
Submitted:
27 April 2023
Posted:
28 April 2023
You are already at the latest version
Abstract
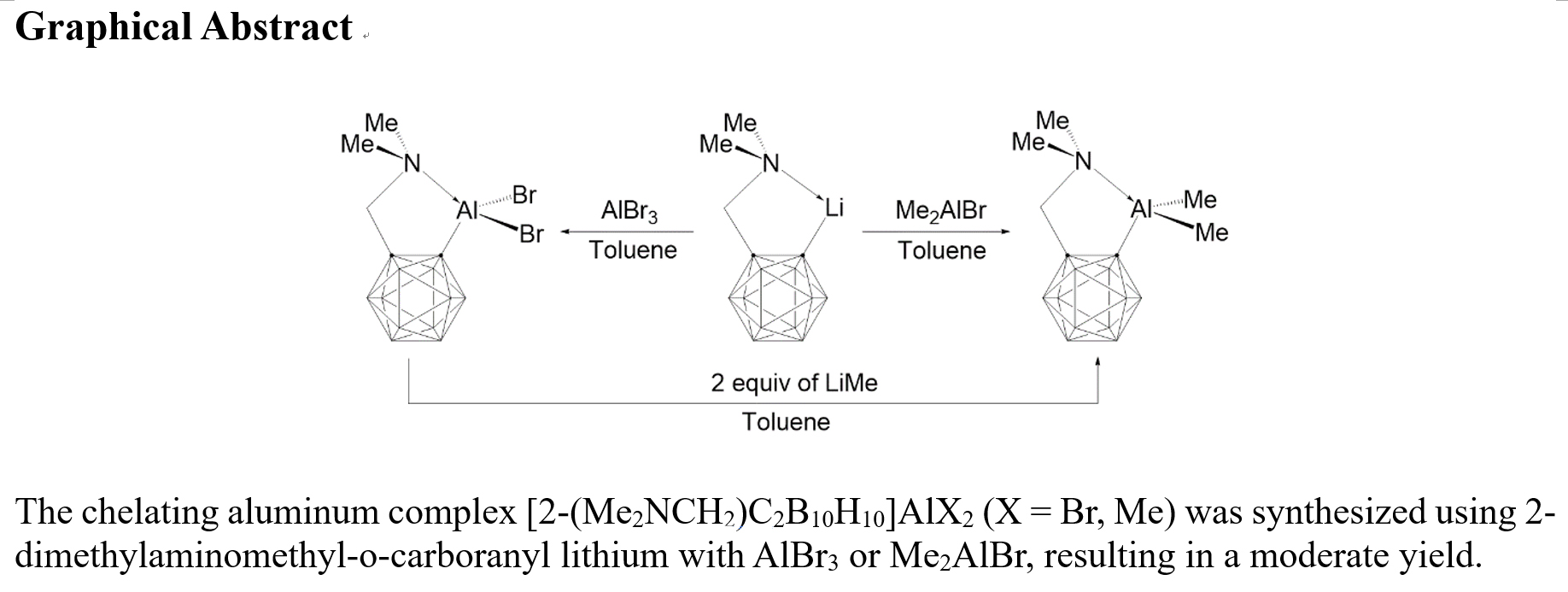
The chelating aluminum complex [2-(Me2NCH2)C2B10H10]AlX2 (X = Br 3, CH3 4) was synthesized using 2-dimethylaminomethyl-o-carboranyl lithium (2) with aluminum tribromide or dimethylaluminum bromide, resulting in a modest yield. Compound 4 was obtained by reacting compound 3 with CH3Li in toluene. All compounds were characterized by infrared (IR) spectroscopy, 1H, 11B, 13C nuclear magnetic resonance (NMR) spectroscopy, and X-ray crystallography. X-ray structural studies of CabNAlBr2 (3) and CabNAlMe2 (4) indicated that the Al atom was located at the center of a distorted tetrahedron. Crystal structures of the CabNAlBr2 (3) [a = 8.9360(3) Å, b = 12.0358(9) Å, c = 14.7730(4) Å, α = β = γ = 90°] and the CabNAlMe2 (4) [a = 8.9551(3) Å, b = 11.9126(9) Å, c = 14.7711(4) Å, α = β = γ = 90°] were obtained. The reactivity of aluminum complexes 3 and 4 with Lewis bases, such as H2O, pyridine, alkylamines, and arylamines, confirmed their rapid decomposition.
Keywords:
C
; N-chelate ligand
; o-carborane
; organoaluminum complexes
; X-ray crystallography
; intramolecular stabilization
1. Introduction
Organometallic compounds with unique properties and specific reactivities can be prepared by utilizing the properties of C,N-chelating ligands1. These properties can be attributed to the extension of their coordination number by the intramolecular coordination of a C,N-chelating ligand. Various organoaluminum compounds with C,N-chelating ligands have been prepared that are bonded to the aluminum atom through an Al‒C covalent bond and an Al‒N dative bond.2-9 Therefore, it is interesting to investigate the possibility of synthesizing such intramolecularly coordinated complexes using the potentially bidentate C,N-chelating o-carboranylamino ligand system CabC,N (1).10,11
This study reports on the preparation and characterization of organoaluminum complexes of the type (CabC,N)AlX2 (X = Br, CH3), in which the aluminum metal center could be regarded as tetracoordinated because of the intramolecular Al‒N coordination. We have previously reported successful synthesis and structural characterization of tetracoordinated gallium complexes of the type (CabC,N)GaX2 (X = Cl or CH3).10 In this respect, we have started investigating the synthesis of intramolecularly coordinated organoaluminum compounds bearing a bulky o-carborane unit12 that might stabilize the tetra-coordinate aluminum metal center.
