Submitted:
13 May 2023
Posted:
17 May 2023
You are already at the latest version
Abstract
The Neurodegenerative Diseases are, according to recent studies, one of the main causes of disability and death worldwide. Interest in molecular genetics has started to have an exponential growth thanks to numerous advancements in tech, shifts in the understanding of the disease as a phenomenon, and the change of perspective regarding gene editing and the upsides of this action. The aim of this paper is to analyse the newest approaches in genetics and molecular sciences regarding four of the most important neurodegenerative disorders: Alzheimer’s Disease, Parkinson’s Disease, Huntington’s Disease and Amyotrophic Lateral Sclerosis. We intend through this review to focus on the newest treatment, diagnosis and predictions plans regarding this large group of diseases, in order to obtain a better accuracy analysing and spotting the emerging signs that could lead to a better outcome in order to increase both the quality and the life span of the patient. Moreover, this review could provide future evidence of possible novelty therapies that target the specific genes and could be useful to be taken into consideration when the classical approaches fail to shed light.
Keywords:
Molecular Genetics
; Neurodegenerative Disease
; Molecular pathology
; Epigenetics
; Gene expression
; Therapeutic targets
; Biomarkers
; Alzheimer’s Disease
; Parkinson’s Disease
; Huntington Disease
; Amyotrphic Lateral Sclerosis
1. Introduction
Neurodegenerative diseases are, according to recent studies, one of the main causes of disability and death worldwide.
Interest in molecular genetics has started to have an exponential growth thanks to numerous advancements in tech, shifts in the understanding of the disease as a phenomenon, and the change of perspective regarding gene editing and the upsides of this action. However, the concept of genes isn’t, as one might consider, a late XXth notion. Aristotle predicted the existence of genes through postulating that the mother had her characteristics encoded inside the menstrual blood, while the father had his inside the semen, Also, Hippocrates’ theory resembled what Charles Darwing later described as “pangenesis” [1]. However, 2 breakthroughs came a few centuries later, firstly when the Czech scientist Johann Gregor Mendel coined the terms “recessive, discrete and dominant factors” by observing his hybridization experiments performed on peas [2]. Later that century, Wilhelm von Waldeyer familiarized the scientific world with the term “Chromosomen”, derived from the work of Theodor Boveri, who coined the notion of “Chromatinelemente” [3]. The Nobel prize for Physiology or Medicine awarded, in 1962, to Francis Crick and James D. Watson , for discovering the key to understanding not only molecular genetics, but also the fundament of life itself- the molecular structure of nucleic acids. This discovery was a huge milestone that lead to understanding the base structure of life- DNA, RNA and the creation of proteins, but also lead to the comprehension of how a small misplacement of some nucleotide subunit can lead to a plethora of changes in the created proteins, further developing the disease.
Neurodegenerative diseases are represented by a group of disorders that are usually associated with protein deposits or misfoldings leading to chemical changes, loss of function as well as apoptosis inside the neurons of the brain and spinal cord [4]. They are chronic, progressive and most important there are many treatments that slow the development of the disease and help manage the symptoms but do not cure the actual problem. Molecular genetics play a very important role in understanding the mechanics of the neurodegenerative disorders as it leads to identifying certain genes that are associated with this type of pathologies and can also be a way of finding more efficient treatments [5]. In addition, molecular genetics played an important role in identifying the specific proteins implicated in forming aggregates inside the cells that lead to the apparition of neurodegenerative disorders.The most common types of proteins that are implicated in forming this aggregates are amyloid-β, tau protein, α-synuclein and prion protein [4,6]. The neurodegenerative disorders have different types of mechanisms behind each one of them, presenting a unique symptomatology, most common neurodegenerative disorders being Alzheimer’s disease, Parkinson’s disease, Huntington’s disease and Amyotrophic lateral sclerosis, each one of them being associated with different genes the common element being the formation of protein aggregates which lead to changing the physical and chemical properties of the nervous cell [4,7].
2. Alzheimer’s disease (AD)
Alzheimer’s disease is the most common neurodegenerative disease that occurs in humans, representing 70% of the total dementia cases [7]. At first, Alois Alzheimer observed, in 1901 the interesting behavior of a 51 year old woman, who suffered from sleep disorders, memory loss, and progressive confusion. During the autopsy, Alois Alzheimer discovered the presence of neurofibrillary tangles and neuritic plaques, concluding that the disease is caused by the agglomeration of these structures. Neurofibrillary tangles - hyperphosphorylated tau proteins and neuritic plaques - aggregated beta amyloids [8]. A study conducted by Gatz et al. found that environmental factors can influence to some degree the chance of developing AD in humans that have the predisposing genes, these factors influencing between 58% to 79% [9]. The perspective of Alzheimer’s Disease was governed for more than 30 years by the amyloid cascade formation, which finished with the formation of the beta amyloids. However, newer studies tend to identify the AD amyloid cascade as a simplified view of the pathophysiology involved in the disease, emphasizing the fact that the glymphatic system, as well as the Lipoprotein receptor-related protein-1; RAGE [10]
3. Amyloid Precursor Protein
The genetic landscape of AD is dominated by mutations of, firstly, the Amyloid precursor protein (APP), mutations that generate autosomal dominant alleles leading to development of early on-set AD [11]. APP can follow either the amyloidogenic or the non-amyloidogenic path, being cleaved by 3 different secretases: alpha, beta or gamma [12]. Physiologically, the APP is cleaved by the alpha or gamma secretases [13], during the non-amyloidogenic path. During the amyloidogenic path, the APP is cleaved by the beta and gamma secretases resulting in Aβ38, Aβ40, Aβ42 [14]. Subsequently, Aβ plaques are formed in a process that starts from Aβ monomers and ends with the development of amyloid plaques. From the three beta amyloids previously mentioned, Aβ42 is the least soluble of them all [15].
Naturally, studies have shown that APP, which is coded on chromosome number 21, is directly correlated with a higher risk of developing early-onset AD in trisomy 21 individuals[16] .
The relevance of amyloid pathogenicity is explainable by taking into consideration a study conducted in 2012 by Jonsson, T., Atwal, J.,Steinberg, S. et al. on a population of 1795 Icelanders. This study proves that the A673T (rs63750847) (or A2T) substitution, also known as the Icelandic mutation, decreases the risk of developing Alzheimer ( and cognitive decline associated with aging) by lowering the overall aggregation and production of Aβ40 and Aβ42 by approximately 40% [17,18]. However, a study conducted in 2014 on a population of 2641 Chinese showed that this gene doesn’t explain the longevity of old Chinese subjects that participated in this study, because A2T isn’t included in all their genomes [19]. Another study performed in 2015 on 3487 Danes shows that the A673T mutation is present in only one subject ( 0,014%) [20], in contrast with 0.43% in the Nordic population [17]. On the other hand, the mutation A673V, a mutation which manifests itself in homozygous state, is a gene that is linked with early-onset AD [21]. However, this mutation leads to a distinctive manifestation of the disease in comparison to dominant genes that determine AD: familial inherited AD usually develops amyloid deposits inside the striatum [22]. In comparison, this mutation tends to avoid the striatum in the incipient phase, focusing the amyloid deposits inside the cerebellum [21].
There are genes that protect against AD, and on the other side there are genes that lead to early-onset AD (Osaka and Arctic mutations) or Cerebral Amyloid Angiopathy (Dutch and Italian mutations). For example, there are 4 mutations that happen on the E693 position on the APP coding gene, exon 17- the Dutch mutation (E693Q), the Osaka mutation (E693del), the Italian mutation (E693K), and the Arctic mutation (E693G), all resulting in a change of the 22nd amino acid in Aβ, thus developing a peptide called E22. E693Q (rs63750579) mutation leads to a peptide called E22Q, E693Q mouse models identify shared features with human Alzheimer’s brain pathology [23]. This mutation’s clinical phenotype is called Hereditary cerebral hemorrhage with amyloidosis (HCHWA-D), patients suffering from recurrent strokes and dementia. Aβ accumulates in the cerebral vessel walls because its overall production increases with this mutation, resulting in the formation of deposits in the meningo cortical vessels that lead to cerebral amyloid angiopathy [24,25]. E693del (dbSNP ID: NA) creates a mutant peptide known as E22Δ that has been shown in a study conducted by Uddin, M.S., Tewari, D., Sharma, G. et al. to lead to increased endoplasmic reticulum stress by increasing the oligomerization of Aβ [26]. Further stress is created, by determining an overexpressed chaperone GRP78 and by the expression of the GFAP (glial fibrillary acidic protein), by E22Δ, thus correlating AD with the glymphatic system [27]. More properties of E22Δ derive from a 2008 study which discovered that this mutant peptide creates less overall amyloid deposits, deposits that are, however, more resistant to proteolysis [28]. Recent studies performed by McKnelly et al. show the destabilizing and cytotoxic effect E22 peptides have on in vitro cell membranes by disturbing the phosphatidylcholine and phosphatidylserine constituting the cell membrane. This analysis showcases that these peptides tend to have a disturbing effect on the cell membrane proportional with the quantity of positive charges in the molecule. Thus, the peptides are, in order from least charged to most charged: E22Δ (2 positive charges), E22G, E22Q (both have 3 positive charges) and E22K (4 positive charges), meaning that the Italian mutation generates the most cytotoxic Aβ [29].

