
Submitted:
02 June 2023
Posted:
02 June 2023
You are already at the latest version
Abstract
β-thalassemia, a congenital genetic hematological disorder characterized by decreased or absence of β-globin chains, leads to decrease in levels of Hemoglobin A. The affected individuals can be categorized into two cohorts based on transfusion dependency: transfusion dependent thalassemia (TDT) and non-transfusion dependent thalassemia (NTDT). Remarkably, despite the primary pathology lying in β-globin chain depletion, β-thalassemia exhibits an intriguing association with iron overload. Iron metabolism, a tightly regulated physiological process, reveals a complex interplay in these patients. Over time, both cohorts of β-thalassemic individuals develop iron overload, albeit through distinct mechanisms.
Addressing the diverse complications arising due to iron overload in β-thalassemic patients, the utilization of iron chelators has gained a lot of significance. With varying efficacies, routes of administration, and modes of action, different iron chelators offer unique benefits to patients. In the Indian context, three commercialized iron chelators have emerged, showcasing a high adherence rate to the iron chelator-based treatment regimens among β-thalassemic individuals. In this review, we explore the intriguing connection between β-thalassemia and iron overload, shedding light on the intricate mechanisms at play. We delve into the intricacies of iron metabolism, unveiling the distinct pathways leading to iron accumulation in these patients. Additionally, we critically evaluate the therapeutic efficacy of different iron chelators, emphasizing their respective advantages in managing iron overload complications. Through this comprehensive analysis, we aim to deepen our understanding of β-thalassemia and iron overload, paving the way for optimized treatment strategies. Ultimately, our findings provide valuable insights into improving the care and outcomes of individuals affected by β-thalassemia.
Keywords:
β-thalassemia
; iron overload
; iron chelators
; TDT
; NTDT
1. Introduction
Thalassemia syndromes are reported as a cluster of multi-genetic inherited hematological diseases that are brought upon by impaired formation of one or more chains of hemoglobin [1]. Globally, around 56,000 infants are annually born with severe thalassemia (alpha or beta) with more than half of them reported to require regular blood transfusions [2]. β-thalassemia is represented by decreased(β+) or absent(β0) synthesis of β-globin chains of the most prevalent form of adult haemoglobin, Hemoglobin A (α2β2), due to one or more mutations in the intronic, exonic and/or promoter region of β-globin (HBB) genes, which are present on chromosome 11 [3,4]. According to the previous data available, β-thalassemia has been described to be majorly an autosomal recessive disorder. β-thalassemic individuals can be divided into three cohorts- β-thalassemia major (TM), β-thalassemia intermedia (TI) and β-thalassemia minor (carrier) [5]. It was estimated that around 10,000-12,000 TM infants are born yearly in India and around 42 million β-thalassemia carriers are present in India. The annual prevalence rate of β-thalassemia was determined to be around 3–4% [6,7]. Due to the quantitative reduction in β-globin, particularly in individuals with TM and TI, excess accumulation of α-globin chains in erythroid precursors have been reported which causes globin chain imbalance, resulting in a state called ‘ineffective erythropoiesis’, where in an attempt to generate more erythrocytes (red blood cells), the maturing nucleated erythroid cells undergo early apoptosis to compensate the imbalance. This ultimately leads to chronic hemolytic anemia, requiring regular blood transfusions and other therapeutic approaches like iron chelation therapies, hematopoietic stem cell transplantation, etc. to alleviate their disease symptoms [8,9]. For the classification of thalassemic disorders, dependence on blood transfusion has also been considered as a parameter and hence, it is considered that there are two types of thalassemia- TDT (transfusion dependent thalassemia) and NTDT (non-transfusion dependent thalassemia) [10]. It is necessary for TDT patients to obtain lifelong, recurrent blood transfusions, whereas the NTDT patients require occasional or infrequent blood transfusions.
Iron is a biometal which is reported as a crucial micronutrient for the survival, growth and sustenance of all organisms involved in various significant biological processes like cellular proliferation, certain redox reactions, cell cycle progression, DNA synthesis, ferroptosis, etc. [11,12] It is a co-factor of multiple enzymes because of its capacity to form complexes with organic ligands [13]. An average human is known to maintain a reserve of 3-5g of iron under physiological conditions (around 55mg/kg in males and around 44mg/kg in females) differentially dispersed across various cell types [14]. Around 80% of the iron pool in the human body is related with hemoglobin present in red blood cells, whereas the rest is contained in macrophages and liver hepatocytes [15]. The fine balance in the iron level in the human body as maintained by iron metabolism is critical for homeostasis. Any disequilibrium on either side, leading to deficiency or overload has been linked with cellular damages and damage to various organs of the body. Remarkably, iron overload is frequently reported as a major consequence of β-thalassemia (both TDT and NTDT). Transfusion dependent β-thalassemic (TDT) individuals who receive regular blood transfusions are predisposed to secondary iron overload in diverse organs like liver, heart, etc. and have a greater propensity towards the development of iron toxicity [16]. In NTDT, ineffective erythropoiesis primarily leads to iron overload in patients. Ineffective erythropoiesis in β-thalassemia, induces an elevated production of erythroid progenitor cells, and this consequently requires increased intestinal iron absorption, which ultimately gets deposited in different organs of the body, instead of formation of more erythrocytes [9,17,18]. Further, ineffective erythropoiesis results in increased serum erythropoietin levels and this is coupled with a decline in serum hepcidin levels (detailed mechanism given in section 3.1. TDT vs NTDT) which ultimately results in enhanced iron uptake and eventually, iron overload in different organs, as well [19,20]. The absence of an effective mechanism for elimination of excess iron from the human body, especially in such conditions leads to a plethora of comorbidities are associated with TDT and NTDT as repercussions of iron overload [21].
This review explores the various mechanisms of overloading of iron in β-thalassemic individuals and discusses the molecular pathways involved in such iron overload conditions. Subsequently, we take a critical look at commercially available iron chelators and the contemporary scenario of their usage in the Indian subcontinent. It is expected that this knowledge will help us gain insights into how the paradigm of management of iron overload conditions in β-thalassemia is evolving in the developing countries.
2. Physiologic Iron Metabolism and Its Regulation:
2.1. Absorption and Cellular Uptake-
Dietary iron is considered to be of two types- heme iron (acquired from hemoglobin, hemoproteins and myoglobin of meat) or non-heme iron (acquired from iron-fortified foods). Heme iron is reported to be easily absorbable when compared to non-heme iron [22]. For its intestinal uptake, iron is converted from its ferric state (Fe3+) to its ferrous state (Fe2+) by the ferric reductase duodenal cytochrome B (DYCTB) [23] at the apical side of the enterocytes facing the intestinal lumen. The proton-coupled divalent metal transporter 1 (DMT1), an iron exporter, conducts the absorption Fe2+ from the gut lumen into the enterocyte cytoplasm [24]. The transport of absorbed iron from the enterocytes to systemic circulation is done by the only-identified mammalian iron exporter, Ferroportin1 (FPN1), expressed on the basolateral side of the enterocytes [25]. When FPN1 facilitates the transport of Fe2+ to the extracellular side of the basolateral membrane, Fe2+ is oxidized to Fe3+ by hephaestin and ceruloplasmin (ferroxidases) for effective binding of Fe3+ with circulatory transferrin (Tf) [26,27]. Most of the Tf molecules is produced by the liver [28].
The assimilation of transferrin-bound iron (TBI) into the cell is facilitated by the transferrin receptor-1 (TfR-1) present on the cell membrane of any normal cell, with the exception of the highly differentiated cells [29]. That is followed by the clathrin mediated endocytosis of the TBI-TfR1 complex [30]. After the release of Fe3+ in the endosome, six transmembrane epithelial antigen of prostrate 2 (STEAP2) reduces it to Fe2+ and this Fe2+ is transferred to the cytoplasm of the cell by Dmt1 [24]. Transferrin receptor 2 (TfR2) is the homologous protein of TfR1 and it is expressed ubiquitously on hepatocytes [31].
Species of Non-transferrin bound iron (NTBI) are also observed in the plasma and it is considered that the major forms of NTBI include Fe3+ bound to citrate or acetate, and its transport is facilitated by zinc transporter Zrt-Irt-like protein 14 (Zip 14), L-type and T-type calcium channels, etc. [32,33,34] NTBI is considered to have the most contribution for the iron loading in the liver hepatocytes when Tf is saturated [35].
2.2. Storage-
For storage of iron in the cells, the major protein responsible is ferritin (Ft), which is reported as a spherical protein nanocage of 24 subunits, consisting of heavy (Ft-H) and light (Ft-L) polypeptide chains [36]. Inside the ferritin sphere, upto 4500 atoms of iron (Fe3+) can be stored by incorporating it into a crystalline solid, called ferrihydrite [FeO(OH)8[FeO(H2PO4)], which restricts reactive oxygen species (ROS) formation [37,38]. Ft is contained in cell cytosol, mitochondria, nucleus as well as in serum. It is observed that mitochondrial Ft (mFt) has the capability to store iron more proficiently than cytosolic Ft [39]. Ferritinophagy is the process which regulates the dissociation of iron from Ft, and it is observed that nuclear receptor co-activator 4 (NCOA4) acts as the cargo receptor by associating with the Ft-L transferring the Ft complex for degradation to the lysosome, making the iron stored in that Ft molecule available for biosynthetic reactions [40,41].