To the best of our knowledge, this is the first example in which a 2-dimethylaminomethyl-o-carboranyl ligand is intramolecularly coordinated to an aluminum atom, thus improving the stability of the tetracoordinated aluminum metal center. This study provides the detailed syntheses and complete characterization of a series of dibromo- and dimethyl-substituted organoaluminum compounds containing potentially bidentate o-carboranylamino ligands.
2. Materials and Methods
2.1. General Procedures
All manipulations were performed under a dry, oxygen-free nitrogen or argon atmosphere using standard Schlenk techniques, or in vacuum in a KK-011AS glove box. Ether and toluene were dried and distilled from sodium benzophenone. Hexane was dried and distilled over CaH2. 1H, 11B, and 13C NMR spectra were recorded on a JEOL 300 MHz NMR spectrometer operating at 300.1, 96.3, and 75.4 MHz, respectively. All the boron-11 chemical shifts were referenced to BF3∙O(C2H5)2 (0.0 ppm), with a negative sign indicating an upfield shift. All proton and carbon chemical shifts were measured relative to the internal residual benzene in the lock solvent (99.5% C6D6) and then referenced to tetramethylsilane (0.00 ppm). IR spectra were recorded using a Bio-Rad FTS-165 spectrophotometer. Elemental analyses were performed using a Carlo Erba Instruments CHNS-O EA1108 analyzer. All melting points were uncorrected. Decaborane and (dimethylamino)-2-propyne were purchased from Katchem and Aldrich, respectively, and used without purification. CabN (1; HCabN, closo-1-[(dimethylamino)methyl]-o-carborane)13 was prepared according to a previously reported method. The starting material AlBr3 was purchased from Strem Chemical and sublimed under dynamic vacuum prior to each use. Dimethylaluminum bromide14 was prepared by conducing stoichiometric ligand redistribution reactions in pentane at room temperature, and then purified by vacuum sublimation at room temperature.
2.2. X-ray Crystallography
Details of the crystal data and a summary of the intensity data collection parameters for 3 and 4 are provided in Table 1. Crystals of 3 and 4 were grown in toluene solution and stored at ‒20 °C. They were then mounted in thin-walled glass capillaries and sealed under an argon atmosphere and transferred to the goniometer of the Bruker SMART 1000 CCD area detector system. The initial unit cell parameters were obtained using the SMART15 software. Data integration, correction for Lorentz and polarization effects, and final cell refinement were performed using SAINTPLUS.16 The data were further corrected for absorption using SADABS.17 The initial structure solutions were obtained by direct methods in SHELXTL.18 Subsequent refinement cycles (based on F2) and Fourier synthesis revealed the positions of all non-hydrogen atoms. The atoms were refined using anisotropic displacement tensors. All hydrogen atoms were added to the calculated positions for the final refinement cycle.
2.3. Synthesis of CabNAlBr2 (3)
To a stirred solution of CabN (1; 0.604 g, 3.0 mmol) in 30 mL of hexane, cooled to ‒10 °C, 1.6 M n-BuLi (2 mL, 3.2 mmol) was added via a syringe. The resulting white suspension was stirred at ‒10 °C for 2 h and then transferred through a cannula to a suspension of AlBr3 (0.80 g, 3.0 mmol) in 50 mL of toluene at ‒78 °C. The reaction temperature was first maintained at ‒78 °C for 1 h, after which the reaction mixture was slowly warmed to room temperature. After being stirred for additional 12 h, the mixture was filtered. The solvent was removed under vacuum, and the resulting residue was taken up in a minimum of toluene and then recrystallized by cooling the solution to ‒20 °C. Compound 3 was isolated from the reaction solution in 82% yield (0.95 g, 2.5 mmol). Anal. Calcd: C, 15.51; H, 4.69; N, 3.62. Found: C, 15.71; H, 4.88; N, 3.72. MP: 231–232 °C. IR (Nujol, cm‒1): ν(C‒H) 3100, 2990; ν(B‒H) 2590. 1H NMR (C6D6, 400 MHz) δ 1.630 (s, N‒CH3), 2.212 (s, N‒CH2). 11B NMR (C6D6, 400 MHz) δ ‒2.31 (d, 1B, JBH = 150 Hz), ‒4.08 (d, 1B, JBH = 175 Hz), ‒5.79 (d, 2B, JBH = 150 Hz), ‒9.72 (d, 1B, JBH = 180 Hz), ‒11.14 (d, 1B, JBH = 125 Hz), ‒11.75 (d, 2B, JBH = 140 Hz), ‒12.95 (d, 2B, JBH = 130 Hz). 13C NMR (C6D6, 400 MHz) δ 52.060 (N‒CH3), 66.517 (N‒CH2), 72.674 (carborane).
