Submitted:
05 June 2023
Posted:
06 June 2023
You are already at the latest version
Abstract
Although there are a large number of databases available for regulatory elements, bottleneck has been created by the lack of bioinformatics tools for predicting types of mechanisms underlying actions of regulatory elements. To reduce the gap, we developed ARabidopsis Transcription regulatory Factor Domain-domain interaction Analysis Tool- Liquid-liquid phase separation (LLPS), Oligomerization, GO analysis (ART FounDATion-LOG), a useful toolkit for protein-nucleic acid interactions (PNI) and protein-protein interactions (PPI) analysis based on domain-domain interaction (DDI). LLPS, protein oligomerization, structural properties of protein domains, and protein modifications are major components in orchestrating spatio-temporal dynamics of PPI and PNI. Our goal is to integrate PPI/PNI information into development of prediction model for identifying important genetic variants in peach. The program unified inter-database relational keys by protein domains for facilitating inference from the model species. Key advantage of the program lies in the integrated information of related features: LOG, structural characterization of domain (e.g. domain linker, intrinsically disordered regions, DDI, domain-motif (peptide) interaction, beta-sheet and transmembrane helices), and post-translational modification. We provided simple tests to demonstrate how to use the program. The program may be applied to other eukaryotic organisms. The program codes and data are freely available for download at and https://sourceforge.net/projects/artfoundation-log/.
Keywords:
Liquid liquid phase separation
; protein oligomerization
; GO
; domain-domain interaction
; domain linker
; intrinsically disordered regions
; domain-peptide interaction
; beta-sheet
; transmembrane helices
; post-translational modification
1. Introduction
Peach (Prunus persica) has been bred for more than 4000 years [1]. Traditional breeding has selected peach cultivars with improved fruit quality and traits over last thousands of years. In the last several decades, marker-assisted breeding has been developed based on advanced next generation sequencing technologies and has gained popularity among breeding scientists [2]. Genome-wide association study (GWAS) have been employed to improve the marker-assisted breeding [2]. However, identifying important functional genetic variants in GWAS data remains challenging due to the complexity of biological systems. There are only limited resources available in peach, compared to model species- Arabidopsis thaliana (A. thaliana). Furthermore, considerable portions of regulatory mechanisms have been conserved across plant species; TF families of A. thaliana are subsets of those of peach. To effectively solve the problem, we took a strategic approach: integration of the immense reservoir of omics data from the model species into the genetic variant analysis in peach; We developed bioinformatics programs based on A. thaliana in order to predict determinative genetic variants using AI and statistics methods.
Well-organized A. thaliana databases can expedite processes of gene regulatory networks (GRN) construction; values of nodes of GRN can be derived from various data such as RNA-seq, transcriptome wide association study (TWAS), epigenome-wide association study (EWAS), phosphorylation site information (proteome), and metabolic pathway in addition to data on genome-wide positions of domain and key regulatory elements [3,4]. However, important pieces of information is missing- interaction modes between regulatory elements/domains, which causes the research bottleneck. For biological processes employ complex regulatory systems, complete annotations of individual interactions are not available. However, integration of LOG, domain-domain interaction (DDI), domain-motif (peptide) interaction (DMI), physicochemical and structural properties of domains, and post-translational modification (PTM) help elucidate mechanisms underlying protein interactions. For example, transcription regulation is triggered by cellular signals and achieved with spatiotemporal coordinations of various protein-nucleic acid interactions (PNI) and protein-protein interactions (PPI) regulated in a number of ways, e.g., protein oligomerization, Liquid liquid phase separation (LLPS) and PTM [5,6]. They have evolved into interwoven regulatory mechanisms in modulating the interactions, for instance, repressing PNI of Histone H1 by inhibiting LLPS formation with phosphorylation, regulating binding activity between scaffold proteins and peptide with oligomerization and LLPS formation, and regulating localization of self-assembled transmembrane protein by phosphorylation of amino acid in domain [7,8,9,10,11,12,13,14].
Here, we introduce ARabidopsis Transcription regulatory Factor Domain-domain interaction Analysis Tool- LLPS, Oligomerization, GO analysis (ART FounDATion-LOG), a usefoul toolkit with integrative resources on key properties modulating PPI and PNI: protein oligomerization, LLPS, protein domain characterization such as structural prerequisites of a particular interaction (e.g. transmembrane, domain linker, intrinsically disordered regions (IDR), DMI, coiled-coil region) and post-translational modification (PTM) (Figure 1) [15,16,17,18,19]. The program consists of seven main modules where each module was built based on existing databases: protein/ protein assembly (prot-assembly) oligomerization module (ProtCAD), features of binding interface of DDI and domain characterization (3did, Plant-PrAS, qPTMplants), LLPS formations (DrLLPS), GO/PO analysis, TF binding profile module (Cis-BP), PPI module (String), TF target module (TF2DNA, Yu et al.), in addition to sequence and structure annotation databases such as UniProt, Pfam, InterPro, PDB, and TAIR [20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36]. Analysis on LOG and domain characterization provides important information on diverse properties of prot assemblies, which help elucidate regulatory mechanisms. Integration of LOG in studying regulation of DDI and domain-nucleotide interaction (DNI) in GRN will significantly enhance prediction power of AI in assessing impact of genetic variants on phenotypic differences. The program is a versatile tool to study a wide spectrum of biological research subjects, for example, gene expression regulation such as TF-target binding activities and prediction on functions of transmembrane proteins within the context of localization.
To demonstrate how to use the program, we performed three simple tests. In the first test, we selected the entries common to oligomerization module and DDI module, divided the data into LLPS and non-LLPS datasets, and developed AI models. The accuracy of the model was 82%, 87%, and 91% in predicting the oligomerization types in LLPS dataset, the LLPS subcellular types, and the oligomerization types in non-LLPS dataset, respectively. In the second test, we selected LLPS factors interacting with TFs and involving in LLPS, merged LOG and TF-target data, and predicted oligomerization types with the accuracy of 97%. Prediction class distribution table was generated with Weka program [37]. In the last test, we selected LLPS factors involved in signaling pathway, made a list of DNA binding motif types of TFs that the factor interacted with, and predicted the motif types with the accuracy of 84%. Association study revealed that DNA binding motif types had strong associations with physicochemical properties, protein secondary structural properties, and important domain features of TFs, while LLPS functional type (scaffold, regulator, client) had strong associations with a category of binding interface of DDI and of DNA binding motif types of TFs. There were no structure data of in-vivo proteins available in PDB. The oligomerization types were predicted based on the prot assemblies grown in-vitro, mainly without LLPS formation. Relatively high associations between categories of DDI and of LLPS functional types suggested that the differences of features of binding interfaces from different domains might reflect the intrinsic properties of domain organization in LLPS formation, which had consistency with correlation analysis in the second test; Domain dimer feature from ProtCAD and DDI interface feature from 3did, LLPS types, LLPS functional types, and number of domains of LLPS factors had relatively high correlation with oligomerization types (corr > 0.6). A considerable portion of regulatory SNPs is related to LOG and domain features [38]. DDI/DNI analysis will provide important clues on what causes phenotypic differences by comparing genetic variants in unknown elements to known regulatory network in the program. The program codes and datasets of ART FounDATion-LOG are available for download at www.artfoundation.kr and sourceforge.
