
Submitted:
13 June 2023
Posted:
14 June 2023
You are already at the latest version
Abstract
Considering Cu(tda) chelate is a receptor for adenine and related 6-aminopurines, now we report the synthesis, molecular and crystal structures, thermal stability, spectral properties and DFT calculations on the [Cu(tda)(9heade)(H2O)]•2H2O (1). N7 is found to be the preferred donor atom of 9heade, that can favor the cooperation of a Cu-N7(9heade) bond and an (9heade)N6-H•••O(tda) interligand interaction. But unprecedentedly, the Cu(tda)-9heade molecular recognition in 1 is essentially featured the Cu-N1(9heade), without any N6-H•••O(carboxyl tda) interligand interaction. Nevertheless, being N1 the most basic donor for N9-substituted adenines, this Cu-N1 bond is now assisted by an O2-water mediated interaction (N6-H•••O2 and O2•••Cu weak contact). Also in the crystal packing, the O-H(ol) of 9heade interacts with its own adenine moiety by an O3-water mediated interaction (O(ol)-H•••O3 plus O3-H36•••π(adenine moiety). In turn these water-mediated interactions seems to be responsible to serious alterations on the physical properties of crystalline or grounded samples. Density functional theory calculations were used to evaluate the interactions energetically. Moreover, the quantum theory of atoms-in-molecules (QTAIM) in combination with the noncovalent interaction plot (NCIPlot) were used to analyze the interactions and rationalize the existence and relative importance of hydrogen bonding, chalcogen bonding and π-stacking interactions.
Keywords:
Copper(II)
; thiodiacetate chelate
; synthetic adenine nucleoside
; molecular and crystal structure
; chelate-nucleoside recognition
; water-mediated interligand interactions
; physical properties
; DFT calculations.
1. Introduction
Staring from a broad point of view, chemical processes can be understood as a favorable consequence of the molecular recognition [1] between two or more reactants. This point of view implies that molecular recognition is always a phenomenon. The ‘favorable conditions’ become determined by the two complementary facets of all chemical process: thermodynamics and kinetics. On this broad basis, we can consider a quasi-infinite number of objectives and experiences aimed at a better understanding of chemical reactions, addressing the factors that determine whether they are or not possible, and whether they end up to be carried out. Currently, these arguments moved many of us to pay attention to the contribution of the so-called ‘weak interactions’ that accompany the formation of more robust chemical bonds. In this context, complex phenomena that emerge in the chemical and biological context [2]) need to be discussed, from much simplest 'model compounds' [3]. In past decades we, as other researchers, become working on mixed-ligand metal complexes formed by reactions between a simple metal salt or a metal chelate with N-bases of nucleic acids, nucleotides, natural or synthetic [4] nucleosides or nucleic bases (see i.e. [5,6]). In in these last cases, efforts in preparing single crystals become essential, because appropriate discussions of their structural results yield detailed information of both their own structures as well as how these compounds recognize themselves to build stable crystals, with or without the implication of solvents [7, 8]. We became recently interested in the synthesis, molecular and/or crystal structures of mixed-ligand metal complexes having a synthetic purine nucleoside, as source of the MBD of these later ligands and of the analysis of the interligand interactions contributing to these ternary complexes at molecular and supramolecular levels. In this regard, the main support concerns to compounds having acyclovir (acv or N9-(2-hydroxyethyl)adenine (9heade) as coligands (see Scheme 1).
The structural ‘status-of art’ for acv-metal complexes was been recently summarized by us [4]. It comprises the following metal binding patterns (hereafter MBP). MBP-1: Complexes with only an M-N7 bond. MBP-2: Cooperation between an M-N7 bond with an H-bond interaction, having O6-acv as H-acceptor. MBP-3: Metal chelates with µ-N7,O6-acv. The only reported compound with this MBP also implies the cooperation of a Cu-N(amino) bond with an (amino)N-H···O6(acv) interligand interaction. The MBP-4: Featured in a compound having acv in the bridging µ-N7,O(ol) role. MBP-5: A case where anionic form (acv-H) displays the bringing (but not chelating) µ-N7,O6 function. And finally MBP-6: For a copper(II) coordination polymer where acv exhibits the highest denticity for a nucleoside, acting as O(e),O(ol)-chelating, µ3-trinucleating function, and a N7,O6,O(e), O(ol)-tetradentate role [9] Interestingly all these MBP enhances that N7 is the preferred donor atom of acv.
In clear contrast, only a few recent reports concerns to nine metal complexes having 9heade as ligand [10-14. Each of they are also here-after referred by its code (with six capital letters) in the Cambridge Structural Database (CSD) along with the corresponding chemical formula, and grouped by the/their MBP(s) following the criteria above-referred for acv metal complexes:
MBP-2. That corresponds to the cooperation of a metal(II)-N7 coordination bond plus an appropriate interligand H-bonding interaction, of the type N-H···O (otherwise indicated below, to N-H···Cl). MII-N7(9heade) bond (M = Co, Cu, Zn or Cd) cooperating with (9heade)N(6)-H···O(coligands) is observed in: trans-[CoII(acac)(9heade)2]·5H2O (RUFBEC, acac = acethylacetonate(2-) [10]), [Cu(pdc)(9heade)(H2O)]·H2O (MOTBOP, pdc = pyridine-2,6-dicarboxylate(2-) [11]), trans-[Cu(pdc)(9heade)(H2O)]·3H2O (MOTBIJ [11], [Cu(pdc)(9heade)(H2O)]·H2O (MOTBOP [11], [Zn(pdc)(9heade) (H2O)] (MOTBUV, [11]), [Cu2(µ2-EDTA)(9heade)2(H2O)2]·5H2O (EDTA = ethylenediaminotetraacetate(4-) (OSEKIJ [12]), {[Cd2(µ2-EDTA)(9heade) (H2O)]·4H2O}n (PUXQEH [13]) and {[Cd(µ-(cis-1,2-chdca)(9heade)(H2O)]·4H2O}n (cis-1,2-chdca = cis-1,2-cyclohexanedicar- boxy-late(2-), ANEHOS [14]). An unusual cooperation, in this kind of compounds, is twice featured in the tetrahedral complex molecule [Zn(9heade)2Cl2] (RUFBAY, [10] involving each Zn-N7(9heade) bond with each (9heade)N6-H···Cl interligand interaction. MBP2+MBP3. Both this MBPs are featured in the dinuclear complex molecule [Cu2(glygly)2(µ2-9hea-de)(H2O)]·8H2O (glygly = glycyl-glicinate(2-), MOTCAC [11]).
The hydrogen bonding (HB) is the most studied and used noncovalent interaction in chemistry, biology, and materials [15-17]. But other forces like σ-hole interactions (e.g. halogen bonding [10]) involving elements of the p-block as electron acceptors (playing the role of the H-atom) are gaining attention among the scientific community, and specially in crystal engineering and supramolecular chemistry [10,18]. Depletion of electron density usually happens on the extension of covalent bonds involving p-block elements that are called σ-holes. Chalcogen bonding (ChB) has been recently recognized by the IUPAC [19] and defined as the “net attractive interaction between an electrophilic region associated with a chalcogen atom in a molecular entity and a nucleophilic region in another, or the same, molecular entity”. Several theoretical works have shown that the origin of ChB is mainly electrostatic [20], though the orbital donor–acceptor can also be very important (LP → σ*) [20]. The strength of the interaction depends on the polarizability of the Ch atom, increasingly for heavier Ch atoms [21]. Noteworthy, it has been recently proved that the metal binding of the chalcogen atom increases its ability as electron donor to participate in this kind of noncovalent interaction [22].
This work aims to report the synthesis, molecular and crystal structures of the title compound [Cu(tda)(9heade)(H2O)]·2H2O (1) as well as the study of its thermal stability, some spectral properties and various theoretical calculations. This last study is focused on the relevance of σ-hole interactions involving the Cu-S(thioether atom) of the tda ligand along with other more conventional interactions like H-bonds and lone pair-π (lp–π) interactions. As above referred, this type of ChB interactions, where σ-hole donor atom is coordinated to transition metals, has been scarcely described and exploited in the literature [22] The ChBs interactions have been characterized energetically using DFT calculations, the quantum theory of atoms-in-molecules (QTAIM), molecular electrostatic potential (MEP) surfaces and the noncovalent interaction plot (NCIPlot) computational tools.
2. Results and Discussions
2.1. About the Strategy of Synthesis
The reported strategy for the synthesis of [Cu(tda)(9heade)(H2O]·2H2O (hereafter compound 1) consists in a two steps reaction (see Section 4.1). First, green basic carbonate of copper(II), malachite, Cu2(CO3)(OH)2 is reacted with H2tda with molar ratio 1:2, corresponding to an equimolar Cu(II):tda ratio) in water, being CO2 the only by-product. It is recommended the filtration without vacuum of the resulting greenish solution to remove as far as possible little amounts of unreacted malachite, which can act as crystallization nucleus. To the Cu(tda) aqueous solution an equimolar amount of 9heade is slowly added, with stirring. Mother liquors of 1 are filtered without vacuum on a crystallizer, which is covered by a plastic film to moderate the evaporation of solvent. This procedure yields long greenish parallelepiped crystals some of them suitable for the crystallographic tools (Figure 1). These crystals were collected by repeat filtrations (up to ~70% of yield) also for thermal and spectral studies, discarding final fractions that can be contaminated with by-products, as appreciated by electron spin resonance (ESR) measurements.
2.2. Molecular and Crystal Structure of Compound 1 and Their Relevant Significance for Molecular and Supra-Molecular Recognition
Relevant crystal data and, structure refinement of [Cu(tda)(9heda)(H2O)]·2H2O (1) are here summarizes in Table 1. Coordination bond lengths and trans-angles for this tentative formula are shown in Table 2. Complete versions of these tables are reported as Supplementary Materials (here after SM) SM.1 and SM.2 Data of H-bonding interactions are also given as SM.3.
2.3. Molecular and Crystal Structures of Compound [Cu(tda)(9heade)(H2O)]·2H2O (1)
In a rather flexible look we can consider that the O2-water atom is at a distance of 3.06 Å from the metal, resulting a very weak Cu(1)···O(2) water contact (the sum of Van der Waals (VdW)radii of Cu (1.40 Å) and O (1.52 Å) is 2.92 Å . Indeed it is well known that these VdW radii are only available by a limited number of elements and they are averaged values, affected by a certain error. From this point of view, coordination bond lengths and angles are reported in Table 2, and Figure 2 represents the crystal structure of the novel compound according to the formula [Cu(tda)(9heda)(H2O)]·2H2O in a very asymmetrically elongated octahedral coordination, type 4+1+1*, where * denotes the above referred Cu···O2(aqua) weak contact (see discussion below).
