
Submitted:
17 May 2023
Posted:
18 May 2023
You are already at the latest version
Abstract
The Paeonia suffruticosa, called as 'Feng Dan', has been used for thousands of years in traditional Chinese medicine. In our chemical investigation on the root bark of the plant, five new phenolic dimers, namely paeobenzofuranone A‒E (1‒5), have been characterized. Their structures were determined by spectroscopic analysis including 1D and 2D NMR, HRESIMS, UV, and IR, as well as ECD calculations. Compounds 2, 4, and 5 showed cytotoxicity against three human cancer cell lines with IC50 values ranging from 6.7 to 25.1 μM. Compounds 1 and 2 showed certain inhibitory activity on NO production. To the best of our knowledge, the benzofuranone dimers and their cytotoxicity of P. suffruticosa are reported for the first time.
Keywords:
Paeonia suffruticosa
; benzofuranones
; cytotoxicity
; NO production inhibition
1. Introduction
Paeonia suffruticosa is a perennial deciduous shrub belonging to the family Paeoniaceae. The dried root bark of P. suffruticosa is called 'Feng Dan' or 'Mudanpi' in China, which is used as a traditional Chinese medicine to clear pathogenic heat from the blood, promot blood circulation to remove blood stasis, as recorded in Chinese Pharmacopoeia (2020 edition). Nowadays, pharmacological studies on P. suffruticosa have demonstrated anti-inflammatory, antioxidant, and anti-tumor activities, as well as central nervous system and cardiovascular system protective activities [1,2,3]. P. suffruticosa has been chemically investigated and is rich in phenolics, monoterpenes and glycosides, flavonoids, triterpenes, sesquiterpenes and lignans. Approximately, 190 constituents have been reported in recent ten years with anti-inflammatory and antitumor properties, representatives are paeonisides A and B, mudanpiosides C and F, and suffruticosol A [4,5,6,7,8].
As part of our ongoing work on bioactive natural products from plants [9,10,11,12,13,14], the study on the chemical constituents of the root bark of P. suffruticosa was carried out. As a result, five new phenolics were isolated, namely paeobenzofuranone A‒E (1‒5). Their structures (Figure 1) were determined by extensive spectroscopic methods. All compounds were evaluated for their cytotoxicity against three human cancer cell lines including breast cancer MDA-MB-231, human myeloid leukemia HL-60, and colon cancer SW480. In addition, their anti-inflammatory activity by inhibiting NO production was also evaluated. Herein, the isolation, structural elucidation, and bio-activities of the compounds from P. suffruticosa are reported.
2. Results and Discussion
2.1. Structural Elucidation of Compounds 1–5
Compound 1 was obtained as colorless oily liquid. Its molecular formula was determined as C20H18O6 by the HR-ESI-MS with a molecular ion peak at m/z 377.09952 ([M + Na]+, calcd. 377.10011). The UV absorption peaks at λmax 290 and 230 nm indicated the presence of a benzene ring and an ester carbonyl. The IR spectrum indicated that 1 possessed hydroxyl (3406 cm-1) and lactone (1716 cm-1) groups. The 1H NMR spectroscopic data (Table 1) of 1 showed two methyl signals at δH 1.67 (s, 3H) and 2.15 (s, 3H), and two singlets for aromatic protons at δH 6.61 (1H, s, H-7) and 7.18 (1H, s, H-4). Interpretation of the 13C NMR (Table 2) and DEPT spectra data displayed 10 carbon signals, which indicated six non-protonated carbons (C-2: δC 179.4, C-3: δC 52.6, C-5: δC 113.3, C-6: δC 153.7, C-8: δC 146.2, C-9: δC 127.4), two CH (C-4: δC 128.1, C-7: δC 110.3) and two CH3 (C-10: δC 18.7, C-11: δC 16.6). Preliminary analysis of these data suggested that 1 should be a benzofuranone derivative with a structure similar to that of 4,6-dihydroxy-3,5-dimethylcoumaran-2-one [15]. The presence of a methyl at δC 18.7 and a carbonyl carbon at δC 179.4 indicated the differences. In the HMBC spectrum (Figure 2), correlations from H3-11 (δH 2.11) to C-4 (δC 128.1), C-5 (δC 113.3), and C-6 (δC 153.7), and from H3-10 (δH 1.67) to C-3 (δC 52.6), C-2 (δC 179.4), and C-9 (δC 127.4) enabled the assignment of the methyl and the carbonyl carbon. Further analysis of 13C NMR data for a quaternary carbon at C-3 (δC 52.6) revealed that 1 should be a symmetric dimer with a linkage by bond of C-3/C-3ʹ. It was also supported by the MS data analysis. The stereochemistry of C-3 and C-3ʹ was identified as 3R and 3ʹS by ECD calculations (Figure 3). Finally, the structure of 1 was identified and trivially named as paeobenzofuranone A.
