Submitted:
31 May 2023
Posted:
31 May 2023
You are already at the latest version
Abstract
N-(diisopropylphosphanyl)benzamide, PhC(O)NHPiPr2, has been synthesized in good yield following two alternative procedures that employ benzamide as the starting material. The first one is a two-step preparation, in which N-(trimetilsilyl)benzamide is reacted with PiPr2Cl, to give the title compound in good yield, whereas the second one is a straightforward synthesis which converts benzamide into N-(diisopropylphosphanyl)benzamide by reaction with PiPr2Cl in the presence of N,N-dimethylpyridin-4-amine (DMAP) and triethylamine. NMR spectroscopy and X-ray diffraction analyses have been performed to characterize the new compound and elucidate its molecular structure in the solid state. N-(diisopropylphosphanyl)benzamide adds to the limited family of amido-substituted phosphines, RC(O)NHPR’2, which can be classified as bidentate hybrid P,O-ligands, both in their neutral and anionic forms, the latter achievable by deprotonation of the NH group.
Keywords:
P ligands
; P(III) compounds
; sterically demanding ligands
; hemilabile ligands
* rpeloso@us.es
1. Introduction
Bidentate hybrid ligands usually combine donor atoms with intrinsically different properties, which can be rationalized, in most cases, in terms of the well-known hard/soft classification proposed by Pearson in the early 1960s [1]. In particular, P,O donor ligands based on P(III) functionalities and O-containing organic groups, such as ketones, ethers, carboxylates, or amides, are representative examples of hemilabile ligands [2], which is easily recognizable when considering, for instance, the type of bonding, and consequent fluxional behavior and reactivity that they are expected to establish with soft metal centers. The interest of these of systems in transition metal catalyzed processes is evident [3,4].
Besides perfectly fitting into the definition of hemilabile hybrid ligand [5], amido-substituted phosphines, RC(O)NHPR’2, can straightforwardly be converted into the corresponding monoanions by selective deprotonation of the C(O)NH group, thus enhancing their versatility in coordination chemistry along with the tendency to act as chelating ligands. Over the last decades, only a few examples of amido-substituted P(III) ligands, generated from acetamide [6], picolinamide [7], benzamide and nicotinamide [8], have been described and used to prepare several transition metal complexes [6,7,8,9,10,11,12,13,14,15,16].
Herein, we report on the synthesis and spectroscopic and structural characterization of the sterically demanding N-(diisopropylphosphanyl)benzamide, PhC(O)NHPiPr2, which incorporates to the class of hemilabile P,O-ligands derived from organic amides.
2. Results and Discussion
Inspired by the reaction strategy reported by Braunstein for the preparation of acetamide-substituted P,O ligands [6], benzamide was firstly converted into N-trimethylsilylbenzamide, which was subsequently reacted with PiPr2Cl in toluene at 70 °C to obtain the title compound, 1, as an air-sensitive colorless material in a ca. 80% yield (Scheme 1, a). In order to optimize the synthesis of PhC(O)NHPiPr2 circumventing the first step of the aforementioned procedure, benzamide was directly treated with an equimolar amount of PiPr2Cl in refluxing toluene in the presence of DMPA and NEt3 (Scheme 1, b) [8]. Although the target compound was actually obtained, the low reaction yield (less than 40%) clearly makes this alternative procedure (b) less convenient than the two-step synthesis that involves the formation of N-trimethylsilylbenzamide (a).
PhC(O)NHPiPr2 was characterized by NMR spectroscopy in solution (CDCl3), with corresponding informative data resumed as follows: i) the 31P{1H} NMR spectrum consists of a singlet at 48.2 ppm; ii) in the 1H NMR spectrum the isopropyl groups give rise to a broad signal at ca. 2.0 ppm assigned to the methyne protons and a multiplet in the range 1.2-1.0 ppm due to the diasterotopic methyl groups; iii) the NH proton resonates as a broad singlet at ca. 6.0 ppm; iv) in the 13C{1H} NMR spectrum the CH3 protons generate two well-defined doublets at 18.8 and 17.9 ppm with 2JCP of 13 and 21 Hz, respectively. In order to provide additional spectroscopic information, the IR spectrum of 1 was recorded and two diagnostic absorptions due to the stretching vibrations of the N-H and C=O bonds were found at 3288 at 1651 cm-1, respectively.