Ignoring the relevance of such very weak Cu···O2(aqua) contact, the copper(II) coordination in 1 can be understood as axially elongated square-based tetrahedral coordination, type 4+1, with the O1-aqua atom as distal donor and chemical formula [Cu(tda)(9heda)(H2O)]·2H2O (Figure 2).
In a simplified approach and more realistic chemical sense, compound 1 agrees well to the formula [Cu(tda)(9heade)(H2O)], where the Cu(II) centre in a rather frequent elongated square-base pyramidal coordination environment, type 4+1 (Figure 3).
The four basal donors are the O(4) and S(1) atoms from the tridentate tda chelators plus the O1-(aqua) and N21-(9heade) atoms, whereas the distal donor atom is O(8)-tda. Note that proximal Cu-O bond distances are always very close to 2 Å. Hence here the O(8) atom will be considered the distal donor atom in such 4+1 Cu(1) coordination, in spite of the Cu(1)-S(1) distance of 2.363(1) Å. Hence the tridentate tda chelator exhibits in 1 a fac-SO+O(distal) conformation. This behaviour has previously reported for three compounds [23-25] including that of [Cu(tda)(Hhyp)(H2O)]·2H2O (Hhyp = hypoxanthine) [24]. In this last compound the metal also exhibits an elongated square-based pyramidal coordination, the N9-unsubstituted Hhyp binds the Cu(II) centre by its most basic N9 donor atom in trans versus the S-tda donor and the O-aqua ligand occupy one among the four closest coordination sites. But note that, in contrast, the O1-aqua ligand of 1 falls in trans versus the O4-tda donor. In all these three previously reported compounds, the Cu-S(tda) bond distance is within the narrow range 2.366-2.373 Å, with an average value of 2.369 Å, very close to the Cu-S(tda) bond of 1 (2.363(1) Å, see Table 2). In clear contrast, eight known Cu(II)-tda compounds with fac-O2+S(distal) conformation have longer Cu-S(distal) distances (2.570-2.742 Å) averaging 2.648 Å exist. This group of compounds includes the 3D-coordination polymer [Cu(µ3-tda)]2 [29] and seven ternary complexes where the four closest coordination bonds are two O-tda donors along with two N-heterocyclic donors from two monodentate ligands (imidazole or 3,5-dimethyl-1H-pyrazole) or a α,α-diimine as bidentate chelator (2,2’-bypiridine or various 1,10-phenantrolines) [25-30].
The tda ligand is also able to play non-chelating roles for Cu(II), without Cu-S(thioether) bond, as documented for the bridging µ2-O,O-tda [23,28] or a µ2-O,O’,O’’,O’’’-tda [31] modes. But no Cu(II) complex with tridentate chelating tda in mer-SO2 conformation has been reported so far!
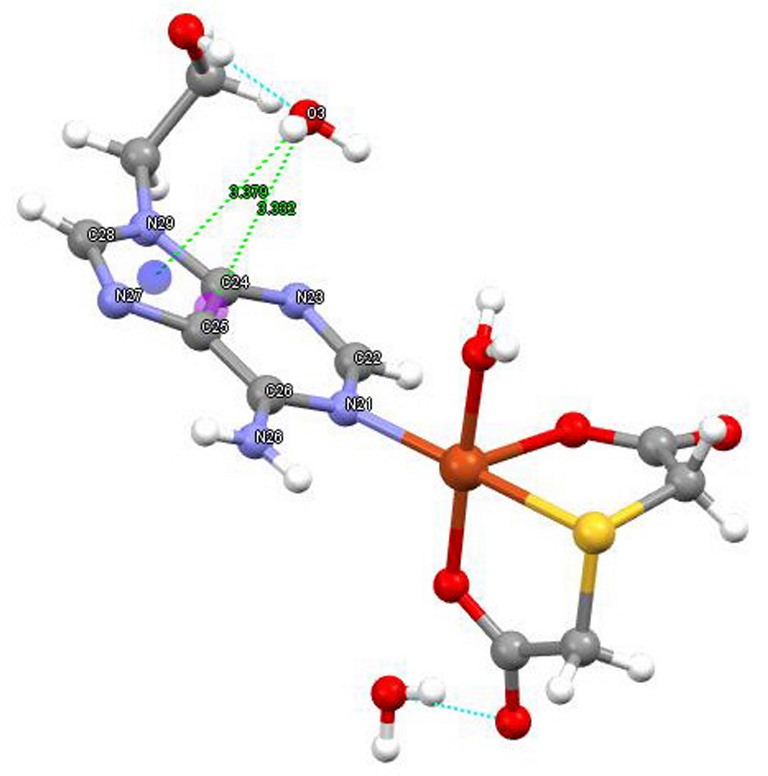
On the other hand, 9heade displays in 1 an unprecedented MBP, using its most basic N-heterocyclic donor instead of its usually preferred N7 donor. Note that the exocyclic -N(6)H2 group falls coplanar to the purine moiety because the N6-lone pair is delocalized into by hiperconjugation with the aromatic purine moiety. That also means a biggest steric hindrance over N1 versus N7. A likely explanation is that, for 6-minopurines, the cooperation of the Cu-N7 bond with a N6-H···O(acceptor) represents a ring motif of seven atoms, whereas the cooperation of the Cu-N1 with a N6-H···O(acceptor) generates a ring motif of six atoms (also taking into account that the involved angles in these motives are also relevant). In other simplest words, the metal binding to the N7 donor of a 6-aminopurine is less hindered that the coordination to N1. Despite limited information on MBPs for 9heade, the µ2-N1,N7-bridging mode has been reported for a coordination polymer having cis-1,2-cyclobutanedicarboxylate(2-) anion and as coligands [14]. But this bridging modes is not the case of compound 1, where the cooperation between the Cu-N1(9heade) with the O2-water mediated H-bonding interaction represents an alternative favouring to the copper(II) binding with the most basic N1-9heade. Note that a cooperation between a Cu-N7 bond and a N6-H···O(carboxylate) in 6-aminopurines generates a seven-membered ring motif, whereas the involvement of Cu-N1 instead of Cu-N6 reduces the dimension of such a ring motif to six atoms.
2.4. Physical Properties
The thermal stability, FT-IR, UV-Vis, and RSE spectroscopies of compound 1 have been investigated, as detailed below.
The registration of the FT-IR spectrum evidences an unusual behaviour in the preparation of sample pellets with dry-IR quality KBr, notably distinct when crystalline samples of 1 are ground more and more (see SM.4-b). Such behaviour is minimized when the pellet is obtained with freshly-prepared microcrystalline samples and minimally grounded with KBr (SM.4-a). The this later spectrum revealed the presence of water/aqua, fac-SO+O(distal)-tda and 9heade, with the following wavenumber (cm-1) bands (those bands with contributions from to two or more IR-chomophores are not referred in this discussion). For water: νas 3446. For tda: νas(CH2) 2952, 2925, νs(CH2) 2884, νs(COO) 1391, 1373 and ν(C-S) 647 (expected at 715-629, and rather weak because it is Raman active) [32,33]. For 9heade: ν(O-H) 3521, νas(NH2) 3313, ν(C-H)arom 2970, skeletal in purines 1686,1571, δ(O-H)ol 1339, ν(C-O)ol 1060, out-of-plane π(C-H)arom 876, 858 (usually 900-860). Relevant insights are the two symmetric stretching bands, νs(COO), related to the fac-SO+O(distal) conformation of tda (instead of a band neat 1385) as well as the loss of intensity of the absorption near 1625 for the grounded sample, where the contribution of the scissoring mode of water, δ(H2O), is overlapped to that of the δ(NH2) for 9heade. On the other hand, the reflectance spectrum, obtained with a grounded sample of 1, shows an anomalously and poorly informative broad band centred at about 750 nm (see SM.5).
The thermal stability of 1 was firstly studied by thermo-gravimetric analysis (TGA, r.t.-950 °C), with FT-IR identifications of the evolved gases (see SM 6), under air-flow, that most frequently yield a metal oxide (CuO for 1) as final residue (R) (Table 3 and Figure 4. Left).
Some comments on TGA are as follows. Non coordinated water are evolved in first step. The loss of the proximal aqua ligand occurs in second step, along with the the beginning of the decarboxylation of tda, which is why the elimination of CO2 is also observed and the weight of the lemonade sample (6.387%) exceeds by 57.7% to the calculated value for the loss of the aqua ligand (4.049%). The differential scanning calorimetry of 1 (DSC, under N2) shows two minima at 129 and 182 °C, respectively (Figure 4. Right). The overlap of burning of tda and 9heade mainly occurs during the third and four steps. During that, in addition of water, CO2 and some CO plus the three typical N-oxides, two peaks at 2955-3000 and 2072-2040 regions strongly suggest the evolution of isothiocianic acid (SCNH) instead of the expected SO2. The residue at 700 °C (~20.08%, not shown in Table 3) and even at 950°C (19.53%) still exceed something to those estimated for a final residue of CuO (17.88%).
It does not seem unlikely that the initial sample used for the TGA experiment contained slightly less than two molecules of uncoordinated water, something assumable given the high environmental temperatures registered in our laboratories during the present work. In this connection, the room temperature ESR spectra, recorded on powdered samples, showed contributions from two Cu(II) species magnetically independents (SM.7). The more intense one (~94%) is characterized by the following principal components of the g tensor: g1 = 2.304, g2 = 2.083 and g3 = 2.055 (<g> = 2.147 ; G = 4.4). These values imply a dx2-y2 ground state and agree with the 4+1 coordination around copper(II), as reported by the crystallographic results. The secondary signal is very narrow and characteristic of an axially symmetric g tensor (g// = 2.204; g⊥ = 2.140; <g> = 2.161; G = 1.5 ). Interestingly, the intensity of this line increases notably when the sample is ground, even if it is done very gently, suggesting that the new signal is originated by the loss of some of the water molecules of the compound. That is the loss of these molecules modifies the magnetic interactions in the compound, as confirmed by the difference between the calculated G values for both signals [34]. Taking into account the two water-mediated H-bonding interactions, above referred, such behavior can be rationalized assuming that a partial loss of any of the two uncoordinated water molecules can motivate the formation of a secondary phase, according to our ESR results.