2. Methods
2.1. Database Modules
ART FounDATion-LOG consists of 7 DB modules containing features extracted from existing databases. It also included a simple model for analyzing TF-target interaction and a rough sketch for detecting nucleotide-containing ligand binding motifs in protein based on NBDB [39].
Oligomerization module: ProtCAD provided prot-assembly information derived from PDB entries. PDB contained multiple plausible in-vitro structures of prot assemblies that formed homo- or hetero-oligomers by oligomerizing by themselves or with other proteins. It provided information such as stoichiometries and symmetries of clusters belonging to a ProtCAD entry (GroupID) with the same Pfam architecture [20]. In this paper, we used the term “homo” to refer to a cluster with the same sequence(s) and had only one letter “A” in stoichiometry while “hetero” to refer to a cluster with the different sequences, e.g. “AB”, in stoichiometry. To distinguish those without symmetry, we used C1_obligate_hetero_single_oligomer_obligate to refer to a cluster with only one type: C1 molecules from multiple sequences (e.g. C1-A2BC) and CMA to a cluster having C1-A1. The term “oligomer” was to refer to a cluster with 2 or higher number, e.g. C2 or D3 in symmetry. We extracted 19 variables which were mainly frequencies of domains (single domains and domain dimers). Pfam dimer was to refer to two domains not necessarily maintaining the sequential order in actual sequence; For example a prot-assembly with Dom1 Dom2 Dom3 in sequence had three possible combinations of domain dimers: Dom1Dom2; Dom1Dom3; Dom2Dom3. Most of variables were vectors containing frequencies in 14 or 11 different oligomerization types, for example, those from an entry with one Pfam assignment or with multiple Pfams, from molecules of the symmetry with an even number (e.g. C2, D2) or an odd number (C3, D3), from a single gene or multiple genes, or from 16 different subcellular locations. Two variables contained a single digit which was the maximum number of symmetries (Table S1).
2.2. DDI Module
3did provided sequences of interfaces from DDI and from DMI [21]. We grouped entries of the same domain members into two clusters, one with the non-redundant (nr) sets (e.g. Dom1Dom2) and the other with redundant sets (e.g. Dom1Dom2Dom1). We extracted 13 features to measure the differences between two clusters. They mainly represented the chemical properties such as the number of interacting domains, the number of interacting motifs, the number of amino acids in each fragment where a fragment indicating sub-region of consecutive amino acids without gaps larger than 3, and ProtCAD value- maximum of symmetries. They also contained information such as mean and standard deviation of values in each cluster as well as a size of memberships of cluster. The ANOVA test was performed regardless of the normality of data because the magnitude of the differences between two clusters was particularly important, but no non-parametric statistics with the capacity was available.
Plant-PrAS provided genome-wide analysis of proteins: the grand average of hydrophobicity (GRAVY), isoelectric point (pI), binary presence/absence values of solubility, low complexity, protein secondary structural properties (b-sheets, IDRs, signal peptide(s), transmembrane helices, disulfide (S–S) bonds and domain linkers), N/O-glycosylation sites, ubiquitination sites, functional regions (PASS), peptide type (chloroplast transit peptide, mitochondrial targeting peptide, secretory pathway signal peptide), and subcellular location (E.R., chlo, mito, cysk, cyto, nucl, plas, extr, golg, pero, and vacu). The domain features derived from Plant-PrAS database will be referred as “Plant-PrAS features”. qPTMplants provided PTM information: Glycation, Lysine, Methylation, N-glycosylation, N-terminal, O-GlcNAcylation, Oxidation, Persulfidation, Phosphorylation, S-cyanylation, S-nitrosylation, S-sulfenylation. The domain features derived from qPTMplants database will be referred as “PTM features”. We mapped them to domains and inter-domain regions based on protein domain positions provided by TAIR [25].
2.3. LLPS Module
DrLLPS included approx. 40 distinct biomolecular condensates (Balbiani body, Cajal body, Centrosome/Spindle pole body, Chromatin, Chromatoid body, Cleavage body, DDX1 body, DNA damage foci, Droplet, Gemini of cajal body, Germ plasm/Polar granule, Histone locus body, Insulator body, Microtubule, Mitochondrial RNA granule, Neuronal granule, Nuage, Nuclear pore complex, Nuclear speckle, Nuclear stress body, Nucleolus, OPT domain, Others, Paraspeckle, P-body, PcG body, Pericentriolar matrix, Perinucleolar compartment, P granule, PML nuclear body, Postsynaptic density, Pyrenoid matrix, Receptor cluster, Sam68 nuclear body, siRNA body, Spindle apparatus, Sponge body, Stress granule, TAM body, U body) [24]. LLPS associated proteins usually were involved in formation of multiple condensates. In the database, a protein was classified according to the associations with the condensates, which resulted in 265 possible LLPS types (e.g. a protein only specialized in PML body formations, a protein involved in a number of LLPS- nucleoli, nuclear speckles, cajal body, centrosome, etc.). In addition, DrLLPS provided functional types of LLPS proteins: Client, Regulator, and Scaffold; In this paper, they were referred to LLPS factors. We created two different levels of variables; one with gene as unit and the other with domain dimer as unit. In gene level, we extracted entire domains belonging to a protein, and counted frequencies of the domains in LLPS types. In the same veins, we extracted the functional type of protein and repeated the process. Considering that we had prot assemblies in different modules and that some of large LLPS factors might have evolved from multiple genes, we created a variable to include partial match to larger molecules in LLPS types and counted the number of Pfam assignments of the larger molecules. In domain dimer level, we made lists of possible domain dimers, and calculated the frequencies of domain dimers. Mainly, variables were vectors containing the frequencies in different LLPS types. RNA binding domain, DNA binding domain (DBD), DMI from 3did, and domains with low complexity regions, with disordered regions, with repeats, with coiled-coil structures, with phosphorylation sites, and with active sites such as residues responsible for catalysis, which will be referred as “special flags”. They were created based on Pfam, D2P2, and DrLLPS [24,33,40].
2.4. GO Analysis Module
TAIR provides GO and PO data [26,27]. We created five categories- GO analysis, signaling pathway, gene association, PO- anatomy gene, and PO- temporal gene. We retrieved 4 types of subcategories; the first one included following attributes involving signaling pathway, for example hormones, response to light, and osmosensing, the second one included 34 major GO terms for GO analysis such as cell communication and response to abiotic stimulus, and the third one included words related to regulatory roles in the annotation, e.g. enhancer, suppressor, chaperone, and activator, and the last sub-category was original attribute of the database, e.g., acts_upstream_of_negative_effect, or part_of. We created vector variables containing frequency information of categories in the same way as those in oligomerization module or LLPS module.
2.5. TF-Target Module
TF information was provided by Cis-BP for human, A. thaliana, and peach [28]. Cis-BP predicted sequence preferences of TFs and measured correlations between DBD sequence similarities and DNA sequence preferences. We counted the number of how many types of DNA motifs TF bound and how many TFs DNA motif bound. The information were incorporated in order to effectively search for TF and TF targets in respect to their relationship to LOG.
2.6. PPI Module
String and TcoF-DB databases provided PPI information for human, A. thaliana, and peach [29,41]. In addition, AtRegNet and Interactome 2.0 provided information for A. thaliana [25,42]. LPInsider and NPInter databases provided interaction between proteins and RNAs [43,44]. The RNAs were grouped according to a type such as lncRNA and miRNA. Pfam assignment into transcript was provided by GenBank, Gencode, TAIR, InterPro, and Pfam [25,33,34,45,46].
