
Submitted:
30 May 2023
Posted:
01 June 2023
You are already at the latest version
Abstract
Autistic spectrum disease (ASD) is an increasingly common diagnosis nowadays with a prevalence of 1-2% in most countries. Its complex causality – a combination of genetic, immune, metabolic and environmental factors - is translated into pleiomorphic developmental disorders of various severity, which have in common two main aspects: repetitive, restrictive behaviors, and difficulties in social interaction varying from awkward habits and verbalization to complete lack of interest from the ASD child for the outside world. The wide variety of ASD causes also makes it very difficult to find a common denominator – a disease biomarker and medication – and currently there is no commonly used diagnostic and therapeutic strategy besides clinical evaluation and psychotherapy.It is known that inflammation is present in a majority of ASD children, and blood inflammatory markers, together with metabolic, electrophysiological markers and imagistics are useful for ASD diagnostic validation and treatment; however, they tend to leave out a sizeable proportion of ASD kids who do not have such modifications. Genetic testing – whole exome sequencing or targeted profiling on about 500 ASD-linked genes- may also provide good insight into pathophysiology, however many times its results are more restrictive rather than offering additional therapeutic options.Here we describe a new biomarker for ASD - the neuron-specific enolase (NSE) - which was elevated above the normal clinical range (less than 16.3 ng/mL) in the vast majority of ASD kids tested in our study (40 of 41, or 97.5%). This finding opens up a new direction for diagnostic confirmation, dynamic evaluation and therapeutic intervention for ASD kids.
Keywords:
ASD
; autism
; autistic spectrum disease
; NSE
; Neuron-specific enolase
; autism biomarker
; neuroinflammation
; neuronal apoptosis
; mTOR
Introduction
Since its first description in a series of 11 children by Leo Kanner 80 years ago (1), ASD is being more frequently diagnosed, so that its estimated direct and indirect economic costs in the USA will likely reach the 500 billion USD mark in 2025, making this pathology more costly than diabetes (2).
The mainstay of ASD treatment is psychotherapy, which typically involves a long an intensive schedule in order to help kids make progress; additionally neurofeedback, antipsychotics such as risperidone and aripiprazole, stimulants such as atomoxetine, other drugs such as oxytocin and bumetanide are prescribed infrequently; more recently stem cell therapies (both hematopoietic and mesenchymal) and mesenchymal – derived exosomes were reported to produce improvements in certain kids (3–7; however, ASD has many aspects yet unknown including pathophysiology and treatment which makes its management challenging.
Abnormal findings related to ASD were reported even before birth, with identification of maternal antibodies to fetal brain protein even mid-pregnancy (8–9). Immune dysfunction seems to be a pervasive aspect in ASD, identified in many studies (10–13) including post-mortem (14), as increased levels of various inflammatory cytokines such as IL-1β, IL-6, IL-8, TNF-α, macrophage chemoattractant protein (MCP)-1, transforming growth factor (TGF)–β1, etc. (15–17); increased GFAP and NFkB specifically underscore neuroinflammation and glial M1 activation in the Central Nervous System (CNS) (18–21); and recently we have more complete and precise profiles of the inflammation present in ASD (22).
Electrophysiological testing such as resting-state electroencephalography (EEG), power spectral densities and the Fitting Oscillations and One-Over-F algorithm can show differences between ASD and typically developing (TD) children (23); dynamic EEG testing involving face perception, visual evoked potentials and perception of biological motion are also specific findings in ASD (24), and successful therapeutic intervention reflected in better social interaction was associated with increased alpha and beta power, decreased theta power (25).
Neuroimaging modifications in ASD is more difficult because on top of the clinical heterogeneity of ASD are added variations in standardization of imaging protocols (26, 27), however specific ASD features were found, such as functional connectivity and gray matter volume; differences of cortical volume in certain areas were shown in ASD patients (28, 29); and also in growth pattern with age (30), an enlarged amygdala and hippocampus in childhood but not adolescence (31–33). The largest imagistic study to date – the ENIGMA ASD study (34) - included a total of 1,571 ASD patients and 1,651 TD controls from 13 countries and found both similar and opposite modifications with other studies: ASD patients had smaller putamen, pallidum, amygdala and the nucleus accumbens, and larger lateral ventricles and mean intracranial volume.
The genetic picture in ASD is also complex given the number of genes involved - about 800 genes with the respective myriad combinations - and even though only 20% -50% of ASD cases can be clearly linked to genetic variants (35–37), the multitude of possible gene alterations: copy number variations, single nucleotide polymorphism (SNPs), inversions, deletions, insertions, duplications, also the epigenetic control of transcription such as modifications of promoter regions (38, 39), and unrelated SNPs converging to alter cellular pathways (40), and finally the diverse phenotypic consequences of genetic alterations – all these factors highlight the difficulties of finding a common denominator in ASD pathology which can be reflected in a universal biomarker.
Adding to the complexity of the genetic determinants of ASD is the influence of environmental factors on vulnerable genetic material, factors which may alter the transcription and translation of the genetic material directly via interaction with the DNA, or indirectly through impairing the DNA repair or oxidative damage (41). Some examples of common types of genes affected are those involved in synapse development – SH3, NLGN, SHANK, CNTNAP2, NRXN, and pathways controlling chromatin remodeling and transcriptional modulation - FMRP, RBFOX, MeCP2, ADNP, etc (42).
Besides sequencing and microarrays, another way of investigating the genetic modifications in ASD is searching for specific plasma or cerebrospinal fluid nucleic acids: circulating, non-coding RNAs (micro RNAs – miRs – and long non-coding RNAs – lncRNAs); these control gene transcription involved in cell signaling, cell adhesion, metabolism, and also cancer (Huang, 2021), and miR-181b-5p, miR-320a, miR-572, miR-130a-3p and miR-19b-3p - are specific to the ASD pathology (43). While some micro RNAs - miR-181b-5p and miR-328 - were down-regulated both in the brain and peripheral blood, miR-106b-5p was shown to be upregulated in the brain of ASD patients in a post-mortem study (44), and downregulated in the peripheral blood (lymphoblastoid lines). A different miRNA profile for ASD was observed by another team (45) and involve miR-197- 5p, miR-3135a, miR-328-3p, miR- 365a-3p, miR-424-5p, miR-500a-5p, miR-619-5p, and miR-664a-3p; these miRNAs target the voltage-dependent calcium channel subunits alpha-1C and beta-1 (CACNA1C and CACNB1 genes) the epigenetic controller gene DICER which codes the type III ribonuclease. Both authors prompt validation of these markers in large patient cohorts.
The difficulty of the ASD biomarker conundrum is underscored by a recent meta-analysis (46) which evaluated 780 studies involving 120.000 ASD patients and 176,000 controls with various biomarkers tested: biochemical (374), neuroimaging (203), neurophysiological (133), neuropsychological [65] and 5 genetic (GWAS), yet an ASD biomarker with at least 80% sensitivity and specificity was still not found. Similar results were obtained by another recent meta-analysis (47) in which were analyzed 940 biomarkers; the least frequent were neuroimaging and neurophysiological biomarkers, and the most frequently studied were molecules such as cytokines, growth factors, oxidative stress indicators, neurotransmitters and hormones; this analysis also showed the low reproducibility of the results of the biomarker studies. In this context it is worthwhile mentioning that even the gold-standard of ASD diagnosis, the Autism Diagnostic Observation Schedule (ADOS) was shown to have a pooled sensitivity of 91% and specificity of 73%, and 52% overall diagnostic accuracy in a meta-analysis involving more than 4000 children (48).
More recent studies integrate data from more than one type of investigations, such the electrophysiological and neurotransmitter data on excitatory/inhibitory balance (E/I on resting EEG), GABA and glutamatergic neurotransmitters, imagistics on cortical thickness and gene expression from various databases, (49–52), and these findings are adding more pieces to the ASD puzzle. When testing a panel of biomarkers, results show much better specificity and sensitivity, so that a panel of 12 inflammatory and immune biomarkers identified 87% of ASD cases (22), and also an 83.3% sensitivity and 84.6% specificity was attained when proteomics testing and a panel of 9 proteins was employed (53).
