
Submitted:
12 June 2023
Posted:
13 June 2023
You are already at the latest version
Abstract
Azapeptides have gained much attention due to their ability to enhance the stability and bioavailability of peptide drugs. Their structural preferences, essential to understanding their function and potential applica-tion in the peptide drug design, remain largely unknown. In this work, we systematically investigated the conformational preferences of three azaamino acid residues in tripeptide models, Ac-azaXaa-Pro-NHMe [Xaa= Asn (4), Asp (5), Ala (6)], using the popular DFT functionals, B3LYP and B3LYP-D3. A solvation model density (SMD) was used to mimic the solvation effect on the conformational behaviors of azapep-tides in water. During the calculation, we considered the impact of the amide bond in the azapeptide mod-els on the conformational preferences of models 4-6. We analyzed the effect of the hydrogen bond be-tween the side-chain main chain and main-chain main-chain on the conformational behaviors of azapep-tides 4-6. We found that the predicted lowest energy conformation for the three models differs depending on the calculation methods. In the gas phase, B3LYP functional indicates that the conformers tttANP-1 and tttADP-1 of azapeptides 4 and 5 correspond to the type I of -turn, the lowest energy conformation with all-trans amide bonds, respectively. Considering the dispersion correction, B3LYP-D3 functional predicts the conformers tctANP-2 and tctADP-3 of azapeptide 4 and 5, which contain the cis amide bond preceding the Pro residue, as the lowest energy conformation in the gas phase. The results imply that azaAsx and Pro residues may involve cis-trans isomerization in the gas phase. In water, the predict-ed lowest energy conformer of azapeptides 4 and 5 differs from the gas phase results and depends on the calculational method. For azapeptide 6, regardless of calculation methods and phases, tttAAP-1 (-I turn) is predicted as the lowest energy conformer. The results imply that the effect of the side chain that can form hydrogen bonds on the conformational preferences of azapeptides 4 and 5 may not be negligible. We compared the theoretical results of azaXaa-Pro models with those of Pro-azaXaa models, showing that in-corporating azaamino acid residue in peptides at different positions can significantly impact the folding patterns and stability of azapeptides.
Keywords:
azapeptide
; foldamer
; conformers
; hydrogen bond
; nbo
1. Introduction
Peptides are promising tools with biological and pharmaceutical applications 1. However, their low metabolic stability because of enzymatic degradation, lack of receptor selectivity, and many other drawbacks made it necessary to search for alternatives with the same function as natural peptides but with better properties 2. These alternatives are called peptidomimetics, in which some structural modifications involve peptides’ backbone or side chain 2, 3. Among peptidomimetics, we focus on azapeptides, a change in the backbone of a peptide by substituting the α-carbon with a nitrogen atom 4-6. This modification confers azapeptides more resistance to physiological degradation and long duration of action 7. Many studies revealed the importance of incorporating an azaamino acid in the peptide’s sequence to enhance the stability and bioavailability of drug candidates 8-10. In addition, synthetic methods 4, 9, 11 and biological studies 5, 8, 9, 12 of azapeptides have been studied. However, the structural behaviors of azaamino acid residue in peptides remain limited.
The conformational preferences of azaamino acid residues have been studied theoretically 13, 14, 15 and experimentally 16, 17, 18, 19. The results showed that the preferred dihedral angles of the backbone of these azaamino acid residues are in the range of ϕ= ±90°±30°, ψ= 0° or ±180°±30°, which is appeared to be the polyproline II (βP), and β-turn motif (αR, αL, δR, or δL conformation) 14, 20. The nomenclature was denoted by Karplus et al. 21. Our groups and others examined the effect of azaamino acid residue on the structures in peptides. For examples, the azaglycine adopted the polyproline II (βP) in azaGly-Pro-Hyp (hydroxyproline) of the collagen 17; the δL conformer (ϕ= 90°±30°, ψ= 0°±30°) in Ac-Phe-azaGly-NH2, forming the βII-turn structure; the αR or αL conformer (ϕ= ±90°±30°, ψ= 0°) in Ac-Aib-azaGly-NH2, adopting βI or βI’-turn 22. Meanwhile, incorporating azaamino acids destabilized the β sheet secondary structure compared to the parent peptide [20]. The results showed that the conformational preference of azaamino acid residue in peptides should be sequence dependence. However, it is still largely unknown of these questions: why azaamino acid residue adopts a specific conformation in the context dependence, the influence of the side chain of azaamino acid residue on its backbone structure, the neighboring effect on the conformational behaviors of azapeptides. As a part of the efforts to answer these questions, we previously examined the conformational properties of tripeptides, Ac-Pro-azaXaa-NHMe [Xaa= Asn (1), Asp(2), Ala(3)] using the DFT method, showing that the azaamino acid in this sequence, regardless of the side chain, adopts βⅡ (βⅡ’) turn structure [15]. And we also suggested the importance of the side-chain and main-chain or main-chain and main-chain hydrogen bonds found in these model peptides in the stability of the azapeptides. In this work, we extended our ongoing efforts in understanding conformational properties of short azapeptides containing three azaamino acid residues, Ac-azaXaa-Pro-NHMe [Xaa = Asn (4), Asp(5), and Ala(6)] (Figure 1). While previous theoretical studies focused on the dipeptide models 14, we wondered the position effect of azaamino acid residue in tripeptides. We used the popular DFT functionals, B3LYP and B3LYP-D3, to compare our previous work 15 in the gas phase and water. The solvation effect was calculated with the SMD method 23. We also investigated the influence of the cis-trans isomerization amide bonds on the conformational preferences of model peptides.
2. Computational Methods
All calculations were performed using the Gaussian 09/16 programs package 24. The starting geometries of the three compounds were from the previous work 15 and were fully optimized in the gas phase at the B3LYP/6-311++G(d, p) level of theory. These geometries combine all possible intramolecular hydrogen bonds formed in the molecule between acceptor groups C=O or Nα and the N-H donor group. The orientation of amide bonds in model azapeptides, which is either cis (ω= 0°) or trans (ω= ±180°) amide bond, was also assessed. All conformations were found to be true local minima, as was indicated by the absence of imaginary frequencies. These conformations were then reoptimized using the B3LYP-D3 25 in the gas phase using the same basis set. The solvent effect was examined using the solvation model density (SMD) 23 with and without the dispersion-correction B3LYP functional in water. Intramolecular hydrogen bond interactions found in the molecules in the gas phase (Figure 2) were also inspected using Natural Bond Orbital (NBO) theory at the two levels of theory using NBO 3.1 program implemented in Gaussian 16 package 24. The strength of the hydrogen bond in azapeptides models was estimated using the second-order perturbation analysis (E(2) value) 26. The NBO overlaps were drawn using the Multiwfn program 27.
3. Results and Discussion
The backbone (ϕ and ψ) dihedral angles of the resulting structures for the three azapeptides 4-6 are denoted by the nomenclature given by Karplus 21 (Supporting information Tables S1–S12). Besides, the side chain of azaAsn and azaAsp are marked g+(gauche+), g-(gauche-), s+(skew+), and s-(skew-) based on the values of χ1’ 28 Relative energies of the most stable conformations of the three model compounds 4-5 calculated at the B3LYP/6-311++G(d,p) and B3LYP-D3/6-311++G(d,p) levels of theory in the gas phase and water are listed in Table 1 and Table 2.
3.1. Cis-Trans Isomerization for Minimum Energy Conformations
Ac-azaAsn-Pro-NHME (4): The conformer tttANP-1 (αRδR(g+)), corresponding to the type of βI-turn, and has a trans-trans orientation of the amide bond preceding both residues, was found as the lowest energy minimum for azaAsn model in the gas phase calculated with the B3LYP method. However, the same conformation is the second energy minimum at the B3LYP-D3 functional with a relative energy of 0.94 kcal/mol. Interestingly, the lowest energy minimum conformer of azapeptide 4 calculated at the B3LYP functional is found in the X-ray structure containing azaAsn-Pro sequence, showing the presence of a β-Ⅰ turn with all-trans amide bonds 18, 19. In inclusion of dispersion correction, B3LYP-D3 functional predicts tctANP-2 as the lowest energy minimum containing a cis amide bond between azaAsn-Pro sequence, which is also predicted to be the most stable conformer in water (Table 2). The second lowest energy minimum in solution calculated with B3LYP functional is ttANP-12 (δLβP(s+)) with a relative energy of 0.94 kcal/mol. This relative energy value was also found for the second energy minimum calculated with B3LYP-D3 functional. Another thing to highlight is the trans amide bond preceding both Pro and azaAsn found for the five lowest energy minima calculated at the B3LYP function in the isolated state, except for the second energy minimum in which the amide bond preceding azaAsn is cis-oriented (Table 1). However, most of the lowest energy minimum conformers calculated with B3LYP-D3 functional in the gas phase are cis-oriented amide bonds preceding either the two residues or at least one. In the solution state, the five lowest energy minima with B3LYP-D3 functional are all trans amide bonds except for the first and the fifth conformers, which have a cis amide bond preceding azaAsn residue. The theoretical results suggest that the azaAsn-Pro sequence may also be involved in cis-trans isomerization in water.