2.7. TF to Target Module
TF binding sites in targets were provided by TF2DNA database for human and by Yu et al (2016) for A. thaliana [25,30]. The binding sites were mapped to gene features by bedmap program [47]. Gencode GFF file and Ensembl GFF file were used for human and for A. thaliana, respectively [46,48]. Gene features included CDS, exon, UTR, intron, upstream-, downstream regions and binding frequencies in each feature were counted.
2.8. Proof of Concept of Search Algorithms
We provided a rough sketch to study interactions between DBD and target motif and between nucleotide-containing ligand and protein's ligand binding motif. To reduce dimensions of variables, amino acids were grouped according to polarity and charge of their side chain (Table 1). Cysteine, glycine, histidine, and proline were considered to have special properties. Each of them made a single membership group.
Using new amino acid group letters, the frequencies of trimers in DBDs were generated: PPP, PPN, PNP, ..., RHR, RRH.
For each DBD, Cis-BP provided ambiguous DNA sequences of the binding motifs in target genes. DNAs and ambiguous DNAs were reassigned to DNA group letters (Table 2). Trimers of DNA group letters and their frequencies in binding motifs were generated.
NBDB provided protein motifs (conserved sequence profiles) that interacted with 24 nucleotide-containing ligands (AMP, ADP, ATP, GMP, GDP, GTP, CTP, CoA, Acelyl-CoA, FMN, F-420, FAD(H), NAD(H), NADP, cyclic nucleotides and dinucleotides, cAMP, cGMP, c-di-AMP, c-di-GMP, and other biologically-relevant cofactors (SAM, PPS, PAP, PLP, ThPP, THD) [39]. We converted NBDB member sequences such as ENAGDTEAPT into new amino acid group letters, and created vector variables containing frequencies of the trimers of the new amino acid group letters per member sequence. Combinations of atoms and moieties belonging to 24 ligands were assigned into 11 groups: RBP, RBPF, RBPN, RBPS, RBPSO, RBSO, TOP, OP, TP, RPF, RPFO (R:ribose, B:base, P:phosphate, F:Flavin, N:Nicotinamide, S:Sulfur, and O:other moiety).
2.9. Demonstration of the Program Usage
2.9.1. Predictions of Important Features- LLPS Subcellular Type, LLPS Factor Type, GO Type, Gene Association Type, Signaling Pathway Type, Subcellular Location, Oligomerization Feature, and Oligomerization Type
We selected entries with multiple Pfam IDs, extracted features from LOG modules except those containing information on oligomerization types of domain dimers or of ProtCAD entries, converted each numeric vector variable to a categorical variable by mapping or clustering algorithms- EM, MakeDensityBasedClusterer, and SimpleKMeans in WEKA program. Most of the vector variables were sparse and mapped to categorical variables without applying clustering methods. The clustering algorithms were applied to three vector variables in LLPS module. Cluster memberships were the values of the categorical variables. We created datasets with entries common to oligomerization and to DDI modules, divided the sets into LLPS and non-LLPS based on LLPS module (Dataset_llps, Dataset_llps2, Dataset_none_llps).
2.9.2. Predictions of Oligomerization Type and Correlations Analysis
We selected TFs with multiple Pfam IDs from TF-target modules, retrieved TF interacting proteins from PPI module, selected only those proteins having LLPS properties, and retrieved information of the LLPS factors from LOG and TF-target modules (Dataset_tf_llps_factor). We excluded variables containing information on oligomerization types of the prot-assembly entries.
2.9.3. Prediction of DNA Binding Motif Types and Association Study
We selected target motif types of TFs that interacted with LLPS factors by physical contacts from the second test, retrieved information from LOG modules as the first and the second test, and additionally domain characterization information from DDI module (Dataset_co_tf_pras_ptm).
To datasets in 1, 2, and 3, we applied classification algorithms in WEKA program (Supplementary Data). We applied FAMD with the package ‘FactoMineR’ in R [49]. FAMD outputs contained information on coordinates of data projected in principal dimensions, cos2, and contrib of variables where cos2 was the quality of representation on principal dimension space and contrib was the contribution to principal dimensions. We applied associations function in dython module from Python to calculate Pearson's correlations [50]. In the third test, we used hotspot algorithm in Weka library to conduct association study to identify the variables having strong associations with DNA binding motif types and with LLPS functional type (scaffold, regulator, client). Detailed information on the algorithms used in Weka library is in the Prediction folder of supplementary data.
3. Results
3.1. Prediction of Important Features
AI models had the average accuracy of 87% in predicting the LLPS types on LLPS dataset (Prediction_llps.llps.type.txt). AI model based on the LLPS dataset without three clustered variables had the accuracy of 82% in predicting oligomerization type, which was higher than that based on all the variables by 25% (Prediction_llps.txt). Three oligomerization types- homo_hetero_moderate_oligomer_obligate, homo_obligate_monomer_oligomer_moderate, and homo_obligate_monomer_obligate- belonged to only prot assemblies in LLPS dataset and not to those in non-LLPS dataset. In the entire dataset, there was only one protein with homo_obligate_monomer_obligate type. Formation and dissolution of LLPS are dynamic and correlate with concentrations of proteins and nucleotides. Therefore, it seemed reasonable to have higher occurrences of the prot assemblies which formed both oligomers and monomers- those ending with “monomer_oligomer_moderate”- in LLPS dataset. Physicochemical and structural properties of LLPS remain elusive. If domain rearrangement occurs within LLPS, it might provide an explanation for higher occurrences of prot assemblies starting with "homo_hetero_moderate", which may have domain sets flexible enough to accommodate domain binding interfaces of both intra-molecule and inter-molecules. We had nested variables belonging to intra_inter_freq that was derived from ProtCAD; they indicated the frequencies of domain sets observed within the same gene and across multiple genes. Those starting with homo_hetero_moderate had non-zero numbers in the nested variables. It should be also noted that 1417 prot assemblies were included in LLPS dataset while only 114 in non-LLPS dataset. AI model developed based on non-LLPS dataset had higher accuracy- 95% (Prediction_none_llps.txt, Figure S1).
3.2. Prediction and Extraction of Important Features from TF-LLPS Factor Data
The accuracy in predicting dominant oligomerization type was 97% (Prediction_tf_llps_factor, Figures S2 and S3). In addition, oligomerization type prediction distributions, in other words probabilities of oligomerization types a prot-assembly formed, were calculated by AI algorithms in Weka library; An example is given in Table S2. Caution should be taken in selecting what prot assemblies to be included in a dataset, if you plan to analyze results from oligomerization type prediction distribution. There were several entries that had the same domain membership but different oligomerization types in the dataset. We selected one oligomerization type at random and included it in the dataset for each of these entries. The distribution had the actual oligomerization types in sequential order from the highest probability. The dataset was made on LLPS factors that interacted with TFs by physical contacts based on String database. However, only a few of them had several entries with multiple oligomerization types. Therefore, an estimation on the credibility of the accuracy needs to be made. Correlation analysis showed that domain dimer feature from ProtCAD, features of binding interface of DDI from 3did, flags, LLPS types, LLPS functional types, and number of domains of LLPS factors had relatively high correlation with oligomerization types (corr > 0.6). We applied FAMD and plotted coordinates of the variables in the first and the second principal dimensions; variables of TF's target motifs were located close to the variables from oligomerization and LLPS modules (Figure 2).