2.4. Synthesis of CabNAlMe2 (4)
A solution of Me2AlBr (0.41 g, 3.0 mmol) in toluene (50 mL) was slowly added to a hexane solution (30 mL) of LiCabN at ‒78 °C. LiBr precipitated from the solution when the mixture was warmed to room temperature. After stirring the mixture overnight at room temperature, the solution was decanted and the remaining solids were washed with toluene. The toluene was then removed in vacuo, leaving a white solid, which was purified by recrystallization from toluene at ‒20 °C. Compound 4 was isolated from the reaction solution in 61% yield (0.47 g, 1.8 mmol). Anal. Calcd: C, 32.67; H, 9.40; N, 5.44. Found: C, 32.74; H, 9.53; N, 5.55. MP: 175–177 °C. IR (Nujol, cm‒1): ν(C‒H) 3100, 2990; ν(B‒H) 2590. 1H NMR (C6D6, 400 MHz) δ 0.520 (s, Al‒CH3), 1.512 (s, N‒CH3), 2.087 (s, N‒CH2). 11B NMR (C6D6, 400 MHz) δ ‒3.18 (d, 1B, JBH = 155 Hz), ‒4.31 (d, 1B, JBH = 137 Hz), ‒10.10 (d, 2B, JBH = 170 Hz), ‒12.88 (d, 6B, JBH = 135 Hz). 13C NMR (C6D6, 400 MHz) δ 10.377 (Al‒CH3), 48.774 (N‒CH3), 63.124 (N‒CH2), 73.800 (carborane).
2.5. Reaction of CabNAlBr2 (3) with LiMe
A 1.4 M solution of LiMe in diethyl ether (1.6 mL, 2.2 mmol) was added to a solution of 3 (0.77 g, 2.0 mmol) in toluene (30 mL) at ‒78 °C. The dry-ice bath was removed, and the solution was stirred for 3 h. Subsequently, all volatile components were removed in vacuo at room temperature. The crude product was then re-dissolved in toluene (40 mL), and the solution was separated by filtration. The volume was then reduced to 20 mL. Crystallization at ‒20 °C afforded pure 4 (0.10 g, 0.40 mmol, 20% yield).
3. Results
3.1. Synthesis of CabNAlBr2 (3)
As shown in Scheme 1, intramolecularly stabilized tetracoordinated dimethylamino-o-carboranyl aluminum compound CabNAlBr2 (3) was synthesized in toluene from aluminum tribromide and dimethylaminomethyl-o-carboranyl lithium compound LiCabN (2). Formation of a five-membered chelate ring affords stabilization to compound 3. Notably, compound 3 was moderately unstable in air and decomposed slowly upon contact with O2 and moisture.
Compound 3 was purified from colorless crystals through low-temperature recrystallization in toluene. Satisfactory elemental analysis was performed for 3. Its 1H, 11B, and 13C NMR spectral data were consistent with the presence of bidentate (dimethylaminomethyl)-o-carboranyl ligand 1 (see Experimental Section). Details of the crystal data and a summary of the intensity data collection parameters of 3 and 4 are provided in Table 1 and Table 2, respectively. The molecular structure of 3, shown in Figure 1, was determined by X-ray diffraction (XRD). The selected interatomic distances and angles are listed in Table 2. Similar to our previously published work,10 the molecule lies on a crystallographic reflection plane and exhibits rigorously imposed reflection symmetry. Atoms Al(1), N(1), and C(13) and cage atoms C(1), C(2), B(7), and B(8) lie on the symmetry plane. The dimethylamine group of the o-carboranyl ligand was coordinated to the aluminum metal in complex 3, resulting in the formation of a five-membered chelate ring, N(1)–Al(1)–C(1)–C(2)–C(13). The Al(1), Br(1), Br(1)*, and C(1) moieties were close to planar, with aluminum 0.509(5) Å above the Br(1)–Br(1)*C(1) plane. The angles around aluminum did not deviate much from the tetrahedral angle, except for the C(1)–Al(1)–N(1) angle in the five-membered ring, which was 92.2(4)°. This also influenced the geometry around the o-carborane cage; the C(2)–C(1)–Al(1) angle (107.9(7)°) within the five-membered ring was similar to that expected at 108°. The Al(1)–N(1) distance in 3 (2.039(1) Å) is longer than that in the corresponding amine-ligated aryl system [o-(Me2NCH2)C6H4]AlBr2 (2.003(5) Å).7 The Al(1)–N(1) distance of 2.039(1) Å is in agreement with the corresponding values observed in adducts formed between X3Al and NR3;19-26 all distances are longer than the sum of the covalent radii for aluminum and nitrogen (1.95 Å).27 The Al(1)–C(1) distance (1.938(1) Å) is similar to that reported for other organoaluminum compounds.6,7,28-31 The Al–Br distance of 2.216(2) Å is in the range of typical covalent Al–Br bond distances (2.310–2.316 Å).7
3.2. Synthesis of CabNAlMe2 (4)
As shown in Scheme 1, CabNAlMe2 (4), an o-carboranyl dimethylaluminum compound, was obtained as a pure product from the reaction of dimethylaluminum bromide with LiCabN (2). This compound also forms colorless crystals, is stable in an inert gas environment, and exhibits slow decomposition upon exposure to air. It is readily soluble in organic solvents, such as pentane, hexane, and aromatic solvents. Its 1H, 11B, and 13C NMR spectra exhibited the expected patterns and shifts. The molecular structure of 4 was determined by XRD. The structure is shown in Figure 2, and the selected bond lengths and angles are listed in Table 2. Similar to compound 3, the molecule lies on a crystallographic reflection plane and thus exhibits rigorously imposed reflection