Compound 2 was obtained as white powder. Its molecular formula was determined as C27H24O6 by HR-ESI-MS (measured at m/z 445.16489 [M + Na]+; calcd. 445.16511). The UV spectrum revealed the benzene ring and ester carbonyl by peaks at λmax 295 and 230 nm. The 1D spectra data of 2 (Table 1 and Table 2) are partially identical to those of 1. Interpretation of the 1H and 13C spectroscopic data of 2 showed two benzofuran parts and an additional benzoyl moiety. The locations of the benzofuran parts were assigned by HMBC correlations from H-10 (δH 1.75) to C-3 (δC 52.6), C-6 (δC 126.3), C-2 (δC 179.3), as well as from H-4 (δH 6.61) to C-5 (δC 126.3) and C-3 (δC 52.6) (Figure 2). Furthermore, the location of the benzoyl was assigned by key HMBC peaks from H-3ʹ (δH 3.81) to C-10ʹ (δC 66.9), from H-2ʹ (δH 4.43) to C-3ʹ (δC 43.5) and C-2ʹ (δC 73.1); from H-10ʹ (δH 4.44) to C-3ʹ (δC 43.5) and C-10ʹ (δC 66.9). The stereochemistry of C-3 and C-3ʹ was established as 3S and 3ʹR by ECD calculations (Figure 3). Then, the structure of 2 was established and named as paeobenzofuranone B.
Compound 3 was obtained as white powder. The IR spectrum indicated that 3 possessed hydroxyl (3394 cm-1 ) and lactone (1712 cm-1) groups. Its molecular formula was determined as C21H22O8 by HR-ESI-MS data analysis (m/z 425.12030 ([M+Na]+, calcd. 425.12124). The 1H and 13C NMR spectra data of 3 (Table 1 and Table 2) are partially the same as those of 1. Interpretation of the 1H and 13C spectroscopic data of 3 revealed one benzofuran part and one benzene ring. The benzofuran part was assigned by HMBC correlations from H-10 (δH 1.75) to C-3 (δC 50.2), C-1ʹ (δC 133.7) and C-9 (δC 126.4), from H-11 (δH 2.02) to C-7 (δC 118.9), C-9 (δC 126.4), C-5 (δC 149.2); and from H-4ʹ (δH 6.88) to C-2ʹ (δC 153.2), C-5ʹ (δC 144.6) (Figure 2). The 1H-1H COSY cross peaks from δH 2.02 (3H, s, H-11) to δH 6.33 (1H, s, H-7), and from δH 1.77 (3H, s, H-11ʹ) to δH 6.88 (1H, s, H-4ʹ ) verified the location of 10-CH3 and 11-CH3. The HMBC correlations verified the benzofuran group attached to C-8 (δC 144.3). Furthermore, from HMBC correlations, the signal of another ester carbonyl group was connected to the benzene ring through the C-7ʹ (δC 75.1), as evidenced from δH 1.63 (3H, s, H-10ʹ ) to δC 75.1 (CH, C-7ʹ ), and from δH 3.73 (OCH3, s, H-9ʹ ) to δC 176.8 (C, C-8ʹ ). The stereochemistry of C-3 and C-7ʹ was established as 3S and 7ʹS by ECD calculations (Figure 3). Eventually, the structure of 3 was elucidated as paeobenzofuranone C.
Compound 4 was obtained as white powder. The UV data absorption showed the ester carbonyl as determined by peaks with λmax at 230 nm. The IR spectrum indicated that 4 possessed hydroxyl (3383 cm-1) and lactone (1708 cm-1) groups. Its molecular formula was determined as C17H16O4 by HR-ESI-MS data analysis (m/z 285.11215 ([M + H]+, calcd. 285.11268). The 1H and 13C spectra data of 4 (Table 1 and Table 2, Supplementary data) are partially the same as those of 1, expect for the benzoyl and hydroxymethyl groups in 4. Interpretation of the 1H and 13C spectroscopic data of 4 implied one benzofuran part and one benzoyl. The locations of the benzofuran parts were assigned by correlations revealed in the HMBC experiment (Figure 2) between the 11-CH3 (δH 2.14) and C-5 (δC 148.9), C-7 (δC 110.6), C-8 (δC 153.4), as well as from H-4 (δH 6.74) to C-5, C-8, C-9 (δC 125.6); and from H-3 (δH 3.81) to C-10 (δC 66.4), H-7 (δH 4.37) to C-1ʹʹ (δC 166.5). The 1H-1H COSY correlations from δH 3.81 (1H, s, H-3) to δH 4.43 (1H, s, H-10), and from δH 4.43 (1H, s, H-10) to δH 6.74 (1H, s, H-4) verified the location of benzofuran part and one benzoyl connecting by C-3 and C-10. The stereochemistry of C-3 was established as 3R by ECD calculations (Figure 4). Therefore, the structure of compound 4 was elucidated as paeobenzofuranone D.
Compound 5 was obtained as white powder. Its molecular formula was determined as C18H18O5 by HR-ESI-MS data analysis (m/z 313.10959 [M-H]-, calcd. 313.10743). The 1H and 13C NMR data resembled those of 4 (Table 1 and Table 2), expect for the presence of an additional methoxy at C-2 in 5, which was confirmed by the key HMBC correlation of H-12 (δH 3.48) with C-2 (δC 111.2). Comprehensive analysis of 2D NMR data indicated that other parts of 5 were the same to those of 4. The stereochemistry of C-2 and C-3 was established as 2S and 3S by ECD calculations (Figure 4). Thus, the structure of compound 5 was established as paeobenzofuranone E.
2.2. Bioactivity Analysis
Five new compounds were tested for their inhibitory activities on nitric oxide production in the model of lipopolysaccharide-activated macrophages. As shown in Table 3, compounds 1 and 2 showed comparable inhibitory activity with the positive control at the concentration of 50 μM. In addition, all compounds were evaluated for their anti-tumor activity against HL-60, SW480, and MDA-MB-231 cell lines. As shown in Table 4, compounds 2, 4 and 5 demonstrated cytotoxicity against three human cancer cell lines. In particular, they exhibited potent cytotoxicity against HL-60 cells, with IC50 of 6.8, 19.1 and 11.1 μM, comparable to those of the positive control. In addition, compounds 4 and 5 showed no cytotoxicity to MDA-MB-231, indicating selectivity to the cancer cell lines.