Note: Attributes in the vertical column in left is Y axis and those in the horizontal row on top is X axis in each scatter plot. Labels of X-axis- A:protcad_llps_one_pfam_groupid_sing_intra_inter_freq_sum; B:protcad_llps_one_pfam_groupid_sing_oli_stat_sum; C:protcad_llps_one_pfam_groupid_sing_even_odd_sum; D:protcad_llps_one_pfam_groupid_sing_all_max_sum; E:protcad_llps_dimer_sing_oli_sum; F:protcad_llps_dimer_sing_intra_inter_freq_sum; G:protcad_llps_dimer_multi_oli_sum; H:protcad_llps_dimer_multi_intra_inter_freq_sum; I:protcad_llps_one_pfam_sing_intra_inter_freq_sum; J:protcad_llps_one_pfam_multi_intra_inter_freq_sum; K:protcad_llps_one_pfam_sing_oli_stat_sum; L:protcad_llps_one_pfam_multi_oli_stat_sum; M:protcad_llps_cell_loc; N:threedid_llps; O:flag_llps; P:ANA_dimer; Q:ANA_onepfam; R:TEMP_dimer; S:TEMP_onepfam; T:CONDEN_FUNC_LLPS_onepfam; U:llpsOrNot; V:llpsChkUniprot; W:llpsType; X:llpsUniprot; Y:co_tf_fam_val; Z:co_tf_motif_fam_vall; A1:plant_prAS_data_0; B1:plant_prAS_data_1; C1:plant_prAS_list_data_0; D1:plant_prAS_list_data_1; E1:plant_prAS_dom_linker_data_0; F1:plant_prAS_dom_linker_data_1; G1:plant_prAS_dom_linker_list_data_0; H1:plant_prAS_dom_linker_list_data_1; I1:ptmfeature_data_0; J1:ptmfeature_data_1; K1:ptmfeature_list_data_0; L1:ptmfeature_list_data_1; M1:ptmfeature_dom_linker_data_0; N1:ptmfeature_dom_linker_list_data_0; O1:query_plant_prAS_data; P1:query_plant_prAS_list_data; Q1:query_plant_prAS_dom_linker_data; R1:query_plant_prAS_dom_linker_list_data; S1:query_ptmfeature_data; T1:query_ptmfeature_list_data; U1:co_tf_per_tf_val_0; V1:co_tf_per_tf_val_1; W1:co_tf_per_tf_val_2; X1:co_tf_per_tf_val_3; Y1:co_tf_per_tf_val_4; Z1:co_tf_per_tf_val_5; A2:co_tf_per_tf_val_6; B2:co_tf_per_tf_val_7; C2:co_tf_per_tf_val_8; D2:co_tf_per_tf_val_9; E2:co_tf_per_tf_val_10; F2:co_tf_per_tf_val_11; G2:co_tf_per_tf_val_12; H2:co_tf_per_tf_val_13; I2:co_tf_per_tf_val_14; J2:co_tf_per_tf_val_15; K2:co_tf_per_tf_val_16; L2:co_tf_per_tf_val_17; M2:co_tf_per_tf_val_18; N2:co_tf_per_tf_val_19; O2:co_tf_per_tf_val_20; P2:co_tf_per_tf_val_21; Q2:co_tf_per_tf_val_22; R2:co_tf_per_tf_val_23; S2:co_tf_per_tf_val_24; T2:co_tf_per_tf_val_25; U2:co_tf_per_tf_val_26; V2:co_tf_per_tf_val_27; W2:co_tf_per_tf_val_28; X2:co_tf_per_tf_val_29; Y2:co_tf_per_tf_val_30; Z2:co_tf_per_tf_val_31; A3:co_tf_per_tf_val_32; B3:co_tf_per_tf_val_33; C3:co_tf_per_tf_val_34; D3:co_tf_per_tf_val_35; E3:co_tf_per_tf_val_36; F3:co_tf_motif_val; Y-AXIS; co_tf_motif_fam_val.
Explanation of variable names:
one_pfam/onepfam: Per one domain; dimer: Per domain dimer (e.g., Dom1 and Dom2); multi: Per Pfam architechture of GroupID (e.g., Dom1, Dom2, Dom3); intra_inter_freq_sum: sum of frequencies of domain(s) in three different groups of prot-assemblies; the first, the second, and the third is where the number of the Uniprot genes is less than, the same as, and the greater than the stoichiometry provided in each GroupID, respectively; oli_stat_sum: sum of frequencies of occurrences in the oligomerization type (e.g., C1_obligate_monomer_obligate, C1_obligate_hetero_single_oligomer_obligate, homo_obligate_monomer_oligomer_moderate, homo_obligate_oligomer_obligate, hetero_obligate_monomer_oligomer_moderate, hetero_obligate_oligomer_obligate, homo_hetero_moderate_monomer_obligate, homo_hetero_moderate_monomer_oligomer_moderate, homo_hetero_moderate_oligomer_obligate); even_odd_sum: sum of frequencies of occurrences in two different groups of symmetries; the first and the second is odd (e.g., C3) and even (e.g., C4), respectively.
cell_loc: presence/absence in 16 different subcellular locations; threedid_llps: differences between non-redundant and redundant sets created based on 13 variables; flag_llps: frequences of special flags; ANA: frequences of 295 different PO IDs; TEMP: frequencies of 64 different PO IDs; CONDEN_FUNC_LLPS_onepfam: frequencies of LLPS functional types: client, regulator, scaffold; llpsType: frequencies in 265 different LLPS types; llpsChkUniprot: if uniprot genes are associated with LLPS or not in each llpsType; llpsUniprot: the maximum number of Pfam assignment of associated uniprot genes in each llpsType; co_tf_fam_val: frequencies of PPI with TF families (BHLH,BZIP,C2H2_ZF,CSD,E2F,HOMEODOMAIN,HSF,MADF,MYB/SANT,NAC/NAM,SOX,TBP,TCP,TCR/CXC,UNKNOWN); co_tf_motif_fam_vall: frequencies of TF motif families (MS02_2.00,MS10_2.00,MS11_2.00,MS13_2.00,MS18_2.00,MS21_2.00,MS27_2.00,MS28_2.00,MS31_2.00,MS33_2.00,MS42_2.00,MS46_2.00,MS56_2.00,MS57_2.00,MS59_2.00,MS62_2.00,MS63_2.00,MS64_2.00); For 37 TFs, (AT1G09770,AT1G55520,AT1G75080,AT2G17870,AT2G23380,AT2G23740,AT2G41130,AT3G02150,AT3G12810,AT3G13445,AT3G17609,AT3G19510,AT3G24140,AT3G24520,AT3G28730,AT3G44460,AT3G47620,AT3G48160,AT3G48430,AT3G52300,AT3G56770,AT3G56850,AT4G02020,AT4G02640,AT4G16780,AT4G29000,AT4G34530,AT4G35580,AT4G37790,AT5G04240,AT5G11260,AT5G22220,AT5G22290,AT5G28770,AT5G46690,AT5G51910,AT5G63420), plant_prAS and ptmfeature data were created; prAS_data: values created based on gravy,pi,local,local2,solblity,ubiqui,glycosyl,lowcomp,beta_sheet,disorder,signal,trans_mem,S–S bond,dom_link,pass,o_glycosyl, where loc1 includes chloroplast transit peptide, mitochondrial targeting peptide, secretory pathway signal peptide and loc2 includes E.R.,chlo,mito, cysk, cyto, nucl, plas, extr, golg, pero, vacu; ptmfeature_data: values created based on Glycation,Lysine,Methylation,N-glycosylation,N-terminal,O-GlcNAcylation,Oxidation,Persulfidation,Phosphorylation,S-cyanylation,S-nitrosylation,S-sulfenylation; query: values of LLPS factor; co_tf_per_tf_val: Per each of 37 TFs, domain associated values were calculated. For details, please refer to the codes provided (Table S1).