We have also used a panel of blood tests while evaluating ASD children for the CORDUS clinical study in our search for a marker which could predict the efficacy of the autologous cord blood treatment administered during this study. Various markers of inflammation, metabolic, and neuroendocrine were performed at an independent clinical laboratory, and we have observed that most children with ASD had persistently elevated plasma values of the neuron-specific enolase (NSE). Patients tested were screened for enrollment in this clinical study; prior to enrolling and testing, Informed Consent was obtained from the children’s parents. The CORDUS Clinical Study is registered on www.clinicaltrials.gov with NCT04007224, and ethics approval was given by the National Bioethics Committee of the Romanian Medicine Agency ANM: IS/4/12.02.2020.
Material and Methods
Patients previously diagnosed with ASD (F84.0 or F84.1 on ICD-11) by a pediatric psychiatrist or psychotherapist were screened for the CORDUS clinical study and tested between April 2022 and April 2023 with a battery of tests which included NSE. The tests analyzed were administered as the initial set of tests for each child, after which the children enrolled were tested again two times; these latter results are not analyzed in this article. During the first set of tests the following plasma and serum parameters were tested: complete blood count with differential (CBC); electrolytes (sodium, potassium, calcium); liver function (AST, ALT); renal function (urea, creatinine); inflammatory panel: erythrocyte sedimentation rate (ESR), serum protein electrophoresis; C-reactive protein ultrasensitive (CRP); tumor necrosis factor alpha (TNF-α); ferritin; also, a panel of nine IgG antineuronal antibodies: Anti Amphiphysin; Anti CV2; Anti PNMA2(Ma2/Ta); Anti Ri; Anti Yo; Anti Hu; Anti Recoverin; Anti SOX1; Anti Titin.
In addition, IL-1 beta, IL-6, IL-8, IL-10, neopterin, procalcitonin, human growth hormone (hGH), thyroid stimulating hormone (TSH), insulin-like growth factor-1 (IGF-1), homocysteine, IgE levels, cationic protein of eosinophils, S100 were performed sporadically, in specific patients based on the clinical evaluation of the study investigator.
Blood was drawn and tests were performed at a clinical laboratory – Bioclinica Laboratory Bucharest – where NSE was determined via the electrochemiluminescence (ECLIA) method, with normal range being considered values of less than 16.3 ng/mL.
We have analyzed the test results in order to compare the frequency of abnormal values for various blood markers and for some of these tests – including NSE - we have repeated the testing after a few weeks for confirmation of the elevated values.
Test results were entered in Microsoft Excel and descriptive statistics was performed also using this software.
Results
Test results were given by the independent clinical laboratory both to patients (parents of children) and to the study investigators; results are summarized in Table 1 and Table 2 below.
We have found that besides TNF- α, inflammatory cytokines which we have tested were not much modified; IL-6 and IL-1β was in normal range for most of the kids tested (increased in 10% of kids tested); and there were similar results with the IL-8 testing. Only two kids tested positive for IgG antineuronal antibodies (one for anti-SOX-1, one for anti-amphyphysin) and upon re-testing within 4 weeks from the initial testing, both kids were negative for the respective antibodies, suggesting crossover reaction to a viral infection (the two kids had cold symptoms 1–3 weeks prior).
We have also considered a set of five blood markers and their abnormal values as positive inflammatory markers (IMs) – ESR, CRP, ferritin, α-2 globulins, TNF-α. We have found that 51 of 55 patients had at least one of these IMs present; most patients (21) had 2 IMs, and 4 patients had no abnormal values on any of the five IMs (data summarized in Figure 1). All these 4 patients with normal IMs had increased NSE values, two had increased eosinophils percentage on CBC, two had slightly lower sodium levels (137 mmol/L) and one had microcytic anemia.
Finally, we have tested homocysteine levels in 30 kids, and of those four had low homocysteine levels (13.3%) and one increased level (3.33%); 2 of 10 tested had low hGH (20%); another 2 kids from the 10 kids tested had low IGF-1, and one kid had high TSH; 5 children from 55 (9.09%) had increased MCV, most likely due to folate/vitamin B12 utilization deficit – one of these children had documented antibodies to folate receptor (results summarized in Table 2).
One patient had increased bilirubin (both total and direct) and this was found by us after observing in this patient’s history an adverse reaction to risperidone (lethargic state) which prompted pharmacogenetic testing which showed SNPs in SLC15A2, SLC22A1, SLCO1B3, ABCB1 and ABCC2 genes (the latter linked to Dubin-Johnson syndrome).
An electrolyte disturbance was observed in a few patients; we have ignored hyperpotassemia (10 instances) on the grounds of possible hemolyzed blood specimens because of difficult venipunctures in some ASD kids, but the observed lowered sodium may be linked to a functional alteration of the NKCC2, a cotransporter protein of Na+, K+ and Cl− encoded by the SLC12A1 gene. This dysfunction is linked to the GABA receptor activity and is improved by administration of low-dose bumetanide – a loop diuretic – which was also shown to ameliorate symptoms of ASD (54); more recently torasemide – another Na+/K+ inhibitor, was proposed to be administered in ASD, with less side effects (55).
Finally, a relatively common occurrence among ASD children was an allergic-type reaction, with increased eosinophils observed in about 1/3 of the kids tested for this study (Table 2). While we have only seldom tested IgE levels and the cationic protein of eosinophils (and found them increased in the few kids tested), it is known that increased levels of IgEs and recurrent infectious episodes are associated with variants of the STAT3 genes and DOCK8 genes, with autosomal dominant transmission, with below are shown to be modified in ASD. STAT pathways can be down-regulated by the administration of luteolin and diosmin, additionally quercetin inhibits the differentiation of Th1 triggered by neural antigens and the IL-12 production (56).
Discussion
Elevated NSE values are known to be present in about 60–80% of small cell lung carcinoma, neuroblastoma, seminoma, and in some patients with benign CNS tumors, neuroendocrine neoplasia – medullary thyroid carcinoma, pancreatic carcinoma, Merkel cell carcinoma, carcinoid tumors; however, there is no published data with the finding of increased NSE values in ASD, which is correlated with increased rates of neuronal apoptosis.
Increased neuronal apoptosis with altered levels of Bcl-2 and p53 in the frontal and cerebellar cortex was observed in a postmortem ASD study (57) and perhaps testing for increased NSE values as a reflection of increased apoptosis was deemed redundant.
The available data from post-mortem studies and imagistics shows that in about 60% of ASD cases there is an increase in synapses (due to excessive formation and/or lack of “pruning” of synapses) and neuron numbers, (30, 58–60) which in most cases lead to non-functional networks; in exceptional cases this zonal neuronal exuberance may explain the rare and very specialized abilities of kids formerly diagnosed with Asperger syndrome. Another observation is that NFkB is necessarily activated in both neuro-inflammation (19) and in synaptic formation (61); besides underscoring the important role of inflammation in many ASD cases (62), it may mean that lowering neuro-inflammation without completely abolishing neuronal NFkB activation is a desirable therapeutic strategy which may reduce neuronal apoptosis and the NSE levels in ASD without diminishing the learning capabilities through synaptic formation.
When we consider markers of systemic inflammation as markers for ASD, we may find decreased specificity because children have frequent infectious episodes, as well as subacute activation of the immune system following vaccination or during prodromal phase of infections, or symptomless infections, so the presence of inflammation is unlikely to be a stand-alone criterion for ASD. Adding NFkB or a more specific marker for neuroinflammation such as GFAP is clearly helpful, but it is likely that these two markers are elevated only in about 2/3 of ASD kids.