3. Experimental
3.1. General Experimental Procedures
UV spectra were obtained on a UH5300 UV-VIS Double Beam Spectrophotometer. IR spectra were accessed by a Shimadzu Fourier transform infrared spectrometer with KBr pellets. HRESIMS were measured on a Thermo Scientific Q Exactive Orbitrap mass spectrometer system. The NMR spectra (1H, 13C, and 2D NMR) were run on a Bruker Avance III NMR instruments at 600 MHz for 1H and 150 MHz for 13C NMR, while tetramethylsilane (TMS) was used as an internal standard. Column chromatography (CC) was executed on silica gel (200−300 mesh, Qingdao Marine Chemical Ltd.), Sephadex LH-20 (Pharmacia Fine Chemical Co., Ltd.), and Reverse phase silica gel (20−45 μm, Fuji Silysia Chemical Ltd.). Medium pressure liquid chromatography (MPLC) was applied to Biotage SP2 equipment, and columns were packed with reverse phase silica gel (C18). An Agilent 1260 series high-performance liquid chromatography (HPLC) system was used for sample analysis (ZORBAX-SB C18 column, 5 μm, 4.6 × 250 mm, flowing rate = 1 mL/min) and preparation (ZORBAX-SB C18 column, 5 μm, 9.4 × 150 mm, flowing rate = 6 mL/min). All fractions were monitored by thin-layer chromatography (TLC) over GF 254 and silica gel 60 plates. The spots were visualized by heating silica gel plates soaked with vanillin-sulfuric acid color component solvent.
3.2. Plant Material
The root barks of P. suffruticosa were collected in August 2021 from Tongling County, Anhui Province, People’s Republic of China. It was identified by Zhenghui Li. (Associate Professor of South-Central Minzu University, Wuhan, Hubei 430074, PR China). A voucher specimen (2021123 FD) is deposited at School of Pharmaceutical Sciences, South-Central Minzu University.
3.3. Extraction and Isolation
The root bark of P. suffruticosa (50 kg) were mechanically crushed and extracted with MeOH/H2O (80:20) at 52℃ for 4 times. The solvent was evaporated in vacuo to give a dark gum (9.3 kg). The latter was dissolved in a liter of water and then respectively extracted with petroleum ether (PE, 8L × 4) and dichloromethane (DCM, 8L × 4) to give PE (1.3 kg) and DCM parts (560 g). DCM part was separated by a silica gel column eluted with PE: acetone (50:1, 40:1, 30:1, 20:1, 10:1) to give eight fractions (TPG-1–8). Fraction TPG-3 (28.5 g) was subjected to ODS silica gel CC and eluted with MeOH/H2O (20:90→90:10, v/v) to yield 10 fractions (Fr. 3-1→3-10). Fr. 3-2 (210 mg) was purified by Sephadex LH-20 (MeOH:DCM = 1:1) to afford three fractions (Fr. 3-2-1, 3-2-2, 3-2-1). Fr. 3-2-3 was purified by preparative HPLC with CH3CN/H2O (30:70→60:40, v/v, 30 min) to give 1 (15.6 mg, tR = 12.5 min), 2 (9.7 mg, tR = 14.8 min), 4 (3.3 mg, tR = 16.9 min), 5 (4.9 mg, tR = 17.8 min), respectively. Fr. 3-2-1 was prepared by HPLC with CH3CN/H2O (37:63→60:40, v/v, 30 min) to give 3 (3.2 mg) at 17.8 min.
3.3.1. Paeobenzofuranone A (1)
3.3.2. Paeobenzofuranone B (2)
3.3.3. Paeobenzofuranone C (3)
3.3.4. Paeobenzofuranone D (4)
3.3.5. Paeobenzofuranone E (5)
3.4. Cytotoxicity Assay
The cytotoxicity for the isolates was evaluated using the MTS assay. Briefly, 1 × 105 cells/mL from three human cancer cell lines, breast cancer MDA-MB-231, human myeloid leukemia HL-60, and colon cancer SW480 were seeded in 96-well plates. After 24 h incubation, cells were treated with test compounds or cisplatin (DDP, positive control) at a given concentrations (40、8、1.6、0.32、0.064 μM) for 48 h. MTS was then added to each well, and the plates were stored for 4 h. Absorbance was read at 490 nm. IC50 (50% concentration of inhibition) was calculated by Reed & Muench method [16,18].
3.5. Anti-Inflammatory Activity Assays
Mouse mononuclear macrophages RAW264.7 were seeded into 96-well plates, induced, and stimulated with 1 μg/mL LPS; at the same time, five new compounds with different concentrations to be tested were added. The drug-free group and the L-NMMA positive drug group were set approximately equal as a comparison. After the cells were cultured overnight, the medium was taken to detect the production of NO, and the absorbance was measured at 570 nm. MTS was added to the remaining medium for cell viability assays to exclude the toxic effects of compounds on cells. The assays were performed as triplicate batch experiments. NO production inhibition rate (%) = (OD570 nm of non-drug treatment group - OD570 nm of sample group)/OD570 nm of non-drug treatment group × 100% [17,19].
