
Submitted:
14 June 2023
Posted:
15 June 2023
You are already at the latest version
Abstract
Nonalcoholic fatty liver disease (NAFLD) has become globally prevalent and is the leading cause of chronic liver disease. Although NAFLD is reversible in the early stage without medical intervention, the condition could be sequentially worsened to nonalcoholic steatohepatitis (NASH) and, in the end, cirrhosis and hepatic cancer. The progression of NAFLD is related to various factors such as genetics, pre-disposed metabolic disorders, and immunologic factors. Because of the complexity of the disease, the treatment options are very limited, and, unfortunately, there is yet no clinically available drug. Thankfully, to date, there have been accumulating research efforts and, as a result, different classes of potent drug candidates have been discovered. Besides, there have also been various attempts to explore pharmaceutical strategies to make the drug candidates to be better drugs. In this review, we provided a brief overview of the drug candidates that have undergone clinical trials. In the latter part, the strategies for developing better drugs are discussed.
Keywords:
Nonalcoholic fatty liver disease
; Nonalcoholic steatohepatitis
; Fibrosis
; Cirrhosis
; Strategies
1. Introduction
NAFLD is a condition featured by hepatic fat accumulation without chronic alcohol consumption or the use of steatogenic drugs. NAFLD is a relatively broad term comprising nonalcoholic fatty liver (NAFL) to stages when chronic fat accumulation results in hepatic inflammation and injury, engendering NASH and cirrhosis [1]. Currently, NAFLD is the most common liver disease worldwide, with a global prevalence of 25%. Various reports have indicated that people with progressing age and conditions such as type 2 diabetes (T2DM), hypertension, obesity, and dyslipidemia are high-risk groups for developing NAFLD [2,3,4]. For example, the prevalence of NAFLD increases to over 90% in obese people and 60% in T2DM patients [5].
NAFLD is diagnosed when hepatic steatosis is detected upon abdominal imaging, which could be accompanied with or without elevation in the levels of alanine and aspartate aminotransferases (ALT and AST) in blood. When upon liver biopsy, if hepatocellular injury (ballooning) and inflammation are detected, in the presence or absence of fibrosis, it is diagnosed as NASH [6,7]. Hepatic steatosis can be detected with the help of noninvasive imaging tests such as magnetic resonance imaging (MRI) and computed tomography (CT). The MRI is more sensitive and expensive than CT in detecting steatosis and can detect steatosis when it is as small as 5% of liver mass [7]. The NAFLD activity score (NAS) can be used to score the histological features upon biopsy by determining the presence of an anomaly. The NAS ≥ 5 has been correlated with the presence of NASH. However, there is a lack of reliable biomarkers for NALFD/NASH in the early stages. The stages of NASH can be determined via the fibrosis-4 index (FIB-4) or NAFLD fibrosis score (NFS). The probability of developing fibrosis can be determined using the NFS. If NFS is < -1.5, it is called “low probability”, between -1.5 - 0.65, “intermediate probability” and > 0.65, “high probability” for fibrosis [8]. Upon histological examination, liver fibrosis can be classified as F0 (absence of fibrosis), F1 (perisinusoidal fibrosis), F2 (portal/periportal fibrosis), F3 (bridging fibrosis), and F4 (cirrhosis) [9]. According to studies, 41.9% of NAFL patients progressed to NASH, and 30% of NASH patients progressed to fibrosis. About 22% of NASH patients with F3 stage fibrosis progressed to cirrhosis [10].
Recently ‘multiple-hit hypothesis’ has been put forward to explain the pathogenesis and progression of NAFLD. Pathogenesis of NAFLD starts with excessive accumulation of free fatty acids (FFAs), cholesterol, and triglycerides (TGs) in the hepatocytes overwhelming the liver’s function in maintaining lipid homeostasis [11]. Generally, the FFAs are metabolized into TGs and transported to the blood. However, in NAFLD, lipid droplets are formed in hepatocytes, a characteristic feature of NAFLD. Dysfunction of adipocytes with uncontrolled hepatic de novo lipogenesis (DNL) and decreased lipolysis lead to hepatic steatosis [12]. The unrestrained hepatic accumulation of TGs and FFAs causes the generation of oxidative stress, mitochondrial dysfunction, endoplasmic reticulum (ER) stress, and hepatocellular damage [11]. The major non-parenchymal cells in the liver, sinusoidal endothelial cells (LSECs) and hepatic stellate cells (HSCs), usually serve for regeneration and recovery of the liver from inflammation and injury. However, capillarization and de-differentiation of LSECs are critical events in the progression of liver fibrosis. The dysfunction of LSECs also promotes inflammatory pathways and steatosis [13]. Compromised hepatocyte integrity and activation of inflammatory pathways such as c-Jun N-terminal kinase-1 (JNK1) and nuclear factor kappa B (NFκB) signaling pathways with suppression of peroxisome proliferator-activated receptor-α (PPAR-α) could induce trans-differentiation of quiescent HSCs into myofibroblast-like activated HSCs (aHSCs) [14,15]. Upon activation, the HSCs lose the storage of vitamin A and become proliferative. They also show increased expression of smooth muscle α-actin (SMA) and transforming growth factor-β1 (TGF-β1), which are primarily responsible for the deposition of type I collagen in the extracellular matrix (ECM) [16]. Hepatocellular ballooning and lobular inflammation with the absence or presence of a certain degree of fibrosis are the primary features of NASH. Long-term injury and inflammation can induce hepatic fibrosis and, in extreme cases, further advance to cirrhosis which is the condition of excessive scarring with impaired liver function.
Other factors contribute to the progression of NAFLD. In the gut, inflammasomes are essential for microbiota homeostasis, expression of microbial peptides, and regulation of immune response. Hence, the lack of inflammasomes in the gut may result in high expression of pro-inflammatory cytokines (e.g., toll-like receptor-4 (TLR-4) and TLR-9) and exacerbate NASH [17]. There are also genetic risk factors that could affect the progression of NASH. The polymorphism in the PNPLA3 gene regulating hepatocellular lipolysis, splice variant (rs72613567:TA) in the HSD17B13 gene, and impairment of TM6SF2 gene are related to increased NAFLD development [7]. Insulin resistance is also a critical factor exacerbating the dysfunction of lipid metabolism and homeostasis. Another pathogenic factor could be the altered gut microbiome and gut-liver axis attributable to the modern diet with high saturated fat, carbohydrate, and sedentary lifestyle. However, the pathological drivers may vary among the patients as multiple molecular pathways are involved in the disease's progression. Because of the involvement of metabolic abnormalities and a broad spectrum of complex phenotypes and epigenetic factors, NAFLD is also called “metabolic-dysfunction associated fatty liver disease (MAFLD) [18].”
The global prevalence of NAFLD and its rapid increase urgently demand the discovery of effective drugs as well as elucidation of the pathological mechanism. The early chapter of this review will introduce a relevant class of drug candidates that have undergone clinical investigation, while the latter part will cover the trends of current pharmaceutical strategies explored to make “better drugs”.
2. Therapeutic drug candidates for the treatment of NAFLD
Unfortunately, there are very limited treatment options for NAFLD therapy [19]. The recommended first-line treatment includes dietary and lifestyle modification. Antioxidants such as vitamin E and glutathione (GSH) are also listed as the first-line treatment for patients without T2DM [20]. In addition, surgical treatments are available. Bariatric surgery could improve the metabolic condition and survival rate, and liver transplantation remains the last treatment option for NASH management [21,22]. However, there is no approved pharmacotherapy for NAFLD [23].
To date, there have been accumulating studies to discover novel drug candidates for treating NAFLD. The main mechanisms of these drugs generally include 1) modulating glucose and lipid homeostasis, 2) reducing hepatic injury and inflammation, and 3) ameliorating fibrosis [19]. In this chapter, we would like to introduce a group of representative drug candidates that have undergone extensive pre-clinical and clinical investigations. The class of drug candidates includes sodium-glucose co-transport protein-2 (SGLT2) inhibitors, glucagon-like peptide-1 receptor (GLP-1R) agonists, peroxisome proliferator-activated receptors (PPAR) agonists, fibroblast growth factors (FGFs), and farnesoid X receptor (FXR) agonists. The schematic illustration of the drugs and their primary mode of action is shown in Figure 1.
2.1. SGLT2 inhibitors
The SGLT2 inhibitors are clinically available anti-hyperglycemic agents that limit renal glucose reabsorption. They were further approved for heart failure and chronic renal diseases [24]. Currently, there are a total of 8 SGLT2 inhibitors approved worldwide: dapagliflozin, canagliflozin, empagliflozin, and ertugliflozin (in U.S), luseogliflozin and topogliflozin (in Japan), ipragliflozin (in Japan and Russia), and remogliflozin etabonate (in India) [24]. Apart from being anti-diabetic drugs, SGLT2 inhibitors are potentially effective drug candidates for the treatment of NAFLD. They can reduce the hepatic lipid content by inhibiting de novo lipogenesis (DNL) [25] and promoting the β-oxidation of FFAs [26]. Moreover, they can reduce bodyweight, and provide anti-oxidative and anti-inflammatory effects [24]. For example, in ob/ob and HFD-fed obese mice, ipragliflozin significantly reduced DNL, and hepatic steatosis, and improved insulin resistance [27]. The effects of ipraglifozin were also revealed in clinical studies. In a clinical trial with T2DM patients with NAFLD, 72-week treatment of ipraglifozin could reverse NAFLD in 67% of the patients, with no development of NASH. Notably, this study’s results implied that ipragliflozin may also help prevent the development of NASH [28]. In another randomized controlled trial, a 24-week treatment of ipragliflozin with pioglitazone and metformin in diabetic NAFLD patients provided a significant reduction of hepatic and whole-body visceral fat contents [29]. According to the meta-analysis of 11 clinical trials, dapagliflozin provided a significantly higher reduction of ALT, AST, γ-glutamyl transferase (GGT), TG, body weight, body mass index (BMI), HbA1c, and fasting plasma glucose levels than the control groups [30]. In a different report, combination therapy with dapagliflozin and omega-3 carboxylic acid more significantly reduced liver fat content than the monotherapy groups [31]. Other SGLT2 inhibitors such as luseogliflozin [32], empagliflozin [33], and canagliflozin [34] were also effective in reducing hepatic fat content, transaminase activity, and HbA1c level. The active clinical studies of SGLT2 for NAFLD are summarized in Table 1.
2.2. GLP-1R agonists
GLP-1 is an incretin hormone released from the enteroendocrine L-cells. It exhibits a broad spectrum of activities, including regulation of insulin signaling, glucose metabolism, satiety signals, and body weight [35]. Regarding NAFLD, GLP-1R agonists are reportedly capable of alleviating hepatic lipotoxicity and inflammation through various cell signaling pathways (e.g., insulin receptor substrate-2 (IRS2)/phosphatidylinositol-3 kinase (PI3K)/Akt [36] and mTORC1 [37] signaling pathways). They can also modulate Ca2+ signaling and reverse hepatic insulin resistance [38].
To date, there have been extensive pre-clinical and clinical study reports of long-activing GLP-1R agonists’ (e.g., dulaglutide, liraglutide, and semaglutide) potential use in the treatment of NAFLD [39]. For example, in a rapid-onset NASH mice model, liraglutide treatment significantly reduced hepatic steatosis, inflammation, and TG levels with improved glucose metabolism and insulin resistance [40]. Also, in methionine and choline-deficient (MCD) diet-fed mice, liraglutide could prevent hepatic inflammation and the development of fibrosis [41]. Similar to liraglutide, semaglutide reduced hepatic DNL and inflammation, improved insulin sensitivity, and promoted β-oxidation of FFAs in obese mice [42]. In clinical trials, liraglutide was well tolerated in NASH patients and reduced hepatic DNL, and improved insulin sensitivity in hepatic and adipose tissues [43,44]. According to a meta-analysis, liraglutide significantly improved the aminotransferase, lipoprotein, and adipose tissue levels among NAFLD patients and reduced BMI and TG levels [45]. Semaglutide also reduced bodyweight, ALT, and high sensitivity C-reactive protein (hsCRP) levels in obese diabetic patients [46]. A systemic review of clinical trials revealed that both liraglutide and semaglutide significantly reduced hepatic fat accumulation and attenuated the worsening of hepatic fibrosis [47]. However, a recent trial result showed that semaglutide (once weekly) is more effective than liraglutide (once daily) in reducing bodyweight in obese diabetic patients [48]. Currently, ongoing clinical trials of GLP-1R agonists for NAFLD are summarized in Table 2.
2.3. PPAR agonists
PPARs are a group of nuclear receptors that serve as transcription factors in lipid metabolism, regulation of inflammation, and glucose homeostasis [49]. Three isoforms of PPARs (PPAR-α, PPAR-β/δ, and PPAR-γ) have been reported. Specifically, the PPAR-α agonists (typically known as “fibrates”) are clinically available anti-hyperlipidemic agents, while the PPAR-γ agonist, thiazolidinedione, is an anti-hyperglycemic agent for T2DM management. Despite differences in tissue distribution, ligands, and physiological characteristics, all three isoforms of PPARs are commonly involved in lipid metabolism [50]. The PPAR-α is the most abundant isoform in metabolically dynamic tissues such as the heart, brown adipose tissues, and the intestinal mucosa (but mainly present in liver and skeletal muscle), where it participates in lipid oxidation and metabolism [50]. The PPAR-γ is predominantly expressed in brown and white adipose tissues and is involved in lipid biosynthesis, adipogenesis, insulin sensitivity, and energy homeostasis [50]. The isoform PPAR-β/δ is ubiquitously present throughout many tissues but primarily expressed in liver and abdominal adipose tissues, where it modulates β-oxidation of FFAs, blood lipid levels, and glucose homeostasis [51].
PPAR agonists have been examined for their effects on NAFLD [49]. Specifically, pioglitazone has been of great interest and there have been a total of 30 clinical studies carried out (or still ongoing). In a clinical trial, pioglitazone improved insulin sensitivity, NAS score, fibrosis score, and TG levels in patients with T2DM and biopsy-proven NASH (ClinicalTrials.gov identifier: NCT00994682). Similarly, according to a meta-analysis of clinical trials, pioglitazone was found able to significantly improve steatosis, inflammation, ballooning grades, fasting glucose, and insulin levels. However, pioglitazone was ineffective for fibrosis [52]. A phase 2 clinical trial is underway (ClinicalTrials.gov identifier: NCT04501406). There have also been clinical trials with rosiglitazone. In a 1-year randomized trial, rosiglitazone improved steatosis and ALT levels in NASH patients. However, additional 2-year treatment with the rosiglitazone could not significantly improve the conditions of steatosis or fibrosis (ClinicalTrials.gov identifier: NCT00492700). Fenofibrate was also clinically investigated but did not meet the criteria (ClinicalTrials.gov identifier: NCT02354976). The active clinical trials of PPAR agonists for NAFLD are summarized in Table 3.
2.4. FGFs
The FGFs are a family of ligands for the transcriptional nuclear FGF receptors (FGFR). Among the 22 known FGFs, specifically, FGF19 and FGF21 are engaged in maintaining lipid and glucose homeostasis [53]. The FGF21 is predominantly present in the liver but also expressed in adipose tissues [54]. In the adipose tissues, the FGF21 induces insulin-independent glucose uptake [54]. Hence, the FGF21 could reduce blood glucose and TG levels in various diabetic mice models. According to Wang et al, the FGF21 treatment for monosodium glutamate (MSG)-induced obesity rats could not only reduce the bodyweight, fat mass, and blood glucose level but also significantly improve liver function and inflammation [55]. Following studies have proven that FGF21 is potentially effective in improving NAFLD through various mechanisms that involve reducing hepatic DNL, inflammation, production of cholesterol and bile acids, and promoting glucose metabolism and glucose uptake by adipocyte tissues [56]. From FGF21-transgenic mice, significant side effects (e.g., stunted growth, female infertility, bone loss, and increased serum glucocorticoid levels) were also recognized, suggesting the necessity of effective yet safer FGFR agonists [57]. In this regard, several bispecific antibodies have been developed against the FGFR-1/β-klotho complex as potential therapeutics for the treatment of obesity-related metabolic diseases. The bFKB1 (also mentioned as “BKFB8488A”), a humanized FGFR-1c/β-klotho complex bispecific antibody developed by Kolumam et al, revealed specific activation of only the FGFR-1c, different from the FGF21 (that activates FGFR-1c, 2c, and 3c) and least interference to the binding of endogenous FGFs (FGF19 and FGF21) to the FGFRs. Notably, sustained metabolic effects (improvement in body weight, hepatic steatosis, hyperlipidemia, and hyperglycemia) were obtained by a single administration of bFKB1, which was equivalent to repeated or continuous infusion of the FGF21 in the tested mice (HFD-fed DIO and db/db) and cynomolgus monkey models. More importantly, side effects observed from FGF21 and FGF19 (such as increased corticosteroid levels and hepatic cell proliferation) were not found in the bFKB1-treated DIO mice [57]. Recently, a 12-week phase 1 clinical trial (ClinicalTrials.gov identifier: NCT03060538) for the bFKB1 has been completed. The bFKB1 was proven safe and could significantly reduce the TG, ALT, AST, and liver fat content in NAFLD patients [58]. The MK-3655 (NGM313) is another humanized monoclonal antibody against the FGFR-1c/β-klotho complex. Compared with pioglitazone, the MK-3655 exhibited a significantly higher reduction in TG, LDL-c, and HAb1c levels and liver fat contents [59]. It is currently under investigation in a phase 2 clinical trial (ClinicalTrials.gov identifier: NCT04583423).
