
Submitted:
08 June 2023
Posted:
08 June 2023
You are already at the latest version
Abstract
Alzheimer’s disease (AD) is characterized by the formation of senile plaques consisting fibrillated amyloid-β (Aβ), dystrophic neurites, and the neurofibrillary tangles of tau. The oligomers/fibrillar Aβ damages the neurons or initiate an intracellular signaling cascade for neuronal cell death leading to Aβ toxicity. The Aβ is a 4 kDa molecular weight peptide originating from the C-terminal region of the amyloid precursor protein via proteolytic cleavage. Apart from the typical AD hallmarks, certain deficits in metabolic alterations have been identified. This study describes the emerging features of AD from the aspect of alternation in the main pathway of carbohydrate metabolism in the human brain. Particularly, the neurons in patients with AD favor glycolysis despite a normal mitochondrial function indicating a Warburg-like effect. In addition, certain dietary patterns are well known for their properties in preventing AD. Among those, a ketogenic diet may substantially improve the symptoms of AD. An effective therapeutic method in the treatment, mitigation, and prevention of AD has not yet been established. Therefore, the researchers pursue the development and establishment of novel therapies effective in suppressing AD symptoms and the elucidation of their underlying protective mechanisms against neurodegeneration aiming for AD therapy in near future.
Keywords:
glucose metabolism
; aerobic glycolysis
; Warburg effect
; Alzheimer’s disease
; amyloid-β
; ketogenic diet
1. Introduction
Neurodegenerative diseases including Alzheimer’s disease (AD) have been affecting public health worldwide, and are estimated to increase three-fold by 2050 [1]. The well-characterized biomarker of neurodegeneration is the accumulation of misfolded and aggregated proteins in the brain. However, the definition of neurodegenerative diseases is descriptive i.e., the formation of specific proteinaceous aggregates which is a key hallmark of a variety of neurodegenerative diseases and often serves for their diagnosis and pathological classification. AD is characterized by the senile plaques (SPs) consisting fibrillated amyloid-β (Aβ) and dystrophic neurites, and the formation of neurofibrillary tangles (NFT) consisting the hyperphosphorylated tau protein. It is hypothesized that the cellular phase of AD involves the aggregation of Aβ that acts as a trigger causing homeostatic imbalance and neuroinflammation [1]. Apart from the typical hallmarks of AD, i.e., aberrant synaptic integrities and progressive neuronal cell death [2], certain deficits in alternative splicing and metabolic alterations have also been identified [3]. Herein, the emerging features of AD from the aspect of alternation of the pivotal pathway of carbohydrate metabolism in the human brain are described.
2. Amyloid aggregation in Alzheimer’s disease
AD is characterized by the deposition of specific peptides such as Aβ extracellularly, and tau intracellularly in the brain [2]. Currently, the established biomarkers are Aβ42, total-tau, and phospho-tau in the cerebrospinal fluid (CSF). The amyloid hypothesis proposes Aβ accumulation to be the major cause of AD. Aβ is the main component of plaques and is derived from a cell surface transmembrane protein, the amyloid precursor protein (APP). It has an approximately 4,000 molecular weight and originates from the C-terminal region of the APP via proteolytic cleavage. The proteolysis of the APP by an α-secretase liberates a soluble APP-α from the surface and leaves a C-terminal APP fragment at the cell surface. The amyloidogenic cleavage of APP is executed through sequential proteolysis by β-secretase and γ-secretase at the N-terminus and C-terminus of the APP, respectively. APP is produced in most human peripheral cells, and amyloid-β is detected in the cerebrospinal fluid and the blood plasma. There are two main isoforms of Aβ peptide in humans: Aβ40 and Aβ42. The Aβ40 is predominantly abundant, while Aβ42 can form fibrils more intensively and considered comparatively more neurotoxic. Aβ is predominantly expressed at the cell membrane and translocated to the extracellular space, where it is deposited as SPs, which are a characteristic feature of AD. The toxicity of Aβ is attributed to oligomers/fibrillar Aβ, which either damage the neurons or initiate an intracellular signaling cascade causing neuronal cell death. The widely recognized model for AD progression suggests that Aβ pathogenesis may be an upstream event in AD and functions as a trigger of downstream pathways, including the tau-mediated toxicity, misfolding of hyperphosphorylated tau isoforms, tau accumulation in tangles, and proliferation of tau proteins leading to cortical neurodegeneration.
