Submitted:
06 June 2023
Posted:
07 June 2023
You are already at the latest version
Abstract
With an increase in energy consumption globally, Fischer-Tropsch (FT) synthesis is a good alternative for producing fuels and chemicals from coal, natural gas or biomass. Among them, coal to liquids has been put into production in countries that have large coal reserves. In this process, Fe-based catalysts are commonly used due to their earth abundance, comparatively wide operation range and ready availability to handle low H2/CO ratio from coal. Despite their extensive applications, the kinetic and mechanistic understandings of Fe carburization and FT reaction on Fe-carbides are relatively limited due to the complexity of phase composition of the applied catalysts. This review summarizes the current state of knowledge of FT synthesis on Fe-carbide with an emphasis on underlying mechanism. Specifically, the employment of model catalyst, such as Raney-Fe, could provide a convenient way to furnish kinetic information regarding Fe carburization and subsequent FT reaction. A major challenge for further understanding catalytic reactions occurring at the Fe-carbide surface is correlating FT activity and selectivity to specific active site. To address this issue, the advancements of both DFT calculations and surface science techniques are highly demanded.
Keywords:
Fischer-Tropsch synthesis
; Fe-carbide
; Mechanism
; Synthesis gas
; Fe carburization
1. Fischer-Tropsch synthesis
Fischer-Tropsch (FT) process has been widely investigated for almost 100 years. In the 1920s, German scientists Franz Fischer and Hans Tropsch first developed this process, in which a mixture of H2 and CO (synthesis gas) can be converted to valuable long-chain hydrocarbons like gasoline, diesel fuel and chemicals (olefins, alcohols or acids) [1,2]. The FT process was first commercialized in Germany prior to the Second World War. It could offer synthetic fuel for the German war machine due to the abundant domestic coal supplies used for producing synthesis gas [3]. After the Second World War, the development of this process stalled because low crude oil prices led to a strong growth and dominance of the petroleum oil industry [4]. The interest in the FT synthesis was revived in South Africa during the Apartheid regime in the 1970s. During that period, supply of oil in South Africa was cut off due to international sanctions, but through the FT synthesis South Africans were still able to produce the required fuels and chemicals from coal. At the same time, the energy crises in 1973 and 1978 have also stimulated the global interest and exploration of alternative fuel production by expanding the commercialization of FT processes [5]. Besides coal, natural gas and biomass are also considered as important alternatives. The process to convert these carbon sources to valuable chemicals is often referred to as X to Liquid (XTL), in which X stands for the feedstock from which synthesis gas is derived, e.g., coal (CTL), natural gas (GTL) or biomass (BTL) [6]. Although most of these embodiments still rely on fossil resources, CTL and GTL enable the production of clean transportation fuels that are free of heavy metals, aromatics or contaminants such as nitrogen and sulfur. Currently, large XTL processes are operated in Malaysia by Shell, in Qatar by Shell and SASOL, in South Africa, Uzbekistan and Nigeria by SASOL. Among them, CTL is of large interest in areas with abundant coal resources, for example China and South Africa. Currently, a large number of CTL demonstration plants and industrialization projects are commissioned in China using Fe-based catalysts [7].
The CTL process generally consists of 4 chemical conversion steps, (Figure 1). In the first step, coal gasification is done in gasifiers to produce synthesis gas, a mixture of CO and H2. Due to the low H/C ratio of coal, the derived synthesis gas has a typical H2/CO ratio below 1 [8]. In the second step, the water-gas shift (WGS) reaction (equation 1) is used to increase the H2/CO ratio tailored for the desired product distribution in the subsequent FT synthesis step. Usually, a constant amount of CO2 is removed in the overall CTL process, either in a WGS step prior to the FT synthesis or in the FT synthesis reactor itself when a catalyst is used that exhibits sufficient WGS activity. CO2 produced in the FT reactor not only decreases CO conversion, but also leads to a higher energy consumption for separating the gas effluent from the reactor. When the main WGS conversion is done in a separate WGS reactor, CO2 capture becomes more viable [9]. After cooling and purification, the synthesis gas is introduced into the FT synthesis reactor and converted into long-chain hydrocarbons (equation 2). Traditionally, these processes are realized in either fixed and fluidized bed reactors or slurry bubble reactors. Fixed beds are suitable for wax production as for instance done in Shell FT plants in Malaysia and Qatar [10]. Separation of products from catalysts are more cost-effective in fixed bed reactors. On the other hand, the pressure drop in such reactors leads to higher operational costs than other reactors. Moreover, as it is costly to replace the catalyst inventory, catalysts should exhibit a long lifespan. As the FT reaction is highly exothermic, it is important to rapidly remove the heat of reaction in order to avoid overheating the catalyst [11]. Compared to fixed bed reactors, fluidized bed reactors can realize a very homogeneous temperature distribution because of the rapid and turbulent gas/liquid movement. Another important advantage of fluidized bed reactors is that deactivated catalyst can be removed continuously, and new catalysts can be added for longer production runs. A drawback of such reactors is the difficulty in separating the catalyst from the products. Fluidized bed reactors are considered to be a promising technology for the production of lower-molecular-weight products on Fe-based catalysts at high temperature [12]. Similar to fluidized bed reactors, slurry bubble reactors can meet the requirement of online removal/addition of catalyst and can be operated under isothermal conditions. Catalysts that feature high mechanical strength and attrition resistance are required for slurry bubble reactors. As the catalysts are suspended in the wax, separating the catalyst from wax is a major challenge [13].
The final step of the overall XTL process is product upgrading. The mixture of products formed during the FT process (e.g., long-chain hydrocarbons or oxygenates) need to be processed in order to obtain high-value transportation fuels and base chemicals using processes such as hydrotreating, hydrocracking and hydro isomerization. In-reactor upgrading of the products of FT reactions by adding zeolites to Fe-based catalysts has also been investigated [14,15]. Among a variety of hydrocarbons, linear α-olefins (LAOs) production and separation is gaining widespread attractions. LAOs are highly valuable intermediates for the chemical industry [16]. Lower olefins (C2-C4 LAOs) are mainly used as building blocks, commonly produced by steam cracking of ethane or naphtha and dehydrogenation of propane [17]. The process to produce lower olefins via FT reaction is referred to as Fischer-Tropsch to olefins (FTO). A lot of work has been conducted to develop highly efficient catalysts to directly convert synthesis gas to lower olefins. [18,19] However, traditional FTO is limited by ASF distribution and suffers from high CH4 selectivity. To address these issues, recently, the development of oxide-zeolite bi-functional catalyst to selectively convert synthesis gas to lower olefins has seen significant progress. The separation of CO activation and C-C coupling onto two different types of active sites can tune C2-C4 LAOs selectivity as high as 80% at a fair CO conversion [20,21]. LAOs with more than 4 carbon atoms, especially in the C5-C10 range, are even more valuable than lower olefins because of their use as co-monomers in polymerization and as feedstocks for lubricants and detergents [22]. Currently, there was no commercial process for directly converting synthesis gas to higher LAO, which couldn’t meet the consumer demand and hamper industrial development globally.
CO + H2O → CO2 + H2
nCO + (2n + 1)H2 → CnH2n+2 + nH2O
2. Catalysts
A general aspect of FT chemistry is the dissociation of CO in atomic C and O on metal surface,[23] although there are also pathways that involve hydrogenation of CO prior to C-O bond cleavage. Group VIII metals with unoccupied d-orbitals are capable for CO dissociation. Based on Brønsted-Evans-Polanyi (BEP) relations, transition metals with a lower d-band filling that will bind the dissociated C and O atoms strongly will lead to low activation barriers for CO dissociation [24]. Termination of the chain-growth reactions are also important as they determine the length of the hydrocarbons obtained [23].
From left to right in the periodic table, the d band of transition metals is filled [25]. Catalysts with lower C and O binding energy to the right will hardly produce long-chain hydrocarbons, because the CHx growth monomers are easily hydrogenated by H2, resulting in high CH4 selectivity. Therefore, CO hydrogenation on Ni mainly produces CH4. For Cu and Rh-based catalysts, the main products for CO hydrogenation are alcohols due to their low CO dissociation ability. Metals such as Ru, Co and Fe bind C and O stronger than Ni, Cu and Rh, resulting in a higher probability for CHx intermediate to couple to long-chain hydrocarbons. Hence, Ru-, Co- and Fe-based catalysts are the most suitable for the FT reaction [26]. The product distribution of CO hydrogenation on transition metals is showed in Figure 2.
Ru-based catalysts display outstanding performance for CO hydrogenation in terms of activity, selectivity and stability. Despite this, Ru cannot serve as a base for catalysts at the industrial scale because of the high price of this cheapest of noble metals [27]. So far, only Co and Fe have been used as the active phase for industrial FT catalysts.
Co-based catalysts outperform Fe-based catalysts at low temperature (200-240 ℃), often referred to as low-temperature Fischer-Tropsch (LTFT). Moreover, the WGS activity of Co is much lower than that of Fe, limiting undesired CO2 formation. On the other hand, the higher CH4 selectivity on Co-based catalysts restricts its application at high temperature (250-350 ℃), which is referred to as high-temperature Fischer-Tropsch (HTFT). In HTFT, Fe-based catalysts are preferred to reduce the amount of CH4 formed [17]. Another advantage of Fe catalysts is that they can handle the low H2/CO ratio of synthesis gas derived from coal and biomass owing to the substantial WGS activity [28]. On the other hand, Co-based catalysts are typically used in combination with natural gas as a source of the synthesis gas feedstock. Overall, the operation conditions for Fe-based catalysts are more flexible and they can also be used in LTFT. Paraffins, such as wax, is the main product for Co-based catalyst in LTFT, whereas Fe is mainly used for producing olefins and oxygenates in HTFT. Metallic Co is regarded as the active phase for the FT reaction, while Fe has a high tendency to form Fe-carbide because of a strong Fe-C bond. The most significant differences between Co- and Fe-based catalysts are shown in Table 1.
In addition to mono-Co or Fe-based catalysts, construction of bimetallic catalysts incorporating both Co and Fe has attracted a wide range of attentions. Yang et al. revealed that adding Co in χ-Fe5C2 enhances the FT performance because Co is more capable of dissociating CO [31]. The synergistic effect of Co-Fe alloy not only leads to higher FT activity, but also conducive to grow long-chain hydrocarbons [32]. It was also reported that bimetallic catalysts were more stable against deactivation compared to pure Co-based catalysts though they still suffered from deactivation at high CO conversion due to high H2O partial pressure [33].
3. Fe-based catalysts
In the Earth’s crust, Fe is the fourth most abundant element, mainly existing in the form of Fe-oxide. This abundance means that Fe is very cheap and an excellent choice for the FT catalysis. For the industrial FT synthesis, precipitated or fused Fe in unsupported form are mainly used as catalyst precursors [34]. Practically, precipitated Fe is employed in fixed bed or slurry bed reactor in LTFT, predominately producing long-chain hydrocarbons e.g., wax [35]. On the other hand, fused Fe is mainly consumed in fluidized bed in HTFT for olefins production [12]. The poor mechanical strength of unsupported catalysts may lead to plugging of the catalyst bed in fixed bed operation or to fouling of downstream equipment in fluidized bed operation. Supported Fe catalysts display enhanced dispersion of the active phase and may withstand the mechanical degradation that threatens unsupported catalysts.
Upon activation, the Fe-oxide precursor is usually converted into a mixture of metallic Fe, Fe-carbides and Fe-oxides and the composition of catalyst depends on many parameters such as the catalyst precursor, catalyst pretreatment and the FT reaction conditions [36]. Despite the complexity of the composition in the working condition, a correlation between Fe-carbide content and FT activity has been widely observed and Fe-carbide formation is believed to be the necessary step to obtain good FT activity [37]. Fe-oxide, on the other hand, is considered to be active for the WGS reaction, which leads to the production of excessive CO2 [38]. In itself, CO2 production represents a loss of valuable carbon product. As some WGS operation is needed in CTL and BTL processes, it can be worthwhile to remove CO2 to reduce the greenhouse gas emissions of the process. A distinct feature of Fe-carbide is that it is air sensitive [39] and readily oxidized in air at room temperature, leading to the formation of Fe-oxide [40]. Thus, activation or carbide formation is preferably done in situ before starting the FT reaction [41]. For research purposes, passivation of the catalyst in a diluted O2 is usually employed [42].
During FT reaction, ε(’)-carbide, χ-Fe5C2, and Θ-Fe3C are commonly observed in Fe-based FT catalysts [43,44]. They are classified as interstitial carbides, because C atoms occupy the interstices of the Fe lattice. According to way C atoms occupy the hexagonally close-packed (hcp) lattice, their structure can be divided into two categories. In ε(´)-carbide, C atoms occupy Fe octahedral interstices, ascribed to octahedral carbides, while the C atoms in Θ-Fe3C and χ-Fe5C2 are situated in trigonal prismatic interstices. The main differences between these three Fe-carbides are listed in Table 2.
Among these three carbides, ε(´)-carbide is the generic term for ε-Fe2C and ε’-Fe2.2C. ε-Fe2C and ε’-Fe2.2C share the same space group and lattice parameter but differ in the chemical environment of Fe atoms [45]. The site occupancy of C in ε-Fe2C and ε’-Fe2.2C is 0.5 and 0.45, respectively. ε(´)-carbide formation is favored at a high carbon chemical potential, which represents the condition of low temperature and high CO partial pressure [36]. However, kinetic factors (lattice deformation, carbon diffusion) can prevent its formation at low temperature. Hence, they are commonly observed in catalysts with relatively small particle and when a support material or chemical promoters are present [46,47]. If temperature exceeds 250 oC, ε(´)-carbide will transform to χ-Fe5C2 [48,49]. χ-Fe5C2 is the most observed carbide phase in the context of FT catalysis. Several works show that χ-Fe5C2 is the main active phase constituent at moderate FT conditions, owing to its relative thermodynamic stability at low carbon chemical potential. When temperature is further increased, χ-Fe5C2 will transform to Θ-Fe3C [50]. The less active Θ-Fe3C can also evolve into more active χ-Fe5C2 [51]. The common feature of carbon-poor Θ-Fe3C is that it can contribute to the buildup of carbonaceous deposits because of its near-metallic nature [36,52]. Excessive carbonaceous deposits like graphite will cause deactivation of the catalyst [53,54].