More specific alterations seen in ASD can be classified as intrinsic or extrinsic in relation to the CNS and the origination site of the possible causal factors: intrinsic factors are primarily affecting the neurons, glia, astrocytes or oligodendrocytes mostly via genetic dysfunctions, while extrinsic factors are primarily affecting the immune system, gut microbiome, metabolic pathways through gene vulnerabilities or exogenous factors (microorganisms, toxicants), and subsequently are altering the CNS function. Some of the extrinsic factors are affecting the CNS after modifying the blood-brain barrier (BBB), however some factors may act simultaneously as both intrinsic and extrinsic factors, one example being the genetic modifications affecting the glutamate receptors mGLuR; in the Figure 2 below we have attempted to summarize those factors.
Among extrinsic CNS factors affecting its functions are the receptors involved in glucose metabolism – the peroxisome proliferator activated receptors ƴ and α (PPAR-Ƴ, PPAR-α) (63) and the receptor for advanced glycation end-products (RAGE), which through NFkB influences neurite outgrowth and neuronal differentiation (64, 65); PPAR-ƴ plays important roles in neuron differentiation and axon polarity (66).
Another extrinsic factor is the intestinal microbiome, which was shown to be different in ASD than in typically developing kids, and its modification through diets including the Nemecek protocol (prebiotic and probiotic administration) has yielded notable results in some ASD cases. Glial over-activation towards the M1 inflammatory phenotype seems to be present in cases of intestinal dysbiosis, with simultaneous involvement of multiple mechanisms, including inflammation via TNF-α, NFkB, increased GFAP and glial activation by LPS (20), alteration of extracellular matrix by metalloproteases (MMPs), increased oxidative stress with lipid peroxidation of membranes, increased BBB permeability due to vascular and/or lymphatic damage identifiable by increased levels of molecules such as thrombospondins (TSP-1) and vascular endothelial growth factors (VEGFs) which alter activity of the endothelium and pericytes.
Propionic acid produced by intestinal bacteria or ingested as food preservative is associated with brain inflammation and ASD traits (67); moreover, fetal exposure to increased maternal lipopolysaccharides or propionic acid resulted in ASD traits in offspring with a sexual dimorphism pattern not seen with other ASD-causing agents (68).
Modifying the gut microbiota has modified levels of acetic, propionic and butyric acids, and improved (decreased) the serotonin levels and dopamine metabolism (normalizing homovanilic acid) in ASD (69). It was also shown that inflammatory cytokines were similarly increased in intestinal and cerebral tissues (hippocampus and amygdala), and the levels of Lactobacillus was correlated negatively with the activation of microglia and astrocytes (70), and also with the activation of purine metabolism in amygdala astrocytes. Comparing urinary metabolites of ASD children with their healthy siblings showed multiple metabolic pathway perturbations, involving tryptophan/serotonin, creatine, glycine, taurine, also pantothenate and melatonin, suggesting a metabolic gut-brain link in ASD (71).
Finally, the intestinal microbiome plays important roles in the synthesis of certain neurotransmitters such as glutamate, serotonin and GABA (72), but through butyrate – which can cross the blood-brain barrier – influences immune regulation, energy metabolism, and transcript and translation of various genes; so that gut microbiota composition was shown to correlate with neurodevelopment; and Bifidobacterium/Enterobacteriaceae vs Bacteroides was associated with differences in toddler and infant temperament and behavior (73).
The increased levels of NSE reflecting increased neuronal apoptosis, triggered by extrinsic pathway (TNF-α activation) or activation of other pathways, and mTOR pathway alone or simultaneously with MAPK and PPAR-ƴ was shown to be activated in many cases, (43, 74) - Figure 2. Simultaneous modifications of MAPK path and the immune system (IgD) were found (53); there are multiple possibilities of E/I modification via disruption of distinct, multiple neurotransmitter paths: glutamatergic – mGLT via ERK, PPAR, NMDAR via PI3K, etc.
ASD causal factors intrinsic to the CNS stem from various dysfunctions of neurons, astrocytes or microglia, a common occurrence being a neurotransmitter synthesis or degradation deficit (GABA, glutamate, NMDA) which besides modifying the excitatory/inhibitory balance, can also alter mitochondria (redox status, energy production) to the point of excitotoxicity, triggering the release of caspases and apoptosis. Another cause may be a membrane alteration (lipid peroxidation, synthesis or degradation of synaptic membranes, etc) with metabolic alterations of astrocytes and glia and M1 glial activation. Glial cells may be over-activated by the failure of astrocytes to recover glutamate from extracellular space, by metabolic residues triggering inflammatory-type reactions, by modifications of glial membrane transporters, etc.
Astrocytes demonstrate unique purinergic-driven activities with specific adenosine receptors, so that A1R activation in astrocytes mediate immunosuppression, while astrocyte A2ARs regulate the glutamate uptake and release by astrocytes, ATP release, activation of Na+/K+ ATPase and synaptic transmission (75).
Astrocytes play important roles in lipid and cholesterol metabolism as it was shown that sterol regulatory element binding proteins (SREBPs) are abundant in hippocampal astrocytes; also, astrocytes control membrane integrity (including synaptic vesicles) by clearing peroxides produced from lipid degradation of membranes, and are involved in synaptic formation and transmission (76).
In this context, lovastatin is known to inhibit ERK and downstream mTOR1 (77), and recently administration of atorvastatin to an animal model of ASD has decreased inflammation and oxidative stress reflected in brain levels of IL-2, IL-17, TNF-α, lactate and malondialdehyde, as well as increased levels of sphingosine-1-phosphate, nerve growth factor (NGF) and number of neurons in hippocampus and cerebellum (78).
Another intrinsic CNS factor is the constitutive activation of mTORC1, frequently a consequence of loss-of function mutations in the phosphatase and tensin homolog (PTEN), fragile X mental retardation protein (FMR1), neurofibromin (NF1) and tuberous sclerosis complex (TSC1/2) genes; lack of inhibitory modulation of these (and other) genes cause the increased activation of the phosphoinositide-dependent kinase (PDK); phosphatidylinositol 3-kinase (PI3K); extracellular signal-regulated kinase (ERK); these in turn can be activated via the NMDA receptor (NMDAR) and also by extrinsic CNS factors via the metabotropic glutamate receptor (mGluR).
After reviewing this data we can infer that the most likely explanation for the pervasive NSE elevation seen in ASD is the single or simultaneous activation of different cellular pathways which are resulting in apoptosis: p38/MAPK; Bcl2-Bax/caspases 3,9; cyclines D, E; PARP/p53; CDK4, ERK/FOS; CREB, β-catenin; pTEN/AKT; Myc, etc, with possibly the most common apoptotic pathways being those involving mTORs.
Studying mTOR inhibitors as pharmacotherapeutic intervention in neurodegenerative pathologies was done previously (77) and so far sirolimus was shown to be safe and effective in 2 year-old children and even better tolerated in kids than adults at full-schedule dosing in other neurodegenerative pathologies linked to mTOR activation (79); it is possible that the lack of efficacy of mTOR inhibitors observed in some studies of tuberous sclerosis was due to specific inhibition of mTOR1 path, leaving mTOR2 functional, or because of simultaneous alteration of mTOR path with others such as Atg7 (59) or simply because the functional alterations seen in ASD are of different intrinsic or extrinsic causalities (metabolic, inflammatory, etc) and neuronal apoptosis is not always mTOR-driven.
Down-modulation of mTOR can be achieved with very low dose tacrolimus (1 mg/week) with lower calcineurin activation and neuro-inflammation; alternatively, mTOR was shown to be down-modulated by naturally-occurring substances which have been used in traditional medicine for centuries so their administration can be viewed as safe – guggulsterone decreased JAK/STAT3 activation and increased / PPAR-ƴ levels (80) and chrysophanol, a naturally-occurring substance extracted from Rheum palmatum extract (67) which inhibits the PI3K/AKT/mTOR pathway, which was shown to be over-active in ASD. Improving the mitochondrial metabolism by administering succinate and/or dichloroacetic acid (DCA) as PDK (and subsequent Akt/mTOR) inhibitor, have shown promising results in ischemic brain and neurodegenerative diseases (81)
Even though we have found that NSE is elevated in the vast majority of ASD kids, it is imperative that the specificity of the NSE is further studied; knowing that NSE values are elevated in other pathologies offers an opportunity to better evaluate and treat these oncological pathologies via the possible differential expression of metabolic (glycolysis, HIF-1α), redox (reduced glutathione) and apoptosis pathways (Bcl/Bax, caspases, etc).