Table 1.
Most proeminent mutations in APP gene.
| Mutation | Pathogenicity | Type of Mutation | Biological Effect | Citation |
| c.-488C>A (rs532314089) |
Alzheimer's Disease | Substitution | Predicted to disrupt binding of transcription factor EGR1. PHRED-scaled CADD = 0.26. Negative regulator in multiple cell types including PC12 neuronal-like rat chromaffin cells, SK-N-SH neuroblastoma cells, C6 glial cells and U373 astroctyoma cells among others |
[30] [31] [32] |
| c.24+38G>A (rs373985746) |
Alzheimer's Disease | Substitution | Predicted benign in silico (PHRED-scaled CADD =10). | [32] |
| c.24+288G>A (rs192348494) |
Alzheimer's Disease | Substitution | Predicted benign in silico (PHRED-scaled CADD =12). | [32] |
| c.-23-377A>G (rs150375400) |
Alzheimer's Disease | Substitution | Predicted benign in silico (PHRED-scaled CADD =10). | [32] |
| A18T | Alzheimer's Disease, Cardiovascular Disease | Substitution | Predicted to disrupt signal peptide cleavage and affect ApoE secretion. PHRED-scaled CADD = 22. | [32,33] |
Presenilin1
Presenilin1, part of the γ-secretase, is encoded by PSEN1, which is located on chromosome 14q24.3 [34]. The change of nucleotides of this gene is responsible for approximately 70 to 80% of manifestations of autosomal dominant Alzheimer’s Disease [35,36]. Naturally, mutations to this gene will determine an unnatural development of Aβ, and in general PSEN1 mutations have a negative effect on the cleavage activity of the γ-secretase, resulting in an increased production in Aβ42 rather than Aβ40 [37]. However, it is important to note that the overall amplification of amyloid production might not be the right answer to explaining why change of nucleotides in the presenilin gene determines AD. Besides the amyloid cascade theory, there is another theory that derives from mouse studies which indicate that presenilin is truly important in the processes of memorizing, learning and nervous cell survivability [38]. Mutations that can happen on exon 4 of the PSEN1 gene include: A79V, M84V, L85P. A79V is an autosomal dominant mutation that determines an amplified proportion of Aβ42 in comparison to Aβ40, by decreasing the latter’s production [39,40]. Koriath et. al discuss in a 2018 study the fact that 4 alleles have been spotted with a frequency of 0.00014% in the gnomAD database, deducting that this mutation has a low penetrance [41]. M84V is another autosomal dominant mutation that determines multiple types of atrophies - temporal and frontal lobe and also cerebellar and cortical atrophy [42]. It determines a greater Aβ42 to Aβ40 ratio, this time by increasing the overall quantity of Aβ42 [43]. Moreover, mutation M84T has also been linked to Alzheimer’s disease. L85P mutant determines a greater ratio of Aβ42 to Aβ40, but is also shown to increase the production of Aβ43 [44]. Autopsies of the people who suffered from this mutation showed an aggregation of amyloid inside the basal ganglia and cortex, but there isn’t yet enough evidence to support theories regarding the pathophysiology of this mutation [45]. Up to this moment only one mutation on the intron 4 of the PSEN1 gene has been identified, that being Int4del (also referred as L113_l114insT) regards to the deletion of a G nucleotide in the splice region of the PSEN1 gene, just after the exon4 therefore transcribing into 3 altered transcripts, one of which coding a protein with an extra threonine addition to the PSEN1 protein; as well as two smaller transcripts due to the apparition of premature stop codons [46]. Studies have shown that this mutant leads to an increase in the Aβ42 to Aβ40 ratio, by decreasing the quantity of Aβ40 & Aβ38 [47,48]. M139V mutation happens on exon 5 of the presenilin 1 gene. Subjects with this mutation develop Alzheimer’s disease without many distinctive clinical and morphological features [49]. Regarding amyloid formation, this mutant will lead to increased Aβ42 & Aβ43, and an overall decrease in Aβ40, Aβ38, Aβ37 [50]. Another mutation of the presenilin 1 gene is seen on exon 7 and is called S212Y. Neuropathologically, this mutation resembles the typical AD neurofibrillary tangles & neuritic plaques [51]. Liu et al determined that in this mutation, production of Aβ42 is increased [47]. Another mutation of the presenilin 1 gene associated with early-onset and late-onset AD is known as A434C . This change of nucleotides lead to accumulation of plaques and amyloids inside the neocortex, accompanied by neurofibrillary tangles, hippocampal and amygdala gliosis [52]. A 2022 study suggests that this mutant leads to a different conformation that results in a particular mode of interaction with the γ-secretase, creating a larger quantity of Aβ42 than normal [53].
Table 2.
Proeminent mutations in PSEN1 gene.
| Mutation | Pathogenicity | Type of Mutation | Biological Effect | Citation |
| A673T(Icelandic) |
Alzheimer’s Disease - Protective | Substitution | This particular type is linked to limited buildup of amyloid and is believed to guard against amyloid-related issues. It results in a decrease of approximately 40 percent in the production of amyloidogenic Aβ peptides, and the Aβ that is produced has a reduced tendency to form clumps. | [17,18,19,20] |
| A673V | Not Classified | Substitution | According to the CERAD criteria, a clear diagnosis of AD was made, as evidenced by substantial Aβ and tau pathology deposits (Braak stage VI) along with cerebral amyloid angiopathy. The deposits found contained elevated levels of Aβ40 and were notably larger, with fewer preamyloid deposits. Perivascular localization was frequently observed. In laboratory studies, it was discovered that A673V caused a shift in β-secretase processing of APP toward the amyloidogenic pathway and amplified Aβ aggregation. | [21,22] |
| E693Q(Dutch) | Hereditary Cerebral Hemorrhage with Amyloidosis- Pathogenic | Substitution | There is a substantial accumulation of amyloid in the cerebral blood vessels, accompanied by hemorrhages and some diffuse plaques in the brain tissue. In laboratory experiments, it was observed that this condition speeds up Aβ aggregation in vitro, leading to greater fibril formation, and may also modify APP processing. | [24,25] |
| E693del (Osaka, E693∆, E693delta | Alzheimer’s Disease - Pathogenic | Deletion | This variant led to an increased oligomerization and nucleation of Aβ aggregates in vitro. Furthermore, it was found that there was no alteration in the Aβ42/Aβ40 ratio, but there was a decrease in both Aβ42 and Aβ40. This variant was also discovered to be more resistant to degradation by neprilysin and insulin-degrading enzyme. Additionally, this variant had a greater inhibitory effect on long-term potentiation (LTP) compared to wild-type Aβ, which suggests a potential negative impact on synaptic plasticity. | [26,27,28,29] |
| E693K(Italian) | Hereditary Cerebral Hemorrhage with Amyloidosis- Pathogenic | Substitution | The observed symptoms include small to large hematomas, subarachnoid bleeding, scars with hemosiderin deposits, small infarcts, and cortical calcifications. Aβ immunoreactivity was observed in vessel walls and neuropil, but there was an absence of neurofibrillary changes and neuritic plaques. Despite a reduction in the Aβ42/Aβ40 ratio and a decrease in Aβ42 levels, the mutant peptide was found to be toxic in cells and aggregates at a faster rate. | [23] |
| E693G (Arctic, E22G) | Alzheimer’s Disease - Pathogenic | Substitution | Several carriers displayed neuropathology that was indicative of AD. Plaques were observed to have a "targetoid" shape, containing heterogeneous truncated Aβ peptides in the center and surrounded by Aβ42. Cell-based assays revealed a reduction in the production of both Aβ40 and Aβ42. Additionally, there was a decrease in proteolytic degradation of Aβ by neprilysin, a type of enzyme that breaks down proteins. | [23,25] |
Presenilin2
Presenilin2, part of the γ-secretase, is encoded by PSEN2. PSEN2 gene is found on chromosome lq42.13 and has a total of 12 exons [53]. Mutations in presenilin genes usually lead to, as stated before, to early-onset Alzheimer’s Disease. However, not all changes in the nucleotides that compose the PSEN2 gene determine AD. For example, a 2018 screening discovered a mutation in a Belgian subject, now known as mutation K82fs (situated on exon 5), who suffered from frontotemporal dementia . This mutation seems to determine a drop in presenilin 2 production in the hippocampal region and in the frontal cortex; further analysis of this pathology revealed Pick’s disease [54]. Another 2 studies performed in 2020, and 2021 revealed a new mutation on this gene: c.*71C>A [55], mutation that happens on exon 13 3’UTR. The fact that this mutation is in the untranslated region is showcased by the fact that it lies where miR-183-5p (an inhibitor of PSEN2 gene) attaches to the gene, inhibiting the suppressing activity of miR-183-5p [56]. Moreover, pathology associated with this mutation is relevant in the diagnosis of AD, for example greater ratio of Aβ42/Aβ40 and hippocampal atrophy [55,56]. Furthermore, M239V is a mutation discovered in an 1995 study, a mutation that lies on exon 8, linked with early-onset AD [57]. Autopsies performed on subjects carrying this substitution showcased beta amyloid and tau aggregations, the golden standard for diagnosing Alzheimer’s Disease. (Figure 1).