2.5. DFT Calculations
The DFT study is basically focused on the analysis of some assemblies observed in the solid state of compound 1. Figure 5-a shows a self-assembled dimer formed in the solid state of 1 via the Hoogsteen side of the adenine. This is the only possibility of self-assembly because the Watson-Crick side is blocked by the coordination of the adenine to Cu(II) by N1. The formation of the two symmetrically equivalent N6–H6···N7 H-bonds generates the R22(10) supramolecular ring. In addition, Figure 5-b shows the interaction of one of the non-coordinated water molecules, that establishes a strong H-bond with the 2-hydroxyethyl group in such a way that one lone-pair (lp) points to the five membered ring of adenine, thus establishing a lp–π interaction (O···Cg distance: 3.38 Å). Both assemblies have been analyzed theoretically in this section.
The MEP surface of compound 1 was initially computed (see Figure 6) to investigate the most electron rich and electron poor regions. The MEP minimum is located at the non-coordinate O-atom of the carboxylate group (–63.5 kcal/mol). The MEP values are also negative at the N3 and N7 atoms of the adenine ring (–9 and –15 kcal/mol, respectively) and hydroxyl O-atom (–17 kcal/mol). The maximum MEP is located at H-atoms of the coordinated water molecule (+75 kcal/mol) as expected due to the strong effect of the coordination to Cu(II) on the acidity of the water protons. The MEP are also large and positive at the H-atoms of the hydroxyl and amino groups, ranging from +53 to +57 kcal/mol. Moreover, the MEPs over the five and six-membered rings of adenine are positive (29 and +28 kcal/mol, respectively) thus revealing that the π-surface is adequate for interacting with electron rich atoms. This MEP analysis also shows that the MEP is positive at the extension of the S-C bonds (+34 kcal/mol, σ-hole), evidencing that the coordinated S-atom of tda ligand is able to act as Lewis acid and establish chalcogen bonds.
Figure 7-a shows the combined QTAIM/NCIPlot analysis of the self-assembled H-bonded dimer commented above, evidencing that each H-bond is characterized by a bond critical point (CP, magenta sphere) and bond path (dashed bonds) interconnecting the H and N-atoms. The N-H··O interactions are also characterized by blue (attractive) NCIplot isosurfaces coincident to the location of the bond CPs. The total dimerization energy is ΔE1 = –9.2 kcal/mol (–4.6 kcal/mol each H-bond), thus confirming their energetic relevance. Figure 7-b shows the combined QTAIM/NCIPlot analysis of the water-adenine complex. The O-H···O interaction is characterized by a bond CP, bond path and dark blue reduced density gradient (RDG) isosurface interconnecting the H and O-atoms. Interestingly, the analysis also shows one bond CP and bond path connecting the O-atom of water to one C–atom of adenine, thus confirming the existence of the lp-π interaction. This contact is also disclosed by the NCIPlot analysis that shows a green and extended RDG isosurface located between the O-atom and the five-membered ring. The total interaction energy is moderately strong, ΔE2 = –5.9 kcal/mol. To estimate the contribution of the lp-π interaction, we have evaluated the strength of the O-H···O interaction by using the simple and reliable methodology recently proposed by Emaniam et al. [35]. In particular, the hydrogen bond strength (ΔE) is calculated using the electron density (ρ) at the bond CP and the equation ΔE = –233.1 × ρ + 0.7. The hydrogen bond strength is indicated in Figure 7b (in red close to the bond CP) revealing that the OH···O interaction is –4.7 kcal/mol and consequently the contribution of the lp-π is only –1.2 kcal/mol. Interestingly, the strength of the NH···N (Figure 5-a) and OH···O interactions (Figure 5-b) are similar in agreement with the color of the NCIplot RDG isosurfaces that characterize those H-bonds.
In the solid state, compound 1 also forms infinite 1D assemblies where the electron rich O-atoms of the thiodiacetate coligand of one monomer are located approximately opposite to the C–S bonds of the adjacent monomer, thus propagating the supramolecular polymer (Figure 8). In addition, an OH···O H-bond between the coordinated water molecule and the carboxylate group is formed that further supports the propagation of the supramolecular polymer. It can be observed that one of both S···O contacts presents an intermolecular distance that is close (0.09 Å longer) than the sum of bonding’s van der Waals radii of O and S, ΣRvdW(O+S) = 3.32 Å. The distance of the other one is 0.3 Å longer than ΣRvdW(O+S) thus the existence of such contact should be supported by theoretical calculations (vide infra).
Figure 9 shows the combined QTAIM/NCIPlot analysis of a dimer extracted from the 1D infinite chain shown in Figure 8. It confirms the existence and attractive nature of both ChBs, each one characterized by a bond CP, bond path and green RDG isosurface connecting the S to the O-atom. The OH··O interaction is characterized by a bond CP, bond path and blue NCIplot isosurface coincident to the location of the bond CP. This reveals that the H-bond is significantly stronger than the ChBs. The QTAIM analysis also discloses the presence of ancillary interactions interconnecting both dimers, like two CH···O H-bonds and other contacts involving the π-systems (π···π, N···π and CH···π). Such intricate combination of interactions provokes a large stabilization of this assembly, as evidenced by the large dimerization energy (ΔE3 = –29.5 kcal/mol), thus confirming the structure directing role of the interactions and their energetic relevance.
3. Concluding Remarks
The molecular and crystal structures of the title compound revealed that in aqueous solution the Cu(tda) chelate and the synthetic nucleoside 9heade recognizes to each other to give a ternary complex molecules, which crystallizes as [Cu(tda)(9heda)(H2O)]·2H2O. The molecular recognition (metal chelate)-(adenine synthetic nucleoside) represents the novel MBP for 9heade, essentially bonded to Cu(II) by N1, its most basic donor atom available for coordination. In addition, two water-mediated H-bonding interactions intra-stabilize the novel compound: 9heade-N6-H···O2(water) with O2···Cu weak contact and 9heade-O(ol)-H···O3(water) with O3-H36···π(centroid of the six-membered ring of adenine moiety). The easy partial loss of these water molecules makes it difficult to interpret the thermal stability and, above all, the spectral properties of the studied compound. However both H-bonded and lp–π assemblies enables to be analyzed energetically using density functional theory (DFT) calculations, reduced density gradient isosurfaces and the topological analysis of bond critical points that was also used to estimate the contribution of the H-bonds. In addition, structurally relevant S···O interactions between the Cu-coordinated S-atom and the carboxylate O-atoms of the tda ligand are rationalized by means of MEP and QTAIM calculations.
4. Materials and Methods
4.1. Reagents and Synthesis of Compound 1
This product has been obtained by reaction between stoichiometric amounts of Cu2CO3(OH)2 (green-malachite, Aldrich), H4EDTA (Aldrich), thiodiacetic acid (TCI) and 9heade (TCI) in water. Various experiments were carried out. In a typical experiment, greenish malachite Cu2CO3(OH)2 (1 mmol, 0.22 g) and H2tda (2 mmol, 0.130 g) were reacted in 200 mL of distilled water, inside a covered Kitasato flask of 500 mL. Its open side outlet allows to remove CO2 (the only by-product!) and prevents possible splashes. The reaction mixture is continuously heated (45-50 °C) and stirred for an hour, until unreacted malachite is not observed. The light green aqueous solution of Cu(tda) chelate was cooled to r.t. and slowly filtered by 250 mL additional glass Büchner funnel with sintered borosilicate plate (No. 3, porosity 16-40 µm) over an Erlenmeyer flask (Cautions: Insufficient reaction times will be avoid and leaves unreacted green on the Büchner plate). Then 9heade (0.36 g, 2 mmol) is added to the chelate solution and the mixture stirred until their solution has been completed. Mother liquor of the ternary system was again filtered, over a proper Büchner r over a 250 mL crystallization flask, being it covered with perforated plastic film to control the evaporation of the solvent. Working in this way, crystals of the desired product 1 appear at three weeks. Handily picked single crystals were uses for crystallographic purposes. Time-by-time samples were checked by FT-IR spectroscopy that revealed that only one product seems to be obtained. The product was be dried at r.t. Overall yields are of ca. 60-75 %. But long standing of samples over 30 °C lose variable amounts of uncoordinated water, causing the problems referred in detail at the discussion of their physical properties. That is also affects variable results of your CNH elemental analysis. Best agreement is as follow: Elemental analysis (%): Calc. for C11H19CuN5O8S, C 29.70, H 4.30, N 15.74. Found, C 29.83, H, 4.18, N, 15.80.
4.2. Physical Measurements
The elemental analysis was performed with a Thermo Scientific Flash 2000 (Thermo Fisher Scientific Inc., Waltham, MA, USA). Infrared spectra (samples in KBr pellets) were recorded using a Jasco FT-IR 6300 spectrometer (Jasco Analítica, Madrid, Spain). Electronic (diffuse reflectance) spectra were obtained in a Varian Cary-5E spectrophotometer (Agilent Scientific Instruments, Santa Clara, CA, USA) from a grinded crystalline sample. Thermogravimetric analyses (TGA) was carried out (10 °C/min) under air-dry flow (100 mL/min) with a thermobalance Mettler-Toledo TGA/DSC1 (Mettler-Toledo, Columbus, OH, USA) and a series of 50 time-spaced FT-IR spectra were recorded to identify evolved gasses throughout the experiment, using a coupled FT-IR Nicolet 550 spectrometer (Thermo Fisher Scientific Inc., Waltham, MA, USA). Differential scanning calorimetry (DSC) measurement was recorded for a sample (6.234 mg) of compound 1, on a DSC-SHIMADZU mod. DSC-50Q instrument (Shimadzu Europe, F.R. Germany GbmH,) under an N2 atmosphere, at 30-400 °C (heating rate 10 °C/min). Electron paramagnetic resonance (EPR) measurements were performed using a Bruker ELEXSYS E500 spectrometer operating at the X-band. The spectrometer was equipped with a super-high-Q resonator ER-4123-SHQ. The magnetic field was calibrated by a NMR probe and the frequency inside the cavity (~9.4 GHz) was determined with an integrated MW-frequency counter. Data was collected and processed using the Bruker Xepr suite.