symmetry. Atoms Al(1), N (1), and C (13) and cage atoms C(1), C(2), B(7), and B(8) lie on the symmetry plane. The geometry of 4 is similar to that of 3 owing to intramolecular Al–N coordination. This tetrahedral distortion, with nearly “normal” angles [C(15)–Al(1)–N(1) (109.1(2)°) and C(15)–Al(1)–C(1) (113.1(2)°] and a significantly different angle [C(1)–Al(1)–N(1) (88.4(2)°], is caused by the transannular Al–N interaction. The Al(1)–N(1) distance (2.090(5) Å) is in agreement with the corresponding values observed in [(AlMe3)3]4[N-tetramethylcyclam] (2.093(3) Å),20 (Me3Al)2[(Me2N)2CH2] (2.104(2), 2.112(2) Å),32 and Me3AlNMe3 (2.099(1) Å)19, but is longer than the sum of the covalent radii for Al and N (1.95 Å).27 The Al(1)–C(1) distance (2.010(6) Å) is slightly longer than the other two Al–C distances (Al(1)–C(15) and Al(1)–C(15)*, 1.947(5) Å). The Al–C distances of the Me2Al group were similar to those of Me3Al in the gas phase.19,33
3.3. Reaction of CabNAlBr2 (3) with LiMe
Compound 4 was synthesized using a second route (Scheme 2). When compound 3 was reacted with 2 equiv of MeLi, the dimethylated compound CabNAlMe2 (4) was obtained. According to Scheme 1, the two bromine atoms of compound 3 can be selectively replaced by organic ligands to produce crystalline compound 4. The NMR spectra of this product were identical to those of 4. The 1H and 13C NMR data, which confirm the proposed stoichiometry, are presented in the Experimental Section.
4. Conclusions
To the best of our knowledge, this study is the first to report intramolecularly coordinated aluminum complexes containing an o-carboranyl C,N-chelating ligand. A combination of X-ray crystallographic and spectroscopic studies, including 1H, 11B, and 13C NMR spectroscopy, confirmed the nature of these compounds. The X-ray crystallographic study of complexes 3 and 4 provided the first structural data for [(dimethylamino)methyl-o-carboranyl]aluminum complexes. In such aluminum bromide complexes, the aluminum atom may be tetracoordinated, depending on the number of C,N-chelating ligands present in the molecule. Moreover, o-carboranyl C,N-chelating ligands can stabilize aluminum compounds, such that they can be isolated as well-defined monomeric species. Additionally, the internal coordination of the nitrogen atom of the o-carboranyl ligand to the aluminum metal center significantly influences the stability of the starting material. This may be the result of the stabilization of the aluminum–carbon bond by the chelate effect, the electronic properties of the aluminum center, and the strength of the aluminum–nitrogen bond. Thus, application of the C,N-chelating o-carboranyl ligand enables the synthesis of stable aluminum(III) compounds. In contrast to previous studies, this study reports that when reacted with a strong Lewis base, such as pyridine, 3 and 4 easily decompose owing to the strong Lewis acid properties of aluminum.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, X-ray structures of Compounds 3 and 4, Tables S1–S6: Detailed information on the structural determinations and structural features of compounds 3and 4are provided in the Supplementary Materials.
Author Contributions
Conceptualization, J.D.L. and H.S.; Designed and performed X-ray crystallography, J. D. L.; Chemical experiments, J.D.L.; Data Curation, J.D.L. and H.S.; Writing—Original Draft Preparation, J.D.L.; Writing—Review & Editing, J.D.L. and H.S.; Supervision, J.D.L.; Project Administration, J.D.L.; Funding Acquisition, J.D.L.
Funding
This research received no external funding.
Acknowledgments
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2022R1F1A1074095).
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A
CCDC 2256552 and 2256553 contains the supplementary crystallographic data of 3 and 4 for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieving.html (or from the Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; or deposit@ccdc.cam.ac.uk). Additional Supporting Information may be found online in the supporting information tab for this article.
References
- Jastrzebski, J. T. B. H.; van Koten, G. Adv. Organomet. Chem. 1993, 35, 241–294, references therein. [Google Scholar]
- Schumann, H.; Hartmann, U.; Wassermann, W.; Dietrich, A.; Görlitz, F. H.; Pohl, L.; Hostalek, M. Chem. Ber. 1990, 123, 2093–2099. [Google Scholar]
- Matsumoto, T.; Takamine, H.; Tanaka, K.; Chujo, Y. Chem. Lett. 2015, 44, 1658–1660. [Google Scholar]
- Zheng, W.; Roesky, H. W.; Mösch-Zanetti, N. C.; Schmidt, H. -G.; Noltemeyer, M. Eur. J. Inorg. Chem. 2002, 1056–1059. [Google Scholar] [CrossRef]
- Kumar, R.; Rahbarnoohi, H.; Heeg, M. J.; Dick, D. G.; Oliver, J. P. Inorg. Chem. 1994, 33, 1103–1108. [Google Scholar]
- Contreras, L.; Cowley, A. H.; Gabbaï, F. P.; Jones, R. A.; Carrano, C. J.; Bond, M. R. J. Organomet. Chem. 1995, 489, C1–C3. [Google Scholar]
- Isom, H. S.; Cowley, A. H.; Decken, A.; Sissingh, F.; Corbelin, S.; Lagow, R. J. Organometallics 1995, 14, 2400-2406.