The molecular structure of 1 in the solid state was ascertained by X-ray diffraction analyses on suitable single crystals, which were grown from toluene solutions by slow evaporation of the solvent at room temperature. Figure 1 shows a representation of a single molecule of compound 1 together with selected bond distances and angles, whereas views of the corresponding packing diagrams are provided in Figure 2. Similarly to what observed for the related phosphanylamide CH3C(O)NHPPh2 [6], the P1, N1, C1, O1, C2 atoms are essentially coplanar, with the P lone pair pointing towards the oxygen atom with a deviation of approximately 15° from the plane. This conformation is expected to enthalpically favor the P,O chelating binding mode in metal complexes. As shown in Figure 2, intermolecular hydrogen bonds involving the amide groups [N∙∙∙O 3.025(5) Å, NH∙∙∙O 2.10(3)] are responsible for the chain disposition of the molecules of PhC(O)NHPiPr2 in the lattice.
4. Materials and Methods
All manipulations were carried out using standard Schlenk and glove-box techniques, under an atmosphere of high-purity nitrogen. Solvents were rigorously dried and degassed before use. N-(trimethylsilyl)benzamide was prepared by a modified version of a previously reported procedure [18]. Other chemicals were purchased from Sigma-Aldrich and used as received. NMR spectra were recorded on a Bruker DRX-400 spectrometer. Solvent peaks were used as the internal reference for 1H and 13C spectra, whereas 31P NMR chemical shifts were referenced to H3PO4. For elemental analyses, a LECO TruSpec CHN elementary analyzer was utilized.
Synthesis of N-(diisopropylphosphanyl)benzamide, 1. Methode (a): triethylamine (0.35 mL, 2.5 mmol) and chlorotrimethylsilylane (0.38 mL, 3.0 mmol) were added to a solution of benzamide (0.30 g, 2.5 mmol) in toluene (20 mL) and allowed to react at room temperature under stirring for 1 h. Solid materials were removed by filtration and the resulting solution was taken to dryness under reduced pressure to afford N-(trimethylsilyl)benzamide as a colorless solid, which can be stored, or entirely used, as the starting material for the following step of this synthesis. A solution of N-(trimethylsilyl)benzamide (0.10 g, 0.52 mmol) in toluene (10 mL) was added with PiPr2Cl (0.083 mL, 0.52 mmol) and allowed to stir for ca. 12 h at 70 °C. The volatiles were removed by evaporation under reduced pressure and the resulting colorless solid substance was washed with pentane (2 × 3 mL) and dried under vacuum (95 mg, 77%). Methode (b): solid samples of benzamide (1.0 g, 8.3 mmol) and DMAP (0.22 g, 1.8 mmol) were dissolved in toluene (20 mL), after which triethylamine (1.2 mL, 8.3 mmol) and PiPr2Cl (1.3 mL, 8.3 mmol) were added. The reaction mixture was heated overnight at 110 °C under stirring. The resulting suspension was filtered to obtain a colorless solution, which was taken to dryness under reduced pressure. The colorless solid residue was washed with pentane (2 × 5 mL) and dried under vacuum (0.70 g, 36%). 1H NMR (400 MHz, CDCl3): δ 7.90-7.70 (m, 2H, o-Ph), 7.50-7.30 (m, 3H, m- and p-Ph), 6.06 (br s, 1H, NH), 1.93 (br s, 2H, CH(CH3)2) 1.20-1.00 (m, 12H, CH(CH3)2) ppm. 31P{1H} NMR (162 MHz, CDCl3): δ 48.2 (s) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 171.2 (s, C=O), 135.1 (s, C-C=O), 131.8 (br s, o-Ph), 128.8 (s, m-Ph), 127.3 (s, p-Ph), 25.9 (d, 1JCP = 13 Hz, CH(CH3)2), 18.8 (d, 2JCP = 21 Hz, CH3), 17.9 (d, 2JCP = 8 Hz, CH3) ppm. IR (neat): 3288 (N—H), 1651 (C=O) cm-1. Anal. Calc. for C13H20NOP: C, 65.8; H, 8.50; N, 5.90. Found: C, 65.9; H, 8.6; N, 5.9.