The hepatocytes, which comprises around 80% of liver mass, act as the major location for storage of absorbed iron due to their ability to produce large number of Ft molecules [28].
The rest of the cellular iron storage occurs within heme-containing proteins like cytochromes, Fe-S cluster-containing proteins like succinate dehydrogenase as well as non-heme/non Fe-S iron-containing proteins like iron- and 2-oxoglutarate dependent dioxygenases [42,43,44].
Figure 1 elucidates the cellular uptake of iron by the enterocytes and the subsequent transfer of transferrin-bound iron to different cells.
2.3. Consumption and Recycling-
The mammalian body has a high iron requirement with the majority of it being used for hemoglobin synthesis by the erythroblasts [45]. Mitochondrion has the most significant role in maintaining cellular iron homeostasis. Iron released from the endosomes is directed to mitochondria in one of the two following ways. 1. iron can be transferred to mitochondria from the endosomes by a cytosolic iron chaperone protein, poly (rC) binding protein 1 (PCBP1) [46]. 2. Iron can also be delivered, without any intermediate, into the mitochondria from the endosomes via a ‘kiss-and-run’ mechanism, as detected in erythroid cells since they have greater demands of iron for hemoglobin synthesis [47]. The transport of iron between the inner membrane of mitochondria is facilitated by mitoferrins 1 and 2 [48]. Inside the mitochondria, iron is utilized for the production of heme and the Fe-S clusters, which in turn, facilitate the biosynthesis of several proteins associated with electron transfer by incorporating into them [49,50].
Senescent erythrocytes show decreased membrane flexibility, presence of membrane phosphatidylserine, alterations on the erythrocyte solute carrier family 4 (anion exchanger) member 1 (SLC4A1), decreased sialic acid and the cluster of differentiation 47 (CD47) antigen [51,52,53]. Hepatic and splenic macrophages scavenge and phagocytose these senescent erythrocytes to free iron from hemoglobin for utilization in another hemoglobin cycle [54].
2.4. Regulation of Iron Metabolism-
The systemic regulation of the intricate metabolism of iron happens in the mammalian body via the Hepcidin-Ferroportin axis. Hepcidin is 25 amino-acid peptide hormone that primarily expressed by the hepatocytes. Reports suggest that binding of hepcidin to FPN1 on any FPN1-expressing cell types leads to rapid ubiqutination, internalization and lysosomal degradation of FPN1, causing disruption of iron export from the cells and retention of iron in them [55]. Hepcidin is expressed as a product of the HAMP gene, which is positioned on Chromosome 19 [56]. Erythropoiesis, anemia, hypoxia and iron deficiency lead to decreased hepcidin production [57,58]. However, infection, inflammation and iron overload results in increased hepcidin production [59,60,61]. A membrane protein Hemojuvelin (Hjv) is reported to vitally regulate the expression of HAMP gene in the liver, that acts via the Bone Morphogenetic Protein (BMP) signaling pathway [62]. A regulatory serine protease called, Matriptase-2 (encoded by TMPRSS6 gene) that is primarily produced by the liver is known to cleave Hjv, and this is eventually found to impede the production and functioning of hepcidin [63].
On the cellular level, the expressions of different iron metabolism proteins like the subunits of Ft, TfR1 and FPN1 are post-transcriptionally regulated by the association of the iron regulatory proteins (IRPs) to different highly conserved iron responsive elements (IREs), located at the untranslated regions (UTRs) of their corresponding mRNA transcripts [64,65]. IREs can be located at either 5′-UTR (FPN1, Ft-H and Ft-L) or the 3′-UTR (TfR1 and DMT1) [64,66,67,68]. IRP1 and IRP2 are RNA-binding proteins that possess the ability to sense cytosolic iron concentration and bind to their corresponding mRNA targets to modify their expression [69]. When an IRP associates with the IREs present at the 5′-UTR, the translation of the mRNA transcript is hindered and the mRNA transcript is degraded. However, when the IRP associates with the IREs present at the 3′-UTR, the mRNA transcript is stabilized and is actively translated [70].
In iron-deficient cells, IRPs bind to IREs present at the 3′-UTR of TfR1 and DMT1 mRNA transcripts, leading to stabilization of their transcripts and subsequent translation, leading to increase of iron import. The IRPs also binds to IREs present at the 5′-UTR of FPN1, Ft-H and Ft-L mRNA transcripts to facilitate the degradation of their mRNA transcripts and hence, facilitate the decrease of iron storage and export [71].
In iron-adequate cells, the IRPs do not bind to the IREs present at 5′-UTRs of the FPN1, Ft-H and Ft-L mRNA transcripts, and hence they are continuously translated. In contrast to this, the mRNA transcripts with IREs in the 3′-UTR (TfR1 and DMT1) are degraded, thus leading to decreased iron import and increased iron storage and export [70].
3. Iron Overload in Beta-Thalassemia:
As previously mentioned, iron overload is seen as inevitability in both TDT and NTDT β-thalassemic patients. The non-transferrin bound plasma iron (labile iron) pool thus formed leads to generation of ROS which causes lipid peroxidation and leads to dysfunction in various organs, like liver, heart and endocrine glands [72]. Hence, β-thalassemic patients are under an increased oxidative stress. The surplus iron also accumulates in the different end-organs and in turn leads to their subsequent dysfunction which ultimately leads to an increased morbidity [72]. However, there lies a difference in the pattern of iron loading and overloading in different organs with respect to transfusion-dependency.
3.1. TDT vs. NTDT-
Around 200-250 mg of elemental iron is present in each unit of transfused packed red blood cells, while the human body is only capable of losing around 1-2mg of iron every day [73]. In TDT patients, around 0.3-0.6 mg/kg of transfusional iron is incorporated into the body daily, considering a monthly transfusion rate of 2 to 4 units of packed red blood cells [74].
The reticuloendothelial macrophages phagocytose the senescent transfused red blood cells and hence, iron is liberated into plasma for binding to Tf [75]. Even after the saturation threshold of the Tf molecules is reached, NTBI is transported into the cardiomyocytes, hepatocytes and endocrine glands via the calcium channels [34]. In the cardiomyocytes, it was reported that uptake of Fe3+ ions was mediated by lipocalin-2 and its receptor, instead of calcium channels [76,77]. This excess iron leads to an irreversible damage in the different organs and hampers the functionality of those organs. Cardiac dysfunction, owing to iron overload in the myocardium, is one of the main comorbidities related to β-thalassemia, and it leads to nearly 71% mortality associated with the disease [78]. In TDT patients, cardiac siderosis, which leads to arrhythmias and heart failure, along with hepatic and endocrine dysfunction, has been reported [79].
In NTDT patients, ineffective erythropoiesis triggers increased iron absorption from the intestines [80,81]. Furthermore, ineffective erythropoiesis also leads to conditions of anemia and hypoxia and thus, as previously mentioned the hepcidin levels decline to facilitate the compensatory iron acquisition for erythropoiesis [82,83]. This results in upregulation of ferroportin which causes iron release from the enterocytes as well as from the reticuloendothelial system into the systemic circulation [58,84,85]. The increased iron burden leads to deposition of iron in a variety of organs (similar to that of TDT, but at a much slower rate), leading to oxidative damage them [86]. Previously, growth differentiation factor- 15 (GDF-15) and twisted gastrulation 1 (TWSG1) were implicated to have important roles in the hepcidin suppression in NTDT patients. However, upregulation of these proteins were not observed in β-thalassemic mice, casting a doubt over their importance [87]. It has been observed that Erythroferrone, a protein expressed by bone marrow and splenic erythroid precursors, has increased production owing to the erythropoietic stimulation for compensation of ineffective erythropoiesis. Erythroferrone is reported to contribute to iron overload as it is seen to cause hepcidin downregulation [88].
Figure 2 shows a concise description of the pathways for iron overload in TDT and NTDT patients.
Interestingly, it has been observed in NTDT that although the patients showed severe liver iron overload, however, they did not show cardiac iron overload. Hence it has been concluded that iron overload distinctly affects the hepatocytes instead of the cardiomyocytes in NTDT patients [89]. TDT has been seen to be correlated with multiple complications like chronic anemia, liver fibrosis, hypothyroidism, growth retardation, diabetes mellitus, etc. It was revealed from the OPTIMAL CARE study that NTDT present a distinct array of comorbidites which are similar to that of TDT. NTDT-related comorbidities generally include osteoporosis, hypogonadism, leg ulcers, etc. whereas TDT-associated complications like heart failure, hypothyroidism and diabetes mellitus occurred at a lower rate in NTDT patients. Young TDT patients have been seen to develop the clinical iron overload after receiving around 10-20 blood transfusions, whereas NTDT patients mostly developed iron overload slowly over the course of 10-15 years [90,91].