Despite extensive research, it is still uncertain which phase is the most active in the FT reaction. The intrinsic activity comparison between ε(´)-carbide and χ-Fe5C2 is widely investigated. Usually, a mixture of several Fe-carbides is obtained during reaction, making it impossible to correlate activity with specific phase [36,56]. Chang et al. found that the intrinsic activity of χ-Fe5C2 is higher than ε(´)-carbide through changing the pretreatment condition [55]. However, Chun et al. found that ε(´)-carbide performs better than χ-Fe5C2 by introducing CO2 in the feed, which increased the amount of ε(´)-carbide [57]. Lu et al. also found that the content of ε(´)-carbide is more active than χ-Fe5C2 [58]. Another exploration by Wezendonk et al. pointed out that the weight-normalized activities (FTY) of χ-Fe5C2 and ε(´)-carbide are virtually identical [59]. There also exist controversial conclusions in terms of the product distribution on different Fe-carbide phases in the FT reaction.[36,55,59,60] The observed differences are probably caused by various factors, such as particle size, exposed facet (phase morphology), surface carbon deposition, support effect and the interference of other phases. To overcome these difficulties, surface-sensitive techniques were applied to in situ measure the chemical and structural composition of well-defined catalysts (single-crystal or flat-model catalysts) under operating conditions [61,62]. These systems were mainly focused on studying time-, pressure-, temperature-dependent Fe carburization kinetics. To further establish structure-activity relationship on Fe-carbide in the FT reaction and study underlying mechanism, synthesizing specific pure Fe-carbide and realizing that the catalyst suffers from no phase change or deactivation during FT reaction is of paramount importance.
Some endeavors have been made to synthesize oxide-free Fe-carbides. By using rapid quenched skeletal iron precursor, Xu et al. prepared a catalyst that mainly consists of ε(´)-carbide and it showed excellent FT activity at relatively low temperature [45]. Peng et al. took advantage of Raney-Fe, which is porous and support-free and can be used as a model catalyst, to successfully synthesize phase-pure ε(´)-carbide by tuning carburization conditions. The obtained ε(´)-carbide showed low CO2 selectivity in LTFT reaction, showed in Figure 3 [9]. By stabilizing ε(´)-carbide into graphene layers, it can also catalyze HTFT reaction without transforming to χ-Fe5C2 [63]. Single-phase χ-Fe5C2 was synthesized by Yang et al. using a facile wet-chemical route with the help of Br agent [64]. In their following work, a range of phase-pure metallic Fe and Fe-carbide nanoparticles were utilized in illustrating the FT mechanism [60].
4. Product distribution
The formation of long-chain hydrocarbons is a key part of the FT chemistry involving a polymerization-like events of C1 monomers. The ideal product distribution follows the Anderson-Schulz-Flory (ASF) statistics [26]. The fraction of the carbon number within the hydrocarbon chain containing n carbon atoms with respect to total carbon numbers is expressed in Equation 3:
where is the chain-growth probability. Assuming it is independent of chain length, it can be expressed as the rate of chain propagation (rp) over the rate of chain termination (rt) plus chain propagation (rp), shown in Equation 4.
Depending on the catalyst and operation conditions, value varies, which in turn results in different product distribution. The theoretical product distribution as a function of chain-growth probability is shown in Figure 4. The lower temperature regime at which the selectivity towards higher hydrocarbons is favorable where mainly Co-based catalysts are used for producing wax as precursor to high-quality transportation fuels. On the other hand, at the high temperatures used in the HTFT process, C5-20 hydrocarbons or lower olefins (C2-4) are the major products and Fe is most often used as a catalyst [12]. Notably, the undesired CH4 selectivity monotonously decreases with increasing . However, there is some deviation from the ASF distribution with regards to CH4 selectivity. The higher CH4 selectivity than predicted by the ASF distribution can have different origins such as a thermodynamic preference to form CH4, the kinetic preference for CHx hydrogenation compared to C-C coupling or the involvement of specific surface sites that are selective to CH4 [65,66,67]. On the other hand, C2 selectivity is often lower than predicted by ASF, which is explained by the strong binding of ethylene with the catalyst surface [68]. Moreover, it has been observed that the chain-growth probability of C7+ products is higher than that of C1-7 ones. The chain-length dependent chain-growth probability has been discussed [69]. It was revealed that compared to higher LAO, lower LAO is more easily to be reinserted into growing chains, leading to the deviation of ASF distribution. Hydrogenolysis, on the other hand, could shorten produced long hydrocarbons by successive demethylation [70]. A high reversibility of chain growth on Co catalysts has been described by Chen et al. [71].
.
5. Reaction mechanism
FT reaction has been widely studied for almost 100 years, but there is still debate with regard to the reaction mechanism. Generally, it can be divided into a sequence of elementary reaction steps, characteristic for a polymerization reaction (i) chain initiation, (ii) chain propagation, and (iii) chain termination.
Chain initiation involves CO dissociative adsorption. Two types of mechanisms were proposed. The adsorbed CO on metal sites can directly dissociate into C and O atoms, referred to as direct CO dissociation. The C atoms will participate following chain-growth reactions as preliminary intermediates, while O atoms will be removed as H2O or CO2 as FT by-products [72]. In addition to direct CO dissociation, hydrogen-assisted CO dissociation pathways were also proposed. Such pathways can provide HCO or COH intermediate originating from the hydrogenation of adsorbed CO before the C-O bond breaks [73]. It is likely that the CO dissociation pathway depends on the surface coverage, the lateral interactions of intermediates and the relative occurrence of different sites on the catalyst surface [74]. On metallic Fe, it is believed that direct CO dissociation is the dominant pathway because of its low CO dissociation barrier [75]. H-assisted CO dissociation mainly proceeds on surface with a large occupancy of sub-surface C atoms while direct CO dissociation takes place on stepped sites of χ-Fe5C2 [43]. On the most stable (510) phase of χ-Fe5C2, the direct CO dissociation is suggested to be the preferred pathway, while the H-assisted CO dissociation mainly take place on other phases [76]. Therefore, it is reasonable to be deduced that both pathways could contribute to the cleavage of CO molecules on Fe-carbide.
During chain propagation, carbide mechanism and CO insertion mechanism are differentiated in terms of whether the chain-growth monomer is CHx or CO. The carbide mechanism, first proposed by Fischer and Tropsch, entails CHx species inserted into the growing hydrocarbon chain [1]. Although Kummer et al. later proved that chemisorbed C is the dominant reactive intermediate rather than bulk carbide,[77] the mechanism is still referred to as the carbide mechanism. However, the formation of oxygenates during the FT reaction is not in keeping with this mechanism. Accordingly, Pichler et al. proposed CO insertion mechanism, in which an HCO intermediate or adsorbed CO can be inserted into the hydrocarbons chain [78]. In this mechanism, the C-O bond scission occurs after the C-C coupling, whereas for the carbide mechanism it is believed that the cleavage of C-O bond precedes the C-C coupling [79]. It has been reported that CO insertion mechanism is not the dominant pathway to the reaction since there is no appreciable molecularly adsorbed CO on Fe surface at 700 mbar [62]. The absence of chemisorbed CO on Fe-carbide was also observable when FT reaction running at relatively low pressure [80]. On the other hand, the high pressure needed for FT reactivity and the resulting high CO coverage indicate the existence of CO insertion mechanism [81]. The rate between oxygenate removal versus C-O bond cleavage determines whether chain growth proceeds via CO or CHx insertion mechanism [82].
In the view of carbide mechanism, chain termination of the adsorbed alkyl group can occur in two different ways. It can either be hydrogenated to form n-paraffin or undergo β-H elimination to form α-olefin [83]. Govender et al. proposed that olefins and paraffins grow on different active sites and do not share the same surface intermediates [84]. The reversibility of β-H elimination could cause the re-insertion of the α-olefin into a different growing chain, thereby deviating from the theoretical product distribution [85]. In addition, re-adsorption of the α-olefins could lead to the formation of more paraffins and isomers by secondary hydrogenation and isomerization reactions, respectively [70]. In the carbide mechanism, termination by CO insertion into a growing hydrocarbon chain is claimed to be the source of oxygenates [82]. In the CO insertion mechanism, the formation of a growing hydrocarbon chain is followed by the removal of the O atom via a hydrogenation step [86]. The formation of alcohols and aldehydes are terminated by hydrogenation and β-H elimination of the CxHyOads species, respectively. Acids are formed via the insertion of CO2 into the growing chains.
6. Fe carburization kinetics and C hydrogenation mechanisms on Fe-carbide
Usually, Fe-carbide is regarded as the active phase for FT reaction. The formation of Fe-carbide is accompanied by hydrocarbon production [87]. It suggests that the C atoms from CO dissociation can either go into the bulk Fe structure or be hydrogenated to CHx, which is either hydrogenated to CH4 or involved into chain-growth reaction. Tuning carburization behaviors of Fe-based catalysts is indispensable to realize optimized FT performances. A higher degree of carburization causes the Fe atoms to be in a more electron deficient state, which enhances the σ donation from CO to the surface and weakens the π back-donation for CO adsorption [88]. Surfaces with more reduced Fe atoms exhibit a lower activation energy for CO dissociation and bind adsorbed C stronger, which is unfavorable for CH4 formation [89,90,91]. Carburization rate is highly dependent on reaction conditions and the composition of the catalyst [56,92]. Ribeiro et al. found that adding alkali promotor could shorten the carburization time span, because it provided higher CO surface coverage for higher carburization rate [93]. The adsorption of Cl- weakened the bonding between Fe and C atoms, thus inhibiting the diffusion of C atoms into the Fe structure [94]. The strong interaction between highly dispersed Fe-oxide and an oxidic support impedes the conversion of Fe-oxide into Fe-carbide [95]. Zhou et al. found that Fe carburization rate was faster when Fe is supported on a silicon substrate than on a silica substrate, because the former could provide an alternative way for O-removal [96]. Butt et al. found that increasing H2 partial pressure promoted carburization of metallic Fe [97]. Similar results were observed by Niu et al. that H2/CO has a higher carburization capability than CO alone on α-Fe [56]. It was further revealed that adding H2 could promote the removal of O atoms, which could free vacancies for CO dissociation. The resulting high C coverage is kinetically favorable for the formation of ε(´)-carbide [75]. The detailed mechanism is showed in Figure 5. The rate of carburization of Fe-oxide is typically controlled by O diffusion from the oxide core to the surface [98]. The pretreatment environment can influence the initial catalytic activity. It was observed that a precipitated Fe-based catalyst pretreated in H2 was more active and reached steady state more rapidly than the corresponding catalyst treated in CO [99]. The initial and steady-state catalyst activities were inextricably correlated with the carburization rates to form active surface carbide nodules [100].
Lohitharn et al. pointed out that the FT activity on Fe-carbide is dependent on the number of active intermediates [101]. Temperature-programmed hydrogenation (TPH) was used to establish the correlation between structural type and reactivity of C species in the Fe-based samples [102]. In order of decreasing reactivity, (i) adsorbed atomic C species and surface carbide, (ii) polymeric, amorphous C species (iii) bulk C, and (iv) graphitic C are distinguished. Xu et al. revealed that the initial catalytic activity of FePtK/SiO2 was positively correlated with the amount of adsorbed atomic C species [103]. An atomic C species could also convert to polymeric carbonaceous species on the surface. Herranz et al. suggested that the polymeric surface carbonaceous species was more closely related to the FT activity than atomic C species [51]. It was revealed that polymeric species, representing C2+ hydrocarbons intermediates has a lower surface coverage than the CH4 intermediates [104]. Ding et al. found that the combination of atomic C species and polymeric surface carbonaceous species resulted in graphitic-type C species, restricting active sites for the FT activity [105]. Reactive adsorbed C, graphitic C, and carbidic C in the bulk of the Fe-carbide could also be distinguished by electron energy loss spectroscopy (EELS) [106].
Govender et al. found two active pools of C on the surface of Fe-based catalysts to be responsible for the formation of CH4. The less active pool occupied the majority of total CHx coverage, while the more active pool was scarce on the surface. The C-C coupling reaction involved both C pools [107]. Graf et al. also suggested that multiple pools existed on Fe-based catalyst for CH4 formation and the addition of K would block the fast channel [108]. It was suggested that the slow pool is from C atoms that diffuse from the interior of Fe-carbide [109]. The involvement of lattice C can be described by a Mars-van Krevelen (MvK) mechanism, which has initially been described for oxidation catalysis by Mars and Van Krevelen [110]. This mechanism was also used for describing the reaction cycle to CH4 on the carbon-terminated Fe5C2 (100) surface [111]. The hypothesis entails the threefold hydrogenation of a lattice carbide, followed by the creation of a surface vacancy (fourfold site) where a CO molecule from the gas phase can dissociate. The reaction pathways are showed in Figure 6.
Kummer et al. first partially carburized a catalyst by 12CO and then fully by radioactive 14CO, leading to a 14C-enirched surface. By carrying out the FT reaction in 12CO/H2, it was found that the hydrocarbons produced at 260 oC contained only 10% 14C as established by measuring their radioactivity. It indicates that the pathway following an MvK mechanism has a minor contribution to hydrocarbons formation [77]. Ordomsky et al. later found that C atoms in Fe-carbide are involved in the chain-growth initiation events of the FT reaction via isotope labeling experiments [112]. The slow evolution of C2+ products in steady-state isotopic transient kinetic analysis (SSITKA) also suggests that less-reactive C pool might be involved in chain growth [84]. The Langmuir-Hinshelwood (L-H) mechanism is currently favored in describing the FT reaction on Co and Ru catalysts [113,114], where the metal-carbon bond is not as strong as on Fe [115]. Catalytic reactions predominantly occur between adsorbed species. Typically, the intrinsic activity of Co and Ru is higher than that of Fe-carbide,[116] suggesting that reaction pathways that follow the L-H mechanism are faster than those involving MvK steps. The presence of two reactive species Cα,ads and Cβ,ads were also proposed on Co-based catalysts by Van Dijk et al., however, with comparable surface coverage and rate constants for CH4 formation [117]. Strikingly, the reaction pathways expand several orders in rate for CH4 formation on Fe-carbide. The fast reaction paths dominate the CH4 formation rate but run over only about 10% of the catalyst surface via an L-H mechanism. The slowest pathway contributes to CH4 formation involving the extraction of lattice C following an MvK mechanism. The comparison of these two pathways on Fe-carbide is illustrated in Figure 7 [80].
7. Promoters
In the preparation of Fe-based catalysts, various promoters are generally required to reasonably modulate the catalyst properties. Depending on the effect of promoters on the FT reaction, they are usually divided into electronic promoters and structural promoters. Electronic-type promoters usually contain alkali metals, transition metals and rare earth metals. Structural promoters generally refer to inorganic oxides that are difficult to be reduced, such as SiO2 or Al2O3.