3. Metabolic reprogramming in the brain
About a hundred years ago, Otto H. Warburg found that the tumor cells tend to utilize aerobic glycolysis to obtain energy and concomitantly produce a large amount of L-lactate [4]. This discovery termed the Warburg effect has been observed in various tumor types including glioblastoma [5]. The Embden-Meyerhof-Parnas pathway (EMP pathway), i.e., glycolysis, and Krebs cycle, also known as tricarboxylic acid cycle (TCA cycle), are the central metabolic pathways providing biochemical precursors for biosynthesis and energy production (Figure 1). Most tumor cells prefer aerobic glycolysis to obtain massive energy, where a large fraction of pyruvate produced by glycolysis is converted into lactate [6]. Although certain genetic impairments in the functioning of the oxidative metabolism enhance aerobic glycolysis in tumors, the aerobic glycolysis does not generally predict the loss of oxidative metabolism [7].
Recently, an accumulating body of evidence suggests that the Warburg effect not only occurs in the tumor cells but also in non-cancerous cells. This indicates that the role of the Warburg effect may go far beyond cancer progression, implying a critical role in non-cancer diseases and physiological activities. It has been reported that aerobic glycolysis and lactic acid production are principal features of astrocytes [8]. The astrocyte-neuron-lactate shuttle (ANLS) hypothesis suggests that the astrocyte-derived lactic acid is assimilated by the neurons and re-converted to pyruvic acid to drive the mitochondrial respiratory chain. Though the ANLS hypothesis is causing controversy, an apparent contradiction exists between the ANLS hypothesis and Warburg effect involving enhanced glucose uptake by the neurons and release of L-lactate.
The human brain, albeit being only a fraction of the total bodyweight, accounts for one-fifth of the total energy utilization of the entire body at its resting state [9]. Two main types of cells exist in the brain — the neurons utilizing ~ 70% of the total brain energy, and glial cells consuming the remaining. The glucose consumption rate in the brain depends upon the type of the cell and expression level of each enzyme. It has been thought that the neurons are metabolically oxidative, and astrocytes are relatively glycolytic [9]. A recent study indicated that the neurons utilize glucose predominantly through glycolysis in vivo and require it for normal functioning [10].
In neurons, glucose is metabolized through either the glycolysis or pentose phosphate pathway (PPP), followed by the Krebs cycle, and oxidative phosphorylation (OXPHOS) system, resulting in the production of water, carbon dioxide, and 30–36 adenosine triphosphate (ATP) molecules (Figure 1). The glycolysis process converts glucose to pyruvate, and then pyruvate is transported to the mitochondria in which pyruvate is used to synthesize acetyl coenzyme A (acetyl-CoA). Further, acetyl-CoA is combined with citrate that then enters into the TCA cycle. These pathways produce nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2), which are subsequently re-oxidized in the electron transport chain (ETC) in the mitochondrial respiratory chain to generate ATP.
4. Metabolic reprogramming and Alzheimer’s disease
The human brain largely depends on glucose as its primary source of energy to fuel the respiratory chain in the mitochondria. Glucose metabolism is enhanced and partially utilized to provide the lipid molecules necessary for neurite growth [11]. The aerobic glycolysis in the brain might support neurite outgrowth, and decreases with age, whose functional significance in the context to the development of AD symptoms remains to be elucidated. According to a recent study the cognitively impaired status was related to a decrease of the typical pattern of aerobic glycolysis in the young adults [12]. On the contrary, the amyloidal deposition with no cognitively impaired features had a linkage to the conservation of aerobic glycolysis, that is stronger than observed even in cognitive impairment-negative and amyloid-negative individuals. These indicate that aerobic glycolysis might be involved in the responses to the early phase of AD pathogenesis, and the age-associated white matter lesions might impair the processes.
With regard to AD, Traxler and colleagues identified the metabolic signature induced by pyruvate kinase M2 (PKM2), an isoform of pyruvate kinases, in the neurons (Figure 1). These neurons prefer glycolysis in spite of a normal mitochondrial function in a manner resembling the Warburg effect using a series of experimental techniques including multimodal-omics on AD patient-derived induced neurons [13]. Unlike pyruvate kinase M1 (PKM1), which forms a tetramer, PKM2 can be translocated into the cell nuclei and trigger the activation of transcription factors including the signal transducer and activator of transcription 3 (STAT3) and hypoxia-inducible factor 1α (HIF1α) to modulate the expression of effector molecules involved in apoptosis [14]. The Warburg effect is associated with escaping apoptosis in many cancer types, while neurons enhance their ability to receive proapoptotic stimuli. The neuronal PKM2 isozyme induced a metabolic shift, which was partially inhibited by a naphthoquinone derivative “shikonin” isolated from the herb Lithospermum erythrorhizon, acting as a chemical inhibitor of PKM2 [13]. These results suggest the potential application of the drug-based targeting of PKM2 for the treatment and/or prevention of age-related neurodegeneration like in AD. A future study concerning the microglial–neuronal interactions might elucidate the molecular mechanisms underlying microglia-regulated neuron dysfunction in AD [15].