3.2. Differential Expression of Different Proteins in Iron Overload Conditions-
The expression of the major apical iron transporter, DMT1 reportedly does not increase in iron overload conditions, indicating that changes in DMT1 levels are not majorly causing iron overload [92]. Higher levels of the ferroxidase ceruloplasmin have been also observed in β-thalassemic patients, which possibly facilitates increased loading of iron onto transferrin and after high transferrin saturation, also onto albumin and citrate to enhance the formation of ROS-generating labile plasma iron, as well [93]. As previously mentioned, there is an augmentation in the FPN1 levels due to suppression of hepcidin to enable increased iron transport from the cells to the plasma. Furthermore, when the iron binding capacity of Tf is saturated and reaches the threshold due to increased iron load, NTBI increases in the plasma in β-thalassemic patients [74]. As β-thalassemia is considered to be associated with ineffective erythropoiesis, the presence of soluble Tfr1 is seen to be increased in β-thalassemic patients, probably to facilitate the transport of iron required for compensation [85]. The serum ferritin levels are greatly enhanced in β-thalassemic patients [94].
3.3. Detection of Iron Overload-
Studies have demonstrated that those β-thalassemic individuals (carriers and patients) who have histidine to aspartic acid substitution at codon 63 (H63D) of Hemochromatosis gene HFE have a greater propensity towards iron overloading, suggesting the modulating ramifications of H63D mutation of HFE gene on iron metabolism [95,96].
In the blood of TDT β-thalassemic patients, the iron overload can be quantified using serum ferritin, hepatic iron content, urinary iron elimination and total iron binding capacity of transferrin (TIBC) levels [94]. Iron toxicity is considered when the serum ferritin level exceeds 2500ng/mL, hepatic iron content levels exceeds 440mmol/g, urinary iron excretion levels exceeds 20mg/day and the transferrin saturation levels are greater than 75% [97]. A serum ferritin level of 1000ng/ml indicates the threshold for starting iron chelation therapies in TDT patients [98]. In NTDT patients, a threshold value of 800ng/ml is reported as the serum ferritin threshold level representative of iron overload in NTDT patients [99].
For evaluation of the liver iron concentrations, R2 or T2* Magnetic resonance imaging (MRI) can be used. For TDT patients, if the concentration of iron in liver surpasses 7mg/g dry weight (dw) of liver iron concentrations (LIC), then there is a higher propensity of iron overload, whereas a LIC greater than 15 mg/g dw increases chances of severe liver fibrosis and mortality. In NTDT patients, LIC exceeding 5mg/g dw is indicative of increased mortality [91,100,101].
T2* MRI is also the gold standard for detecting cardiac iron overload in milliseconds in β-thalassemic patients. The T2* is observed to get shorter when iron deposition in the myocardium increases [102]. Previous reports indicate that there is an intensifying impairment in the Left Ventricular Ejection Fraction(LVEF) when the T2* value < 20 milliseconds and there is detoriation in the functioning of right and left ventricles when T2* values <14 milliseconds in β-thalassemic patients [103,104]. Severe iron overload is associated with cardiac T2* values < 10 milliseconds [105]. However, a non-significant relationship has been demonstrated between cardiac T2* and serum ferritin levels [103].
4. Iron Chelators:
Since all β-thalassemic patients regardless of their transfusion dependency or non-dependency acquire iron overload, it is imperative to employ iron chelators to eliminate the excess of toxic iron in them and thus, alleviate the symptoms of iron overload. However, for the utilization of iron chelators for a patient, a specific treatment regime is chosen which benefits the individual, taking into consideration the chelating medication’s long-term efficacy, safety and cost.
The three iron chelators commonly used are- Deferoxamine, Deferiprone and Deferasirox. The chemical structures of these three iron chelators have been shown in Figure 3.
Deferoxamine (DFO) is reported to bind to iron at a 1:1 molar ratio [106]. DFO is observed to decrease serum ferritin level and hepatic iron overload in β-thalassemic patients [107]. However, since the plasma half-life of DFO is low, continual injections are required for the iron overloaded patients, either subcutaneously or intravenously [108].
Deferiprone (DFP) is reported to bind to iron at a 3:1 molar ratio. It is administered orally and dosages of 75-120 mg/kg/day of DFP are seen to be usually sufficient to induce a negative iron balance [109]. DFP treatment regime has a high percentage of adherence rate (79-80%) compared to that of Deferoxamine (59-78%); however, it is used as a secondary alternative in β-thalassemic patients only when Deferoxamine is unavailable, since not much about its pharmacodynamics is still unknown to us [110].
Deferasirox (DFX) is reported to bind to iron at a 2:1 molar ratio. DFX is now used by millions of TDT patients with iron overload. DFX is administered orally and it is seen to only increase fecal iron excretion. It has been reported that DFX lowers liver iron content and serum ferritin levels, along with increase in fecal iron excretion in iron overloaded patients at the prescribed dosage of 10-40 mg/kg/day [111,112].
Table 1 elucidates the different characteristics of these three iron chelators.
5. Guidelines for Usage of Iron Chelators in India:
Blood transfusion is reported to be one of the prevalent clinical interventions in modern medicine, alleviating the severe symptoms of patients with chronic anemias such as thalassemia, sickle cell disease, myelodysplastic syndromes, etc, where patients require regular blood transfusions for survival or to improve the quality of life. Previous reports suggested that each millilitre of red cells are packed with approximately 0.8 mg iron, and our physiological mechanism limits us to effectively eliminate approximately 1-2mg of accumulating iron per day only through desquamated oral and intestinal epithelia [119]. Therefore, it is evident that patients who require multiple blood transfusions are often prone to develop rapid iron-overload in the body. Effective management of iron-overload requires efficient monitoring of bodily iron storage. The iron status of the body is readily evaluated by different methods as previously mentioned. Previous reports suggested that serum ferritin level can be regarded as a reliable, cost efficient and readily detectable indicator of bodily iron storage. It has been extensively used to monitor the iron status of the body in recent times. With the advent of modern medicine, iron-chelation has become an effective strategy to alleviate symptoms associated with iron-overload in patients with transfusion-dependent chronic anemias. According to the guidelines of the Ministry of Health and Family Welfare, Government of India, once the serum ferritin levels exceed 1000 mg/L after approximately 10th-12th blood transfusion, the recommended dosages of the three iron chelators as a part of the iron overload treatment regimes are-
- DFO- Continuous subcutaneous injection over 8-12 hours or more with the help of infusion pump, dispersed in water, dosage- 25-50mg/kg/day
- DFP- Orally, in 2-3 divided dosages, dosage- 50-100mg/kg/day
- DFX- Orally, dispersed in water or juice, dosage- 20-40mg/kg/day
- Combination therapy- When patients no longer respond to monotherapies, it is advisable to shift to combined regimes of DFX and DFO [120].
It is absolutely essential for iron-overloaded patients to adhere to the iron chelation therapies for decreased mortality as well as decreased comorbidities. However, in one study from India, it was observed that non-adherence to the iron chelation therapies was found in 10.7% of the iron overloaded patients. The levels of serum ferritin were reported to be exacerbated significantly in non-adherent patients in comparison to that of the adherent patients and higher rate of cardiac and hepatic iron overload was also observed in them.It was also observed that adherence to the treatment regime was highest with DFX, followed by DFP, and finally, DFO [121]. ]. In 2017, Bhattacharyya et al. have reported that DFX is found to be an effective iron chelator that can reduce serum ferritin level efficiently in Indian HbE/β-thalassemia patients with minimum or no adverse effect [122].
In another study, Naithani et al. 2005, assessed the safety of oral iron chelator DFP in young(<6years) thalassemia patients in India. This study reported Thrombocytopenia as the major side effect of the drug and requires frequent monitoring of blood counts [123].
In 2021, Chandra et al. evaluated the risk of development of neutropenia between two thalassemicgroups (patients on combined DFP and DFX and patients with DFX alone). No significant correlations (p=0.87) were found [124]. The adherence to the iron chelation therapies in India has been found to be significantly higher than that of the adolescents of other South Asian countries like Malaysia (51.4%) [125]. Various reasons have been elucidated for the non-adherence to the iron chelation therapies including poor family support, low family income as well as side effects of the iron chelators [121].
All these reports elucidated the present scenario of iron chelators used in treating the patients with iron overload in India. Iron chelation therapy is being looked as one of the feasible options to partially combat the complications associated with regular blood transfusions in severe chronic cases of anemias in India. However, further studies are required to find out the most effective and safe usage of these drugs in patients having heterogeneous clinical symptoms associated with iron overload. Till such time, strict monitoring is required to administer the optimal dosages of these iron chelators used in different clinical settings in India.
6. Conclusions
Iron overload is an indispensible comorbidity associated with β-thalassemia. So, iron chelation therapy is an absolute necessity when it comes to holistic treatment of β-thalassemic patients. Despite the high adherence rate mentioned in the previous-mentioned study, it is difficult to quantify the adherence to the iron chelation therapies from all over India. More research is crucial to understand the entire picture of iron chelation regimes and adherence to them across the whole demographic of India. The β-thalassemia trait is unequally distributed across the Indian subcontinent, so it is necessary to provide adequate treatment to all the iron overloaded or to those who have the higher propensity to develop iron overload. Hence, affordable healthcare options are very necessary for effective clinical management of iron overload, as the medications are currently quite costly, and these might benefit a higher number of people affected with β-thalassemia all over India.
Author Contributions
S.B., M.R. and N.C. were involved in original draft preparation; P.C.S., T.K.D. and N.C. have reviewed and edited the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the Department of Biotechnology (DBT), Ministry of Science and Technology, Government of India (Project Title: “Micro-RNA based reprogramming of fetal hemoglobin in beta-thalassemia”, Sanction no: BT/PR32054/MED/97/454/2019).