3.6. ECD Calculations
The conformers of the five calculated compounds were generated by MMFF in ChemDraw. The ECD were calculated at the B3LYP/6-31+G(d,p) level in methanol with the PCM model. The calculated ECD curves and weighted ECD were all generated using SpecDis 1.71 based on the Boltzmann distribution theory, the simulated spectra of all the predominant conformers were averaged to obtain the final conformation-ally averaged data [20]. All of the Density functional theory (DFT) calculations were implemented by the Gaussian 16 software package with Gaussian 09 default keyword. For computational details of compounds 1–5, see the Supporting Information.
4. Conclusions
In present study, the chemical investigation on Paeonia suffruticosa resulted in the isolation of five new benzofuran ring compounds, containing rare dimers (compounds 1–3) and hetero-dimers (compounds 4 and 5). Their structures were determined by extensive spectroscopic methods. These kinds of constituents were rarely reported from natural resources. This work represents the first report of five new benzofuran dimer compounds of P. suffruticosa and their cytotoxicity. This research broadens the horizon of the structural diversity of P. suffruticosa.
Supplementary Materials
The following supporting information can be downloaded at XXX. Figures S1-S35: HRESIMS, 1D & 2D NMR, ECD data of compounds 1–5.
Author Contributions
T.F. and J.K.L. designed and guided the experiment; Q.Q.M. performed the isolation and identification of the compounds, and wrote the manuscript; S.Y.T and Y.Q.Z. contributed to the isolation of these compounds; X.R.P. reviewed the manuscript; Z.H.L. plant material and identification; T.F. and J.K.L. reviewed the manuscript. All authors have agreed to the published version of this manuscript.
Funding
This work was financially supported by the National Natural Science Foundation of China (22177138, 21961142008).
Acknowledgments
The authors thank the Bioactivity Screening Center, Kunming Institute of Botany, Chinese Academy of Sciences for screening the bioactivity of the compounds.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ding, L.; Zhao, F.; Chen, L.; Jiang, Z.; Liu, Y.; Li, Z.; Qiu, F.; Yao, X. New monoterpene glycosides from Paeonia suffruticosa Andrews and their inhibition on NO production in LPS-induced RAW 264.7 cells. 7 cells. Bioorg. Med. Chem. Lett. 2012, 22(23), 7243–7247. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.C.; Tang, S.W.; Lam, S.H.; Wang, C.C.; Liu, Y.H.; Lin, H.Y.; Lee, S.S.; Lin, J.Y. Aqueous extract of Paeonia suffruticosa inhibits migration and metastasis of renal cell carcinoma cells via suppressing VEGFR-3 pathway. Evid. Based Compl. Alt. 2012, 409823. [Google Scholar] [CrossRef]
- Qiu, H.; Zhang, L.; Zhu, M.; Zhang, M.; Chen, J.; Feng, L.; Jia, X.; Jacob, J.A. Capture of anti-coagulant active ingredients from Moutan Cortex by platelet immobilized chromatography and evaluation of anticoagulant activity in rats. Biomed. Pharmacother. 2017, 95, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Pei, L.; Jie, S.; Chunnian, H.; Pei, X. Genus Paeonia: A comprehensive review on traditional uses, phytochemistry, pharmacological activities, clinical application, and toxicology. J. Ethnopharmacol. 2021, 269:113708. [CrossRef]