Table 3.
Recent proeminent mutations in PSEN2 gene.
| Mutation | Pathogenicity | Type of Mutation | Biological Effect | Citation |
| A79V | Alzheimer’s Disease - Pathogenic | Substitution | The observed neuropathology was in line with that of AD. It was observed that this variant led to an increase in the Aβ42/Aβ40 ratio and a decrease in the Aβ37/Aβ42 ratios in cells. | [39,40,41] |
| M84V | Alzheimer’s Disease - Pathogenic | Substitution | In two cases, the observed neuropathology was consistent with AD. Additionally, MRI scans revealed cortical and cerebellar atrophy in these two cases. In the third case, frontal and temporal lobe atrophy was observed. Cell studies revealed an increase in both Aβ42 and the Aβ42/Aβ40 ratio. | [42,43] |
| L85P | Alzheimer’s Disease - Pathogenic | Substitution | SPECT and PET scans showed bilateral hypoperfusion and hypometabolism in the occipital and temporal lobes. Cell studies revealed an increase in the Aβ42/Aβ40 ratio as well as increased Aβ42 levels in transfected cells. In vitro studies indicated a decrease in Aβ42 production and the complete absence of Aβ40 production. | [44,45] |
| L113_I114insT (int4del) |
Alzheimer’s Disease - Pathogenic | Substitution | The observed neuropathology was consistent with AD, and included neuron loss in the hippocampus and entorhinal cortex, the presence of neuritic plaques and neurofibrillary tangles in the hippocampus, and amyloid angiopathy which was particularly evident in the cerebellum. The identified mutation involved a deletion of a G in the splice donor site of intron 4, resulting in the production of three aberrant transcripts. Further investigations indicated an increase in both Aβ42 and the Aβ42/Aβ40 ratio, as well as a reduction in Aβ40 and Aβ38 production in patient brain membranes. | [46,47,48] |
| M139V | Alzheimer’s Disease - Pathogenic | Substitution | Decrease in the levels of Aβ40, Aβ38, and Aβ37, and an increase in the levels of Aβ42 and Aβ43. In iPSC-derived neurons, the levels of mutant protein were found to be variable, suggesting protein instability. | [49,50,51] |
Thus, correlating the anatomopathological features discovered during autopsies with the familial information and heritage of these patients determine the classification of this gene as a gene that is linked with early-onset Alzheimer’s Disease [58].
Apolipoprotein E
However, early-onset Alzheimer’s Disease counts for approximately 1-2% of the total cases of AD. The majority of genetical cases of AD reported are a cause of mutations of APOE (apolipoprotein E). Apolipoprotein E is a glycoprotein that consists of 299 amino acids, and is created inside the central nervous system by a great number of glial cells, including microglias, astrocytes, the cells of the choroid plexus, mural vascular cells and neurons that undergo stress. ApoE is abundantly expressed both peripherally and centrally; however, due to the BBB, it exists as separate pools. Therefore, it is crucial to understand what roles each pool may play in AD pathogenesis as well as therapeutic opportunities they present. Peripherally, apoE is produced primarily by the liver and plays an essential role in redistribution and metabolism of lipids such as triglycerides, cholesterol, cholesteryl esters and phospholipids through lipoprotein particles [59]. ApoE isoforms are associated with different lipoprotein particles in peripheral circulation; for instance, ApoE4 tends to preferentially associate itself with triglyceride-rich particles while ApoE2 and ApoE3 prefer high density lipoproteins (HDLs). ApoE-mediated cholesterol and lipid transport is critical for proper CNS formation and repair. ApoE3 shows a greater effect than ApoE4 at stimulating neurite outgrowth after injury; hence its prevalence among astrocytes. ApoE4 alters structural reorganization of neurons, decreases expression of key synaptic proteins, and inhibits glutamatergic signaling critical for neuronal plasticity and network maintenance. ApoE's effects vary by cell type; for instance, it is expressed by astrocytes, microglia, pericytes and oligodendrocytes under various circumstances. Therefore, in order to gain a comprehensive understanding of its role within the brain it is crucial to examine its structure, lipidation status and biochemical properties among different cell types that express it [60].Study has demonstrated that ApoE, a protein found in the brain, plays an integral role in Alzheimer's Disease by its interaction with amyloid-beta protein. ApoE was discovered co-depositing with Aβ in amyloid plaques; thus contributing directly to AD risk. Knocking out ApoE in amyloid model mice alters their Aβ plaque morphology significantly indicating it plays a crucial role in fibrilization and amyloid deposition processes.
Studies of apoE's effects on amyloid pathology have demonstrated isoform-dependent effects; with apoE4 having the greatest impact, followed by apoE3 and then apoE2. Studies indicate that those carrying an increased level Aβ Ab compared to apoE3 carriers and earlier deposition, with greater overall deposition, wider cortical distribution and earlier deposition onset than its APOE2 counterpart, while delaying deposition, less severe pathology, and protecting cognitive function were seen with these carriers [61].
Studies have also demonstrated that ApoE4 stabilizes soluble, cytotoxic Aβ fragments and enhances fibrillogenesis to speed early amyloid pathology seeding. Thus, their interaction may serve as a potential target for therapeutic intervention at early stages of amyloid disease progression [62,63].
ApoE plays an essential role in clearing away antibodies via several mechanisms, including receptor-mediated clearance and proteolytic degradation. Neurons utilize LRP1 receptors to absorb Aβ /apoE complexes from neurons via LRP1, with this process impaired for carriers of APOE4 due to reduced complex stability between apoE4 and Aβ. Soluble Aβ can also be removed by proteolytic enzymes; however apoE4 proves less effective at this than apoE2 or apoE3, leading to reduced clearance overall [64].
ApoE and tau
One defining characteristic of Alzheimer's disease (AD) pathology is the formation of Neural Focal Thresholds (NFTs). NFTs consist of hyperphosphorylated tau aggregates as well as Ab plaques. Studies have demonstrated that carrying the APOE4 allele increases tau phosphorylation more than either APOE2 or APOE3, particularly when exposed to Ab oligomers. Furthermore, PET imaging studies on humans with this allele reveal greater tau deposition regardless of plaque presence. Additionally, neuronal APOE4 was found to promote tau phosphorylation and cell death more effectively than APOE3 in induced pluripotent stem cell cultures; animal models indicate that this genotype was also associated with higher total tau and phospho-tau levels, exacerbating tau-mediated neurodegeneration through modulating microglial activation [65]. Recently published research has demonstrated that deletion of Astrocytic ApoE4 can significantly decrease tau-related synaptic degeneration and disease-associated gene signatures, protecting against microglial phagocytosis as well as providing protection from tau. One study using AAV-tau delivery found that APOE2 may cause tau phosphorylation and aggregation to increase, potentially due to formation of tau/apoE complexes primarily produced when non-lipidated APOE2 was present. Recent genome-wide association study (GWAS) results indicate that APOE2 may offer protection from AD risk by differentially regulating protein phosphatase 2A (PP2A), an important tau phosphatase in the human brain, unlike the detrimental impact of APOE4 [66,67]. Taken together, these results show how impact of ApoE on tauopathy pathogenesis and tau-mediated neurotoxicity depends on which isoform is chosen.ApoE's role in tau pathology has drawn much interest both within AD research and among researchers studying related tau-related diseases like FTD (Frontotemporal Dementia), CTE (Chronic traumatic encephalopathy), and CBD (Corticobasal degeneration). For instance, FTD patients carrying the APOE4 genotype display earlier onset tau pathology, more severe neurodegeneration, and greater cognitive decline than non-APOE4 carriers, suggesting apoE may influence tau pathology independently of Ab pathology. Therefore, understanding its molecular mechanisms within tauopathy may provide important insight for developing strategies against AD and related neurodegenerative neurodegenerative conditions like FTD or CTE/CBD [68].