Ac-azaAsp-Pro-NHMe (5): The results of Ac-azaAsp-Pro-NHMe are similar to those of the AzaAsn model. B3LYP functional predict tttADP-1 (αRδR(g+)) with all-trans amide bonds as the lowest energy minimum in the gas phase. The second most stable conformation, tttADP-2 (αRγ’(g+)) (ΔE= 1.00 kcal/mol), is also oriented to all-trans amide bonds. However, B3LYP-D3 functional predicts the conformer tctADP-3 as the lowest energy minimum and the conformer tctADP-4 as the second energy minimum. The results imply that the dispersion interaction may be essential in stabilizing the cis amide bond in the azaAsp-Pro sequence. Contrary to the isolated state, both SMD/B3LYP and SMD/B3LYP functionals predict the conformer tttADP-9 (δLβP(s+)) as the lowest energy conformer (Table 2). Similarly, the second most stable conformation in water with the two methods is tttADP-7, with all-trans amide bonds. However, the third lowest energy minimum tctADP-3 (βδR(g+)) (ΔE=1.07 kcal/mol) has a cis orientation of amide bond preceding Pro residue. The results also imply that the azaAsp-Pro sequence may be involved in cis-trans isomerization in water.
Ac-azaAla-Pro-NHMe (6): The results of azapeptide 6 significantly differ from those of azapeptides 4 and 5. Both B3LYP and B3LYP-D3 functionals predict that the conformer tttAAP-1(αRδR), related to the β-Ⅰ turn structure, is the lowest energy minimum in the gas and water. In addition, the B3LYP function predicts that the four most stable conformers are trans-trans amide bonds in the gas phase. However, the third and the fourth energy minimum have trans-cis amide bonds with B3LYP-D3 functional. The two most stable conformations calculated by SMD/B3LYP in water have all trans amide bonds, while. In contrast,he most stable conformation calculated by SMD/B3LYP-D3 was found to be all trans amide followed by two trans-cis-trans or trans-cis-trans conformers (Table 2). The results imply that the side chain of azaAsn and azaAsp may play a vital role in cis-trans isomer. At the same time,n while the conformer of azalea residue containing a cis amide bond is found in the higher energy minima.
3.2. Hydrogen Bonds in Ac-azaXaa-Pro-NHMe
Optimized structures of Ac-azaXaa-Pro-NHMe are presented with the denotation of hydrogen bonds (Supporting Information Tables S1–S12 and Figures S1–S12. NBO orbital interactions corresponding to hydrogen bonds are depicted in the Supporting information Tables S13–S17, calculated with the B3LYP method. We used the E(2) value to describe the strength of the hydrogen bond 26.
The most stable conformation in the gas phase at the B3LYP method is tttANP-1, which is stabilized with two intramolecular hydrogen bonds (HBs) closing C10 and C6 pseudo cycles between carbonyl and N-H groups C2=O1….H2-N4 and C8=O4….H1-N1, respectively (Figure 3). The C10 pseudo cycle is a HB interaction related to the β-Ⅰ turn which is found in X-ray structure of Z-azaAsn(Me)-Pro-NHiPr 19. The HB is made up of three interactions: πCO → σ*NH of 1.43 kcal/mol, nO→σ*NH of 1.98 kcal/mol, and n’O → σ*NH of 0.81 kcal/mol (Figure 3). The sum ΣEHB of the individual E(2) stabilization energies of the three H.B. interactions is 4.22 kcal/mol. It is not the strongest interaction, yet tttANP-1 is the most stable conformation. Moreover, the strongest interaction is that related to the C8 pseudo cycle in tctANP-7 conformation in which the sum of the individual E(2) stabilization energies, associated with πCO, nO, and n’O / σ*NH overlaps involved in the C.O….HN HB is 9.34 kcal/mol. The same result was found with the B3LYP-D3 method (Supporting information Table S16) since the tctANP-7 conformation has the highest value of the sum of E(2) (9.98 kcal/mol) for the C8 HB pseudo cycle. However, the most stable conformation, tttANP-1, has a sum value of 5.39 kcal/mol related to the C10 pseudo cycle. We notice a slight increase in the values of the sum of the individual E(2) energies calculated with B3LYP-D3 compared to those found with B3LYP.
The opposite conclusions are drawn in water; the SMD/B3LYP method predicted the tttANP-12 (δLβP) conformer to be the lowest energy minimum in water. This conformer is stabilized with one H.B. closing a C8 pseudo cycle (Supporting information Figure S7) between the C=O of the main chain and the N-H of the azaAsn side chain. The length of the hydrogen bond is calculated to be 1.92Å. Unlike the gas phase, results in water showed that the second most stable conformation is the conformer tttANP-1, stabilized with an H.B. interaction closing a C10 pseudo cycle, and the H.B. length is equal to 2.30Å. We observed the absence of the C6 pseudo cycle found in the gas phase for the tttANP-1 conformer. Also, the HBs length becomes longer than that in the gas phase.
Unlike the B3LYP functional, the B3LYP-D3 functional predicted the conformer tctANP-2 to be the lowest energy minimum in gas and solution states. This conformer is stabilized with two intramolecular HBs in the gas phase. One is a backbone-side chain HB closing a C6 pseudo cycle, and a backbone HB closing a C5 ring, with the length of HBs estimated to be 2.26Å for both interactions. The C6 ring was not observed in the solution for this same conformer. However, the length of the C5 HB was slightly larger than that found in the gas phase (2.33Å). Our result indicates that the side chain of azaAsn in solution may be involved in an intermolecular interaction with water.
The results of compound (5) showed few similarities with those of compound (4). The most stable conformer in the gas phase with the B3LYP functional is tttADP-1, with two H.B.s interactions closing C10 and C6 rings. The C10 HB is related to the βⅠ turn motif for which the sum of the individual E(2) stabilization energies (Supporting information Table S14), associated with πCO, nO and n’O / σ*NH overlaps (Figure 4) involved in the CO….HN HB is 3.71 kcal/mol. This turn motif was also found in the X-ray structure of Z-azaAsp(OEt)-Pro-NhiPr19. Comparing these results with those observed with B3LYP-D3 functional, we see clearly that the dispersion-corrected B3LYP-D3 functional might not predict the lowest energy minimum; since it is tctADP-3 conformer with one H.B. closing a C5 pseudo cycle (Supporting information Figure S5) with a sum of E(2) is 1.22 kcal/mol related to the n’N→ σ*NH NBO overlap of the N.H…..N backbone H.B. 29(Figure 4).
The tttADP-9 conformer was found as the most stable conformation in water with SMD/B3LYP and SMD/B3LYP-D3 methods. This conformer is stabilized with one H.B. interaction, a C=O…H-O backbone-side chain HB. We also notice that the length of this type of H.B. is slightly larger with the dispersion-corrected functional (1.75Å) than that found with the traditional B3LYP functional (1.64Å).
The most stable conformation of compound (3) in the gas phase with and without dispersion functional is ttAAP-1, which is stabilized with an H.B. closing a C10 pseudo cycle related to βⅠ turn (Supporting information Figures S3 and S6). The length of such bond is predicted to be 2.16 and 2.08 Å with B3LYP and B3LYP-D3 functional, respectively. The HB length has slightly decreased from B3LYP to B3LYP-D3, contrary to azaAsn and azaAsp models. The NBO overlaps of the C10 HB are shown in Figure 5. The sum of E(2) associated with πCO, nO, and n’O / σ*NH overlaps calculated with B3LYP functional is 3.62 kcal/mol (Supporting Information Table S15). While that calculated with B3LYP-D3 functional is 4.15 kcal/mol (Supporting information Table S18), which is only made of two πCO and nO / σ*NH overlaps.
Results of the azaAla model in the water show significant similarities with those of the gas phase. The ttAAP-1 conformer is the lowest energy minimum and stabilized with a C10 backbone C=O…H-N HB. The length of this H.B. is found to be 2.32Å with B3LYP functional and 2.14Å with B3LYP-D3 functional, which is the same finding as in the gas phase.
3.3. Asx Turn Vs βⅠ-tun in Ac-azaAsx-Pro-NHMe (x= n or p)
Hydrogen-bonded Asx turns are similar to β turns since both have a C10 pseudo cycle. However, in Asx turns, it is the side chain’s carbonyl group of Asn or Asp at the i+1 position, which is hydrogen bonded to NH of the backbone of residue at the i+3 position. Thus, the nomenclature of Asx-turns is the same as β-turns. Four categories were found: type-Ⅰ, type-Ⅱ, type-Ⅰ′, and type-Ⅱ′ Asx turn (Table 3). This nomenclature is based on the dihedral angles ϕ and ψ of residues i+1 and i+2. Since Asx turns are mimicry of β turns, the dihedral angles ϕe and ψe of Asx turn (Figure 6) should resemble those of i+1 in β turns, while ϕ and ψ of residue i+1 of Asx turns should resemble those of residue i+2 in β turns 30, 31.