5. Oxidative stress and its connection to Alzheimer’s disease
Oxidative stress is an imbalance between the generation of reactive oxygen species (ROS) and the antioxidants, and has been shown to contribute markedly to the pathogenesis and progression of AD [16]. The ROS represents reactive chemicals derived from molecule O2, and which are continuously produced and catabolized in the cells of aerobic organisms. The free radical types of ROS include superoxide anion radicals and hydroxyl radicals; the non-free radical ROS forms include H2O2 and O3 [17]. The excessive ROS production participate in the pathogenesis of a variety of disorders; the dysregulation of ROS metabolism enhances the neurodegenerative diseases including AD (Figure 2).
ROS also play a role as secondary messengers in the redox-mediated signaling transduction pathways including the cascade of mitogen-activated protein kinases (MAPKs), p38 MAPKs, and c-Jun N-terminal kinases (JNKs). Therefore, ROS exhibits bivalent functions, with the other being the induction of the Akt pathway [18] through the inhibition of the counteracting phosphatase and tensin homolog (PTEN). The events triggered via the phosphatidylinositol-3 kinase (PI3K) pathway involve the activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and production of ROS. Thus, ROS are crucial to intracellular redox regulation and positively regulate the PI3K signal pathway via mechanisms regulating the reversible oxidation and inactivation of PTEN and other enzymes that negatively regulate this pathway.
6. Microbiome-microglia interactions in Alzheimer’s disease
The study of the gut microbiome is gaining prominence in the fields of biology and medicine and has particularly emerged as a key regulator of brain pathophysiology [19]. The maintenance of a healthy gut microbiota is one of the critical factors for supporting the homeostasis of the immune system and cognitive–emotional balance via the production of several bioactive metabolites, thus giving rise to the gut microbiota–brain axis [8]. The composition of the gut microbiome is generally variable among individuals, and once the profile of the commensal microbiota is established to a certain extent during childhood, it exhibits strong resilience, i.e., its composition and activity subsequently remains substantially stable. The resilience of the health-promoting microbiota protects the host from a variety of dysbiosis related pathologies. Thus, the interventions targeting them might be promising strategies for treating diseases and promoting health.
The mechanisms underlying the effects of intestinal microbiome on the brain partially involve neuronal-immune associations [20]. The microbiome-derived metabolites such as short-chain fatty acids (SCFAs), e.g., acetate, propionate, and butyrate are the commonly identified signaling metabolites that affect microglia. In addition to serving as energy sources for neurons and affecting the maturation of microglia, these SCFAs may influence the physiological function of the neurons. SCFAs are also able to regulate the levels of secretory neurotransmitters and neurotrophic factors. For example, acetate has previously demonstrated to regulate the release of neurotransmitters including glutamate, glutamine, and γ-amino butyric acid in the hypothalamus and increase the neuropeptide expression. Propionate and butyrate exert a distinct influence on the intracellular K+ levels, implying the involvement of SCFAs in intracellular signaling regulation. Microglia receive the local environmental cues in the brain and respond to signals from the distal regions of the body, e.g., from the gastrointestinal (GI) tract. Therefore, the analysis of the physiological role of the GI microbiome in regulating the maturation of microglia and their proper function in the central nervous system is gaining momentum, because the gut microbe-derived SCFAs function as mediators of the gut–brain axis [21]. However, the molecular mechanism underlying this crosstalk remains largely uncovered. A recent study identified one of the microbiota-derived SCFAs, acetate, as a signaling metabolite that can enhance the maturation of microglia, maintain the homeostatic metabolic state, and regulate microglial phagocytosis and AD pathological progression during neurodegeneration [22] (Figure 2). This study further demonstrated that the supplementation of acetate to the diet of a mouse model for AD induced the pro-inflammatory phenotype of microglia with elevated cytokine expression, which was previously shown to suppress the phagocytosis by microglia in the presence of Aβ. These observations suggest that acetate is a crucial, GI microbe-derived signaling molecule that drives the metabolic pathways of microglia in AD. This study may pave the way for the development of microbiota-targeted therapeutics to modulate brain microglia in patients with AD [23].
7. Amyloid-β and aerobic glycolysis in Alzheimer’s disease
It is important to note that the dysregulation of glucose assimilation in neurodegenerative diseases is not limited to the neurons but to the glial cells, particularly the astrocytes which can survive on aerobic glycolysis without mitochondrial respiration, and thereby might support neuronal physiological activities [24]. However, recently Aβ has shown induction of metabolism shift from OXPHOS to aerobic glycolysis in microglia [25] (Figure 2). Under such situation, the two metabolic systems might be dysregulated, which resulted in microglial dysfunction. The mechanism behind this involved phosphorylation of the mammalian target of rapamycin (mTOR) protein and enhanced expression of HIF-1α in the microglia by Aβ, thereby activating the pro-inflammatory signals. The higher rate of mTOR and HIF-1α signals occurred with a reduction in O2 consumption and an enhancement in extracellular acidification, thus shifting the glycolytic balance in microglia [26]. This metabolic reprogramming of the microglia may drive Alzheimer’s pathogenesis, since enhanced generation of lactic acid results in epigenetical regulations that triggers glycolysis-associated genes’ expression via a positive loop. Further, amyloid triggers immunological tolerance and causes dysregulation in the glycolysis in the microglia. Overall, these findings suggest a significant neuro-immune dysregulation which can mediate the AD-associated pathologic conditions.