3.3. Prediction and Association Study on LLPS Factor Interacting with TFs
The accuracy in predicting DNA binding motif types of TFs that the LLPS factor interacted with was 97% (Prediction_co_tf_pras_ptm, Figure 2). Association study with Hotspot algorithm showed that DNA binding motif types had strong associations with Plant-PrAS features of TFs, which is a higher ranked variable created based on all of the Plant-PrAS features. Those serving as both scaffold and regulator had coiled-coil regions. All of the presence/absence value of multiple oligomerization type had a perfect match with binary variable- DMI flag. It should be noted that some LLPS factors had the same domain architectures, which might have biased the data.
4. Discussions
The Plant-PrAS features had associations with properties of different functional types of LLPS. For example, LLPS factors that contained WD40 domains served as scaffold. Some of their partner TFs had domain linker, S–S bond, IDR, beta-sheet, low complexity regions, glycosylation, ubiquitination, all of which also belonged to the LLPS factors (scaffold) themselves, except domain linker. Interestingly, TFs interacting with the LLPS factors all had phosphorylation. TFs containing WD40 involved in transcription activation of anthocyanin synthesis related structural genes in barley [51]. A number of different LLPS scaffolds and regulators seemed to manage coordinated interactions for anthocyanin synthesis, transport, and storage where Natural Deep Eutectic Solvent (NADES) was speculated to be used as inert solvent, which suggested highly complex regulatory processes [52]. As physicochemical properties of liquid condensates remain largely unknown, in-vitro experiments from liquid condensates may encounter problems of partial information. As interaction of liquid condensate is relatively new research topic, standard methods in molecular biology and downstream analysis may require implementation of new protocols and algorithms. Although current technology may have limitations in providing complete information, it may offer practical information for biomarker development. Structural properties contributing to LLPS formation or satisfying constraints imposed by the LLPS, which pose an impact on DNA binding sites, may be roughly estimated with comparative study using AI models based on a large number of factors indicating cellular processes retrieved from numerous databases. GO analysis showed that proteins containing WD40 are part of histone deacetylase complex, nuclear pore, vesicle coat, ubiquitin ligase complex, preribosome, and spliceosome, and enable the following: DNA-binding transcription factor, histone binding, kinase binding, protein heterodimerization and homodimerization activity, kinase activity, phosphatase regulator, ribosome binding, signaling receptor complex, structural molecule activity and transcription cis-regulatory region in A. thaliana (Table S3). WD40 may form important structural platforms for proteins involving in epigenetic activities, which are strongly correlated with genetic variants leading to diseases and phenotype diversity [53,54,55,56].
The Plant-PrAS features and PTM features had supplemented the limited representational power of flags. For instance, homeobox, bZIP, and TCR TF families included gene members that interacted with LLPS factors and had domains with coiled-coil regions. Coiled-coil regions showed strong associations with DBD domains and domains containg Intero annotation called "activity". The canonical coiled-coil has structural motif- heptad repeat [57]. The domains involving in oligomerization such as leucine zipper, N-terminal of homeobox, and helix-loop-helix (HLH) also contain repeats. Although a considerable portion of TF families has various repeats in oligomerization domains, repeats flag is only equipped with the capacity to detect domains defined as repeats. Addition of flags with detection capacity for domains containing such repeats and half sites will improve the program performance. Considering cellular in-vivo environment is dynamic with a large number of factors constantly changing, it will increase accuracy if we make predictions based on multiple variables from modules rather than a single variable such as oligomerization type (e.g. homo_obligate_monomer_oligomer_moderate) in predicting interaction modes in PPI. The proof of concept of search algorithms in the method section can be implemented in PNI analysis with additions of structural elements of nucleotides such as repeats and G-quadruplex, which remains for further research [58,59,60,61,62].
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. All datasets and program codes can be downloaded at www.artfoundation.kr. Figure S1: Attribute matrix of the non-LLPS dataset; Figure S2: Attribute matrix of the TF-LLPS factor dataset; Figure S3: FAMD output- the projected coordinates of the variables from LOG, DDI, and TF-target modules in TF-LLPS factor data; Table S1: Variable and feature lists; Table S2: Prediction class distribution table; Table S3. Lists of functions from GO analysis of proteins containing WD40.
Author Contributions
JEK: Conceptualization, Methodology, Software, Formal analysis, Writing - Original Draft. JHJ: Conceptualization, Funding acquisition, Resources. JHK: Conceptualization, Writing - Review & Editing. JHL: Conceptualization. KH: Conceptualization. SK: Conceptualization, Project administration. NJ: Conceptualization, Funding acquisition, Project administration, Resources, Writing - Review & Editing.
Funding
This work was carried out with the support of “Cooperative Research Program for Agriculture Science and Technology Development (Project No. PJ01604401)” Rural Development Administration, Republic of Korea. This study was supported by 2023 the RDA Fellowship Program of National Institute of Horticultural and Herbal Science, Rural Development Administration, Republic of Korea.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
ART FounDATion-LOG were written in Java programming language. The program codes, datasets, models, and outputs from AI models are available for download at www.artfoundation.kr and https://sourceforge.net/projects/artfoundation-log/.
Acknowledgments
We thank Yoo Song-i for helping data processing.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Verde, I., Abbott, A. et al. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat Genet. 2013. 45, 487–494. [CrossRef]
- Cao, K., Zhou, Z., Wang, Q. et al. Genome-wide association study of 12 agronomic traits in peach. Nat Commun. 2016. 7, 13246. [CrossRef]
- Mark W E J Fiers, Liesbeth Minnoye, Sara Aibar, Carmen Bravo González-Blas, Zeynep Kalender Atak, Stein Aerts, Mapping gene regulatory networks from single-cell omics data, Briefings in Functional Genomics. 2018. Volume 17, Issue 4, Pages 246–254. [CrossRef]
- Li, Y., Li, Q., Beuchat, G., Zeng, H., Zhang, C., and Chen, L.Q. Combined analyses of translatome and transcriptome in Arabidopsis reveal new players responding to magnesium deficiency. J. Integr. Plant Biol. 2021. 63: 2075– 2092.
- Qi Su, Sohum Mehta, Jin Zhang. Liquid-liquid phase separation: Orchestrating cell signaling through time and space. Molecular Cell. 2021. Volume 81, Issue 20. Pages 4137-4146. [CrossRef]
- Peng PH, Hsu KW, Wu KJ. Liquid-liquid phase separation (LLPS) in cellular physiology and tumor biology. Am J Cancer Res. 2021. 15;11(8):3766-3776. 7.
- Yan G. Zhao, Hong Zhang. Phase Separation in Membrane Biology: The Interplay between Membrane-Bound Organelles and Membraneless Condensates. Developmental Cell. Volume 55. Issue 1. 200. Pages 30-44.