4.3. Crystallography
A Green block crystal of [Cu(tda)(9heade)(H2O)]·2H2O (1) was mounted on a glass fiber and used for data collection. Crystal data were collected at 298(2) K, using a Bruker D8 VENTURE diffractometer. Graphite monochromatic CuK(α) radiation (λ = 1.54184 Å) was used throughout. The data were processed with APEX3 [35] and corrected for absorption using SADABS (transmissions factors: 1.000 - 0.708) [36]. The structure was solved by direct methods using the program XT [37] and refined by full-matrix least-squares techniques against F2 using XL [37]. Positional and anisotropic atomic displacement parameters were refined for all non-hydrogen atoms. Hydrogen atoms were located in difference maps and included as fixed contributions riding on attached atoms with isotropic thermal parameters 1.2/1.5 times those of their carrier atoms. Criteria of a satisfactory complete analysis were the ratios of root mean squares shift to standard deviation less than 0.001 and no significant features in final difference maps. Atomic scattering factors taken of the International Tables for Crystallography [38]. Molecular graphics plotted with PLATON [39]. A summary of the crystal data, experimental details and refinement results are listed in Table 1.
4.4. Computational Details
The calculations of the non-covalent interactions were carried out using the Gaussian-16 [40] and the PBE0-D3/def2-TZVP level of theory [41,42]. To evaluate the interactions in the solid state, the crystallographic coordinates have been used. The interaction energies have been computed by calculating the difference between the energies of isolated monomers and their assembly. The interaction energies were calculated with correction for the basis set superposition error (BSSE) by using the Boys–Bernardi counterpoise technique [43]. The Bader’s "Atoms in molecules" theory (QTAIM) [44] has been used to study the interactions discussed herein by means of the AIMAll calculation package [45]. The molecular electrostatic potential surfaces (isosurface 0.001 a.u.) have been computed using the Gaussian-16 software [46].
In order to assess the nature of interactions in terms of being attractive or repulsive and revealed them in real space, we have used NCIPLOT index, which is a method for plotting non-covalent interaction regions [48] based on the NCI (Non-Covalent Interactions) visualization index derived from the electronic density [48] The reduced density gradient (RDG), coming from the density and its first derivative, is plotted as a function of the density (mapped as isosurfaces) over the molecule of interest. The sign of the second Hessian eigenvalue times the electron density [i.e. sign(λ2)ρ in atomic units] enables the identification of attractive/stabilizing (blue-green colored isosurfaces) or repulsive (yellow-red colored isosurfaces) interactions using 3D-Plots. For the plots shown in Figure 3 and Figure 4 the NCIplot index parameters are: RGD = 0.5; ρ cut off = 0.04 a.u.; color range: –0.035 a.u. ≤ sign(λ2)ρ ≤ 0.035 a.u.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org., Table SM.1. Crystal data and structure refinement for 1; Table SM.2. Selected bond lengths [Å] and angles [°] for 1; Table SM.3. Hydrogen bonds for 1; Figure SM.4: Infrared spectrum of 1; Figure SM.5: Electronic (Reflectance) spectrum of 1; Figure SM.6: Termogravimetric analysis (TGA) of 1 with FT-IR spectra for identification of evolved gases and Figure SM.7. ESR spectra.
Author Contributions
Conceptualization, A.M.-H. and J.N.-G.; methodology, all authors; software, A.M.-H., A.C. and A.F.; investigation, C.R.-M. and R.N.-C.; writing—original draft preparation, all authors, writing—review and editing, all authors; visualisation, A.C., D.Ch.-L., A.F. and J.N.-G. project administration, A.C., A.F., A.M.-H. and J.N.-G.; funding acquisition, A.C., A.F., D.Ch.-L and J.N.-G. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by MICIU/AEI of Spain project PID2020-115637GB-I00 FEDER, MICINN of Spain project PGC2018-102047-B-I00, Project B-FQM-478-UGR20 (FEDER-Universidad de Granada, Spain) which support is acknowledge. Founds and instrumental support of Research groups FQM-283 (Junta de Andalucía, Spain) are also acknowledge.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lehn, J.M. Perspectives in Supramolecular Chemistry-From Molecular Recognition towards Molecular Information Processing and Self-Organization. Angew. Chem. Int. Ed. 1990, 29, 1304–1319. [Google Scholar] [CrossRef]
- Persch, E.; Dumele, O.; Diederich, F. Molecular Recognition in Chemical and Biological Systems. Angew. Chem. Int. Ed. 2015, 54, 3290–3327. [Google Scholar] [CrossRef] [PubMed]
- Rebek, J. Molecular Recognition with Model Systems. Angew. Chem. Int. Ed. 1990, 29, 245–255. [Google Scholar] [CrossRef]
- Belmont-Sánchez, J.C.; Choquesillo-Lazarte, D.; Navarrete-Casas, R.; Frontera, A.; Castiñeiras, A.; Niclós-Gutiérrez, J.; Matilla-Hernández, A. A tetranuclear Ni(II)-cubane cluster molecule build by four µ3-O-methanolate (MeO) ligands, externally cohesive by four unprecedented bridging µ2-N7,O6-acyclovirate (acv-H) anions. Crystals 2023, 13, 7. [Google Scholar] [CrossRef]
- Domínguez-Martín, A.; Brandi-Blanco, M.P.; Matilla-Hernández, A.; El Bakkali, H.; Nurchi, V.M.; González-Párez, J.M.; Castiñeiras, A.; Niclós-Gutiérrez, J. Unravelling the versatile metal binding modes of adenine: Looking at the molecular recognition patterns of deaza- and aza-adenines in mixed ligand metal complexes. Coord. Chem. Rev. 2013, 257, 2814–2838. [Google Scholar] [CrossRef]
- Amo-Ochoa, P.; Zamora, F. Coordination polymers with nucleobases: From structural aspects to potential applications. Coord. Chem. Rev. 2014, 276, 34–58. [Google Scholar] [CrossRef]
- Brandi-Blanco, M.P.; Choquesillo-Lazarte, D. , Domínguez-Martín, A.; Matilla-Hernández, A.; González-Pérez, J.M.; Castiñeiras, A.; Niclós-Gutiérrez, J. Molecular recognition modes between adenine or adeniniun(1+) ion and binary MII(pdc) chelates (M = Co-Zn; pdc = pyridine-2,6-dicarboxylate(2-) ion). J. Inorg. Biochem. 2013, 127, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Khaled Hassanein, K.; Castillo, O.; Gómez-García, C.J.; Zamora, F.; Amo-Ochoa, P. Asymmetric and symmetric dicopper(II) paddle-wheel units with modified nucleobases. Cryst. Growth Des. 2015, 15, 5485–5494. [Google Scholar] [CrossRef]
- Pérez-Toro, I.; Domínguez-Martín, A.; Choquesillo-Lazarte, D.; González-Pérez, J.M.; Castiñeiras, A.; Niclós-Gutiérrez, J. Highest reported denticity of a synthetic nucleoside in the unprecedented tetradentate mode of acyclovir. Cryst. Growth Des. 2018, 18, 4282–4286. Cryst. Growth Des. 2018, 18, 4282–4286. [Google Scholar] [CrossRef]
- Hammud, H.H.; Travis Holman, K.; Al-Noaimi, M.; Sadiq Sheikh, N.; Ghannoum, A.M.; Bouhadir, K.H.; Masoud, M.S.; Karnati, R.K. Structures of selected transition metal complexes with 9-(2-hydroxyethyl)adenine: Potentiometric complexation and DFT studies. J. Mol. Struct. 2020, 1205, 127548. [Google Scholar] [CrossRef]
- Ruiz-González, N.; García-Rubiño, M.E.; Domínguez-Martín, A.; Choquesillo-Lazarte, D.; Franconetti, A.; Frontera, A.; Castiñeiras, A.; González-Pérez, J.M.; Niclós-Gutiérrez, J. Molecular and supra-molecular recognition patterns in ternary copper(II) or zinc(II) complexes with selected rigid-planar chelators and a synthetic adenine-nucleoside. J. Inorg. Biochem. 2020, 203, 110920. [Google Scholar] [CrossRef]