Conclusion
NSE is proposed as an important new biomarker for ASD which can greatly improve the diagnostic and therapeutic capabilities ASD and possibly other neurodegenerative disorders, and adding pathway-specific biomarkers such as GFAP, reduced glutathione, vanilmandelic acid (VMA), homovanilic acid (HVA), quinolinic acid (QA) through dual mass spectroscopy testing (MS/MS) of blood or urine can not only improve its specificity, but also direct treatment towards decreasing neuro-inflammation or improving the mitochondrial function and neuronal red-ox status with specific agents. Normal NSE values in a child with autistic traits but no neurotoxicity can direct treatment towards a genetic or growth factor deficiency.
Clinical evaluation offers the most important diagnostic elements of ASD, as well as practical landmarks for therapeutic intervention and thus is indispensable, however NSE adds a material and more specific component for ASD diagnostic and treatment. NSE testing enables earlier therapeutic interventions and better evaluation of their efficacy, allowing the child to acquire and develop functional neuronal networks earlier in life, with better integration in the regular educational schedule. Late therapeutic interventions in ASD are usually followed by less, more limited progress because of different developmental modifications in the CNS, which in ASD were shown to be age-specific (82).
Finally, after communicating this important finding, we need to study and replicate our results on a larger scale, as well as test NSE in conjunction with one or more markers suggested above (GFAP, HVA, VMA, QA, etc) before NSE can be recommended for routine clinical use in the diagnostic and treatment of ASD.
Authors’ Contribution
SF – design of study and writing the manuscript; BR and DR – testing patients, reviewing the data and the manuscript and critical analysis and additions.
Informed Consent Statement
Informed consent was obtained from each child’s parents prior to enrolling and testing.
Conflicts of Interest
The authors report no conflict of interest related to this work.
References
- Kanner, L. Autistic disturbances of affective contact. Nerv. Child 1943, 2, 217–250. [Google Scholar]
- Wong, R.S.Y. Neuroinflammation in autism spectrum disorders: potential target for mesenchymal stem cell-based therapy. Egypt. J. Neurol. Psychiatry Neurosurg. 2022, 58, 1–13. [Google Scholar] [CrossRef]
- Gesundheit, B.; Ashwood, P.; Keating, A.; Naor, D.; Melamed, M.; Rosenzweig, J.P. Therapeutic properties of mesenchymal stem cells for autism spectrum disorders. Med Hypotheses 2015, 84, 169–177. [Google Scholar] [CrossRef]
- Dawson, G.; Sun, J.M.; Davlantis, K.S.; Murias, M.; Franz, L.; Troy, J.; Simmons, R.; Sabatos-DeVito, M.; Durham, R.; Kurtzberg, J. Autologous Cord Blood Infusions Are Safe and Feasible in Young Children with Autism Spectrum Disorder: Results of a Single-Center Phase I Open-Label Trial. STEM CELLS Transl. Med. 2017, 6, 1332–1339. [Google Scholar] [CrossRef]
- Sun, J.M.; Dawson, G.; Franz, L.; Howard, J.; McLaughlin, C.; Kistler, B.; Waters-Pick, B.; Meadows, N.; Troy, J.; Kurtzberg, J. Infusion of human umbilical cord tissue mesenchymal stromal cells in children with autism spectrum disorder. STEM CELLS Transl. Med. 2020, 9, 1137–1146. [Google Scholar] [CrossRef]
- Murias, M.; Major, S.; Compton, S.; Buttinger, J.; Sun, J.M.; Kurtzberg, J.; Dawson, G. Electrophysiological Biomarkers Predict Clinical Improvement in an Open-Label Trial Assessing Efficacy of Autologous Umbilical Cord Blood for Treatment of Autism. STEM CELLS Transl. Med. 2018, 7, 783–791. [Google Scholar] [CrossRef]
- Gopalarethinam, J.; Nair, A.P.; Iyer, M.; Vellingiri, B.; Subramaniam, M.D. Advantages of mesenchymal stem cell over the other stem cells. Acta Histochem. 2023, 125, 152041. [Google Scholar] [CrossRef]
- Croen, L.A.; Braunschweig, D.; Haapanen, L.; Yoshida, C.K.; Fireman, B.; Grether, J.K.; Kharrazi, M.; Hansen, R.L.; Ashwood, P.; Van de Water, J. Maternal Mid-Pregnancy Autoantibodies to Fetal Brain Protein: The Early Markers for Autism Study. Biol. Psychiatry 2008, 64, 583–588. [Google Scholar] [CrossRef]
- Braunschweig, D.; Duncanson, P.; Boyce, R.; Hansen, R.; Ashwood, P.; Pessah, I.N.; Hertz-Picciotto, I.; Van de Water, J. Behavioral Correlates of Maternal Antibody Status Among Children with Autism. J. Autism Dev. Disord. 2011, 42, 1435–1445. [Google Scholar] [CrossRef]
- Li, X.; Chauhan, A.; Sheikh, A.M.; Patil, S.; Chauhan, V.; Li, X.-M.; Ji, L.; Brown, T.; Malik, M. Elevated immune response in the brain of autistic patients. J. Neuroimmunol. 2009, 207, 111–116. [Google Scholar] [CrossRef]
- Morgan, J.T.; Chana, G.; Pardo, C.A.; Achim, C.; Semendeferi, K.; Buckwalter, J.; Courchesne, E.; Everall, I.P. Microglial Activation and Increased Microglial Density Observed in the Dorsolateral Prefrontal Cortex in Autism. Biol. Psychiatry 2010, 68, 368–376. [Google Scholar] [CrossRef]
- Suzuki, K.; Sugihara, G.; Ouchi, Y.; Nakamura, K.; Futatsubashi, M.; Takebayashi, K.; Yoshihara, Y.; Omata, K.; Matsumoto, K.; Tsuchiya, K.J.; et al. Microglial Activation in Young Adults With Autism Spectrum Disorder. JAMA Psychiatry 2013, 70, 49–58. [Google Scholar] [CrossRef]
- Usui, N.; Kobayashi, H.; Shimada, S. Neuroinflammation and Oxidative Stress in the Pathogenesis of Autism Spectrum Disorder. Int. J. Mol. Sci. 2023, 24, 5487. [Google Scholar] [CrossRef]
- Vargas, D.L.; Nascimbene, C.; Krishnan, C.; Zimmerman, A.W.; Pardo, C.A. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol. 2004, 57, 67–81. [Google Scholar] [CrossRef]
- Molloy, C.; Morrow, A.; Meinzenderr, J.; Schleifer, K.; Dienger, K.; Manningcourtney, P.; Altaye, M.; Willskarp, M. Elevated cytokine levels in children with autism spectrum disorder. J. Neuroimmunol. 2006, 172, 198–205. [Google Scholar] [CrossRef]
- Gesundheit, B.; Rosenzweig, J.P.; Naor, D.; Lerer, B.; Zachor, D.A.; Procházka, V.; Melamed, M.; Kristt, D.A.; Steinberg, A.; Shulman, C.; et al. Immunological and autoimmune considerations of Autism Spectrum Disorders. J. Autoimmun. 2013, 44, 1–7. [Google Scholar] [CrossRef]
- Hughes, H.; R. J.Moreno; Ashwood, P. Innate immune dysfunction and neuroinflammation in autism spectrum disorder (ASD). Brain, Behav. Immun. 2023, 108, 245–254. [Google Scholar] [CrossRef]
- Laurence, J.A.; Fatemi, S.H. Glial fibrillary acidic protein is elevated in superior frontal, parietal and cerebellar cortices of autistic subjects. Cerebellum 2005, 4, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Malik, M.; Tauqeer, Z.; Sheikh, A.M.; Wen, G.; Nagori, A.; Yang, K.; Brown, W.T.; Li, X. NF-κB Signaling in the Brain of Autistic Subjects. Mediat. Inflamm. 2011, 2011, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wang, K.K. Glial fibrillary acidic protein: from intermediate filament assembly and gliosis to neurobiomarker. Trends Neurosci. 2015, 38, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Vakilzadeh, G.; Martinez-Cerdeño, V. Pathology and Astrocytes in Autism. Neuropsychiatr. Dis. Treat. 2023, ume 19, 841–850. [Google Scholar] [CrossRef]
- Gesundheit, B.; Zisman, P.D.; Hochbaum, L.; Posen, Y.; Steinberg, A.; Friedman, G.; Ravkin, H.D.; Rubin, E.; Faktor, O.; Ellis, R. Autism spectrum disorder diagnosis using a new panel of immune- and inflammatory-related serum biomarkers: A case-control multicenter study. Front. Pediatr. 2023, 11, 967954. [Google Scholar] [CrossRef]
- Levin, A.R.; Naples, A.J.; Scheffler, A.W.; Webb, S.J.; Shic, F.; Sugar, C.A.; Murias, M.; Bernier, R.A.; Chawarska, K.; Dawson, G.; et al. Day-to-Day Test-Retest Reliability of EEG Profiles in Children With Autism Spectrum Disorder and Typical Development. Front. Integr. Neurosci. 2020, 14, 21. [Google Scholar] [CrossRef]
- Webb, S.J.; Naples, A.J.; Levin, A.R.; Hellemann, G.; Borland, H.; Benton, J.; Carlos, C.; McAllister, T.; Santhosh, M.; Seow, H.; et al. The Autism Biomarkers Consortium for Clinical Trials: Initial Evaluation of a Battery of Candidate EEG Biomarkers. Annual Meeting of the International-Society-for-Autism-Research. LOCATION OF CONFERENCE, COUNTRYDATE OF CONFERENCE; pp. 41–49.