Furthermore, the azaAsx-Pro chain contains another C10 pseudo-cycle similar to an Asx turn, which is related to the H.B. involving the NδH or Oδ-H of the side chain of Asn or Asp, respectively, and carbonyl group two residues ahead which will be noted (C10SC). Results of the azaAsn model compound in the gas phase calculated with the B3LYP functional showed that all three conformations adopting the βⅠ-turn motif are more stable than those adopting the Asx turn. However, the most stable Asx turn conformation tttANP-9 is only 2.89 kcal/mol higher in energy, which means there is a competition between Asx and βⅠ turn secondary structures. The C10 C=O….H-N backbone side chain HB with the sum of E(2) being 5.76 kcal/mol which is associated with nO and n’O / σ*NH overlaps (Supporting information Table S13). On the other hand, the most stable Asx turn conformation with B3LYP-D3 functional is cctANP-17, which has a sum of E(2) value of 1.88 kcal/mol related to nO and n’O / σ*NH overlaps (Supporting information Table S16). Moreover, the majority of βⅠ-turn conformations are more stable than the Asx turn, and the cctANP-16 conformation related to the C10SC interaction is more stable than the Asx turn. The sum of the E(2) value (3.05 kcal/mol) related to the C10 HB of this conformer is higher than that of cccANP-17, which means a stronger HB bond interaction. The Asx turn was not reported in the azaAsn-Pro sequence yet. Instead, the Nδ-H of the side chain of azaAsn is bounded to Nα of the same residue 18, 19. Moreover, based on the dihedral angles of a typical Asx turn (Table 3), the most stable Asx turn conformation tttANP-9 (ΔE =2.89 kcal/mol for B3LYP; ΔE=4.52 kcal/mol for B3LYP-D3) is of type-Ⅰ Asx turn. This Asx turn was also found in Asn-Pro studied with NMR and IR spectroscopies 30. On the contrary, a study of a database of 500 proteins found that the frequency of occurrence of Asx turn is type-Ⅱ′, while the most common type is βI turn 30. These findiA recent study of Asx also supported these findingsn-Ala and Asn-Gly in crystallized protein structures in the PDB 31, revealing that the most common Asx turn in these two sequences are type-Ⅱ′ and type-Ⅱ, respectively. In sum, our results suggest that azaAsn and azaAsp residues in models 4 and 5 could have different folding patterns than natural peptides and the,y could also enhance the formation of a type-Ⅰ Asx turn instead of type-Ⅱ′ mostly found in peptides. Even though the Asx turn is more stable than the C10S,C which is similar to the Asx turn, most conthe formers with C10SC are more stable than those adopting the Asx turn, especially with B3LYP-D3 functional.
In solution, results showed few similarities regarding the most stable Asx turn conformation with B3LYP functional, which was found to be the conformer tttANP-9. However, the tctANP-14 conformer related to the C10SC turn also has the same relative energy as tttANP-9 (∆E=2.95 kcal/mol). The C10SC turn was found to be more stable than Asx turn in solution with the B3LYP-D3 functional since the tctANP-14 conformer adopting the C10SC turn has a relative energy of 2.07 kcal/mol and the tttANP-9 conformer related to the Asx turn is 4.20 kcal/mol energy. The result indicates that the side chain of azaAsn in the azaAsn-Pro sequence acts as a proton donor in water.
Similarly, the βⅠ turn structure in the azaAsp-Pro sequence with B3LYP functional is more stable than Asx turn in the isolated state. This is observed when comparing tttADP-1 and tttADP-8 conformers. The latter conformer related to the type-Ⅰ Asx turn is 3.70 kcal/mol higher in energy than the first one, which adopts the βⅠ turn. It is also noticed that the Asx turn in this sequence is less stable than that found for the azaAsn-Pro sequence. In addition, conformers cctADP-15 and tctADP-16 adopt an undefined type of Asx turn stabilized with the C10 cycle. Like the azaAsn model compound, the Asx turn in the azaAsp model compound is more stable than the C10SC motif as we compare the most stable Asx turn conformer tttADP-8 and the most stable C10SC conformer cctADP-19 (ΔE=7.40 Kcal/mol). Also, most Asx turn conformations are energetically more favorable than C10SC conformations. Like the B3LYP functional, the B3LYP-D3 functional predicted that the Asx turn is more stable than the C10SC turn in the isolated state. The B3LYP-D3 functional showed that the tctADP-16 conformer is the most stable Asx turn with a relative energy of 3.83 kcal/mol, and the sum of E(2) value is equal to 2.47 kcal/mol and associated with nO and n’O / σ*NH overlaps.
Similar results were found in the solution state since the βⅠ turn is more stable than C10SC and Asx turn conformations. However, the C10SC is more stable than Asx turn with B3LYP functional, conformation tcADP-22 is the most stable C10SC turn conformation with a relative energy value of 5.15 kcal/mol while conformation cctADP-15 adopting the Asx turn has a relative energy value of 9.29 kcal/mol. The B3LYP-D3 functional reveals that the C10SC turn is more stable than the Asx turn, with the conformer tctADP-22 being the most stable C10SC conformer in water with a relative energy of 3.45 kcal/mol.
4. Conclusions
In summary, conformational preferences of three azapeptides models Ac-azaXaa-Pro-NHMe [Xaa= Asn, Asp, Ala] were studied using the B3LYP/6-311++G(d,p) and B3LYP-D3/6-311++G(d,p) levels of theory in isolated and water to examine the effect of side chain’s nature and position effect on the folding patterns of azapeptides. Our result demonstrates that the position of azaamino acid residue largely affects the folding patterns of these compounds. Another finding is the orientation of amide bonds preceding azaamino acid residue and Pro. The most stable conformations of all three compounds in gas and solution states calculated with B3LYP functional are all trans amide bonds. Moreover, the stability order of the three secondary structures stabilized with a 10-membered HB in azaAsn and azaAsp models is βⅠ ˃ C10S ˃Asx for azaAsn-Pro and azaAsp-Pro sequences in water calculated by SMD/B3LYP and SMD/B3LYP-D3, and for athe zaAsp-Pro model in the gas phase also calculated by two methods. In contrast, the stability order for the azaAsn-Pro in the gas phase calculated with the two functionals is βⅠ ˃ Asx ˃ C10SC. Note the hat azaXaa-Pro sequence prefers βI-turn whereas Pro-azaXaa sequence prefers βII-turn structure 15. Our computational investigation of the conformational properties of short azapeptides demonstrates that the nature of the side chain of azapeptides could largely influence the folding patterns of these components.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org.
Author Contributions
Mouna El Khabchi performed the calculations, contributed to the interpretation of the results, and wrote the manuscript. Mohammed Mcharfi and Mohammed Benzakour supervised the findings of this work, contributed to the interpretation of the results, and oversaw overall direction and planning. Asmae Fitri and Adil Touimi Benjelloun: contributed to the final version of the manuscript. Jong-Wong Song reviewed and edited the manuscript. Ho-Jin Lee performed the calculations and reviewed and edited the manuscript.
Acknowledgments
JWS and HJL would like to acknowledge the partial financial support from the Korea Ministry of Science and ICT (2020R1A2C1102741).