Institutional Review Board Statement
Not Applicable.
Informed Consent Statement
Not Applicable.
Data Availability Statement
Not Applicable.
Acknowledgments
Subhangi Basu acknowledges the University Grants Commission (UGC), Government of India, and New Delhi for providing a research fellowship, The authors would like to thank the Indian Institute of Technology Kharagpur for providing infrastructural support. The authors would like to acknowledge the Department of Biotechnology (DBT), Ministry of Science and Technology, Government of India for providing funding.
Conflicts of Interest
The authors have no relevant financial or non-financial interests to disclose.
References
- A. J. Marengo-Rowe, “The thalassemias and related disorders.,” Proc. (Bayl. Univ. Med. Cent)., vol. 20, no. 1, pp. 27–31, Jan. 2007. [CrossRef]
- B. Modell and M. Darlison, “Global epidemiology of haemoglobin disorders and derived service indicators.,” Bull. World Health Organ., vol. 86, no. 6, pp. 480–487, Jun. 2008. [CrossRef]
- R. Galanello and R. Origa, “Beta-thalassemia.,” Orphanet J. Rare Dis., vol. 5, p. 11, May 2010. 11. [CrossRef]
- B. Giardine et al., “Systematic documentation and analysis of human genetic variation in hemoglobinopathies using the microattribution approach,” Nat. Genet., vol. 43, no. 4, pp. 295–302, Mar. 2011. [CrossRef]
- R. Origa, “β-Thalassemia.,” Genet. Med. Off. J. Am. Coll. Med. Genet., vol. 19, no. 6, pp. 609–619, Jun. 2017. [CrossRef]
- R. Aggarwal, A. Prakash, and M. Aggarwal, “Thalassemia: An overview YR - 2014/1/1,” J. Sci. Soc., no. 1 UL-https://www.jscisociety.com/article.asp?issn=0974-5009;year=2014;volume=41;issue=1;spage=3;epage=6;aulast=Aggarwal;t=5, pp. 3 OP-6 VO – 41. [CrossRef]
- M. of H. & F. W. G. of India, “National Health Mission - Guidelines on Hemoglobinopathies in India: Prevention and control of hemoglobinopathies in india.,” pp. 1–138, 2016, [Online]. Available: http://nhm.gov.in/images/pdf/programmes/RBSK/Resource_Documents/Guidelines_on_Hemoglobinopathies_in India.pdf.
- S. L. Thein, “Pathophysiology of beta thalassemia--a guide to molecular therapies.,” Hematol. Am. Soc. Hematol. Educ. Progr., pp. 31–37, 2005. [CrossRef]
- S. Rivella, “β-thalassemias: paradigmatic diseases for scientific discoveries and development of innovative therapies.,” Haematologica, vol. 100, no. 4, pp. 418–430, Apr. 2015. [CrossRef]
- K. M. Musallam, S. Rivella, E. Vichinsky, and E. A. Rachmilewitz, “Non-transfusion-dependent thalassemias.,” Haematologica, vol. 98, no. 6, pp. 833–844, Jun. 2013. [CrossRef]
- J. Beard and O. Han, “Systemic iron status,” Biochim. Biophys. Acta, vol. 1790, pp. 584–588, 2008. [CrossRef]
- X. Chen, C. Yu, R. Kang, and D. Tang, “Iron Metabolism in Ferroptosis.,” Front. cell Dev. Biol., vol. 8, p. 590226, 2020. [CrossRef]
- P. A. Frey and G. H. Reed, “The ubiquity of iron.,” ACS Chem. Biol., vol. 7, no. 9, pp. 1477–1481, Sep. 2012. [CrossRef]
- D. Mancardi, M. Mezzanotte, E. Arrigo, A. Barinotti, and A. Roetto, “Iron Overload, Oxidative Stress, and Ferroptosis in the Failing Heart and Liver.,” Antioxidants (Basel, Switzerland), vol. 10, no. 12, Nov. 2021. [CrossRef]
- A. Lawen and D. J. R. Lane, “Mammalian iron homeostasis in health and disease: uptake, storage, transport, and molecular mechanisms of action.,” Antioxid. Redox Signal., vol. 18, no. 18, pp. 2473–2507, Jun. 2013. [CrossRef]
- L. Malcovati, “Impact of transfusion dependency and secondary iron overload on the survival of patients with myelodysplastic syndromes.,” Leuk. Res., vol. 31 Suppl 3, pp. S2-6, Dec. 2007. [CrossRef]
- R. Gupta, K. M. Musallam, A. T. Taher, and S. Rivella, “Ineffective Erythropoiesis: Anemia and Iron Overload.,” Hematol. Oncol. Clin. North Am., vol. 32, no. 2, pp. 213–221, Apr. 2018. [CrossRef]
- P. R. Oikonomidou and S. Rivella, “What can we learn from ineffective erythropoiesis in thalassemia?,” Blood Rev., vol. 32, no. 2, pp. 130–143, Mar. 2018. [CrossRef]
- L. Melchiori, S. Gardenghi, and S. Rivella, “beta-Thalassemia: HiJAKing Ineffective Erythropoiesis and Iron Overload.,” Adv. Hematol., vol. 2010, p. 938640, 2010. [CrossRef]
- S. Gardenghi, R. W. Grady, and S. Rivella, “Anemia, ineffective erythropoiesis, and hepcidin: interacting factors in abnormal iron metabolism leading to iron overload in β-thalassemia.,” Hematol. Oncol. Clin. North Am., vol. 24, no. 6, pp. 1089–1107, Dec. 2010. [CrossRef]
- R. Colah, A. Gorakshakar, and A. Nadkarni, “Global burden, distribution and prevention of β-thalassemias and hemoglobin E disorders.,” Expert Rev. Hematol., vol. 3, no. 1, pp. 103–117, Feb. 2010. [CrossRef]
- P. A. Sharp, “Intestinal iron absorption: regulation by dietary & systemic factors.,” Int. J. Vitam. Nutr. Res. Int. Zeitschrift fur Vitamin- und Ernahrungsforschung. J. Int. Vitaminol. Nutr., vol. 80, no. 4–5, pp. 231–242, Oct. 2010. [CrossRef]
- B. Silva and P. Faustino, “An overview of molecular basis of iron metabolism regulation and the associated pathologies.,” Biochim. Biophys. Acta, vol. 1852, no. 7, pp. 1347–1359, Jul. 2015. [CrossRef]
- H. Gunshin et al., “Cloning and characterization of a mammalian proton-coupled metal-ion transporter.,” Nature, vol. 388, no. 6641, pp. 482–488, Jul. 1997. [CrossRef]
- N. T. V Le and D. R. Richardson, “Ferroportin1: a new iron export molecule?,” Int. J. Biochem. Cell Biol., vol. 34, no. 2, pp. 103–108, Feb. 2002. [CrossRef]
- K.-Y. Yeh, M. Yeh, L. Mims, and J. Glass, “Iron feeding induces ferroportin 1 and hephaestin migration and interaction in rat duodenal epithelium.,” Am. J. Physiol. Gastrointest. Liver Physiol., vol. 296, no. 1, pp. G55-65, Jan. 2009. [CrossRef]
- G. M. Brittin and Q. T. Chee, “Relation of ferroxidase (ceruloplasmin) to iron absorption.,” J. Lab. Clin. Med., vol. 74, no. 1, pp. 53–59, Jul. 1969.
- G. J. Anderson and D. M. Frazer, “Hepatic iron metabolism.,” Semin. Liver Dis., vol. 25, no. 4, pp. 420–432, Nov. 2005. [CrossRef]
- P. Ponka and C. N. Lok, “The transferrin receptor: role in health and disease.,” Int. J. Biochem. Cell Biol., vol. 31, no. 10, pp. 1111–1137, Oct. 1999. [CrossRef]
- C. E. Herbison et al., “The role of transferrin receptor 1 and 2 in transferrin-bound iron uptake in human hepatoma cells.,” Am. J. Physiol. Cell Physiol., vol. 297, no. 6, pp. C1567-75, Dec. 2009. [CrossRef]
- D. Trinder and E. Baker, “Transferrin receptor 2: a new molecule in iron metabolism.,” Int. J. Biochem. Cell Biol., vol. 35, no. 3, pp. 292–296, Mar. 2003. [CrossRef]
- C. Hershko, G. Graham, G. W. Bates, and E. A. Rachmilewitz, “Non-specific serum iron in thalassaemia: an abnormal serum iron fraction of potential toxicity.,” Br. J. Haematol., vol. 40, no. 2, pp. 255–263, Oct. 1978. [CrossRef]
- M. Grootveld, J. D. Bell, B. Halliwell, O. I. Aruoma, A. Bomford, and P. J. Sadler, “Non-transferrin-bound iron in plasma or serum from patients with idiopathic hemochromatosis. Characterization by high performance liquid chromatography and nuclear magnetic resonance spectroscopy.,” J. Biol. Chem., vol. 264, no. 8, pp. 4417–4422, Mar. 1989.