Studies conducted to date have demonstrated that ApoE binds to regions of tau that contribute to pathogenic NFT formation, and one potential mechanism is that ApoE may bind tau and block its phosphorylation sites. This interaction has been shown to be isoform-specific, with apoE3 showing stronger binding affinity to tau's microtubule-binding region than apoE4. According to research, reduced binding affinity of apoE4 to tau may increase GSK3-mediated tau hyperphosphorylation and subsequent formation of NFTs. Alternately, some experts hypothesize that apoE4 inhibits Wnt signaling pathway through LRP5/6 receptors by increasing GSK3 activity and leading to tau phosphorylation. Current research is exploring these potential mechanisms further so as to understand apoE's contributions in tau pathogenesis [69].
ApoE and neuroinflammation
Recent literature indicates that inflammation is an integral component of neurodegeneration, with its modulation by ApoE gaining increasing attention. ApoE may contribute to AD pathogenesis through various pathways; however, evidence is mounting suggesting they converge into neuroinflammation . Microglia cells often surround plaques found in postmortem brain tissue and play an active role in orchestrating an inflammatory response and clearing out amyloid plaques from memory cells. Studies conducted on mice lacking ApoE have demonstrated decreased microglial response to plaques, suggesting it may be necessary for proper microglial activation in response to amyloid aggregation. Emerging research has also demonstrated that disease-associated microglia (DAM) or microglial neurodegenerative (MGnD) phenotypes exhibit a consistent transcriptional signature across Alzheimer's mouse models, with ApoE serving as a central regulator. ApoE's effect on microglial function appears to vary depending on its isoform, with recent research showing that apoE3 induces more effective microglial responses to Aβ injection than its isoform apoE4. This observation could be explained by Triggering Receptor Expressed on Myeloid Cells 2 (TREM2), which interacts with ApoE with high affinity to modulate microglial responses. Evidence indicates that binding of apoE to TREM2 depends on both its isoform and lipidation status, potentially explaining differences in microglial function between isoforms. ApoE4 may impair homeostatic microglial functions due to reduced lipidation or affinity with TREM2, possibly accounting for its less potent homeostatic microglial responses compared with other isoforms [70,71]. Studies have demonstrated that C-reactive protein (CRP), produced by hepatocytes and released into plasma or serum, can be modulated by an individual's APOE genotype in their peripheral immune system. CRP is an inflammatory protein produced in response to inflammation or injury and its levels vary accordingly. Proteomic analysis of cerebrospinal fluid has demonstrated lower CRP levels among APOE4 carriers compared with individuals carrying either APOE3 or APOE2. CSF samples also reveal reduced concentrations of CRP and complement cascade proteins among these carriers, in comparison with individuals who carry either APOE2 or APOE3 [72]. However, in spite of this tendency in genotype, AD prevalence increases sharply with increasing serum CRP levels - with its greatest impact seen among APOE4 carriers. However, in a longitudinal cohort APOE haplotype but not CRP haplotype was associated with life-long cognitive decline - disproving any association between CRP and cognitive decline. Therefore, those carrying the APOE4 allele may experience abnormal immune reactions in response to pathological development that lead to injury responses and cognitive deficits [73]. Therefore, targeting apoE-mediated inflammatory responses as part of therapeutic approaches for Alzheimer's Disease or neurodegeneration could prove useful and should be explored further as a potential solution.
4. Important ApoE Mutations involved in AD onset
c.-488C>A
The biological impact of this variant found within the APOE promoter remains unknow [30]. It falls within the functional domain of HuD protein that spans nucleotides -651 to -366 [31] which has been shown to act as a negative regulator in multiple cell types including PC12 neuronal-like rat chromaffin cells, SK-N-SH neuroblastoma cells, C6 glial cells and U373 astroctyoma cells among others. Furthermore, substitution of nucleotide 488C will remove potential binding sites used by transcription factor EGR1 which would interact directly with this transcription factor [32].
c.-24+38G>A
Yee et al. conducted a study that sequenced the APOE genes of 257 Southern Chinese individuals spanning 69 AD patients, 83 subjects with mild cognitive impairment (MCI) and 105 cognitively healthy controls in South China; two AD patients (1.4%), one control (0.5%) and no MCI patients (0%) carried this variant; it was detected globally at an incidence frequency of 0.00033 in gnomAD variant database and most carriers having East Asian heritage (0.0037 frequency; 43 heterozygotes) [32].
c.-24+288G>A
Yee et al. conducted a study where this variant was identified in 257 Southern Chinese individuals spanning Alzheimer's Disease (AD), mild cognitive impairment (MCI) and cognitively healthy controls from Southern China. One AD patient (0.7%), one MCI patient (0.6%) and one control (0.5%) carried it [32]; gnomAD reported the variant c.-24+288G>A as having a worldwide frequency of 0.00016 with only five heterozygote carriers of East Asian origin worldwide; conversely it was significantly more prevalent among East Asians with an East Asian ancestry ancestry population with an incidence rate of 0.015 according to gnomAD v2.1.1 (Oct 2022); out of 22 carriers listed there, all were of East Asian ancestry with at least one homozygote from this region.
c.-23-377A>G
This variant was identified in a study which involved sequencing the APOE genes of 257 individuals of Southern Chinese origin, comprising 69 AD patients, 83 subjects with MCI, and 105 cognitively healthy individuals - including six AD patients (4.3%), three MCI patients (1.8%), and three controls (1.4%) [32].
In the gnomAD variant database, this variant was reported at an overall frequency of 0.00073 and at much higher frequency among individuals of East Asian ancestry - 22.2 carriers were identified from East Asia alone with one being homozygous for it.
A18T
Yee et al. conducted an in-depth analysis of 257 Southern Chinese individuals' APOE genes, comprising of AD 69 patients, mild cognitive impairment in 83 subjects and 105 healthy controls and found one AD patient - 0.7%, three MCI patients - 18% , four controls - 1.9% [32].
Zhou et al. identified this variant as one of six APOE variants with potential clinical relevance and functional consequences due to its high prevalence in at least one population and predicted deleterious effects by in silico algorithms. They performed whole genome and exome sequencing analyses from 138,632 individuals from different populations and discovered that this variation alters ApoE signal peptide sequence at its cleavage site, potentially hindering secretory efficiency by disrupting recognition at this spot [33].
Table 4.
Recently discovered new ApoE gene mutations.