References
- Barman, P.; Joshi, S.; Sharma, S.; Preet, S.; Sharma, S.; Saini, A. Strategic Approaches to Improvise Peptide Drugs as Next Generation Therapeutics. Int J Pept Res Ther 2023, 29 (4), 61. From NLM. Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic peptides: current applications and future directions. Signal Transduction and Targeted Therapy 2022, 7 (1), 48. [CrossRef]
- Wątły, J.; Miller, A.; Kozłowski, H.; Rowińska-Żyrek, M. Peptidomimetics—An infinite reservoir of metal binding motifs in metabolically stable and biologically active molecules. J Inorg Biochem 2021, 217, 111386. [Google Scholar] [CrossRef]
- Avan, I.; Hall, C. D.; Katritzky, A. R. Peptidomimetics via modifications of amino acids and peptide bonds. Chemical Society Reviews 2014, 43(10), 3575–3594. [Google Scholar] [CrossRef]
- Fan Cheng, K.; VanPatten, S.; He, M.; Al-Abed, Y. Azapeptides -A History of Synthetic Milestones and Key Examples. Curr Med Chem 2022, 29(42), 6336–6358. [Google Scholar] [CrossRef]
- Zega, A. Azapeptides as pharmacological agents. Curr Med Chem 2005, 12(5), 589–597. [Google Scholar] [CrossRef]
- Arujõe, M.; Järv, J.; Mastitski, A.; Ploom, A.; Troska, A. Aza-peptides: expectations and reality. Proceedings of the Estonian Academy of Sciences 2022. [Google Scholar]
- Härk, H. H.; Troska, A.; Arujõe, M.; Burk, P.; Järv, J.; Ploom, A. Kinetic study of aza-amino acid incorporation into peptide chains: Influence of the steric effect of the side chain. Tetrahedron 2022, 129, 133161. [Google Scholar] [CrossRef]
- Proulx, C.; Zhang, J.; Sabatino, D.; Chemtob, S.; Ong, H.; Lubell, W. D. Synthesis and Biomedical Potential of Azapeptide Modulators of the Cluster of Differentiation 36 Receptor (CD36). Biomedicines 2020, 8(8). [Google Scholar] [CrossRef] [PubMed]
- Bourguet, C. B.; Boulay, P. L.; Claing, A.; Lubell, W. D. Design and synthesis of novel azapeptide activators of apoptosis mediated by caspase-9 in cancer cells. Bioorg Med Chem Lett 2014, 24(15), 3361–3365. [Google Scholar] [CrossRef] [PubMed]
- Mhidia, R.; Melnyk, O. Selective cleavage of an azaGly peptide bond by copper(II). Long-range effect of histidine residue. J Pept Sci 2010, 16 (3), 141-147. https://doi.org/10.1002/psc.1211. Ludwig, C.; Desmoulins, P. O.; Driancourt, M. A.; Goericke-Pesch, S.; Hoffmann, B. Reversible downregulation of endocrine and germinative testicular function (hormonal castration) in the dog with the GnRH-agonist azagly-nafarelin as a removable implant “Gonazon”; a preclinical trial. Theriogenology 2009, 71 (7), 1037-1045. Ho, T. L.; Nestor, J. J., Jr.; McCrae, G. I.; Vickery, B. H. Hydrophobic, aza-glycine analogues of luteinizing hormone-releasing hormone. Int J Pept Protein Res 1984, 24 (1), 79-84. DOI: 10.1111/j.1399-3011.1984.tb00931.x. Dutta, A. S.; Furr, B. J.; Giles, M. B.; Valcaccia, B.; Walpole, A. L. Potent agonist and antagonist analogues of luliberin containing an azaglycine residue in position 10. Biochem Biophys Res Commun 1978, 81 (2), 382-390. [CrossRef]
- owles, M.; Proulx, C. Solid phase submonomer azapeptide synthesis. Methods Enzymol 2021, 656, 169-190. https://doi.org/10.1016/bs.mie.2021.04.020. Dai, C.; Ma, J.; Li, M.; Wu, W.; Xia, X.; Zhang, J. Diversity-oriented submonomer synthesis of azapeptides mediated by the Mitsunobu reaction. Organic Chemistry Frontiers 2019, 6 (14), 2529-2533, 10.1039/C9QO00296K. DOI: 10.1039/C9QO00296K. Chingle, R. M.; Proulx, C.; Lubell, W. D. Azapeptide Synthesis Methods for Expanding Side-Chain Diversity for Biomedical Applications. Accounts of chemical research 2017, 50 7, 1541-1556. Doan, N. D.; Zhang, J.; Traoré, M.; Kamdem, W.; Lubell, W. D. Solid-phase synthesis of C-terminal azapeptides. J Pept Sci 2015, 21 (5), 387-391. DOI: 10.1002/psc.2711 From NLM. Ollivier, N.; Besret, S.; Blanpain, A.; Melnyk, O. Silver-catalyzed azaGly ligation. Application to the synthesis of azapeptides and of lipid-peptide conjugates. Bioconjug Chem 2009, 20 (7), 1397-1403. DOI: 10.1021/bc9000195. Bowles, M.; Proulx, C. Late-Stage N-Alkylation of Azapeptides. Organic letters 2022. Luo, Z.; Xu, L.; Tang, X.; Zhao, X.; He, T.; Lubell, W. D.; Zhang, J. Synthesis and biological evaluation of novel all-hydrocarbon cross-linked aza-stapled peptides. Organic & Biomolecular Chemistry 2022, 20 (40), 7963-7971, 10.1039/D2OB01496C. [CrossRef]
- Singh, S.; Shrivastava, R.; Singh, G.; Ali, R.; Sankar Ampapathi, R.; Bhadhuria, S.; Haq, W. AzaGly-Appended Peptidomimetics Structurally Related to PTR6154 as Potential PKB/Akt Inhibitors. Chembiochem 2017, 18 (12), 1061-1065. https://doi.org/10.1002/cbic.201700031. Sabatino, D.; Proulx, C.; Pohankova, P.; Ong, H.; Lubell, W. D. Structure-activity relationships of GHRP-6 azapeptide ligands of the CD36 scavenger receptor by solid-phase submonomer azapeptide synthesis. J Am Chem Soc 2011, 133 (32), 12493-12506. DOI: 10.1021/ja203007u From NLM. Altiti, A. S.; He, M.; VanPatten, S.; Cheng, K. F.; Ahmed, U.; Chiu, P. Y.; Mughrabi, I. T.; Jabari, B. A.; Burch, R. M.; Manogue, K. R.; et al. Thiocarbazate building blocks enable the construction of azapeptides for rapid development of therapeutic candidates. Nature Communications 2022, 13. [CrossRef]
- Lee, H.-J.; Kim, J. H.; Jung, H. J.; Kim, K.-Y.; Kim, E.-J.; Choi, Y.-S.; Yoon, C.-J. Computational study of conformational preferences of thioamide-containing azaglycine peptides. Journal of Computational Chemistry 2004, 25 (2), 169-178. https://doi.org/10.1002/jcc.10364. Lee, H.-J.; Jung, H. J.; Kim, J. H.; Park, H.-M.; Lee, K.-B. Conformational preference of azaglycine-containing dipeptides studied by PCM and IPCM methods. Chemical Physics 2003, 294 (2), 201-210. DOI: https://doi.org/10.1016/j.chemphys.2003.06.001. Lee, H.; Song, J.; Choi, Y.; Ro, S.; Yoon, C. The energetically favorable cis peptide bond for the azaglycine-containing peptide: For-AzGly-NH2 model. Physical Chemistry Chemical Physics 2001, 3 (9), 1693-1698. DOI: 10.1039/b009651m. Kang, Y. K.; Byun, B. J. Conformational preferences and cis-trans isomerization of azaproline residue. J Phys Chem B 2007, 111 (19), 5377-5385. DOI: 10.1021/jp067826t. [CrossRef]
- Lee, H. J.; Song, J. W.; Choi, Y. S.; Park, H. M.; Lee, K. B. A theoretical study of conformational properties of N-methyl azapeptide derivatives. J Am Chem Soc 2002, 124(40), 11881–11893. [Google Scholar] [CrossRef] [PubMed]
- El Khabchi, M.; Lahlou, H.; El Adnani, Z.; McHarfi, M.; Benzakour, M.; Fitri, A.; Benjelloun, A. T. Conformational preferences of Ac-Pro-azaXaa-NHMe (Xaa = Asn, Asp, Ala) and the effect of intramolecular hydrogen bonds on their stability in gas phase and solution. J Mol Model 2021, 27(12), 368. [Google Scholar] [CrossRef] [PubMed]
- Lee, H. J.; Park, H. M.; Lee, K. B. The beta-turn scaffold of tripeptide containing an azaphenylalanine residue. Biophys Chem 2007, 125 (1), 117-126. https://doi.org/10.1016/j.bpc.2006.05.028 From NLM. Melton, S. D.; Smith, M. S.; Chenoweth, D. M. Incorporation of Aza-Glycine into Collagen Peptides. The Journal of Organic Chemistry 2020, 85 (3), 1706-1711. DOI: 10.1021/acs.joc.9b02539. Etzkorn, F. A.; Ware, R. I.; Pester, A. M.; Troya, D. Conformational Analysis of n→π* Interactions in Collagen Triple Helix Models. The Journal of Physical Chemistry B 2019, 123 (2), 496-503. DOI: 10.1021/acs.jpcb.8b08384. Melton, S. D.; Chenoweth, D. M. Variation in the Yaa position of collagen peptides containing azaGlycine. Chem Commun (Camb) 2018, 54 (84), 11937-11940. DOI: 10.1039/c8cc06372a. Zhang, Y.; Malamakal, R. M.; Chenoweth, D. M. Aza-Glycine Induces Collagen Hyperstability. J Am Chem Soc 2015, 137 (39), 12422-12425. DOI: 10.1021/jacs.5b04590. [CrossRef]
- Harris, T.; Chenoweth, D. M. Sterics and Stereoelectronics in Aza-Glycine: Impact of Aza-Glycine Preorganization in Triple Helical Collagen. J Am Chem Soc 2019, 141 (45), 18021-18029. https://doi.org/10.1021/jacs.9b05524. Kasznel, A. J.; Harris, T.; Porter, N. J.; Zhang, Y.; Chenoweth, David M. Aza-proline effectively mimics l-proline stereochemistry in triple helical collagen. Chemical Science 2019, 10 (29), 6979-6983, 10.1039/C9SC02211B. DOI: 10.1039/C9SC02211B. Kasznel, A. J.; Zhang, Y.; Hai, Y.; Chenoweth, D. M. Structural Basis for Aza-Glycine Stabilization of Collagen. J Am Chem Soc 2017, 139 (28), 9427-9430. DOI: 10.1021/jacs.7b03398. [CrossRef]
- André, F.; Vicherat, A.; Boussard, G.; Aubry, A.; Marraud, M. Aza-peptides. III. Experimental structural analysis of aza-alanine and aza-asparagine-containing peptides. J Pept Res 1997, 50, 372–381. [Google Scholar] [CrossRef] [PubMed]
- André, F.; Boussard, G.; Bayeul, D.; Didierjean, C.; Aubry, A.; Marraud, M. Aza-peptides. II. X-ray structures of aza-alanine and aza-asparagine-containing peptides. J Pept Res 1997, 49(6), 556–562. [Google Scholar] [CrossRef] [PubMed]
- Song, J. W.; Lee, H. J.; Choi, Y. S.; Yoon, C. J. Origin of rotational barriers of the N-N bond in hydrazine: NBO analysis. J Phys Chem A 2006, 110(5), 2065–2071. [Google Scholar] [CrossRef] [PubMed]
- Karplus, P. A. Experimentally observed conformation-dependent geometry and hidden strain in proteins. Protein Sci 1996, 5(7), 1406–1420. [Google Scholar] [CrossRef] [PubMed]
- Ro, S.; Lee, H.; Ahn, I.; Shin, D.; Lee, K.; Yoon, C.; Choi, Y. Torsion angle based design of peptidomimetics: A dipeptidic template adopting beta-I turn (Ac-Aib-AzGly-NH2). Bioorganic & Medicinal Chemistry 2001, 9(7), 1837–1841. [Google Scholar] [CrossRef]
- Marenich, A. V.; Cramer, C. J.; Truhlar, D. G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. The Journal of Physical Chemistry B 2009, 113(18), 6378–6396. [Google Scholar] [CrossRef]
- Gaussian 16, Revision C.01; Gaussian, Inc.: 2019. (accessed.