- Nesterov SV, Ilyinsky NS, Uversky VN. Liquid-liquid phase separation as a common organizing principle of intracellular space and biomembranes providing dynamic adaptive responses. Biochim Biophys Acta Mol Cell Res. 2021. 1868(11):119102.
- Li J, Zhang M, Ma W, Yang B, Lu H, Zhou F, Zhang L. Post-translational modifications in liquid-liquid phase separation: a comprehensive review. Mol Biomed. 2022. 11;3(1):13. [CrossRef]
- Stoyle CL, Stephens PE, Humphreys DP, Heywood S, Cain K, Bulleid NJ. IgG light chain-independent secretion of heavy chain dimers: consequence for therapeutic antibody production and design. Biochem J. 2017. 7;474(18):3179-3188. [CrossRef]
- Tan, W., Cheng, S., Li, Y. et al. Phase separation modulates the assembly and dynamics of a polarity-related scaffold-signaling hub. Nat Commun. 2022. 13, 7181. [CrossRef]
- Oliver AW, Swift S, Lord CJ, Ashworth A, Pearl LH. Structural basis for recruitment of BRCA2 by PALB2. EMBO Rep. 2009. 10(9):990-6. [CrossRef]
- Koehler Lydia C., Grese Zachary R., Bastos Alliny C. S., Mamede Lohany D., Heyduk Tomasz, Ayala Yuna M., TDP-43 Oligomerization and Phase Separation Properties Are Necessary for Autoregulation, Frontiers in Neuroscience. 2022. (16).
- Stein A, Aloy P. Novel peptide-mediated interactions derived from high-resolution 3-dimensional structures. PLoS Comput Biol. 2010. 20;6(5):e1000789. [CrossRef]
- Liu J, Perumal NB, Oldfield CJ, Su EW, Uversky VN, Dunker AK. Intrinsic disorder in transcription factors. Biochemistry. 2006. 6;45(22):6873-88.
- Puranik S, Acajjaoui S, Conn S, Costa L, Conn V, Vial A, Marcellin R, Melzer R, Brown E, Hart D, Theißen G, Silva CS, Parcy F, Dumas R, Nanao M, Zubieta C. Structural basis for the oligomerization of the MADS domain transcription factor SEPALLATA3 in Arabidopsis. Plant Cell. 2014. 26(9):3603-15. [CrossRef]
- Sayou, C., Nanao, M., Jamin, M. et al. A SAM oligomerization domain shapes the genomic binding landscape of the LEAFY transcription factor. Nat Commun. 2016. 7, 11222.
- Kato M, Hata N, Banerjee N, Futcher B, Zhang MQ. Identifying combinatorial regulation of transcription factors and binding motifs. Genome Biol. 2004. 5(8):R56. [CrossRef]
- Sanchez-Burgos I, Espinosa JR, Joseph JA, Collepardo-Guevara R. RNA length has a non-trivial effect in the stability of biomolecular condensates formed by RNA-binding proteins. PLoS Comput Biol. 2022. 2;18(2):e1009810.
- Qifang Xu, Roland?L Dunbrack, Jr., The protein common assembly database (ProtCAD) a comprehensive structural resource of protein complexes, Nucleic Acids Res. 2022. gkac937. [CrossRef]
- Roberto Mosca, Arnaud Céol, Amelie Stein, Roger Olivella, Patrick Aloy, 3did: a catalog of domain-based interactions of known three-dimensional structure, Nucleic Acids Res. 2014. Volume 42, Issue D1, 1.Pages D374–D379. [CrossRef]
- Kurotani A, Yamada Y, Shinozaki K, Kuroda Y, Sakurai T. Plant-PrAS: a database of physicochemical and structural properties and novel functional regions in plant proteomes. Plant Cell Physiol. 2015 Jan;56(1):e11. [CrossRef]
- Xue H, Zhang Q, Wang P, Cao B, Jia C, Cheng B, Shi Y, Guo WF, Wang Z, Liu ZX, Cheng H. qPTMplants: an integrative database of quantitative post-translational modifications in plants. Nucleic Acids Res. 2022 Jan 7;50(D1):D1491-D1499. [CrossRef]
- Ning W, Guo Y, Lin S, Mei B, Wu Y, Jiang P, Tan X, Zhang W, Chen G, Peng D, Chu L, Xue Y. DrLLPS: a data resource of liquid-liquid phase separation in eukaryotes. Nucleic Acids Res. 2020. 8;48(D1):D288-D295. [CrossRef]
- Rhee SY, Beavis W, Berardini TZ, Chen G, Dixon D, Doyle A, Garcia-Hernandez M, Huala E, Lander G, Montoya M, Miller N, Mueller LA, Mundodi S, Reiser L, Tacklind J, Weems DC, Wu Y, Xu I, Yoo D, Yoon J, Zhang P. The Arabidopsis Information Resource (TAIR): a model organism database providing a centralized, curated gateway to Arabidopsis biology, research materials and community. Nucleic Acids Res. 2003. 1;31(1):224-8. [CrossRef]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000. 25(1):25-9.
- R. L. Walls*, Cooper*, L. D., Elser, J. L., Gandolfo, M. A., Mungall, C. J., Smith, B., Stevenson, D. W., and Jaiswal, P., “The Plant Ontology Facilitates Comparisons of Plant Development Stages Across Species”, Frontiers in Plant Science. 2019. vol. 10.
- Weirauch MT, Yang A, Albu M, Cote AG, Montenegro-Montero A, Drewe P, Najafabadi HS, Lambert SA, Mann I, Cook K, Zheng H, Goity A, van Bakel H, Lozano JC, Galli M, Lewsey MG, Huang E, Mukherjee T, Chen X, Reece-Hoyes JS, Govindarajan S, Shaulsky G, Walhout AJ, Bouget FY, Ratsch G, Larrondo LF, Ecker JR, Hughes TR. Determination and inference of eukaryotic transcription factor sequence specificity. Cell. 2014. 11;158(6):1431-43. [CrossRef]
- Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, Jensen LJ, Mering CV. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019. 8;47(D1):D607-D613. [CrossRef]
- Pujato M, Kieken F, Skiles AA, Tapinos N, Fiser A. Prediction of DNA binding motifs from 3D models of transcription factors; identifying TLX3 regulated genes. Nucleic Acids Res. 2014. 42(22) : 13500-12.
- Yu, CP., Lin, JJ. & Li, WH. Positional distribution of transcription factor binding sites in Arabidopsis thaliana. Sci Rep. 2016. 6, 25164. [CrossRef]
- Wang Y, Wang Q, Huang H, Huang W, Chen Y, McGarvey PB, Wu CH, Arighi CN, UniProt Consortium. A crowdsourcing open platform for literature curation in UniProt. Plos Biology. 2021. 19(12):e3001464.