- M. D. Knutson, “Non-transferrin-bound iron transporters.,” Free Radic. Biol. Med., vol. 133, pp. 101–111, Mar. 2019. [CrossRef]
- R. E. Fleming and P. Ponka, “Iron overload in human disease.,” N. Engl. J. Med., vol. 366, no. 4, pp. 348–359, Jan. 2012. [CrossRef]
- X. Liu and E. C. Theil, “Ferritin as an iron concentrator and chelator target.,” Ann. N. Y. Acad. Sci., vol. 1054, pp. 136–140, 2005. [CrossRef]
- P. Arosio, L. Elia, and M. Poli, “Ferritin, cellular iron storage and regulation.,” IUBMB Life, vol. 69, no. 6, pp. 414–422, Jun. 2017. [CrossRef]
- S. Dev and J. L. Babitt, “Overview of iron metabolism in health and disease.,” Hemodial. Int., vol. 21 Suppl 1, no. Suppl 1, pp. S6–S20, Jun. 2017. [CrossRef]
- S. Levi et al., “A human mitochondrial ferritin encoded by an intronless gene.,” J. Biol. Chem., vol. 276, no. 27, pp. 24437–24440, Jul. 2001. [CrossRef]
- J. D. Mancias, X. Wang, S. P. Gygi, J. W. Harper, and A. C. Kimmelman, “Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy.,” Nature, vol. 509, no. 7498, pp. 105–109, May 2014. [CrossRef]
- D. C. Fuhrmann, A. Mondorf, J. Beifuß, M. Jung, and B. Brüne, “Hypoxia inhibits ferritinophagy, increases mitochondrial ferritin, and protects from ferroptosis.,” Redox Biol., vol. 36, p. 101670, Sep. 2020. [CrossRef]
- H. Senn and K. Wüthrich, “Amino acid sequence, haem-iron co-ordination geometry and functional properties of mitochondrial and bacterial c-type cytochromes.,” Q. Rev. Biophys., vol. 18, no. 2, pp. 111–134, May 1985. [CrossRef]
- R. Lill, “Function and biogenesis of iron-sulphur proteins.,” Nature, vol. 460, no. 7257, pp. 831–838, Aug. 2009. [CrossRef]
- C. C. Philpott and M.-S. Ryu, “Special delivery: distributing iron in the cytosol of mammalian cells.,” Front. Pharmacol., vol. 5, p. 173, 2014. [CrossRef]
- M. J. Koury and P. Ponka, “New insights into erythropoiesis: the roles of folate, vitamin B12, and iron.,” Annu. Rev. Nutr., vol. 24, pp. 105–131, 2004. [CrossRef]
- C. C. Philpott, “The flux of iron through ferritin in erythrocyte development.,” Curr. Opin. Hematol., vol. 25, no. 3, pp. 183–188, May 2018. [CrossRef]
- A. Hamdi, T. M. Roshan, T. M. Kahawita, A. B. Mason, A. D. Sheftel, and P. Ponka, “Erythroid cell mitochondria receive endosomal iron by a ‘kiss-and-run’ mechanism.,” Biochim. Biophys. Acta, vol. 1863, no. 12, pp. 2859–2867, Dec. 2016. [CrossRef]
- P. N. Paradkar, K. B. Zumbrennen, B. H. Paw, D. M. Ward, and J. Kaplan, “Regulation of mitochondrial iron import through differential turnover of mitoferrin 1 and mitoferrin 2.,” Mol. Cell. Biol., vol. 29, no. 4, pp. 1007–1016, Feb. 2009. [CrossRef]
- S. SANO, S. INOUE, Y. TANABE, C. SUMIYA, and S. KOIKE, “Significance of mitochondria for porphyrin and heme biosynthesis.,” Science, vol. 129, no. 3344, pp. 275–276, Jan. 1959. [CrossRef]
- G. Kispal, P. Csere, C. Prohl, and R. Lill, “The mitochondrial proteins Atm1p and Nfs1p are essential for biogenesis of cytosolic Fe/S proteins.,” EMBO J., vol. 18, no. 14, pp. 3981–3989, Jul. 1999. [CrossRef]
- P. S. Low, S. M. Waugh, K. Zinke, and D. Drenckhahn, “The role of hemoglobin denaturation and band 3 clustering in red blood cell aging.,” Science, vol. 227, no. 4686, pp. 531–533, Feb. 1985. [CrossRef]
- D. Bratosin et al., “Molecular mechanisms of erythrophagocytosis. Characterization of the senescent erythrocytes that are phagocytized by macrophages.,” C. R. Acad. Sci. III., vol. 320, no. 10, pp. 811–818, Oct. 1997. [CrossRef]
- G. J. C. G. M. Bosman, F. L. A. Willekens, and J. M. Werre, “Erythrocyte aging: a more than superficial resemblance to apoptosis?,” Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol., vol. 16, no. 1–3, pp. 1–8, 2005. [CrossRef]
- J. L. V. Corrons, L. B. Casafont, and E. F. Frasnedo, “Concise review: how do red blood cells born, live, and die?,” Ann. Hematol., vol. 100, no. 10, pp. 2425–2433, Oct. 2021. [CrossRef]
- K. Yeh, M. Yeh, and J. Glass, “Hepcidin regulation of ferroportin 1 expression in the liver and intestine of the rat.,” Am. J. Physiol. Gastrointest. Liver Physiol., vol. 286, no. 3, pp. G385-94, Mar. 2004. [CrossRef]
- C. H. Park, E. V Valore, A. J. Waring, and T. Ganz, “Hepcidin, a urinary antimicrobial peptide synthesized in the liver.,” J. Biol. Chem., vol. 276, no. 11, pp. 7806–7810, Mar. 2001. [CrossRef]
- M. Vokurka, J. Krijt, K. Sulc, and E. Necas, “Hepcidin mRNA levels in mouse liver respond to inhibition of erythropoiesis.,” Physiol. Res., vol. 55, no. 6, pp. 667–674, 2006. [CrossRef]
- G. Nicolas et al., “The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation.,” J. Clin. Invest., vol. 110, no. 7, pp. 1037–1044, Oct. 2002. [CrossRef]
- P. J. Schmidt, P. T. Toran, A. M. Giannetti, P. J. Bjorkman, and N. C. Andrews, “The transferrin receptor modulates Hfe-dependent regulation of hepcidin expression.,” Cell Metab., vol. 7, no. 3, pp. 205–214, Mar. 2008. [CrossRef]
- L. Lin, E. V Valore, E. Nemeth, J. B. Goodnough, V. Gabayan, and T. Ganz, “Iron transferrin regulates hepcidin synthesis in primary hepatocyte culture through hemojuvelin and BMP2/4.,” Blood, vol. 110, no. 6, pp. 2182–2189, Sep. 2007. [CrossRef]
- Y. Kanamori, M. Murakami, M. Sugiyama, O. Hashimoto, T. Matsui, and M. Funaba, “Hepcidin and IL-1β.,” Vitam. Horm., vol. 110, pp. 143–156, 2019. [CrossRef]
- J. L. Babitt et al., “Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression.,” Nat. Genet., vol. 38, no. 5, pp. 531–539, May 2006. [CrossRef]
- L. Silvestri, A. Pagani, A. Nai, I. De Domenico, J. Kaplan, and C. Camaschella, “The serine protease matriptase-2 (TMPRSS6) inhibits hepcidin activation by cleaving membrane hemojuvelin.,” Cell Metab., vol. 8, no. 6, pp. 502–511, Dec. 2008. [CrossRef]
- C. P. Anderson, M. Shen, R. S. Eisenstein, and E. A. Leibold, “Mammalian iron metabolism and its control by iron regulatory proteins.,” Biochim. Biophys. Acta, vol. 1823, no. 9, pp. 1468–1483, Sep. 2012. [CrossRef]
- Z. D. Zhou and E.-K. Tan, “Iron regulatory protein (IRP)-iron responsive element (IRE) signaling pathway in human neurodegenerative diseases.,” Mol. Neurodegener., vol. 12, no. 1, p. 75, Oct. 2017. [CrossRef]
- J. L. Casey et al., “Iron-responsive elements: regulatory RNA sequences that control mRNA levels and translation.,” Science, vol. 240, no. 4854, pp. 924–928, May 1988. [CrossRef]
- D.-L. Zhang, R. M. Hughes, H. Ollivierre-Wilson, M. C. Ghosh, and T. A. Rouault, “A ferroportin transcript that lacks an iron-responsive element enables duodenal and erythroid precursor cells to evade translational repression.,” Cell Metab., vol. 9, no. 5, pp. 461–473, May 2009. [CrossRef]
- D. Tchernitchko, M. Bourgeois, M.-E. Martin, and C. Beaumont, “Expression of the two mRNA isoforms of the iron transporter Nramp2/DMTI in mice and function of the iron responsive element.,” Biochem. J., vol. 363, no. Pt 3, pp. 449–455, May 2002. [CrossRef]
- G. J. Anderson and D. M. Frazer, “Current understanding of iron homeostasis.,” Am. J. Clin. Nutr., vol. 106, no. Suppl 6, pp. 1559S-1566S, Dec. 2017. [CrossRef]
- T. A. Rouault, “The role of iron regulatory proteins in mammalian iron homeostasis and disease.,” Nat. Chem. Biol., vol. 2, no. 8, pp. 406–414, Aug. 2006. [CrossRef]
- N. Sukhbaatar and T. Weichhart, “Iron Regulation: Macrophages in Control.,” Pharmaceuticals (Basel)., vol. 11, no. 4, Dec. 2018. [CrossRef]
- O. Kakhlon and Z. I. Cabantchik, “The labile iron pool: characterization, measurement, and participation in cellular processes(1).,” Free Radic. Biol. Med., vol. 33, no. 8, pp. 1037–1046, Oct. 2002. [CrossRef]
- A. W. Nienhuis and D. G. Nathan, “Pathophysiology and Clinical Manifestations of the β-Thalassemias.,” Cold Spring Harb. Perspect. Med., vol. 2, no. 12, p. a011726, Dec. 2012. [CrossRef]
- K. Leecharoenkiat, P. Lithanatudom, W. Sornjai, and D. R. Smith, “Iron dysregulation in beta-thalassemia.,” Asian Pac. J. Trop. Med., vol. 9, no. 11, pp. 1035–1043, Nov. 2016. [CrossRef]
- T. D. Coates, “Physiology and pathophysiology of iron in hemoglobin-associated diseases.,” Free Radic. Biol. Med., vol. 72, pp. 23–40, Jul. 2014. [CrossRef]
- S. Kumfu, S. Chattipakorn, S. Fucharoen, and N. Chattipakorn, “Ferric iron uptake into cardiomyocytes of β-thalassemic mice is not through calcium channels.,” Drug Chem. Toxicol., vol. 36, no. 3, pp. 329–334, Jul. 2013. [CrossRef]
- S. Kumfu, S. C. Chattipakorn, and N. Chattipakorn, “Silencing of lipocalin-2 and its receptor improved cardiomyocytes viability via decreasing iron uptake, mitochondrial fission, mitophagy and apoptosis under iron overload condition,” Eur. Heart J., vol. 41, no. Supplement_2, p. ehaa946.3392, Nov. 2020. [CrossRef]
- G. M. Brittenham et al., “Efficacy of deferoxamine in preventing complications of iron overload in patients with thalassemia major.,” N. Engl. J. Med., vol. 331, no. 9, pp. 567–573, Sep. 1994. [CrossRef]
- D. Farmakis, J. Porter, A. Taher, M. Domenica Cappellini, M. Angastiniotis, and A. Eleftheriou, “2021 Thalassaemia International Federation Guidelines for the Management of Transfusion-dependent Thalassemia.,” HemaSphere, vol. 6, no. 8, p. e732, Aug. 2022. [CrossRef]
- T. Tanno and J. L. Miller, “Iron Loading and Overloading due to Ineffective Erythropoiesis.,” Adv. Hematol., vol. 2010, p. 358283, 2010. [CrossRef]
- P. Pootrakul et al., “The effect of erythroid hyperplasia on iron balance.,” Blood, vol. 71, no. 4, pp. 1124–1129, Apr. 1988.