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J Chem Phys 2010, 132(15), 154104. [Google Scholar] [CrossRef]
- Brenner, V.; Gloaguen, E.; Mons, M. Rationalizing the diversity of amide–amide H-bonding in peptides using the natural bond orbital method. Physical Chemistry Chemical Physics 2019, 21(44), 24601–24619. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: a multifunctional wavefunction analyzer. J Comput Chem 2012, 33(5), 580–592. [Google Scholar] [CrossRef]
- Alemán, C.; Puiggalí, J. Conformational Preferences of the Asparagine Residue. Gas-Phase, Aqueous Solution, and Chloroform Solution Calculations on the Model Dipeptide. The Journal of Physical Chemistry B 1997, 101(17), 3441–3446. [Google Scholar] [CrossRef]
- Baruah, K.; Sahariah, B.; Sakpal, S. S.; Deka, J. K. R.; Bar, A. K.; Bagchi, S.; Sarma, B. K. Stabilization of Azapeptides by N(amide)···H-N(amide) Hydrogen Bonds. Org Lett 2021, 23(13), 4949–4954. [Google Scholar] [CrossRef] [PubMed]
- Abbadi, A.; McHarfi, M.; Aubry, A.; Premilat, S.; Boussard, G.; Marraud, M. Involvement of side functions in peptide structures: the Asx turn. Occurrence and conformational aspects. Journal of the American Chemical Society 1991, 113(7), 2729–2735. [Google Scholar] [CrossRef]
- D’Mello V, C.; Goldsztejn, G.; Rao Mundlapati, V.; Brenner, V.; Gloaguen, E.; Charnay-Pouget, F.; Aitken, D. J.; Mons, M. Characterization of Asx Turn Types and Their Connate Relationship with β-Turns. Chemistry 2022, 28(25), e202104328. [Google Scholar] [CrossRef]
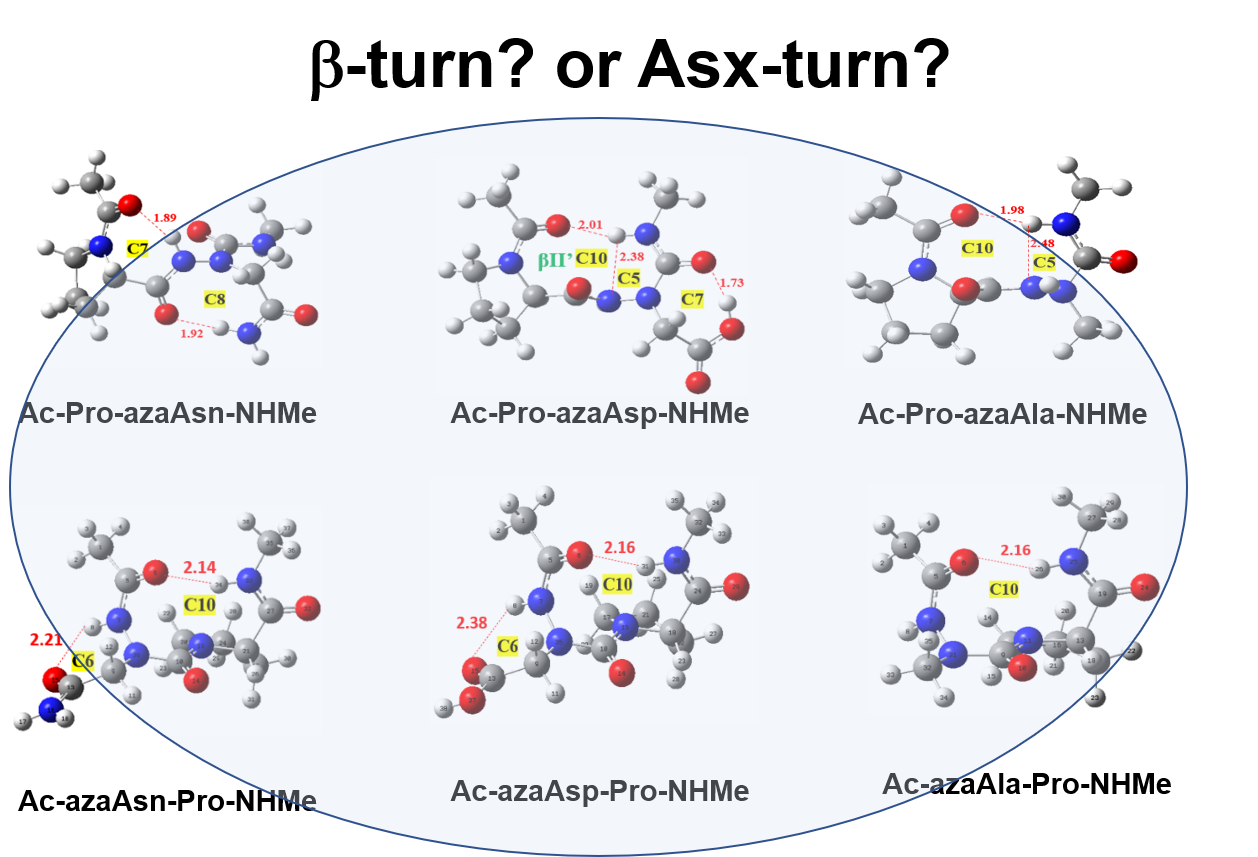
Figure 1.
Definition of torsional angle parameters of azaamino acid residue-containing tripeptide models, Ac-azaXaa-Pro-NHMe [ Xaa = Asn (4), Asp (5), Ala (6)].
Figure 1.
Definition of torsional angle parameters of azaamino acid residue-containing tripeptide models, Ac-azaXaa-Pro-NHMe [ Xaa = Asn (4), Asp (5), Ala (6)].
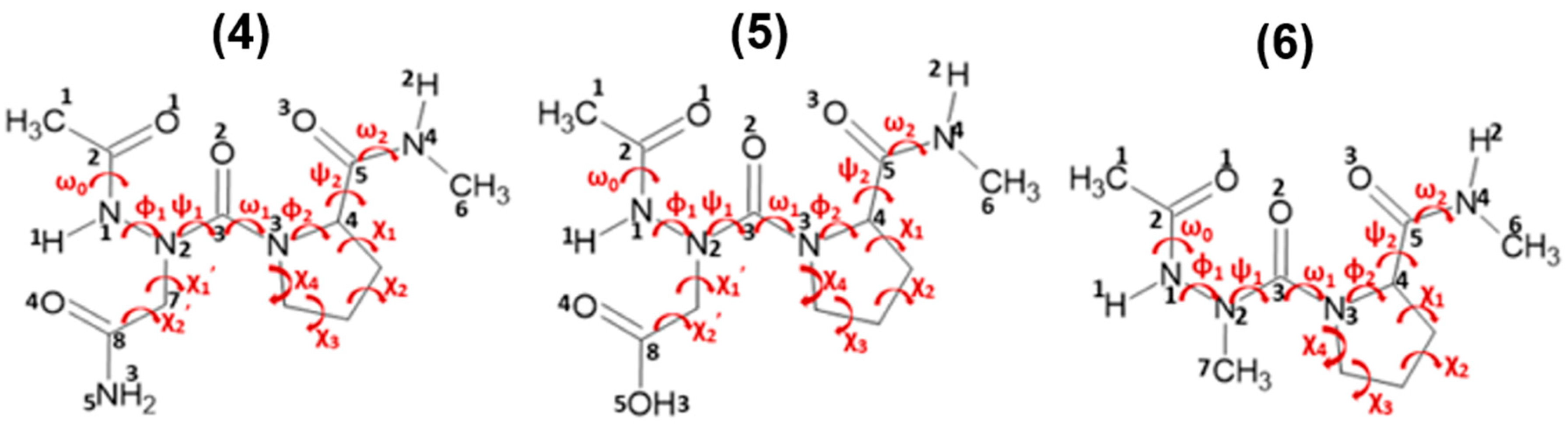
Figure 2.
Cyclic motifs formed by hydrogen bonds in Ac-azaXaa-Pro-NHMe [Xaa = Asn (4), Asp (5), Ala (6)].
Figure 2.
Cyclic motifs formed by hydrogen bonds in Ac-azaXaa-Pro-NHMe [Xaa = Asn (4), Asp (5), Ala (6)].