| Mutation | Pathogenicity | Type of Mutation | Biological Effect | Citation |
| K82fs | Tauopathy and Pick’s Disease | Deletion | The neuropathological findings were consistent with Pick's disease. A frameshift was identified to start at K82, and the mutant protein was found to be reduced in the frontal cortex and hippocampus. | [54] |
| c.*71C>A | Alzheimer’s Disease - Pathogenic | Substitution | In one case, an MRI scan revealed widening of the sulcus, fissure, and temporal horn, along with a decrease in hippocampal volume. Additionally, FDG-PET showed hypometabolism in the bilateral frontal, parietal, and temporal lobes. Among the five affected carriers, CSF analysis showed Aβ42, total tau, and phospho-tau levels consistent with AD. The study suggests a possible reduction in the binding of PSEN2 expression suppressor miR-183-5p, which may lead to an increased Aβ42/Aβ40 ratio. | [55,56] |
| M239V | Alzheimer’s Disease - Pathogenic | Substitution | The brain pathology showed diffuse cerebral atrophy, senile plaques, neurofibrillary tangles (Braak and Braak stage VI), ectopic neurons in subcortical white matter, and extracellular "ghost" neurofibrillary tangles. In cell-based assays, there was an increase in the Aβ42/Aβ40 ratio and an increase in Aβ42 levels. However, there was no change in the proteolytic products PSEN2-CTF and PSEN2-NTF. | [57,58] |
Microtubule-associated protein tau
The discovery that microtubule-associated protein tau (MAPT) gene mutations caused frontotemporal dementia with Parkinsonism linked to chromosome 17 (FTLD-17) was an historic moment, providing genetic evidence of dysfunction within tau alone as sufficient cause of neurodegeneration independent of Aβ. Abnormal accumulation of tau is seen across various central nervous system disorders such as AD, Pick's disease (PiD), progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), and argyrophilic grain disease - thus targeting tau offers the possibility not only of treating AD itself but also of treating many other tauopathies associated with Aβ. Human brains produce six distinct isoforms of tau through alternative splicing of the MAPT gene located on chromosome 17q21. The different isoforms result from alternatively splicing exons 2 and 3, leading to variants with zero (0N), one (1N), or two (2N) N-terminus inserts [74]. Exon 10 can also affect protein production, leading to three (3R) or four (4R) microtubule-binding domains residing on C-terminal tau proteins based on whether they contain three (3R) or four (4R). 3R tau binds less tightly than 4R tau, so that six tau isoforms could exist: 3R0N, 3R1N, 3R2N and 4R0N are all possible isoforms. An average brain contains equal levels of 3R and 4R tau. However, in certain tauopathies - for instance those linked to frontotemporal dementia with parkinsonism linked to mutations near exon 10 on chromosome 17 (FTDP-17), such as some families suffering from frontotemporal dementia with parkinsonism linked to mutations near exon 10 - there may be an increase in 4R tau, increasing interaction with microtubules [75,76]. Tau undergoes various posttranslational modifications during both normal physiological processes and stress-induced responses, such as glycosylation, ubiquitination, glycation, nitration, and oxidation processes; with phosphorylation being the most widely studied. When exposed to healthy brain environments such as Alzheimer's Disease or other tauopathies such as multiple myeloma or Parkinsonism the levels of tau phosphorylation vary; in healthy brain tissue around two or three residues while neurodegenerative conditions such as Alzheimer's or tauopathies involve much higher phosphorylation with nine or phosphates per molecule being created by imbalanced activity between tau kinases and phosphatases resulting in hyperphosphorylated tau being localized within its environment resulting in multiple serine/threonine/tyrosine residues on different places on its protein structure due to imbalance between its kinases/phosphatases activity [77].Glycosylation, ubiquitination, glycation, nitration and oxidation are among the many posttranslational modifications that play a role in controlling tau during both normal and stress-induced responses. Of these modifications, phosphorylation has been widely studied . Tau is typically phosphorylated on two to three residues in healthy brains. But in AD and other tauopathies, its hyperphosphorylation occurs at nine phosphates per molecule. This imbalance results from disruptions in the activity of tau kinases and tau phosphatases, leading to decreased affinity of tau for microtubules as well as resistance against degradation by both ubiquitin-proteasome pathway degradation as well as calcium activated neutral proteases. Hyperphosphorylated tau forms fibrils and aggregates into NFTs over time. Major tau kinases include GSK-3b, CDK5, PKA, MAPK CaMK II MARK [78,79]; while PP2A has been identified as the primary dephosphorylation enzyme for abnormal tau. Changes in tau kinases and phosphatases have long been documented as markers of AD and related conditions, with expression and activation rates of tau kinases and phosphatases often increasing over time [80]. A variety of processes, including Aβ, impaired brain glucose metabolism, inflammation and infection all play a part in abnormal tau hyperphosphorylation; therefore identifying pathways governing posttranslational modifications of tau may prove extremely valuable when searching for therapeutic targets.
Research has demonstrated that an abnormal hyperphosphorylation of tau occurs prior to its accumulation in Alzheimer's disease-affected neurons. This hyperphosphorylated tau has been identified both within neurofibrillary tangles as well as within the cytosols of AD brains. Utilizing mAb Tau-1 for immunocytochemical studies has demonstrated that abnormally phosphorylated tau (not normal tau) accumulates in neurons without tangles (stage "0" tangles) in Alzheimer's and aged hippocampi [81]. At present, tau found in neurofibrillary tangles is known to be ubiquitinated while abnormally hyperphosphorylated tau isolated from AD brain cytosol does not display this property, indicating abnormal hyperphosphorylation occurs prior to its accumulation into neurofibrillary tangles. Vincent et al. demonstrated that tau phosphorylation occurs prior to PHF formation in AD brain by employing monoclonal antibodies targeting mitotic phospho epitopes. One possible explanation for abnormal hyperphosphorylation of tau is conformational changes occurring within diseased brains that make it an even more favorable substrate for phosphorylation and/or dephosphorylation, respectively. Davies and his colleagues have developed monoclonal antibodies to detect conformational changes of tau, and have demonstrated that tau indeed undergoes conformational changes both in AD patients and transgenic mice that overexpress human tau. As in FTDP-17, which is caused by certain missense mutations of tau, these mutations make tau more susceptible to hyperphosphorylation by brain protein kinases and lead to its hyperphosphorylation. However, in AD it is less likely that tau mutations alone are responsible for hyperphosphorylation; several neuronal proteins become over phosphorylated due to an imbalance between protein phosphorylation and dephosphorylation processes. Biochemical analyses have demonstrated that Alzheimer's brain tissue contains excessive levels of tubulin and neurofilaments that have become hyperphosphorylated; immunocytochemical analysis shows neurofilaments and MAP1B to also be affected. Furthermore, both PHF-abnormally hyperphosphorylated taus and its cytosolic counterpart are readily dephosphorylated by in vitro phosphatases [82].
5. Important MAPT Mutations involved in AD onset
MAPT IVS10+12 C>T
This mutation was identified as the causative mutation in the Kumamoto pedigree, a Japanese kindred with frontotemporal dementia [83]. Primary clinical symptoms included parkinsonism and dementia manifesting during their fifth decade, with an average onset age of 53 years and length of illness lasting seven years (n=6).
Brain tissue from affected individuals was observed to contain elevated exon 10 tau transcripts and 4R tau isoforms, with elevated exon 10 tau aggregates observed both in neurons and glial cells; isolated tau filaments displayed twisted ribbon-like morphologies made up of hyperphosphorylated 4R tau. Yasuda et al reported neuropathological findings for one member of this pedigree while Takamatsu et al discussed neuropathological findings for one individual within this pedigree [83,84].
MAPT A152T
In a large series of American and European people, the A152T variant was found to be associated with an increased risk of DLB, but not PD [85]. The variant was found in 10 out of 2456 controls (minor allele frequency 0.20 percent). Among PD patients, 18 out of 3229 carried the variant (MAF 0.28 percent) and amoung DLB patients, six out of 442 patients carried the variant (MAF 0.68 percent). In addition, two out of 181 patients with multiple system atrophy carried the variant (MAF 0.55 percent), a non-significant increase in frequency.
Consistent with the variable clinical presentations associated with this variant, neuropathological reports are similarly diverse. Abnormal tau accumulation appears to be the unifying feature in all cases for which postmortem findings are available. In some cases prominent Lewy body pathology is seen [86]. In other cases, the pathology is indicative of PNLA, as indicated by the prominent neuronal loss and tau deposition in the globus pallidus, subthalamic nucleus, and substantia nigra with lower levels of pathology in the motor cortex, striatum, pontine nuclei and cerebellum.
This variant has been shown to impair tau's ability to bind microtubules, resulting in less-efficient microtubule assembly and impaired microtubule stability. In addition, although the mutant protein appears to aggregate with lower efficiency than wild-type protein overall, it is more prone to oligomer formation [87]. Isogenic human iPSCs generated from fibroblasts of an A152T carrier showed that the mutant tau is predisposed to proteolysis by caspases and other proteases and leads to greater tau pathology.
MAPT K257T
Autopsy analysis revealed Pick's disease, a subtype of FTD characterized by severe frontotemporal atrophy in particular in the temporal lobes. Neocortex, hippocampus, and some subcortical regions displayed numerous tau-positive Pick bodies while diffuse hyperphosphorylated tau was detected in certain cell bodies [88]. Recombinant tau protein with K257T mutation displayed reduced capacity to facilitate microtubule assembly [88].