FGF19 is mainly involved in the regulation of bile acid synthesis. The luminal bile salt in the ileum induces the FGF19 expression. The FGF19 signals through its binding to the β-Klotho-FGFR4 and inhibits bile acid synthesis by suppressing the expression of cholesterol 7α-hydroxylase (CYP7A1) [60]. In the report by Wunsch et al, the levels of FGF19 in the serum and liver were found to correlate with the severity of cholestatic liver diseases such as primary biliary cirrhosis [61]. FGF19 dysregulation has also been reported in NAFLD, but interestingly, a reduced fasting FGF19 was correlated with the development of steatosis in obese adolescents [62]. Hence, FGF19 has been considered a potential drug candidate for treating NAFLD. However, the pro-tumorigenic activity of FGF19 remained a major concern [61]. To address this issue, the development of non-tumorigenic FGF19 analogs has been actively pursued. For example, a genetically engineered FGF19-M52 protein constructed by Gadaleta et al retained the intrinsic properties of FGF19 without being tumorigenic. The FGF19-M52 treatment of Abcb4-/- and fxr-/- mice could protect mice from hepatic collagen deposition, liver damage, and spontaneous fibrosis [63]. Aldafermin (NGM282), another non-tumorigenic FGF19 analog, induced a significant reduction in hepatic fat content (≥ 30%) and improvement of NAS score (with two or more points) in patients by 12-week treatment without worsening of NASH and fibrosis [64]. Two 24-week clinical studies with aldafermin (ClinicalTrials.gov identifier: NCT03912532 and NCT02443116) showed a significant reduction of hepatic fat, ALT, AST, and Pro-C3 levels with improvement in hepatic fibrosis. However, an increase in cholesterol level was also observed as a side effect. In this regard, co-administration of a cholesterol-modulating agent (such as statins) was considered beneficial [65]. The FGF analogs in active clinical trials for NAFLD are summarized in Table 4.
2.5. FXR agonists
The nuclear receptor FXR is a transcription factor highly expressed in the liver and intestine. It serves as a bile acid sensor and is involved in bile acid metabolism and regulation of cholesterol and TGs [66]. The deficiency of FXR in mice was related to NASH-like pathology with increased hepatic steatosis, ballooning, and inflammation [67]. The activation of FXR can 1) indirectly suppress the transcription of CYP7A1 via regulation of the FGF19 gene [66], 2) induce inhibition of the SREBP1-c regulated fatty acid synthesis by activation of an atypical nuclear receptor heterodimer partner, SHP (small heterodimer partner; alternative name: NR0B2) [68], and 3) promote the expression of PPARα, leading to the promotion of FFA oxidation [69]. Thus, the FXR agonists are likely to exhibit a dual action to improve NAFLD by inhibiting lipogenesis (via the crosstalk with SREBP-1c) and promoting lipolysis (via activation of the FXR-PPARα pathway) [70]. Owing to these beneficiary actions through the FXR, many FXR agonists have been investigated for their potential use in NAFLD treatment.
The recent trend in the development of FRX agonists is to discover potent molecules that cause less adverse effects, as the treatment of early FXR agonists (e.g., bile acids and primary bile acid analogs) were often accompanied by pruritus, the elevation of LDL cholesterol levels, and liver toxicity. It was found that the pruritus might be caused by the activation of GPBAR1 (alternative names: TGR5, M-BAR, BG-37, hGPCR19, and AXOR 109) [71,72]. Hence, many research efforts have been made to develop more FXR-selective bile acid-based agonists. A representative example is the obeticholic acid (OCA, alternatively, 6α-ethyl-chenodeoxycholic acid or INT-747), a semi-synthetic chenodeoxycholic acid (CDCA) variant. It is a potent and selective FXR agonist (EC50: 99 nM) [73] with 100 times higher affinity than the endogenous CDCA [74]. In pre-clinical studies with NAFLD mice models (melanocortin 4-receptor deficient (MC4R-KO) NASH mice and western diet-fed, CCl4-treated mice), the OCA improved insulin resistance, liver steatosis, and fibrosis [75,76]. OCA's therapeutic efficacy for NAFLD has been further investigated in clinical trials. In a 72-week study (ClinicalTrials.gov identifier: NCT01265495), the OCA treatment for NASH patients improved hepatic steatosis, NAS (> 2 points), lobular inflammation, and ballooning. However, the OCA treatment worsened hepatic insulin resistance and increased LDL cholesterol levels. Hence, in another phase 2 clinical trial (ClinicalTrials.gov identifier: NCT02633956), a combined treatment of OCA and atorvastatin was carried out and the results showed that atorvastatin could attenuate the OCA-induced increase in LDL cholesterol levels. A phase 3 clinical trial (ClinicalTrials.gov identifier: NCT02548351) to assess the effects of OCA in patients with NASH F2-F3 stage fibrosis has recently been completed, while another phase 3 study (ClinicalTrials.gov identifier: NCT03439254) is currently underway. The INT-767 is another semi-synthetic FXR agonist. It was reportedly 300 times more potent than the natural homolog [77]. Eight-week treatment of the INT-767 with Lepob/ob NASH mice showed improvement in hepatic steatosis, histopathology, ballooning, and inflammation. Hepatic TG content, ALT, and AST levels were also reduced. Notably, the INT-767 revealed superior effects to the OCA in improving the fibrosis stages (36 vs. 82%) [78]. In another pre-clinical study with a high-fat diet (HFD)-rabbit model, the INT-767 attenuated insulin resistance in the visceral adipose tissue and reduced fatty acid synthesis and steatosis [79]. These results supported that the INT-767 may also be considered a potential drug candidate for the treatment of NAFLD. The EDP-305 is a steroidal FXR agonist lacking the carboxylic group associated with the formation of hepatotoxic metabolites (via conjugation with taurine or glycine). It has been reported to possess anti-steatotic, anti-inflammatory, anti-fibrotic, and hepatocyte protective activities. In 2 NASH mice models, dietary-induced NASH and streptozotocin-HFD mice, the EDP-305 decreased lipogenic gene (SBREP1c and SCD1) levels and hepatic lipid profiles with significant improvement in the NAS [80]. In a phase 2 trial (ClinicalTrials.gov identifier: NCT03421431), 12-week treatment with the EDP-305 led to a 7% reduction in liver fat, but pruritus was reported in about half of the participants. Another phase 2 study (ClinicalTrials.gov identifier: NCT04378010) is currently underway.
There have also been developed non-steroidal FXR agonists. For example, cilofexor (GS-9674) is an FXR agonist that does not go through enterohepatic circulation. A pre-clinical study has shown that cilofexor could reduce hepatic fibrosis (with a 37% reduction of Col1α1 expression) and portal hypertension in a NASH rat model [81]. The effects of combination therapy of cilofexor and selonsertib (apoptosis signal-regulating kinase-1 inhibitor) or firsocostat (an acetyl-CoA carboxylase inhibitor) have been clinically investigated in NASH patients with F3-F4 stage compensated cirrhosis (ClinicalTrials.gov identifier: NCT03449446). While cilofexor alone could not meet the trial's primary outcomes, a significant decrease in NAS (≥ 2 points), steatosis, lobular inflammation, and ballooning was reported by the cilofexor/firsocostat combination therapy. In addition, improvement in ALT, AST, bile acid, insulin levels, and liver stiffness was also observed. Currently, another phase 2 clinical trial is underway to evaluate the effects of cilofexor/firsocostat and semaglutide combined therapy on NASH patients with F4 compensated cirrhosis (ClinicalTrials.gov identifier: NCT04971785). Tropifexor (LJN452) is also a non-bile acid FXR agonist. A phase 2 clinical trial has been conducted to evaluate the effects on NASH (ClinicalTrials.gov identifier: NCT02855164). Unfortunately, it also did not meet the histological endpoints, and an uncontrolled increase in serum LDL levels was observed as a main side effect. As a different approach, a phase 2 clinical trial with the combination of tropifexor and an SGLT1/2 dual inhibitor, licogliflozin, is currently ongoing to assess the effects on NASH and fibrosis (ClinicalTrials.gov identifier: NCT04065841). Similarly, a combination of MET409 and empagliflozin is also in a phase 2 clinical trial (ClinicalTrials.gov identifier: NCT04702490). Nidufexor (LMB763) is an FXR partial agonist. It serves as a potent modulator of the FXR-dependent genes which eventually leads to a significant reduction in hepatic steatosis, inflammation, and fibrosis with an improvement of the NAS in the STAMTM NASH mice [82]. It was also evaluated in phase 2 clinical trial (ClinicalTrials.gov identifier: NCT02913105). The active clinical trials of FXR agonists for NAFLD are summarized in Table 5.
3. Strategies to enhance the efficacy of NAFLD drug candidates
Because of the complexity of NAFLD, the journey to discover an effective drug is yet far from reaching. However, along the arduous way to find a new entity drug, there has also been growing interest and continuous efforts to develop highly efficient strategies to make “better drugs” to achieve the goal. Some of the representative strategies may include 1) developing multiple receptor agonists that could provide synergistic effects, 2) engineering long-acting derivatives that could allow higher drug accumulation in the liver, and 3) devising ligand-mediated hepatic drug targeting systems. The schematic illustration of the strategies is depicted in Figure 2.
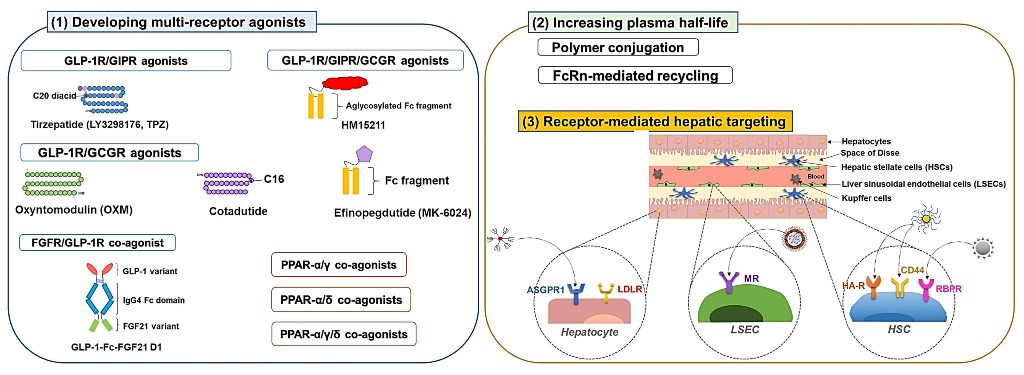
3.1. Developing multiple receptor agonists
Developing multi-receptor agonists has been a widely adopted strategy for enhanced therapeutic effects in treating metabolic disorders (e.g., obesity, T2DM, and NAFLD). This strategy is believed to provide synergistic or at least additive therapeutic effects by activating multiple receptor signaling. To date, there have been engineered various co-agonists that could simultaneously target critical receptors involved in the NAFLD, such as GLP-1R, glucose-dependent insulinotropic polypeptide (GIP) receptor (GIPR), glucagon receptor (GCGR) [83], PPARα, PPARβ/δ, and PPARγ [84,85].
3.1.1. GLP-1R co-agonists
GIP is a 42 amino acid length peptide (YAEGTFISDYSIAMDKIHQQDFVNWLLAQKG KKNDWKHNITQ) synthesized by the K cells present in the duodenum and the jejunum [86]. Like GLP-1, the GIP serves as an incretin and induces insulin secretion from the pancreatic β cells in response to high glucose levels in the duodenum [87]. The GIP also protects the β cells from apoptosis and promotes their proliferation [87]. Furthermore, the GIP can regulate the appetite and reduce food intake through its action in the hypothalamus [88]. However, unlike GLP-1, GIP stimulates glucagon secretion and possesses a lipid storage action in the adipose tissue [89]. The GIPRs are present in the CNS and many peripheral organs and tissues (e.g., pancreas, gut, heart, and adipose tissues). There appears to exist a positive correlation between the GIPR level and resistance to obesity. Also, T2DM patients often have reduced postprandial levels of GIP secretion [90].
According to the report by Finan, the GLP-1R/GIPR dual-agonists could provide more significant hypoglycemic, hyperlipidemic, and anti-obesity effects compared with single agonists with attenuated gastrointestinal discomfort and nausea (typically associated with GLP-1R agonists) [47]. The most successful case of the dual GLP-1R and GIPR agonist would be the tirzepatide (LY3298176, TPZ) which is a C18 fatty diacid moiety-conjugated 39-mer peptide [YXEGTFTSDYSIXLDKIAQKAFVQWLIAGGPSSGAPPPS; X is α-aminoisobutyric acid (Aib), K20 is acylated with a γGlu-2×OEG linker and the C18 fatty diacid moiety] [91,92]. The TPZ has been extensively investigated in 45 clinical trials and was recently approved by the FDA for the treatment of T2DM (MounjaroTM, Eli Lilly and company). In clinical studies, the TPZ more significantly reduced bodyweight and food intake than the dulaglutide [89]. Other than T2DM, a phase 2 trial is ongoing to investigate the effect of NASH treatment (ClinicaTrials.gov identifier: NCT04166773).
Glucagon is a 29-mer peptide hormone (HSQGTFTSDYSKYLDSRRAQDFVQWLMNT) secreted from the pancreatic α cells [93]. Its production is regulated by various factors (stimulated by hypoglycemia) or substances (inhibited by amylin and insulin) [94]. The primary role of glucagon is to elevate the blood glucose level by promoting gluconeogenesis and glycogenolysis [95]. Besides, glucagon reduces fatty acid synthesis, promotes lipolysis, and regulates cholesterol biosynthesis [96]. Due to these activities, compared with GLP-1R mono-agonists, the GLP-1R/GCGR dual agonists could offer enhanced lipolysis and energy expenditure (via GCGR) as well as improved glucose homeostasis and insulin sensitization (mediated via GLP-1R). Currently, several dual agonists of GLP-1R/GCGR are under investigation for their use in the management of NAFLD.
Oxyntomodulin (OXM), a 37-mer peptide hormone (HSQGTFTSDYSKYLDSRRAQDF VQWLMNTKRNRNNIA) secreted by the intestinal L-cells, is an endogenous dual agonist for the GLP-1R and the GCGR. The OXM is known to play a crucial role in bodyweight control and glycemic regulation [97], and the treatment of OXM has shown potential effects on bodyweight loss. However, the bioactivity of the OXM in activating the GLP-1R and GCGR is lower than both the endogenous GLP-1 and GCG (18 and 50-fold lower in EC50 levels, respectively) [98]. In addition, because of the difference in the activity toward the GLP-1R and the GCGR, although known as a dual agonist, the metabolic effects of OXM are mainly attributed to the GLP-1R agonism [98,99]. As dual-agonism may provide superior glucoregulatory and anti-obesity effects to mono-agonists [100], there have been attempts to modify the peptide sequence of OXM for more balanced dual agonism for the GLP-1R and GCGR. The study by Ma et al hints at how to modify the OXM [98]. Based on the native OXM sequence, they introduced oppositely charged α-helical favoring amino acid residues [Glu (E) and Lys (K)] in the mid-region of the peptide sequence (S16E, R17K, Q20K, and D21E) to allow them to form salt bridges among themselves to stabilize the conformation. This modification improved activity by 18.9- and 6.4-fold for GLP-1R and GCGR, respectively.
Further, they substituted the C-terminal 8 amino acid residues of the intervening peptide-1 to a truncated exendin-4 sequence, which led to an extra increase in the GLP-1R and GCGR activity (1.7- and 1.8-fold, respectively). Overall, by these modifications, the activity of OXM could improve to about 64% activity of both the GLP-1 and GCG. Apart from these substitution works for improving the receptor affinity, they also introduced an unnatural amino acid residue (Aib) replacing the DPP-4-hydrolysis liable Ser residue at position 2 and did a Q24C substitution for PEG conjugation. These modifications did not significantly affect the OXM activity. However, conjugation of a 40 kDa size PEG to this modified OXM led to a large decrease in the activity (6 and 4-fold reduction of GLP-1R and GCGR activity compared with the non-PEGylated modified OXM).
Other examples of dual GLP-1R/GCGR agonists include cotadutide (MEDI0382) and efinopegdutide (MK-6024, HM12525A). The cotadutide is a palmitoylated glutamate-extended peptide (1'-[palmtoyl-Glu]; HSQGTFTSDKSEYLDSERARDFVAWLEAGG [amide bridge: Glu1'-Lys10]) modified from the native glucagon with balanced agonism toward GLP-1R and GCGR (but still have a 5-fold bias toward the GLP-1R activation than the GCGR) [101]. The activity for GLP-1R and GCGR was 14.7- and 1.8-fold higher than the OXM. Henderson et al reported that, in DIO mice, the cotadutide produced greater weight loss and comparable hypoglycemic effects to liraglutide. The authors hypothesized that this might be due to the dual action of the drug via GCGR-mediated increased energy expenditure and GLP-1R-mediated reduced food intake. The significant weight loss effect was also found in cynomolgus monkeys [101]. According to Boland et al, the cotadutide improved hepatic lipid profiles as well as glucose tolerance and insulin sensitivity. Moreover, in obese trans-fat-containing amylin liver (AMLN) NASH mice, it could improve hepatic fibrosis and inflammation [102]. In a 52-week phase 2 clinical trial (ClinicaTrials.gov identifier: NCT03235050), significant improvement in lipid profiles, HbA1c, bodyweight, and NAS was observed in cotadutide-treated patients with T2DM and obesity. In the case of the efinopegdutide, detailed information on the structure is not informed, but it was reported to possess a balanced activity toward the GLP-1R and GCGR [83]. A phase 2 clinical trial (ClinicaTrials.gov identifier: NCT04944992) was recently completed and currently there is another active clinical trial onging (NCT05364931).
HM15211 is a triple agonist for the GLP-1R/GIPR/GCGR. The FDA has granted it a fast-track designation for treating NASH and fibrosis based on promising pre-clinical study results. The efficacy of HM15211 was evaluated in various mice models (DIO, AMLN, or MCD-diet mice). Commonly from these studies, significant bodyweight loss and improvement in hepatic steatosis were observed [103]. In the NASH mice models, the HM15211 revealed superior anti-steatosis and anti-fibrotic effects compared to single agonists (liraglutide, OCA, and selonsertib) [103]. The HM15211 was also more effective in reducing bodyweight and hepatic lipid content than liraglutide [104]. In a fructose-fed hamster model, the HM15211 showed significant effects on obesity and dyslipidemia. Cell studies suggested that higher clearance of LDL may be the main cause for this improvement, as a significant increase (4.1-fold) of LDLR was observed from the HM15211-treated HepG2 cells [105]. Notably, the HM15211 also prevented HSC activation and fibrosis by reducing the production of transforming growth factor- β (TGF-β) and collagen in the HSCs [106]. A phase 1 clinical trial has been completed (ClinicaTrials.gov identifier: NCT03744182), and a phase 2 clinical trial (ClinicaTrials.gov identifier: NCT04505436) is currently underway.