K, Na, Mn, Cu and S are the most reported chemical promoters for Fe-based catalysts. Among them, K, and Na could suppress CH4 selectivity and enhance olefins selectivity because they can donate electrons to Fe to inhibit hydrogenation of C-containing intermediates and hamper readsorption of olefins [93,118]. K is also known to facilitate carburization during catalyst activation due to increased CO adsorption [93]. S might lower H coverage on Fe-carbide thereby suppressing CH4 formation [17]. Cu could promote Fe-oxide reduction, which is favorable for carburization and shorten the pretreatment period [119]. Mn promoted catalyst also enhances olefins selectivity and suppresses CH4 selectivity [120]. Mn was also found to promote the dispersion of Fe and increase the active surface area [121]. Solid solution compound is formed when Mn was added, similar to the role of Zn、La、Zr、V、Cr or Ce. The mixed oxide phase tends to inhibit crystallite growth and thus, smaller Fe particles are retained. Lohitharn et al. reported that the introduction of Mn had no influence on the intrinsic activity of Fe rather than increasing the number of active sites with SSITKA [122]. The same holds true for K-promoted catalyst [123]. Jensen et al. found that Mn-containing catalyst was 2 times as active as Fe on a basis of specific rate, whereas it was less than half as active on a basis of weight [124]. They proposed that MnO can electronically or chemically alter the nature of Fe as a electron donor. Liu et al. however found that the adding of Mn will lead to lower degree of carburization, accordingly, relatively lower activity [125]. Ribeiro et al. also found that Mn hinders Fe carburization and the carburized catalyst displays higher Fe3O4 content than the catalyst without Mn [126]. Strikingly, increasing Mn content led to higher CH4 and lower light olefins selectivity. They attributed this trend to higher WGS rates observed on the FeMn catalysts because of high oxidation degree. Zhang et al. suggested that Mn could also migrate to the catalyst surface and the enrichment of MnO on the catalyst surface would retard Fe reduction [127].
SiO2 and Al2O3 are used as structural promoters to increase surface area of the catalyst and enhance the dispersion of the active site. On the other hand, the strong interaction of Si or Al hydroxyls with Fe could inhibit Fe reduction or carburization [128].
8. Deactivation of Fe-based catalysts
As Fe-based catalysts are usually used at high temperature for the FT reaction, they will inevitably face deactivation because of catalyst sintering, particle agglomeration and attrition [129]. To avoid physical deterioration, a support is needed to disperse and stabilize the active phase [130]. Strong support-metal interactions can inhibit sintering of the catalyst [131]. Some structural promoters are also added to enhance such interactions [132]. The change in the chemical state of the catalyst also poses a negative effect on the catalytic activity. Oxidation of metallic Fe and/or Fe-carbide phases is believed to be one of the factors for catalyst deactivation, especially at high CO conversion [133]. The formation of Fe-oxide will cause higher CO2 selectivity owing to WGS reaction [9]. Apart from oxidation, formation of coke will also lead to a decrease in FT activity. The accumulation of graphite-like carbonaceous species on the catalyst surfaces will restrict the availability of active sites and block the pores [36]. It may also lead to undesired side reactions [66]. A slight increase in CH4 selectivity during deactivation was observed [134]. It was revealed that the deposition of C during the ongoing FT reaction has a higher tendency to cover fast sites, which is mainly responsible for long-chain hydrocarbons, than slow sites, which mainly produces CH4. Therefore, a decreases in FT activity is accompanied with an increases in CH4 selectivity [135]. The influence of C deposits on Fe-carbide for the FT reaction is showed in Figure 8.
Eliason et al. proposed two deactivation paths occurring in parallel and/or coupled. First, the transformation of atomic C species to amorphous polymeric C species followed by the formation of graphitic C. Second, the transformation of high activity χ-Fe5C2 to less active ε(´)-carbide [103]. Jung et al. found that the transformation of ε(´)-carbide to χ-Fe5C2 is accompanied by deactivation, because the decomposition of metastable ε(´)-carbide can lead to a buildup of coke [47]. The detached C from ε(´)-carbide seems to serve as the nucleation site for the Boudouard reaction (2CO → C + CO2) [136]. The transformation between reactive C species and graphitic C was reported to be reversible [66,137]. Increasing H coverage by increasing H2 partial pressure or decreasing CO partial pressure will constrain the formation of amorphous/graphitic C on Fe-carbide [138]. Only elevated total pressure in combination with a high H2/CO ratio was found to provide a sufficient H coverage to restrict C deposition. When the H2/CO ratio is as low as unity, increasing the total pressure can increase C deposition rate [139]. Rising temperature can retard C deposition thermodynamically, but accelerate carburization kinetically [91]. Increasing H2O vapor content could also inhibit C deposition by inhibiting the formation of Boudouard-type carbonaceous species [140,141]. However, local high H2O vapor partial pressure formed by the WGS reaction may also irreversibly oxidize the catalyst, leading to the deactivation of the catalyst [142]. Smaller particles showed a lower tendency to build up inactive surface carbonaceous species on the catalyst surface [143]. The introduction of promoters (Na + S) could result in significant C deposition by facilitating Fe carburization over the initial hours of the FT reaction [109]. However, the use of S, in the absence of Na, could increase the resistance against C deposition. The support effect on carbonaceous deposits formation was also studied by Galuszka et al. [53]. It suggested that strong metal-support interaction might counter deactivation by maintaining a balance between active and inactive C species.
9. Structure-activity relationships
As in all heterogeneous catalysts in which nanoparticles are used, there is a profound interest in establishing structure-activity relationship for catalyst optimization. For Co- and Ru-based catalysts, the turnover rate of the CO dissociation is dependent on particle size [144,145]. It has been observed that the turnover rate increases with particle size and shows maximum at intermediate size. This structure-sensitive phenomenon is usually interpreted in terms of geometric effects of the surface metal atoms. It is predicated that below a particle size of 6 nm the density of step-edge sites decreases. The metal atoms on step-edge sites are more reactive to CO dissociation owing to lower coordination number. This can be understood in terms of an electronic effect, because the decrease in coordination number of metal atoms will back-donate electrons to the anti-bonding orbital of CO, thus lowering the activation energy for CO dissociation. Geometrically, the CO molecule can align with step-edge sites without bending, which is more favorable for CO dissociation than on planar surface. CO dissociation is usually regarded as the rate-limiting step for smaller Co particle because low-coordination sites at corners and edges are poisoned by CO [146]. Hence, the enhanced CO dissociation ability on larger Co particle size will subsequently increase the overall FT activity. Generally, larger Co particles favor chain growth, producing heavier hydrocarbons. However, a reversed phenomenon was reported on small Co particle size confined in mesoporous SiO2 supports [147].
For Fe-carbides, controversy also exists in terms of particle size effect on FT activity. Some publications presented that smaller particles feature lower TOF and higher CH4 selectivity than large particles [148,149]. Torres Galvis et al. observed an opposite particle size effect. In the initial state, when the surfaces are relatively clean, smaller particle size presents higher surface-specific activity because more corner and edge atoms reside on small promoted Fe-carbide particles, which are beneficial for CH4 formation [118]. The formation of C2+ hydrocarbons is independent of particle size, whereas the TOF for CH4 formation decreases when larger particle is used. However, small particle suffers more from deactivation because of the loss of active surface area from sintering or C deposition. For unpromoted Fe-carbide, the apparent TOF increases with decreasing particle size, albeit with no difference in terms of selectivity. The fact that not all published work finds the same particle size effect for Fe may be due to the sensitivity of carbon over-layers built up during activation. If the interference of C deposits could be excluded, compared to Co and Ru, the opposite structure-activity relationship on Fe-carbide is probably caused by the intrinsic nature of chemical bonding of metal carbide. On the Fe catalysts, the binding strength of C atom is stronger than Co or Ru. The weaker Fe-C bond corresponds to higher activity on Fe-carbide because it requires less energy needed for C removal through hydrogenation [68,115]. It indicates that the removal of C atoms by hydrogenation is a rate-limiting step for Fe-carbide rather than CO dissociation [135,141].
Theoretical work pointed out that H adsorption also plays an important role in determining product selectivity [150]. Xie et al. observed that CH4 formation occurs equally fast on edges and terraces sites for unprompted Fe-based catalysts, but it slows down on the terraces sites of promoted catalysts. There is a linear relationship between apparent TOF and CHx coverage, the latter is more abundant on small particle size. In addition, an increase in particle size leads to an increase in H surface residence time and a decrease of H coverage, indicating hydrogenation is suppressed at large particle size [109].
10. Summary and outlook
FT synthesis is an increasingly important approach for producing sulfur-free and aromatic-free liquid fuels and valuable chemicals via synthesis gas, generated from coal, natural gas, or biomass. Fe and Co are the only viable transition metals used in commercial FT catalysts. Although Fe-based catalysts are not as active as Co-based catalysts, they show a broader range of operation windows (pressure, temperature, and feedstock composition), and more importantly, they are much cheaper than Co. Fe-carbides are the active phases of Fe-based catalysts in the FT reaction while Fe-oxide is responsible for the formation of CO2 via WGS reaction. Due to the complex changes of Fe phases during pretreatment and FT reaction, the kinetic and mechanistic understandings of Fe carburization and FT reaction on Fe-carbide are of paramount importance in guiding the design of well-performed catalysts. Apart from catalyst modulation, the optimization of reaction conditions also plays a critical role in realizing excellent FT performance. Depending on the reaction conditions, the chain-growth probability () changes, which in turn results in a different product distribution. The reaction mechanism of the FT reaction has been the subject of many research projects since the discovery of the reaction itself. The involvement of chain initiation, chain propagation and chain termination renders FT reaction network as one of the most sophisticated yet intriguing system to be studied. On top of that, the general reaction mechanism and elementary reaction steps usually suitable to describe FT reaction on Co or Ru-based catalysts do not fully hold true for Fe-based catalysts. The bond strength between C and Fe is much higher than the other mentioned transition metals. Therefore, the dissociated C further reacts with Fe to form Fe-carbide, in which lattice C can take part in FT reaction via an MvK mechanism. It was hypothesized that high-active sites are related to L-H type mechanism, whereas the low-active sites follow MvK like kinetics. Due to the fact that Fe-carbide surface is largely terminated by lattice C, it can offer a plausible explanation for why the intrinsic activity of Fe-carbide is lower than Co. C deposition is an integral part of the Fe catalyzed FT reaction, making it difficult to correlate the intrinsic activity of a specific Fe-carbide to its FT performance. Another interfering factor is the existence of Fe-oxide, which can produce extra CO2. Running FT reaction at high pressure appears to reverse the deactivation caused by the buildup of C deposits at low pressure and realizes a stable FT activity. However, the high-pressure synthesis gas could induce the surface oxidization of Fe. To some extent, the build-up of C deposits can protect surface Fe from oxidization. The particle size effect of Fe-carbide on the FT reaction remains to be further explored by excluding aspects such as phase evolution of Fe-carbide, the presence of Fe-oxide and the influence of C deposits during reaction.
To gain more insight on the mechanistic information of FT reaction on Fe-based catalysts, synthesizing phase-pure Fe-carbide and preserving their physical and chemical integrity during reaction is highly demanded. Therefore, the employment of model catalyst, such as Raney-Fe, which features higher reducibility compared to conventional industrial catalysts, enables studying Fe carburization and FT mechanism in the absence of perturbing oxidic phases. It could also be interesting to compare the performance of phase-pure Fe-carbide with a catalyst that is a mixture of Fe-carbide and Fe-oxide. The direct measurement of the contribution of lattice C in FT reaction is still a challenge, especially when considering chain growth. More carefully designed isotopic transient experiments are needed to address this issue. The accurate interpretation of the occurrence of multiple L-H reaction pathways is not yet possible and likely requires integration of explicit DFT calculations, which suggests CO activation and CH4 formation rates are strongly dependent on the exposed crystal facets and active site geometry. Moreover, the correlation between each active site on Fe-carbide and the corresponding FT activity needs to be studied in more detail by using advanced surface science techniques, if required, on single-crystal or flat-model catalysts. The evolution of C and O intermediates on atomically defined carbide surfaces should be tracked by time-resolved spectroscopy to establish structure-activity relationships. Another approach to systematically studying the facet-dependent FT activity and the interaction of the Fe-C bond on different carbides is the inclusion of DFT calculations. The origin of CO2 is also worth more investigations, considering its detrimental effect on both carbon efficiency and climate change.
Funding
This work was supported by the National Key Research and Development Program of China (project no. 2022YFB4101400).
Conflicts of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- F. Fischer, H. F. Fischer, H. Tropsch, The Synthesis of Petroleum at Atmospheric Pressures from Gasification Products of Coal, Brennst. Chem. 7 (1926) 97-104.
- F. Fischer, H. F. Fischer, H. Tropsch, Development of Noval Catalysts for Fischer-Tropsch Synthesis, Brennst. Chem. 4 (1923) 276-285.
- Office of Fossil Energy and Carbon Management, 2000, https://www.energy.gov/fecm/early-days-coal-research.
- Zhang, Q.; Kang, J.; Wang, Y. Development of Novel Catalysts for Fischer–Tropsch Synthesis: Tuning the Product Selectivity. ChemCatChem 2010, 2, 1030–1058. [Google Scholar] [CrossRef]
- E. de Smit, B. M. E. de Smit, B. M. Weckhuysen, The Renaissance of Iron-based Fischer-Tropsch Synthesis: on the Multifaceted Catalyst Deactivation Behaviour, Chem. Soc. Rev., 37 (2008) 2758-2781.
- D. L. King, A. D. L. King, A. de Klerk, Overview of Feed-to-Liquid (XTL) Conversion, ACS Symp. Ser., 1084 (2011) 1-24.
- Hao, X.; Dong, G.; Yang, Y.; Xu, Y.; Li, Y. Coal to Liquid (CTL): Commercialization Prospects in China. Chem. Eng. Technol. 2007, 30, 1157–1165. [Google Scholar] [CrossRef]
- Y. Cao, Z. Y. Cao, Z. Gao, J. Jin, H. Zhou, M. Cohron, H. Zhao, H. Liu, W. Pan, Synthesis Gas Production with an Adjustable H2/CO Ratio through the Coal Gasification Process: Effects of Coal Ranks And Methane Addition, Energy Fuels, 22 (2008) 1720-1730.
- P. Wang, W. P. Wang, W. Chen, F. K. Chiang, A. I. Dugulan, Y. Song, R. Pestman, K. Zhang, J. Yao, B. Feng, P. Miao, W. Xu, E. J. M. Hensen, Synthesis of Stable and Low-CO2 Selective Iron carbide Fischer-Tropsch Catalysts, Sci. Adv., 4 (2018) eaau2947.
- M. E. Dry, The Fischer-Tropsch process: 1950-2000, Catal. Today, 71 (2002) 227-241.