- Jaina Mistry, Sara Chuguransky, Lowri Williams, Matloob Qureshi, Gustavo A Salazar, Erik L L Sonnhammer, Silvio C E Tosatto, Lisanna Paladin, Shriya Raj, Lorna J Richardson, Robert D Finn, Alex Bateman, Pfam: The protein families database in 2021, Nucleic Acids Res. 2021. Volume 49, Issue D1, 8, Pages D412–D419. [CrossRef]
- Paysan-Lafosse T, Blum M, Chuguransky S, Grego T, Pinto BL, Salazar GA, Bileschi ML, Bork P, Bridge A, Colwell L, Gough J, Haft DH, Letunić I, Marchler-Bauer A, Mi H, Natale DA, Orengo CA, Pandurangan AP, Rivoire C, Sigrist CJA, Sillitoe I, Thanki N, Thomas PD, Tosatto SCE, Wu CH, Bateman A. InterPro in 2022. Nucleic Acids Res. 2022. [CrossRef]
- H.M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T.N. Bhat, H. Weissig, I.N. Shindyalov, P.E. Bourne, The Protein Data Bank. Nucleic Acids Res. 2000. 28: 235-242 . [CrossRef]
- Berardini, T.Z., Reiser, L., Li, D., Mezheritsky, Y., Muller, R., Strait, E. and Huala, E. The arabidopsis information resource: Making and mining the “gold standard” annotated reference plant genome. genesis. 2015. 53: 474-485. [CrossRef]
- Eibe Frank, Mark A. Hall, and Ian H. Witten. The WEKA Workbench. Online Appendix for "Data Mining: Practical Machine Learning Tools and Techniques", Morgan Kaufmann, Fourth Edition, 2016.
- Degtyareva AO, Antontseva EV, Merkulova TI. Regulatory SNPs: Altered Transcription Factor Binding Sites Implicated in Complex Traits and Diseases. Int J Mol Sci. 2021. 16;22(12):6454. [CrossRef]
- Zheng Z, Goncearenco A, Berezovsky IN. Nucleotide binding database NBDB--a collection of sequence motifs with specific protein-ligand interactions. Nucleic Acids Res. 2016. 4;44(D1):D301-7. [CrossRef]
- Oates ME, Romero P, Ishida T, Ghalwash M, Mizianty MJ, Xue B, Dosztányi Z, Uversky VN, Obradovic Z, Kurgan L, Dunker AK, Gough J. D²P²: database of disordered protein predictions. Nucleic Acids Res. 2013. 41(Database issue):D508-16. [CrossRef]
- Schaefer U, Schmeier S, Bajic VB. TcoF-DB: dragon database for human transcription co-factors and transcription factor interacting proteins. Nucleic Acids Res. 2011. 39(Database issue):D106-10. [CrossRef]
- Palaniswamy SK, James S, Sun H, Lamb RS, Davuluri RV, Grotewold E. AGRIS and AtRegNet. a platform to link cis-regulatory elements and transcription factors into regulatory networks. Plant Physiol. 2006. 140(3):818-29. [CrossRef]
- Li Y, Wei L, Wang C, Zhao J, Han S, Zhang Y, Du W. LPInsider: a webserver for lncRNA-protein interaction extraction from the literature. BMC Bioinformatics. 2022. 23(1):135. [CrossRef]
- Yuan J, Wu W, Xie C, Zhao G, Zhao Y, Chen R. NPInter v2.0: an updated database of ncRNA interactions. Nucleic Acids Res. 2014. 42(Database issue):D104-8. [CrossRef]
- Benson DA, Cavanaugh M, Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW. GenBank. Nucleic Acids Res. 2013. 41(Database issue):D36-42. [CrossRef]
- Adam Frankish, Mark Diekhans, Anne-Maud Ferreira, Rory Johnson, Irwin Jungreis, Jane Loveland, Jonathan M Mudge, Cristina Sisu, James Wright, Joel Armstrong, If Barnes, Andrew Berry, Alexandra Bignell, Silvia Carbonell Sala, Jacqueline Chrast, Fiona Cunningham, Tomás Di Domenico, Sarah Donaldson, Ian T Fiddes, Carlos García Girón, Jose Manuel Gonzalez, Tiago Grego, Matthew Hardy, Thibaut Hourlier, Toby Hunt, Osagie G Izuogu, Julien Lagarde, Fergal J Martin, Laura Martínez, Shamika Mohanan, Paul Muir, Fabio C P Navarro, Anne Parker, Baikang Pei, Fernando Pozo, Magali Ruffier, Bianca M Schmitt, Eloise Stapleton, Marie-Marthe Suner, Irina Sycheva, Barbara Uszczynska-Ratajczak, Jinuri Xu, Andrew Yates, Daniel Zerbino, Yan Zhang, Bronwen Aken, Jyoti S Choudhary, Mark Gerstein, Roderic Guigó, Tim J P Hubbard, Manolis Kellis, Benedict Paten, Alexandre Reymond, Michael L Tress, Paul Flicek, GENCODE reference annotation for the human and mouse genomes, Nucleic Acids Res. 2019. Volume 47, Issue D1, Pages D766–D773.
- Shane Neph, M. Scott Kuehn, Alex P. Reynolds, Eric Haugen, Robert E. Thurman, Audra K. Johnson, Eric Rynes, Matthew T. Maurano, Jeff Vierstra, Sean Thomas, Richard Sandstrom, Richard Humbert, John A. Stamatoyannopoulos, BEDOPS: high-performance genomic feature operations, Bioinformatics. 2012. Volume 28, Issue 14. Pages 1919–1920. [CrossRef]
- Flicek P, Amode MR, Barrell D, Beal K, Brent S, Chen Y, Clapham P, Coates G, Fairley S, Fitzgerald S, Gordon L, Hendrix M, Hourlier T, Johnson N, Kähäri A, Keefe D, Keenan S, Kinsella R, Kokocinski F, Kulesha E, Larsson P, Longden I, McLaren W, Overduin B, Pritchard B, Riat HS, Rios D, Ritchie GR, Ruffier M, Schuster M, Sobral D, Spudich G, Tang YA, Trevanion S, Vandrovcova J, Vilella AJ, White S, Wilder SP, Zadissa A, Zamora J, Aken BL, Birney E, Cunningham F, Dunham I, Durbin R, Fernández-Suarez XM, Herrero J, Hubbard TJ, Parker A, Proctor G, Vogel J, Searle SM. Ensembl 2011. Nucleic Acids Res. 2011. 39(Database issue):D800-6. [CrossRef]
- Lê S, Josse J, Husson F. “FactoMineR: A Package for Multivariate Analysis.” Journal of Statistical Software. 2008. 25(1), 1–18. [CrossRef]
- Van Rossum, G., & Drake, F. L. Python 3 Reference Manual. Scotts Valley, CA: CreateSpace. 2009.