8. Nutrition-based interventions for the control of Alzheimer’s disease and amyloid pathology
An accumulating body of evidence indicates that dietary choices have certain role in protecting against the neurodegeneration associated with AD, but the exact association between the nutritional profile of the diet and its neuroprotective effects remains largely unknown [27]. In addition, it is quite difficult to examine the distinct effects of each diet plan involved. Although a variety of lifestyle factors may have an influence on the central nervous system (CNS), regulation of diet-related facets may be a possible method for prevention of CNS dysfunction. The current understanding of the mechanisms involved in AD suggests the function of oxidative stress, which occurs when the generation of ROS exceeds the capability of the antioxidant-mediated sequestering system especially with aging, which may contribute to the progression of age-related neurodegeneration including AD. Thus, nutraceuticals with antioxidant activity, such as dietary plant polyphenols may benefit the prevention of AD [28,29] (Figure 2).
Certain dietary patterns in Japan are well known for their ability to prevent AD. The Ogimi diet rich in L-serine which is similar to the Okinawa diet, has been approved for Phase II clinical trials in patients of AD [30]. A high-ketogenic diet exhibited the potential for alleviating the symptoms of AD, suggesting the applicability of ketone-based nutritional therapeutics as a promising strategy for the complementation of the energy levels in the brain in patients with AD [31] (Figure 2). The Mediterranean diet had been associated with an improvement in memory, an increase in Aβ42:Aβ40, and pTau181 [32]. Among these, the high-ketogenic diet is the most promising therapeutic method for mitigating AD because it may restore the metabolic imbalance caused due to the occurrence of aerobic glycolysis in the neurons and the microglia. The proposed high-ketogenic diet with low carbohydrates enhanced the supply of ketone bodies biosynthesized in the liver using free fatty acids as a substitute for those cells with a high demand for glucose (Figure 1). A well-formulated diet that is adequately ketogenic was regarded to be nutritionally safe, although prone to being deficient in micronutrients [33]. Additionally, in a recent meta-analysis, a healthy diet comprised of higher consumption of fruits, vegetables, legumes, and fish but a minimal intake of saturated fat and sodium, has been identified to demonstrate potential protective effects against AD. The plausible mechanisms through which a high-ketogenic diet could improve Aβ levels in AD are an alteration in the glycolytic metabolism, a decrease in the generation of ROS, assimilation of ketones as an alternative source of energy in the neurons, and an amelioration in neuronal inflammation [34].
9. Conclusions and future directions
AD is characterized by the SPs consisting fibrillated Aβ and dystrophic neurites, and the formation of tau NFT. The Aβ toxicity is attributed to the formation of oligomers/fibrillar Aβ, which either damages the neurons or initiates an intracellular signaling cascade leading to neuronal cell death. Aβ is a peptide with approximately a 4,000 of molecular weight originating from the C-terminal region of the APP by proteolytic cleavage. Other than the typical hallmarks of AD, certain defects such as metabolic alterations have been identified. Herein, the emerging features of AD from the aspect of alteration in the central pathway of carbohydrate metabolism in the human brain are described. In particular, the neurons in patients with AD favor glycolysis in a manner like Warburg effect in spite of the normal functionality in mitochondrion.
A therapeutic method that is significantly effective in the treatment, mitigation, and prevention of AD has not yet been established. Therefore, the establishment of novel therapies including stem cell therapy [35] effective in suppressing the symptoms of AD and elucidation of the mechanisms behind their protective effects against neurodegeneration is being pursued by researchers. Recent studies suggest that the manipulation of glucose metabolism may result in significant mitigation of Aβ-related AD pathology and neurodegeneration. They also imply that the modulation of the intestinal microbiome may be a promising therapeutic option to prevent AD. The potentially practical approaches for the therapeutic intervention of metabolic disorders might be diet-based interventions. Such studies might lead to the establishment of novel therapeutic interventions for AD in near future.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article..