- Murias, M.; Major, S.; Compton, S.; Buttinger, J.; Sun, J.M.; Kurtzberg, J.; Dawson, G. Electrophysiological Biomarkers Predict Clinical Improvement in an Open-Label Trial Assessing Efficacy of Autologous Umbilical Cord Blood for Treatment of Autism. STEM CELLS Transl. Med. 2018, 7, 783–791. [Google Scholar] [CrossRef]
- Uddin, L.Q.; Dajani, D.R.; Voorhies, W.; Bednarz, H.; Kana, R.K. Progress and roadblocks in the search for brain-based biomarkers of autism and attention-deficit/hyperactivity disorder. Transl. Psychiatry 2017, 7, e1218–e1218. [Google Scholar] [CrossRef]
- Sussman, D.; Leung, R.; Vogan, V.; Lee, W.; Trelle, S.; Lin, S.; Cassel, D.; Chakravarty, M.; Lerch, J.; Anagnostou, E.; et al. The autism puzzle: Diffuse but not pervasive neuroanatomical abnormalities in children with ASD. NeuroImage: Clin. 2015, 8, 170–179. [Google Scholar] [CrossRef]
- Khundrakpam, B.S.; Lewis, J.D.; Kostopoulos, P.; et al. Cortical thickness abnormalities in autism spectrum disorders through late childhood, adolescence, and adulthood: a large-scale MRI study. Cereb Cortex 2017, 27, 1721–1731. [Google Scholar] [CrossRef]
- Zielinski, B.A.; Prigge, M.B.D.; Nielsen, J.A.; Froehlich, A.L.; Abildskov, T.J.; Anderson, J.S.; Fletcher, P.T.; Zygmunt, K.M.; Travers, B.G.; Lange, N.; et al. Longitudinal changes in cortical thickness in autism and typical development. Brain 2014, 137, 1799–1812. [Google Scholar] [CrossRef]
- Courchesne, E.; Campbell, K.; Solso, S. Brain growth across the life span in autism: age-specific changes in anatomical pathology. Brain Res 2011, 1380, 138–145. [Google Scholar] [CrossRef]
- 31. Schumann, C.M.; Hamstra, J.; Goodlin-Jones, B.L.; et al. The amygdala is enlarged in children but not adolescents with autism, the hippocampus is enlarged at all ages. J Neurosci 2004, 24, 6392–640. [Google Scholar] [CrossRef]
- Nordahl, C.W.; Scholz, R.; Yang, X.; Buonocore, M.H.; Simon, T.; Rogers, S.; Amaral, D.G. Increased Rate of Amygdala Growth in Children Aged 2 to 4 Years With Autism Spectrum Disorders. Arch. Gen. Psychiatry 2012, 69, 53–61. [Google Scholar] [CrossRef]
- Groen, W.; Teluij, M.; Buitelaar, J.; et al. Amygdala and hippocampus enlargement during adolescence in autism. J Am Acad Child Adolesc Psychiatry 2010, 49, 552–560. [Google Scholar]
- van Rooij, D.; Anagnostou, E.; Arango, C.; Auzias, G.; Behrmann, M.; Busatto, G.F.; et al. Cortical and subcortical brain morphometry differences between patients with autism spectrum disorder and healthy individuals across the lifespan: tesults from the ENIGMA ASD working group. Am J Psychiatry. 2018, 175, 359–69. [Google Scholar] [CrossRef]
- Rylaarsdam, L.E.; Guemez-Gamboa, A. Genetic Causes and Modifiers of Autism Spectrum Disorder. Front. Cell. Neurosci. 2019, 13, 385. [Google Scholar] [CrossRef]
- Wiśniowiecka-Kowalnik, B.; Nowakowska, B.A. Genetics and epigenetics of autism spectrum disorder—current evidence in the field. J. Appl. Genet. 2019, 60, 37–47. [Google Scholar] [CrossRef]
- Genovese, A.; Butler, M.G. The Autism Spectrum: Behavioral, Psychiatric and Genetic Associations. Genes 2023, 14, 677. [Google Scholar] [CrossRef]
- Havdahl, A.; Niarchou, M.; Starnawska, A.; Uddin, M.; van der Merwe, C.; Warrier, V. Genetic contributions to autism spectrum disorder. Psychol. Med. 2021, 51, 2260–2273. [Google Scholar] [CrossRef]
- Koesterich, J.; An, J.-Y.; Inoue, F.; Sohota, A.; Ahituv, N.; Sanders, S.J.; Kreimer, A. Characterization of De Novo Promoter Variants in Autism Spectrum Disorder with Massively Parallel Reporter Assays. Int. J. Mol. Sci. 2023, 24, 3509. [Google Scholar] [CrossRef]
- Anney, R.; Klei, L.; Pinto, D.; Regan, R.; Conroy, J.; Magalhaes, T.R.; Correia, C.; Abrahams, B.S.; Sykes, N.; Pagnamenta, A.T.; et al. A genome-wide scan for common alleles affecting risk for autism. Hum. Mol. Genet. 2010, 19, 4072–4082. [Google Scholar] [CrossRef]
- Pugsley, K.; Scherer, S.W.; Bellgrove, M.A.; Hawi, Z. Environmental exposures associated with elevated risk for autism spectrum disorder may augment the burden of deleterious de novo mutations among probands. Mol. Psychiatry 2021, 27, 710–730. [Google Scholar] [CrossRef]
- Apte, M.; Kumar, A. Correlation of mutated gene and signalling pathways in ASD. IBRO Neurosci. Rep. 2023, 14, 384–392. [Google Scholar] [CrossRef]
- Vasu, M.M.; Anitha, A.; Thanseem, I.; Suzuki, K.; Yamada, K.; Takahashi, T.; Wakuda, T.; Iwata, K.; Tsujii, M.; Sugiyama, T.; et al. Serum microRNA profiles in children with autism. Mol. Autism 2014, 5, 40–40. [Google Scholar] [CrossRef]
- Abu-Elneel, K.; Liu, T.; Gazzaniga, F.S.; Nishimura, Y.; Wall, D.P.; Geschwind, D.H.; Lao, K.; Kosik, K.S. Heterogeneous dysregulation of microRNAs across the autism spectrum. neurogenetics 2008, 9, 153–161. [Google Scholar] [CrossRef]
- Kichukova, T.M.; Popov, N.T.; Ivanov, I.S.; Vachev, T.I. Profiling of Circulating Serum MicroRNAs in Children with Autism Spectrum Disorder using Stem-loop qRT-PCR Assay. Folia Medica 2017, 59, 43–52. [Google Scholar] [CrossRef]