Other than the peptidyl agonists, various small molecule-based GLP-1R agonists have recently been discovered. Many are designed to be bispecific for GLP-1R and GIPR or GCGR. Currently, CT-388 is in phase 1 clinical trial for indications of T2DM and obesity (ClinicaTrials.gov identifier: NCT04838405), while CT-868 is under a phase 2 trial for T2DM and obesity (ClinicaTrials.gov identifier: NCT05110846). DD01, a long-acting dual agonist for GLP-1R/GCGR, is also in a phase 1 clinical trial to investigate the effects on NASH, T2DM, and obesity (ClinicaTrials.gov identifier: NCT04812262). The GLP-1R dual agonists in active clinical trials for NAFLD are summarized in Table 6.
3.1.2. PPAR dual and pan agonists
The PPARs are transcription factors that regulate critical genes involved in the homeostasis of glucose and lipids. Despite different tissue distribution and physiological effects, the 3 isoforms (PPAR-α, PPAR-β/δ, and PPAR-γ) commonly play critical roles in the regulation of lipid and glucose metabolism [107], which could benefit the treatment of NAFLD [108]. Recently, expecting synergistic (or additive) effects, many PPAR dual and pan agonists have been developed and investigated for their potential use in NAFLD therapy. The PPAR multi-receptor agonists in clinical trials for NAFLD are summarized in Table 7.
Glitazar is a class of dual agonists with varying degrees of agonism for PPAR-α/γ. Through pre-clinical and clinical studies, their combined effects on dyslipidemia (by PPAR-α) and insulin resistance (by PPAR- γ) have proven their utility in treating metabolic disorders [109]. A representative drug in this class is the saroglitazar. The saroglitazar is a marketed drug for treating T2DM and dyslipidemia in India (trade name: Lipaglyn) [110]. It is a PPAR-α/γ dual agonist but with predominant PPAR-α activity. As expected, in clinical studies, the saroglitazar could reduce TG, VLDL, and LDL cholesterols, increase HDL cholesterol and improve blood glucose profiles [111]. Regarding NAFLD, pre-clinical studies by Kumar et al revealed that saroglitazar could provide superior therapeutic effects to pioglitazone on improving not only hepatic steatosis and inflammation but also fibrosis in the high-fat western diet and lib sugar water (WDSW)-induced NAFLD mice [112]. Based on the successes of pre-clinical studies, there have been 8 clinical studies (keyword: “NAFLD” and “saroglitazar”) conducted or currently ongoing (ClinicalTrials.gov identifier: NCT03639623, NCT03617263, 04193982, NCT03061721, NCT02265276, NCT05211284, NCT03863574, NCT05011305). In phase 2 clinical trial, 16-week treatment of saroglitazar improved the markers of fibrosis and hepatocellular injury and reduced hepatic fat content (ClinicalTrials.gov identifier: NCT03061721) [85]. Unfortunately, other than saroglitazar, most glitazars have failed during development because of relevant adverse events, including potential carcinogenic or cardiovascular, renal, or bone marrow toxicity issues [109].
Elafibranor (GFT505) is a PPAR-α/δ dual agonist with preferential activity on the PPAR-α. It was investigated early for potential use in T2DM and hyperlipidemia but recently has drawn more interest in treating NAFLD. According to the pre-clinical studies by Staels et al, the elafibranor induced a significant reduction in hepatic gene expression related to inflammation (IL-1β, TNF-α, and F4/80) and fibrogenesis (TGF-β and TIMP-2), leading to reduction of steatosis, inflammation, and fibrosis [113]. In a phase 2b clinical trial, the elafibranor could resolve NASH in NAS over 4 patients with no worsening of hepatic fibrosis [114]. Unfortunately, a phase 3 trial for NASH was recently terminated due to limited efficacy (ClinicaTrials.gov identifier: NCT02704403).
Lanifibranor (IVA337) is a PPAR-α/γ/δ pan agonist. According to the pre-clinical study by Lefere et al, the lanifibranor could provide a superior anti-NAFLD activity than single agonists [fenofibrate (PPAR-α), pioglitazone (PPAR-γ) and GW501516 (PPAR-δ)] in different NAFLD mice models [115]. Notably, synergistically combined effects of the single agonists were realized in the lanifibranor-treated mice. The study results suggested improvement of hepatic steatosis by PPAR-α activation, reduction of hepatic inflammation and macrophage activation by PPAR-δ, and deactivation of HSCs by PPAR-γ [115]. In a completed phase 2 clinical trial with active NASH patients, lanifibranor treatment significantly improved the steatosis activity fibrosis (SAF) score and decreased liver enzyme levels and biomarkers of lipid, inflammation, and fibrosis [84]. Currently, 3 clinical trials (including 2 phase 3) are ongoing with lanifibranor to evaluate the therapeutic efficacy in NAFLD (ClinicalTrials.gov identifier: NCT05232071 NCT03459079, and NCT04849728). Active clinical trials of PPAR multi-receptor agonists are summarized in Table 7.
3.1.3. FGFR dual agonists
Recently, FGFR/GLP-1R dual agonists are gaining interest as potential NAFLD therapeutics. According to Liu et al, there appears to be a synergistic interplay loop among the FGFR and GLP-1R agonists. It was found that FGF21 mediates GLP-1R’s effects on inhibiting the hepatic glucose output, while the hepatic production of FGF21 could be upregulated by GLP-1R agonists (e.g., exenatide and liraglutide) [116]. The YH25724 and GLP-1-Fc-FGF21 D1 are long-acting dual agonists against the FGFR and GLP-1R. Both of them have shown promising effects in pre-clinical studies. In MCD-diet mice, the YH25724 more significantly alleviated hepatic fibrosis and inflammation than single agonists (Fc-GLP-1, Fc-FGF21, and dulaglutide). Consistently, in DIO mice, a significantly higher reduction in hepatic steatosis, plasma TG levels, and bodyweight was achieved by YH25724, compared with the single agonists [117]. The GLP-1-Fc-FGF21 D1, developed by Pan et al. is a long-acting GLP-1R/FGFR dual agonist that consists of an FGF21 with improved β-klotho binding property [118]. The HFD-fed ob/ob mice treated with the GLP-1-Fc-FGF21 D1 provided a significantly higher hypoglycemic effect with a more significant bodyweight reduction than single agonists. Furthermore, the GLP-1-Fc-FGF21 D1 also offered more significant therapeutic effects in attenuating NASH progression regarding hepatic function, lipid profiles, and NAS scores than the single agonists.
3.2. Development of long-acting derivatives
A bottleneck challenge for the clinical application of active proteins (or peptides) is their too-short plasma half-lives. Different strategies have been adopted to improve their pharmacokinetic (PK) profiles, including polymer conjugation and fusion with albumin or immunoglobulin G (IgG) Fc domain.
3.2.1. Polymer conjugation
The polymer conjugation strategy mainly relies on increasing the protein’s hydrodynamic radius to retard their glomerular filtration as well as physically interfere with the uptake by phagocytic or endothelial cells. Various polymers have been explored for modifying protein therapeutics. The most widely adopted polymer is polyethylene glycol (PEG), and there are also intrinsically disordered inert large polypeptide repeats such as XTEN, PAS (proline, alanine, Serine), and ELP (elastin-like polypeptide) [119].
Despite possessing potential anti-obesity activity, the short plasma half-life (12 min) of OXM remains a challenge to fully realize its effects [120]. Ma et al also reported the study of a PEGylated OXM analog with amino acid substituted for increased GCGR and GLP-1R activity (by 2- and 8-fold, respectively). The 40 kDa size PEG conjugation extended the plasma half-life of the OXM analog to 19.9 - 25.2 h (in mice). Seven weeks of once-weekly treatment of the modified OXM analog in db/db mice could provide more significant improvement in insulin sensitivity and glucose tolerance with reduced plasma TG and cholesterol levels than liraglutide. Furthermore, in diet-induced obese (DIO) mice, a more significant reduction in body fat mass and hepatic lipid content was also achieved compared to the liraglutide [98].
There have also been many efforts to develop PEGylated FGF21, as the short plasma half-life of FGF21 (0.7 - 1.1 h in mice) limits its therapeutic use [54]. A PEGylated variant of FGF21, B1344, was developed by Ye et al. [121]. To increase the potency of human FGF-21, they carried out an interesting approach, replacing a functional domain of the human FGF-21 with that of the mouse FGF21 and insertion of an alanine residue at the N-terminal site [121]. The FGF-21 mutant (ahmFGF-21) was further PEGylated at the N-terminal site with a 20 kDa mPEG-propionaldehyde. The PEGylated-ahmFGF-21 revealed an extended plasma half-life, reduced immunogenicity, and improved hypoglycemic and hyperlipidemic effects [121]. When the B1344 was tested in MCD-diet-induced NASH mice and obese cynomolgus monkeys, significant effects were observed regarding reduced hepatic fat content, bodyweight, plasma lipid profiles, hepatic steatosis, inflammation, and fibrosis [122].
Notably, there are reports of different approaches to achieve site-specific PEGylation of the FGF21. Xu et al reported a systematic study to engineer site-specific mono- and dual-PEGylated FGF21 that retain good activity but cause minimal vacuole formation (a potential risk caused by PEG). To achieve this goal, they produced FGF21 mutants by introducing cysteine residues at selected positions in the protein sequence (based on an FGFR1c binding model) and chemically conjugated them with sulfhydryl-reactive PEG. The results revealed that the PEGylation site could significantly affect the activity of FGF21, and the PEG configuration and number could also influence the vacuologenesis [123].
Pegbelfermin (BMS-986036) is another example of a long-acting FGF21, synthesized by site-specific conjugation of a linear 30 KDa size PEG to the FGF21 mutant introduced with an unnatural p-acetyl phenylalanine (pAcF) residue. The PEGylation could significantly extend the plasma half-life of FGF21 to 14.7 - 33.9 h in rats. In a pre-clinical study, twice-weekly treatment of the PEGylated FGF21 improved lipid profiles, pancreatic function, and insulin sensitivity [124]. The pegbelfermin has also been investigated in a series of clinical studies.[125] In a 16-week phase 2a clinical trial, once-a-week treatment with pegbelfermin significantly reduced hepatic fat content in NASH patients (ClinicalTrials.gov identifier: NCT03486912) [126]. In a different phase 2 clinical trial, pegbelfermin significantly improved the high-density lipoprotein (HDL) cholesterol, markers of fibrogenesis neoepitope-specific N-terminal pro-peptide of type III collagen (PRO-C3) in patients with diabetes and obesity (ClinicalTrials.gov identifier NCT02097277) [127]. Recently, 2 phase 2b clinical trials (ClinicalTrials.gov identifier: NCT03486912 and NCT03486899) have been carried out to evaluate the therapeutic effects of pegbelfermin in NASH and liver cirrhosis [128].
Pegozafermin (BIO89-100) is a site-specific glycoPEGylated recombinant FGF21 analog with an extended half-life of 55-100 h (in human), potentially allowing once weekly or every 2-week dosing [129,130]. The pegozafermin is prepared by chemically conjugating a 20 kDa size linear PEG to an FGF21 mutant, mediated by a glycosyl linker. In a phase 1b/2a trial, 12-week treatment with the pegozafermin significantly reduced hepatic volume and fat fraction and improved NAFLD-related markers (TG, LDL-C, HDL-C, non-HDL-C, adiponectin, bodyweight, ALT, and PRO-C3) [131]. A 24-week phase 2 clinical trial (ClinicalTrials.gov identifier: NCT04929483) is ongoing with NASH patients in F2/F3 fibrosis stage.
Apart from the PEGylation, conjugation of ELPs has also been employed for extending the plasma half-life of FGF21. The ELPs are peptide polymers composed of a repetitive (VPGXaaG) sequence (Xaa: any amino acid residues besides proline) [132]. Notably, they are thermally responsive and possess a tunable transition temperature that could be modulated by their Xaa residue and molecular size. By elaborately designing the ELP-drug sequence to have an adequate transition temperature (27 - 32℃), Gilroy et al could develop a GLP-1-ELP-FGF21 fusion protein that forms a depot after subcutaneous injection and released in a sustained-release fashion (absorption half-life: 7.6 days) [133]. The GLP-1-ELP-FGF21 fusion protein design consisted of 3 components: GLP-1 at the N-terminus, FGF21 at the C-terminus, and ELP in the middle. The presence of ELP between the GLP-1 and FGF21 provided flexibility, ensuring dual receptor activity. The GLP1-ELP-FGF21 showed comparable activity for GLP-1R and FGFR to the ELP-GLP1 and ELP-FGF21 [133]. However, probably due to additive (or synergistic) effects, treating GLP1-ELP-FGF21 to db/db mice provided a more significant glycemic control and bodyweight reduction compared to monotherapy or the treatment of GLP1/FGF21 mixture.
3.2.2. Exploiting neonatal Fc receptor (FcRn)-mediated recycling
Serum proteins such as albumin and IgG have a relatively long plasma half-life (generally several weeks) attributed to the FcRn-mediated recycling. The FcRn shares structural similarities with the MHC class I molecule and is expressed in many tissues and organs, such as the brain, heart, kidneys, and gut. The FcRn was first recognized as the carrier molecule for the transport of IgG from the mother to the fetus [134], but later, it was found that the FcRn plays a crucial role in extending the plasma half-life of the IgG and albumin [135]. The FcRn-mediated recycling relies on the pH-dependent binding of IgG (via the Fc region) and albumin with the FcRn. When the IgG or albumin in the bloodstream internalizes endothelial cells, high-affinity binding with the FcRn occurs at acidic pH (< 6.5) in the endosomes. The FcRn binding allows protection from enzymatic degradation and lets the IgG or albumin get recycled back to circulation (or transcytose to the other side) while other molecules unbound to FcRn become degraded in the lysosomes [134]. This FcRn-mediated recycling has been successfully exploited in many protein therapeutics by directly conjugating them with albumin or IgG (specifically the Fc region). An alternative way is to couple the proteins with albumin-binding moieties (e.g., lipids), which allows the formation of non-covalent complexes with circulating albumins after administration.
Human serum albumin (HSA), produced from the liver, is the most abundant plasma protein (accounts for around 50%) [136]. It comprises 3 homologous domains, with each domain made up of 2 subdomains. The HSA exhibits a strong pH-dependent binding affinity toward the FcRn, enabling it to utilize the FcRn-mediated recycling effectively [137]. The FcRn binding occurs at the hydrophobic cavities located between subdomains IIA and IIIA of the HSA. The high plasma concentration and an extended plasma half-life render albumin to become an effective carrier. Furthermore, the HSA can bind to various plasma substances, including fatty acids. The crystallographic investigation confirmed albumin binding with different size fatty acids and the presence of multiple binding sites [138]. These findings led to the development of long-acting lipid-conjugated protein therapeutics.
For the GLP-1R agonists, different approaches have been attempted to utilize the FcRn-mediated recycling, and some of the long-acting modified agonists have successfully reached clinical approval. A straightforward strategy to achieve the goal was directly fusing human serum albumin (HSA) to the drug candidate. For example, albiglutide is an approved long-acting GLP-1 fusion protein consisting of a modified GLP-1(7-36) dimer and human serum albumin. For improved resistance against DDP-4, the albiglutide has A8G substitutions for both the GLP-1s. The plasma half-life of albiglutide is 5 days (in human) [139]. Recently, a more widely adopted strategy has been to develop lipid-conjugated drugs. Liraglutide and semaglutide are good examples of lipid-conjugated long-acting GLP-1 analogs with high albumin binding efficacy. Liraglutide has a similar peptide backbone to the GLP-1(7-37) but differs by a K34R substitution and covalent conjugation of a palmitic acid (C16) to the acylated K26 position via a γ-glutamic acid spacer [140]. In the case of semaglutide, compared with liraglutide, it has an additional substitution at A8 with an unnatural 2-aminobutyric acid (Aib) for protection from DDP-4-induced degradation. The semaglutide also possesses an octadecanoic (C18) diacid moiety conjugated at K26 via a “γGlu-2xOEG” linker [141]. The reported plasma half-lives of liraglutide and semaglutide are 11-15 h and 1 week, respectively (plasma half-life of GLP-1: 1.5 - 5 min) [141,142,143]. The GLP-1R/GCGR dual agonist, cotadutide, is also made up of a palmitoylated 31-mer peptide, and the its plasma half-life in T2DM patients was 12.9 h [144].
Conjugation of proteins to the IgG Fc domain is also a widely adopted strategy to exploit FcRn-mediated recycling. The FcRn-binding site of the IgG Fc domain, positioned at the interface of CH2/CH3 domains, is conserved among different species [145]. The notable pH dependency of the IgG/FcRn binding is primarily attributable to the histidine residues on both sides. The histidine residues on the FcRn contribute to greater stability at acidic pH, while those on the IgG (e.g., H310 and H433 for IgG4) are directly engaged in salt bridge formation with the counterparts of FcRn [146]. On the FcRn, the IgG has a distinct binding site that is non-overlapping with albumin [137]. Dulaglutide is a good example of the class of IgG Fc-coupled drugs. Dulaglutide, an FDA-approved T2DM therapeutics, is a recombinant fusion protein consisting of 2 identical DPP-4-resistant modified GLP-1 (7-37) separately fused to each arm of modified IgG4 Fc domain via peptide linkers. The modified GLP-1 (7-37) composing the dulaglutide has an amino acid substitution at 3 positions (A8G, G22E, and R36G). Its long plasma half-life (90 – 95 h) makes it suitable for once-weekly s.c. administration [147]. Efinopegdutide (MK-6024, HM12525A) is a chemical conjugate of a synthetic GLP-1/GCG peptide with a human IgG Fc via a PEG linker [83]. This efinopegdutide also has a long plasma half-life of 112.5 – 276.2 h (with s.c. administration in human). Similarly, HM15211 is an IgG Fc domain conjugate of GLP-1R/GIPR/GCGR triple co-agonist linked by a flexible PEG linker. Its plasma half-life is 72.09 – 142.10 h (with s.c. administration in human) [148].