MAPT L266V
Kobayashi et al. reported on one case that underwent autopsy that revealed severe frontotemporal atrophy with Pick-like pathology, evident by prominent atrophy of both frontal and temporal cortices as well as caudate nucleus and substantia nigra, prominent neuronal threads, coiled bodies, and ballooned neurons throughout all layers of cortex and the brainstem; abundant tau-positive inclusions within neurons and astrocytes with high concentration of tau-positive inclusions present throughout all cortical layers and brainstem; there were abundant tau-positive inclusions found among neurons and astrocytes with high concentration found throughout all cortical layers as well as high concentration found within caudate nucleus; numerous neuronal threads threads as well as neuronal threads, coiled bodies, ballooned neurons were observed; Additionally neuronal threads, and ballooned neurons were observed - all present and noticeable for its severity [89].
Hogg et al. reported another case characterized by severe frontotemporal atrophy with Pick-like pathology, as well as significant atrophy of the hippocampus and parietal lobe. Neuronal loss was extreme across cortex and substantia nigra with severe atrophy in these regions; there was also significant gliosis present, while Tau-positive inclusions were widely distributed, including within hippocampus, striatum and substantia nigra; Pick bodies were Gallyas silver positive and contained straight filaments distributed randomly throughout layers of cortex [90].
In vitro, this mutation alters exon 10's splicing, leading to higher levels of tau transcripts with four microtubule binding repeat domains (4R tau). This leads to decreased rates of microtubule assembly induced by tau and lower tubulin polymerization levels - more specifically with 3R tau isoforms being more likely to assemble than their 4R counterparts.
Table 5.
Most relevant mutations in MAPT gene leading to AD.
| Mutation | Pathogenicity | Type of Mutation | Biological Effect | Citation |
| IVS10+12 C>T | Familial Danish Dementia - Pathogenic | Substitution | The mutant protein leads to the formation of tau aggregates in both neurons and glia, and isolated tau filaments exhibit a twisted, ribbon-like morphology and consist of hyperphosphorylated 4-repeat (4R) tau isoforms. The mutation also causes a destabilization of a stem-loop structure that regulates the alternative splicing of exon 10, resulting in a higher frequency of inclusion of exon 10 and an increased proportion of 4R tau isoforms. | [83,84] |
| A152T | Alzheimer’s Disease - Risk | Substitution | The presence of tau pathology is a common feature, often accompanied by Lewy bodies, amyloid plaques, or TDP-43 pathology. The mutant tau has a decreased ability to bind to microtubules, leading to less efficient microtubule assembly and impaired microtubule stability. Additionally, it has an increased propensity to form tau oligomers and is more susceptible to proteolysis by caspases. | [85,86,87] |
| K257T | Tauopathy and Frontotemporal - Pathogenic |
Substitution | The patient exhibited frontotemporal atrophy with significant temporal lobe involvement. Tau-positive Pick bodies were found in the neocortex, hippocampus, and subcortical regions similar to those seen in sporadic Pick's disease. Some cell bodies showed diffuse hyperphosphorylated tau. In vitro analysis showed that recombinant tau protein with the K257T mutation had a decreased ability to promote microtubule assembly. | [88] |
| L266V | Frontotemporal - Pathogenic |
Substitution | The patient had severe atrophy of the frontal and temporal lobes, with extensive neuronal loss and gliosis. Tau-positive inclusions, including Pick bodies, and tau-positive argyrophilic astrocytes with stout filaments and round or irregular argyrophilic inclusions were also observed. In molecular studies, there were increased levels of exon 10+ tau mRNA and soluble four-repeat (4R) tau. The patient showed a decreased rate and extent of tau-induced microtubule assembly, as well as a specific increase in tau self-assembly for the 3R isoform. | [89,90] |
6. The evolving landscape of Alzheimer's disease treatments: exploring current and future perspectives
Donanemab, a humanized anti-body that targets the N truncated pyroglutamate-amyloid-b peptide (pGlu3Ab, AbpE3) has shown potential to reduce cerebral amyloid depositions in Alzheimer's disease, constituting a promising treatment option for AD. This therapy aims either to reduce pGlu3 Aβ formation at glutamyl cyclase, or to clear pGluAb after formation and/or block aggregation [91,92,93]. Donanemab was found to be highly active against amyloid, especially cored plaques within the CNS. However, its efficacy as a treatment of AD is still uncertain. The binding properties of antibodies targeting AbpE3 are different against the soluble and aggregated forms of AbpE3-42 [94,95].
Lowe et. al. proved that Donanemab shows good tolerance to dosages up to 10mg/kg, with a terminal half-life mean of four days following a single dose of 0.1-3mg/kg. The half-life was increased to 10 day with a dosage of 10 mg/kg. A 40-50% decrease in amyloid levels was seen at 24 weeks with a Standardized Uptake Ratio (SUVR), which decreased from 1.65 to 0.36, and a change in Centiloids (CL), which decreased from -44.4 (SD 14.2) from baseline. Moreover, 90 % of the subjects developed antidrug antibodies 3 months after a single dose.
In a separate report by Lowe et al [96]. It was shown that Donanemab caused rapid amyloid decreases even after just one dose [97]. The mean reduction in PET amyloid was -16.5 CL at 10 mg/kg, -40.0 CL at 20 mg/kg, and -50.6% with 40 mg/kg. The multiple-dosage groups at week 24 showed a mean reduction of amyloid levels in the 10mg/kg Q4weekly arm, a 50.2 CL for the 10mg/kg Q2weekly arm, and a 58.4 CL for the 20mg/kg Q4weekly arm. In both the single-dose and multi-dose cohorts, some patients had an amyloid clearance level below 24.2. Donanemab was effective in treating all but one patient (97.8%) [97].
In another cohort, nearly 40% of participants (46 of 115) receiving Donanemab reached the full amyloid clearing threshold (24.1 CL), and their baseline amyloid levels were lower than the group as a whole. In the first 24 week period, there was a moderately negative correlation (r=-0.54) between the baseline amyloid level and that of the plaques removed. The amyloid clearing was sustained, with a very low rate of reaccumulation (0.02 mean rate over one year). Participants who had an amyloid concentration of =11 CL by week 24 but discontinued treatment, would require about 3.9 to accumulate amyloid up to 24.1 CL. The overall tau accumulation was reduced by 34% in the Donanemab groups compared to placebos at week 76 [98].
In another study by Lowe et al. two patients experienced asymptomatic amyloid imaging abnormalities as a result of cerebral microhemorrhages. One patient discontinued treatment because of these reactions. In a second study by the same authors, seven serious adverse effects were reported in six patients. Only one patient died from a non-drug related myocardial ischemia. In another study by the same authors [97], seven serious adverse events were reported among six patients, with only one patient dying due to a non-drug-related myocardial infarction. Two patients (4%) from the interventional arms stopped taking Donanemab because of adverse reactions.
Another investigation relates that the rate of mortality from all causes was lower for participants who received Donanemab (0.76%) compared to those who received placebo (1.6%). The study also found that there were no differences between the groups in terms of the rates of serious adverse reactions, which were respectively 19.85% and 20.0% in the Donanemab group and the placebo group [98].
As a conclusion, Donanemab is now being tested as an alternative therapy for Alzheimer's. It has been a long time since the urgent need to slow the disease's progression was met. Alzheimer's disease treatments have been approved by the FDA, but there has been controversy over this approval. There is also a need for better and more effective treatments. According to a systematic review of phase III trials for preclinical Alzheimer's, the first anti-amyloid therapies were performed. It is important to plan carefully, to perform longitudinal assessments and to store and manage data effectively as clinical trials and new therapeutics are developed. The research conducted will determine Donanemab’s effectiveness for a diverse population, increasing the retention of the treatment and improving referrals to clinical trials.
7. Parkinson’s disease (PD)
Parkinson's disease (PD) is a progressive neurodegenerative condition, most often seen among elderly individuals worldwide. It's estimated to affect between 0.3% of the general population and 1-3% of those over age 65. By 2030, its numbers are expected to climb from 8.7 million to 9.3 million. James Parkinson first described PD symptoms in 1817 and they typically include dysfunctions of the somatomotor system including rigidity, bradykinesia, postural instability, gait dysfunction and tremors .
Disease progression leads to progressive degeneration of the nigrostriatal dopaminergic pathway, leading to significant neuron loss in substantia nigra pars compacta (SNpc) neurons and depletion of dopamine (DA). Non-motor dysfunctions such as dementia, hyposmia and gastrointestinal abnormalities often accompany disease progression.