- Cortese, S.; Solmi, M.; Michelini, G.; Bellato, A.; Blanner, C.; Canozzi, A.; Eudave, L.; Farhat, L.C.; Højlund, M.; Köhler-Forsberg, O.; et al. Candidate diagnostic biomarkers for neurodevelopmental disorders in children and adolescents: a systematic review. World Psychiatry 2023, 22, 129–149. [Google Scholar] [CrossRef]
- Parellada, M.; Andreu-Bernabeu, Á.; Burdeus, M.; Cáceres, A.S.J.; Urbiola, E.; Carpenter, L.L.; Kraguljac, N.V.; McDonald, W.M.; Nemeroff, C.B.; Rodriguez, C.I.; et al. In Search of Biomarkers to Guide Interventions in Autism Spectrum Disorder: A Systematic Review. Am. J. Psychiatry 2023, 180, 23–40. [Google Scholar] [CrossRef]
- Ansel, A.; Posen, Y.; Ellis, R.; Deutsch, L.; Zisman, P.D.; Gesundheit, B. Biomarkers for Autism Spectrum Disorders (ASD): A Meta-analysis. Rambam Maimonides Med J. 2019, 10, e0021. [Google Scholar] [CrossRef]
- Hollestein, V.; Poelmans, G.; Forde, N.J.; Beckmann, C.F.; Ecker, C.; Mann, C.; Schäfer, T.; Moessnang, C.; Baumeister, S.; Banaschewski, T.; et al. Excitatory/inhibitory imbalance in autism: the role of glutamate and GABA gene-sets in symptoms and cortical brain structure. Transl. Psychiatry 2023, 13, 1–9. [Google Scholar] [CrossRef]
- French, L.; Paus, T. A FreeSurfer view of the cortical transcriptome generated from the Allen Human Brain Atlas. Front. Neurosci. 2015, 9, 323. [Google Scholar] [CrossRef]
- Hawrylycz, M.J.; Lein, E.S.; Guillozet-Bongaarts, A.L.; Shen, E.H.; Ng, L.; Miller, J.A.; Van De Lagemaat, L.N.; Smith, K.A.; Ebbert, A.; Riley, Z.L.; et al. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature 2012, 489, 391–399. [Google Scholar] [CrossRef]
- Buch, A.M.; Vértes, P.E.; Seidlitz, J.; Kim, S.H.; Grosenick, L.; Liston, C. Molecular and network-level mechanisms explaining individual differences in autism spectrum disorder. Nat. Neurosci. 2023, 26, 650–663. [Google Scholar] [CrossRef]
- Hewitson, L.; Mathews, J.A.; Devlin, M.; Schutte, C.; Lee, J.; German, D.C. Blood biomarker discovery for autism spectrum disorder: A proteomic analysis. PLOS ONE 2021, 16, e0246581. [Google Scholar] [CrossRef]
- Delpire, E.; Ben-Ari, Y. A Wholistic View of How Bumetanide Attenuates Autism Spectrum Disorders. Cells 2022, 11, 2419. [Google Scholar] [CrossRef]
- Dogan, M.; Company, I.H.M.; Albayrak, Y.; Erbas, O. Torasemide Improves the Propionic Acid-Induced Autism in Rats: A Histopathological and Imaging Study. Anatol. J. Psychiatry 2023, 24, 22–31. [Google Scholar] [CrossRef]
- Singh, R.; Kisku, A.; Kungumaraj, H.; Nagaraj, V.; Pal, A.; Kumar, S.; Sulakhiya, K. Autism Spectrum Disorders: A Recent Update on Targeting Inflammatory Pathways with Natural Anti-Inflammatory Agents. Biomedicines 2023, 11, 115. [Google Scholar] [CrossRef]
- Araghi-Niknam, M.; Fatemi, S.H. Levels of Bcl-2 and P53 Are Altered in Superior Frontal and Cerebellar Cortices of Autistic Subjects. Cell. Mol. Neurobiol. 2003, 23, 945–952. [Google Scholar] [CrossRef]
- Hazlett, H.C.; Poe, M.D.; Gerig, G.; Styner, M.; Chappell, C.; Smith, R.G.; Vachet, C.; Piven, J. Early Brain Overgrowth in Autism Associated With an Increase in Cortical Surface Area Before Age 2 Years. Arch. Gen. Psychiatry 2011, 68, 467–76. [Google Scholar] [CrossRef]
- Tang, G.; Gudsnuk, K.; Kuo, S.-H.; Cotrina, M.L.; Rosoklija, G.; Sosunov, A.; Sonders, M.S.; Kanter, E.; Castagna, C.; Yamamoto, A.; et al. Loss of mTOR-Dependent Macroautophagy Causes Autistic-like Synaptic Pruning Deficits. Neuron 2014, 83, 1131–1143. [Google Scholar] [CrossRef]
- Kim, H.-J.; Cho, M.-H.; Shim, W.H.; Kim, J.K.; Jeon, E.-Y.; Kim, D.-H.; Yoon, S.-Y. Deficient autophagy in microglia impairs synaptic pruning and causes social behavioral defects. Mol. Psychiatry 2016, 22, 1576–1584. [Google Scholar] [CrossRef]
- Gutierrez, H.; Hale, V.A.; Dolcet, X.; Davies, A. NF-κB signalling regulates the growth of neural processes in the developing PNS and CNS. Development 2005, 132, 1713–1726. [Google Scholar] [CrossRef]
- Liao, X.; Li, Y. Nuclear Factor Kappa B in Autism Spectrum Disorder: A Systematic Review. Pharmacol. Res. 2020, 159, 104918. [Google Scholar] [CrossRef]
- Nabetani, M.; Mukai, T.; Taguchi, A. Cell Therapies for Autism Spectrum Disorder Based on New Pathophysiology: A Review. Cell Transplant. 2023, 32. [Google Scholar] [CrossRef]
- Wang, L.; Li, S.; Jungalwala, F.B. Receptor for advanced glycation end products (RAGE) mediates neuronal differentiation and neurite outgrowth. J. Neurosci. Res. 2007, 86, 1254–1266. [Google Scholar] [CrossRef]
- Juranek, J.; Mukherjee, K.; Kordas, B.; Załęcki, M.; Korytko, A.; Zglejc-Waszak, K.; Szuszkiewicz, J.; Banach, M. Role of RAGE in the Pathogenesis of Neurological Disorders. Neurosci. Bull. 2022, 38, 1248–1262. [Google Scholar] [CrossRef]
- Kumar, S.; Mehan, S.; Narula, A.S. Therapeutic modulation of JAK-STAT, mTOR, and PPAR-γ signaling in neurological dysfunctions. J. Mol. Med. 2022, 101, 9–49. [Google Scholar] [CrossRef]
