaya, G.; Smirnova, O.; Malev, E.; Khromov-Borisov, N.; Klokova, E.; Karpenko, M.; Papayan, L.; Petrishchev, N. Thrombin generation test for evaluation of antiplatelet treatment in patients with coronary artery disease after percutaneous coronary intervention. Platelets 2018, 29, 185–191. [Google Scholar] [CrossRef]

Table 1.
Effects of antiplatelet drugs on platelet-dependent fibrin formation in blood plasma (recalcification assay).
Table 1.
Effects of antiplatelet drugs on platelet-dependent fibrin formation in blood plasma (recalcification assay).
| No exogenous agonists | ||
| Lag phase, min | Vmax, %А450/min | |
| Control (n = 15) |
6.4 ± 1.7 | 11.8 ± 2.5 |
| ASA (n = 11) |
6.1 ± 1.8 | 12.1 ± 4.3 |
| Ticagrelor (n = 15) |
8.0 ± 1.7*** | 7,9 ± 2,3*** |
| ASA + Ticagrelor (n = 10) |
7.2 ± 1.2*** | 8.6 ± 2.4** |
| Ruciromab (n = 7) |
9.8 ± 1.5** | 5.7 ± 0.8*** |
| PGE1 (n = 7) |
10.1 ± 1.0*** | 4.8 ± 0.7*** |
| + Collagen | ||
| Control (n = 15) |
5.1 ± 1.2 | 18.7 ± 3.8*** |
| ASA (n = 9) |
5.1 ± 1.1 | 17.8 ± 4.5 |
| Ticagrelor (n = 15) |
6.3 ± 1.4*** | 12.6 ± 3.8*** |
| ASA + Ticagrelor (n = 8) |
6.0± 1.1** | 11.8 ± 3.0** |
| Ruciromab (n = 6) |
7.1 ± 1.7** | 11.4 ± 2.2** |
| PGE1 (n = 6) |
7.5± 1.9** | 10.2 ± 2.3*** |
Recalcification assay was performed without exogenous platelet agonists or after pretreatment of platelets with collagen (10 µg/ml). Means ± SD are presented (n – number of experiments). **р < 0.01, ***р < 0.001 – significance of differences from “Control” (no antiplatelet drugs) (paired t-test) . Parameters of fibrin formation without platelets: lag phase – 23.1 ± 7.0 min, Vmax – 6.4 ± 1.2 %А450/min (n = 15).
Table 2.
Effects of antiplatelet drugs on thrombin generation in blood plasma (TGT).
| No exogenous agonists | ||||
| Lag phase, min |
ETP1, nM х min |
Peak, nM |
Vmax, nM/min | |
| Control (n = 9) |
3.9 ± 0.3 | 1137 ± 146 | 84 ± 23 | 17 ± 7 |
| ASA (n = 5) |
3.7 ± 0.1 | 1177 ± 204 | 93 ± 28 | 20 ± 8 |
| Ticagrelor (n = 7) |
3.8 ± 0.2 | 1058 ± 164 | 60 ± 17** | 10 ± 4** |
| ASA + Ticagrelor (n = 5) |
3.6 ± 0.3 | 1103 ± 236 | 67 ± 26* | 12 ± 6* |
| Ruciromab (n = 6) |
3.7 ± 0.3 | 1115 ± 190 | 62 ± 27* | 12 ± 6* |
| PGE1 (n = 5) |
3.5 ± 0.3* | 1007 ± 186* | 62 ± 18* | 10 ± 3* |
| + Collagen | ||||
| Lag phase, min |
ETP1, nM х min |
Peak, nM |
Vmax, nM/min | |
| Сontrol (n = 8) |
3.8 ± 0.3 | 1234 ± 140 | 137 ± 24 | 36 ± 10 |
| ASA (n = 7) |
3.8 ± 0.2 | 1257 ± 125 | 131 ± 15 | 33 ± 5 |
| Ticagrelor (n = 7) |
3.8 ± 0.3 | 1277 ± 193 | 109 ± 30** | 25 ± 12** |
| ASA + Ticagrelor (n = 7) |
3.8 ± 0.3 | 1284 ± 145 | 111 ± 21* | 25 ± 6** |
| Ruciromab (n = 7) |
3.7 ± 0.3 | 1327 ± 193 | 115 ± 17* | 26 ± 5* |
| PGE1 (n = 7) |
3.9 ± 0.2 | 1222 ± 183 | 105 ± 24** | 24 ± 8** |
Thrombin generation test (TGT) was performed without exogenous platelet agonists or after pretreatment of platelets with collagen (10 µg/ml). 1ETP – endogenous thrombin potential. Means ± SD are presented (n – number of experiments). *р < 0.05, **р < 0.01 – – significance of differences from “Control” (no antiplatelet drugs) (paired t-test). Thrombin generation parameters without platelets: lag phase – 4.3 ± 0.3 min, ETP – 302 ± 49 nM х min, peak – 20 ± 3 nM, Vмакс – 3.7 ± 0.6 nM/min (n = 9).
Table 3.
Phosphatidylserine (PS) exposure on platelets from healthy volunteers free of any medications and patients with ACS receiving dual antiplatelet therapy (ASA + ticagrelor). Whole blood.
Table 3.
Phosphatidylserine (PS) exposure on platelets from healthy volunteers free of any medications and patients with ACS receiving dual antiplatelet therapy (ASA + ticagrelor). Whole blood.
| Healthy volunteers (no medications) |
Patients with ACS (ASA + ticagrelor) |
|||
|---|---|---|---|---|
| PS+ platelets, % |
PS++ platelets, % | PS+ platelets, % |
PS++ platelets, % | |
| No agonists | 1.9 ± 1.3 (n = 30) |
0.4 ± 0.4 (n = 30) |
1.2 ± 1.0 (n = 25) p = 0.022 |
0.2 ± 0.2 (n = 25) p = 0.014 |
| TRAP | 10.9 ± 5.1 (n = 20) |
3.1 ± 2.8 (n = 20) |
5.1 ± 2.8 (n = 19) p < 0.001 |
1.2 ± 1.3 (n = 19) p = 0.011 |
| TRAP + collagen | 52.0 ± 7.6 (n = 28) |
36.4 ± 10.2 (n = 28) |
41.7 ± 10.2 (n = 26) p < 0.001 |
26.6 ± 11.9 (n = 26) p = 0.002 |
Phosphatidylserine (PS) exposure on platelets was evaluated in whole blood. Platelets were not activated (“No agonists”) or activated with TRAP (20 µM) or TRAP (20 µM) + collagen (10 µg/ml). Means ± SD are presented (n – umber of analyses). р – Significance of differences from “Healthy volunteers” (t-test for means).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