- García-Rubiño, M.E.; Matilla-Hernández, A.; Frontera, A.; Lezama, L.; Niclós-Gutiérrez, J.; Choquesillo-Lazarte, D. Dicopper(II)-EDTA Chelate as a bicephalic receptor model for a synthetic adenine nucleoside. Pharmaceuticals 2021, 14, 426. [Google Scholar] [CrossRef] [PubMed]
- Belmont-Sánchez, J.C.; García-Rubiño, M.E.; Frontera, A.; Matilla-Hernández, A.; Castiñeiras, A.; Niclós-Gutiérrez, J. Novel Cd(II) coordination polymers afforded with EDTA or trans-1,2-CDTA chelators and imidazole, adenine, or 9-(2-hydroxyethyl)adenine coligands. Crystals 2020, 10, 391. [Google Scholar] [CrossRef]
- Sushrutha, S.R.; Hota, R.; Natarajan, S. Adenine-Based Coordination Polymers: Synthesis, Structure, and Properties. Eur. J. Inorg. Chem. 2016, 2962–2964. [Google Scholar] [CrossRef]
- Pimentel, G.C.; McClellan, A.L. The Hydrogen Bond; W.H. Freeman & Co.: San Francisco, CA, USA, 1960. [Google Scholar]
- Desiraju, G.R.; Steiner, T. The Weak Hydrogen Bond. In Structural Chemistry and Biology; International Union of Crystallography, Oxford Science Publications: Oxford, UK, 1999. [Google Scholar]
- Gilli, G.; Gilli, P. The Nature of the Hydrogen Bond: Outline of a Comprehensive Hydrogen Bond Theory; International Union of Crystallography, Oxford Science Publications: Oxford, UK, 2009. [Google Scholar]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. The bright future of unconventional σ/π-hole interactions. ChemPhysChem 2015, 16, 2496–2517. [Google Scholar] [CrossRef] [PubMed]
- Aakeröy, C.B.; Bryce, D.L.; Desiraju, G.R.; Frontera, A.; C. Legon, A.; Nicotra, F.; Rissanen, K.; Scheiner, S.; Terraneo, G.; Metrangolo, P.; et al. Definition of the chalcogen bond (IUPAC Recommendations 2019). Pure Appl. Chem., 2019, 91, 1889–1892. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T.; Resnati, G. The σ-hole revisited. Phys. Chem. Chem. Phys. 2017, 19, 32166–32178. [Google Scholar] [CrossRef]
- Pascoe, D.J.; Ling, K.B.; Cockroft, S.L. The origin of chalcogen-bonding interactions. J. Am. Chem. Soc. 2017, 139, 15160–15167. [Google Scholar] [CrossRef]
- Gurbanov, A.V.; Kuznetsov, M.L.; Mahmudov, K.T.; Pombeiro, A.J.L.; Resnati, G. Resonance assisted chalcogen bonding as a new synthon in the design of dyes. Chem.-Eur. J. 2020, 26, 14833–14837. [Google Scholar] [CrossRef]
- Priimagi, A.; Cavallo, G.; Metrangolo, P.; Resnati, G. The halogen bond in the design of functional supramolecular materials: Recent advances. Acc. Chem. Res. 2013, 46, 2686–2695. [Google Scholar] [CrossRef]
- Riley, K.E.; Murray, J.S.; Fanfrlík, J.; Řezáč, J.; Solá, R.J.; Concha, M.C.; Ramos, F.M.; Politzer, P. Halogen bond tunability I: The effects of aromatic fluorine substitution on the strengths of halogen-bonding interactions involving chlorine, bromine, and iodine. J. Mol. Model. 2011, 17, 3309–3318. [Google Scholar] [CrossRef]
- Gomila, R.M.; Bauzá, A.; Frontera, A. Enhancing chalcogen bonding by metal coordination. Dalton Trans. 2022, 51, 5977–5982. [Google Scholar] [CrossRef]
- Frontera, A.; Bauzá, A. Metal coordination enhances chalcogen bonds: CSD survey and theoretical calculations. Int. J. Mol. Sci., 2022, 23, 4188. [Google Scholar] [CrossRef] [PubMed]
- Kopel, P.; Travnicek, Z.; Marek, J.; Korabik, M.; Mrozinski, J. Polyhedron 2003, 22, 411–418. 22. [CrossRef]
- Patel, D.K.; Choquesillo-Lazarte, D.; Domínguez-Martín, A.; Brandi-Blanco, M.P.; González-Pérez, J.M.; Castiñeiras, a.; Niclós-Gutiérrez, J. Inorg Chem. 2011, 50, 10549–10551. [CrossRef]
- Abbaszadeh, A.; Safari, N.; Amani, V.; Notash, B.; Raei, F.; Eftekhar, F. ; Eftekhar, F. Iran J. Chem. Chem. Eng. 2014, 33, 1–13. [Google Scholar] [CrossRef]
- Baggio, R.; Garland, M.T.; Manzur, J.; Peña, O.; Perec, M.; Spodine, E.; Vega, A. A dinuclear copper(II) complex involving monoatomic O-carboxylate bridging and Cu–S(thioether) bonds: [Cu(tda)(phen)]2·H2tda (tda-thiodiacetate, phen-phenanthroline). Inorg. Chim. Acta 1999, 286, 74–79. [Google Scholar] [CrossRef]
- Shahwaz Ahmad, M.; Khalid, M.; Shahnawaz Khan, M.; Shahid, M.; Ahmad, M.; Monika; Ansaric, A. ; Ashafaq, M. Exploring catecholase activity in dinuclear Mn(II) and Cu(II) complexes: An experimental and theoretical approach. New J. Chem. 2020, 44, 7998–8009. [Google Scholar] [CrossRef]
- Bonomo, R.P.; Rizzarelli, E.; Bresciani-Pahor, N.; Nardin, G. Properties and X-ray crystal structures of copper(II) mixed complexes with thiodiacetate and 2,2'-bipyridyl or 2,2':6'.2''-terpyridyl. J. Chem. Soc. Dalton Trans. 1982, 681–685. [Google Scholar] [CrossRef]
- Alarcón-Payer, C.; Pivetta, T.; Choquesillo-Lazarte, D.; González-Pérez, J.M.; Crisponi, G.; Castiñeiras, A.; Niclós-Gutiérrez, J. Thiodiacetato-copper(II) chelates with or without N-heterocyclic donor ligands: Molecular and/or crystal structures of [Cu(tda)]n, [Cu(tda)(Him)2(H2O)] and [Cu(tda)(5Mphen)]·2H2O (Him = imidazole, 5Mphen = 5-methyl-1,10-phenanthroline). Inorg. Chim. Acta 2005, 358, 1918–1926. [Google Scholar] [CrossRef]
- Hong, Y. HUMPEN. Private communication in CSD, 2015.
- Khullar, S.; Mandal, S.K. Effect of spacer atoms in the dicarboxylate linkers on the formation of coordination architecture. Molecular rectangles vs 1D coordination polymers: Synthesis, crystal structures, vapor/gas adsorption studies, and magnetic properties. Cryst. Growth Des. 2014, 14, 6433–6444. [Google Scholar] [CrossRef]
- Coates, J. Interpretation of infrared spectra, A practical approach. In Encyclopedia of analytical chemistry; Meyers, R.A. John Wiley & Sons Ltd.: Chichester, 2000; pp. 10815–10837. [Google Scholar]
- Coates, J.P. The interpretation of infrared spectra: Published reference sources. Appied Spectroscopy Reviews, 2006, 31, 179–192. [Google Scholar] [CrossRef]
- Hathaway, B.J.; Billing, D.E. The Electronic Properties and Stereochemistry of Mono-Nuclear Complexes of the Copper(II) Ion. Coord. Chem. Rev. 1970, 5, 143–207. [Google Scholar] [CrossRef]
- Bruker, APEX3 Software, Bruker AXS Inc. v2018.7-2, Madison, Wisconsin, USA. 2019.
- Sheldrick, G.M. SADABS. Program for Empirical Absorption Correction of Area Detector Data. University of Goettingen, Germany, 1997.
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.J.C. International Tables for Crystallography. Vol. C, Kluwer Academic Publishers: Dordrecht, The Netherlands, 1995.
- A. L. Spek. Structure validation in chemical crystallography. Acta Crystallogr. 2009, D65, 148–155. [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01. Gaussian, Inc., Wallingford CT, 2016.
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu . J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys., 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Bader, R.F.W. J. J. Phys. Chem. A 1998, 102, 7314–7323. [CrossRef]
- Keith, T.A. AIMAll (Version 13.05.06), TK Gristmill Software, Overland Park, KS, 2013.
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A program for plotting noncovalent interaction regions, J. Chem. Theory Comput., 2011, 7, 625–632. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed]
- Emamian, S.; Lu, T.; Kruse, H.; Emamian, H. Exploring nature and predicting strength of hydrogen bonds: A correlation analysis between atoms-in-molecules descriptors, binding energies, and energy components of symmetry-adapted perturbation theory. J. Comput. Chem. 2019, 40, 2868–2881. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
Formulas of the purine-synthetic nucleosides acyclovir (acv, left) and N9-(2-hydroxyethyl)adenine (9heade, right).
Scheme 1.
Formulas of the purine-synthetic nucleosides acyclovir (acv, left) and N9-(2-hydroxyethyl)adenine (9heade, right).
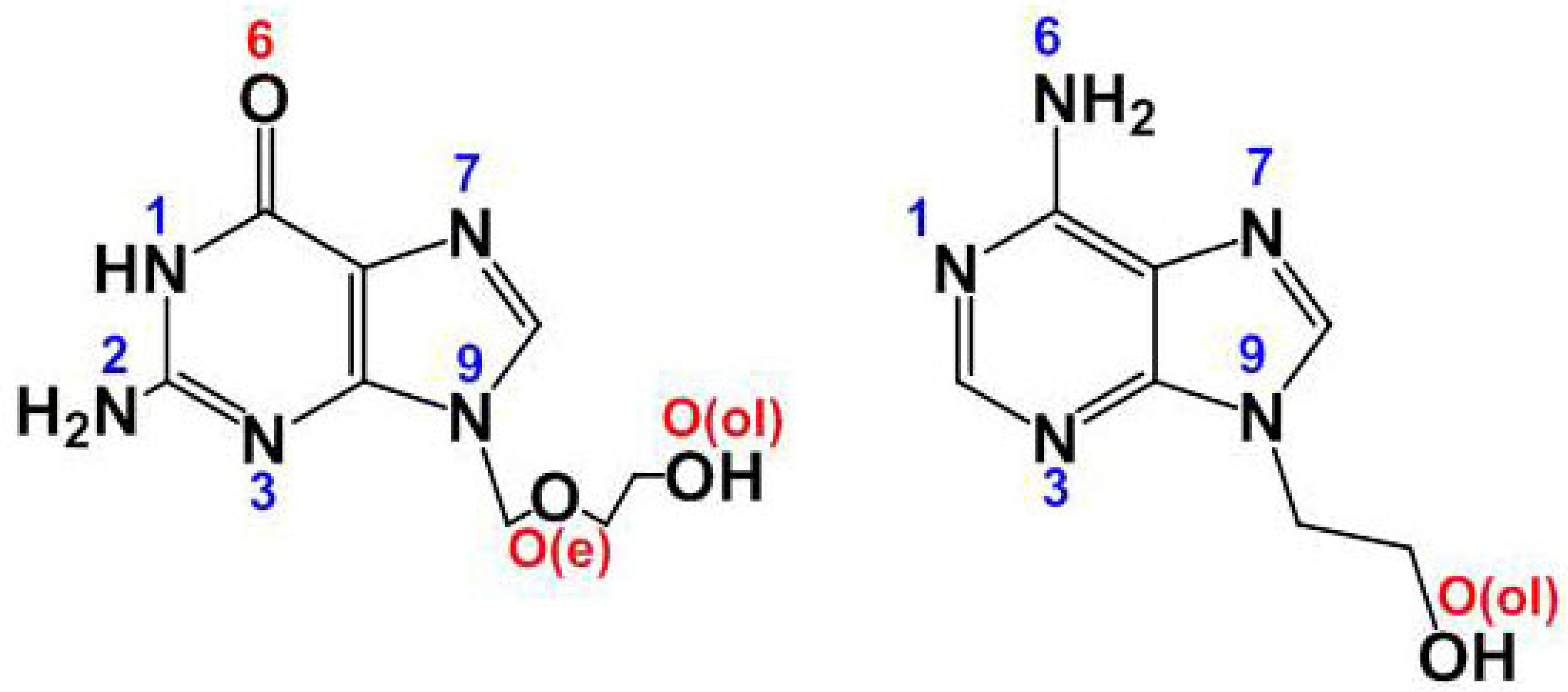
Figure 1.
Representative pictures of crystals of [Cu(tda)(9heade)(H2O)]·2H2O (1).
Figure 2.