- Mencherini, T.; Picerno, P.; Festa, M.; Russo, P.; Capasso, A.; Aquino, R. Triterpenoid constituents from the roots of Paeonia rockii ssp. rockii. J. Nat. Prod., 2011, 74, 2116–2121. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Ohno, O.; Suenaga, K.; Miyamoto, K. Apoptosis-inducing activity and antiproliferative effect of Paeoniflorigenone from moutan cortex. Biosci. Biotechnol. Biochem. 2017, 81, 1106–1113. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Wang, Y.; Gao, J.; Lu, Z.; Yin, W.; Deng, R. Resveratrol trimers from seed cake of Paeonia rockii. Molecules. 2014, 19, 19549–19556. [Google Scholar] [CrossRef] [PubMed]
- Furuya, R.; Hu, H.; Zhang, Z.; Shigemori, H. Suffruyabiosides A and B, two new monoterpene diglycosides from Moutan Cortex. Molecules. 2012, 17, 4915–4923. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.K.; Ma, Y.B.; Wu, D.G.; Lu, Y.; Shen, Z.Q.; Zheng, Q.T.; Chen, Z.H. Paeonilide, a novel anti-PAF active monoterpenoid derived metabolite from Paeonia delavayi. Biosci. Biotech. Biochem. 2000, 64, 1511–1514. [Google Scholar] [CrossRef]
- Yang, X.Y.; Feng, T.; Li, Z.H.; Sheng, Y.; Yin, X.; Leng, Y.; Liu, J.K. Conosilane A, an unprecedented sesquiterpene from the Cultures of Basidiomycete Conocybe siliginea. Org. Lett. 2012, 14, 5382–5384. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Gao, Y.; Zhang, L.; Liu, J.K. Bi-linderone, a highly modified methyl-linderone dimer from Lindera aggregata with activity toward improvement of insulin sensitivity in vitro. Org. Lett. 2010, 12, 2354–2357. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.L.; Hsieh, K.L.; Liu, J.K. Guajadial: an unusual meroterpenoid from guava leaves Psidium guajava. Org. Lett. 2007, 9, 5135–5138. [Google Scholar] [CrossRef] [PubMed]
- Feng, T.; Su, J.; Ding, Z.H.; Zheng, Y.T.; Li, Y.; Leng, Y.; Liu, J.K. Chemical constituents and their bioactivities of Tongling White Ginger (Zingiber officinale). J. Agric. Food Chem. 2011, 59, 11690–11695. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.G.; Cheng, C.Q.; Liu, J.K. X-Ray Crystal Structure of Angulatusine A, a new sesquiterpene alkaloid from Celastrus Angulatus. J. Nat. Prod. 1992, 55, 982–985. [Google Scholar] [CrossRef]
- Ha, D.T.; Ngoc, T.M.; Lee, I.S.; Lee, Y. M.; Kim, J.S.; Jung, H.J. Inhibitors of aldose reductase and formation of advanced glycation end-products in moutan cortex (Paeonia suffruticosa). J. Nat. Prod. 2009, 72, 1465–1470. [Google Scholar] [CrossRef]
- Yu, H. L.; Long, Q.; Yi, W.F.; Yang, B. J.; Ding, X.; Hao, X. J. Two new C21 steroidal glycosides from the roots of Cynanchum paniculatum. Natural Products and Bioprospecting. 2019, 04, 26. [Google Scholar] [CrossRef] [PubMed]
- Reed, L.J.; Muench, H. A simple method of estimating fifty percent endpoints. Am. J. Epidemiol. 1938, 27, 493–497. [Google Scholar] [CrossRef]
- Anh, H. L. T.; Cuc, N. T.; Tai, B. H.; Yen, P. H.; Nhiem, N. X.; ThaoD. T.; Nam, N. H.; Minh, C. V.; Kiem, P. V.; Kim, Y. H. Synthesis of chromonylthiazolidines and their cytotoxicity tohuman cancer cell Lines. Molecules. 2015, 20, 1151–1160. [Google Scholar] [CrossRef] [PubMed]
- Snene, A.; Mokni, R.E.; Jmii, H.; Jlassi, I.; Jadane, H.; Falconieri, D.; Piras, A.; Dhaouadi, H.; Porcedda, S; Hammami, S. In vitro antimicrobial, antioxidant and antiviral activities of the essential oil and various extracts of wild (Daucus virgatus (Poir.) Maire) from Tunisia. Ind Crops Prod. 2017, 109, 109–115. [Google Scholar] [CrossRef]
- Bruhn, T.; Schaumloeffel, A.; Hemberger, Y.; Bringmann, G.; SpecDis: quantifying the comparison of calculated and experimental electronic circular dichroism spectra, Chirality. 2013, 25, 243-249. [CrossRef]
Figure 1.
Structures of compounds 1‒5.
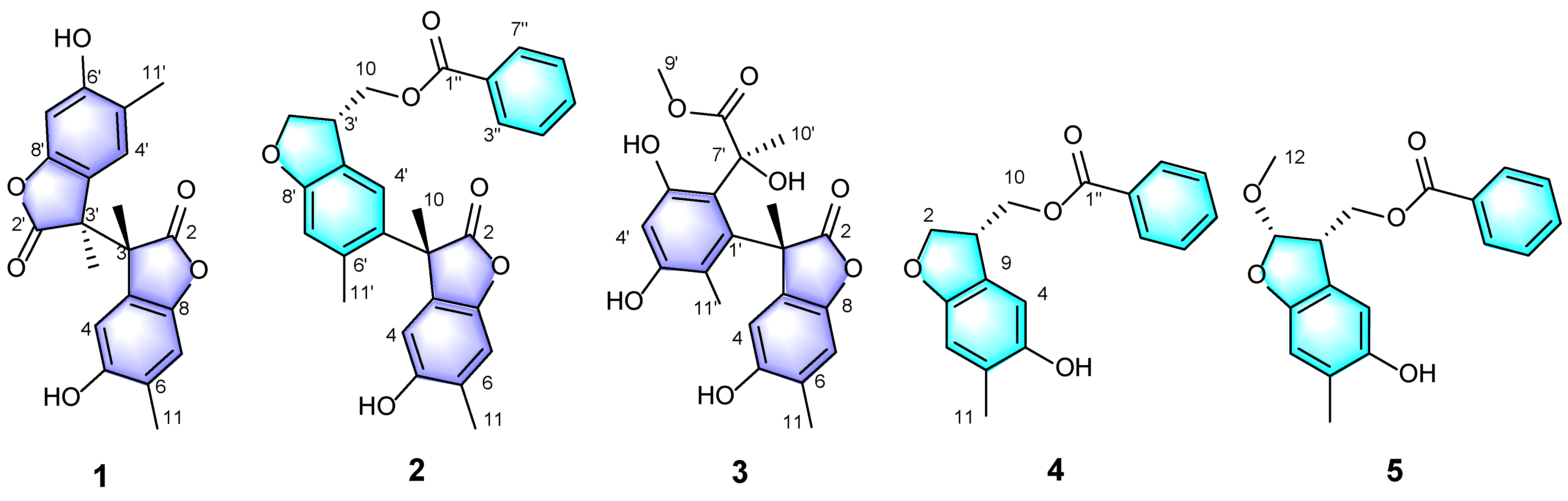
Figure 2.
Selected HMBC and 1H–1H COSY correlations of compounds 1–5.