Efruxifermin (AR001, AMG876, Fc-FGF21[RGE]) is a fusion protein composed of a human IgG1 Fc domain and 2 modified FGF21s that have amino acid substitutions at 3 positions (L98R, P171G, and A180E) for reduced aggregation, protection from proteolytic degradation and increased binding affinity to β-klotho [149]. The reported plasma half-life of efruxifermin is 3 – 3.5 days (in human) [150]. The efruxifermin was clinically investigated for NASH therapy [150]. Sixteen-week of fruxifermin treatment (ClinicalTrials.gov identifier: NCT03976401) provided a significant reduction in the hepatic fat fraction (HFF), serum ALT levels and improvement in NAS (≥ 2 scores) without worsening of fibrosis. Reduction in fibrosis and hepatic injury markers by efruxifermin treatment was also reported [151]. Currently, two phase 2 trials (ClinicalTrials.gov identifier: NCT05039450 and NCT04767529) are underway. Pan et al developed a group of mutant FGF21 fusion proteins [118]. While screening for mutants with enhanced affinity to β-klotho, they also constructed long acting-FGF21 fusion proteins (composed of IgG4 Fc and the FGF21 mutant) and, further, GLP-1-Fc-FGF21 fusion proteins. In vitro assay results confirmed GLP-1-Fc-FGF21 fusion proteins possessing higher binding affinity toward the β-klotho and greater potency for downstream signal activation than a single agonist or a mixture. Animal studies also revealed more potent anti-diabetic and anti-obesity activity by the GLP-1-Fc-FGF21 fusion protein than single agonists. Notably, the plasma half-life of the GLP-1-Fc-FGF21 was 30.3 h (in mice) and 25.9 h (in rats).
3.3. Receptor-mediated hepatic targeting
A major challenge of hepatic drug delivery for the treatment of NAFLD is that only a small portion of the administered drug may reach the liver. An effective strategy to overcome this issue may be exploiting a nano-carrier, as the major distribution/elimination site of most of the NPs is the liver [152]. However, even though the NPs could successfully reach the liver, they would generally end up cleared by the Kupffer cells (KCs). Phagocytosed NPs by KCs would be eliminated from the system without eliciting any effects [153]. Other factors (e.g., size, surface charge, pathological status, etc.) could significantly affect the fate of the delivered NPs in the liver [154]. For example, the fenestrae of the LSECs allow the penetration of substances smaller than 200 nm [155]. When Park et al investigated the hepatic distribution of PLGA NPs (particle size: 271 nm and zeta potential: -28.3 mV), most of the NPs were found in non-parenchymal cells (KCs and LSECs), with 15% of the NPs accumulated in KCs, 20% in LSECs, 1% in HSCs, and only 4% in hepatocytes [156]. For cell-specific targeting, to date, there have been continuous research efforts to develop drug-loaded NPs decorated with ligands that could selectively bind to receptors expressed on different liver cell types. Such receptors that have been widely adopted for recent studies include the asialoglycoprotein receptor (ASGPR), hyaluronan receptor (HA-R), low-density lipoprotein receptor (LDLR), mannose receptor (MR), and retinol-binding protein receptors (RBPR).
ASGPR is a C-lectin-type scavenger receptor mainly present in hepatocytes [157]. The ASGPR clears up desialylated serum glycoproteins, and ligands with exposed terminal N-acetylgalactosamine (GalNAc) or galactose (Gal) residues could also be taken up by the ASGPR [157]. Owing to the selectivity toward galactose derivatives, high expression in hepatocytes, and induction of robust internalization, ASGPR-mediated targeting has been considered an appealing strategy for hepatic drug delivery [158]. Indeed, Bon et al reported a significantly higher hepatic distribution of anti-ASGPR antibody (42% I.D./g tissue at 8 h post-administration) over the control antibody (4.8% I.D./g tissue) [159]. Consistently, Sharma et al reported that a novel galactose dendrimer (GAL-24) could be preferentially localized to the liver [160]. Upon administration of GAL-24 to the mice, significant liver accumulation of 20% ID in 1 h and 80% ID in 24 h was reported. Notably, about 80% of the hepatocytes were positive for GAL-24, with minimal off-target accumulation and systemic toxicity.
Hyaluronan (HA) is a polymer that is an essential component of the extracellular matrix. The HA is mainly cleared by the lymph node but could also be cleared through HA-R-mediated hepatic clearance [161]. The HA-R has two isoforms, stablin-1 (Stab1) and stabilin-2 (Stab2). Among them, Stab2 is the primary receptor involved in HA clearance. This Stab2 is highly expressed in the liver, lymph nodes, and spleen [162]. Apart from the HA-R, the HA could also bind CD44 and mediate targeting to CD44 over-expressed activated HSCs. Exploiting the HA-R/CD44 for liver targeting, Yang et al developed a conjugate of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and HA (HA-TRAIL) to treat liver fibrosis. When administered to N-nitrosodimethylamine-induced hepatic fibrosis SD rat model, the HA-TRAIL showed a significantly higher delivery and a prolonged residency in the liver, compared to the unmodified TRAIL (> 4 days) after single i.v. administration [163]. Li et al developed a deoxycholic acid-modified HA-conjugated micelle loaded with silibinin [164]. Compared with the nonspecific micelle, the HA-conjugated micelle showed an HSC-specific delivery in the fibrotic liver and led to a more significant improvement of hepatic fibrosis condition in a CCl4-induced fibrosis rat model.
Retinol binding protein 4 (RBP4) is a transporter protein belonging to the lipocalin family [165]. This RBP4 is mainly produced from the liver (esp. in HSCs) and circulates through the blood. It serves a significant role in vitamin A (VA) transportation and cellular uptake [166]. Of note, the HSCs are a major site for retinol storage. About 80% of the total retinol is taken up from the blood circulation and stored in HSCs via the help of retinol-binding protein receptors (RBPRs) [167]. Considering the presence of RBPRs in the HSCs, retinol has often been exploited as a ligand for HSC targeting. For example, Qiao et al developed a retinol-modified polymeric micelle, poly(lactide-co-glycolide)-polyspermine-poly(ethylene glycol)-retinol (PLGA-PSPE-PEG-VA) loaded with silibinin and Col1α1 siRNA (siCol1α1) (“CGPVM”). The CGPVM highly accumulated in the liver (esp. for the fibrotic liver) and showed the greatest anti-fibrotic effects among the experimental groups [168]. Hassan et al developed a retinol-modified chitosan NP loaded with JQ1 and atorvastatin. They prepared low (0.59 retinol/nm2) and high (0.86 retinol/nm2) density retinol-tagged NPs and examined their HSC uptake and liver accumulation profiles in healthy and CCl4-induced hepatic fibrosis mice. Interestingly, compared with the unmodified NP, not the high-density retinol-tagged NP but the low-density retinol-tagged NP showed significantly higher (2.7-fold) uptake in HSC than the HEK 293 cells. Furthermore, only the low-density retinol-tagged NP revealed significantly higher (2-fold) liver accumulation in the fibrotic mice compared with the healthy mice. The authors suggested that the higher density of retinol on the surface of the NP might have caused steric hindrance to the interaction between the NPs and RBPs [169].
MR, also known as cluster of differentiation 206 (CD206), is a type I transmembrane receptor belonging to the family of C-type lectin family. The MR is present on the surface of various cells (e.g., macrophages, dendritic cells, and LSECs). Specifically, it was reported that there are present 20,000-25,000 MRs per cell on the LSECs [170]. The representative ligands of the MR include mannose, N-acetylglucosamine, and N-acetylgalactosamine (GalNAc) [171]. Because of the presence of MR in LSECs, MR has been considered a favorite target for hepatic drug delivery. Kim et al recently reported interesting data regarding cell-specific targeting in the liver. They evaluated the cell uptake and transfection of mRNA-loaded mannose-decorated LNPs. The results suggested that the lipid-PEG content, particle size, and a ligand (mannose) could be critical in targeting efficiency. With 1.5% PEG-lipid but without mannose, the mRNA transfected cells were 80, 40, and 10% for hepatocytes, LSECs, and Kupffer cells, respectively. With increasing the PEG-lipid content, the difference in transfected cell population became smaller between the hepatocytes and LSECs, while it got larger at lower PEG-lipid content. They mentioned that the higher PEG content may have reduced the apolipoprotein E (ApoE)-mediated cellular uptake of the LNPs by interfering with the adsorption of ApoE. Notably, with the decoration of mannose on the particles, the transfected cell ratios dramatically changed for the hepatocytes and LSECs (hepatocytes: 70 to 15%, and LSECs: 15 to 70%). The results further implicated that MR-mediated targeting could be a powerful tool for selective drug delivery to the LSECs [172].
LDLR is a cell surface endocytic receptor that mediates the uptake of low-density lipoproteins (LDL) rich in cholesterol. It recognizes apolipoprotein B100 (ApoB100) and ApoE, which are embedded in LDL particles and chylomicron/very low-density lipoprotein remnants, respectively. The LDLR is highly expressed in the liver and regulated by sterol regulatory element binding protein-2 (SREBP-2) [173]. Various approaches have been attempted to exploit the high affinity of LDLR to the ApoB100 and ApoE for hepatic drug delivery. Wang et al developed an ApoB100-decorated lipid NP loaded with anticancer drugs sorafenib and dihydroartemisinin to target hepatic cancer cells. The ApoB100-decorated NPs showed significantly higher uptake in the liver cancer cells than the control NPs [174]. Through cellular analysis on ldlr -/- hepatocytes, Akinc et al. demonstrated that siRNA-loaded lipid NP (LNP) could internalize hepatocytes by the ApoE-LDLR pathway. An ionizable lipid, DLinMCD3MA (MC3), was also reported to have hepatocyte homing properties mediated by the affinity of ApoE to the LDLR [175]. In 2018, the FDA approved an siRNA NP formulation based on a second-generation MC3-LNP for treating polyneuropathies named ONPATTRO (ALN-TTR01).
4. Conclusion and future perspectives
To date, extensive research has been conducted to discover effective therapeutics for treating NAFLD. A broad spectrum of drug candidates has been studied, and a group has entered clinical investigations. On the other hand, various pharmaceutical strategies, including (the development of co-agonists or long-acting drugs and devising active hepatic targeting techniques) have been explored to improve the therapeutic efficacy of the drugs. There has yet been no significant success in clinical, but there are growing expectations for the advent of effective and safe drugs for treating NAFLD in the near future.
Author Contributions
Conceptualization, K.A.M. and M.C.S.; investigation, R.A., D.L., K.A.M. and M.C.S.; resources, K.A.M. and M.C.S.; visualization, R.A. and M.C.S.; project administration, K.A.M. and M.C.S.; funding acquisition, K.A.M. and M.C.S; supervision, K.A.M. and M.C.S.; data curation, R.A., D.L., K.A.M., and M.C.S.; writing—original draft preparation, R.A., D.L., K.A.M. and M.C.S.; writing—review and editing, R.A., K.A.M., and M.C.S. All authors have read and agreed to the published version of the manuscript.
Funding
This work was financially supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT; Ministry of Science and ICT) (NRF-2021R1F1A1058214 to M.C.S., NRF-2021R1F1A1064206 to K.A.M.) and also supported by the NRF funded by the Ministry of Education, Science and Technology (NRF-2018R1D1A1A02047809 to M.C.S., and NRF-2018R1D1A1B07048818 to K.A.M.).
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Clark, J.M.; Brancati, F.L.; Diehl, A.M. Nonalcoholic fatty liver disease. Gastroenterology 2002, 122, 1649–1657. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nature reviews Gastroenterology & hepatology 2018, 15, 11–20. [Google Scholar]
- Afolabi, B.I.; Ibitoye, B.O.; Ikem, R.T.; Omisore, A.D.; Idowu, B.M.; Soyoye, D.O. The relationship between glycaemic control and non-alcoholic fatty liver disease in Nigerian type 2 diabetic patients. Journal of the National Medical Association 2018, 110, 256–264. [Google Scholar] [CrossRef]
- Liu, M.; Wang, J.; Zeng, J.; Cao, X.; He, Y. Association of NAFLD with diabetes and the impact of BMI changes: a 5-year cohort study based on 18,507 elderly. The Journal of Clinical Endocrinology & Metabolism 2017, 102, 1309–1316. [Google Scholar]
- Younossi, Z.M. Non-alcoholic fatty liver disease–a global public health perspective. Journal of hepatology 2019, 70, 531–544. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-h.; Cho, Y.; Lee, B.-W.; Park, C.-Y.; Lee, D.H.; Cha, B.-S.; Rhee, E.-J. Nonalcoholic fatty liver disease in diabetes. Part I: epidemiology and diagnosis. Diabetes & metabolism journal 2019, 43, 31–45. [Google Scholar]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nature medicine 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Treeprasertsuk, S.; Björnsson, E.; Enders, F.; Suwanwalaikorn, S.; Lindor, K.D. NAFLD fibrosis score: a prognostic predictor for mortality and liver complications among NAFLD patients. World journal of gastroenterology: WJG 2013, 19, 1219. [Google Scholar] [CrossRef]
- Eren, F.; Kaya, E.; Yilmaz, Y. Accuracy of Fibrosis-4 index and non-alcoholic fatty liver disease fibrosis scores in metabolic (dysfunction) associated fatty liver disease according to body mass index: failure in the prediction of advanced fibrosis in lean and morbidly obese individuals. European journal of gastroenterology & hepatology 2022, 34, 98–103. [Google Scholar]
- Sanyal, A.J.; Harrison, S.A.; Ratziu, V.; Abdelmalek, M.F.; Diehl, A.M.; Caldwell, S.; Shiffman, M.L.; Aguilar Schall, R.; Jia, C.; McColgan, B. The natural history of advanced fibrosis due to nonalcoholic steatohepatitis: data from the simtuzumab trials. Hepatology 2019, 70, 1913–1927. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Duan, Q.; Wu, R.; Harris, E.N.; Su, Q. Pathophysiological communication between hepatocytes and non-parenchymal cells in liver injury from NAFLD to liver fibrosis. Advanced Drug Delivery Reviews 2021, 176, 113869. [Google Scholar] [CrossRef]
- Hammoutene, A.; Rautou, P.-E. Role of liver sinusoidal endothelial cells in non-alcoholic fatty liver disease. Journal of hepatology 2019, 70, 1278–1291. [Google Scholar] [CrossRef]
- Lang, A.; Schoonhoven, R.; Tuvia, S.; Brenner, D.A.; Rippe, R.A. Nuclear factor κB in proliferation, activation, and apoptosis in rat hepatic stellate cells. Journal of hepatology 2000, 33, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Marra, F.; Efsen, E.; Romanelli, R.G.; Caligiuri, A.; Pastacaldi, S.; Batignani, G.; Bonacchi, A.; Caporale, R.; Laffi, G.; Pinzani, M. Ligands of peroxisome proliferator-activated receptor γ modulate profibrogenic and proinflammatory actions in hepatic stellate cells. Gastroenterology 2000, 119, 466–478. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. Journal of Biological Chemistry 2000, 275, 2247–2250. [Google Scholar] [CrossRef]