Two complex molecules of 1, assuming an asymmetrically elongated Cu(II) a coordination, type 4+1+1*. Water mediated interactions of the N6-H···O2(water) with weak O2···Cu contact plus O32-H···O3(water) with the lone pair-pi (lp-π) are depicted. A symmetry related pair of complex molecules connects by two N6-H···N7.
Figure 2.
Two complex molecules of 1, assuming an asymmetrically elongated Cu(II) a coordination, type 4+1+1*. Water mediated interactions of the N6-H···O2(water) with weak O2···Cu contact plus O32-H···O3(water) with the lone pair-pi (lp-π) are depicted. A symmetry related pair of complex molecules connects by two N6-H···N7.
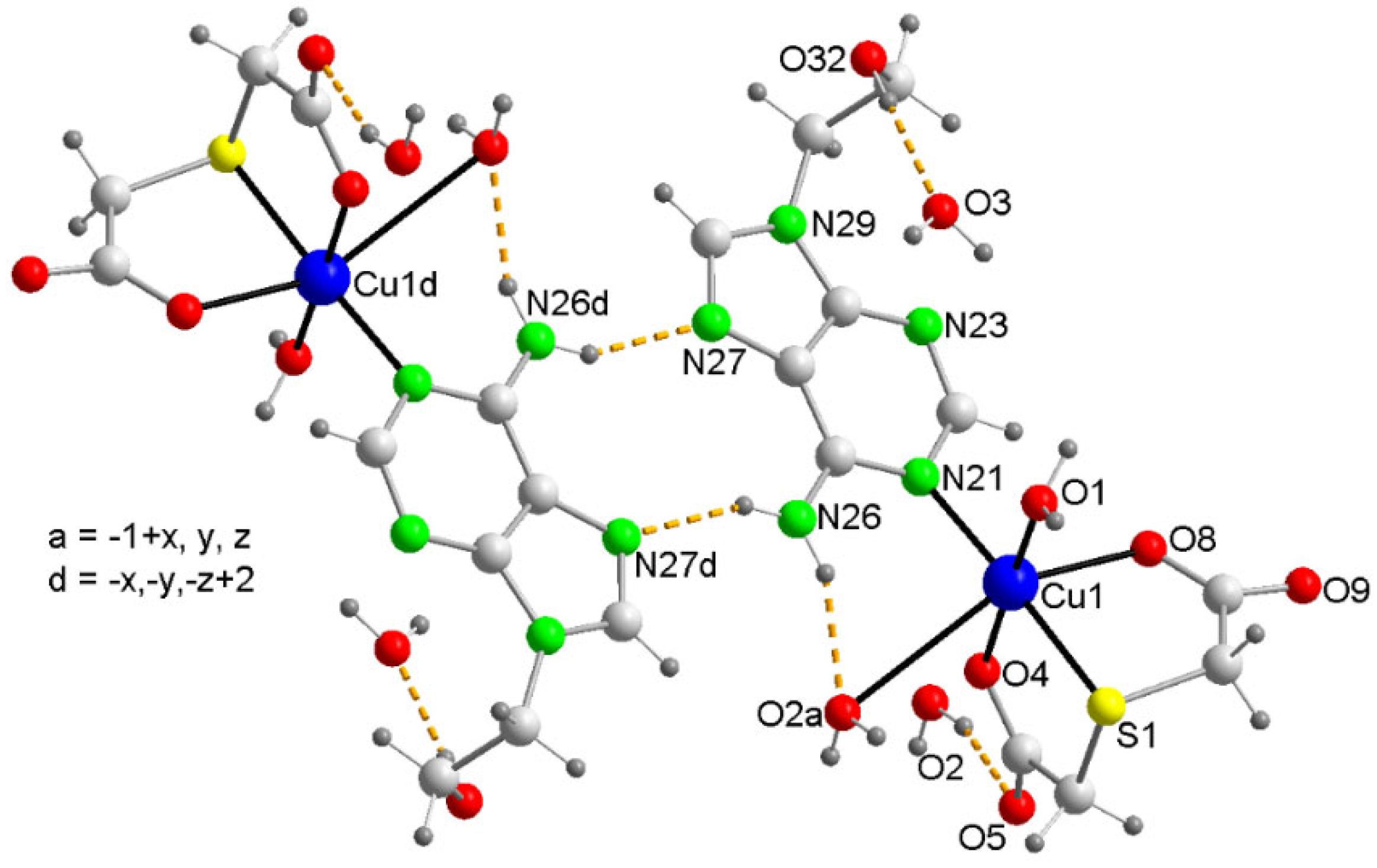
Figure 3.
Structure of compound 1 in the more realistic chemical formula [Cu(tda)(9heda)(H2O)]·2H2O. By this way compound 1 gains stability by means of two water-mediated interactions: N6-H···O2(water) and O2···Cu weak contact plus 9heade-O(ol)-H···O3(water) and O3-H36···π(six-membered ring of adenine moiety).
Figure 3.
Structure of compound 1 in the more realistic chemical formula [Cu(tda)(9heda)(H2O)]·2H2O. By this way compound 1 gains stability by means of two water-mediated interactions: N6-H···O2(water) and O2···Cu weak contact plus 9heade-O(ol)-H···O3(water) and O3-H36···π(six-membered ring of adenine moiety).
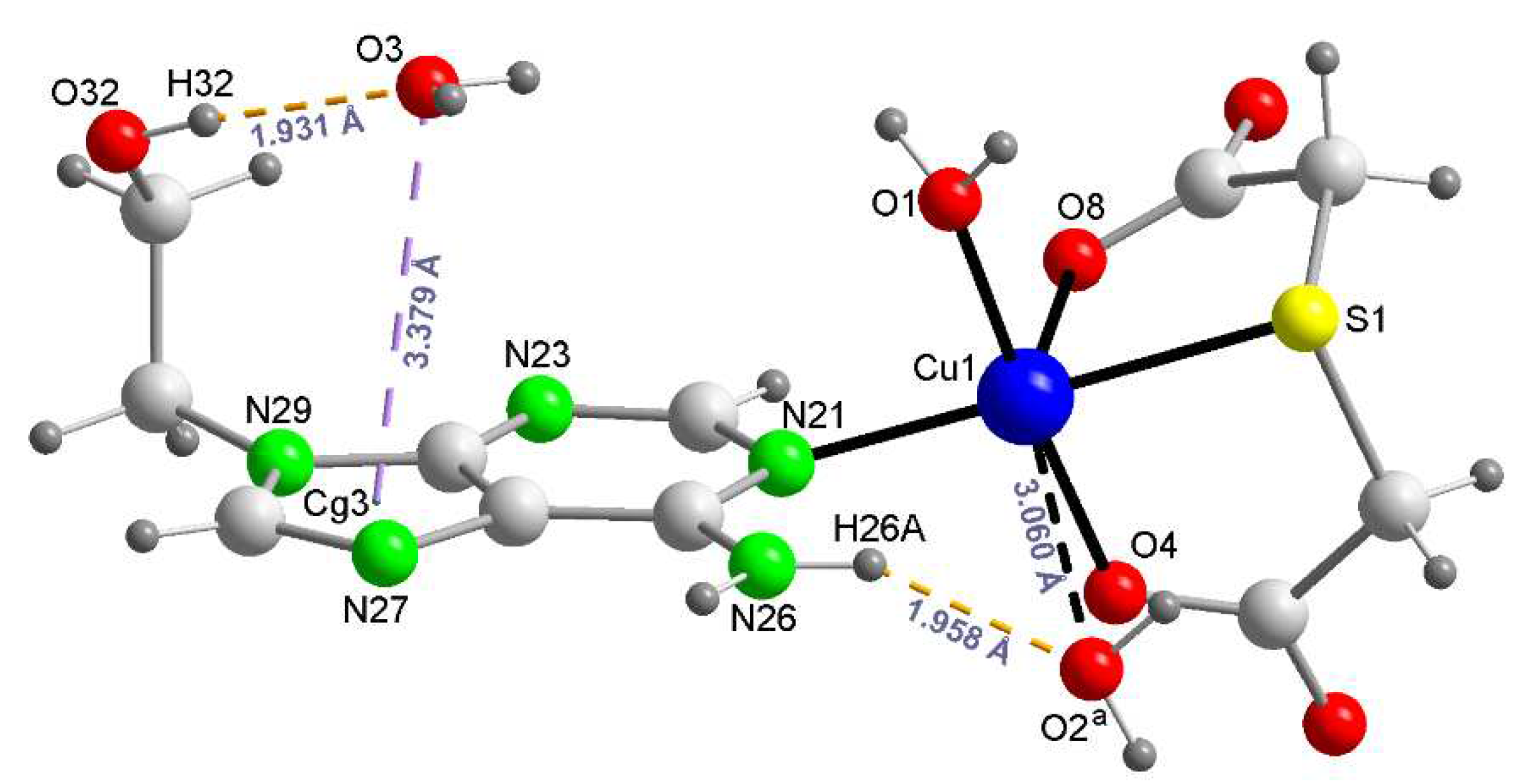
Figure 4.
Left: TGA curve of 1 (weight sample 11.61 mg). Right: DSC curve of 1.