- Dry, M.E.; Hoogendoorn, J.C. Technology of the Fischer-Tropsch Process. Catal. Rev. 1981, 23, 265–278. [Google Scholar] [CrossRef]
- Steynberg, A.; Espinoza, R.; Jager, B.; Vosloo, A. High temperature Fischer–Tropsch synthesis in commercial practice. Appl. Catal. A: Gen. 1999, 186, 41–54. [Google Scholar] [CrossRef]
- M. E. Dry, Practical and Theoretical Aspects of the Ccatalytic Fischer-Tropsch Process, Appl. Catal. A: Gen., 138 (1996) 319-344.
- Karre, A.V.; Kababji, A.; Kugler, E.L.; Dadyburjor, D.B. Effect of addition of zeolite to iron-based activated-carbon-supported catalyst for Fischer–Tropsch synthesis in separate beds and mixed beds. Catal. Today 2012, 198, 280–288. [Google Scholar] [CrossRef]
- Martínez, A.; López, C. The influence of ZSM-5 zeolite composition and crystal size on the in situ conversion of Fischer–Tropsch products over hybrid catalysts. Appl. Catal. A: Gen. 2005, 294, 251–259. [Google Scholar] [CrossRef]
- Tobisch, S.; Ziegler, T. Catalytic Oligomerization of Ethylene to Higher Linear α-Olefins Promoted by Cationic Group 4 Cyclopentadienyl-Arene Active Catalysts: Toward the Computational Design of Zirconium- and Hafnium-Based Ethylene Trimerization Catalysts. Organometallics 2004, 24, 256–265. [Google Scholar] [CrossRef]
- Galvis, H.M.T.; Bitter, J.H.; Khare, C.B.; Ruitenbeek, M.; Dugulan, A.I.; de Jong, K.P. Supported Iron Nanoparticles as Catalysts for Sustainable Production of Lower Olefins. Science 2012, 335, 835–838. [Google Scholar] [CrossRef]
- Y. Xu, X. Y. Xu, X. Li, J. Gao, J. Wang, G. Ma, X. Wen, Y. Yang, Y. Li, M. Ding, A Hydrophobic FeMn@Si Catalyst Increases Olefins from Syngas by Suppressing C1 by-products, Science, 371 (2021) 610-613.
- J. Xie, P. P. J. Xie, P. P. Palalanen, T. W. van Deelen, B. M. Wechhuysen, M. J. Louwerse, K. P. de Jong, Promoted Cobalt Metal Catalysts Suitable for the Production of Lower Olefins from Natural Gas, Nat. Commun., 10 (2019) 167.
- Jiao, F.; Li, J.; Pan, X.; Xiao, J.; Li, H.; Ma, H.; Wei, M.; Pan, Y.; Zhou, Z.; Li, M.; et al. Selective conversion of syngas to light olefins. Science 2016, 351, 1065–1068. [Google Scholar] [CrossRef]
- Cheng, K.; Gu, B.; Liu, X.; Kang, J.; Zhang, Q.; Wang, Y. Direct and Highly Selective Conversion of Synthesis Gas into Lower Olefins: Design of a Bifunctional Catalyst Combining Methanol Synthesis and Carbon–Carbon Coupling. Angew. Chem. Int. Ed. 2016, 55, 4725–4728. [Google Scholar] [CrossRef]
- Belov, G.P. Tetramerization of ethylene to octene-1 (a review). Pet. Chem. 2012, 52, 139–154. [Google Scholar] [CrossRef]
- Filot, I.A.W.; van Santen, R.A.; Hensen, E.J.M. Quantum chemistry of the Fischer–Tropsch reaction catalysed by a stepped ruthenium surface. Catal. Sci. Technol. 2014, 4, 3129–3140. [Google Scholar] [CrossRef]
- Zhao, Z.-J.; Liu, S.; Zha, S.; Cheng, D.; Studt, F.; Henkelman, G.; Gong, J. Theory-guided design of catalytic materials using scaling relationships and reactivity descriptors. Nat. Rev. Mater. 2019, 4, 792–804. [Google Scholar] [CrossRef]
- J. A. van Bokhoven, J. T. J. A. van Bokhoven, J. T. Miller, d Electron Density and Reactivity of the d Band as a Function of Particle Size in Supported Gold Catalysts, J. Phys. Chem. C, 111 (2007) 9245-9249.
- Schulz, H. Short history and present trends of Fischer–Tropsch synthesis. Appl. Catal. A Gen. 1999, 186, 3–12. [Google Scholar] [CrossRef]
- W. Ma, A. K. W. Ma, A. K. Dalai, Effects of Structure and Particle Size of Iron, Cobalt and Ruthenium Catalysts on Fischer-Tropsch Synthesis, Reactions, 2 (2021) 62-77.
- G. P. van der Laan, A. A. C. M. G. P. van der Laan, A. A. C. M. Beenackers, Kinetics and Selectivity of the Fischer-Tropsch Synthesis: A Literature Review, Catal. Rev., 41 (1999) 255-318.
- Khodakov, A.Y.; Chu, W.; Fongarland, P. Advances in the Development of Novel Cobalt Fischer−Tropsch Catalysts for Synthesis of Long-Chain Hydrocarbons and Clean Fuels. Chem. Rev. 2007, 107, 1692–1744. [Google Scholar] [CrossRef] [PubMed]
- H. Schulz, Comparing Fischer-Tropsch Synthesis on Iron-and Cobalt Catalysts: The Dynamics of Structure and Function, Fischer-Tropsch Sythesis, Catalysts and Catalysis, 163 (2007) 177-199.
- C. Yang, B. C. Yang, B. Zhao, R. Gao, S. Yao, P. Zhai, S. Li, J. Yu, Y. Hou, D. Ma, Construction of Synergistic Fe5C2/Co Heterostructured Nanoparticles as an Enhanced Low Temperature Fischer-Tropsch Synthesis Catalyst, ACS Catal., 7 (2017) 5661-5667.
- S. M. Ismail, M. S. M. Ismail, M. Casavola, B. Liu, A. Gloter, T. W. van Deelen, M. Versluijs, J. D. Meeldijk, O. Stephan, ́ K. P. de Jong, F. M. F. de Groot, Atomic-Scale Investigation of the Structural and Electronic Properties of Cobalt-Iron Bimetallic Fischer-Tropsch Catalysts, ACS Catal., 9 (2019) 7998-8011.
- V. R. Calderone, N. R. V. R. Calderone, N. R. Shiju, D. C. Ferre´, G. Rothenberg, Bimetallic catalysts for the Fischer-Tropsch reaction, Green Chem., 13 (2011) 1950-1959.
- Zhang, J.; Abbas, M.; Chen, J. The evolution of Fe phases of a fused iron catalyst during reduction and Fischer–Tropsch synthesis. Catal. Sci. Technol. 2017, 7, 3626–3636. [Google Scholar] [CrossRef]
- Espinoza, R.; Steynberg, A.; Jager, B.; Vosloo, A. Low temperature Fischer–Tropsch synthesis from a Sasol perspective. Appl. Catal. A: Gen. 1999, 186, 13–26. [Google Scholar] [CrossRef]
- E. de Smit, F. E. de Smit, F. Cinquini, A. M. Beale, O. V. Safonova, W. van Beek, P. Sautet, B. M. Weckhuysen, Stability and Reactivity of ϵ-χ-θ Iron Carbide Catalyst Phases in Fischer-Tropsch Synthesis: Controlling μC, J. Am. Chem. Soc., 132 (2010) 14928-14941.
- J. W. Niemantsverdriet, A. M. J. W. Niemantsverdriet, A. M. van der Kraan, Behavior of Metallic Iron Catalysts during Fischer-Tropsch Synthesis Studied with Mössbauer Spectroscopy, X-ray Diffraction, Carbon Content Determination, and Reaction Kinetic Measurements, J. Phys. Chem. B, 84 (1980) 3363-3370.
- Chen, W.; Fan, Z.; Pan, X.; Bao, X. Effect of Confinement in Carbon Nanotubes on the Activity of Fischer−Tropsch Iron Catalyst. J. Am. Chem. Soc. 2008, 130, 9414–9419. [Google Scholar] [CrossRef]
- Janbroers, S.; Louwen, J.; Zandbergen, H.; Kooyman, P. Insights into the nature of iron-based Fischer–Tropsch catalysts from quasi in situ TEM-EELS and XRD. J. Catal. 2009, 268, 235–242. [Google Scholar] [CrossRef]
- D. M. Shroff, A. K. D. M. Shroff, A. K. Datye, The Importance of Passivation in the Study of Iron Fischer-Tropsch Catalysts, Catal. Lett., 37 (1996) 101-106.
- Moyer, M.M.; Karakaya, C.; Kee, R.J.; Trewyn, B.G. In Situ Formation of Metal Carbide Catalysts. ChemCatChem 2017, 9, 3090–3101. [Google Scholar] [CrossRef]
- Y. Zhang, N. Y. Zhang, N. Sirimanothan, R. J. O’Brien, H. H. Hamdeh, B. H. Davis, Study of Deactivation of Iron-Based Fischer-Tropsch Synthesis Catalysts, Stud. Surf. Sci. Catal.,139 (2001) 125-132.
- R. J. P. Broos, B. R. J. P. Broos, B. Zijlstra, I. A. W. Filot, E. J. M. Hensen, Quantum-Chemical DFT Study of Direct and H- and C-Assisted CO Dissociation on the χ-Fe5C2 Hägg Carbide, J. Phys. Chem. C, 122 (2018) 9929-9938.
- Liu, X.-W.; Cao, Z.; Zhao, S.; Gao, R.; Meng, Y.; Zhu, J.-X.; Rogers, C.; Huo, C.-F.; Yang, Y.; Li, Y.-W.; et al. Iron Carbides in Fischer–Tropsch Synthesis: Theoretical and Experimental Understanding in Epsilon-Iron Carbide Phase Assignment. J. Phys. Chem. C 2017, 121, 21390–21396. [Google Scholar] [CrossRef]
- K. Xu, B. K. Xu, B. Sun, J. Lin, W. Wen, Y. Pei, S. Yan, M. Qiao, X. Zhang, B. Zong, ε-Iron Carbide as a Low-Temperature Fischer-Tropsch Synthesis Catalyst, Nat. Commun., 5 (2014) 5783.
- Bukur, D.; Okabe, K.; Rosynek, M.; Li, C.; Wang, D.; Rao, K.; Huffman, G. Activation Studies with a Precipitated Iron Catalyst for Fischer-Tropsch Synthesis: I. Characterization Studies. J. Catal. 1995, 155, 353–365. [Google Scholar] [CrossRef]
- Jung, H.; Thomson, W.J. Dynamic X-ray diffraction study of an unsupported iron catalyst in Fischer-Tropsch synthesis. J. Catal. 1992, 134, 654–667. [Google Scholar] [CrossRef]
- Nagakura, S. Study of Metallic Carbides by Electron Diffraction Part III. Iron Carbides. J. Phys. Soc. Jpn. 1959, 14, 186–195. [Google Scholar] [CrossRef]
- Wezendonk, T.A.; Santos, V.P.; Nasalevich, M.A.; Warringa, Q.S.; Dugulan, A.I.; Chojecki, A.; Koeken, A.C.J.; Ruitenbeek, M.; Meima, G.R.; Islam, H.-U.; et al. Elucidating the Nature of Fe Species during Pyrolysis of the Fe-BTC MOF into Highly Active and Stable Fischer–Tropsch Catalysts. ACS Catal. 2016, 6, 3236–3247. [Google Scholar] [CrossRef]
- Königer, A.; Hammerl, C.; Zeitler, M.; Rauschenbach, B. Formation of metastable iron carbide phases after high-fluence carbon ion implantation into iron at low temperatures. Phys. Rev. B 1997, 55, 8143–8147. [Google Scholar] [CrossRef]
- Herranz, T.; Rojas, S.; Perezalonso, F.; Ojeda, M.; Terreros, P.; Fierro, J.L.G. Genesis of iron carbides and their role in the synthesis of hydrocarbons from synthesis gas. J. Catal. 2006, 243, 199–211. [Google Scholar] [CrossRef]
- de Smit, E.; Beale, A.M.; Nikitenko, S.; Weckhuysen, B.M. Local and long range order in promoted iron-based Fischer–Tropsch catalysts: A combined in situ X-ray absorption spectroscopy/wide angle X-ray scattering study. J. Catal. 2009, 262, 244–256. [Google Scholar] [CrossRef]
- Galuszka, J.; Sang, T.; Sawicki, J. Study of carbonaceous deposits on fischer-tropsch oxide-supported iron catalysts. J. Catal. 1992, 136, 96–109. [Google Scholar] [CrossRef]
- Hazemann, P.; Decottignies, D.; Maury, S.; Humbert, S.; Meunier, F.C.; Schuurman, Y. Selectivity loss in Fischer-Tropsch synthesis: The effect of carbon deposition. J. Catal. 2021, 401, 7–16. [Google Scholar] [CrossRef]
- Q. Chang, C. Q. Chang, C. Zhang, C. Liu, Y. Wei, A. V. Cheruvathur, A. I. Dugulan, J. W. Niemantsverdriet, X. Liu, Y. He, M. Qing, L. Zheng, Y. Yun, Y. Yang, Y. Li, Relationship between Iron Carbide Phases (ε-Fe2C, Fe7C3, and χ-Fe5C2) and Catalytic Performances of Fe/SiO2 Fischer-Tropsch Catalysts, ACS Catal., 8 (2018) 3304-3316.
- L. Niu, X. L. Niu, X. Liu, J. Liu, X. Liu, X. Wen, Y. Yang, J. Xu, Y. Li, Tuning Carburization Behaviors of Metallic Iron Catalysts with Potassium Promoter and CO/syngas/C2H4/C2H2 Gases, J. Catal., 371 (2019) 333-345.