- Tian, X.; Woski, M.; Lustig, C.; Pape, T.; Fröhlich, R.; Le Van, D.; Bergander, K.; Mitzel N., W. Organometallics 2005, 24, 82-88.
- Cowley, A. H.; Gabbaï, F. P.; Isom, H. S.; Decken, A. J. Organomet. Chem. 1995, 500, 81–88. [Google Scholar]
- Lee, J.-D.; Baek, C.-K.; Ko, J.; Park, K.; Cho, S.; Min, S.-K.; Kang, S. O. Organometallics 1999, 18, 2189-2197.
- Lee, J.-D.; Kim, S.-J.; Yoo, D.; Ko, J.; Cho, S.; Kang, S. O. Organometallics 2000, 19, 1695-1703.
- Beall, H. In Boron Hydride Chemistry, Muetterties, E., Ed.; Academic Press: New York, 1975; Chapter 9.
- Heying, T. L.; Ager, J. W., Jr.; Clark, S. L.; Mangold, D. J.; Goldstein, H. L.; Hillman, M.; Polak, R. J.; Szymanski, J. W. Inorg. Chem. 1963, 2, 1089–1092. [Google Scholar]
- Grosse, A. V.; Mavity, J. M. J. Org. Chem. 1940, 5, 106–121. [Google Scholar]
- SMART V5. 05 Software for the CCD Dector System; Bruker Analytical X-ray Systems, Inc.: Madison, WI, 1998. [Google Scholar]
- SAINTPLUS, V5. 00 Software for the CCD Dector System; Bruker Analytical X-ray Systems, Inc.: Madison, WI, 1998. [Google Scholar]
- SADABS. Program for absorption correction using SMART CCD based on the method of: Blessing, R. H. SADABS. Program for absorption correction using SMART CCD based on the method of: Blessing, R. H. Acta Crystallogr. 1995, A51, 33.
- SHELXTL, Version 5. 03; Siemens Industrial Automation, Inc.: Madison, WI, 1994. [Google Scholar]
- Anderson, G. A.; Forgaard, F. R.; Haaland, A. Acta Chem. Scand. 1972, 26, 1947–1954. [Google Scholar] [CrossRef]
- Robinson, G. H.; Zhang, H.; Atwood, J. L. J. Organomet. Chem. 1987, 331, 153–160. [Google Scholar]
- Atwood, J. L.; Bennett, F. R.; Elms, F. M.; Jones, C.; Raston, C. L.; Robinson, K. D. J. Am. Chem. Soc. 1991, 113, 8183–8185. [Google Scholar]
- Atwood, J. L.; Bennett, F. R.; Jones, C.; Koutsantonis, G. A.; Raston, C. L.; Robinson, K. D. J. Chem. Soc, Chem. Commun. 1992, 541–543. [Google Scholar]
- Atwood, J. L.; Jones, C. Raston, C. L.; Robinson, K. D. Main Group Chem. 1996, 1, 345–347. [Google Scholar] [CrossRef]
- Andrews, P. C.; Gardiner, M. G.; Raston, C. L.; Tolhurst, V. -A. Inorg. Chim. Acta 1997, 259, 249–255. [Google Scholar] [CrossRef]
- Alexander, S. G.; Cole, M. L.; Forsyth, C. M. Chem. Eur. J. 2009, 15, 9201–9214. [Google Scholar] [CrossRef]
- Carmalt, C. J.; King, S. J.; Mileham, J. D.; Sabir, E.; Tocher, D. A. Organometallics 2004, 23, 2939-2943.
- Bondi, A. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar]
- Evans, W. J.; Chamberlain, L. R.; Ziller, J. W. J. Am. Chem. Soc. 1987, 109, 7209–7211. [Google Scholar]
- Li, X. -W.; Su, J.; Robinson, G. H. H. Chem. Commun. 1998, 1281–1282. [CrossRef]
- Cui, C.; Roesky, H. W.; Noltemeyer, M.; Schmidt, H. -G. Organometallics 1999, 18, 5120–5123. [Google Scholar] [CrossRef]
- Hatop, H.; Roesky, H. W.; Labahn, T.; Fischer, A.; Schmidt, H. -G. ; Noltemeyer, M. Organometallics 2000, 19, 937–940. [Google Scholar] [CrossRef]
- Mitzel, N. W.; Lustig, C. Z. Naturforsch. 2004, 59B, 1532-1539.
- Almenningen, A.; Halvorsen, S.; Haaland, A. Chem. Commun. 1969, 644. [Google Scholar]
Scheme 1.
Synthesis of CabNAlBr2 (3) and CabNAlMe2 (4).