- Sharma, A.R.; Batra, G.; Saini, L.; Sharma, S.; Mishra, A.; Singla, R.; Singh, A.; Singh, R.S.; Jain, A.; Bansal, S.; et al. Valproic Acid and Propionic Acid Modulated Mechanical Pathways Associated with Autism Spectrum Disorder at Prenatal and Neonatal Exposure. CNS Neurol. Disord. - Drug Targets 2022, 21, 399–408. [Google Scholar] [CrossRef]
- Foley, K.A.; MacFabe, D.F.; Vaz, A.; Ossenkopp, K.; Kavaliers, M. Sexually dimorphic effects of prenatal exposure to propionic acid and lipopolysaccharide on social behavior in neonatal, adolescent, and adult rats: Implications for autism spectrum disorders. Int. J. Dev. Neurosci. 2014, 39, 68–78. [Google Scholar] [CrossRef]
- Wang, Y.; Li, N.; Yang, J.-J.; Zhao, D.-M.; Chen, B.; Zhang, G.-Q.; Chen, S.; Cao, R.-F.; Yu, H.; Zhao, C.-Y.; et al. Probiotics and fructo-oligosaccharide intervention modulate the microbiota-gut brain axis to improve autism spectrum reducing also the hyper-serotonergic state and the dopamine metabolism disorder. Pharmacol. Res. 2020, 157, 104784. [Google Scholar] [CrossRef]
- Ji, S.; Han, S.; Yu, L.; Du, L.; You, Y.; Chen, J.; Wang, M.; Wu, S.; Li, S.; Sun, X.; et al. Jia Wei Xiao Yao San ameliorates chronic stress-induced depression-like behaviors in mice by regulating the gut microbiome and brain metabolome in relation to purine metabolism. Phytomedicine 2022, 98, 153940. [Google Scholar] [CrossRef]
- Liang, Y.; Xiao, Z.; Ke, X.; Yao, P.; Chen, Y.; Lin, L.; Lu, J. Urinary Metabonomic Profiling Discriminates Between Children with Autism and Their Healthy Siblings. J. Pharmacol. Exp. Ther. 2020, 26, e926634-1–e926634-8. [Google Scholar] [CrossRef]
- Amin, N.; Liu, J.; Bonnechere, B.; MahmoudianDehkordi, S.; Arnold, M.; Batra, R.; Chiou, Y.-J.; Fernandes, M.; Ikram, M.A.; Kraaij, R.; et al. Interplay of Metabolome and Gut Microbiome in Individuals With Major Depressive Disorder vs Control Individuals. JAMA Psychiatry 2023, 80, 597–609. [Google Scholar] [CrossRef]
- Aatsinki, A.-K.; Lahti, L.; Uusitupa, H.-M.; Munukka, E.; Keskitalo, A.; Nolvi, S.; O'Mahony, S.; Pietilä, S.; Elo, L.L.; Eerola, E.; et al. Gut microbiota composition is associated with temperament traits in infants. Brain, Behav. Immun. 2019, 80, 849–858. [Google Scholar] [CrossRef]
- Thomas, S.D.; Jha, N.K.; Ojha, S.; Sadek, B. mTOR Signaling Disruption and Its Association with the Development of Autism Spectrum Disorder. Molecules 2023, 28, 1889. [Google Scholar] [CrossRef]
- Agostinho, P.; Madeira, D.; Dias, L.; Simões, A.P.; Cunha, R.A.; Canas, P.M. Purinergic signaling orchestrating neuron-glia communication. Pharmacol. Res. 2020, 162, 105253. [Google Scholar] [CrossRef]
- van Deijk, A.F.; Camargo, N.; Timmerman, J.; Heistek, T.; Brouwers, J.F.; Mogavero, F.; Mansvelder, H.D.; Smit, A.B.; Verheijen, M.H. Astrocyte lipid metabolism is critical for synapse development and function in vivo. Glia 2017, 65, 670–682. [Google Scholar] [CrossRef]
- Sato, A. mTOR, a Potential Target to Treat Autism Spectrum Disorder. CNS Neurol. Disord. - Drug Targets 2016, 15, 533–543. [Google Scholar] [CrossRef]
- Durankuş, F.; Budak, K.; Albayrak, Y.; Sever, I.H.; Özkul, B.; Uyanıkgil, Y.; Albayrak, N.; Erbas, O. Atorvastatin Improves the Propionic Acid-Induced Autism in Rats: The Roles of Sphingosine-1-Phosphate and Anti-inflammatory Action. Cureus 2023, 15. [Google Scholar] [CrossRef]
- Śmiałek, D.; Jóźwiak, S.; Kotulska, K. Safety of Sirolimus in Patients with Tuberous Sclerosis Complex under Two Years of Age—A Bicenter Retrospective Study. J. Clin. Med. 2023, 12, 365. [Google Scholar] [CrossRef]
- Khera, R.; Mehan, S.; Bhalla, S.; Kumar, S.; Alshammari, A.; Alharbi, M.; Sadhu, S.S. Guggulsterone Mediated JAK/STAT and PPAR-Gamma Modulation Prevents Neurobehavioral and Neurochemical Abnormalities in Propionic Acid-Induced Experimental Model of Autism. Molecules 2022, 27, 889. [Google Scholar] [CrossRef]
- Afridi, R.; Kim, J.-H.; Rahman, H.; Suk, K. Metabolic Regulation of Glial Phenotypes: Implications in Neuron–Glia Interactions and Neurological Disorders. Front. Cell. Neurosci. 2020, 14, 20. [Google Scholar] [CrossRef]
- Chow, M.L.; Pramparo, T.; Winn, M.E.; Barnes, C.C.; Li, H.-R.; Weiss, L.; Fan, J.-B.; Murray, S.; April, C.; Belinson, H.; et al. Age-Dependent Brain Gene Expression and Copy Number Anomalies in Autism Suggest Distinct Pathological Processes at Young Versus Mature Ages. PLOS Genet. 2012, 8, e1002592. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.-X.; Chen, Y.; Guo, H.-R.; Chen, G.-F. Systematic Review and Bioinformatic Analysis of microRNA Expression in Autism Spectrum Disorder Identifies Pathways Associated With Cancer, Metabolism, Cell Signaling, and Cell Adhesion. Front. Psychiatry 2021, 12. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Number of Inflammation markers (IM) detected simultaneously in patients.
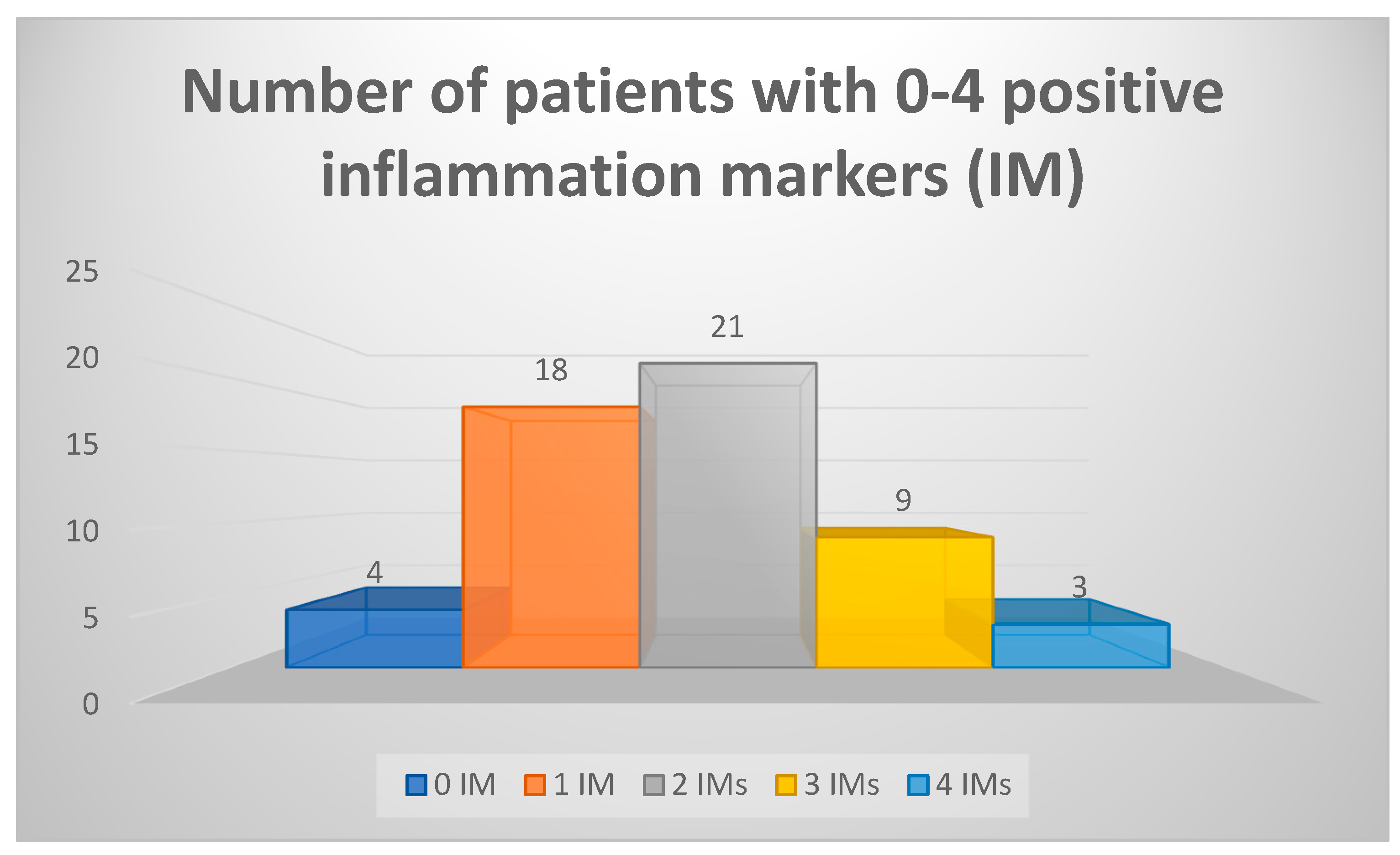
Figure 2.