- Chen L, Cui Y, Yao Y, An L, Bai Y, Li X, Yao X, Wu K. Genome-wide identification of WD40 transcription factors and their regulation of the MYB-bHLH-WD40 (MBW) complex related to anthocyanin synthesis in Qingke (Hordeum vulgare L. var. nudum Hook. f.). BMC Genomics. 2023. 4;24(1):166. [CrossRef]
- Buhrman K, Aravena-Calvo J, Ross Zaulich C, Hinz K, Laursen T. Anthocyanic Vacuolar Inclusions: From Biosynthesis to Storage and Possible Applications. Front Chem. 2022. 28;10:913324. [CrossRef]
- Ma M, Ru Y, Chuang LS, Hsu NY, Shi LS, Hakenberg J, Cheng WY, Uzilov A, Ding W, Glicksberg BS, Chen R. Disease-associated variants in different categories of disease located in distinct regulatory elements. BMC Genomics. 2015.16 Suppl 8(Suppl 8):S3. [CrossRef]
- Terrile MC, Tebez NM, Colman SL, Mateos JL, Morato-López E, Sánchez-López N, Izquierdo-Álvarez A, Marina A, Calderón Villalobos LIA, Estelle M, Martínez-Ruiz A, Fiol DF, Casalongué CA, Iglesias MJ. S-Nitrosation of E3 Ubiquitin Ligase Complex Components Regulates Hormonal Signalings in Arabidopsis. Front Plant Sci. 2022. 4;12:794582. [CrossRef]
- Zhu S, Gu J, Yao J, Li Y, Zhang Z, Xia W, Wang Z, Gui X, Li L, Li D, Zhang H, Liu C. Liquid-liquid phase separation of RBGD2/4 is required for heat stress resistance in Arabidopsis. Dev Cell. 2022. 14;57(5):583-597.e6. [CrossRef]
- Feng C, Cai XW, Su YN, Li L, Chen S, He XJ. Arabidopsis RPD3-like histone deacetylases form multiple complexes involved in stress response. J Genet Genomics. 2021. 20;48(5):369-383. [CrossRef]
- Truebestein L, Leonard TA. Coiled-coils: The long and short of it. Bioessays. 2016. 38(9):903-16. [CrossRef]
- Dang M, Li T, Song J. ATP and nucleic acids competitively modulate LLPS of the SARS-CoV2 nucleocapsid protein. Commun Biol. 2023. 21;6(1):80. [CrossRef]
- Dang M, Li T, Zhou S, Song J. Arg/Lys-containing IDRs are cryptic binding domains for ATP and nucleic acids that interplay to modulate LLPS. Commun Biol. 2022. 1;5(1):1315. [CrossRef]
- Yueying Zhang, Minglei Yang, Susan Duncan, Xiaofei Yang, Mahmoud A S Abdelhamid, Lin Huang, Huakun Zhang, Philip N Benfey, Zoë A E Waller, Yiliang Ding, G-quadruplex structures trigger RNA phase separation, Nucleic Acids Res. 2019. Volume 47, Issue 22, Pages 11746–11754. [CrossRef]
- Erin M. Langdon, Amy S. Gladfelter, Chapter Four - Probing RNA Structure in Liquid–Liquid Phase Separation Using SHAPE-MaP, Editor(s): Elizabeth Rhoades, Methods in Enzymology, Academic Press, Volume 611, Pages 67-79, 2018. [CrossRef]
- Haibo Zhu, Hao Fu, Tianyu Cui, Lin Ning, Huaguo Shao, Yehan Guo, Yanting Ke, Jiayi Zheng, Hongyan Lin, Xin Wu, Guanghao Liu, Jun He, Xin Han, Wenlin Li, Xiaoyang Zhao, Huasong Lu, Dong Wang, Kongfa Hu, Xiaopei Shen, RNAPhaSep: a resource of RNAs undergoing phase separation, Nucleic Acids Res. 2022. Volume 50, Issue D1. Pages D340–D346. [CrossRef]
Figure 1.
Main Database modules in ART FounDATion-LOG.
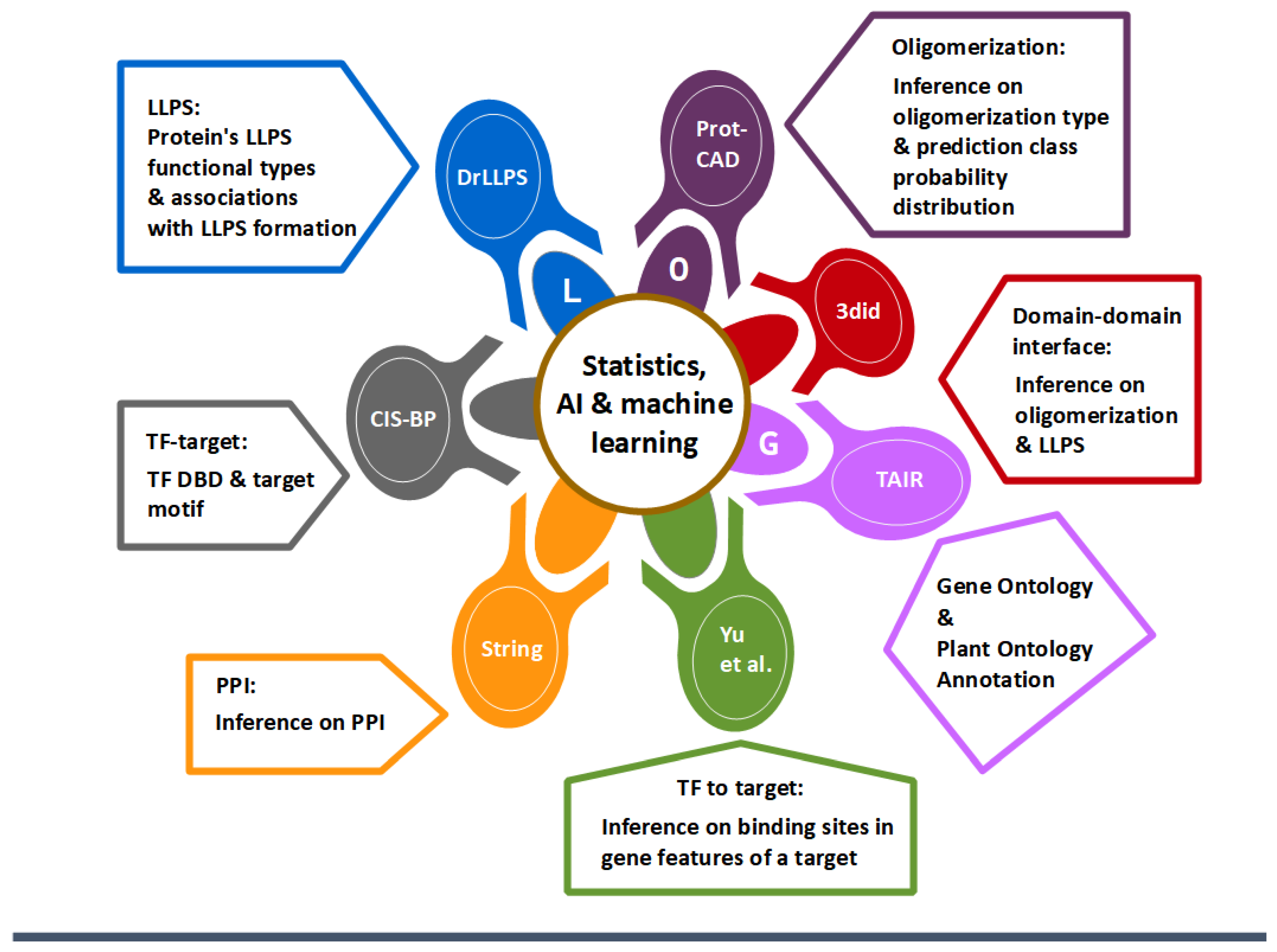
Figure 2.
Attribute matrix of the co_tf_pras_ptm dataset.


Table 1.
Conversion table of amino acids.
| Amino acid group letter | Amino acid | Amino acid features |
| P | R,K,S,T | Positive or polar-uncharged |
| N | D,E,N,Q | Negative or polar-uncharged |
| H | A,V,I,L,M | Hydrophobic |
| R | F,W,Y | Ring structures |
| S | C,G,P,H | Special properties |
Table 2.
Conversion table of DNAs/ambiguous DNAs.
| DNA group letter | DNA/ambiguous DNA |
| G | G |
| Z | R,S,K,B,D,V |
| X | A,C,T,Y,W,M,H |
| N | N |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