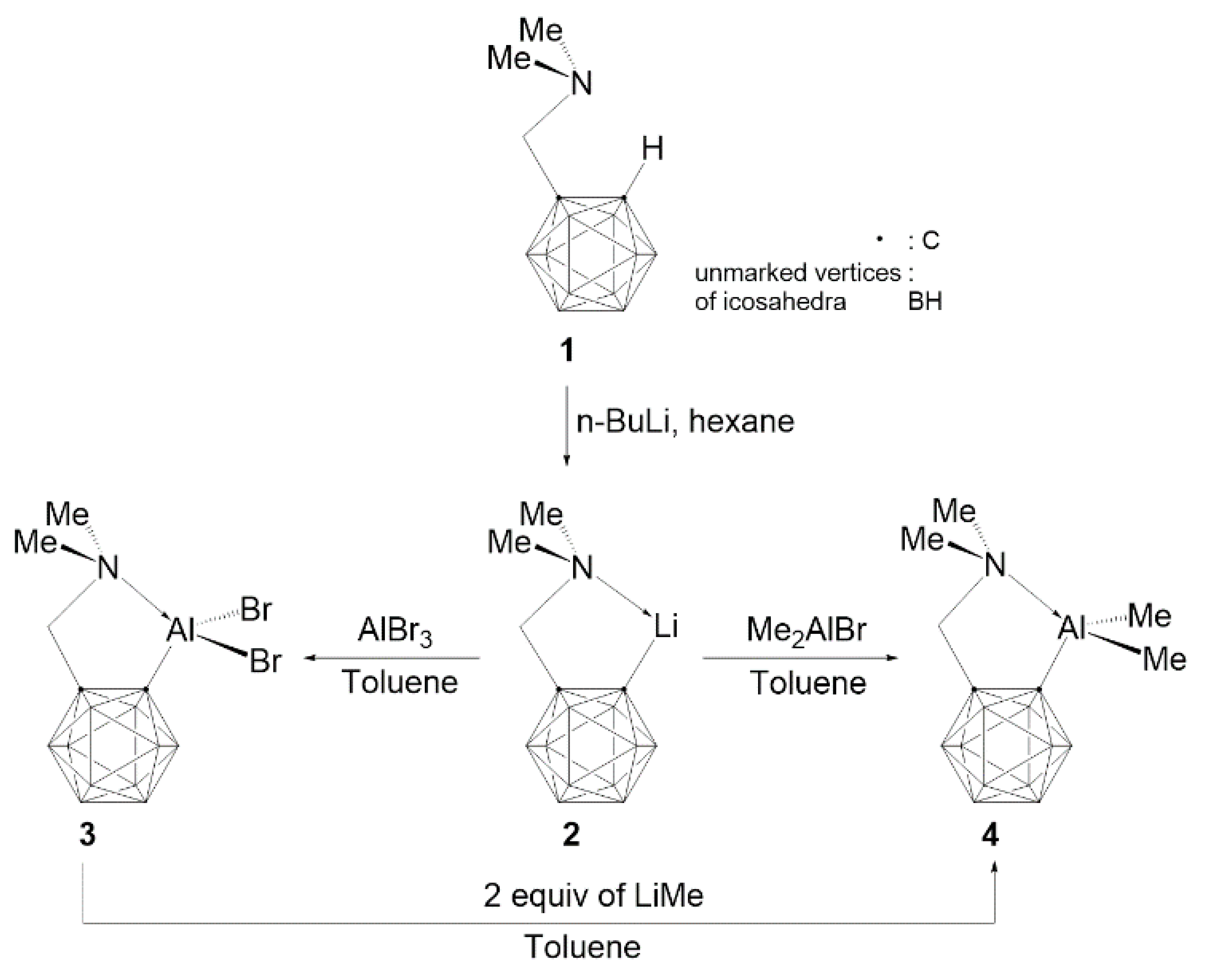
Figure 1.
Molecular structure of CabNAlBr2 (3). The thermal ellipsoids are drawn at the 30% probability level.
Figure 1.
Molecular structure of CabNAlBr2 (3). The thermal ellipsoids are drawn at the 30% probability level.
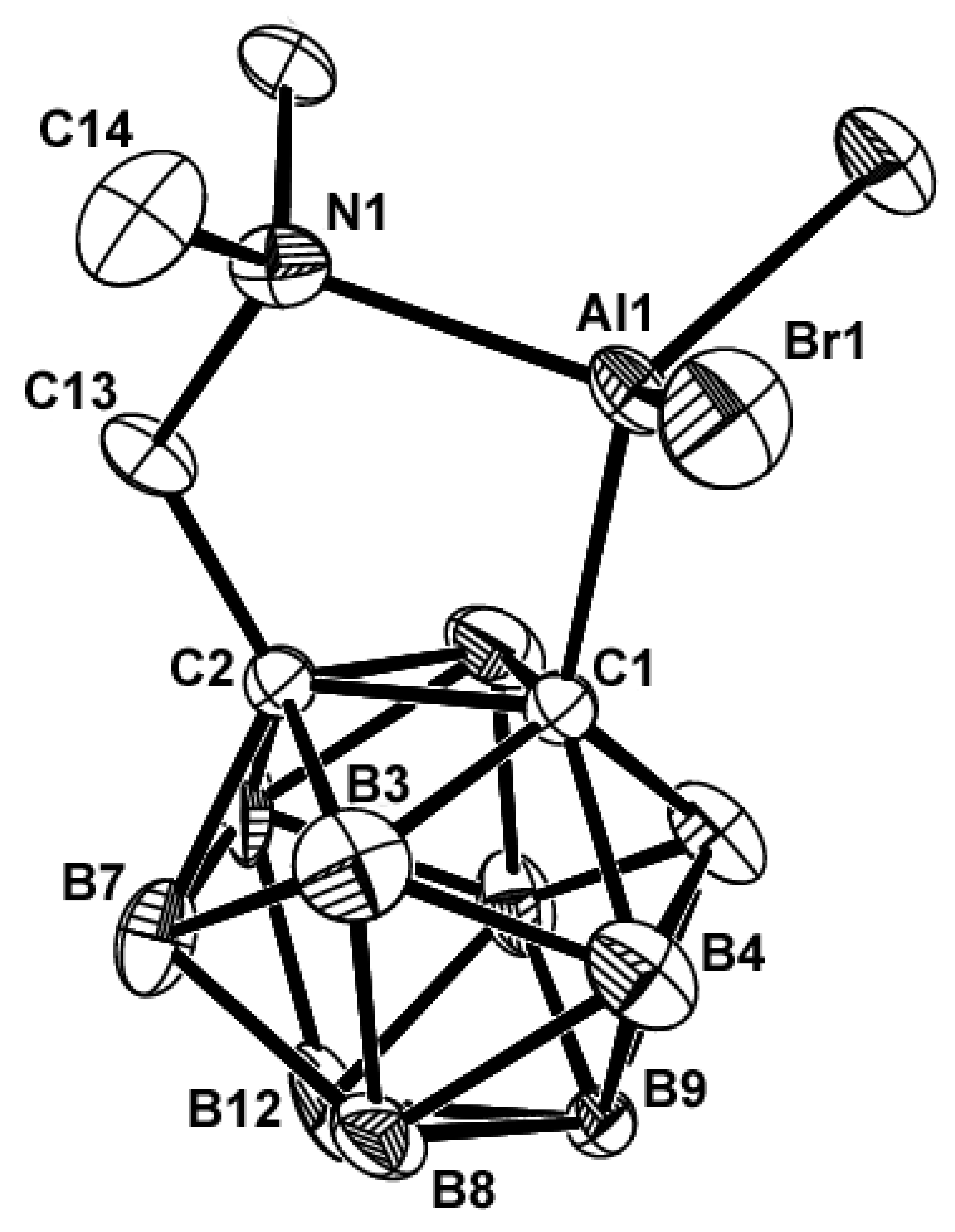