- Pierantonelli, I.; Rychlicki, C.; Agostinelli, L.; Giordano, D.M.; Gaggini, M.; Fraumene, C.; Saponaro, C.; Manghina, V.; Sartini, L.; Mingarelli, E. Lack of NLRP3-inflammasome leads to gut-liver axis derangement, gut dysbiosis and a worsened phenotype in a mouse model of NAFLD. Scientific reports 2017, 7, 12200. [Google Scholar] [CrossRef]
- Tilg, H.; Effenberger, M. From NAFLD to MAFLD: when pathophysiology succeeds. Nature reviews Gastroenterology & hepatology 2020, 17, 387–388. [Google Scholar]
- Finotti, M.; Romano, M.; Auricchio, P.; Scopelliti, M.; Brizzolari, M.; Grossi, U.; Piccino, M.; Benvenuti, S.; Morana, G.; Cillo, U. Target therapies for NASH/NAFLD: from the molecular aspect to the pharmacological and surgical alternatives. Journal of Personalized Medicine 2021, 11, 499. [Google Scholar] [CrossRef]
- Sumida, Y.; Naito, Y.; Tanaka, S.; Sakai, K.; Inada, Y.; Taketani, H.; Kanemasa, K.; Yasui, K.; Itoh, Y.; Okanoue, T. Long-term (>= 2 yr) efficacy of vitamin E for non-alcoholic steatohepatitis. Hepatogastroenterology 2013, 60, 1445–1450. [Google Scholar]
- Sasaki, A.; Nitta, H.; Otsuka, K.; Umemura, A.; Baba, S.; Obuchi, T.; Wakabayashi, G. Bariatric surgery and non-alcoholic Fatty liver disease: current and potential future treatments. Frontiers in endocrinology 2014, 5, 164. [Google Scholar] [CrossRef]
- Saeed, N.; Glass, L.; Sharma, P.; Shannon, C.; Sonnenday, C.J.; Tincopa, M.A. Incidence and risks for nonalcoholic fatty liver disease and steatohepatitis post-liver transplant: systematic review and meta-analysis. Transplantation 2019, 103, e345–e354. [Google Scholar] [CrossRef]
- Cardoso, A.C.; de Figueiredo-Mendes, C.A.; Villela-Nogueira, C.; A. Current management of NAFLD/NASH. Liver International 2021, 41, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Androutsakos, T.; Nasiri-Ansari, N.; Bakasis, A.-D.; Kyrou, I.; Efstathopoulos, E.; Randeva, H.S.; Kassi, E. SGLT-2 inhibitors in NAFLD: expanding their role beyond diabetes and cardioprotection. International journal of molecular sciences 2022, 23, 3107. [Google Scholar] [CrossRef] [PubMed]
- Jojima, T.; Tomotsune, T.; Iijima, T.; Akimoto, K.; Suzuki, K.; Aso, Y. Empagliflozin (an SGLT2 inhibitor), alone or in combination with linagliptin (a DPP-4 inhibitor), prevents steatohepatitis in a novel mouse model of non-alcoholic steatohepatitis and diabetes. Diabetology & metabolic syndrome 2016, 8, 1–11. [Google Scholar]
- Wallenius, K.; Kroon, T.; Hagstedt, T.; Löfgren, L.; Sörhede-Winzell, M.; Boucher, J.; Lindén, D.; Oakes, N.D. The SGLT2 inhibitor dapagliflozin promotes systemic FFA mobilization, enhances hepatic β-oxidation, and induces ketosis. Journal of lipid research 2022, 63. [Google Scholar] [CrossRef]
- Komiya, C.; Tsuchiya, K.; Shiba, K.; Miyachi, Y.; Furuke, S.; Shimazu, N.; Yamaguchi, S.; Kanno, K.; Ogawa, Y. Ipragliflozin improves hepatic steatosis in obese mice and liver dysfunction in type 2 diabetic patients irrespective of body weight reduction. PloS one 2016, 11, e0151511. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Kessoku, T.; Kawanaka, M.; Nonaka, M.; Hyogo, H.; Fujii, H.; Nakajima, T.; Imajo, K.; Tanaka, K.; Kubotsu, Y. Ipragliflozin improves the hepatic outcomes of patients with diabetes with NAFLD. Hepatology communications 2022, 6, 120–132. [Google Scholar] [CrossRef]
- Han, E.; Lee, Y.-h.; Lee, B.-W.; Kang, E.S.; Cha, B.-S. Ipragliflozin additively ameliorates non-alcoholic fatty liver disease in patients with type 2 diabetes controlled with metformin and pioglitazone: a 24-week randomized controlled trial. Journal of clinical medicine 2020, 9, 259. [Google Scholar] [CrossRef]
- He, K.; Li, J.; Xi, W.; Ge, J.; Sun, J.; Jing, Z. Dapagliflozin for nonalcoholic fatty liver disease: a systematic review and meta-analysis. Diabetes research and clinical practice 2022, 109791. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, J.W.; Lundkvist, P.; Jansson, P.-A.; Johansson, L.; Kvarnström, M.; Moris, L.; Miliotis, T.; Forsberg, G.-B.; Risérus, U.; Lind, L. Effects of dapagliflozin and n-3 carboxylic acids on non-alcoholic fatty liver disease in people with type 2 diabetes: a double-blind randomised placebo-controlled study. Diabetologia 2018, 61, 1923–1934. [Google Scholar] [CrossRef] [PubMed]
- Sumida, Y.; Murotani, K.; Saito, M.; Tamasawa, A.; Osonoi, Y.; Yoneda, M.; Osonoi, T. Effect of luseogliflozin on hepatic fat content in type 2 diabetes patients with non-alcoholic fatty liver disease: a prospective, single-arm trial (LEAD trial). Hepatology Research 2019, 49, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Kuchay, M.S.; Krishan, S.; Mishra, S.K.; Farooqui, K.J.; Singh, M.K.; Wasir, J.S.; Bansal, B.; Kaur, P.; Jevalikar, G.; Gill, H.K. Effect of empagliflozin on liver fat in patients with type 2 diabetes and nonalcoholic fatty liver disease: a randomized controlled trial (E-LIFT Trial). Diabetes care 2018, 41, 1801–1808. [Google Scholar] [CrossRef] [PubMed]
- Itani, T.; Ishihara, T. Efficacy of canagliflozin against nonalcoholic fatty liver disease: a prospective cohort study. Obesity Science & Practice 2018, 4, 477–482. [Google Scholar]
- Andersen, A.; Lund, A.; Knop, F.K.; Vilsbøll, T. Glucagon-like peptide 1 in health and disease. Nature Reviews Endocrinology 2018, 14, 390–403. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Liang, Y.; Luo, Y.; Li, Z.; Wen, Y.; Shen, J.; Li, R.; Zheng, H.; Gu, H.F.; Xia, N. Liraglutide ameliorates nonalcoholic fatty liver disease in diabetic mice via the IRS2/PI3K/Akt signaling pathway. Diabetes, Metabolic Syndrome and Obesity: Targets and Therapy 2019, 12, 1013. [Google Scholar] [CrossRef]
- Ao, N.; Ma, Z.; Yang, J.; Jin, S.; Zhang, K.; Luo, E.; Du, J. Liraglutide ameliorates lipotoxicity-induced inflammation through the mTORC1 signalling pathway. Peptides 2020, 133, 170375. [Google Scholar] [CrossRef]
- Ali, E.S.; Girard, D.; Petrovsky, N. Impaired Ca2+ signaling due to hepatic steatosis mediates hepatic insulin resistance in Alström syndrome mice that is reversed by GLP-1 analog treatment. American Journal of Physiology-Cell Physiology 2021, 321, C187–C198. [Google Scholar] [CrossRef]
- Chen, Y.; Xu, Y.-n.; Ye, C.-y.; Feng, W.-b.; Zhou, Q.-t.; Yang, D.-h.; Wang, M.-w. GLP-1 mimetics as a potential therapy for nonalcoholic steatohepatitis. Acta Pharmacologica Sinica 2022, 43, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- Duparc, T.; Briand, F.; Trenteseaux, C.; Merian, J.; Combes, G.; Najib, S.; Sulpice, T.; Martinez, L.O. Liraglutide improves hepatic steatosis and metabolic dysfunctions in a 3-week dietary mouse model of nonalcoholic steatohepatitis. American Journal of Physiology-Gastrointestinal and Liver Physiology 2019, 317, G508–G517. [Google Scholar] [CrossRef] [PubMed]
- Somm, E.; Montandon, S.A.; Loizides-Mangold, U.; Gaïa, N.; Lazarevic, V.; De Vito, C.; Perroud, E.; Bochaton-Piallat, M.-L.; Dibner, C.; Schrenzel, J. The GLP-1R agonist liraglutide limits hepatic lipotoxicity and inflammatory response in mice fed a methionine-choline deficient diet. Translational Research 2021, 227, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Pontes-da-Silva, R.M.; de Souza Marinho, T.; de Macedo Cardoso, L.E.; Mandarim-de-Lacerda, C.A.; Aguila, M.B. Obese mice weight loss role on nonalcoholic fatty liver disease and endoplasmic reticulum stress treated by a GLP-1 receptor agonist. International Journal of Obesity 2022, 46, 21–29. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Hull, D.; Guo, K.; Barton, D.; Hazlehurst, J.M.; Gathercole, L.L.; Nasiri, M.; Yu, J.; Gough, S.C.; Newsome, P.N. Glucagon-like peptide 1 decreases lipotoxicity in non-alcoholic steatohepatitis. Journal of hepatology 2016, 64, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Gaunt, P.; Aithal, G.P.; Barton, D.; Hull, D.; Parker, R.; Hazlehurst, J.M.; Guo, K.; Abouda, G.; Aldersley, M.A. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. The Lancet 2016, 387, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Song, T.; Jia, Y.; Li, Z.; Wang, F.; Ren, L.; Chen, S. Effects of liraglutide on nonalcoholic fatty liver disease in patients with type 2 diabetes mellitus: a systematic review and meta-analysis. Diabetes Therapy 2021, 12, 1735–1749. [Google Scholar] [CrossRef] [PubMed]
- Newsome, P.; Francque, S.; Harrison, S.; Ratziu, V.; Van Gaal, L.; Calanna, S.; Hansen, M.; Linder, M.; Sanyal, A. Effect of semaglutide on liver enzymes and markers of inflammation in subjects with type 2 diabetes and/or obesity. Alimentary pharmacology & therapeutics 2019, 50, 193–203. [Google Scholar]
- Mantovani, A.; Petracca, G.; Beatrice, G.; Csermely, A.; Lonardo, A.; Targher, G. Glucagon-like peptide-1 receptor agonists for treatment of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis: an updated meta-analysis of randomized controlled trials. Metabolites 2021, 11, 73. [Google Scholar] [CrossRef]
- Rubino, D.M.; Greenway, F.L.; Khalid, U.; O’Neil, P.M.; Rosenstock, J.; Sørrig, R.; Wadden, T.A.; Wizert, A.; Garvey, W.T.; Arauz-Pacheco, C. Effect of weekly subcutaneous semaglutide vs daily liraglutide on body weight in adults with overweight or obesity without diabetes: the STEP 8 randomized clinical trial. Jama 2022, 327, 138–150. [Google Scholar] [CrossRef]
- Choudhary, N.S.; Kumar, N.; Duseja, A. Peroxisome proliferator-activated receptors and their agonists in nonalcoholic fatty liver disease. Journal of Clinical and Experimental Hepatology 2019, 9, 731–739. [Google Scholar] [CrossRef]
- Gross, B.; Pawlak, M.; Lefebvre, P.; Staels, B. PPARs in obesity-induced T2DM, dyslipidaemia and NAFLD. Nature Reviews Endocrinology 2017, 13, 36–49. [Google Scholar] [CrossRef]
- Grygiel-Górniak, B. Peroxisome proliferator-activated receptors and their ligands: nutritional and clinical implications-a review. Nutrition journal 2014, 13, 1–10. [Google Scholar] [CrossRef]
- Lian, J.; Fu, J. Pioglitazone for NAFLD patients with prediabetes or type 2 diabetes mellitus: a meta-analysis. Frontiers in endocrinology 2021, 12, 615409. [Google Scholar] [CrossRef]
- Powers, C.; McLeskey, S.; Wellstein, A. Fibroblast growth factors, their receptors and signaling. Endocrine-related cancer 2000, 7, 165–197. [Google Scholar] [CrossRef] [PubMed]
- Kharitonenkov, A.; Shiyanova, T.L.; Koester, A.; Ford, A.M.; Micanovic, R.; Galbreath, E.J.; Sandusky, G.E.; Hammond, L.J.; Moyers, J.S.; Owens, R.A. FGF-21 as a novel metabolic regulator. The Journal of clinical investigation 2005, 115, 1627–1635. [Google Scholar] [CrossRef]
- Wang, W.-F.; Li, S.-M.; Ren, G.-P.; Zheng, W.; Lu, Y.-J.; Yu, Y.-H.; Xu, W.-J.; Li, T.-H.; Zhou, L.-H.; Liu, Y. Recombinant murine fibroblast growth factor 21 ameliorates obesity-related inflammation in monosodium glutamate-induced obesity rats. Endocrine 2015, 49, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Keinicke, H.; Sun, G.; Mentzel, C.M.J.; Fredholm, M.; John, L.M.; Andersen, B.; Raun, K.; Kjaergaard, M. FGF21 regulates hepatic metabolic pathways to improve steatosis and inflammation. Endocrine connections 2020, 9, 755–768. [Google Scholar] [CrossRef]
- Kolumam, G.; Chen, M.Z.; Tong, R.; Zavala-Solorio, J.; Kates, L.; van Bruggen, N.; Ross, J.; Wyatt, S.K.; Gandham, V.D.; Carano, R.A. Sustained brown fat stimulation and insulin sensitization by a humanized bispecific antibody agonist for fibroblast growth factor receptor 1/βKlotho complex. EBioMedicine 2015, 2, 730–743. [Google Scholar] [CrossRef]
- Wong, C.; Dash, A.; Fredrickson, J.; Lewin-Koh, N.; Chen, S.; Yoshida, K.; Liu, Y.; Gutierrez, J.; Kunder, R. Fibroblast growth factor receptor 1/Klothoβ agonist BFKB8488A improves lipids and liver health markers in patients with diabetes or NAFLD: A phase 1b randomized trial. Hepatology 2022. [Google Scholar] [CrossRef] [PubMed]
- Depaoli, A.; Phung, V.; Bashir, M.R.; Morrow, L.; Beysen, C.; Yan, A.; Ling, L.; Baxter, B.; Luskey, K.L.; Olefsky, J.M. 140-LB: NGM313, a novel activator of b-Klotho/FGFR1c, improves insulin resistance and reduces hepatic fat in obese, nondiabetic subjects. Diabetes 2019, 68. [Google Scholar] [CrossRef]
- Kurosu, H.; Choi, M.; Ogawa, Y.; Dickson, A.S.; Goetz, R.; Eliseenkova, A.V.; Mohammadi, M.; Rosenblatt, K.P.; Kliewer, S.A.; Kuro-o, M. Tissue-specific expression of βKlotho and fibroblast growth factor (FGF) receptor isoforms determines metabolic activity of FGF19 and FGF21. Journal of Biological Chemistry 2007, 282, 26687–26695. [Google Scholar] [CrossRef]
- Wunsch, E.; Milkiewicz, M.; Wasik, U.; Trottier, J.; Kempińska-Podhorodecka, A.; Elias, E.; Barbier, O.; Milkiewicz, P. Expression of hepatic fibroblast growth factor 19 is enhanced in primary biliary cirrhosis and correlates with severity of the disease. Scientific reports 2015, 5, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wojcik, M.; Janus, D.; Dolezal-Oltarzewska, K.; Kalicka-Kasperczyk, A.; Poplawska, K.; Drozdz, D.; Sztefko, K.; Starzyk, J.B. A decrease in fasting FGF19 levels is associated with the development of non-alcoholic fatty liver disease in obese adolescents. Journal of Pediatric Endocrinology and Metabolism 2012, 25, 1089–1093. [Google Scholar] [CrossRef] [PubMed]
- Gadaleta, R.M.; Scialpi, N.; Peres, C.; Cariello, M.; Ko, B.; Luo, J.; Porru, E.; Roda, A.; Sabbà, C.; Moschetta, A. Suppression of hepatic bile acid synthesis by a non-tumorigenic FGF19 analogue protects mice from fibrosis and hepatocarcinogenesis. Scientific Reports 2018, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Rossi, S.J.; Paredes, A.H.; Trotter, J.F.; Bashir, M.R.; Guy, C.D.; Banerjee, R.; Jaros, M.J.; Owers, S.; Baxter, B.A. NGM282 improves liver fibrosis and histology in 12 weeks in patients with nonalcoholic steatohepatitis. Hepatology 2020, 71, 1198–1212. [Google Scholar] [CrossRef]
- Rinella, M.E.; Trotter, J.F.; Abdelmalek, M.F.; Paredes, A.H.; Connelly, M.A.; Jaros, M.J.; Ling, L.; Rossi, S.J.; DePaoli, A.M.; Harrison, S.A. Rosuvastatin improves the FGF19 analogue NGM282-associated lipid changes in patients with non-alcoholic steatohepatitis. Journal of Hepatology 2019, 70, 735–744. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, F.; Guo, G.L. Tissue-specific function of farnesoid X receptor in liver and intestine. Pharmacological research 2011, 63, 259–265. [Google Scholar] [CrossRef]
- Bjursell, M.; Wedin, M.; Admyre, T.; Hermansson, M.; Böttcher, G.; Göransson, M.; Lindén, D.; Bamberg, K.; Oscarsson, J.; Bohlooly-Y, M. Ageing Fxr deficient mice develop increased energy expenditure, improved glucose control and liver damage resembling NASH. PloS one 2013, 8, e64721. [Google Scholar] [CrossRef]
- Cariou, B.; Staels, B. FXR: a promising target for the metabolic syndrome? Trends in pharmacological sciences 2007, 28, 236–243. [Google Scholar] [CrossRef]
- Zhou, S.; You, H.; Qiu, S.; Yu, D.; Bai, Y.; He, J.; Cao, H.; Che, Q.; Guo, J.; Su, Z. A new perspective on NAFLD: Focusing on the crosstalk between peroxisome proliferator-activated receptor alpha (PPARα) and farnesoid X receptor (FXR). Biomedicine & Pharmacotherapy 2022, 154, 113577. [Google Scholar]
- Miyazaki, T.; Honda, A.; Ikegami, T.; Iida, T.; Matsuzaki, Y. Human-specific dual regulations of FXR-activation for reduction of fatty liver using in vitro cell culture model. Journal of Clinical Biochemistry and Nutrition 2019, 64, 112–123. [Google Scholar] [CrossRef]