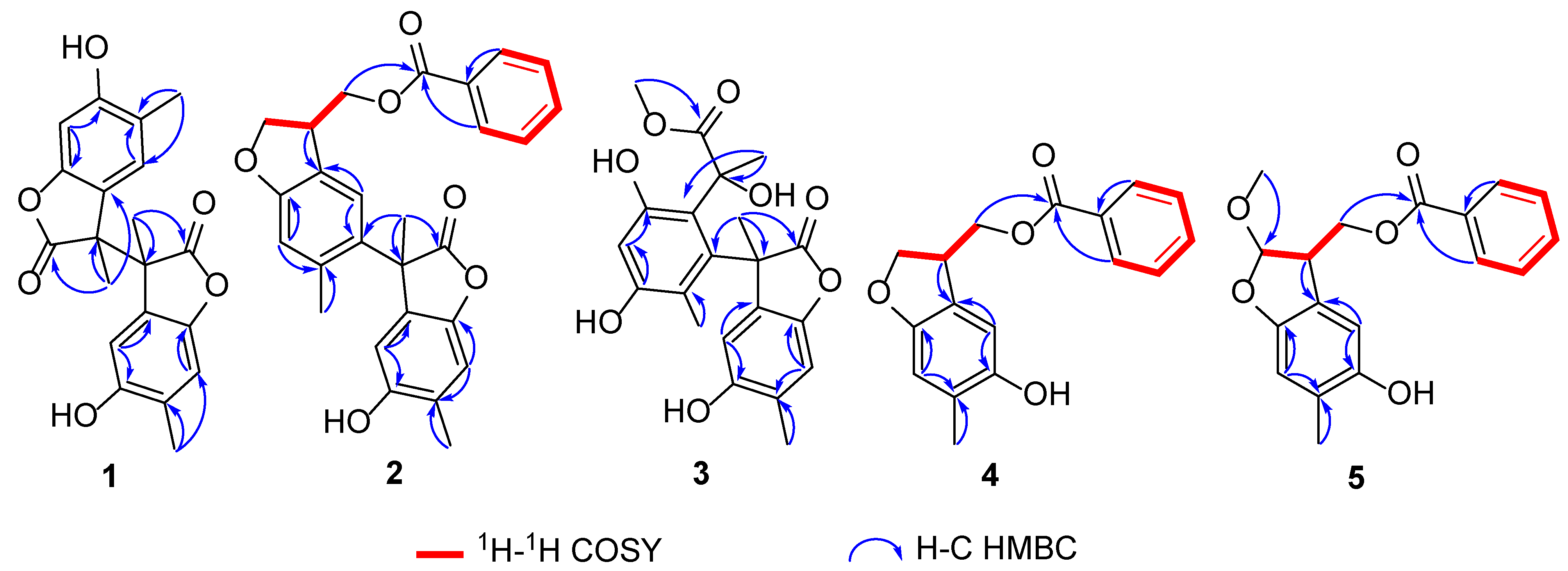
Figure 3.
ECD calculations for compounds 1–4.
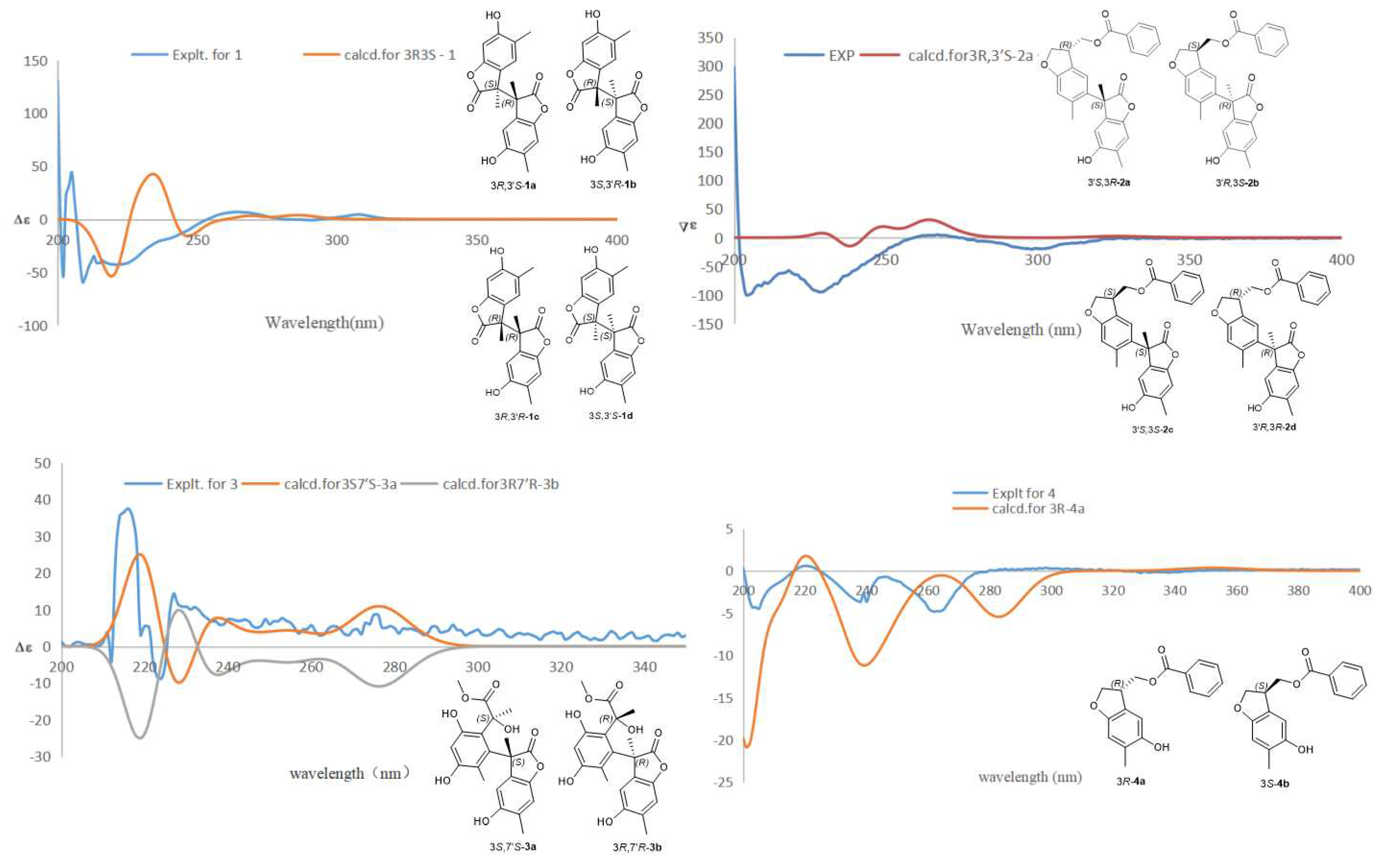
Figure 4.