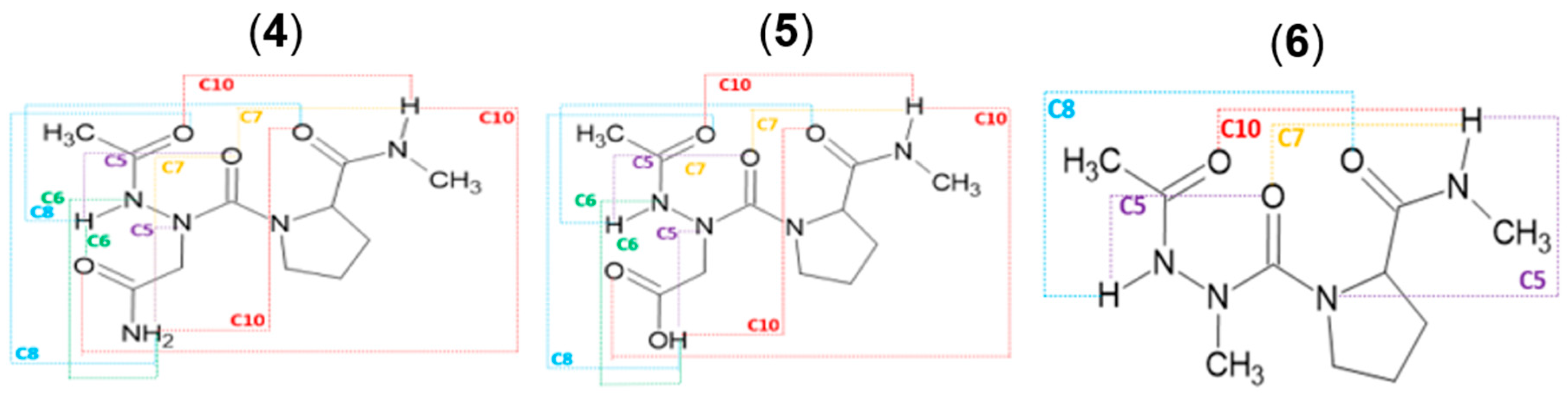
Figure 3.
NBO overlaps of tttANP-1 and tctANP-2 in the gas phase. ‘t’ and ‘c’ represents ‘trans’ and ‘cis’ amide bond, respectively. The E(2) value is shown in blue color for B3LYP and red color for B3LYP-D3 functional.
Figure 3.
NBO overlaps of tttANP-1 and tctANP-2 in the gas phase. ‘t’ and ‘c’ represents ‘trans’ and ‘cis’ amide bond, respectively. The E(2) value is shown in blue color for B3LYP and red color for B3LYP-D3 functional.
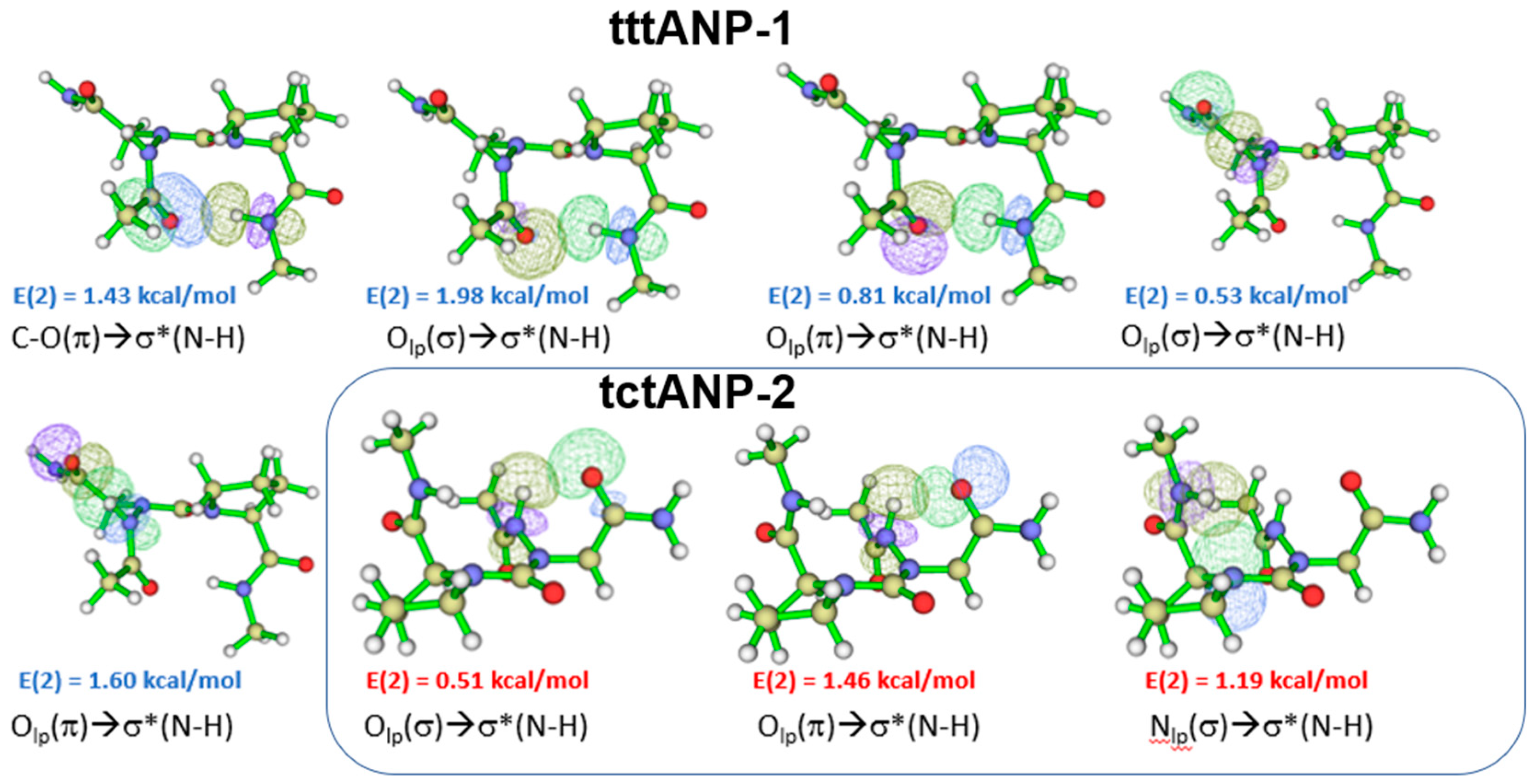
Figure 4.
NBO overlaps of the lowest energy conformer tttADP-1 (@B3LYP) and tctADP-3 (@B3LYP-D3) in the gas phase. The E(2) value is shown (See Supporting Information Tables S14 and S17).
Figure 4.
NBO overlaps of the lowest energy conformer tttADP-1 (@B3LYP) and tctADP-3 (@B3LYP-D3) in the gas phase. The E(2) value is shown (See Supporting Information Tables S14 and S17).
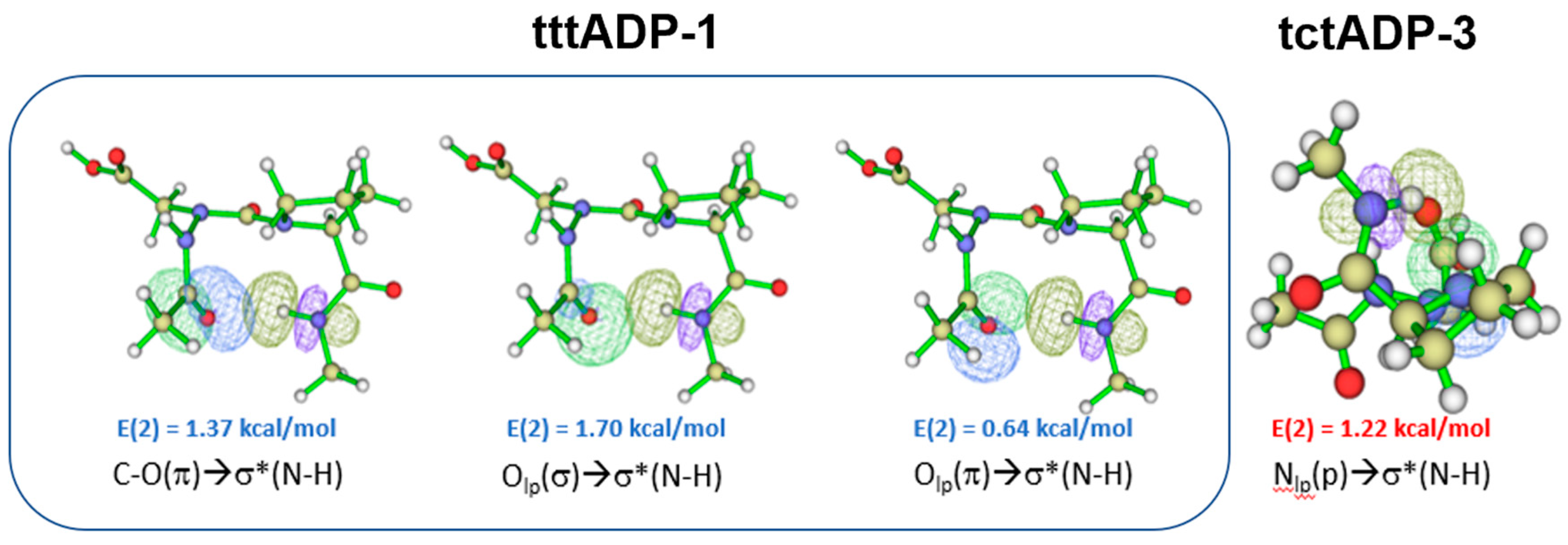
Figure 5.
NBO overlaps of tttAAP-1 in the gas phase. The E(2) value is shown in blue color for B3LYP and red color for B3LYP-D3 functional.
Figure 5.