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
Aβ, amyloid-β
AD, Alzheimer’s disease
ANLS, astrocyte-neuron-lactate shuttle
APP, amyloid precursor protein
ATP, adenosine 5′-triphosphate
CoA , coenzyme A
CSF, cerebrospinal fluid
DHAP, dihydroxyacetone phosphate
ETC, electron transport chain
FADH2, flavin adenine dinucleotide
GI tract, gastrointestinal tract
JNK, c-Jun N-terminal kinase
MAPK, mitogen-activated protein kinase
mTOR , mammalian target of rapamycin
NADH , nicotinamide adenine dinucleotide
NADPH, nicotineamide adenine dinucleotide phosphate
NFT, neurofibrillary tangles
OXPHOS, oxidative phosphorylation
PEP, phosphoenolpyruvate
PI3K, phosphatidylinositol-3 kinase
PKM1, pyruvate kinase M1
PKM2, pyruvate kinase, muscle, M2 isoform
PPP, pentose phosphate pathway
PTEN, phosphatase and tensin homolog
SCFA, short-chain fatty acid
SP, senile plaque
STAT3, signal transducer and activator of tran-scription 3
TCA, tricarboxylic acid
References
- Balusu, S.; Praschberger, R.; Lauwers, E.; De Strooper, B.; Verstreken, P. Neurodegeneration cell per cell. Neuron 2023, 111, 767–786. [Google Scholar] [CrossRef]
- Wilson, D.M. 3rd; Cookson, M.R.; Van Den Bosch, L.; Zetterberg, H.; Holtzman, D.M.; Dewachter, I. Hallmarks of neurodegenerative diseases. Cell 2023, 186, 693–714. [Google Scholar] [CrossRef]
- Johnson, E.C.B.; Dammerm, E.B.; Duongm, D.M.; Pingm, L.; Zhoum, M.; Yinm, L.; Higginbothamm, L.A.; Guajardom, A.; Whitem, B.; Troncosom, J.C.; et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat. Med. 2020, 26, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. 100 years of the Warburg effect: a historical perspective. Endocrine-Related Cancer 2022, 29, T1–T13. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. We need to talk about the Warburg effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef] [PubMed]
- Bas-Orth, C.; Tan, Y.-W.; Lau, D.; Bading, H. Synaptic Activity Drives a Genomic Program That Promotes a Neuronal Warburg Effect. J. Biol. Chem. 2017, 292, 5183–5194. [Google Scholar] [CrossRef] [PubMed]
- Camandola, S.; Mattson, M.P. Brain metabolism in health, aging, and neurodegeneration. EMBO J. 2017, 36, 1474–1492. [Google Scholar] [CrossRef]
- Li, H.; Guglielmettim, C.; Seim, Y.J.; Zilberterm, M.; Le Pagem, L.M.; Shieldsm, L.; Yangm, J.; Nguyenm, K.; Tiretm, B.; Gaom, X.; et al. Neurons require glucose uptake and glycolysis in vivo. Cell Rep. 2023, 42, 112335. [Google Scholar] [CrossRef]
- Segarra-Mondejar, M.; Casellas-Díaz, S.; Ramiro-Pareta, M.; Müller-Sánchez, C.; Martorell-Riera, A.; Hermelo, I.; Reina, M.; Aragonés, J.; Martínez-Estrada, O.M.; Soriano, F.X. Synaptic activity-induced glycolysis facilitates membrane lipid provision and neurite outgrowth. EMBO J. 2018, 37, e97368. [Google Scholar] [CrossRef] [PubMed]
- Goyal, M.S.; Blazey, T.; Metcalf, N.V.; McAvoy, M.P.; Strain, J.F.; Rahmani, M.; Durbin, T.J.; Xiong, C.; Benzinger, T.L.-S.; Morris, J.C.; et al. Brain aerobic glycolysis and resilience in Alzheimer disease. Proc. Natl. Acad. Sci. 2023, 120. [Google Scholar] [CrossRef]
- Traxler, L.; Herdy, J.R.; Stefanoni, D.; Eichhorner, S.; Pelucchi, S.; Szücs, A.; Santagostino, A.; Kim, Y.; Agarwal, R.K.; Schlachetzki, J.C.; et al. Warburg-like metabolic transformation underlies neuronal degeneration in sporadic Alzheimer’s disease. Cell Metab. 2022, 34, 1248–1263. [Google Scholar] [CrossRef]
- Victor, M.B.; Tsai, L.-H. Walking the high wire: How neurons maintain stability in the crossline of neurodegeneration. Cell Metab. 2022, 34, 1227–1229. [Google Scholar] [CrossRef]
- Murdock, M.H.; Tsai, L.-H. Insights into Alzheimer’s disease from single-cell genomic approaches. Nat. Neurosci. 2023, 26, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Butterfieldm, D.A.; Halliwellm, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Murai, T.; Matsuda, S. Pleiotropic Signaling by Reactive Oxygen Species Concerted with Dietary Phytochemicals and Microbial-Derived Metabolites as Potent Therapeutic Regulators of the Tumor Microenvironment. Antioxidants 2023, 12, 1056. [Google Scholar] [CrossRef]