Pathological hallmarks of Parkinson disease (PD) include accumulations of a-synuclein aggregates known as Lewy bodies or neurites in certain areas of the central nervous system, such as basal ganglia, dorsal motor nucleus of vagus (DMV), olfactory bulb (OB), locus coeruleus (LC), intermediolateral nucleus in spinal cord (IML), celiac ganglia, enteric nervous system (ENS) [99].
New research indicates that Parkinson's Disease (PD) neuropathology could be caused by environmental stressors and the natural process of aging itself. Exposure to environmental toxins, drugs of abuse or the stress of aging may lead to chronic low-level inflammation in the brain leading to something known as "inflammaging," leading to neuron cellular senescence.
Pathologically, Parkinson's patients typically display damage in the substantia nigra pars compacta and pontine locus ceruleus regions of their brains characterized by depigmentation, neuronal loss and gliosis. By the time symptoms manifest themselves, approximately 60-70% of neurons from this region have already been lost [100].
Genetic factors have been estimated to account for roughly 25% of the risk associated with Parkinson's disease, and genetic variants associated with it vary both in terms of frequency and risk . While rare mutations within individual genes (known as monogenic causes) may contribute to its development (known as monogenic causes), these were generally discovered through linkage analysis in affected families using linkage analysis; some common genetic variants that only contribute a small amount to risk are also discovered via genome-wide association studies (GWAS), including many common genetic variants that contribute an intermediate risk, such as GBA or LRRK2 variants.
Genetic classification of Parkinson's can lead to various treatment approaches and prognoses for each subgroup, often depending on age of onset, family history and pathogenic variant presence; age at onset, family history and presence of pathogenic variants are frequently used as criteria for stratifying this form of PD; monogenic forms may or may not represent typical forms of idiopathic PD. Importantly, some genes involved in monogenic PD have also been identified through GWAS studies as common variants. One such gene, SNCA, which was discovered through these analyses to have common variants is also implicated in monogenic PD pathogenesis; supporting the role of a-synuclein. Other pathways may also play a part in its pathogenesis such as tau aggregation; linked with other neurodegenerative conditions like Alzheimer's and frontotemporal dementia [101].
Familial Parkinson's, also referred to as Mendelian or monogenic PD, is characterized by rare yet high-penetrance genetic variants that increase risk. Autosomal dominant (e.g., SNCAA53T and VPS35D620N) and recessive forms of familial Parkinson disease have been identified using linkage analysis in families and next-generation sequencing technologies, though only 5-10% of cases fall under these single gene variants. Conversely, low-penetrance genetic variants with more frequent associations to sporadic Parkinson's Disease have been identified through genome-wide association studies (GWAS). At first glance, distinguishing familial from sporadic disease may help with diagnosis, prognosis, and genetic counseling for at-risk family members; however, such classification may obscure shared genetic or biological mechanisms that underlie them both.
Example: Both rare and common genetic variants associated with SNCA have been shown to increase Parkinson's risk, underscoring its role as an aSyn-mediated disease mechanism. Missense variants in SNCA such as p.A53T, p.A30P and p.E46K cause autosomal dominant familial Parkinson disease while the common risk variant SNCArs356168 occurs in approximately 40% of European-ancestry populations and has only modest effects on disease risk [102].
SNCA, or synuclein complex A, is a 14.5 kDa protein consisting of 140 amino acids encoded by five exons and having a transcript length of 3041bps. Located on 4q21.3-q22 of human chromosome 4, this synuclein protein family also includes SNCB (5q35) and SNCG (10q23.2-q23.3). The structure of SNCA protein comprises an N-terminal region with incomplete KXKEGV motifs, an extremely hydrophobic NAC domain and an acidic C-terminal domain. Under physiological conditions, it appears as either an intrinsically disordered monomer or helically folded tetramer structure; although previously thought to be toxic in this form. Recent observations have refuted this idea.
Over the last two decades, various hypotheses have been put forward regarding the toxic structural form of SNCA; none has yet been unanimously agreed upon. What is known is that its neurotoxic form accumulates within neurons before disseminating throughout anatomically interconnected regions in Parkinson's disease brain through interneuronal transmission using various mechanisms.
Although SNCA is most abundantly expressed in the brain, it also appears in heart, skeletal muscle and pancreas cells. While its exact function remains undetermined, several hypotheses have been proposed based on its structure, physical properties and interactions with interacting partners. SNCA may play an essential role in regulating dopamine release and transport, inducing microtubule-associated protein tau fibrillization and exerting a neuroprotective phenotype in non-dopaminergic neurons by modulating both p53 expression and transactivation of proapoptotic genes leading to decreased caspase-3 activation.
Given SNCA's central role in neurodegenerative processes, its essentiality may suggest that selective forces among sarcopterygians play a vital role in modulating its molecular and cellular mechanisms. Fine-tuning of these mechanisms through minute changes to protein activity could have contributed to evolutionary adaptations that meet different environmental and ecological needs. Current evidence supports that amino acids 32 to 58 of SNCA's N-terminal lipid binding domain are critical to its normal cellular functioning and disease pathogenesis. Lineage-specific substitutions could have led to structural remodeling and functional adaptation in SNCA over generations, and any mutation affecting its critical regions is likely to be harmful. These discoveries provide the framework for investigating their critical roles through various interaction studies as well as targeting them with drug discovery efforts to treat FPD [103].
Mutations in LRRK2 account for 5-12% of familial Parkinsonism cases and 1-5% of sporadic cases. So far, seven missense LRRK2 mutations have been identified as pathogenic: R1441G, R1441C and R1441H were all found to be pathogenic; these variants can be found within different functional domains of LRRK2, including R1441G located on R1441C which affect R1441H; Y1699C was also involved, as were G2019S, R1628P, G2385R and I2020T variants specific to certain populations. G2019S mutation, which leads to constitutive activation of the kinase, is one of the most prevalent. It accounts for an estimated 36% of familial and sporadic Parkinson's cases among North African Arabs; approximately 30% among Ashkenazi Jewish populations; up to 6% among familial cases in Europe and North America, as well as up to 3% among apparently sporadic cases; however it does not occur among Asian populations. Various other LRRK2 mutations such as G2385R R1628P S1647T R1398H N551K have also been associated with Parkinsonism within certain Asian populations. Studies conducted among Asian populations spanning Singapore, Taiwan, and mainland China have established that LRRK2 variants G2385R or R1628P may increase risk for Parkinson disease . Furthermore, G2385R variant has been found to increase risk for Parkinson's Disease among Japanese and Korean populations; these variants were not seen among Indians and Caucasians . Although LRRK2 mutations exist in familial PD, no differences exist in clinical features or neurochemical differentiation between idiopathic and familial forms of Parkinsonism. Both forms of Parkinson disease (PD) involve profound dopaminergic neuronal degeneration and gliosis in the SNpc, decreased dopamine levels in the caudate putamen and Lewy body pathology in the brainstem; therefore understanding LRRK2 plays an essential role for all forms of PD [104].
Mutations in the PINK1 gene are an important cause of early-onset Parkinson's disease (EOPD), accounting for 1-9% of genetic cases and 15% of early-onset cases - second only to Parkin mutations. First identified by Unoki and Nakamura in 2001, its 18 Kb span contains eight exonic regions that encode for an essential serine/threonine protein kinase essential for mitochondrial functioning and metabolism .
As reported by MDSGene database, worldwide there have been 151 PINK1 mutation carriers who carry 62 different disease-causing sequence variants involved with both sporadic and familial Parkinson disease cases; 13 definitely pathogenic mutations exist alongside 44 possibly pathogenic variants (13 definitely pathogenic mutations and 44 possibly pathogenic variants).
PINK1, an encoded protein from the PINK1 gene, primarily localizes to mitochondria where it serves as a serine/threonine-type protein kinase that regulates mitochondrial quality control (mitoQC). MitoQC involves maintaining respiring mitochondrial networks while selectively eliminating damaged ones through mitophagy, an essential process critical for cell homeostasis. Furthermore, in addition to mitoQC functions PINK1 also plays an anti-death pro-survival role under various forms of stress conditions preventing neuronal cell death under various stress conditions. Additionally its protein contains an N-terminal mitochondrial targeting sequence (MTS or TMD), transmembrane sequence (TMS or TMD), and C-terminal domain [105].
8. Perspectives of Treatment
The metal-based hypothesis of neurodegeneration is an attractive explanation for the pathophysiology behind Parkinson's Disease. This hypothesis proposes that reactive oxygen species are generated by redox-active metals, particularly iron. ROS (reactive oxygen species) cause membrane phospholipids to be peroxidized, resulting in the production of reactive aldehydes. Both ROS, and reactive aldehydes, modify a synuclein causing it to aggregate. Aggregated α-synuclein causes mitochondrial dysfunction, resulting in a vicious cycle of increased ROS production and decreased ATP synthesis. In order to provide a more effective treatment of PD, a multi-task strategy targeting these events is needed [106].