X-ray diffraction analyses. A summary of the crystallographic data and the structure refinement results for compound 1 are given in Table S1 (Supplementary Materials). A crystal of suitable size for X-ray diffraction analysis was coated with dry perfluoropolyether and mounted on glass fibers and fixed in a cold nitrogen stream (T = 213 K) to the goniometer head. Data collection was carried out on a Bruker-Nonius X8kappa APEX II CCD area detector, using monochromatic radiation λ(Mo Kα) = 0.71073 Å, by means of ω and φ scans with a width of 0.50 degrees. The data were reduced (SAINT) [19] and corrected for absorption effects by the multi-scan method (SADABS) [19,20]. The structures were solved by direct methods (SIR-2002) [21] and refined against all F2 data by full-matrix least-squares techniques (SHELXTL-2018/3) [22,23] minimizing w[Fo2-Fc2]2. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were included from calculated positions and refined riding on their respective carbon atoms with isotropic displacement parameters. CCDC 2263762 (1) contains the supplementary crystallographic data for this paper. The data can be obtained free of charge from The Cambridge Crystallographic Data Centre via: https://www.ccdc.cam.ac.uk/structures/.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: title; Table S1: title; Video S1: title.
Author Contributions
conceptualization, R.P.; investigation, M.M.A, D.G.G. (synthesis, NMR), E.A. (X-ray), and R.P; formal analysis, E.A. and R.P; writing—original draft preparation, R.P.; writing—review and editing, E.A and R.P.; supervision, R.P. All authors have read and agreed to the published version of the manuscript
Funding
This research was funded by Ministerio de Ciencia e Innovación (MICINN/AEI/10.13039/501100011033/, “ERDF A way of making Europe”, Grant CTQ2017-82893-C2-2-R, Grants CTQ2016-75193-P and PID2019-110856GA-I00), and Junta de Andalucía (Grants US126226 and P18-FR-4688).
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Pearson, R.G. Hard and soft acid and bases. J. Am. Chem. Soc. 1963, 85, 3533–3539. [Google Scholar] [CrossRef]
- Jeffrey, J.C.; Rauchfuss, T.B. Metal complexes of hemilabile ligands. Reactivity and structure of dichlorobis(o-(diphenylphosphino)anisole)ruthenium(II). Inorg. Chem. 1979, 18, 2658–2666. [Google Scholar] [CrossRef]
- Bader, A.; Lindner, E. Coordination chemistry and catalysis with hemilabile oxygen-phosphorus ligands. Coord. Chem. Rev. 1991, 108, 27–110. [Google Scholar] [CrossRef]
- Braunsten, P. Functional ligands and complexes for new structures, homogeneous catalysts and nanomaterials. J. Organomet. Chem. 2004, 689, 3953–3967. [Google Scholar] [CrossRef]
- Braunstein, P.; Naud, F. Hemilability of Hybrid Ligands and the Coordination Chemistry of Oxazoline-Based Systems. Angew. Chem. Int. Ed. 2001, 40, 680–699. [Google Scholar] [CrossRef]
- Braunstein, P.; Frison, C.; Morise, X.; Adams, R.D. Coordination properties of novel hemilabile acetamide-derived P,O phosphine ligands. Crystal structures of Ph2PNHC(O)Me and [PdMe{PPh2NHC(O)Me}{PPh2NHC(O)Me}][O3SCF3. Dalton Trans. 2000, 2205–2214. [Google Scholar] [CrossRef]