- Matsuda, S.; Nakagawa, Y.; Tsuji, A.; Kitagishi, Y.; Nakanishi, A.; Murai, T. Implications of PI3K/AKT/PTEN Signaling on Superoxide Dismutases Expression and in the Pathogenesis of Alzheimer’s Disease. Diseases 2018, 6, 28. [Google Scholar] [CrossRef]
- Mossad, O.; Erny, D. The microbiota–microglia axis in central nervous system disorders. Brain Pathol. 2020, 30, 1159–1177. [Google Scholar] [CrossRef]
- Silpe, J.E.; Balskus, E.P. Deciphering Human Microbiota–Host Chemical Interactions. ACS Central Sci. 2020, 7, 20–29. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Sierra, A.; Stevens, B.; Tremblay, M.-E.; Aguzzi, A.; Ajami, B.; Amit, I.; Audinat, E.; Bechmann, I.; Bennett, M.; et al. Microglia states and nomenclature: A field at its crossroads. Neuron 2022, 110, 3458–3483. [Google Scholar] [CrossRef]
- Erny, D.; Dokalis, N.; Mezö, C.; Castoldi, A.; Mossad, O.; Staszewski, O.; Frosch, M.; Villa, M.; Fuchs, V.; Mayer, A.; et al. Microbiota-derived acetate enables the metabolic fitness of the brain innate immune system during health and disease. Cell Metab. 2021, 33, 2260–2276. [Google Scholar] [CrossRef]
- Lynch, C.M.; Clarke, G.; Cryan, J.F. Powering up microbiome-microglia interactions. Cell Metab. 2021, 33, 2097–2099. [Google Scholar] [CrossRef]
- Tang, B.L. Glucose, glycolysis, and neurodegenerative diseases. J. Cell Physiol. 2020, 235, 7653–7662. [Google Scholar] [CrossRef]
- Baik, S.H.; Kang, S.; Lee, W.; et al. A breakdown in metabolic reprogramming causes microglia dysfunction in Alzheimer’s disease. Cell Metab. 2019, 30, 493–507e496. [Google Scholar] [CrossRef]
- Mitra, S.; Banik, A.; Saurabh, S.; Maulik, M.; Khatri, S.N. Neuroimmunometabolism: A New Pathological Nexus Underlying Neurodegenerative Disorders. J. Neurosci. 2022, 42, 1888–1907. [Google Scholar] [CrossRef]
- Díaz, G.; Lengele, L.; Sourdet, S.; Soriano, G.; Barreto, P.d.S. Nutrients and amyloid β status in the brain: A narrative review. Ageing Res. Rev. 2022, 81, 101728. [Google Scholar] [CrossRef]
- Murai, T.; Matsuda, S. The Chemopreventive Effects of Chlorogenic Acids, Phenolic Compounds in Coffee, against Inflammation, Cancer, and Neurological Diseases. Molecules 2023, 28, 2381. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Ikeda, Y.; Nakagawa, Y.; Tsuji, A.; Kitagishi, Y.; Matsuda, S. Special bioactive compounds and functional foods may exhibit neuroprotective effects in patients with dementia (Review). Biomed. Rep. 2020, 13, 1–1. [Google Scholar] [CrossRef] [PubMed]
- Cox, P.A.; Metcalf, J.S. Traditional Food Items in Ogimi, Okinawa: l-Serine Content and the Potential for Neuroprotection. Curr. Nutr. Rep. 2017, 6, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Myette-Côté, É.; Soto-Mota, A.; Cunnane, S.C. Ketones: potential to achieve brain energy rescue and sustain cognitive healtduring ageing. Br. J. Nutr. 2022, 128, 407–423. [Google Scholar] [CrossRef] [PubMed]
- Ballarini, T.; Melo van Lent, D.; Brunner, J.; Schröder, A.; Wolfsgruber, S.; Altenstein, S.; Brosseron, F.; Buerger, K.; Dechent, P. ; DELCODE study group. Mediterranean diet, Alzheimer disease biomarkers and brain atrophy in old age. Neurology 2021, 96, e2920–2932. [Google Scholar]
- Yassine, H.N.; Self, W.; Kerman, B.E.; Santoni, G.; Shanmugam, N.N.; Abdullah, L.; Golden, L.R.; Fonteh, A.N.; Harrington, M.G.; Gräff, J.; et al. Nutritional metabolism and cerebral bioenergetics in Alzheimer’s disease and related dementias. Alzheimer’s Dement. 2022, 19, 1041–1066. [Google Scholar] [CrossRef]