ECD calculations for compound 5.
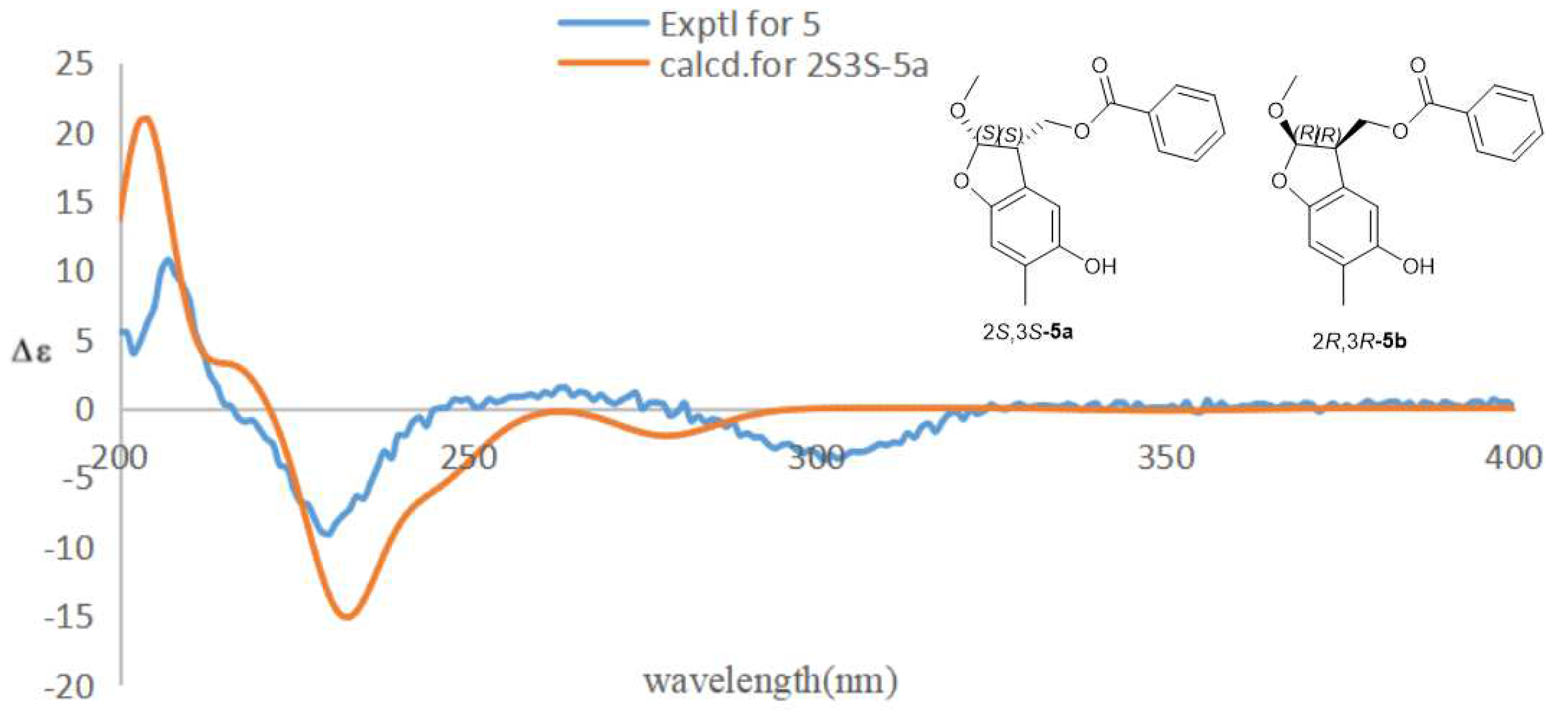

Table 1.