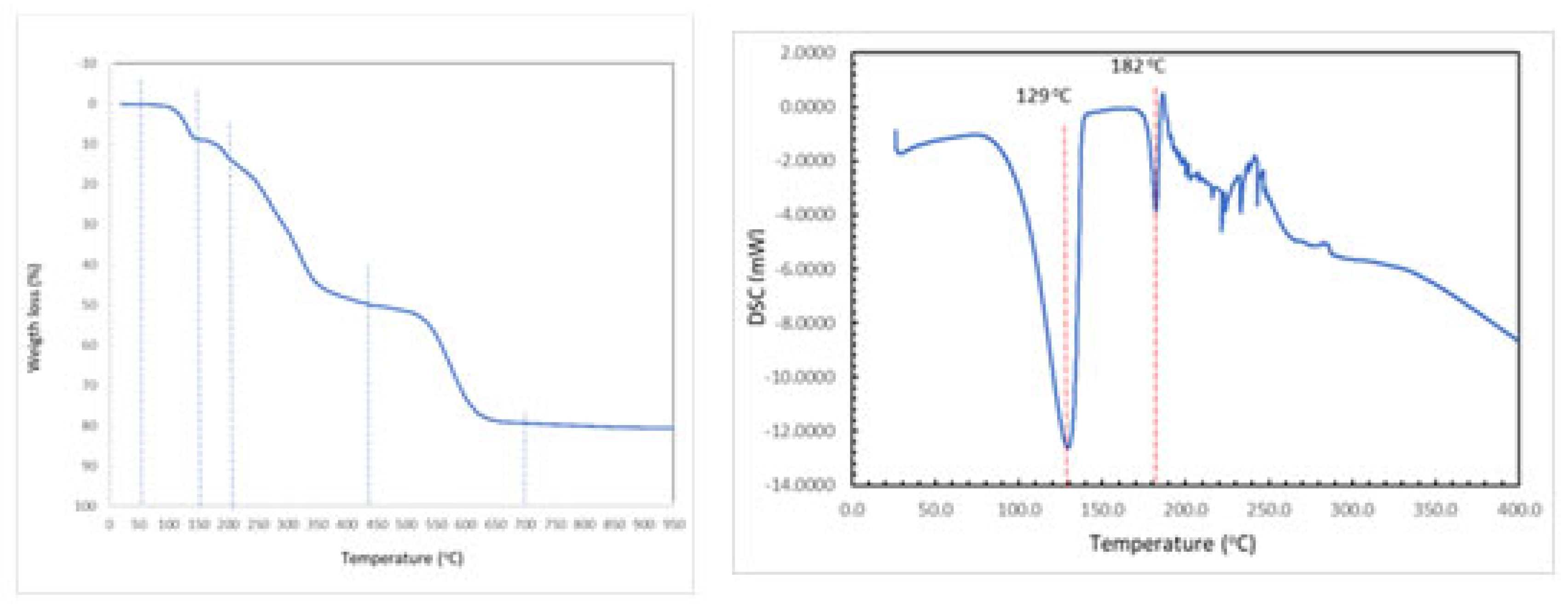
Figure 5.
Partial view of the X-ray structure of 1 with indication of the H-bonds (a) and lp–π interactions (b). Distances in Å.
Figure 5.
Partial view of the X-ray structure of 1 with indication of the H-bonds (a) and lp–π interactions (b). Distances in Å.
Figure 6.
MEP surface 1 at the PBE0-D3/def2-TZVP level of theory (isosurface 0.001 a.u.). The energies at selected points of the surface are given in kcal/mol.
Figure 6.
MEP surface 1 at the PBE0-D3/def2-TZVP level of theory (isosurface 0.001 a.u.). The energies at selected points of the surface are given in kcal/mol.
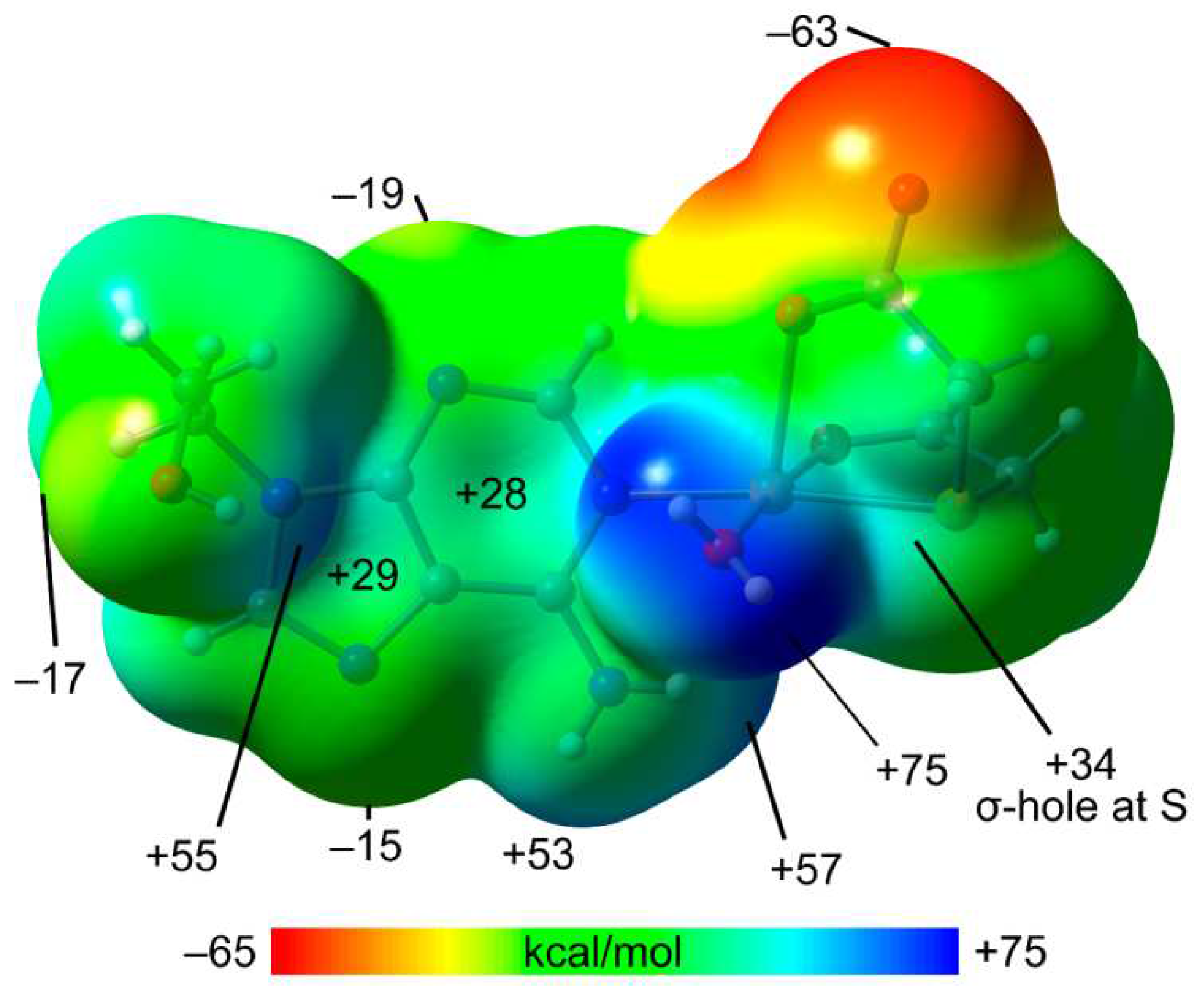
Figure 7.
QTAIM/NCIPlot analysis of intermolecular bond CPs (pink spheres), bond paths and RDG isosurfaces of the H-bond dimer (a) and water complex (b) observed in the X-ray of 1.
Figure 7.
QTAIM/NCIPlot analysis of intermolecular bond CPs (pink spheres), bond paths and RDG isosurfaces of the H-bond dimer (a) and water complex (b) observed in the X-ray of 1.
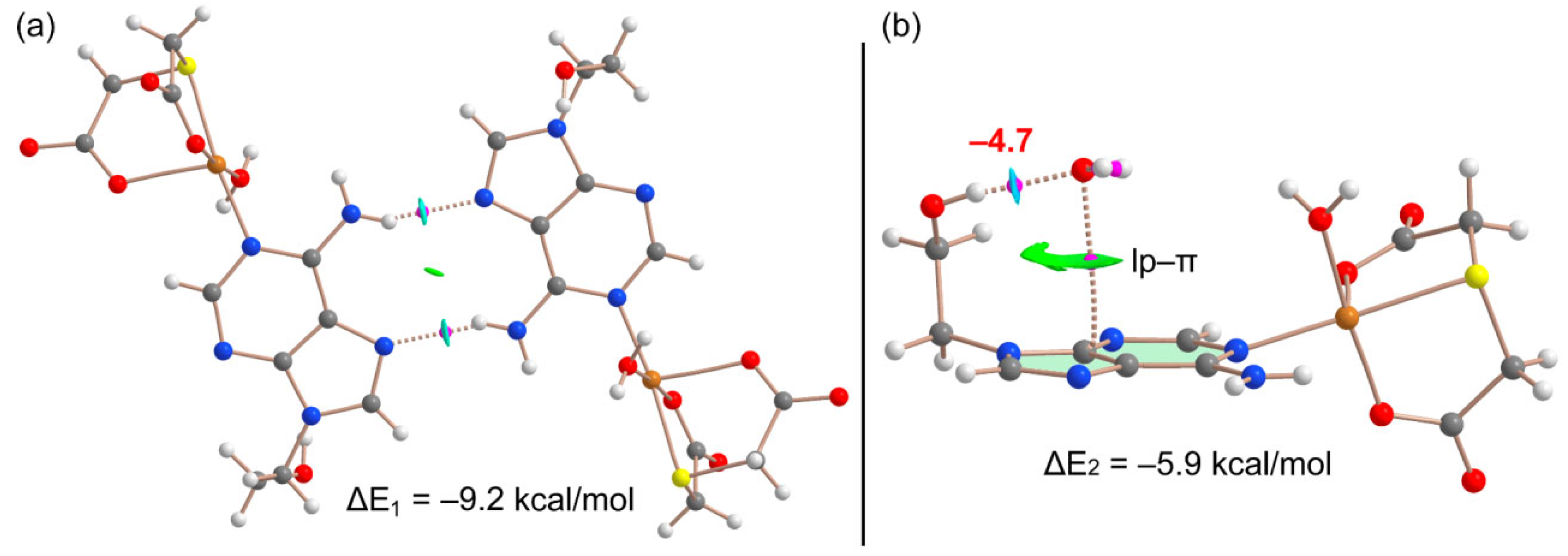
Figure 8.
Partial view of the 1D assembly of 1 with indication of the ChB and H-bonds. Distances in Å. H-atoms omitted apart from those of the coordinated aqua molecule.
Figure 8.
Partial view of the 1D assembly of 1 with indication of the ChB and H-bonds. Distances in Å. H-atoms omitted apart from those of the coordinated aqua molecule.