- Neth, B.J.; Mintzm, A.; Whitlowm, C.; Jungm, Y.; Solingapuram Saim, K.; Registerm, T.C.; Kellarm, D.; Lockhartm, S.N.; Hoscheidtm, S.; et al. Modified ketogenic diet is associated with improved cerebrospinal fluid biomarker profile, cerebral perfusion, and cerebral ketone body uptake in older adults at risk for Alzheimer’s disease: a pilot study. Neurobiol. Aging 2020, 86, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Salewski, R.P.; Eftekharpour, E.; Fehlings, M.G. Are induced pluripotent stem cells the future of cell-based regenerative therapies for spinal cord injury? J. Cell Physiol. 2010, 222, 515–521. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The central glucose metabolic pathways in cells of the nervous system. The Embden-Meyerhof-Parnas pathway (EMP pathway), i.e., glycolysis, and Krebs cycle, also known as tricarboxylic acid cycle (TCA cycle), are the central metabolic pathways providing biochemical precursors for biosynthesis and energy production. In neurons, glucose is metabolized through either the glycolysis or pentose phosphate pathway (PPP), followed by the Krebs cycle, and oxidative phosphorylation (OXPHOS) system, resulting in the production of water, CO2, and 30–36 ATP molecules. The glycolysis process converts glucose to pyruvate, and then pyruvate is transported to the mitochondria in which pyruvate is used to synthesize acetyl-CoA. Further, acetyl-CoA is combined with citrate that then enters into the Krebs cycle. These pathways produce nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2), which are subsequently re-oxidized in the electron transport chain in the mitochondrial respiratory chain to generate ATP. With regard to Alzheimer’s disease (AD), the metabolic signature induced by pyruvate kinase M2 (PKM2), an enzyme that converts phosphoenolpyruvate (PEP) to pyruvate, causes a metabolic rewiring towards glycolysis in a manner resembling the Warburg effect in which pyruvate produced by glycolysis is intensively converted into lactate. A high-ketogenic diet exhibited the potential for alleviating the symptoms of AD, suggesting the applicability of ketone-based nutritional therapeutics as a promising strategy for the complementation of the energy levels in the brain in patients with AD. The high-ketogenic diet with low carbohydrates enhanced the supply of ketone bodies biosynthesized in the liver using free fatty acids as a substitute for those cells with a high demand for glucose.
Figure 1.
The central glucose metabolic pathways in cells of the nervous system. The Embden-Meyerhof-Parnas pathway (EMP pathway), i.e., glycolysis, and Krebs cycle, also known as tricarboxylic acid cycle (TCA cycle), are the central metabolic pathways providing biochemical precursors for biosynthesis and energy production. In neurons, glucose is metabolized through either the glycolysis or pentose phosphate pathway (PPP), followed by the Krebs cycle, and oxidative phosphorylation (OXPHOS) system, resulting in the production of water, CO2, and 30–36 ATP molecules. The glycolysis process converts glucose to pyruvate, and then pyruvate is transported to the mitochondria in which pyruvate is used to synthesize acetyl-CoA. Further, acetyl-CoA is combined with citrate that then enters into the Krebs cycle. These pathways produce nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2), which are subsequently re-oxidized in the electron transport chain in the mitochondrial respiratory chain to generate ATP. With regard to Alzheimer’s disease (AD), the metabolic signature induced by pyruvate kinase M2 (PKM2), an enzyme that converts phosphoenolpyruvate (PEP) to pyruvate, causes a metabolic rewiring towards glycolysis in a manner resembling the Warburg effect in which pyruvate produced by glycolysis is intensively converted into lactate. A high-ketogenic diet exhibited the potential for alleviating the symptoms of AD, suggesting the applicability of ketone-based nutritional therapeutics as a promising strategy for the complementation of the energy levels in the brain in patients with AD. The high-ketogenic diet with low carbohydrates enhanced the supply of ketone bodies biosynthesized in the liver using free fatty acids as a substitute for those cells with a high demand for glucose.