- Pigeon et al., “A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload.,” J. Biol. Chem., vol. 276, no. 11, pp. 7811–7819, Mar. 2001. [CrossRef]
- T. Ganz, “Hepcidin and iron regulation, 10 years later.,” Blood, vol. 117, no. 17, pp. 4425–4433, Apr. 2011. [CrossRef]
- E. Nemeth et al., “Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization.,” Science, vol. 306, no. 5704, pp. 2090–2093, Dec. 2004. [CrossRef]
- R. Origa et al., “Liver iron concentrations and urinary hepcidin in beta-thalassemia.,” Haematologica, vol. 92, no. 5, pp. 583–588, May 2007. [CrossRef]
- A. T. Taher, V. Viprakasit, K. M. Musallam, and M. D. Cappellini, “Treating iron overload in patients with non-transfusion-dependent thalassemia.,” Am. J. Hematol., vol. 88, no. 5, pp. 409–415, May 2013. [CrossRef]
- C. Camaschella and A. Nai, “Ineffective erythropoiesis and regulation of iron status in iron loading anaemias.,” Br. J. Haematol., vol. 172, no. 4, pp. 512–523, Feb. 2016. [CrossRef]
- L. Kautz et al., “Erythroferrone contributes to hepcidin suppression and iron overload in a mouse model of β-thalassemia.,” Blood, vol. 126, no. 17, pp. 2031–2037, Oct. 2015. [CrossRef]
- A. T. Taher, K. M. Musallam, J. C. Wood, and M. D. Cappellini, “Magnetic resonance evaluation of hepatic and myocardial iron deposition in transfusion-independent thalassemia intermedia compared to regularly transfused thalassemia major patients.,” American journal of hematology, vol. 85, no. 4. United States, pp. 288–290, Apr. 2010. [CrossRef]
- A. T. Taher et al., “Overview on practices in thalassemia intermedia management aiming for lowering complication rates across a region of endemicity: the OPTIMAL CARE study.,” Blood, vol. 115, no. 10, pp. 1886–1892, Mar. 2010. [CrossRef]
- A. T. Taher and A. N. Saliba, “Iron overload in thalassemia: different organs at different rates.,” Hematol. Am. Soc. Hematol. Educ. Progr., vol. 2017, no. 1, pp. 265–271, Dec. 2017. [CrossRef]
- F. Canonne-Hergaux, J. E. Levy, M. D. Fleming, L. K. Montross, N. C. Andrews, and P. Gros, “Expression of the DMT1 (NRAMP2/DCT1) iron transporter in mice with genetic iron overload disorders.,” Blood, vol. 97, no. 4, pp. 1138–1140, Feb. 2001. [CrossRef]
- S. M. Awadallah, N. A. Nimer, M. F. Atoum, and S. A. Saleh, “Association of haptoglobin phenotypes with ceruloplasmin ferroxidase activity in β-thalassemia major.,” Clin. Chim. Acta., vol. 412, no. 11–12, pp. 975–979, May 2011. [CrossRef]
- A. K. Mishra and A. Tiwari, “Iron overload in Beta thalassaemia major and intermedia patients.,” Maedica (Buchar)., vol. 8, no. 4, pp. 328–332, Sep. 2013.
- M. A. Melis, M. Cau, F. Deidda, S. Barella, A. Cao, and R. Galanello, “H63D mutation in the HFE gene increases iron overload in beta-thalassemia carriers.,” Haematologica, vol. 87, no. 3, pp. 242–245, Mar. 2002.
- O. R. Zekavat, M. Zareian Jahromi, S. Haghpanah, Z. Kargar Jahromi, and N. Cohan, “Association of HFE Gene Mutations With Serum Ferritin Level and Heart and Liver Iron Overload in Patients With Transfusion-dependent Beta-Thalassemia.,” J. Pediatr. Hematol. Oncol., vol. 43, no. 1, pp. e26–e28, Jan. 2021. [CrossRef]
- C. Hershko, “Pathogenesis and management of iron toxicity in thalassemia.,” Ann. N. Y. Acad. Sci., vol. 1202, pp. 1–9, Aug. 2010. [CrossRef]
- C. Borgna-Pignatti et al., “Survival and complications in patients with thalassemia major treated with transfusion and deferoxamine.,” Haematologica, vol. 89, no. 10, pp. 1187–1193, Oct. 2004.