1H NMR Spectroscopic Data of Compounds 1–5 in Methanol-d4 (600 MHz, J in Hz).
| Position | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| 2 | 4.46, m | 5.48, t (1.8) | |||
| 3 | 3.81, t (7.4) | 3.80, t (7.5) | |||
| 4 | 7.18, s | 6.61, s | 6.96, s | 6.74, s | 6.72, s |
| 7 | 6.61, s | 6.52, s | 6.33, s | 4.37, dd (18.0, 9.1) 4.44, dd (10.7, 5.9) | 4.46, dd (11.1, 5.5) 4.33, dd (11.1, 7.8) |
| 10 | 1.67, s, 3H | 1.75, s, 3H | 1.72, s, 3H | 4.43, m | 4.48, m |
| 11 | 2.15, s, 3H | 2.13, s, 3H | 2.02, s, 3H | 2.14, s, 3H | 2.15, s, 3H |
| 12 | 3.48, s | ||||
| 2′ | 4.43, m; 4.06, m | 8.00, d (1.5) | 7.97, d (1.5) | ||
| 3′ | 3.81, t (7.5) | 7.44, m | 7.45, m | ||
| 4′ | 7.18, s | 6.63, s | 6.88, s | 7.58, m | 7.61, m |
| 5′ | 6.61, s | 6.62, s | 7.49, m | 7.48, m | |
| 6′ | 1.67, s, 3H | 8.01, d (1.3) | 7.99, d (1.3) | ||
| 7′ | 2.15, s, 3H | 6.85, s | |||
| 9′ | 3.73, s | ||||
| 10′ | 4.60, dd (18.0, 9.1); 4.44, dd (10.8, 5.9) | 1.63, s, 3H | |||
| 11′ | 2.05, s, 3H | 1.77, s, 3H | |||
| 3′′ | 8.01, d (1.2) | ||||
| 4′′ | 7.48, t (7.8) | ||||
| 5′′ | 7.56, m | ||||
| 6′′ | 7.48, t (7.8) | ||||
| 7′′ | 8.00, d (1.4) |
Table 2.
13C NMR Spectroscopic Data of Compounds 1–5 in Methanol-d4 (150 MHz).
| Position | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| 2 | 179.4, C | 179.3, C | 182.7, C | 73.5, CH2 | 111.2, CH |
| 3 | 52.6, C | 52.6, C | 50.2, C | 42.0, CH | 50.5, CH |
| 4 | 128.1, CH | 112.2, CH | 115.6, CH | 110.8, CH | 112.6, CH |
| 5 | 113.3, C | 149.3, C | 149.2, C | 148.9, C | 150.9, C |
| 6 | 153.7, C | 126.3, C | 125.0, C | 128.5, CH | 124.4, CH |
| 7 | 110.3, CH | 112.4, CH | 118.9, CH | 110.6, CH | 112.3, CH |
| 8 | 146.2, C | 125.9, C | 144.3, C | 153.4, C | 153.2, C |
| 9 | 127.4, C | 127.4, C | 126.4, C | 125.6, C | 129.8, C |
| 10 | 18.7, CH3 | 18.6, CH3 | 22.3, CH3 | 66.4, CH2 | 66.3, CH2 |
| 11 | 16.6, CH3 | 16.6, CH3 | 15.9, CH3 | 15.6, CH3 | 17.0, CH3 |
| 12 | 56.2, OCH3 | ||||
| 1′ | 133.7, C | ||||
| 2′ | 179.4, C | 73.1, CH2 | 153.2, C | ||
| 3′ | 52.6, C | 43.5, CH | 122.0, C | ||
| 4′ | 128.1, CH | 111.7, CH | 115.9, CH | ||
| 5′ | 113.3, C | 126.4, C | 144.6, C | ||
| 6′ | 153.7, C | 131.4, CH | 125.7, C | ||
| 7′ | 110.3, CH | 110.3, CH | 75.1, C | ||
| 8′ | 146.2, C | 154.9, C | 176.8, C | ||
| 9′ | 127.4, C | 124.4, C | 53.2, OCH3 | ||
| 10′ | 18.7, CH3 | 66.9, CH2 | 26.0, CH3 | ||
| 11′ | 16.6, CH3 | 16.8, CH3 | 10.4, CH3 | ||
| 1′′ | 168.1, C | 166.5, C | 167.9, C | ||
| 2′′ | 130.7, C | 129.7, C | 131.2, C | ||
| 3′′ | 129.7, CH | 129.2, CH | 130.4, CH | ||
| 4′′ | 128.1, CH | 128.2, CH | 129.5, CH | ||
| 5′′ | 134.5, CH | 132.9, CH | 130.7, CH | ||
| 6′′ | 129.7, CH | 128.2, CH | 129.5, CH | ||
| 7′′ | 128.1, CH | 129.1, CH | 130.4, CH |
Table 3.
Inhibitory activities of compounds 1–5 on NO production.
| Compound | Concentration (μM) | Inhibition Activity (100%) |
|---|---|---|
| L-NMMAa | 50 | 52.0 ± 1.96 |
| 1 | 50 | 43.9 ± 2.07 |
| 2 | 50 | 44.6 ± 0.52 |
| 3 | 50 | 13.0 ± 1.59 |
| 4 | 50 | 33.7 ± 2.24 |
| 5 | 50 | 30.9 ± 1.56 |
aL-NMMA (NG-monomethyl-L-arginine, monoacetate salt) was use as the positive control.
Table 4.
Cytotoxicity of compounds 2, 4 and 5 (IC50 ± SD, μM).
| Compound | HL-60 | MDA-MB-231 | SW480 |
|---|---|---|---|
| 2 | 6.8 ± 0.11 | 20.9 ± 0.46 | 12.6 ± 0.73 |
| 4 | 19.1 ± 0.32 | – | 8.9 ± 0.40 |
| 5 | 11.1 ± 1.61 | – | 10.7 ± 0.43 |
| DDPa | 23.5 ± 0.77 | 16.9 ± 1.19 | 25.1 ± 1.26 |
aDDP (Cisplatin) was used as the positive control.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