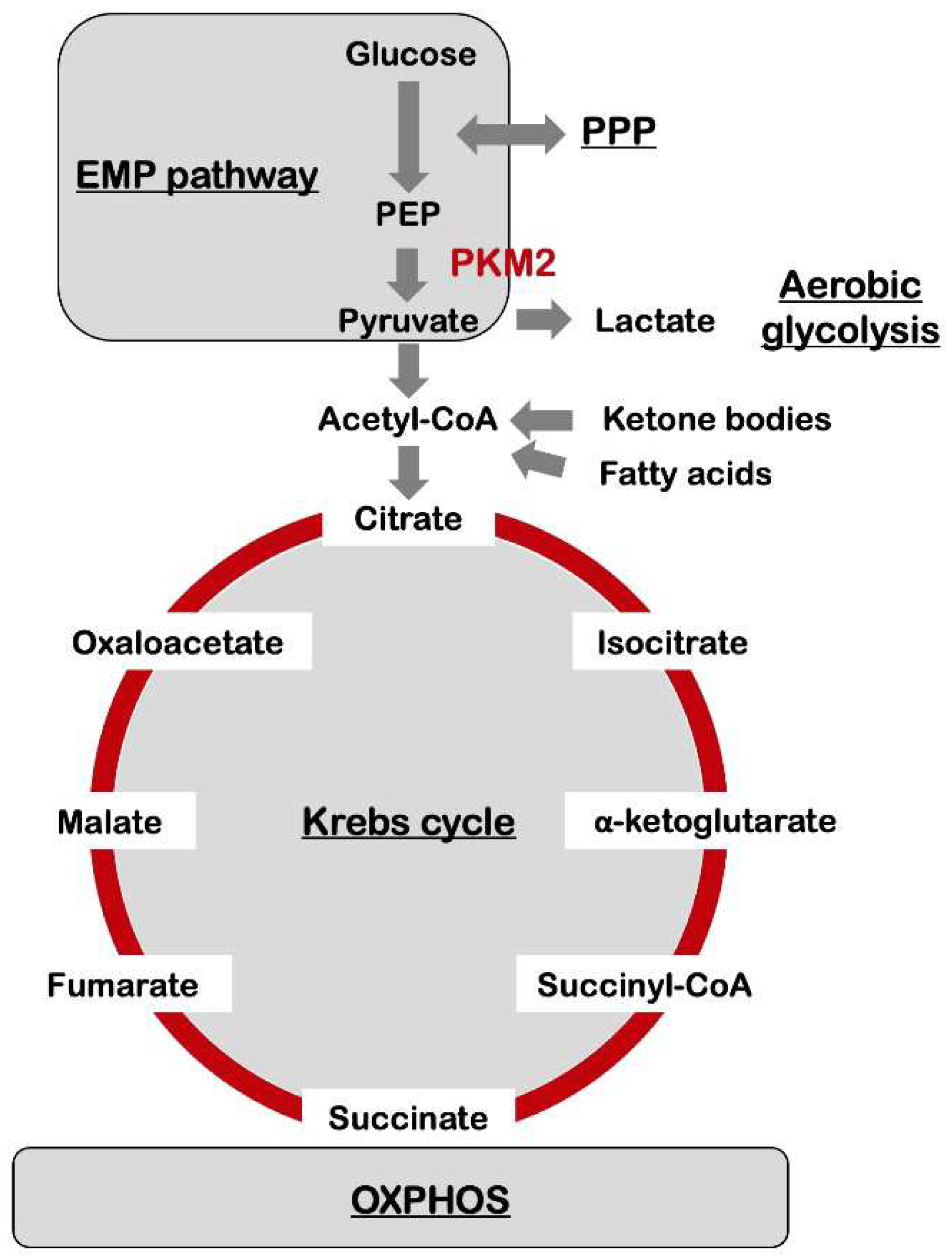
Figure 2.
The multiple factors that may contribute to the pathogenesis and progression of Alzheimer’s disease. Alzheimer’s disease (AD) is characterized by the deposition of specific peptides such as amyloid-β (Aβ) extracellularly, and tau intracellularly in the brain. Aβ has shown induction of metabolic reprogramming from OXPHOS to aerobic glycolysis in microglia, which may drive AD pathology. The oxidative stress is an imbalance between the generation of reactive oxygen species (ROS) and the antioxidants, and has been shown to contribute markedly to the pathogenesis and progression of AD. Gut microbiota-derived SCFA, acetate, acts as a signaling metabolite that can enhance the maturation of microglia, maintain the homeostatic metabolic state, and regulate microglial phagocytosis and AD pathological progression during neurodegeneration. Nutraceuticals with antioxidant activity, such as dietary plant polyphenols may benefit the prevention of AD. A high-ketogenic diet exhibited the potential for alleviating the symptoms of AD, suggesting the applicability of ketone-based nutritional therapeutics as a promising strategy for the complementation of the energy levels in the brain in patients with AD.
Figure 2.
The multiple factors that may contribute to the pathogenesis and progression of Alzheimer’s disease. Alzheimer’s disease (AD) is characterized by the deposition of specific peptides such as amyloid-β (Aβ) extracellularly, and tau intracellularly in the brain. Aβ has shown induction of metabolic reprogramming from OXPHOS to aerobic glycolysis in microglia, which may drive AD pathology. The oxidative stress is an imbalance between the generation of reactive oxygen species (ROS) and the antioxidants, and has been shown to contribute markedly to the pathogenesis and progression of AD. Gut microbiota-derived SCFA, acetate, acts as a signaling metabolite that can enhance the maturation of microglia, maintain the homeostatic metabolic state, and regulate microglial phagocytosis and AD pathological progression during neurodegeneration. Nutraceuticals with antioxidant activity, such as dietary plant polyphenols may benefit the prevention of AD. A high-ketogenic diet exhibited the potential for alleviating the symptoms of AD, suggesting the applicability of ketone-based nutritional therapeutics as a promising strategy for the complementation of the energy levels in the brain in patients with AD.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