- Milton, H.L.; Wheatley, M.V; Slawin, A.M.Z.; Woollins, J.D. Synthesis and coordination of 2-diphenylphosphinopicolinamide. Polyhedron 2004, 23, 3211–3220. [Google Scholar] [CrossRef]
- Ly, T.Q.; Slawin, A.M.Z.; Woollins, J.D. Synthesis and coordination chemistry of RC(E)NHPPh2 (E = O or S; R = H2N, Ph, or Py). X-ray structure of [Pt{H2NC(S)NHPPh2}2] 2Cl 0.5MeOH. Polyhedron 1999, 18, 1761–1766. [Google Scholar] [CrossRef]
- Morise, X.; Green, M.L.H.; Braunstein, P.; Rees, L.H.; Vei, I.C. Monocyclopentadienyl complexes of niobium, tantalum and tungsten containing heterodifunctional P,O ligands. New J. Chem. 2003, 27, 32–38. [Google Scholar] [CrossRef]
- Braunstein, P.; Frison, C.; Morise, X. Synthesis and crystal structure of [PdMe{PPh2NHC(O)Me}{O=C(NH2)Me}][BF4], a palladium complex containing the acetamide ligand. C. R. Chimie 2002, 5, 131–135. [Google Scholar] [CrossRef]
- Oberbeckmann-Winter, N.; Braunstein, P.; Welter, R. Palladium Phosphino-Iminolate Complexes as Metalloligands for Coinage Metals: A Versatile, Ambivalent Behavior. Organometallics 2005, 24, 3149–3157. [Google Scholar] [CrossRef]
- Jones, N.G.; Green, M.L.H.; Vei, I.; Cowley, A.; Morise, X.; Braunstein, P. Synthesis of molybdenum arene complexes containing amide-derived heterodifunctional P,O ligands. J. Chem. Soc., Dalton Trans. 2002, 1487–1493. [Google Scholar] [CrossRef]
- Agostinho, M.; Rosa, V.; Avilés, T.; Welter, R.; Braunstein, P. Synthesis and characterization of Co and Ni complexes stabilized by keto- and acetamide-derived P,O-type phosphine ligands. Dalton Trans. 2009, 814–822. [Google Scholar] [CrossRef]
- Navratil, M.; Cisarova, I.; Stepnicka, P. Intermolecular interactions in the crystal structures of chlorogold(I) complexes with N-phosphinoamide ligands. Inorg. Chim. Acta 2021, 516, 120138. [Google Scholar] [CrossRef]
- Bhattacharyya, P.; Ly, T.Q.; Slawin, A.M.Z.; Woollins, J. D. P- versus P,O- coordination in complexes of N-(diphenyl phosphino)arylamide ligands ArC(O)NHPPh2 (Ar = 3-pyridyl, phenyl). X-ray crystal structures of [RhCl2(η5-C5Me5)L2] and [NiCl(EtOH)L22]·Cl·[NiCl2L22] (L2= C6H5CONHPPh2). Polyhedron 2001, 20, 1803–1808. [Google Scholar] [CrossRef]
- Braunstein, P.; Frison, C.; Morise, X. Stepwise ethene and/or methyl acrylate/CO insertions into the Pd-C bond of cationic palladium(II) complexes stabilized by a (P,O) chelate. Angew. Chem. Int. Ed. 2000, 39, 2867–2870. [Google Scholar] [CrossRef]
- T. -Y; Liang, X.; Wu, C.; Xue, L.-S.; Xu, Q.-L.; Zhang, S.; Liu, X.; Zheng, Y.-X; Wang, X.-Q. Syntheses, crystal structure, photophysical property and theoretical study of a new series of iridium complexes with N-(diphenylphosphoryl)benzamide derivatives as the ancillary ligands. J. Organomet. Chem. 2014, 755, 110–119. [Google Scholar]
- Blondiaux, E.; Cantat, T. Efficient metal-free hydrosilylation of tertiary, secondary and primary amides to amines. Chem. Commun. 2014, 50, 9349–9352. [Google Scholar] [CrossRef]
- Bruker. SAINT. APEX2 2007, Bruker AXS Inc., Madison, Wisconsin, USA.
- Sheldrick, G.M. SADABS, Programs for Scaling and Absorption Correction of Area Detector Data. SADABS, Programs Scaling Absorpt. Correct. Area Detect. Data 1997, University of Göttingen: Göttingen, Germany; (b) Bruker. SADABS. APEX2 2007, Bruker AXS Inc., Madison, Wisconsin, USA.