- Chun, D.H.; Park, J.C.; Hong, S.Y.; Lim, J.T.; Kim, C.S.; Lee, H.-T.; Yang, J.-I.; Hong, S.; Jung, H. Highly selective iron-based Fischer–Tropsch catalysts activated by CO2-containing syngas. J. Catal. 2014, 317, 135–143. [Google Scholar] [CrossRef]
- Lu, F.; Chen, X.; Lei, Z.; Wen, L.; Zhang, Y. Revealing the activity of different iron carbides for Fischer-Tropsch synthesis. Appl. Catal. B: Environ. 2020, 281, 119521. [Google Scholar] [CrossRef]
- Wezendonk, T.A.; Sun, X.; Dugulan, A.I.; van Hoof, A.J.; Hensen, E.J.; Kapteijn, F.; Gascon, J. Controlled formation of iron carbides and their performance in Fischer-Tropsch synthesis. J. Catal. 2018, 362, 106–117. [Google Scholar] [CrossRef]
- Zhao, H.; Liu, J.-X.; Yang, C.; Yao, S.; Su, H.-Y.; Gao, Z.; Dong, M.; Wang, J.; Rykov, A.I.; Wang, J.; et al. Synthesis of Iron-Carbide Nanoparticles: Identification of the Active Phase and Mechanism of Fe-Based Fischer–Tropsch Synthesis. CCS Chem. 2021, 3, 2712–2724. [Google Scholar] [CrossRef]
- Moodley, P.; Scheijen, F.; Niemantsverdriet, J.; Thüne, P. Iron oxide nanoparticles on flat oxidic surfaces—Introducing a new model catalyst for Fischer–Tropsch catalysis. Catal. Today 2010, 154, 142–148. [Google Scholar] [CrossRef]
- M. Shipilin, D. M. Shipilin, D. Degerman, P. Lömker, C. M. Goodwin, G. L. S. Rodrigues, M. Wagstaffe, J. Gladh, H. Wang, A. Stierle, C. Schlueter, L. G. M. Pettersson, A. Nilsson, P. Amann, In Situ Surface-Sensitive Investigation of Multiple Carbon Phases on Fe(110) in the Fischer-Tropsch Synthesis, ACS Catal., 12 (2022) 7609-7621.
- Lyu, S.; Wang, L.; Li, Z.; Yin, S.; Chen, J.; Zhang, Y.; Li, J.; Wang, Y. Stabilization of ε-iron carbide as high-temperature catalyst under realistic Fischer–Tropsch synthesis conditions. Nat. Commun. 2020, 11, 1–8. [Google Scholar] [CrossRef] [PubMed]
- C. Yang, H. C. Yang, H. Zhao, Y. Hou, D. Ma, Fe5C2 Nanoparticles: A Facile Bromide-Induced Synthesis and as an Active Phase for Fischer-Tropsch Synthesis, J. Am. Chem. Soc., 134 (2012) 15814-15821.
- Schulz, H. Major and Minor Reactions in Fischer–Tropsch Synthesis on Cobalt Catalysts. Top. Catal. 2003, 26, 73–85. [Google Scholar] [CrossRef]
- Chen, W.; Kimpel, T.F.; Song, Y.; Chiang, F.-K.; Zijlstra, B.; Pestman, R.; Wang, P.; Hensen, E.J.M. Influence of Carbon Deposits on the Cobalt-Catalyzed Fischer–Tropsch Reaction: Evidence of a Two-Site Reaction Model. ACS Catal. 2018, 8, 1580–1590. [Google Scholar] [CrossRef]
- Zijlstra, B.; Broos, R.J.; Chen, W.; Filot, I.A.; Hensen, E.J. First-principles based microkinetic modeling of transient kinetics of CO hydrogenation on cobalt catalysts. Catal. Today 2019, 342, 131–141. [Google Scholar] [CrossRef]
- Filot, I.A.W.; van Santen, R.A.; Hensen, E.J.M. The Optimally Performing Fischer–Tropsch Catalyst. Angew. Chem. Int. Ed. 2014, 53, 12746–12750. [Google Scholar] [CrossRef]
- Kuipers, E.; Vinkenburg, I.; Oosterbeek, H. Chain Length Dependence of α-Olefin Readsorption in Fischer-Tropsch Synthesis. J. Catal. 1995, 152, 137–146. [Google Scholar] [CrossRef]
- Kuipers, E.; Scheper, C.; Wilson, J.; Vinkenburg, I.; Oosterbeek, H. Non-ASF Product Distributions Due to Secondary Reactions during Fischer–Tropsch Synthesis. J. Catal. 1996, 158, 288–300. [Google Scholar] [CrossRef]
- Chen, W.; Filot, I.A.W.; Pestman, R.; Hensen, E.J.M. Mechanism of Cobalt-Catalyzed CO Hydrogenation: 2. Fischer–Tropsch Synthesis. ACS Catal. 2017, 7, 8061–8071. [Google Scholar] [CrossRef] [PubMed]
- T. Zubkov, G. A. T. Zubkov, G. A. Morgan, J. T. Yates, O. Kühlert, M. Lisowski, R. Schillinger, D. Fick, H. J. Jänsch, The Effect of Atomic Steps on Adsorption and Desorption of CO on Ru(109), Surf. Sci., 526 (2003) 57-71.
- Mitchell, W.J.; Xie, J.; Jachimowski, T.A.; Weinberg, W.H. Carbon Monoxide Hydrogenation on the Ru(001) Surface at Low Temperature Using Gas-Phase Atomic Hydrogen: Spectroscopic Evidence for the Carbonyl Insertion Mechanism on a Transition Metal Surface. J. Am. Chem. Soc. 1995, 117, 2606–2617. [Google Scholar] [CrossRef]
- Chen, W.; Zijlstra, B.; Filot, I.A.W.; Pestman, R.; Hensen, E.J.M. Mechanism of Carbon Monoxide Dissociation on a Cobalt Fischer–Tropsch Catalyst. ChemCatChem 2017, 10, 136–140. [Google Scholar] [CrossRef]
- Chai, J.; Pestman, R.; Chen, W.; Dugulan, A.I.; Feng, B.; Men, Z.; Wang, P.; Hensen, E.J. The role of H2 in Fe carburization by CO in Fischer-Tropsch catalysts. J. Catal. 2021, 400, 93–102. [Google Scholar] [CrossRef]
- T. H. Pham, X. T. H. Pham, X. Duan, G. Qian, X. Zhou, D. Chen, CO Activation Pathways of Fischer-Tropsch Synthesis on χ-Fe5C2 (510): Direct versus Hydrogen-Assisted CO Dissociation, J. Phys. Chem. C, 118 (2014) 10170-10176.
- Kummer, J.T.; DeWitt, T.W.; Emmett, P.H. Some Mechanism Studies on the Fischer-Tropsch Synthesis Using C14. J. Am. Chem. Soc. 1948, 70, 3632–3643. [Google Scholar] [CrossRef]
- Pichler, H.; Schulz, H. Neuere Erkenntnisse auf dem Gebiet der Synthese von Kohlenwasserstoffen aus CO und H2. Chem. Ing. Tech. 1970, 42, 1162–1174. [Google Scholar] [CrossRef]
- M. A. Petersen, W. J. M. A. Petersen, W. J. van Rensburg, CO Dissociation at Vacancy Sites on Hägg Iron Carbide: Direct Versus Hydrogen-Assisted Routes Investigated with DFT, Top. Catal., 58 (2015) 665-674.
- Chai, J.; Pestman, R.; Chen, W.; Donkervoet, N.; Dugulan, A.I.; Men, Z.; Wang, P.; Hensen, E.J.M. Isotopic Exchange Study on the Kinetics of Fe Carburization and the Mechanism of the Fischer–Tropsch Reaction. ACS Catal. 2022, 12, 2877–2887. [Google Scholar] [CrossRef]
- J. Schweicher, A. J. Schweicher, A. Bundhoo, A. Frennet, N. Kruse, H. Daly, F. C. Meunier, DRIFTS/MS Studies during Chemical Transients and SSITKA of the CO/H2 Reaction over Co-MgO Catalysts, J. Phys. Chem. C, 114 (2010) 2248-2255.
- van Santen, R.A.; Markvoort, A.J.; Filot, I.A.W.; Ghouri, M.M.; Hensen, E.J.M. Mechanism and microkinetics of the Fischer–Tropsch reaction. Phys. Chem. Chem. Phys. 2013, 15, 17038–17063. [Google Scholar] [CrossRef]
- M. Claeys, E. M. Claeys, E. van Steen, Basic studies, Stud. Surf. Sci. Catal., 152 (2004) Chapter 8.
- Govender, N.S.; Botes, F.G.; de Croon, M.H.; Schouten, J.C. Mechanistic pathway for C2+ hydrocarbons over an Fe/K catalyst. J. Catal. 2014, 312, 98–107. [Google Scholar] [CrossRef]
- Bhatelia, T.; Li, C.; Sun, Y.; Hazewinkel, P.; Burke, N.; Sage, V. Chain length dependent olefin re-adsorption model for Fischer–Tropsch synthesis over Co-Al2O3 catalyst. Fuel Process. Technol. 2014, 125, 277–289. [Google Scholar] [CrossRef]
- Filot, I.A.W.; van Santen, R.A.; Hensen, E.J.M. Quantum chemistry of the Fischer–Tropsch reaction catalysed by a stepped ruthenium surface. Catal. Sci. Technol. 2014, 4, 3129–3140. [Google Scholar] [CrossRef]
- Niemantsverdriet, J.; van der Kraan, A. On the time-dependent behavior of iron catalysts in Fischer-Tropsch synthesis. J. Catal. 1981, 72, 385–388. [Google Scholar] [CrossRef]
- Liu, X.; Liu, J.; Yang, Y.; Li, Y.-W.; Wen, X. Theoretical Perspectives on the Modulation of Carbon on Transition-Metal Catalysts for Conversion of Carbon-Containing Resources. ACS Catal. 2021, 11, 2156–2181. [Google Scholar] [CrossRef]
- C. Huo, Y. C. Huo, Y. Li, J. Wang, H. Jiao, Insight into CH4 Formation in Iron-Catalyzed Fischer-Tropsch Synthesis, J. Am. Chem. Soc., 131 (2009) 14713-14721.
- Wang, J.; Huang, S.; Howard, S.; Muir, B.W.; Wang, H.; Kennedy, D.F.; Ma, X. Elucidating Surface and Bulk Phase Transformation in Fischer–Tropsch Synthesis Catalysts and Their Influences on Catalytic Performance. ACS Catal. 2019, 9, 7976–7983. [Google Scholar] [CrossRef]
- S. Zhao, X. S. Zhao, X. Liu, C. Huo, Y. Li, J. Wang, H. Jiao, Determining Surface Structure and Stability of ε-Fe2C, χ-Fe5C2, θ-Fe3C and Fe4C Phases under Carburization Environment from Combined DFT and Atomistic Thermodynamic Studies, Catal. Struct. React., 1 (2014) 44-60.
- Asano, R.; Sasaki, Y.; Ishii, K. Carburization of Iron by Ar-CO-H2 at 1523 K. ISIJ Int. 2002, 42, 121–126. [Google Scholar] [CrossRef]
- Ribeiro, M.C.; Jacobs, G.; Davis, B.H.; Cronauer, D.C.; Kropf, A.J.; Marshall, C.L. Fischer−Tropsch Synthesis: An In-Situ TPR-EXAFS/XANES Investigation of the Influence of Group I Alkali Promoters on the Local Atomic and Electronic Structure of Carburized Iron/Silica Catalysts. J. Phys. Chem. C 2010, 114, 7895–7903. [Google Scholar] [CrossRef]
- Yang, Z.; Zhao, T.; Huang, X.; Chu, X.; Tang, T.; Ju, Y.; Wang, Q.; Hou, Y.; Gao, S. Modulating the phases of iron carbide nanoparticles: from a perspective of interfering with the carbon penetration of Fe@Fe3O4 by selectively adsorbed halide ions. Chem. Sci. 2016, 8, 473–481. [Google Scholar] [CrossRef]
- M. D. Shroff, D. S. M. D. Shroff, D. S. Kalakkad. K. E. Coulter, S. D. Köhler, M. S. Harrington, N. B. Jackson, A. G. Sault, A. K. Dayte, Activation of Precipitated Iron Fischer-Tropsh Synthesis Catalysts, J. Catal., 156 (1995) 185-207.
- Zhou, X.; Mannie, G.J.A.; Yin, J.; Yu, X.; Weststrate, C. (.-J.; Wen, X.-D.; Wu, K.; Yang, Y.; Li, Y.-W.; Niemantsverdriet, J.W.(. Iron Carbidization on Thin-Film Silica and Silicon: A Near-Ambient-Pressure X-ray Photoelectron Spectroscopy and Scanning Tunneling Microscopy Study. ACS Catal. 2018, 8, 7326–7333. [Google Scholar] [CrossRef]
- J. B. Butt, Carbide Phases on Iron-Based Fischer-Tropsch Synthesis Catalysis Part 1: Characterization Studies, Catal. Lett., 7 (1990) 83-106.
- Li, S.; Ding, W.; Meitzner, G.D.; Iglesia, E. Spectroscopic and Transient Kinetic Studies of Site Requirements in Iron-Catalyzed Fischer−Tropsch Synthesis. J. Phys. Chem. B 2001, 106, 85–91. [Google Scholar] [CrossRef]
- Bukur, D.B.; Lang, X.; Ding, Y. Pretreatment effect studies with a precipitated iron Fischer–Tropsch catalyst in a slurry reactor. Appl. Catal. A: Gen. 1999, 186, 255–275. [Google Scholar] [CrossRef]
- S. Li, G. D. S. Li, G. D. Meitzner. E. Iglesia, Structure and Site Evolution of Iron Oxide Catalyst Precursors during the Fischer-Tropsch Synthesis, J. Phys. Chem. B, 105 (2001) 5743-5750.
- Lohitharn, N.; Goodwin, J.G. An investigation using SSITKA of Chain growth on Fe and FeMnK Fischer–Tropsch synthesis catalysts. Catal. Commun. 2009, 10, 758–762. [Google Scholar] [CrossRef]
- S. A. Eliason, C. H. S. A. Eliason, C. H. Bartholomew, Temperature-Programmed Reaction Study of Carbon Transformations on Iron Fischer-Tropsch Catalysts during Steady-State Synthesis, Stud. Surf. Sci. Catal., 111 (1997) 517-526.