Figure 2.
Molecular structure of CabNAlMe2 (4). The thermal ellipsoids are drawn at the 30% probability level.
Figure 2.
Molecular structure of CabNAlMe2 (4). The thermal ellipsoids are drawn at the 30% probability level.
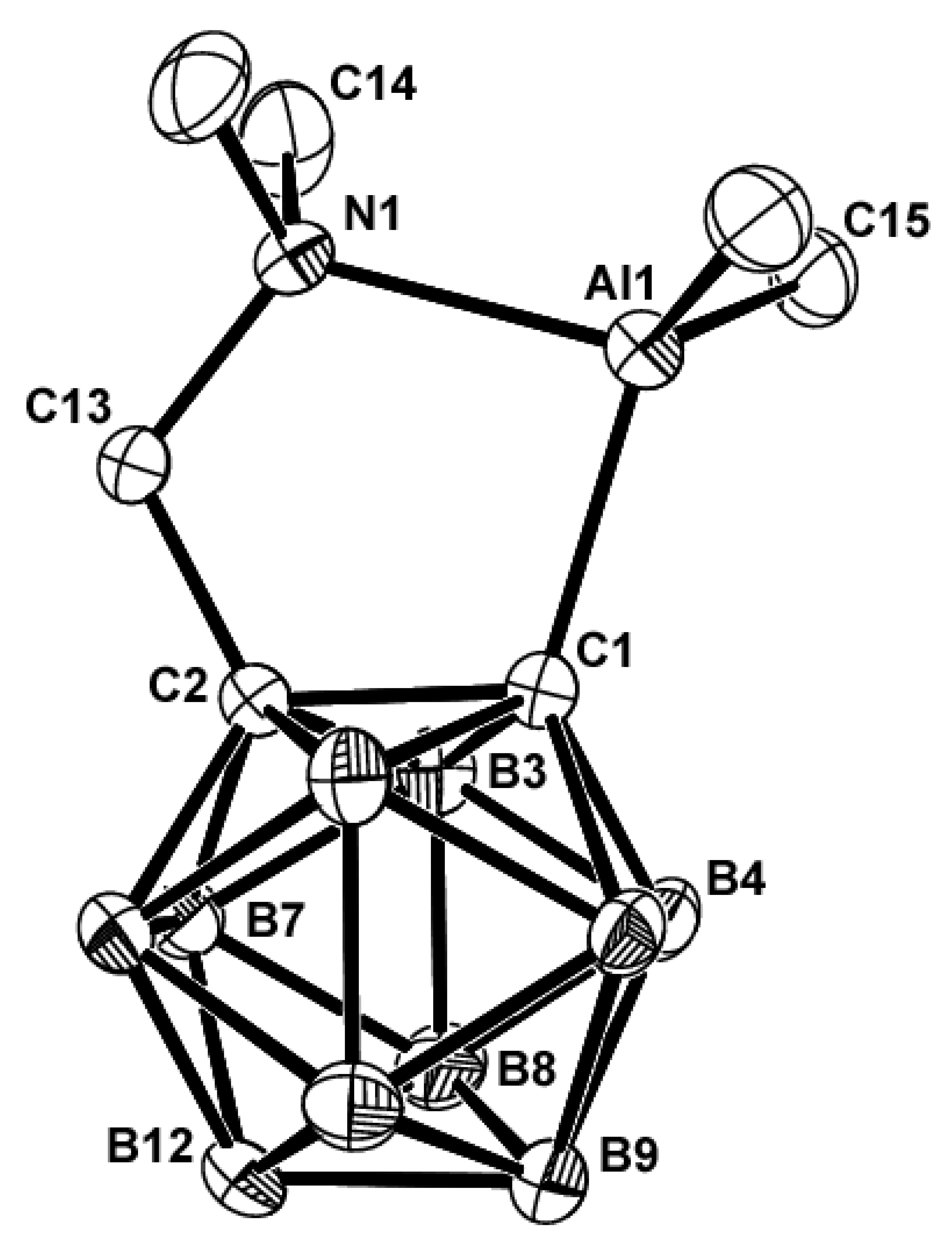

Table 1.