- Burla, M. C.; Camalli, M.; Carrozzini, B.; Cascarano, G. L.; Giacovazzo, C.; Polidori, G.; Spagna, R. SIR2002 : The Program. J. Appl. Cryst. 2003, 36, 1103–1103. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A Short History of SHELX. Acta Cryst. 2008, A64, 112-122; b) Sheldrick, G.M. SHELXT - Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar]
Scheme 1.
Two alternative syntheses of N-(diisopropylphosphanyl)benzamide (1).
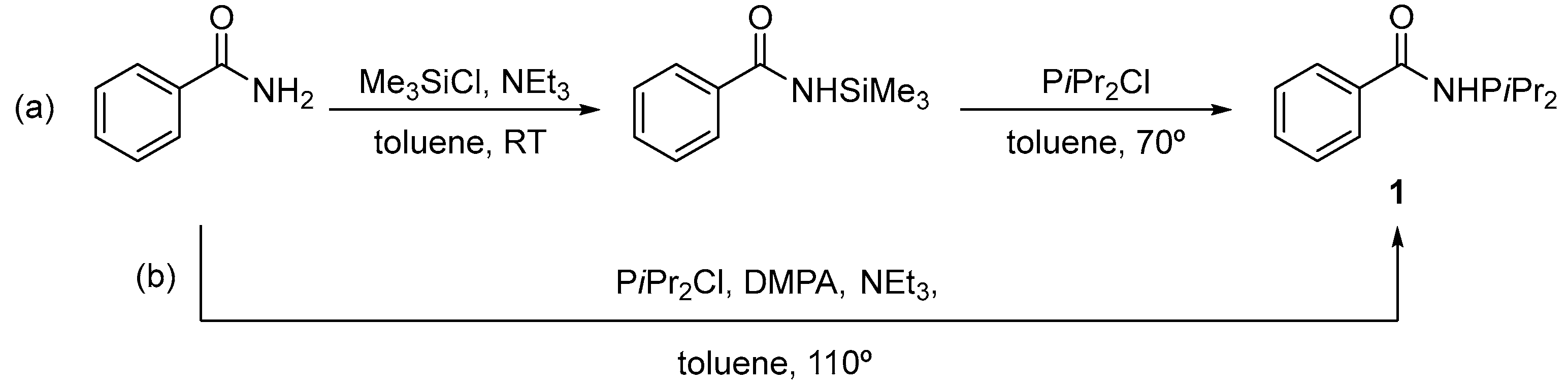
Figure 1.
ORTEP drawing of compound 1 with thermal ellipsoids at the 30% probability level and showing the positional disorder of the phenyl group at two refined extreme positions, both of them with coefficients of 0.5. Hydrogen atoms, except H1, were omitted for clarity. Selected bond distances (Å) and angles (°): C1-O1 1.231(6), C1-N1 1.363(6), P1-N1 1.735(3), P1-C8 1.851(5), P1-C11 1.829(5); O1-C1-N1 122.3(5), C1-N1-P1 122.4(3), N1-P1-C8 100.1(2), N1-P1-C11 98.6(2), C8-P1-P11 102.8(2).
Figure 1.
ORTEP drawing of compound 1 with thermal ellipsoids at the 30% probability level and showing the positional disorder of the phenyl group at two refined extreme positions, both of them with coefficients of 0.5. Hydrogen atoms, except H1, were omitted for clarity. Selected bond distances (Å) and angles (°): C1-O1 1.231(6), C1-N1 1.363(6), P1-N1 1.735(3), P1-C8 1.851(5), P1-C11 1.829(5); O1-C1-N1 122.3(5), C1-N1-P1 122.4(3), N1-P1-C8 100.1(2), N1-P1-C11 98.6(2), C8-P1-P11 102.8(2).