- A. T. Taher et al., “Deferasirox reduces iron overload significantly in nontransfusion-dependent thalassemia: 1-year results from a prospective, randomized, double-blind, placebo-controlled study.,” Blood, vol. 120, no. 5, pp. 970–977, Aug. 2012. [CrossRef]
- P. T. Telfer, E. Prestcott, S. Holden, M. Walker, A. V Hoffbrand, and B. Wonke, “Hepatic iron concentration combined with long-term monitoring of serum ferritin to predict complications of iron overload in thalassaemia major.,” Br. J. Haematol., vol. 110, no. 4, pp. 971–977, Sep. 2000. [CrossRef]
- C. Borgna-Pignatti et al., “Hepatocellular carcinoma in the thalassaemia syndromes.,” Br. J. Haematol., vol. 124, no. 1, pp. 114–117, Jan. 2004. [CrossRef]
- D. Ansah et al., “Cardiac Magnetic Resonance Strain in Beta Thalassemia Major Correlates with Cardiac Iron Overload.,” Child. (Basel, Switzerland), vol. 10, no. 2, Jan. 2023. [CrossRef]
- L. J. Anderson et al., “Cardiovascular T2-star (T2*) magnetic resonance for the early diagnosis of myocardial iron overload.,” Eur. Heart J., vol. 22, no. 23, pp. 2171–2179, Dec. 2001. [CrossRef]
- C. Liguori et al., “Relationship between myocardial T2 values and cardiac volumetric and functional parameters in β-thalassemia patients evaluated by cardiac magnetic resonance in association with serum ferritin levels.,” Eur. J. Radiol., vol. 82, no. 9, pp. e441-7, Sep. 2013. [CrossRef]
- E. Gammella, S. Recalcati, I. Rybinska, P. Buratti, and G. Cairo, “Iron-induced damage in cardiomyopathy: oxidative-dependent and independent mechanisms.,” Oxid. Med. Cell. Longev., vol. 2015, p. 230182, 2015. [CrossRef]
- H.-J. Cui et al., “Efficacy of deferoxamine in animal models of intracerebral hemorrhage: a systematic review and stratified meta-analysis.,” PLoS One, vol. 10, no. 5, p. e0127256, 2015. [CrossRef]
- V. Uygun and E. Kurtoglu, “Iron-chelation therapy with oral chelators in patients with thalassemia major.,” Hematology, vol. 18, no. 1, pp. 50–55, Jan. 2013. [CrossRef]
- K. H. M. Kuo and M. Mrkobrada, “A systematic review and meta-analysis of deferiprone monotherapy and in combination with deferoxamine for reduction of iron overload in chronically transfused patients with β-thalassemia.,” Hemoglobin, vol. 38, no. 6, pp. 409–421, 2014. [CrossRef]
- G. J. Kontoghiorghes et al., “Effective chelation of iron in beta thalassaemia with the oral chelator 1,2-dimethyl-3-hydroxypyrid-4-one.,” Br. Med. J. (Clin. Res. Ed)., vol. 295, no. 6612, pp. 1509–1512, Dec. 1987. [CrossRef]
- J. Caro, K. F. Huybrechts, and T. C. Green, “Estimates of the effect on hepatic iron of oral deferiprone compared with subcutaneous desferrioxamine for treatment of iron overload in thalassemia major: a systematic review.,” BMC Blood Disord., vol. 2, no. 1, p. 4, Nov. 2002. [CrossRef]
- E. Nisbet-Brown et al., “Effectiveness and safety of ICL670 in iron-loaded patients with thalassaemia: a randomised, double-blind, placebo-controlled, dose-escalation trial.,” Lancet (London, England), vol. 361, no. 9369, pp. 1597–1602, May 2003. [CrossRef]
- H. Nick et al., “Development of tridentate iron chelators: from desferrithiocin to ICL670.,” Curr. Med. Chem., vol. 10, no. 12, pp. 1065–1076, Jun. 2003. [CrossRef]
- S. Entezari et al., “Iron Chelators in Treatment of Iron Overload.,” J. Toxicol., vol. 2022, p. 4911205, 2022. [CrossRef]
- C. Hershko and D. J. Weatherall, “Iron-chelating therapy.,” Crit. Rev. Clin. Lab. Sci., vol. 26, no. 4, pp. 303–345, 1988. [CrossRef]
- F. N. al-Refaie, L. N. Sheppard, P. Nortey, B. Wonke, and A. V Hoffbrand, “Pharmacokinetics of the oral iron chelator deferiprone (L1) in patients with iron overload.,” Br. J. Haematol., vol. 89, no. 2, pp. 403–408, Feb. 1995. [CrossRef]
- M. Jain, A. Jitani, and T. Dolai, “Current Practices in the Management of Beta-Hemoglobinopathies,” 2020.
- R. C. Hider, X. Kong, V. Abbate, R. Harland, K. Conlon, and T. Luker, “Deferitazole, a new orally active iron chelator.,” Dalton Trans., vol. 44, no. 11, pp. 5197–5204, Mar. 2015. [CrossRef]
- A. T. Taher et al., “Safety and pharmacokinetics of the oral iron chelator SP-420 in β-thalassemia.,” Am. J. Hematol., vol. 92, no. 12, pp. 1356–1361, Dec. 2017. [CrossRef]
- T. D. Coates, “Iron overload in transfusion-dependent patients,” pp. 337–344, 2019.
- G. of I. Ministry of Health and Family welfare, “Prevention and control of Hemoglobinopathies in India- Thalassemia, Sickle cell disease and other variant emoglobinsH.” p. 146, 2016.
- S. Sidhu, S. Kakkar, P. Dewan, N. Bansal, and P. C. Sobti, “Adherence to Iron Chelation Therapy and Its Determinants.,” Int. J. Hematol. stem cell Res., vol. 15, no. 1, pp. 27–34, Jan. 2021. [CrossRef]
- D. Bhattacharyya, R. Chowdhury, S. Choudhuri, P. Ghosh, and M. Bhattacharyya, “Efficacy and Safety of Deferasirox in Chelation Naïve HbEβ Thalassemia Patients: Initial Experience from a Tertiary Centre of Eastern India,” Blood, vol. 130, no. Supplement 1, p. 4760, Dec. 2017. [CrossRef]
- J. Chandra et al., “Efficacy and Safety of Thalidomide in Patients With Transfusion-Dependent Thalassemia.,” Indian Pediatr., vol. 58, no. 7, pp. 611–616, Jul. 2021.
- R. Naithani, J. Chandra, and S. Sharma, “Safety of oral iron chelator deferiprone in young thalassaemics.,” Eur. J. Haematol., vol. 74, no. 3, pp. 217–220, Mar. 2005. [CrossRef]
- R. Mohamed, A. H. Abdul Rahman, F. Masra, and Z. Abdul Latiff, “Barriers to adherence to iron chelation therapy among adolescent with transfusion dependent thalassemia.,” Front. Pediatr., vol. 10, p. 951947, 2022. [CrossRef]
Figure 1.
The Absorption and Cellular Uptake of Iron in Enterocyte and the transport of Transferrin-bound iron to other cells for Utilization. (DYCB- Duodenal Cytochrome B, DMT1- Divalent Metal Transporter 1, Ft- ferritin, FPN1- Ferroportin1, Tf- Transferrin, TfR1- Transferrin Receptor 1).
Figure 1.
The Absorption and Cellular Uptake of Iron in Enterocyte and the transport of Transferrin-bound iron to other cells for Utilization. (DYCB- Duodenal Cytochrome B, DMT1- Divalent Metal Transporter 1, Ft- ferritin, FPN1- Ferroportin1, Tf- Transferrin, TfR1- Transferrin Receptor 1).
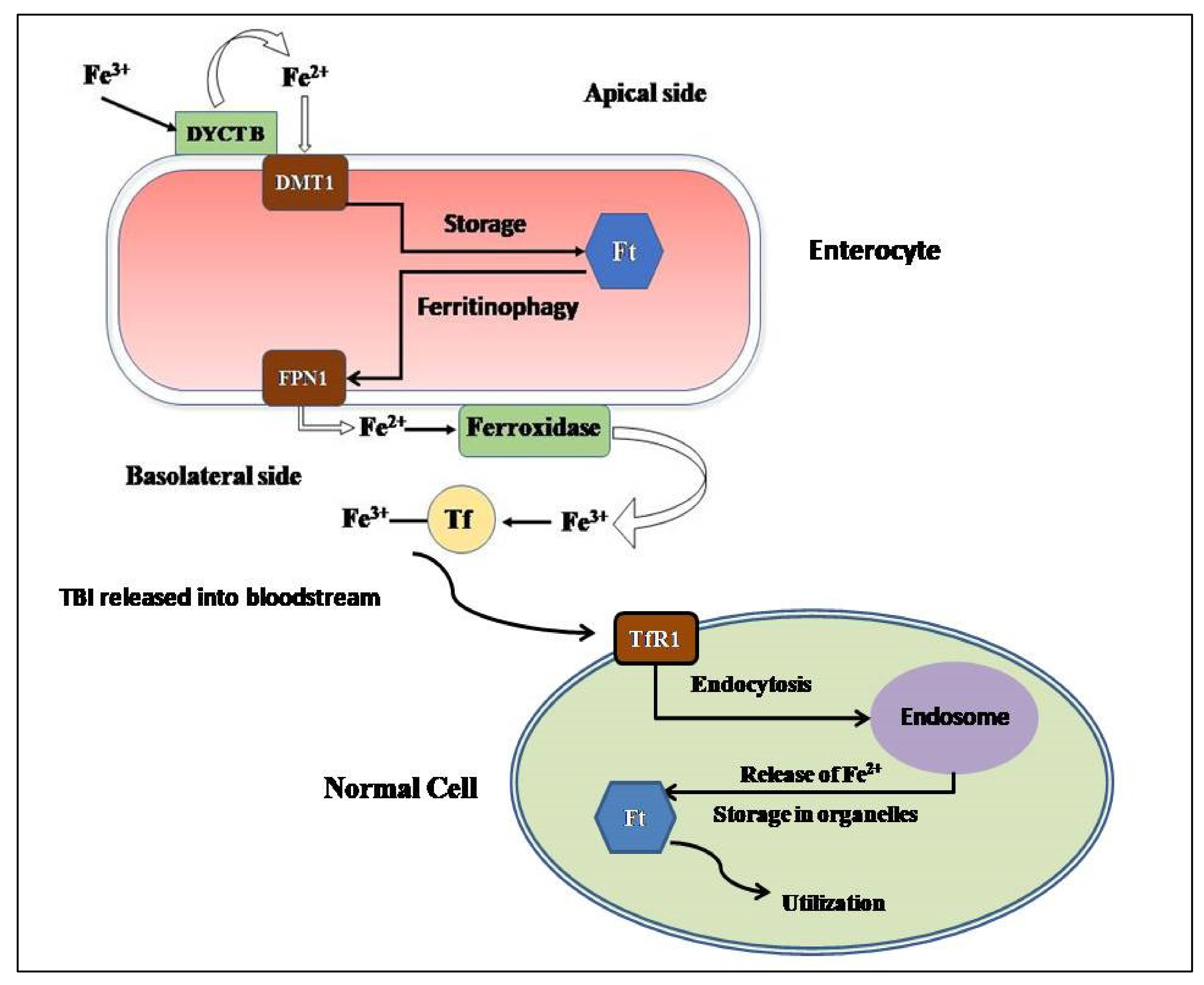
Figure 2.