- Xu, J.; Bartholomew, C.H. Temperature-Programmed Hydrogenation (TPH) and in Situ Mössbauer Spectroscopy Studies of Carbonaceous Species on Silica-Supported Iron Fischer−Tropsch Catalysts. J. Phys. Chem. B 2004, 109, 2392–2403. [Google Scholar] [CrossRef] [PubMed]
- Govender, N.; de Croon, M.; Schouten, J. Reactivity of surface carbonaceous intermediates on an iron-based Fischer–Tropsch catalyst. Appl. Catal. A: Gen. 2010, 373, 81–89. [Google Scholar] [CrossRef]
- Ding, M.; Yang, Y.; Wu, B.; Wang, T.; Ma, L.; Xiang, H.; Li, Y. Transformation of carbonaceous species and its influence on catalytic performance for iron-based Fischer–Tropsch synthesis catalyst. J. Mol. Catal. A: Chem. 2011, 351, 165–173. [Google Scholar] [CrossRef]
- Jin, Y.; Xu, H.; Datye, A.K. Electron Energy Loss Spectroscopy (EELS) of Iron Fischer–Tropsch Catalysts. Microsc. Microanal. 2006, 12, 124–134. [Google Scholar] [CrossRef]
- Govender, N.S.; Botes, F.G.; de Croon, M.H.; Schouten, J.C. Mechanistic pathway for methane formation over an iron-based catalyst. J. Catal. 2008, 260, 254–261. [Google Scholar] [CrossRef]
- Graf, B.; Schulte, H.; Muhler, M. The formation of methane over iron catalysts applied in Fischer–Tropsch synthesis: A transient and steady state kinetic study. J. Catal. 2010, 276, 66–75. [Google Scholar] [CrossRef]
- Xie, J.; Yang, J.; Dugulan, A.I.; Holmen, A.; Chen, D.; de Jong, K.P.; Louwerse, M.J. Size and Promoter Effects in Supported Iron Fischer–Tropsch Catalysts: Insights from Experiment and Theory. ACS Catal. 2016, 6, 3147–3157. [Google Scholar] [CrossRef]
- Mars, P.; van Krevelen, D. Oxidations carried out by means of vanadium oxide catalysts. Chem. Eng. Sci. 1954, 3, 41–59. [Google Scholar] [CrossRef]
- Gracia, J.M.; Prinsloo, F.F.; Niemantsverdriet, J.W. Mars-van Krevelen-like Mechanism of CO Hydrogenation on an Iron Carbide Surface. Catal. Lett. 2009, 133, 257–261. [Google Scholar] [CrossRef]
- Ordomsky, V.V.; Legras, B.; Cheng, K.; Paul, S.; Khodakov, A.Y. The role of carbon atoms of supported iron carbides in Fischer–Tropsch synthesis. Catal. Sci. Technol. 2015, 5, 1433–1437. [Google Scholar] [CrossRef]
- Keyvanloo, K.; Lanham, S.J.; Hecker, W.C. Kinetics of Fischer-Tropsch synthesis on supported cobalt: Effect of temperature on CO and H 2 partial pressure dependencies. Catal. Today 2016, 270, 9–18. [Google Scholar] [CrossRef]
- Mohammad, N.; Bepari, S.; Aravamudhan, S.; Kuila, D. Kinetics of Fischer–Tropsch Synthesis in a 3-D Printed Stainless Steel Microreactor Using Different Mesoporous Silica Supported Co-Ru Catalysts. Catalysts 2019, 9, 872. [Google Scholar] [CrossRef]
- J. Cheng, P. J. Cheng, P. Hu, P. Ellis, S. French, G. Kelly, C. M. Lok, Chain Growth Mechanism in Fischer-Tropsch Synthesis: A DFT Study of C−C Coupling over Ru, Fe, Rh, and Re Surfaces, J. Phys. Chem. C, 112 (2008) 6082-6086.
- M. J. Valero-Romero, M. M. J. Valero-Romero, M. Á. Rodríguez-Cano, J. Palomo, J. Rodríguez-Mirasol, T. Cordero, Carbon-Based Materials as Catalyst Supports for Fischer-Tropsch Synthesis: A Review, Front. Mater. Sci., 7 (2021) 617432.
- H. A. J. van Dijk, J. H. B. J. H. A. J. van Dijk, J. H. B. J. Hoebink, J. C. Schouten, A Mechanistic Study of the Fischer-Tropsch Synthesis Using Transient Isotopic Tracing. Part 2: Model Quantification, Catal. Today, 26 (2003) 163-171.
- Galvis, H.M.T.; Bitter, J.H.; Davidian, T.; Ruitenbeek, M.; Dugulan, A.I.; de Jong, K.P. Iron Particle Size Effects for Direct Production of Lower Olefins from Synthesis Gas. J. Am. Chem. Soc. 2012, 134, 16207–16215. [Google Scholar] [CrossRef] [PubMed]
- E. de Smit, F. M. F. E. de Smit, F. M. F. de Groot, R. Blume, M. Havecker, A. K. Gericke, B. M. Weckhuysen, The Role of Cu on the Reduction Behavior and Surface Properties of Fe-Based Fischer-Tropsch Catalysts, Phys. Chem. Chem. Phys., 12 (2010) 667-680.
- Hindermann, J.P.; Hutchings, G.J.; Kiennemann, A. Mechanistic Aspects of the Formation of Hydrocarbons and Alcohols from CO Hydrogenation. Catal. Rev. 1993, 35, 1–127. [Google Scholar] [CrossRef]
- Campos, A.; Lohitharn, N.; Roy, A.; Lotero, E.; Goodwin, J.G.; Spivey, J.J. An activity and XANES study of Mn-promoted, Fe-based Fischer–Tropsch catalysts. Appl. Catal. A: Gen. 2010, 375, 12–16. [Google Scholar] [CrossRef]
- N. Lohitharn, J. G. N. Lohitharn, J. G. Goodwin, Impact of Cr, Mn and Zr Addition on Fe Fischer-Tropsch Synthesis Catalysis: Investigation at the Active Site Level Using SSITKA, J. Catal., 257 (2008) 142-151.
- Lohitharn, N.; Goodwin, J.G., Jr.; Synthesis, F. ; Olefins; Hydrocarbon Effect of K promotion of Fe and FeMn Fischer–Tropsch synthesis catalysts: Analysis at the site level using SSITKA. J. Catal. 2008, 260, 7–16. [Google Scholar] [CrossRef]
- Jensen, K.; Massoth, F. Studies on iron-manganese oxide carbon monoxide catalysts II. Carburization and catalytic activity. J. Catal. 1985, 92, 109–118. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, J.-F.; Bao, J.; Zhang, Y. Manganese-Modified Fe3O4 Microsphere Catalyst with Effective Active Phase of Forming Light Olefins from Syngas. ACS Catal. 2015, 5, 3905–3909. [Google Scholar] [CrossRef]
- Ribeiro, M.C.; Jacobs, G.; Pendyala, R.; Davis, B.H.; Cronauer, D.C.; Kropf, A.J.; Marshall, C.L. Fischer−Tropsch Synthesis: Influence of Mn on the Carburization Rates and Activities of Fe-Based Catalysts by TPR-EXAFS/XANES and Catalyst Testing. J. Phys. Chem. C 2011, 115, 4783–4792. [Google Scholar] [CrossRef]
- Yang, Z.; Zhang, Z.; Liu, Y.; Ding, X.; Zhang, J.; Xu, J.; Han, Y. Tuning direct CO hydrogenation reaction over Fe-Mn bimetallic catalysts toward light olefins: Effects of Mn promotion. Appl. Catal. B: Environ. 2020, 285, 119815. [Google Scholar] [CrossRef]
- F. H. Wielers, A. J. H. M. F. H. Wielers, A. J. H. M. Kock, C. E. C. A. Hop, J. W. Geus, A. M. van Kraan, The Reduction Behavior of Silica-Supported and Alumina-Supported Iron Catalysts: A Mössbauer and Infrared Spectroscopic Study, J. Catal., 117 (1989) 1-18.
- Lin, Q.; Cheng, M.; Zhang, K.; Li, W.; Wu, P.; Chang, H.; Lv, Y.; Men, Z. Development of an Iron-Based Fischer—Tropsch Catalyst with High Attrition Resistance and Stability for Industrial Application. Catalysts 2021, 11, 908. [Google Scholar] [CrossRef]
- H. J. Schulte, B. H. J. Schulte, B. Graf, W. Xia, M. Muhler, Nitrogen- and Oxygen-Functionalized Multiwalled Carbon Nanotubes Used as Support in Iron-Catalyzed, HighTemperature Fischer-Tropsch Synthesis, ChemCatChem, 4 (2012) 350-355.
- Lokteva, E.S.; Golubina, E.V. Metal-support interactions in the design of heterogeneous catalysts for redox processes. Pure Appl. Chem. 2019, 91, 609–631. [Google Scholar] [CrossRef]
- R. Zhao, J. G. R. Zhao, J. G. Goodwin Jr. K. Jothimurugesan, S. K. Gangwal, J. J. Spivey, Spray-Dried Iron Fischer-Tropsch Catalysts. 2. Effect of Carburization on Catalyst Attrition Resistance, Ind. Eng. Chem. Res., 40 (2001) 1320-1328.
- Thüne, P.; Moodley, P.; Scheijen, F.; Fredriksson, H.; Lancee, R.; Kropf, J.; Miller, J.; Niemantsverdriet, J.W. (. The Effect of Water on the Stability of Iron Oxide and Iron Carbide Nanoparticles in Hydrogen and Syngas Followed by in Situ X-ray Absorption Spectroscopy. J. Phys. Chem. C 2012, 116, 7367–7373. [Google Scholar] [CrossRef]
- Eliason, S.; Bartholomew, C. Reaction and deactivation kinetics for Fischer–Tropsch synthesis on unpromoted and potassium-promoted iron catalysts. Appl. Catal. A: Gen. 1999, 186, 229–243. [Google Scholar] [CrossRef]
- Chai, J.; Pestman, R.; Chiang, F.-K.; Men, Z.; Wang, P.; Hensen, E.J. Influence of carbon deposits on Fe-carbide for the Fischer-Tropsch reaction. J. Catal. 2022, 416, 289–300. [Google Scholar] [CrossRef]
- Pour, A.N.; Housaindokht, M.R.; Tayyari, S.F.; Zarkesh, J. Fischer-Tropsch synthesis by nano-structured iron catalyst. J. Nat. Gas Chem. 2010, 19, 284–292. [Google Scholar] [CrossRef]
- Nakamura, J.; Tanaka, K.-I.; Toyoshima, I. Reactivity of deposited carbon on Co$z.sbnd;Al2O3 catalyst. J. Catal. 1987, 108, 55–62. [Google Scholar] [CrossRef]
- Warringham, R.; Davidson, A.L.; Webb, P.B.; Tooze, R.P.; Ewings, R.A.; Parker, S.F.; Lennon, D. Examining the temporal behavior of the hydrocarbonaceous overlayer on an iron based Fischer–Tropsch catalyst. RSC Adv. 2019, 9, 2608–2617. [Google Scholar] [CrossRef] [PubMed]
- Koeken, A.C.J.; Galvis, H.M.T.; Davidian, T.; Ruitenbeek, M.; de Jong, K.P. Suppression of Carbon Deposition in the Iron-Catalyzed Production of Lower Olefins from Synthesis Gas. Angew. Chem. Int. Ed. 2012, 51, 7190–7193. [Google Scholar] [CrossRef] [PubMed]
- M. Sarkari, F. M. Sarkari, F. Fazlollahi, H. Ajamein, H. Atashi, W. C. Hecker, L. L. Baxter, Fischer-Tropsch Synthesis: Development of Kinetic Expression for a Sol-Gel Fe-Ni/Al2O3 Catalyst, Fuel Process. Technol., 127 (2012) 163-170.
- M. Claeys, E. M. Claeys, E. van Steen, On the Effect of Water during Fischer-Tropsch Synthesis with a Ruthenium Catalyst, Catal. Today, 71 (2002) 419-427.
- Ning, W.; Koizumi, N.; Chang, H.; Mochizuki, T.; Itoh, T.; Yamada, M. Phase transformation of unpromoted and promoted Fe catalysts and the formation of carbonaceous compounds during Fischer–Tropsch synthesis reaction. Appl. Catal. A: Gen. 2006, 312, 35–44. [Google Scholar] [CrossRef]
- McDonald, M.; Storm, D.; Boudart, M. Hydrocarbon synthesis from CO$z.sbnd;H2 on supported iron: Effect of particle size and interstitials. J. Catal. 1986, 102, 386–400. [Google Scholar] [CrossRef]
- Liu, J.-X.; Su, H.-Y.; Sun, D.-P.; Zhang, B.-Y.; Li, W.-X. Crystallographic Dependence of CO Activation on Cobalt Catalysts: HCP versus FCC. J. Am. Chem. Soc. 2013, 135, 16284–16287. [Google Scholar] [CrossRef] [PubMed]
- J. M. G. Carballo, J. J. M. G. Carballo, J. Yang, A. Holmen, S. García-Rodríguez, S. Rojas, M. Ojeda, J. L. G. Fierro, Catalytic Effects of Ruthenium Particle Size on the Fischer-Tropsch Synthesis, J. Catal., 284 (2011) 102-108.
- Den Breejen, J.P.; Radstake, P.B.; Bezemer, G.L.; Bitter, J.H.; Frøseth, V.; Holmen, A.; de Jong, K.P. On the Origin of the Cobalt Particle Size Effects in Fischer−Tropsch Catalysis. J. Am. Chem. Soc. 2009, 131, 7197–7203. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Tian, Y.; Lyu, S.; Zhao, N.; Ma, K.; Ding, T.; Jiang, Z.; Wang, L.; Zhang, J.; Zheng, L.; et al. Confined small-sized cobalt catalysts stimulate carbon-chain growth reversely by modifying ASF law of Fischer–Tropsch synthesis. Nat. Commun. 2018, 9, 3250. [Google Scholar] [CrossRef]
- Barkhuizen, D.; Mabaso, I.; Viljoen, E.; Welker, C.; Claeys, M.; van Steen, E.; Fletcher, J.C.Q. Experimental approaches to the preparation of supported metal nanoparticles. Pure Appl. Chem. 2006, 78, 1759–1769. [Google Scholar] [CrossRef]
- Park, J.-Y.; Lee, Y.-J.; Khanna, P.K.; Jun, K.-W.; Bae, J.W.; Kim, Y.H. Alumina-supported iron oxide nanoparticles as Fischer–Tropsch catalysts: Effect of particle size of iron oxide. J. Mol. Catal. A: Chem. 2010, 323, 84–90. [Google Scholar] [CrossRef]
- Yin, J.; Liu, X.; Liu, X.-W.; Wang, H.; Wan, H.; Wang, S.; Zhang, W.; Zhou, X.; Teng, B.-T.; Yang, Y.; et al. Theoretical exploration of intrinsic facet-dependent CH4 and C2 formation on Fe5C2 particle. Appl. Catal. B: Environ. 2020, 278, 119308. [Google Scholar] [CrossRef]
Figure 1.
Schematic representation of the CTL process.