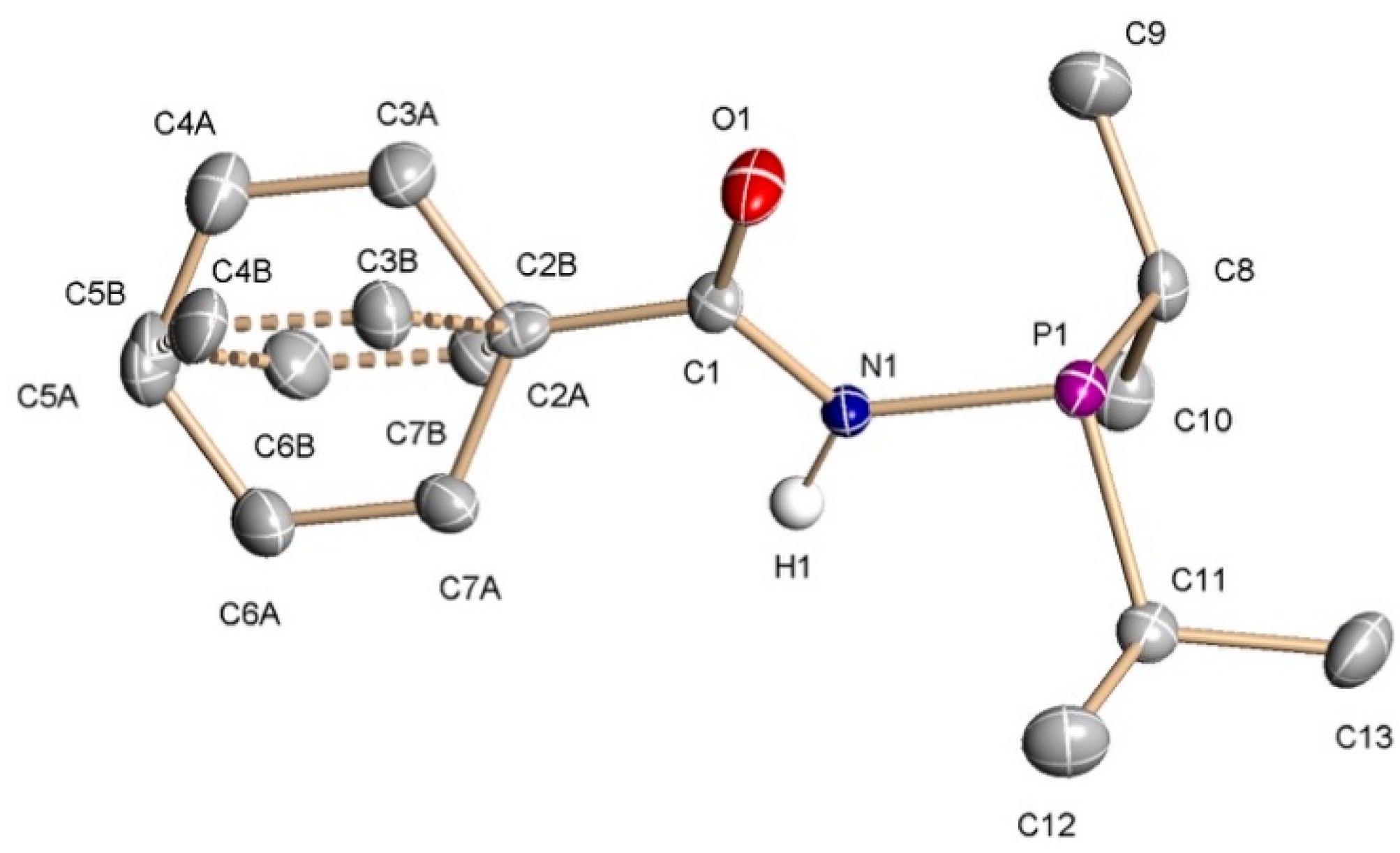
Figure 2.
Partial packing diagrams for 1: (a) view along the crystallographic a-axis; (b) view along the crystallographic b-axis. Hydrogen bonds are represented as dotted lines connecting N—H and C=O fragments.
Figure 2.
Partial packing diagrams for 1: (a) view along the crystallographic a-axis; (b) view along the crystallographic b-axis. Hydrogen bonds are represented as dotted lines connecting N—H and C=O fragments.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