- Fiorucci, S.; Mencarelli, A.; Palladino, G.; Cipriani, S. Bile-acid-activated receptors: targeting TGR5 and farnesoid-X-receptor in lipid and glucose disorders. Trends in pharmacological sciences 2009, 30, 570–580. [Google Scholar] [CrossRef] [PubMed]
- Alemi, F.; Kwon, E.; Poole, D.P.; Lieu, T.; Lyo, V.; Cattaruzza, F.; Cevikbas, F.; Steinhoff, M.; Nassini, R.; Materazzi, S. The TGR5 receptor mediates bile acid–induced itch and analgesia. The Journal of clinical investigation 2013, 123, 1513–1530. [Google Scholar] [CrossRef] [PubMed]
- Pellicciari, R.; Fiorucci, S.; Camaioni, E.; Clerici, C.; Costantino, G.; Maloney, P.R.; Morelli, A.; Parks, D.J.; Willson, T.M. 6α-ethyl-chenodeoxycholic acid (6-ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. Journal of medicinal chemistry 2002, 45, 3569–3572. [Google Scholar] [PubMed]
- Zhang, Y.; Jackson, J.P.; St. Claire III, R.L.; Freeman, K.; Brouwer, K.R.; Edwards, J.E. Obeticholic acid, a selective farnesoid X receptor agonist, regulates bile acid homeostasis in sandwich-cultured human hepatocytes. Pharmacology Research & Perspectives 2017, 5, e00329. [Google Scholar]
- Goto, T.; Itoh, M.; Suganami, T.; Kanai, S.; Shirakawa, I.; Sakai, T.; Asakawa, M.; Yoneyama, T.; Kai, T.; Ogawa, Y. Obeticholic acid protects against hepatocyte death and liver fibrosis in a murine model of nonalcoholic steatohepatitis. Scientific reports 2018, 8, 1–13. [Google Scholar] [CrossRef]
- Zhang, G.; Wang, X.; Chung, T.-Y.; Ye, W.; Hodge, L.; Zhang, L.; Chng, K.; Xiao, Y.-F.; Wang, Y.J. Carbon tetrachloride (CCl4) accelerated development of non-alcoholic fatty liver disease (NAFLD)/steatohepatitis (NASH) in MS-NASH mice fed western diet supplemented with fructose (WDF). BMC gastroenterology 2020, 20, 1–13. [Google Scholar] [CrossRef]
- Rizzo, G.; Passeri, D.; De Franco, F.; Ciaccioli, G.; Donadio, L.; Rizzo, G.; Orlandi, S.; Sadeghpour, B.; Wang, X.X.; Jiang, T. Functional characterization of the semisynthetic bile acid derivative INT-767, a dual farnesoid X receptor and TGR5 agonist. Molecular pharmacology 2010, 78, 617–630. [Google Scholar] [CrossRef]
- Roth, J.D.; Feigh, M.; Veidal, S.S.; Fensholdt, L.K.; Rigbolt, K.T.; Hansen, H.H.; Chen, L.C.; Petitjean, M.; Friley, W.; Vrang, N. INT-767 improves histopathological features in a diet-induced ob/ob mouse model of biopsy-confirmed non-alcoholic steatohepatitis. World Journal of Gastroenterology 2018, 24, 195. [Google Scholar] [CrossRef]
- Comeglio, P.; Cellai, I.; Mello, T.; Filippi, S.; Maneschi, E.; Corcetto, F.; Corno, C.; Sarchielli, E.; Morelli, A.; Rapizzi, E. INT-767 prevents NASH and promotes visceral fat brown adipogenesis and mitochondrial function. Journal of Endocrinology 2018, 238, 107–127. [Google Scholar] [CrossRef]
- Chau, M.; Li, Y.; Roqueta-Rivera, M.; Garlick, K.; Shen, R.; Wang, G.; Or, Y.S.; Jiang, L.-J. Characterization of EDP-305, a highly potent and selective farnesoid X receptor agonist, for the treatment of non-alcoholic steatohepatitis. Int J Gastroenterol 2019, 3, 4–16. [Google Scholar] [CrossRef]
- Schwabl, P.; Hambruch, E.; Budas, G.R.; Supper, P.; Burnet, M.; Liles, J.T.; Birkel, M.; Brusilovskaya, K.; Königshofer, P.; Peck-Radosavljevic, M. The non-steroidal FXR agonist cilofexor improves portal hypertension and reduces hepatic fibrosis in a rat NASH model. Biomedicines 2021, 9, 60. [Google Scholar] [CrossRef] [PubMed]
- Chianelli, D.; Rucker, P.V.; Roland, J.; Tully, D.C.; Nelson, J.; Liu, X.; Bursulaya, B.; Hernandez, E.D.; Wu, J.; Prashad, M. Nidufexor (LMB763), a novel FXR modulator for the treatment of nonalcoholic steatohepatitis. Journal of medicinal chemistry 2020, 63, 3868–3880. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Kim, J.-H.; Yi, J.; Han, O.; Kim, Y.; Baek, E.; Jung, S.Y.; Kwon, S.C.; Trautmann, M.E.; Hompesch, M. The ultra-long acting LAPS GLP/GCG dual agonist, HM12525A, demonstrated safety and prolonged pharmacokinetics in healthy volunteers: a phase 1 first-in-human study. Diabetologia 2015, 58. [Google Scholar]
- Francque, S.M.; Bedossa, P.; Ratziu, V.; Anstee, Q.M.; Bugianesi, E.; Sanyal, A.J.; Loomba, R.; Harrison, S.A.; Balabanska, R.; Mateva, L. A randomized, controlled trial of the pan-PPAR agonist lanifibranor in NASH. New England Journal of Medicine 2021, 385, 1547–1558. [Google Scholar] [PubMed]
- Gawrieh, S.; Noureddin, M.; Loo, N.; Mohseni, R.; Awasty, V.; Cusi, K.S.; Kowdley, K.; Lai, M.; Schiff, E.; Parmar, D. a PPAR-α/γ Agonist, for Treatment of Nonalcoholic Fatty Liver Disease: A Randomized Controlled Double-Blind Phase 2 Trial. Hepatology 2021, 74, 1809–1824. [Google Scholar] [PubMed]
- Gabe, M.B.N.; van der Velden, W.J.; Smit, F.X.; Gasbjerg, L.S.; Rosenkilde, M.M. Molecular interactions of full-length and truncated GIP peptides with the GIP receptor–A comprehensive review. Peptides 2020, 125, 170224. [Google Scholar] [CrossRef]
- Bastin, M.; Andreelli, F. Dual GIP–GLP1-receptor agonists in the treatment of type 2 diabetes: a short review on emerging data and therapeutic potential. Diabetes, Metabolic Syndrome and Obesity: Targets and Therapy 2019, 1973–1985. [Google Scholar] [CrossRef]
- Seino, Y.; Fukushima, M.; Yabe, D. GIP and GLP-1, the two incretin hormones: similarities and differences. Journal of diabetes investigation 2010, 1, 8–23. [Google Scholar] [CrossRef]
- Yamada, Y.; Seino, Y. Physiology of GIP-a lesson from GIP receptor knockout mice. Hormone and metabolic research 2004, 36, 771–774. [Google Scholar]
- Nauck, M.A.; Meier, J.J. The incretin effect in healthy individuals and those with type 2 diabetes: physiology, pathophysiology, and response to therapeutic interventions. The lancet Diabetes & endocrinology 2016, 4, 525–536. [Google Scholar]
- Hartman, M.L.; Sanyal, A.J.; Loomba, R.; Wilson, J.M.; Nikooienejad, A.; Bray, R.; Karanikas, C.A.; Duffin, K.L.; Robins, D.A.; Haupt, A. Effects of novel dual GIP and GLP-1 receptor agonist tirzepatide on biomarkers of nonalcoholic steatohepatitis in patients with type 2 diabetes. Diabetes Care 2020, 43, 1352–1355. [Google Scholar] [PubMed]
- Zhao, F.; Zhou, Q.; Cong, Z.; Hang, K.; Zou, X.; Zhang, C.; Chen, Y.; Dai, A.; Liang, A.; Ming, Q. Structural insights into multiplexed pharmacological actions of tirzepatide and peptide 20 at the GIP, GLP-1 or glucagon receptors. Nature Communications 2022, 13, 1057. [Google Scholar]
- Unger, R.H.; Orci, L. Glucagon and the A cell: physiology and pathophysiology. New England Journal of Medicine 1981, 304, 1575–1580. [Google Scholar] [CrossRef]
- Zhang, X.-X.; Pan, Y.-H.; Huang, Y.-M.; Zhao, H.-L. Neuroendocrine hormone amylin in diabetes. World journal of diabetes 2016, 7, 189. [Google Scholar] [CrossRef]
- Habegger, K.M.; Heppner, K.M.; Geary, N.; Bartness, T.J.; DiMarchi, R.; Tschöp, M.H. The metabolic actions of glucagon revisited. Nature Reviews Endocrinology 2010, 6, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Galsgaard, K.D. The vicious circle of hepatic glucagon resistance in non-alcoholic fatty liver disease. Journal of Clinical Medicine 2020, 9, 4049. [Google Scholar] [PubMed]
- Cohen, M.A.; Ellis, S.M.; Le Roux, C.W.; Batterham, R.L.; Park, A.; Patterson, M.; Frost, G.S.; Ghatei, M.A.; Bloom, S.R. Oxyntomodulin suppresses appetite and reduces food intake in humans. The Journal of Clinical Endocrinology & Metabolism 2003, 88, 4696–4701. [Google Scholar]
- Ma, T.; Huo, S.; Xu, B.; Li, F.; Wang, P.; Liu, Y.; Lei, H. A novel long-acting oxyntomodulin analogue eliminates diabetes and obesity in mice. European journal of medicinal chemistry 2020, 203, 112496. [Google Scholar] [CrossRef] [PubMed]
- Holst, J.J. Enteroglucagon. Annual Review of Physiology 1997, 59, 257–271. [Google Scholar] [CrossRef]
- Pocai, A.; Carrington, P.E.; Adams, J.R.; Wright, M.; Eiermann, G.; Zhu, L.; Du, X.; Petrov, A.; Lassman, M.E.; Jiang, G. Glucagon-like peptide 1/glucagon receptor dual agonism reverses obesity in mice. Diabetes 2009, 58, 2258–2266. [Google Scholar] [CrossRef]
- Henderson, S.; Konkar, A.; Hornigold, D.; Trevaskis, J.; Jackson, R.; Fritsch Fredin, M.; Jansson-Löfmark, R.; Naylor, J.; Rossi, A.; Bednarek, M. Robust anti-obesity and metabolic effects of a dual GLP-1/glucagon receptor peptide agonist in rodents and non-human primates. Diabetes, Obesity and Metabolism 2016, 18, 1176–1190. [Google Scholar] [CrossRef]
- Boland, M.L.; Laker, R.C.; Mather, K.; Nawrocki, A.; Oldham, S.; Boland, B.B.; Lewis, H.; Conway, J.; Naylor, J.; Guionaud, S. Resolution of NASH and hepatic fibrosis by the GLP-1R and GCGR dual-agonist cotadutide via modulating mitochondrial function and lipogenesis. Nature metabolism 2020, 2, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Lee, J.S.; Park, E.; Kim, D.J.; Kim, Y.H.; Choi, I.Y. Therapeutic effect of a novel long-acting GLP-1/GIP/Glucagon triple agonist (HM15211) in NASH and fibrosis animal models. In.EASD annual meeting; 2018.
- Choi, I.Y.; Lee, J.S.; Kim, J.K.; Park, Y.J.; Jung, S.Y.; Kim, Y.H.; Kwon, S.C. Potent body weight loss and efficacy in a NASH animal model by a novel long-acting GLP-1/Glucagon/GIP triple-agonist (HM15211). American Diabetes Association’s 77th Scientific Session. 2017. [Google Scholar]
- Choi, J.; Jo, H.; Kim, J.K.; Lee, S.D.; Lee, S.H.; Choi, I.Y. 996-P: Therapeutic Effect of a Novel Long-Acting GLP-1/GIP/Glucagon Triple Agonist (HM15211) in a Dyslipidemia Animal Model. Diabetes 2019, 68. [Google Scholar] [CrossRef]
- Kim, J.K.; Lee, J.S.; Kwon, H.; Lee, J.; Kim, D.; Bae, S.; Lee, S.H.; Choi, I.Y. 1803-P: Antifibrotic Effect of a Novel Long-Acting GLP-1/GIP/Glucagon Triple Agonist (HM15211) in BDL-Induced Liver Fibrosis Mice. Diabetes 2020, 69. [Google Scholar] [CrossRef]
- Zarei, M.; Aguilar-Recarte, D.; Palomer, X.; Vázquez-Carrera, M. Revealing the role of peroxisome proliferator-activated receptor β/δ in nonalcoholic fatty liver disease. Metabolism 2021, 114, 154342. [Google Scholar] [CrossRef] [PubMed]
- Fajas, L.; Auboeuf, D.; Raspé, E.; Schoonjans, K.; Lefebvre, A.-M.; Saladin, R.; Najib, J.; Laville, M.; Fruchart, J.-C.; Deeb, S. The organization, promoter analysis, and expression of the human PPARγ gene. Journal of Biological Chemistry 1997, 272, 18779–18789. [Google Scholar] [CrossRef]
- Kalliora, C.; Drosatos, K. The glitazars paradox: Cardiotoxicity of the metabolically beneficial dual PPARα and PPARγ activation. Journal of cardiovascular pharmacology 2020, 76, 514. [Google Scholar] [CrossRef]
- Agrawal, R. The first approved agent in the Glitazar’s class: saroglitazar. Current drug targets 2014, 15, 151–155. [Google Scholar] [CrossRef]
- Munigoti, S.P.; Harinarayan, C. Role of Glitazars in atherogenic dyslipidemia and diabetes: Two birds with one stone? Indian Journal of Endocrinology and Metabolism 2014, 18, 283. [Google Scholar] [CrossRef]
- Kumar, D.P.; Caffrey, R.; Marioneaux, J.; Santhekadur, P.K.; Bhat, M.; Alonso, C.; Koduru, S.V.; Philip, B.; Jain, M.R.; Giri, S.R. The PPAR α/γ agonist saroglitazar improves insulin resistance and steatohepatitis in a diet induced animal model of nonalcoholic fatty liver disease. Scientific Reports 2020, 10, 9330. [Google Scholar] [CrossRef]
- Staels, B.; Rubenstrunk, A.; Noel, B.; Rigou, G.; Delataille, P.; Millatt, L.J.; Baron, M.; Lucas, A.; Tailleux, A.; Hum, D.W. Hepatoprotective effects of the dual peroxisome proliferator-activated receptor alpha/delta agonist, GFT505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 2013, 58, 1941–1952. [Google Scholar] [CrossRef] [PubMed]
- Sumida, Y.; Okanoue, T.; Nakajima, A.; NAFLD, J.S.G.o. Phase 3 drug pipelines in the treatment of non-alcoholic steatohepatitis. Hepatology Research 2019, 49, 1256–1262. [Google Scholar] [CrossRef] [PubMed]
- Lefere, S.; Puengel, T.; Hundertmark, J.; Penners, C.; Frank, A.K.; Guillot, A.; De Muynck, K.; Heymann, F.; Adarbes, V.; Defrêne, E. Differential effects of selective-and pan-PPAR agonists on experimental steatohepatitis and hepatic macrophages☆. Journal of hepatology 2020, 73, 757–770. [Google Scholar] [CrossRef]
- Liu, J.; Yang, K.; Yang, J.; Xiao, W.; Le, Y.; Yu, F.; Gu, L.; Lang, S.; Tian, Q.; Jin, T. Liver-derived fibroblast growth factor 21 mediates effects of glucagon-like peptide-1 in attenuating hepatic glucose output. EBioMedicine 2019, 41, 73–84. [Google Scholar]
- Hong, H.; Kim, J.; Choi, H.; Kim, D.; Kim, T.; Tølbøl, K.; Feigh, M.; Illemann, M.; Rigbolt, K.; Vrang, N. YH25724, a novel long-acting GLP-1/FGF21 dual agonist lowers both non-alcoholic fatty liver disease activity score and fibrosis stage in a diet-induced obese mouse model of biopsy-confirmed non-alcoholic steatohepatitis. Journal of Hepatology 2017, 1, S16–S17. [Google Scholar] [CrossRef]
- Pan, Q.; Lin, S.; Li, Y.; Liu, L.; Li, X.; Gao, X.; Yan, J.; Gu, B.; Chen, X.; Li, W. A novel GLP-1 and FGF21 dual agonist has therapeutic potential for diabetes and non-alcoholic steatohepatitis. EBioMedicine 2021, 63, 103202. [Google Scholar] [CrossRef] [PubMed]
- Zaman, R.; Islam, R.A.; Ibnat, N.; Othman, I.; Zaini, A.; Lee, C.Y.; Chowdhury, E.H. Current strategies in extending half-lives of therapeutic proteins. Journal of Controlled Release 2019, 301, 176–189. [Google Scholar] [CrossRef]
- Schjoldager, B.; Baldissera, F.; Mortensen, P.; Holst, J.; Christiansen, J. Oxyntomodulin: a potential hormone from the distal gut. Pharmacokinetics and effects on gastric acid and insulin secretion in man. European journal of clinical investigation 1988, 18, 499–503. [Google Scholar] [CrossRef]
- Ye, X.; Qi, J.; Sun, G.; Ren, G.; Zhu, S.; Wu, Y.; Yu, D.; Zhao, J.; Liu, M.; Li, D. Enhancement of the pharmacological efficacy of FGF-21 by genetic modification and PEGylation. Current pharmaceutical biotechnology 2013, 14, 1287–1298. [Google Scholar] [CrossRef]
- Cui, A.; Li, J.; Ji, S.; Ma, F.; Wang, G.; Xue, Y.; Liu, Z.; Gao, J.; Han, J.; Tai, P. The effects of B1344, a novel fibroblast growth factor 21 analog, on nonalcoholic steatohepatitis in nonhuman primates. Diabetes 2020, 69, 1611–1623. [Google Scholar]
- Xu, J.; Bussiere, J.; Yie, J.; Sickmier, A.; An, P.; Belouski, E.; Stanislaus, S.; Walker, K.W. Polyethylene glycol modified FGF21 engineered to maximize potency and minimize vacuole formation. Bioconjugate chemistry 2013, 24, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Mu, J.; Pinkstaff, J.; Li, Z.; Skidmore, L.; Li, N.; Myler, H.; Dallas-Yang, Q.; Putnam, A.-M.; Yao, J.; Bussell, S. FGF21 analogs of sustained action enabled by orthogonal biosynthesis demonstrate enhanced antidiabetic pharmacology in rodents. Diabetes 2012, 61, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Verzijl, C.R.; Van De Peppel, I.P.; Struik, D.; Jonker, J.W. Pegbelfermin (BMS-986036): an investigational PEGylated fibroblast growth factor 21 analogue for the treatment of nonalcoholic steatohepatitis. Expert opinion on investigational drugs 2020, 29, 125–133. [Google Scholar] [CrossRef]
- Sanyal, A.; Charles, E.D.; Neuschwander-Tetri, B.A.; Loomba, R.; Harrison, S.A.; Abdelmalek, M.F.; Lawitz, E.J.; Halegoua-DeMarzio, D.; Kundu, S.; Noviello, S. Pegbelfermin (BMS-986036), a PEGylated fibroblast growth factor 21 analogue, in patients with non-alcoholic steatohepatitis: a randomised, double-blind, placebo-controlled, phase 2a trial. The Lancet 2018, 392, 2705–2717. [Google Scholar] [CrossRef]