Figure showing schematic diagram illustrating the main pathways for Iron Overload in TDT and NTDT patients. Although ineffective erythropoiesis happens in all β- thalassemic patients, blood transfusions lead to faster iron overload in TDT patients.
Figure 2.
Figure showing schematic diagram illustrating the main pathways for Iron Overload in TDT and NTDT patients. Although ineffective erythropoiesis happens in all β- thalassemic patients, blood transfusions lead to faster iron overload in TDT patients.
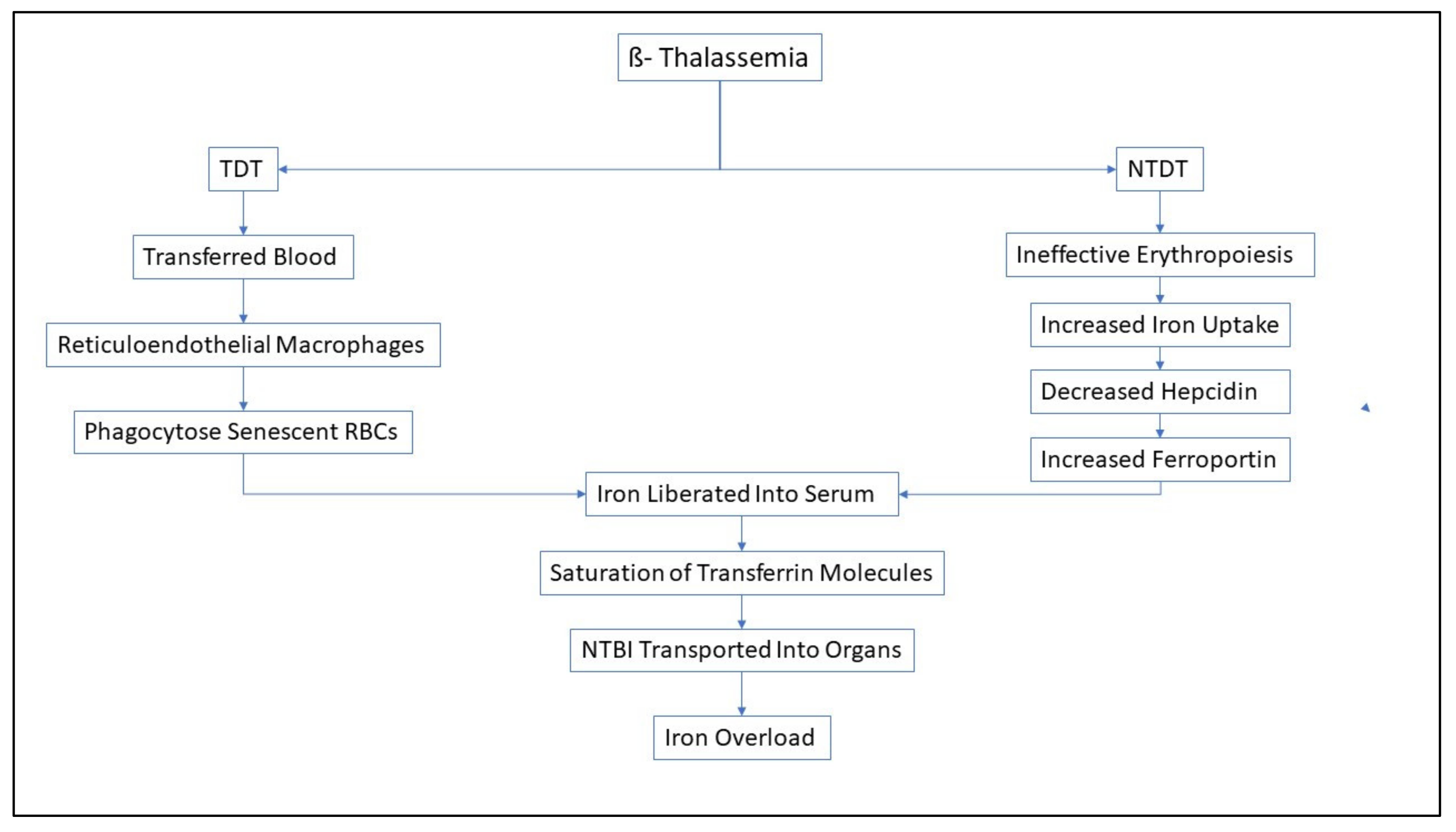
Figure 3.
The Chemical Structures of the three clinically-approved iron chelators used to treat iron overload.
Figure 3.
The Chemical Structures of the three clinically-approved iron chelators used to treat iron overload.
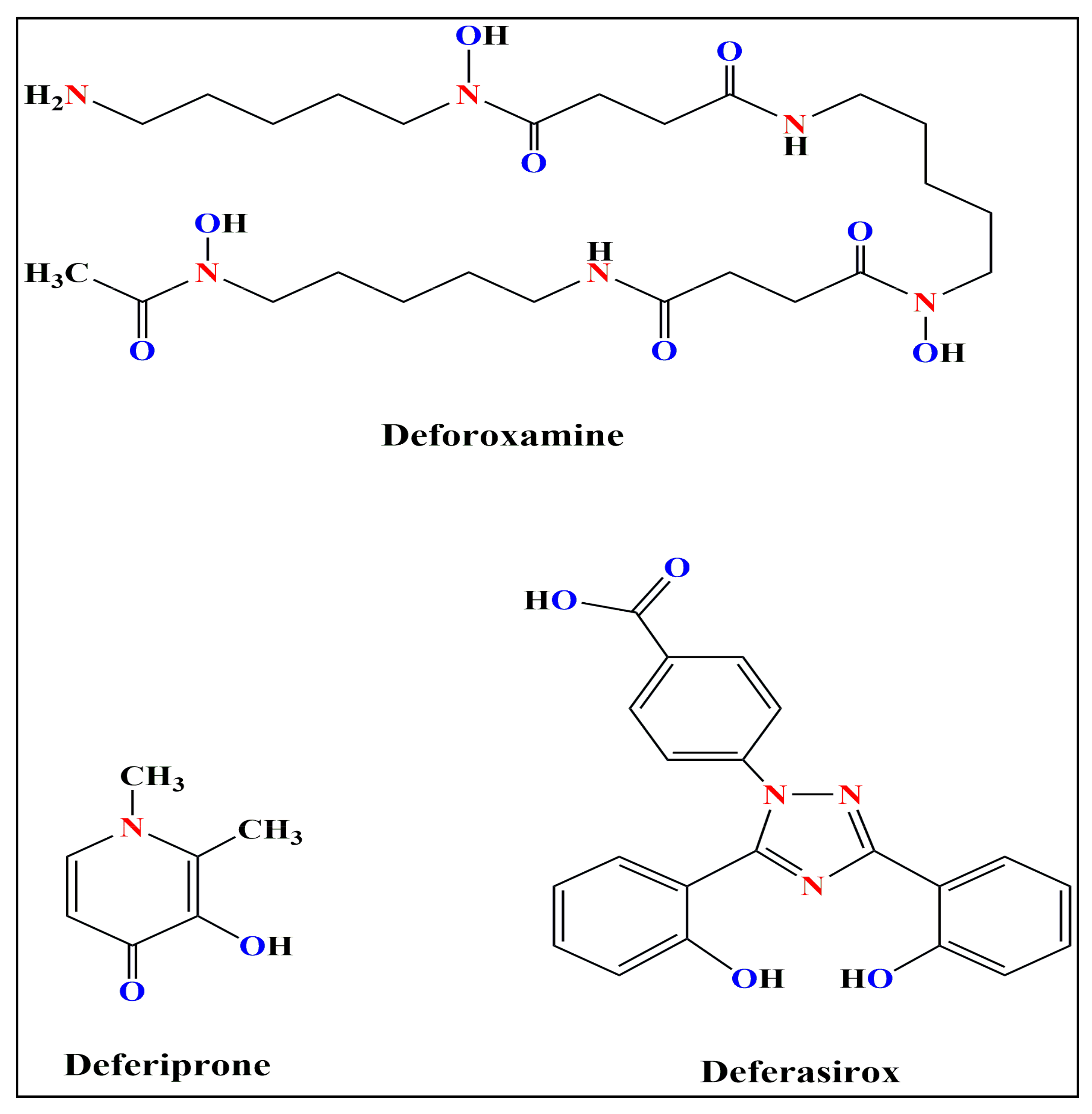

Table 1.
A list of of different commercially available iron chelators used to treat iron overload.
| Characteristics | Deferoxamine | Deferiprone | Deferasirox |
| Structure | Hexadentate | Bidentate | Tridentate |
| Route of Administration | Subcutaneous or Intravenous Injections | Oral | Oral |
| Mechanism of Action | Chelates NTBI, Ft-bound iron; promotes ferritinophagy [113] | Chelate labile iron in cytosol [113] | Chelate labile iron in cytosol; increase hepcidin levels [113] |
| Route of Excretion | Biliary and Urinary [114] | Urinary [115] | Fecal |
| Adverse Effects | Hearing disorders, Growth Retardation, Lung/Renal Toxicity, Bone Abnormalities, Visual disorders, Pain at site of injection [116]. |
Severe Agranulocytosis, Gastrointestinal problems, Arthritis [116]. |
Rash, Renal disorders, Gastrointestinal problems [116] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