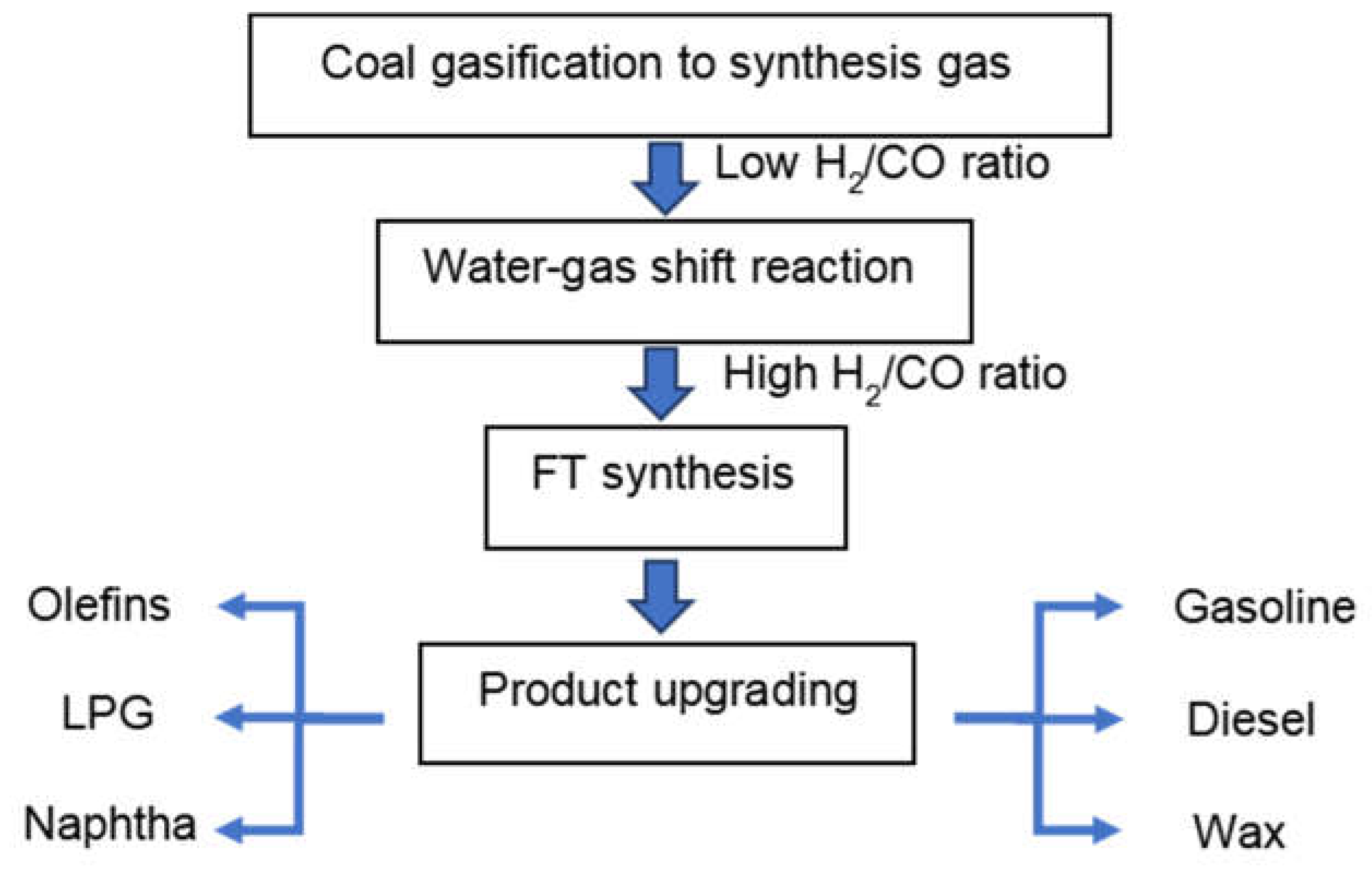
Figure 2.
The product distribution of CO hydrogenation on transition metals.
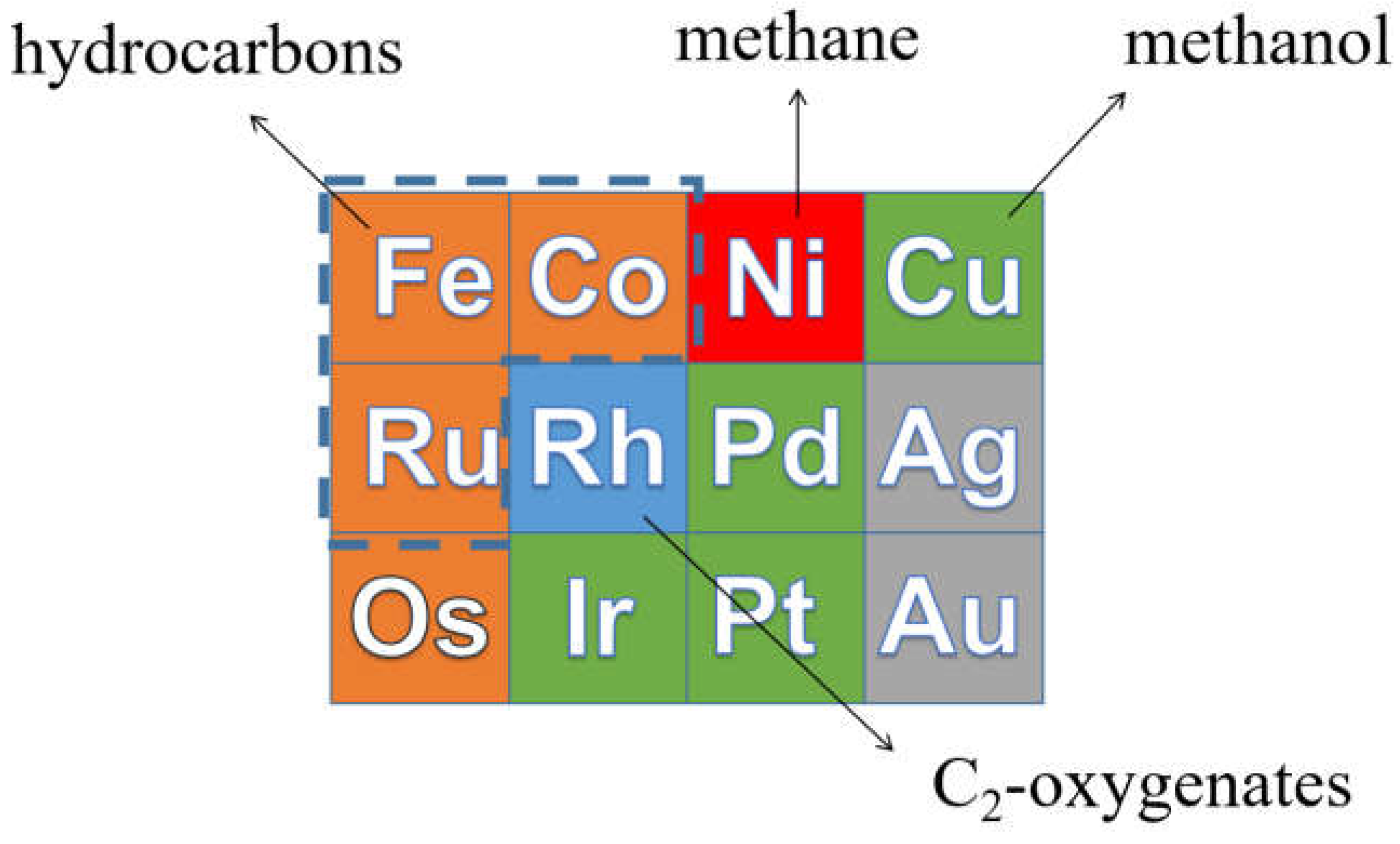
Figure 3.
FT performance of the Raney-Fe catalyst as a function of time on stream. (A) H2/CO = 1.5, 23 bar, 235 oC, GHSV of 18,000 hour−1. (B) After 175-hour time on stream, the reaction temperature was increased to 250 oC and kept there for 24 hours followed by a decrease to 235 oC [9]..
Figure 3.
FT performance of the Raney-Fe catalyst as a function of time on stream. (A) H2/CO = 1.5, 23 bar, 235 oC, GHSV of 18,000 hour−1. (B) After 175-hour time on stream, the reaction temperature was increased to 250 oC and kept there for 24 hours followed by a decrease to 235 oC [9]..
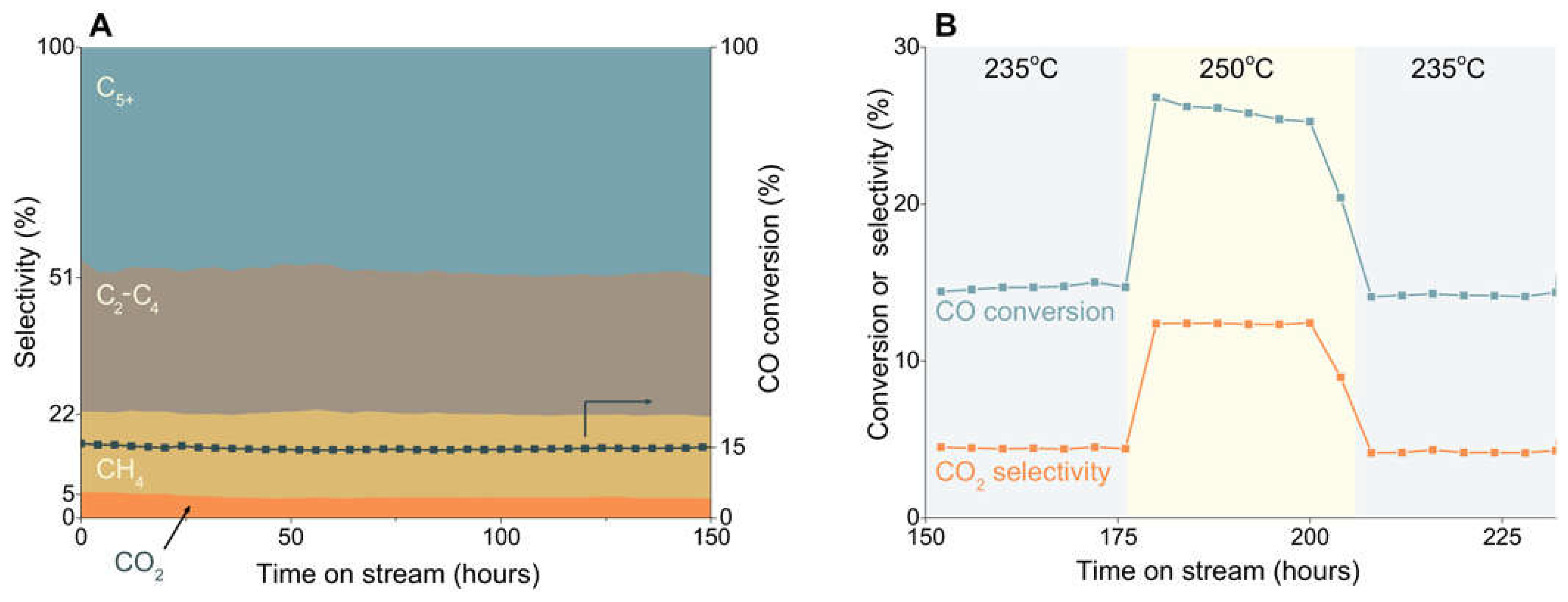
Figure 4.
Theoretical product distribution as a function of chain-growth probability
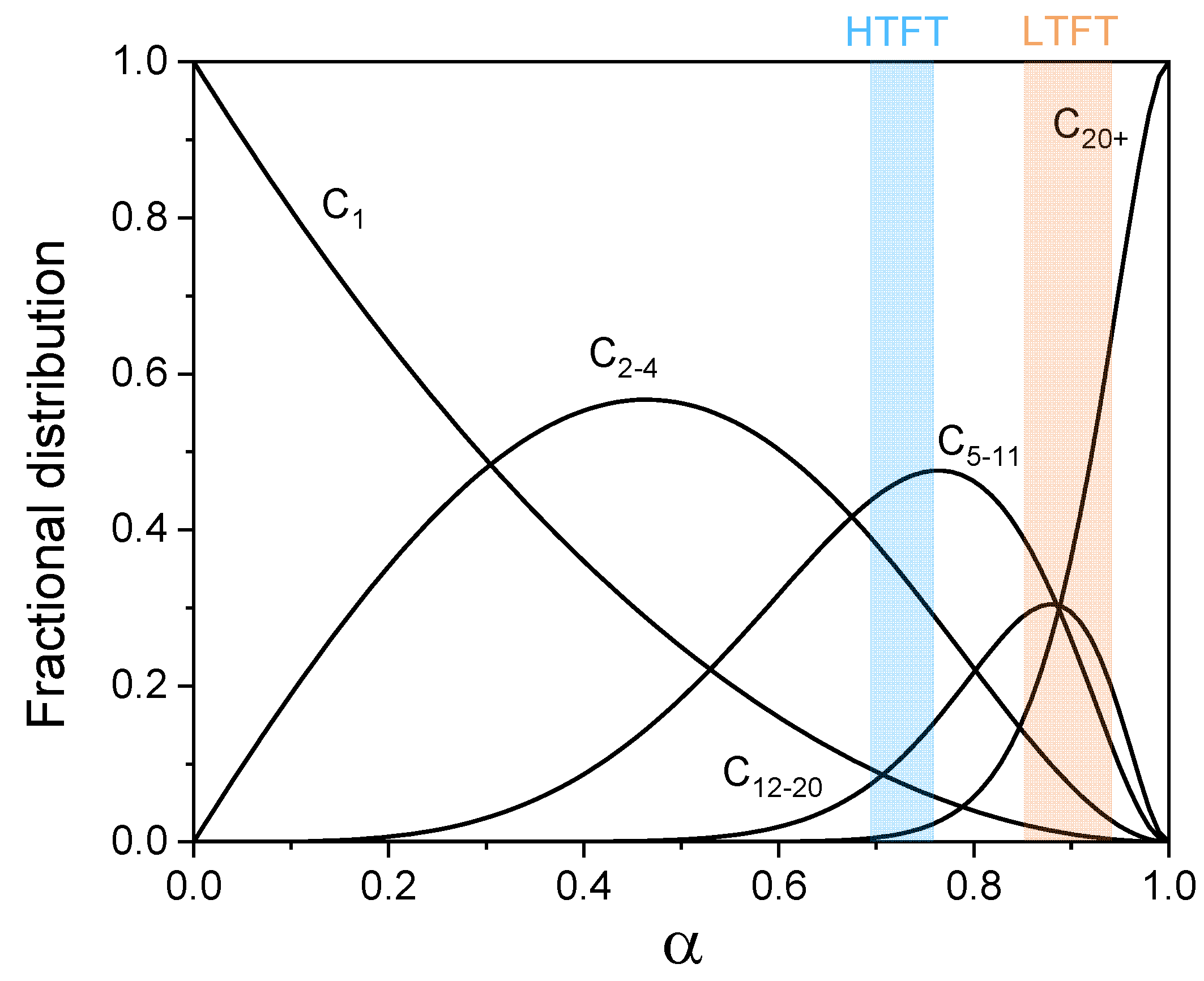
Figure 5.
Schematic representation of the role of H2 on Fe-carbide formation [75].
Figure 5.
Schematic representation of the role of H2 on Fe-carbide formation [75].