- Charles, E.D.; Neuschwander-Tetri, B.A.; Pablo Frias, J.; Kundu, S.; Luo, Y.; Tirucherai, G.S.; Christian, R. Pegbelfermin (BMS-986036), PEGylated FGF21, in patients with obesity and type 2 diabetes: results from a randomized phase 2 study. Obesity 2019, 27, 41–49. [Google Scholar] [CrossRef]
- Abdelmalek, M.F.; Charles, E.D.; Sanyal, A.J.; Harrison, S.A.; Neuschwander-Tetri, B.A.; Goodman, Z.; Ehman, R.A.; Karsdal, M.; Nakajima, A.; Du, S. The FALCON program: Two phase 2b randomized, double-blind, placebo-controlled studies to assess the efficacy and safety of pegbelfermin in the treatment of patients with nonalcoholic steatohepatitis and bridging fibrosis or compensated cirrhosis. Contemporary Clinical Trials 2021, 104, 106335. [Google Scholar] [CrossRef] [PubMed]
- Pierce, A.; Rosenstock, M.; Margalit, M.; Mansbach, H. BIO89-100, a novel glycoPEGylated FGF21 Analog, Demonstrates Triglyceride Reduction and Broad Metabolic Effects in Spontaneously Diabetic Obese Cynomolgus Monkeys. Journal of Clinical Lipidology 2020, 14, 584–585. [Google Scholar] [CrossRef]
- Rosenstock, M.; Ayalon, M.; Mansbach, H.; Liu, Y.; Margalit, M. LBP-29-BIO89-100, a novel PEG-FGF21 analogue, is efficacious following weekly and every 2-week subcutaneous dosing in spontaneous diabetic cynomolgus monkeys. Journal of Hepatology 2019, 70, e155. [Google Scholar] [CrossRef]
- Loomba, R.; Lawitz, E.J.; Frias, J.P.; Ortiz-Lasanta, G.; Johansson, L.; Franey, B.B.; Morrow, L.; Rosenstock, M.; Hartsfield, C.L.; Chen, C.-Y. Safety, pharmacokinetics, and pharmacodynamics of pegozafermin in patients with non-alcoholic steatohepatitis: a randomised, double-blind, placebo-controlled, phase 1b/2a multiple-ascending-dose study. The Lancet Gastroenterology & Hepatology 2023, 8, 120–132. [Google Scholar]
- Saxena, R.; Nanjan, M.J. Elastin-like polypeptides and their applications in anticancer drug delivery systems: a review. Drug delivery 2015, 22, 156–167. [Google Scholar] [CrossRef]
- Gilroy, C.; Capozzi, M.; Varanko, A.; Tong, J.; D'alessio, D.; Campbell, J.; Chilkoti, A. Sustained release of a GLP-1 and FGF21 dual agonist from an injectable depot protects mice from obesity and hyperglycemia. Science advances 2020, 6, eaaz9890. [Google Scholar] [CrossRef] [PubMed]
- Roopenian, D.C.; Akilesh, S. FcRn: the neonatal Fc receptor comes of age. Nature reviews immunology 2007, 7, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Kontermann, R.E. Strategies for extended serum half-life of protein therapeutics. Current opinion in biotechnology 2011, 22, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Fanali, G.; Di Masi, A.; Trezza, V.; Marino, M.; Fasano, M.; Ascenzi, P. Human serum albumin: from bench to bedside. Molecular aspects of medicine 2012, 33, 209–290. [Google Scholar]
- Chaudhury, C.; Brooks, C.L.; Carter, D.C.; Robinson, J.M.; Anderson, C.L. Albumin binding to FcRn: Distinct from the FcRn− IgG interaction. Biochemistry 2006, 45, 4983–4990. [Google Scholar]
- Bhattacharya, A.A.; Grüne, T.; Curry, S. Crystallographic analysis reveals common modes of binding of medium and long-chain fatty acids to human serum albumin. Journal of molecular biology 2000, 303, 721–732. [Google Scholar] [CrossRef]
- Brønden, A.; Knop, F.K.; Christensen, M.B. Clinical pharmacokinetics and pharmacodynamics of albiglutide. Clinical pharmacokinetics 2017, 56, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, L.B.; Nielsen, P.F.; Huusfeldt, P.O.; Johansen, N.L.; Madsen, K.; Pedersen, F.Z.; Thøgersen, H.; Wilken, M.; Agersø, H. Potent derivatives of glucagon-like peptide-1 with pharmacokinetic properties suitable for once daily administration. Journal of medicinal chemistry 2000, 43, 1664–1669. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.; Bloch, P.; Schaffer, L.; Pettersson, I.; Spetzler, J.; Kofoed, J.; Madsen, K.; Knudsen, L.B.; McGuire, J.; Steensgaard, D.B. Discovery of the once-weekly glucagon-like peptide-1 (GLP-1) analogue semaglutide. Journal of medicinal chemistry 2015, 58, 7370–7380. [Google Scholar] [CrossRef]
- Elbrønd, B.; Jakobsen, G.; Larsen, S.; Agersø, H.; Jensen, L.B.; Rolan, P.; Sturis, J.; Hatorp, V.; Zdravkovic, M. Pharmacokinetics, pharmacodynamics, safety, and tolerability of a single-dose of NN2211, a long-acting glucagon-like peptide 1 derivative, in healthy male subjects. Diabetes care 2002, 25, 1398–1404. [Google Scholar] [CrossRef]
- Hui, H.; Farilla, L.; Merkel, P.; Perfetti, R. The short half-life of glucagon-like peptide-1 in plasma does not reflect its long-lasting beneficial effects. European journal of endocrinology 2002, 146, 863–869. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Ly, N.; Li, J.; Arends, R.H. Population Pharmacokinetics of Cotadutide in Subjects with Type 2 Diabetes. Clinical Pharmacokinetics 2022, 61, 833–845. [Google Scholar] [CrossRef]
- Monnet, C.; Jorieux, S.; Urbain, R.; Fournier, N.; Bouayadi, K.; De Romeuf, C.; Behrens, C.K.; Fontayne, A.; Mondon, P. Selection of IgG variants with increased FcRn binding using random and directed mutagenesis: impact on effector functions. Frontiers in Immunology 2015, 6, 39. [Google Scholar] [CrossRef]
- Vaughn, D.E.; Bjorkman, P.J. Structural basis of pH-dependent antibody binding by the neonatal Fc receptor. Structure 1998, 6, 63–73. [Google Scholar] [CrossRef]
- Naver, S.V.; Jimenez-Solem, E.; Christensen, M.; Andersen, J.T.; Knop, F.K. Dulaglutide: a novel once-weekly glucagon-like peptide-1 receptor agonist. Clinical investigation 2014, 4, 729–743. [Google Scholar] [CrossRef]
- Choi, J.D.; Baek, S.; Kim, Y.; Eun, K.; Kwon, S.C.; Morrow, L.; Hompesch, M.; Kang, J. 982-P: A Double-Blinded, Placebo Controlled, Single Ascending Dose Study for Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics after Subcutaneous Administration of Novel Long-Acting GLP-1/GIP/Glucagon Triple Agonist (HM15211) in Healthy Obese Subjects. Diabetes 2019, 68. [Google Scholar]
- Stanislaus, S.; Hecht, R.; Yie, J.; Hager, T.; Hall, M.; Spahr, C.; Wang, W.; Weiszmann, J.; Li, Y.; Deng, L. A novel Fc-FGF21 with improved resistance to proteolysis, increased affinity toward β-klotho, and enhanced efficacy in mice and cynomolgus monkeys. Endocrinology 2017, 158, 1314–1327. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, A.; Abuqayyas, L.; Denney, W.S.; Tillman, E.J.; Rolph, T. AKR-001, an Fc-FGF21 analog, showed sustained pharmacodynamic effects on insulin sensitivity and lipid metabolism in type 2 diabetes patients. Cell Reports Medicine 2020, 1, 100057. [Google Scholar] [CrossRef]
- Harrison, S.A.; Ruane, P.J.; Freilich, B.L.; Neff, G.; Patil, R.; Behling, C.A.; Hu, C.; Fong, E.; de Temple, B.; Tillman, E.J. Efruxifermin in non-alcoholic steatohepatitis: a randomized, double-blind, placebo-controlled, phase 2a trial. Nature medicine 2021, 27, 1262–1271. [Google Scholar] [CrossRef]
- Boey, A.; Ho, H.K. All roads lead to the liver: metal nanoparticles and their implications for liver health. Small 2020, 16, 2000153. [Google Scholar] [CrossRef]
- Sadauskas, E.; Wallin, H.; Stoltenberg, M.; Vogel, U.; Doering, P.; Larsen, A.; Danscher, G. Kupffer cells are central in the removal of nanoparticles from the organism. Particle and fibre toxicology 2007, 4, 1–7. [Google Scholar] [CrossRef]
- Alexis, F.; Pridgen, E.; Molnar, L.K.; Farokhzad, O.C. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Molecular pharmaceutics 2008, 5, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Xin, X.; Ma, J.; Tan, C.; Osna, N.; Mahato, R.I. Therapeutic targets, novel drugs, and delivery systems for diabetes associated NAFLD and liver fibrosis. Advanced Drug Delivery Reviews 2021, 176, 113888. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-K.; Utsumi, T.; Seo, Y.-E.; Deng, Y.; Satoh, A.; Saltzman, W.M.; Iwakiri, Y. Cellular distribution of injected PLGA-nanoparticles in the liver. Nanomedicine: Nanotechnology, Biology and Medicine 2016, 12, 1365–1374. [Google Scholar] [CrossRef]
- Monestier, M.; Charbonnier, P.; Gateau, C.; Cuillel, M.; Robert, F.; Lebrun, C.; Mintz, E.; Renaudet, O.; Delangle, P. ASGPR-Mediated Uptake of Multivalent Glycoconjugates for Drug Delivery in Hepatocytes. ChemBioChem 2016, 17, 590–594. [Google Scholar] [CrossRef] [PubMed]
- Petrov, R.A.; Maklakova, S.Y.; Ivanenkov, Y.A.; Petrov, S.A.; Sergeeva, O.V.; Yamansarov, E.Y.; Saltykova, I.V.; Kireev, I.I.; Alieva, I.B.; Deyneka, E.V. Synthesis and biological evaluation of novel mono-and bivalent ASGP-R-targeted drug-conjugates. Bioorganic & medicinal chemistry letters 2018, 28, 382–387. [Google Scholar]
- Bon, C.; Hofer, T.; Bousquet-Mélou, A.; Davies, M.R.; Krippendorff, B.-F. Capacity limits of asialoglycoprotein receptor-mediated liver targeting. In.MAbs: Taylor & Francis. 2017; 1360–1369. [Google Scholar]
- Sharma, R.; Porterfield, J.E.; An, H.-T.; Jimenez, A.S.; Lee, S.; Kannan, S.; Sharma, A.; Kannan, R.M. Rationally designed galactose dendrimer for hepatocyte-specific targeting and intracellular drug delivery for the treatment of liver disorders. Biomacromolecules 2021, 22, 3574–3589. [Google Scholar] [CrossRef] [PubMed]
- Fraser, J.; Laurent, T.C.; Pertoft, H.; Baxter, E. Plasma clearance, tissue distribution and metabolism of hyaluronic acid injected intravenously in the rabbit. Biochemical Journal 1981, 200, 415–424. [Google Scholar] [CrossRef]
- Fraser, J.R.E.; Appelgren, L.-E.; Laurent, T.C. Tissue uptake of circulating hyaluronic acid: a whole body autoradiographic study. Cell and tissue research 1983, 233, 285–293. [Google Scholar] [CrossRef]
- Yang, J.-A.; Kong, W.H.; Sung, D.K.; Kim, H.; Kim, T.H.; Lee, K.C.; Hahn, S.K. Hyaluronic acid–tumor necrosis factor-related apoptosis-inducing ligand conjugate for targeted treatment of liver fibrosis. Acta biomaterialia 2015, 12, 174–182. [Google Scholar] [CrossRef]
- Li, W.; Zhou, C.; Fu, Y.; Chen, T.; Liu, X.; Zhang, Z.; Gong, T. Targeted delivery of hyaluronic acid nanomicelles to hepatic stellate cells in hepatic fibrosis rats. Acta Pharmaceutica Sinica B 2020, 10, 693–710. [Google Scholar] [CrossRef]
- Newcomer, M.E.; Ong, D.E. Plasma retinol binding protein: structure and function of the prototypic lipocalin. Biochimica et Biophysica Acta (BBA)-Protein Structure and Molecular Enzymology 2000, 1482, 57–64. [Google Scholar] [CrossRef]
- Uchio, K.; Tuchweber, B.; Manabe, N.; Gabbiani, G.; Rosenbaum, J.; Desmouliere, A. Cellular retinol-binding protein-1 expression and modulation during in vivo and in vitro myofibroblastic differentiation of rat hepatic stellate cells and portal fibroblasts. Laboratory Investigation 2002, 82, 619–628. [Google Scholar] [CrossRef]
- Lee, Y.S.; Jeong, W.I. Retinoic acids and hepatic stellate cells in liver disease. Journal of gastroenterology and hepatology 2012, 27, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.-B.; Fan, Q.-Q.; Xing, L.; Cui, P.-F.; He, Y.-J.; Zhu, J.-C.; Wang, L.; Pang, T.; Oh, Y.-K.; Zhang, C. Vitamin A-decorated biocompatible micelles for chemogene therapy of liver fibrosis. Journal of Controlled Release 2018, 283, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Hassan, R.; Tammam, S.N.; El Safy, S.; Abdel-Halim, M.; Asimakopoulou, A.; Weiskirchen, R.; Mansour, S. Prevention of hepatic stellate cell activation using JQ1-and atorvastatin-loaded chitosan nanoparticles as a promising approach in therapy of liver fibrosis. European Journal of Pharmaceutics and Biopharmaceutics 2019, 134, 96–106. [Google Scholar] [CrossRef]
- Magnusson, S.; Berg, T. Extremely rapid endocytosis mediated by the mannose receptor of sinusoidal endothelial rat liver cells. Biochemical Journal 1989, 257, 651–656. [Google Scholar] [CrossRef]
- Schlesinger, P.H.; Doebber, T.W.; Mandell, B.F.; White, R.; DeSchryver, C.; Rodman, J.; Miller, M.; Stahl, P. Plasma clearance of glycoproteins with terminal mannose and N-acetylglucosamine by liver non-parenchymal cells. Studies with β-glucuronidase, N-acetyl-β-d-glucosaminidase, ribonuclease B and agalacto-orosomucoid. Biochemical Journal 1978, 176, 103–109. [Google Scholar] [CrossRef]
- Kim, M.; Jeong, M.; Hur, S.; Cho, Y.; Park, J.; Jung, H.; Seo, Y.; Woo, H.; Nam, K.; Lee, K. Engineered ionizable lipid nanoparticles for targeted delivery of RNA therapeutics into different types of cells in the liver. Science Advances 2021, 7, eabf4398. [Google Scholar] [CrossRef]
- Go, G.-w.; Mani, A. Low-density lipoprotein receptor (LDLR) family orchestrates cholesterol homeostasis. The Yale journal of biology and medicine 2012, 85, 19. [Google Scholar]
- Wang, Z.; Duan, X.; Lv, Y.; Zhao, Y. Low density lipoprotein receptor (LDLR)-targeted lipid nanoparticles for the delivery of sorafenib and Dihydroartemisinin in liver cancers. Life sciences 2019, 239, 117013. [Google Scholar] [CrossRef] [PubMed]
- Akinc, A.; Querbes, W.; De, S.; Qin, J.; Frank-Kamenetsky, M.; Jayaprakash, K.N.; Jayaraman, M.; Rajeev, K.G.; Cantley, W.L.; Dorkin, J.R. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Molecular Therapy 2010, 18, 1357–1364. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic representation of crosstalk between the organs and major regulatory pathways in NAFLD. A group of NAFLD drug candidates (e.g., SGLT2 inhibitors, GLP-1R agonists, PPAR agonists, FGF19 and FGF21 analogs, and FXR agonists) have been developed and have undergone (or are still ongoing) clinical trials. The action mechanisms of these compounds involve various and multiple pathways related to 1) regulation of glucose and lipid homeostasis in the network of adipose tissues, brain, pancreas, and liver, 2) modulating de novo lipogenesis, lipolysis, and β-oxidation of free fatty acids, 3) reducing food intake and body weight, 4) improving insulin resistance in liver and adipose tissues, and 5) regulating bile acid production. (CYP7α1, cholesterol 7-α-hydroxylase; FFA, free fatty acid; FGF, fibroblast growth factors; FGFR, fibroblast growth factor receptor; FXR, farnesoid X receptor; GLP-1R, glucagon-like peptide-1 receptor; PPAR, peroxisome proliferator-activator receptor; KBL, β-klotho; SGLT2, sodium-glucose co-transporter 2; SREBP-1, sterol regulatory element-binding protein-1; TGs, triglycerides; VLDL, very low density lipoprotein).
Figure 1.
Schematic representation of crosstalk between the organs and major regulatory pathways in NAFLD. A group of NAFLD drug candidates (e.g., SGLT2 inhibitors, GLP-1R agonists, PPAR agonists, FGF19 and FGF21 analogs, and FXR agonists) have been developed and have undergone (or are still ongoing) clinical trials. The action mechanisms of these compounds involve various and multiple pathways related to 1) regulation of glucose and lipid homeostasis in the network of adipose tissues, brain, pancreas, and liver, 2) modulating de novo lipogenesis, lipolysis, and β-oxidation of free fatty acids, 3) reducing food intake and body weight, 4) improving insulin resistance in liver and adipose tissues, and 5) regulating bile acid production. (CYP7α1, cholesterol 7-α-hydroxylase; FFA, free fatty acid; FGF, fibroblast growth factors; FGFR, fibroblast growth factor receptor; FXR, farnesoid X receptor; GLP-1R, glucagon-like peptide-1 receptor; PPAR, peroxisome proliferator-activator receptor; KBL, β-klotho; SGLT2, sodium-glucose co-transporter 2; SREBP-1, sterol regulatory element-binding protein-1; TGs, triglycerides; VLDL, very low density lipoprotein).