NBO overlaps of tttAAP-1 in the gas phase. The E(2) value is shown in blue color for B3LYP and red color for B3LYP-D3 functional.
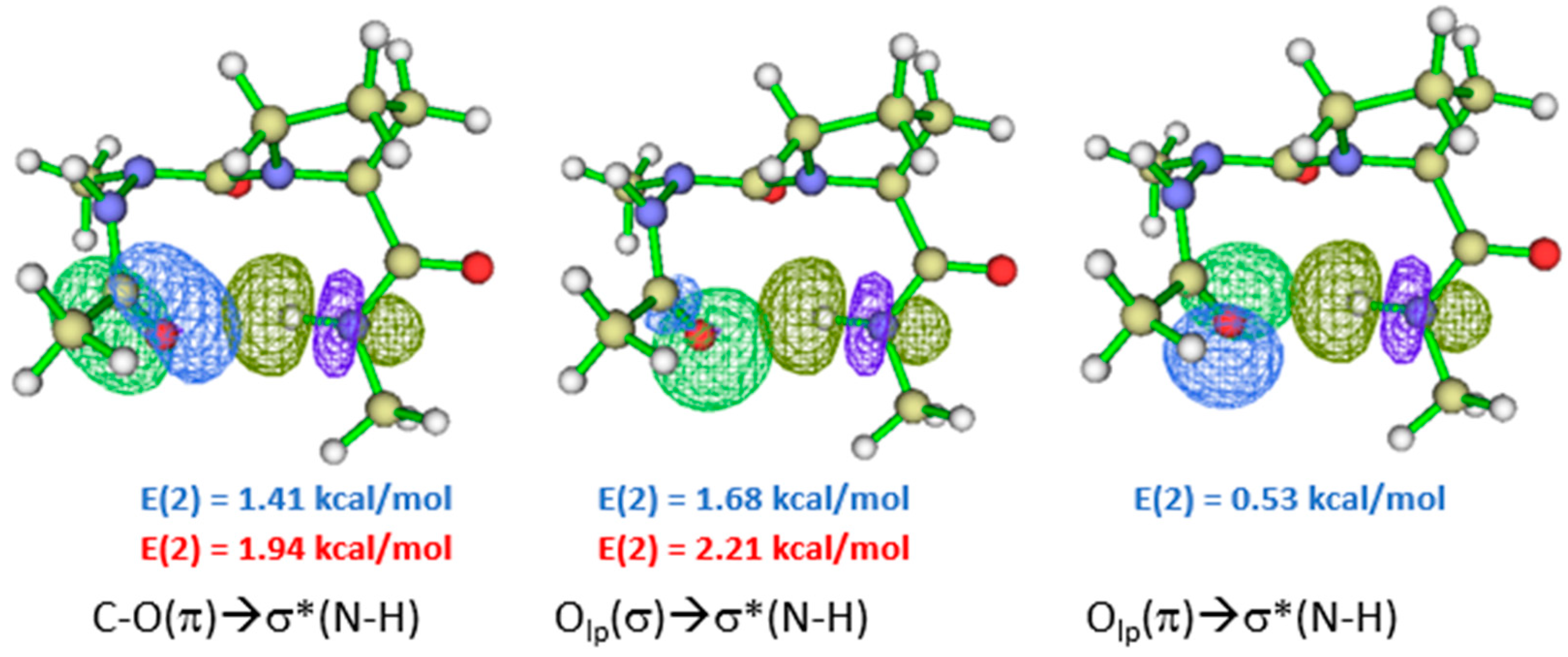
Figure 6.
Definition of torsional angles of Asx turns in azaAsx-Pro with R= NH2 or O.H.
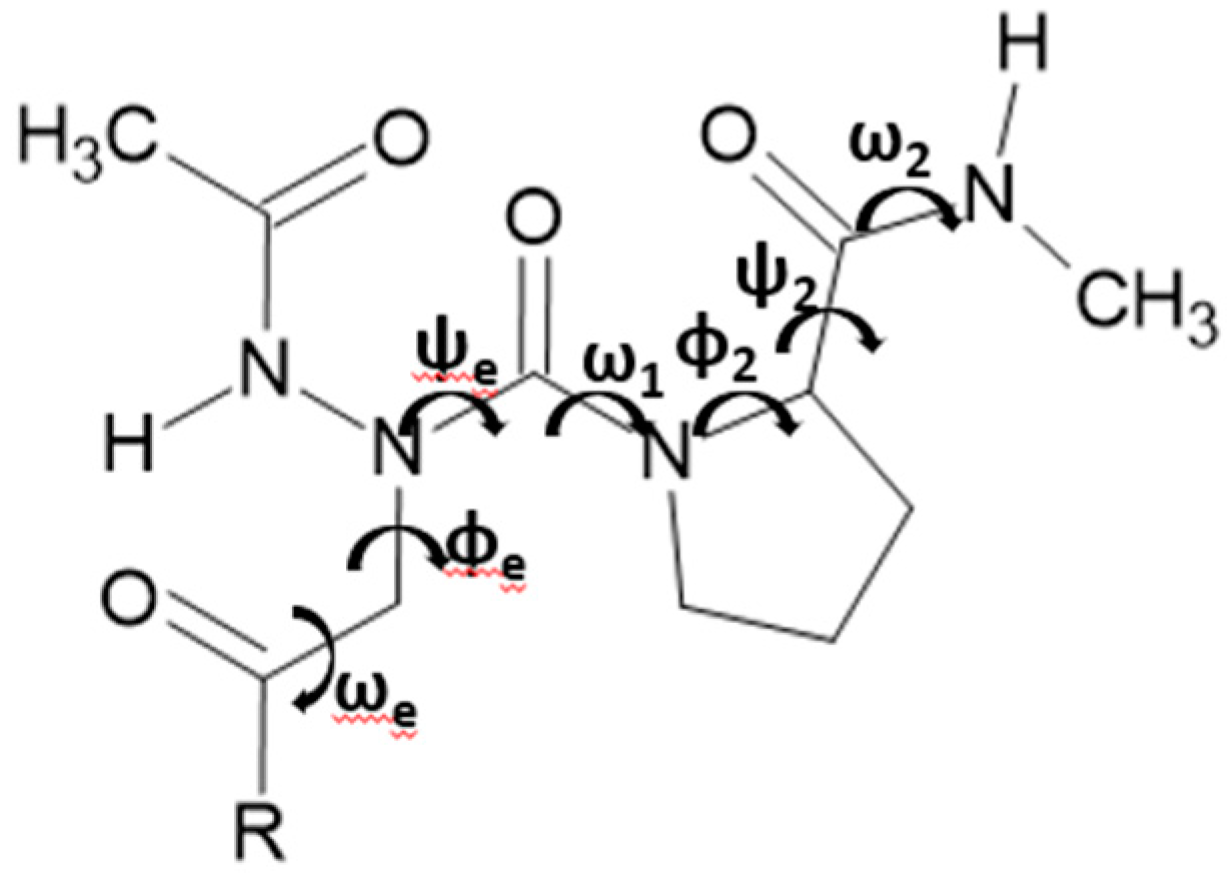

Table 1.
Relative energies in (kcal/mol) of model compounds (4), (5), and (6) at B3LYP (M1) and B3LYP-D3 (M2) functionals with the 6-311++G (d, p) basis set in the isolated form.
Table 1.
Relative energies in (kcal/mol) of model compounds (4), (5), and (6) at B3LYP (M1) and B3LYP-D3 (M2) functionals with the 6-311++G (d, p) basis set in the isolated form.