X-ray crystallographic data and processing parameters for compounds 3 and 4.
| Identification code Empirical formula Formula weight Temperature Wavelength Crystal system, space group Unit cell dimensions Volume Z, Dcalc Absorption coefficient F(000) Crystal size θ range for data collection Limiting indices Reflections collected / unique Completeness to θ = 25.96 Refinement method Data / restraints / parameters Goodness-of-fit on F2 Final R indices [I>2σ (I)] R indices (all data) Extinction coefficient Largest diff. peak and hole |
KOR103 C5 H18 B10 N1 Br2 Al1 387.08 293(2) K 0.71073 Å Orthorhombic, Cmc 21 a = 8.9360(3) Å, α = 90.00º b = 12.0358(9) Å, β = 90.00º c = 14.7730(4) Å, γ = 90.00º 1588.9(1) Å−3 4, 1.618 g/cm3 5.132 mm−1 752 0.2 × 0.16 × 0.14 mm 2.76 to 25.97º ‒8≤h≤10, ‒8≤k≤14, ‒18≤l≤0 1693 / 853 [R(int) = 0.0891] 99.9 % Full-matrix least-squares on F2 853 / 1 / 98 0.923 aR1 = 0.0394, bwR2 = 0.0834 aR1 = 0.1020, bwR2 = 0.0881 0.19(3) 0.713 and –0.700 e·Å−3 |
KOR106 C7 H24 B10 N1 Al1 257.37 293(2) K 0.71073 Å Orthorhombic, Cmc 21 a = 8.9551(3) Å, α = 90.00º b = 11.9126(9) Å, β = 90.00º c = 14.7711(4) Å, γ = 90.00º 1575.8(1) Å−3 4, 1.085 g/cm3 0.153 mm−1 544 0.18 × 0.14 × 0.10 mm 2.76 to 25.95º ‒8h≤11, ‒8≤k≤14, ‒8≤l≤18 1748 / 845 [R(int) = 0.0703] 100.0 % Full-matrix least-squares on F2 845 / 1 / 105 1.005 aR1 = 0.0448, bwR2 = 0.1110 aR1 = 0.0876, bwR2 = 0.1066 ‒0.3(6) 0.193 and –0.227 e·Å−3 |
aR1 = ∑||Fo|-|Fc|| (based on reflections with Fo2>2σF 2), bwR2 = [∑[w(Fo2-Fc2)2]/∑[w(Fo2)2]]1/2; w = 1/[σ2(Fo2)+(0.095P)2]; and P = [max(Fo2, 0)+2Fc2]/3(also with Fo2>2σF 2)
Table 2.
Selected interatomic distances (Å) and angles (°) for 3 and 4.
| Compound 3 | |||
| Al1−C1 | 1.938(1) | Al1−N1 | 2.039(1) |
| Al1−Br1 | 2.216(2) | N1−C13 | 1.518(2) |
| Al1−Br#1 | 2.216(2) | N1‒C14 | 1.476(1) |
| C1‒C2 | 1.649(2) | N1‒C#14 | 1.476(1) |
| C2‒C13 | 1.516(2) | ||
| C1−Al1−N1 | 92.2(4) | C13−N1−Al1 | 109.4(8) |
| C1−Al1−Br1 | 113.7(2) | C2−C1−Al1 | 107.9(7) |
| C1−Al1−Br#1 | 113.7(2) | Br#1−Al1−Br1 | 115.7(2) |
| N1−Al1−Br1 | 109.5(2) | C#14−N1−C14 | 108.6(1) |
| N1−Al1−Br#1 | 109.5(2) | C14‒N1‒C13 | 109.2(8) |
| Compound 4 | |||
| Al1−C1 | 2.010(6) | Al1−N1 | 2.090(5) |
| Al1−C15 | 1.947(5) | N1−C13 | 1.507(7) |
| Al1−C#15 | 1.947(5) | N1‒C14 | 1.488(5) |
| C1‒C2 | 1.652(8) | N1‒C#14 | 1.488(5) |
| C2‒C13 | 1.532(8) | ||
| C1−Al1−N1 | 88.4(2) | C13−N1−Al1 | 113.2(3) |
| C1−Al1−C15 | 113.1(2) | C2−C1−Al1 | 108.9(3) |
| C1−Al1−C#15 | 113.1(2) | C#15−Al1−C15 | 119.4(3) |
| N1−Al1−C15 | 109.1(2) | C#14−N1−C14 | 108.0(6) |
| N1−Al1−C#15 | 109.1(2) | C14‒N1‒C13 | 108.3(3) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