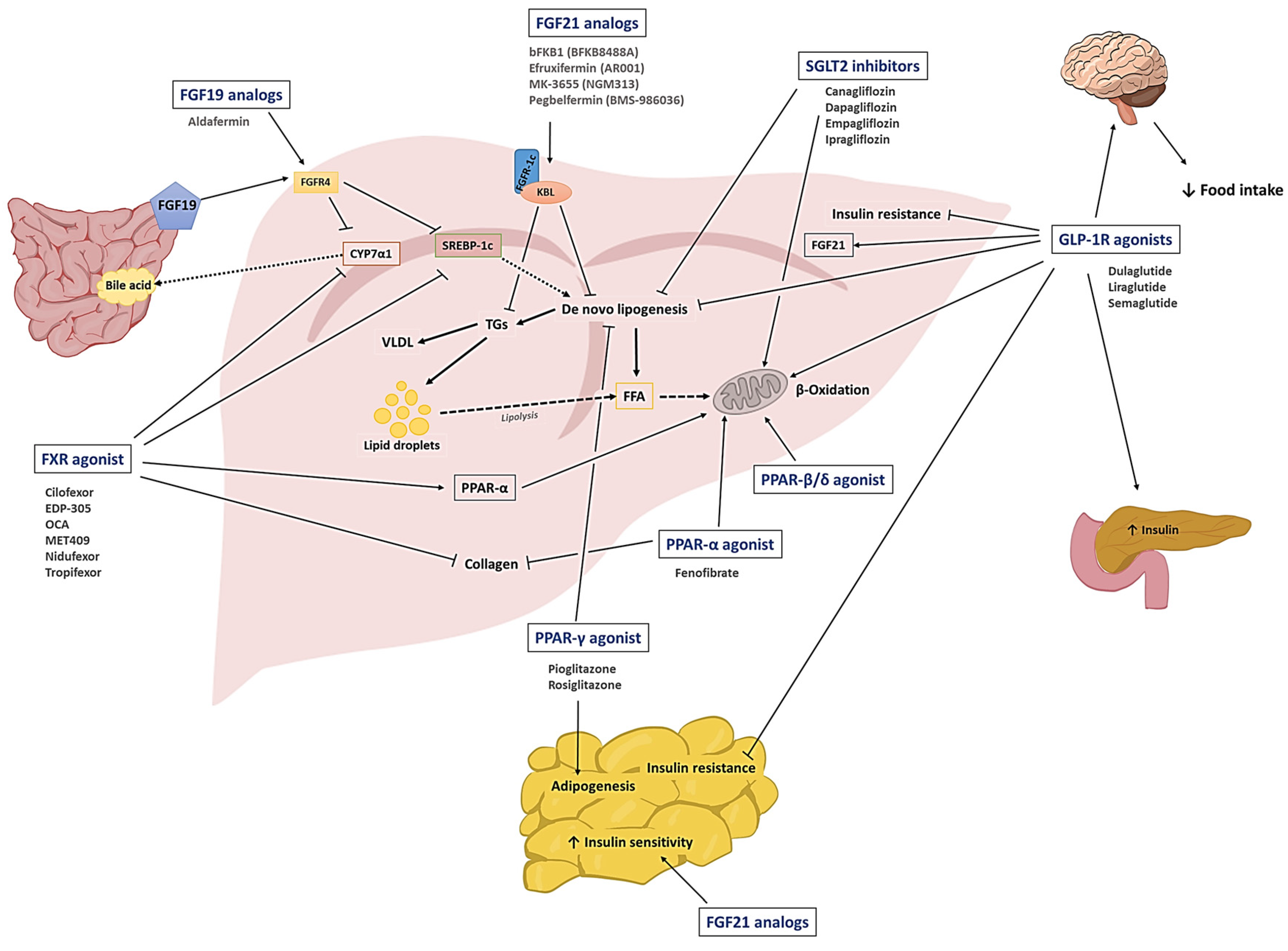
Figure 2.
Schematic diagram of the pharmaceutical strategies to make “better drugs” for NAFLD therapy. To enhance the therapeutic efficacy of NALFD drug candidates, the 3 strategies widely explored include 1) the development of multi-receptor agonists, 2) engineering long-acting derivatives, and 3) devising ligand-decorated nanoparticle-based hepatic targeting systems. The increase of plasma half-life could be accomplished by conjugation of polymers (e.g. PEG) or fusion of IgG Fc or human serum albumin (HSA). The latter enables the drug compound to go through FcRn-mediated recycling. Liver targeting could be achieved by utilizing ligands selective for receptors expressed on the membranes of liver-composing cells. (ASGPR, asialoglycoprotein; FcRn, neonatal Fc receptor; FGFR, fibroblast growth factor receptor; GIPR, glucose-dependent insulinotropic polypeptide receptor; GCGR, glucagon receptor; GLP-1R, glucagon-like peptide-1 receptor; HA-R: hyaluronan receptor; HSA, human serum albumin; HSCs, hepatic stellate cells; LDLR, low-density lipoprotein receptor; LSECs, liver sinusoidal endothelial cells; MR, mannose receptor; PEG, polyethylene glycol; PPAR, peroxisome proliferator-activator receptor; RBPR, retinol-binding protein receptor).
Figure 2.
Schematic diagram of the pharmaceutical strategies to make “better drugs” for NAFLD therapy. To enhance the therapeutic efficacy of NALFD drug candidates, the 3 strategies widely explored include 1) the development of multi-receptor agonists, 2) engineering long-acting derivatives, and 3) devising ligand-decorated nanoparticle-based hepatic targeting systems. The increase of plasma half-life could be accomplished by conjugation of polymers (e.g. PEG) or fusion of IgG Fc or human serum albumin (HSA). The latter enables the drug compound to go through FcRn-mediated recycling. Liver targeting could be achieved by utilizing ligands selective for receptors expressed on the membranes of liver-composing cells. (ASGPR, asialoglycoprotein; FcRn, neonatal Fc receptor; FGFR, fibroblast growth factor receptor; GIPR, glucose-dependent insulinotropic polypeptide receptor; GCGR, glucagon receptor; GLP-1R, glucagon-like peptide-1 receptor; HA-R: hyaluronan receptor; HSA, human serum albumin; HSCs, hepatic stellate cells; LDLR, low-density lipoprotein receptor; LSECs, liver sinusoidal endothelial cells; MR, mannose receptor; PEG, polyethylene glycol; PPAR, peroxisome proliferator-activator receptor; RBPR, retinol-binding protein receptor).
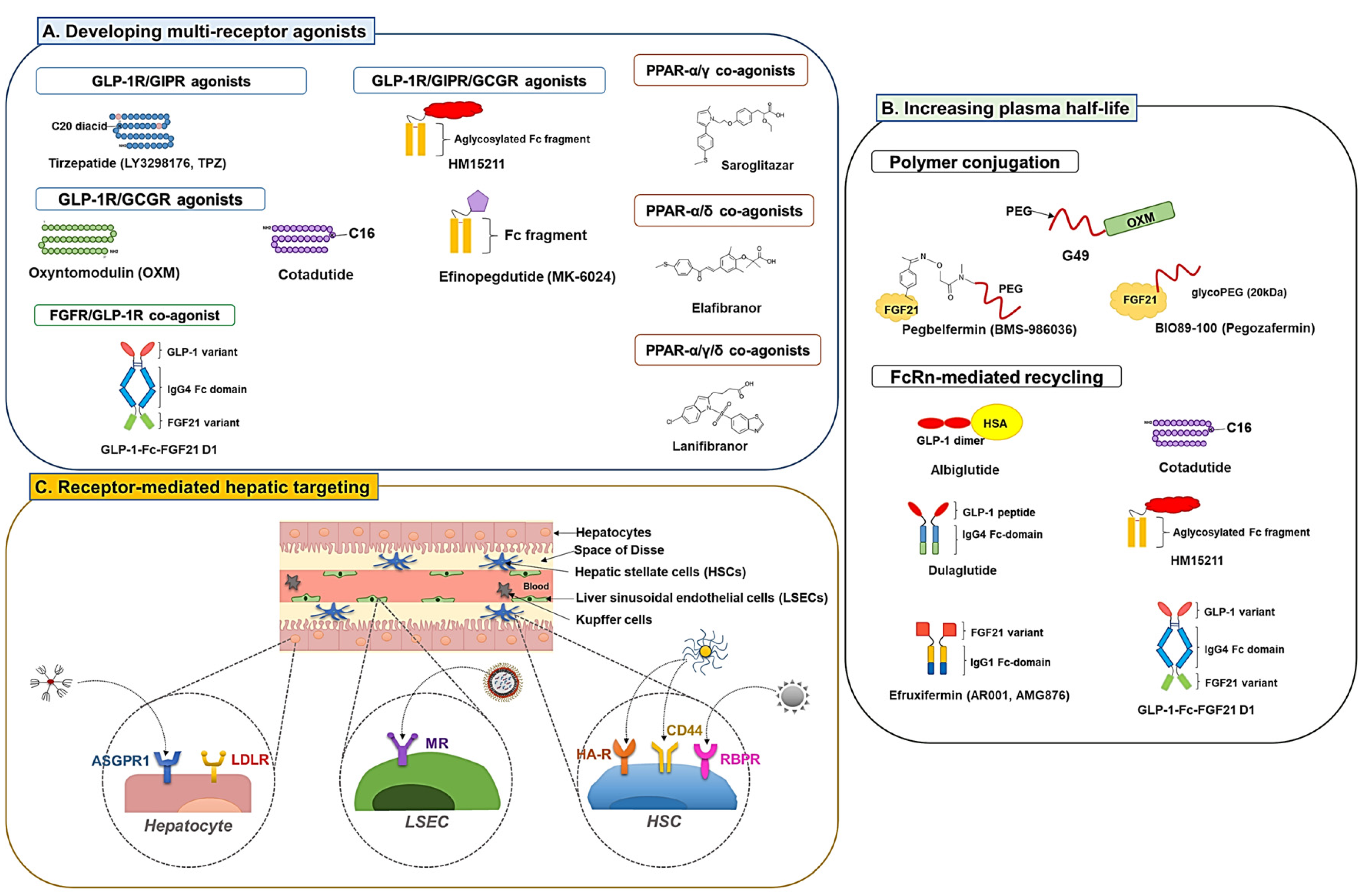

Table 1.
Active clinical trials of SGLT2 inhibitors for NAFLD.
| No. | Molecules | Development stage | Clinical trial identification | Year of completion |
|---|---|---|---|---|
| 1 | Canagliflozin | N/A | NCT05422092 | 2023 |
| N/A | NCT05513729 | 2024 | ||
| 2 | Dapagliflozin | Phase 3 | NCT03723252 | 2022 |
| NCT05308160 | 2024 | |||
| Phase 4 | NCT05140694 | 2025 | ||
| 3 | Empagliflozin | Phase 2 | NCT03867487 | 2024 |
| NCT05140694 | 2025 | |||
| Phase 4 |
NCT03646292 | 2021 | ||
| NCT04642261 | 2022 | |||
| NCT04976283 | 2023 | |||
| NCT05140694 | 2025 | |||
| 4 | Ipragliflozin | Phase 4 | NCT02875821 | 2017 |
Table 2.
Active clinical trials of GLP-1R agonists for NAFLD.
| No. | Molecules | Development stage | Clinical trial identification | Year of completion |
|---|---|---|---|---|
| 1 | Dulaglutide | N/A | NCT03590626 | 2020 |
| Phase 4 | NCT03648554 | 2024 | ||
| 2 | Liraglutide | Phase 4 | NCT02147925 | 2017 |
| NCT03068065 | 2017 | |||
| 3 | Semaglutide | Phase 1 | NCT03357380 | 2020 |
| Phase 2 | NCT03987074 | 2020 | ||
| NCT03987451 | 2021 | |||
| NCT02970942 | 2021 | |||
| NCT03884075 | 2023 | |||
| NCT04216589 | 2023 | |||
| NCT04971785 | 2024 | |||
| NCT05016882 | 2024 | |||
| Phase 3 | NCT04822181 | 2028 | ||
| Phase 4 | NCT04639414 | 2023 | ||
| NCT05195944 | 2024 |
Table 3.
Active clinical trials of PPAR agonists for NAFLD.
| No. | Molecules | Target | Development stage | Clinical trial identification | Year of completion |
|---|---|---|---|---|---|
| 1 | Fenofibrate | PPAR-α | Phase 2 | NCT02354976 | 2016 |
| NCT02781584 | 2022 | ||||
| 2 | Pioglitazone | PPAR-γ | Phase 4 | NCT00227110 | 2006 |
| NCT00994682 | 2014 | ||||
| NCT02875821 | 2017 | ||||
| NCT03796975 | 2019 | ||||
| NCT04976283 | 2023 | ||||
| 3 | Rosiglitazone | PPAR-γ | Phase 2 | NCT02285205 | 2015 |
| Phase 4 | NCT01406704 | 2013 |
Table 4.
Active clinical trials of fibroblast growth factor (FGF) analogs for NAFLD.
| No. | Molecules | Structure | Development stage | Clinical trial identification | Year of completion |
|---|---|---|---|---|---|
| 1 | Aldafermin (NGM282) | FGF19 analog | Phase 2 | NCT02443116 | 2020 |
| NCT03912532 | 2021 | ||||
| NCT04210245 | 2023 | ||||
| 2 | bFKB1 (BFKB8488A) | FGFR1c/β-klotho bispecific antibody | Phase 1 | NCT03060538 | 2019 |
| Phase 2 | NCT04171765 | 2023 | |||
| 3 | Efruxifermin (AR001) | Fc-FGF21 analog | Phase 2 | NCT03976401 | 2022 |
| NCT05039450 | 2024 | ||||
| NCT04767529 | 2024 | ||||
| 4 | MK-3655 (NGM313) | FGFR1c/β-klotho bispecific antibody | Phase 2 | NCT04583423 | 2024 |
| 5 | Pegbelfermin (BMS-986036) | PEG conjugated FGF21 analog | Phase 2 | NCT03486899 | 2021 |
| NCT03486912 | 2021 |
Table 5.
Active clinical trials of FXR agonists for NAFLD.
| No. | Molecules | Development stage | Clinical trial identification | Year of completion |
|---|---|---|---|---|
| 1 | Cilofexor (GS-9674) Combination with selonsertib and firsocostat | Phase 2 | NCT03449446 | 2019 |
| Cilofexor + selonsertib and firsocostat | NCT02781584 | 2020 | ||
| Cilofexor + semaglutide and firsocostat | NCT03987074 | 2020 | ||
| Cilofexor + semaglutide and firsocostat | NCT04971785 | 2024 | ||
| 2 | EDP-305 | Phase 2 | NCT03421431 | 2019 |
| NCT04378010 | 2022 | |||
| 3 | OCA, 6-α-ethyl-chenodeoxycholic acid (6-ECDCA, INT-747) | Phase 3 | NCT03439254 | 2022 |
| NCT02548351 | 2025 | |||
| 4 | MET409 | Phase 2 | NCT04702490 | 2022 |
| 5 | Nidufexor (LMB763) | Phase 2 | NCT02913105 | 2018 |
| 6 | Tropifexor (LJN452) + cenicriviroc | Phase 2 | NCT03517540 | 2020 |
| 7 | Tropifexor +licogliflozin | Phase 2 | NCT04065841 | 2024 |
Table 6.
Active clinical trials of GLP-1R dual/triple agonists for NAFLD.
| No. | Molecules | Target | Development stage | Clinical trial identification | Year of completion |
|---|---|---|---|---|---|
| 1 | Cotadutide (MEDI0382) | GLP-1R /GCGR |
Phase 1 | NCT03625778 | 2019 |
| NCT04208620 | 2020 | ||||
| NCT04091373 | 2020 | ||||
| Phase 2 | NCT03235050 | 2019 | |||
| NCT04019561 | 2021 | ||||
| NCT03555994 | 2021 | ||||
| 2 | CT-388 | GLP-1R /GIPR | Phase 1 | NCT04838405 | 2022 |
| 3 | CT-868 | GLP-1R /GIPR | Phase 2 | NCT05110846 | 2022 |
| Phase 1 | NCT04973111 | 2022 | |||
| 4 | DD01 | GLP-1R /GCGR |
Phase 1 | NCT04812262 | 2022 |
| 5 | Efinopegdutide (MK-6024) | GLP-1R/ GCGR | Phase 2 | NCT04944992 | 2022 |
| 6 | HM15211 | GLP-1R /GIPR/ GCGR |
Phase 1 | NCT03744182 | 2021 |
| Phase 2 | NCT04505436 | 2024 | |||
| 7 | Tirzepatide (LY3298176, TPZ) | GLP-1R/ GIPR |
N/A | NCT05078255 | 2023 |
| Phase 1 | NCT03951753 | 2021 | |||
| NCT04407234 | 2021 | ||||
| NCT04050553 | 2022 | ||||
| Phase 2 | NCT03131687 | 2018 | |||
| NCT03311724 | 2021 | ||||
| NCT04166773 | 2023 | ||||
| Phase 3 | NCT03954834 | 2020 | |||
| NCT03861039 | 2021 | ||||
| NCT03861052 | 2021 | ||||
| NCT05024032 | 2022 | ||||
| NCT04660643 | 2023 | ||||
| NCT04657016 | 2023 | ||||
| NCT04657003 | 2023 | ||||
| NCT04184622 | 2024 |
Table 7.
Active clinical trials of PPAR multi-receptor agonists for NAFLD.
| No. | Molecules | Target | Development stage | Clinical trial identification | Year of completion |
|---|---|---|---|---|---|
| 1 | Elafibranor | PPAR α/δ agonist | Phase 2 |
NCT03883607 | 2020 |
| NCT03953456 | 2020 | ||||
| Phase 3 | NCT02704403 | 2020 | |||
| 2 | Lanifibranor | PPAR-α/γ/δ agonist | Phase 2 | NCT03008070 | 2020 |
| NCT03459079 | 2022 | ||||
| NCT05232071 | 2022 | ||||
| Phase 3 | NCT04849728 | 2028 | |||
| 3 | Saroglitazar | PPAR-α/γ agonist | Phase 2 | NCT03061721 | 2020 |
| NCT03863574 | 2020 | ||||
| NCT03617263 | 2023 | ||||
| NCT05011305 | 2023 | ||||
| NCT03639623 | 2022 | ||||
| NCT05211284 | 2025 | ||||
| Phase 3 | NCT02265276 | 2015 | |||
| NCT04193982 | 2021 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