| Ac-azaAsn-Pro-NHMe | Ac-azaAsp-Pro-NHMe | Ac-azaAla-Pro-NHMe | ||||||
| ΔE(M1) | ΔE(M2) | ΔE(M1) | ΔE(M2) | ΔE(M1) | ΔE(M2) | |||
| tttANP-1 | a 0.00 | 0.94 | tttADP-1 | c 0.00 | 0.69 | tttAAP-1 | e 0.00 | f 0.00 |
| tctANP-2 | 0.94 | b 0.00 | tttADP-2 | 1.00 | 2.70 | tttAAP-2 | 0.75 | 1.13 |
| tttANP-3 | 1.32 | 2.64 | tctADP-3 | 1.07 | d 0.00 | tttAAP-3 | 1.00 | 2.51 |
| tttANP-4 | 1.38 | 1.95 | tctADP-4 | 1.44 | 0.38 | tttAAP-4 | 1.26 | 3.70 |
| tttANP-5 | 1.96 | 2.89 | cttADP-5 | 2.07 | 2.57 | tctAAP-5 | 1.44 | 1.82 |
| cttANP-6 | 2.45 | 3.07 | tttADP-6 | 2.95 | 3.33 | cttAAP-6 | 1.51 | 2.26 |
| tctANP-7 | 2.64 | 2.01 | tttADP-7 | 3.33 | 4.39 | cttAAP-7 | 1.63 | 2.20 |
| tttANP-8 | 2.64 | 3.20 | tttADP-8 | 3.70 | 5.46 | tctAAP-8 | 2.26 | 1.26 |
| tttANP-9 | 2.89 | 4.52 | tttADP-9 | 4.52 | 5.33 | tttAAP-9 | 2.70 | 4.89 |
| cttANP-10 | 3.07 | 3.26 | cttADP-10 | 4.52 | 4.58 | tctAAP-10 | 2.76 | 2.20 |
| cttANP-11 | 3.26 | 2.95 | cttADP-11 | 4.58 | 4.83 | cttAAP-11 | 3.51 | 3.01 |
| tttANP-12 | 4.08 | 4.89 | cctADP-12 | 4.83 | 3.39 | tctAAP-12 | 3.70 | 5.27 |
| tctANP-13 | 4.27 | 3.20 | tctADP-13 | 5.08 | 4.89 | cctAAP-13 | 3.77 | 2.01 |
| tctANP-14 | 4.46 | 3.33 | tttADP-14 | 5.65 | 7.53 | tttAAP-14 | 3.89 | 4.64 |
| tttANP-15 | 4.46 | 5.65 | cctADP-15 | 6.02 | 4.89 | cctAAP-15 | 4.33 | 2.95 |
| cctANP-16 | 4.52 | 2.32 | tctADP-16 | 6.34 | 3.83 | cttAAP-16 | 4.96 | 6.46 |
| cctANP-17 | 4.64 | 3.20 | tctADP-17 | 6.78 | 7.22 | ctcAAP-17 | 5.52 | 5.02 |
| cttANP-18 | 4.83 | 4.96 | cctADP-18 | 6.97 | 4.96 | ttcAAP-18 | 6.28 | 5.58 |
| tctANP-19 | 5.02 | 3.26 | cctADP-19 | 7.40 | 5.40 | cctAAP-19 | 6.34 | 5.46 |
| cttANP-20 | 5.40 | 5.71 | tctADP-20 | 7.66 | 6.21 | cccAAP-20 | 8.47 | 6.15 |
| cctANP-21 | 5.46 | 4.46 | tttADP-21 | 7.72 | 9.04 | tccAAP-21 | 10.10 | 7.66 |
| cctANP-22 | 5.58 | 3.26 | tctADP-22 | 8.03 | 6.97 | cccAAP-22 | 11.92 | 9.73 |
| cttANP-23 | 5.96 | 5.77 | cttADP-23 | 8.03 | 7.97 | |||
| tctANP-24 | 6.71 | 7.22 | ttcADP-24 | 9.29 | 8.84 | |||
| tctANP-25 | 6.90 | 5.52 | tccADP-25 | 10.10 | 7.47 | |||
| cttANP-26 | 7.59 | 7.53 | cttADP-26 | 10.86 | 11.11 | |||
| tttANP-27 | 7.59 | 9.66 | cttADP-27 | 11.30 | 10.73 | |||
| ttcANP-28 | 7.97 | 7.59 | cccADP-28 | 13.43 | 9.22 | |||
| tccANP-29 | 9.16 | 6.21 | cctADP-29 | 13.43 | 12.61 | |||
| tttANP-30 | 9.79 | 11.36 | cccADP-30 | 16.13 | 12.74 | |||
| cctANP-31 | 9.98 | 9.48 | ||||||
| cccANP-32 | 10.35 | 6.34 | ||||||
| cccANP-33 | 14.75 | 11.80 | ||||||
aE = -1005.6114 a.u ; bE = -1005.6582 a.u; cE = -1025.4846 a.u ; dE = -1025.5303 a.u; eE = -836.8502 a.u ;.fE = -836.8898 a.u.
Table 2.
Relative energies in (kcal/mol) of model compounds 4, 5, and 6 at the SMD/B3LYP/6-311++G (d, p), M3, and the SMD/B3LYP-D3/6-311++G(d, p), M4, levels of theory in water.
Table 2.
Relative energies in (kcal/mol) of model compounds 4, 5, and 6 at the SMD/B3LYP/6-311++G (d, p), M3, and the SMD/B3LYP-D3/6-311++G(d, p), M4, levels of theory in water.
| Ac-azaAsn-Pro-NHMe | Ac-azaAsp-Pro-NHMe | Ac-azaAla-Pro-NHMe | ||||||
| ΔE(M3) | ΔE(M4) | ΔE(M3) | ΔE(M4) | ΔE(M3) | ΔE(M4) | |||
| tttANP-1 | 1.13 | 1.51 | tttADP-1 | 3.14 | 2.70 | tttAAP-1 | e 0.00 | f 0.00 |
| tctANP-2 | 1.38 | b 0.00 | tttADP-2 | 5.21 | 5.71 | tttAAP-2 | 2.01 | 3.07 |
| tttANP-3 | 3.07 | 4.52 | tctADP-3 | 3.64 | 1.38 | tttAAP-3 | 2.76 | 4.27 |
| tttANP-4 | 3.01 | 3.07 | tctADP-4 | 5.15 | 3.58 | tttAAP-4 | 2.70 | 6.46 |
| tttANP-5 | 2.76 | 3.45 | cttADP-5 | 6.84 | 6.28 | tctAAP-5 | 3.26 | 3.95 |
| cttANP-6 | 4.64 | 4.96 | tttADP-6 | 4.27 | 3.45 | cttAAP-6 | 4.96 | 5.46 |
| tctANP-7 | 4.27 | 3.70 | tttADP-7 | 2.82 | 2.45 | cttAAP-7 | 5.15 | 5.71 |
| tttANP-8 | 1.51 | 1.57 | tttADP-8 | 5.40 | 5.77 | tctAAP-8 | 2.01 | 0.88 |
| tttANP-9 | 2.95 | 4.20 | tttADP-9 | c 0.00 | d 0.00 | tttAAP-9 | 2.64 | 4.64 |
| cttANP-10 | 6.78 | 6.59 | cttADP-10 | 5.65 | 5.27 | tctAAP-10 | 1.82 | 2.26 |
| cttANP-11 | 7.15 | 6.34 | cttADP-11 | 7.66 | 6.90 | cttAAP-11 | 2.82 | 2.32 |
| tttANP-12 | a 0.00 | 0.82 | cctADP-12 | 6.02 | 3.45 | tctAAP-12 | 5.02 | 6.34 |
| tctANP-13 | 3.33 | 4.89 | tctADP-13 | 5.84 | 4.33 | cctAAP-13 | 4.46 | 3.07 |
| tctANP-14 | 2.95 | 2.07 | tttADP-14 | 6.09 | 6.90 | tttAAP-14 | 1.69 | 6.34 |
| tttANP-15 | 4.64 | 5.84 | cctADP-15 | 9.29 | 7.78 | cctAAP-15 | 4.58 | 3.01 |
| cctANP-16 | 3.64 | 2.38 | tctADP-16 | 6.02 | 5.08 | cttAAP-16 | 3.77 | 4.96 |
| cctANP-17 | 3.77 | 2.07 | tctADP-17 | 4.71 | 4.64 | ctcAAP-17 | 5.52 | 5.02 |
| cttANP-18 | 3.51 | 4.02 | cctADP-18 | 7.15 | 4.58 | ttcAAP-18 | 4.33 | 7.22 |
| tctANP-19 | 3.07 | 2.13 | cctADP-19 | 6.02 | 4.14 | cctAAP-19 | 4.46 | 3.95 |
| cttANP-20 | 4.33 | 4.58 | tctADP-20 | 7.40 | 5.15 | cccAAP-20 | 7.34 | 6.21 |
| cctANP-21 | 6.78 | 5.96 | tttADP-21 | 5.08 | 6.84 | tccAAP-21 | 7.91 | 5.58 |
| cctANP-22 | 6.02 | 4.08 | tctADP-22 | 5.15 | 3.45 | cccAAP-22 | 11.04 | 9.29 |
| cttANP-23 | 5.58 | 4.02 | cttADP-23 | 5.90 | 4.83 | |||
| tctANP-24 | 2.70 | 3.51 | ttcADP-24 | 5.15 | 10.42 | |||
| tctANP-25 | 4.14 | 4.20 | tccADP-25 | 10.17 | 6.46 | |||
| cttANP-26 | 3.89 | 3.45 | cttADP-26 | 8.03 | 7.53 | |||
| tttANP-27 | 4.08 | 5.84 | cttADP-27 | 7.59 | 6.21 | |||
| ttcANP-28 | 10.17 | 9.79 | cccADP-28 | 10.73 | 6.40 | |||
| tccANP-29 | 9.35 | 6.15 | cctADP-29 | 9.91 | 8.22 | |||
| tttANP-30 | 3.07 | 5.71 | cccADP-30 | 12.80 | 7.72 | |||
| cctANP-31 | 6.34 | 5.52 | ||||||
| cccANP-32 | 8.22 | 4.64 | ||||||
| cccANP-33 | 12.56 | 10.10 | ||||||
aE = -1005.6567 a.u ; bE = -1005.7028 a.u; cE = -1025.5295 a.u ; dE = -1025.5733 a.u; eE = -836.8844 a.u ; fE = -836.9238 a.u.
Table 3.
Backbone dihedral angles of residues at the i+1 and the i+2 positions for the major types of β turns.
Table 3.
Backbone dihedral angles of residues at the i+1 and the i+2 positions for the major types of β turns.
| turn | ϕi+1 (°) Asx ϕe |
ψi+1 (°) Asx ψe |
ϕi+2 (°) | ψi+2 (°) |
|---|---|---|---|---|
| βⅠ | -60 | -30 | -90 | 0 |
| βⅠ’ | 60 | 30 | 90 | 0 |
| βⅡ | -60 | 120 | 80 | 0 |
| βⅡ’ | 60 | -120 | -80 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