Elevated NSE levels resulting from neuronal apoptosis induced by intrinsic and extrinsic CNS factors.
Figure 2.
Elevated NSE levels resulting from neuronal apoptosis induced by intrinsic and extrinsic CNS factors.
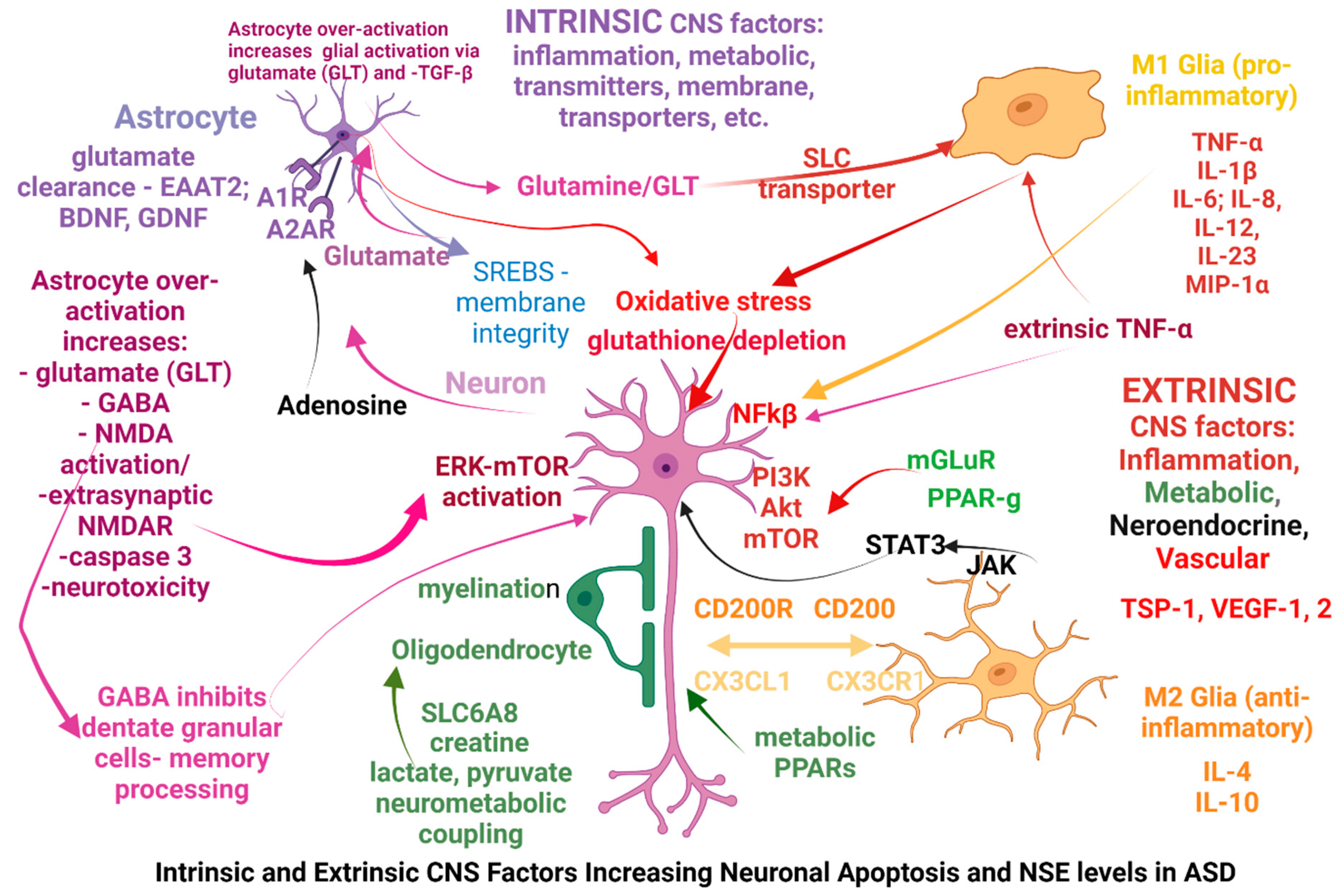

Table 1.
Results of Inflammatory markers (IM) testing, and NSE.
| ESR | Gamma-globulins | ferritin | CRP | Eosino-phils | TNF-α | α-2 globulins | NSE | |
|---|---|---|---|---|---|---|---|---|
| Normal laboratory range | < 16 mm/h | 11.1–18.8% | 5.3 – 99.9 ng/mL | < 0,1 mg/dL | 0–4.5% | < 8,1 pg/mL | < 11.8% | <16,3 ng/mL |
| Patients tested (n=) | 55 | 55 | 51 | 51 | 55 | 52 | 55 | 41 |
| Number abnormal values (n=; % =) | 11; 20% | 15, 27.3% |
14; 27.4% |
15; 29.41% |
18; 32.72% |
36; 69.2% |
39; 70.9% |
40; 97.5% |
| Mean +/- SD | 11+/- 9.9 | N/A | N/A | N/A | N/A | 9.22 +/– 3.98 | 13.1 +/- 4.39 | 24.0 +/- 10.4 |
| Number (n=) of Low/Higher values | 11 High | 15 Low | 10 Low/ 4 High | 15 High | 18 high | 36 High | 39 High | 40 High |
Table 2.
Other levels tested in blood: electrolyte, hematologic, metabolic, endocrine (RBC – red blood cells; MCV – mean corpuscular volume of RBCs; MHC – mean hemoglobin concentration; Na – sodium; IGF-1 – insulin like growth factor -1; hGH – human growth hormone); d- direction of modification.
Table 2.
Other levels tested in blood: electrolyte, hematologic, metabolic, endocrine (RBC – red blood cells; MCV – mean corpuscular volume of RBCs; MHC – mean hemoglobin concentration; Na – sodium; IGF-1 – insulin like growth factor -1; hGH – human growth hormone); d- direction of modification.
| Low MCV, low MHC | High RBC, low MCV (thalassemia) | Low Na (<138 mmol/L) | High MCV | Urea | Liver function tests | Homo-cysteine | IGF-1; hGH | |
|---|---|---|---|---|---|---|---|---|
| #; (%) | 1/55; (1.81%) | 2/55; (3.63%) | 4/55; (7.27%) | 5/55; (9.09%) | 5/55; (9.09%) | 6/55; (10.9%) | 5/30; (16.6%) | 2/10; (20%) |
| d | Low/Low | High/Low | Low | High | High | High | 4 Low, 1 High | Low |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