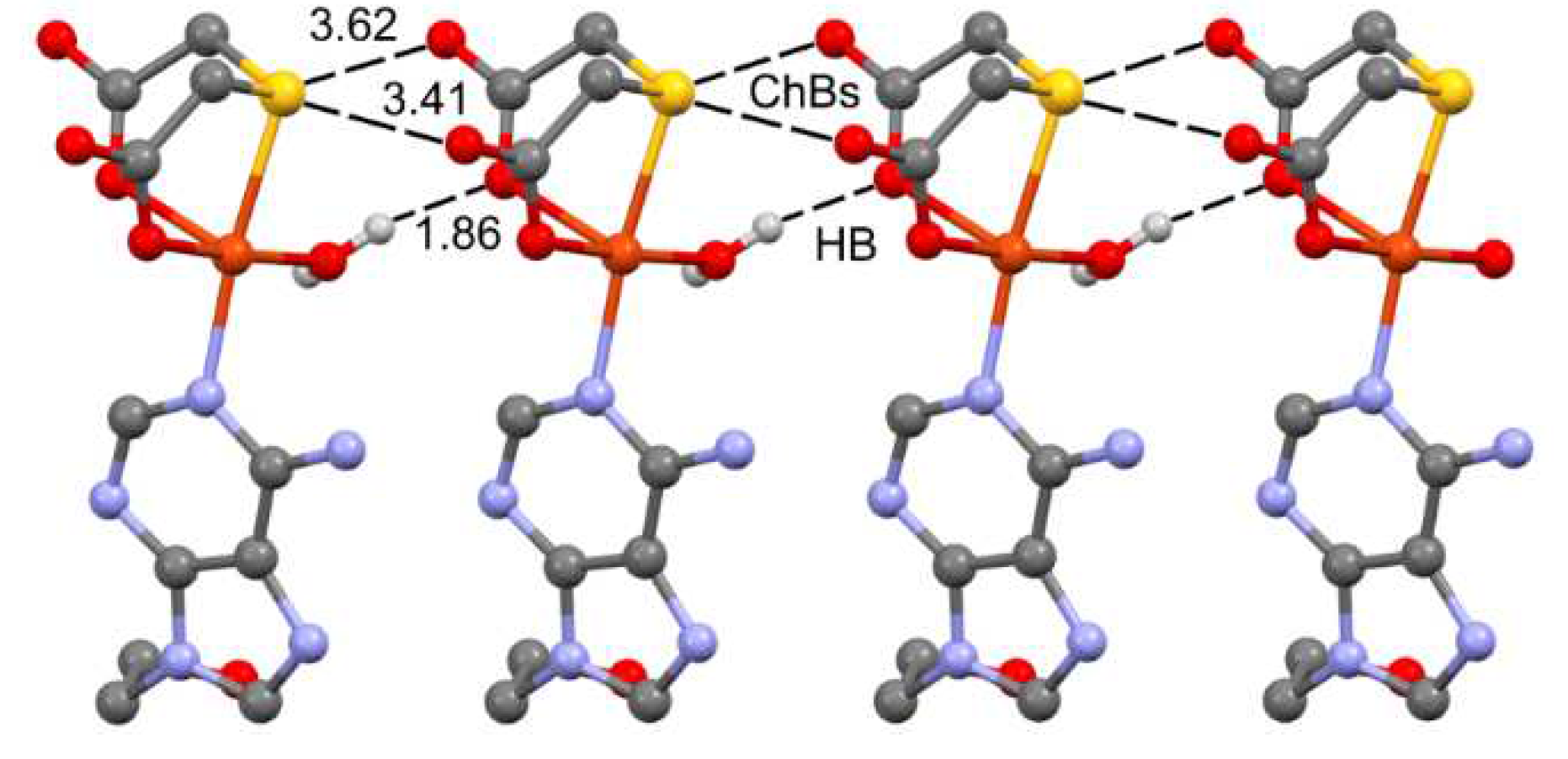
Figure 9.
QTAIM/NCIPlot analysis of intermolecular bond CPs (red spheres), bond paths and RDG isosurfaces of a dimeric assembly of compound 1.
Figure 9.
QTAIM/NCIPlot analysis of intermolecular bond CPs (red spheres), bond paths and RDG isosurfaces of a dimeric assembly of compound 1.
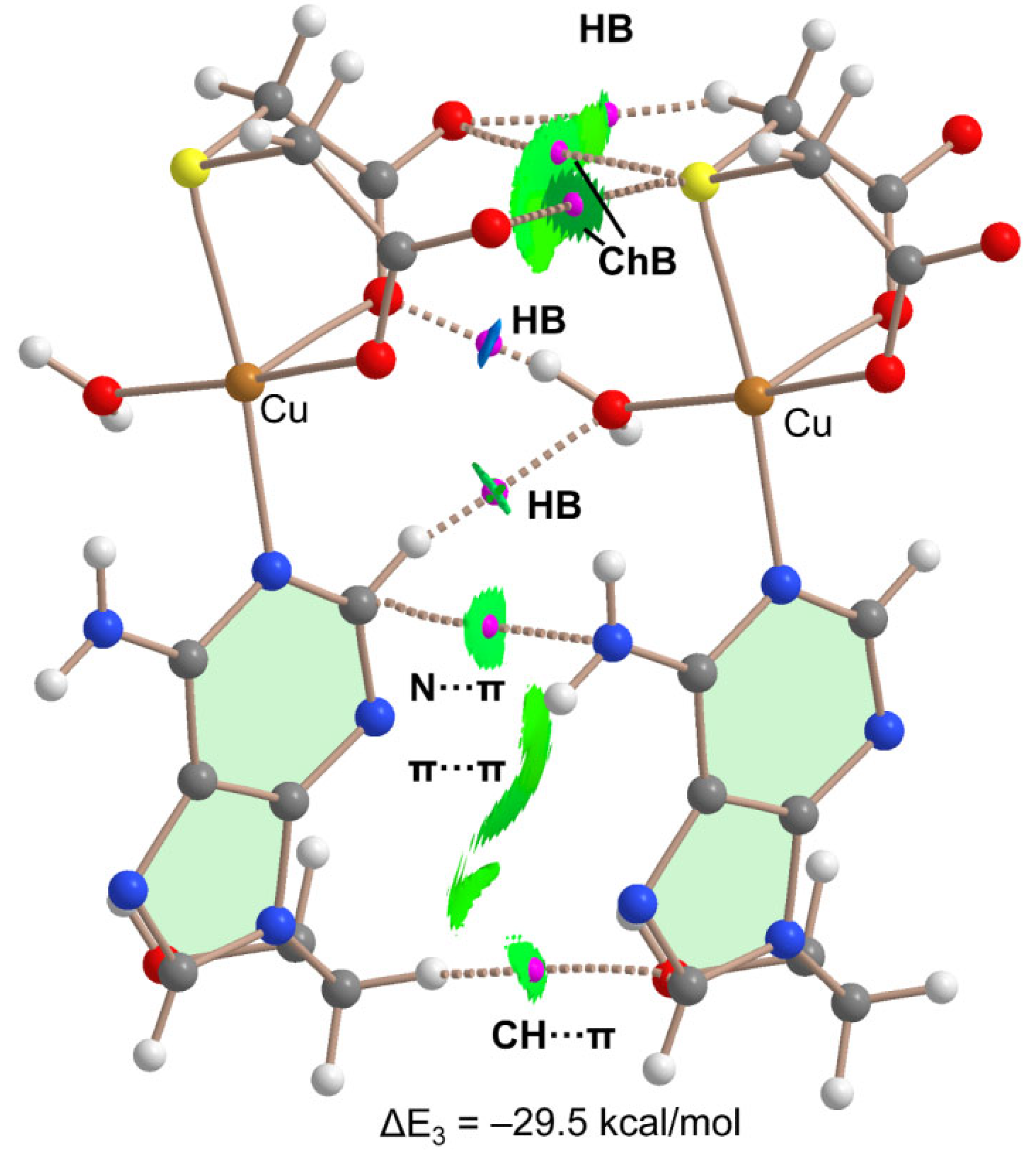

Table 1.
Crystal data and structure refinement for [Cu(tda)(9heade)(H2O)]·2H2O (1).
| Empirical formula | C11H19CuN5O8S |
| Formula weight | 444.91 |
| Temperature | 298(2) K |
| Wavelength | 1.54178 Å |
| Crystal system, space group | Triclinic, |
| Unit cell dimensions | a = 5.3908(3) Å |
| b = 12.9325(7) Å | |
| c = 13.4804(7) Å | |
| α = 62.939(3)° | |
| = 86.260(3)° | |
| γ = 81.195(4)° | |
| Volume | 827.05(8) Å3 |
| Z, Calculated density | 2, 1.787 Mg/m3 |
| Reflections collected / unique | 10657 / 2881 |
| Data / parameters | 2881 / 238 |
| Goodness-of-fit on F2 | 1.132 |
| Final R indices [I>2σ(I)] |
R1 = 0.0415, wR2 = 0.1173 |
| R indices (all data) |
R1 = 0.0447, wR2 = 0.1198 |
| Largest diff. peak and hole | 0.323 and −0.452 e.Å−3 |
| CCDC number | 2267019 |
Table 2.
Coordination bond lengths (Å) and trans-angles (°) for compound 1 for the tentative formula [Cu(tda)(9heda)(H2O)]·2H2O, assuming a distorted elongated octahedral coordination of copper(II), type 4+1+1*, being * a very weak contact.
Table 2.
Coordination bond lengths (Å) and trans-angles (°) for compound 1 for the tentative formula [Cu(tda)(9heda)(H2O)]·2H2O, assuming a distorted elongated octahedral coordination of copper(II), type 4+1+1*, being * a very weak contact.
| Cu(1)-O(4) | 1.933(2) |
| Cu(1)-O(1) | 1.962(2) |
| Cu(1)-N(21) | 2.025(2) |
| Cu(1)-O(8) | 2.262(2) |
| Cu(1)-S(1) | 2.3625(8) |
| Cu(1)-O2a | 3.060(4) |
| O(4)-Cu(1)-O(1) | 175.04(10) |
| N(21)-Cu(1)-S(1) | 173.58(7) |
Symmetry transformation: a = x-1,y,z.
Table 3.
Summary of results obtained from the TGA analysis of compound 1.
| Step or R | Temp. (°C) | Time (min) | Weight (%)Exp. Cal. | Evolved gases or residue (R) | |
|---|---|---|---|---|---|
| 1 | ~50-155 | 2-13 | 8.911 | 8.098 | 2 H2O*, CO2 (t*) |
| 2 | 155-220 | 13-20 | 6.387 | 4.049 | 1 H2O, CO2 |
| 3 | 220-440 | 20-43 | 34.784 | - | CO2, H2O, SCNH(t), N2O (t*), |
| 4 | 440-700 | 43-42 | 14.307 | - | CO2, H2O, CO, SCNH,N2O, NO, NO2 |
| 5 | 700-950 | 42-67 | 29.077 | - | CO2, H2O, CO, N2O, NO, NO2 |
| R | 950 | 95 | 19.532 | 17.879 | CuO |
*t = trace amounts.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