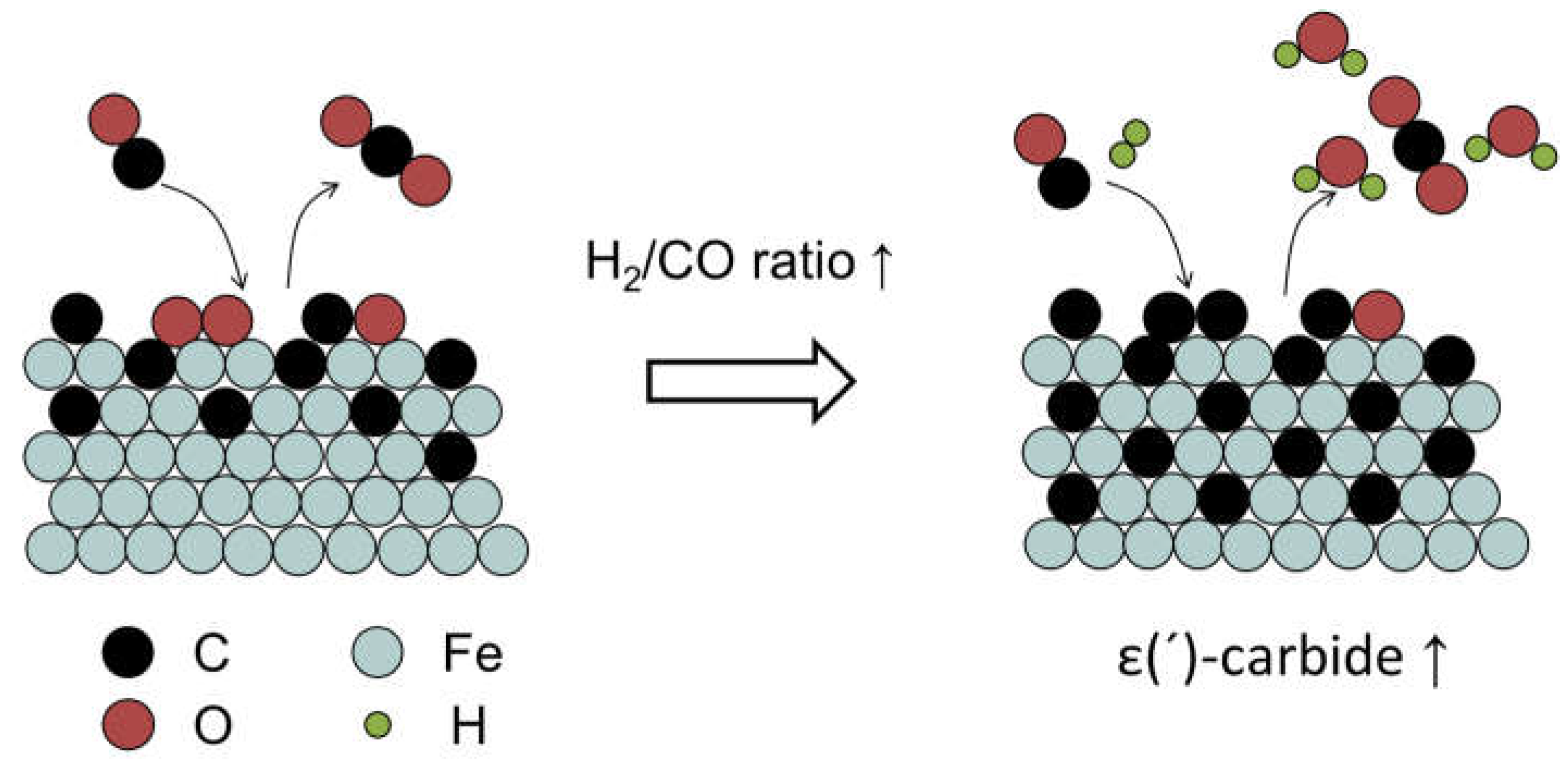
Figure 6.
Schematic representation of the MvK mechanism for FT reaction towards CH4 on Fe-carbide catalysts [111].
Figure 6.
Schematic representation of the MvK mechanism for FT reaction towards CH4 on Fe-carbide catalysts [111].
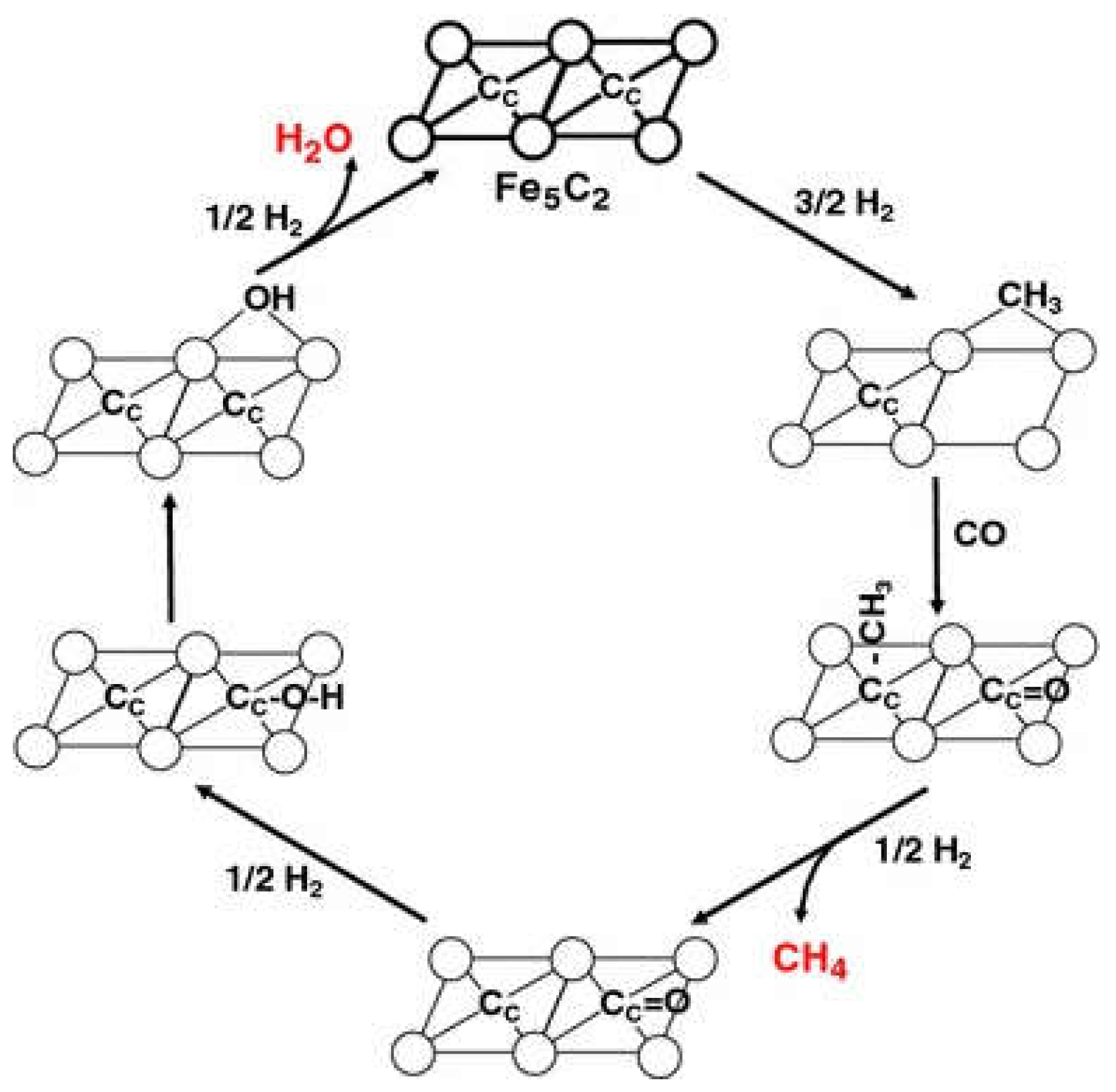
Figure 7.
The comparison of the L-H mechanism and MvK mechanism on Fe-carbide for CH4 formation [80].
Figure 7.
The comparison of the L-H mechanism and MvK mechanism on Fe-carbide for CH4 formation [80].
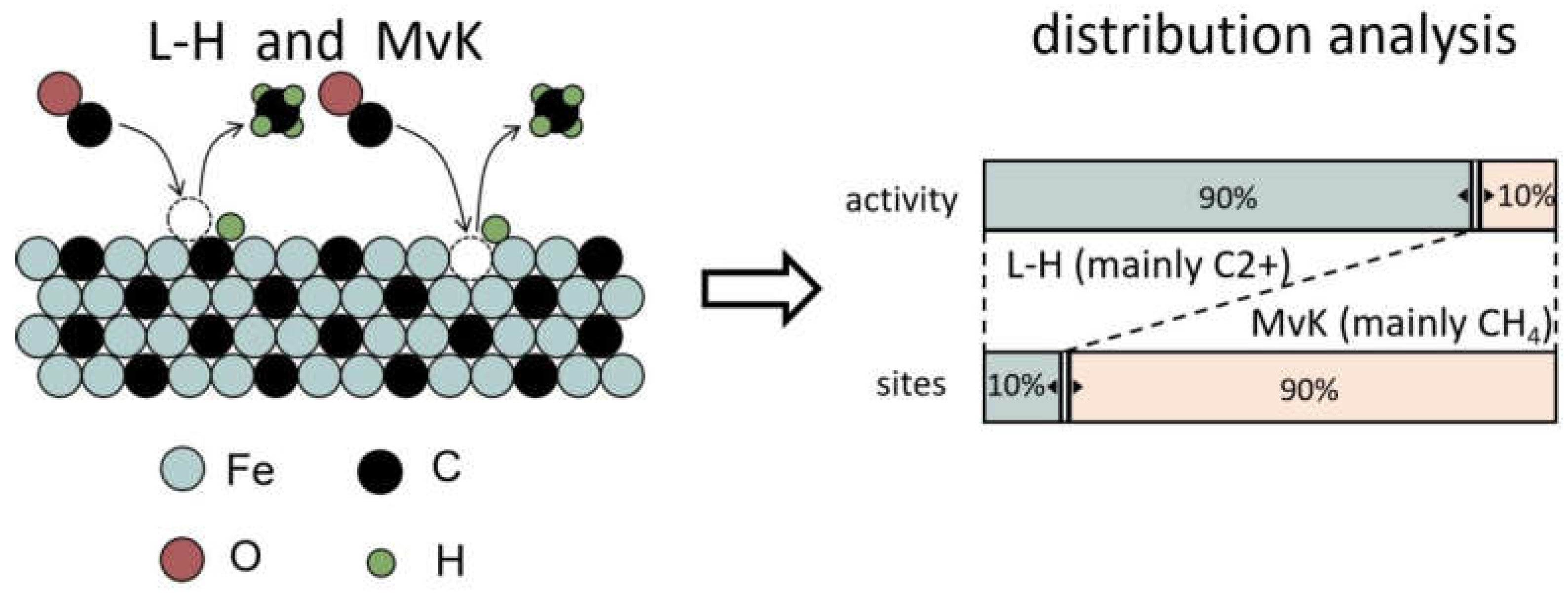
Figure 8.
The mechanism underlying the influence of C deposits on Fe-carbide for the FT reaction [135].
Figure 8.
The mechanism underlying the influence of C deposits on Fe-carbide for the FT reaction [135].
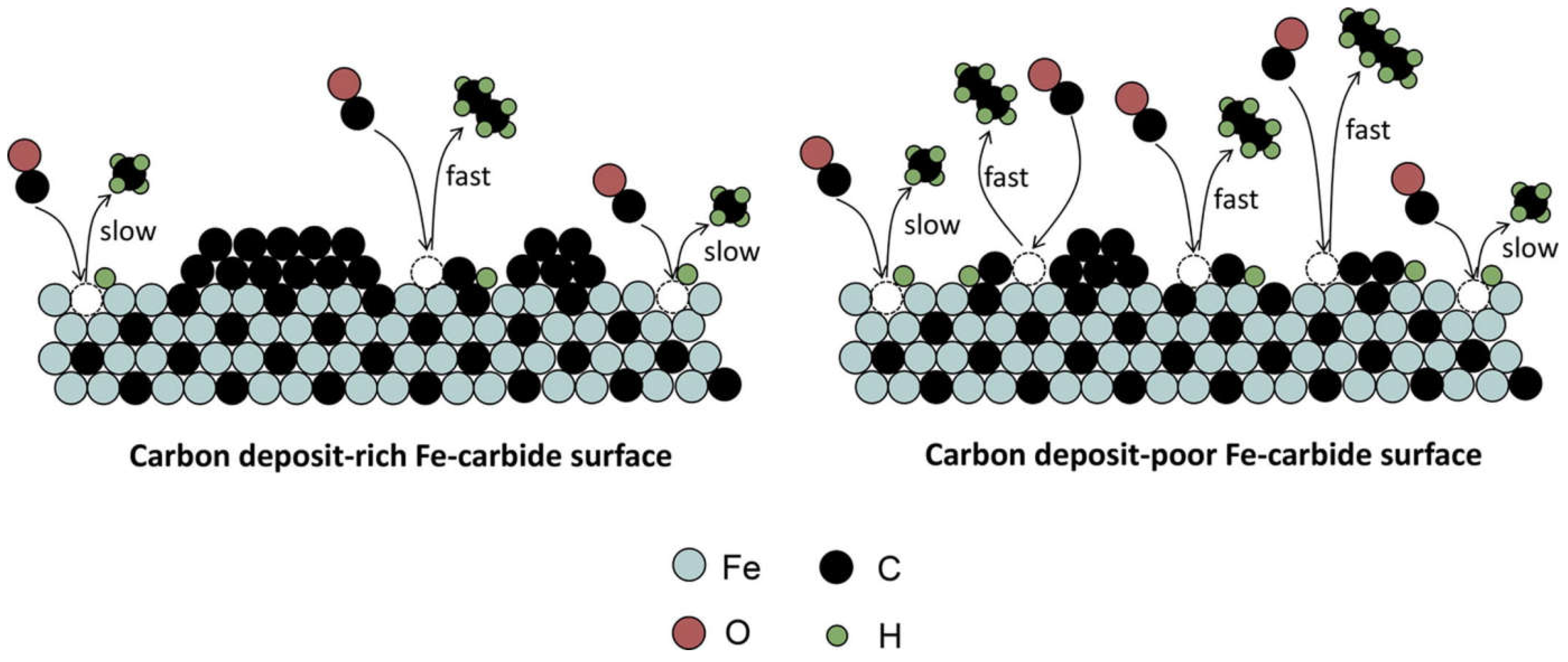


Table 2.
Comparison of ε(´)-carbide, χ-Fe5C2 and Θ-Fe3C.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