Coenzyme Q10 is a vital antioxidant that is important in reducing oxidative stresses, a factor implicated in Parkinson's disease and other neurodegenerative diseases. In order to establish their potential as a marker of disease, a number of studies have investigated the levels of CoQ10 found in different tissues of people with PD or other parkinsonian disorders. Several studies have also explored the therapeutic potential of CoQ10 for the treatment of PD or PS.Several clinical studies have examined the ability of ubiquinol (the antioxidant form of Coenzyme 10 or CoQ10) to reduce oxidative damages observed in PD. CoQ10 can restore mitochondrial function by bypassing Complex I dysfunction, which is a feature of sporadic PD. A meta-analysis of eight controlled trials involving 899 patients found that CoQ10 is well tolerated, safe and does not improve motor symptoms compared to placebo. The study authors do not recommend CoQ10 as a routine treatment for PD except in cases where levodopa is wearing off [107].
Recent clinical trials have shown that iron chelation therapy is a promising approach to treating Parkinson's Disease. Due to the multifactorial nature PD, targeting a specific factor, such as iron, may not be enough for complete neuroprotection. It may be necessary to develop and test multifunctional drugs that combine the iron chelation process with other protective properties.
A growing global population is aging, and central nervous system disorders such as Parkinson's disease and Alzheimer's are becoming more prevalent. These disorders are linked to iron accumulation in certain areas of the mind. Finding effective treatments for these conditions is therefore crucial to improving the longevity and quality of life of elderly people [108]. DFO (deferrioxamine) was administered intramuscularly in early studies of Alzheimer's patients. DFP (deferiprone) was the first oral chelator used to treat Friedreich's Ataxia [109]. This condition is characterized by frataxin deficiency, which is the mitochondrial chaperone for iron. Animal studies have shown that DFO or DFP can reduce iron in different brain regions, and also provide neuroprotection for an animal model of Parkinson’s disease. In two clinical trials, oral DFP was administered to PD patients in cohorts. MRI measurements showed that the iron content of the substantia nigra, as measured by DFP, decreased. UPDRS scores also improved. Iron chelation was not effective in patients who had high levels of inflammatory marker IL-6. DFP has a major problem with agranulocytosis, and neutropenia. This requires testing of white blood counts every week and complicates logistics. DFP is currently being tested in phase II clinical trials on early-stage PD [106,110].
α-Synuclein is another point of interest regarding the treatment of PD. Although clinical trials using monoclonal antibodies to treat α-synuclein aggregates have failed to show any improvement in Parkinson's symptoms, other studies in progress or recruiting participants may prove that targeting α synucleinopathies as a therapeutic option is possible.
9. Huntington’s disease (HD)
Huntington’s disease represents a neurodegenerative disorder that bears the name of the American physiologist George Huntington (1850-1916). He was the first one to observe the main manifestation of this disease, characterised by uncontrolled movements that tend to be compared to dance-like movements of the body, named chorea, as well as abnormalities regarding both the personality and patient’s way of thinking. The motor disturbance can be split in 2 different stages: the incipient stage, characterised by a hyperkinetic syndrome, due to the loss of the medium spiny neurons (MSNs) of the indirect pathway, followed in the later stages of the disease by the loss of the MSNs of the direct pathway, which lead to a hypokinetic syndrome [111]. Moreover, he was the one to observe this pathology, has both a hereditary nature as well as a progressive on-set, saying “Once it begins it clings to the bitter end” [112].
However, Huntington's disease represents a rare pathology, with an incidence of 10.6 to 13.7 out of 100.000 individuals. The statistics vary across the ethnic groups due to the differences of the HTT gene. According to a study conducted by Bates et. al 2015 [113], the average length of repeated CAG trinucleotide sequences varies from 18.4-18.7 in European population and 17.5-17.7 in East Asian population.
As it was more and more studied, it was shown that Huntington’s Disease represents an autosomal dominant progressive neurodegenerative disorder, consisting of a repetitive set of (CAG)n trinucleotide sequence in a gene found on the chromosome 4p16.3, between D4S10 and D4S98 [114] (Gusella et al 1983), called huntingtin (HTT), leading to a polyglutamine expansion . This repetitive sequence of trinucleotides, leads to a mutation of the huntingtin gene (mHTT).
However, the number of CAG units repeated in an allele has a strong significance when it comes to predicting whether the allele is a disease generating one or not. The normal range for the healthy population is between 6-35 units. Between 36-39 units, the disease is not guaranteed to occur, but there are also chances of developing it. Over 40 units, the mutation is regarded as highly penetrant and it will generate a phenotype along the adult population [115].
On the other side, the length of the CAG repeted sequence is not only correlated with the chances of developing the Huntington’s disease, but also with the on-set age of this pathology. The study conducted by Persichetti et. al (1994), showed that the bigger the sequence of trinucleotides is, the earlier is the beginning of the neurological symptoms [116].
Additionally, other variations have been identified in the HTT gene beyond its polymorphic/expanded CAG repeat. These include modifications in both its coding sequence (such as an expanded CCG repeat after CAG repeat and deletion polymorphism at codon 2642) as well as untranslated sequences, intron sequences and those flanking its centromeric and telomeric ends. These variations have been used to define HTT haplotypes, which represent groups of sequence variants on specific chromosomes that tend to remain relatively unchanged between generations due to limited recombination events in this relatively small segment of genome [117].
These haplotypes, carrying expanded alleles in HD patients, have revealed that approximately 50% of Europeans with HD have one common ancestor while multiple independent mutations on different chromosomal backbones account for the rest. However, none of the most frequent haplotypes found either on HD chromosomes or among HD heterozygotes appear to significantly alter motor diagnosis age. Therefore, while natural sequence variation at HTT might occasionally serve as a source of disease modification in HD, its contribution is not significant enough [118].
The mHTT gene leads to the formation of an abnormal huntingtin protein with an extended polyglutamine tail at the NH2-terminal end.
The toxicity of the mHTT gene is generated through the formation of 2 kinds of mRNA. First one is represented by the HTT mRNA while the second one is a HTT mRNA exon 1, that encodes only the first exon due to the CAG repeated sequence [119]. Therefore, from the first type of mRNA will result a full-length huntingtin protein made of huntingtin fragments, loops that are used for proteolytic cleavage which will later represent the site for complex post-translational modifications that will lead to the HTT exon 1 fragment, as well as some individual boxes. The HTT mRNA exon 1 on the other side, translates itself with the help of the ribosome just in a huntingtin fragment, which is arranged from 3 parts. First one is described as a mixed sequence of 17 amino-acids, the second part is the polyglutamine sequence, also called polyQ, while the last part of the exon 1 fragment is characterized by a proline-rich domain (PRD) (Figure 2).
Upon translation, either the full-length huntingtin protein or the HTT exon1 protein is generated. The HTT exon1 fragment comprises the HTTNT sequence, the polyglutamine sequence encoded by the CAG repeat, and a proline-rich domain. On the other hand, the full-length huntingtin protein includes the HTT exon1 sequence, as well as ordered and disordered protein segments represented by boxes and loops, respectively.
Proteolytic cleavage, which occurs at recognition sequences located in the disordered segments, leads to the formation of various products, including HTT exon1-like fragments. Fragments with expanded polyQ segments play an important role in the development of Huntington's disease through molecular mechanisms that have yet to be fully understood.
Huntington's disease begins early, before any symptoms have emerged, with transcriptional dysregulation taking place due to mutations of HTT that disrupt transcriptional machinery via interactions with transcription factors and molecular mediators such as CBP (cAMP response element-binding protein).
Recent research has demonstrated changes to chromatin remodeling through impaired histone activity, and reductions in mitogen-activated and stress-activated protein kinase 1 (MSK-1) activity among striatal neurons of Huntington's disease patients and animal models. Overexpression of MSK-1 resulted in increased expression of peroxisome proliferator-activated receptor gamma coactivator alpha (PGC-1a); a transcriptional co-activator involved in mitochondrial biogenesis that may also protect against neuronal death [120].
Histone deacetylase (HDAC) inhibitors have shown promising results in animal models of Huntington's disease, and may represent a viable therapeutic target. Class III HDACs (sirtuins) have demonstrated promise as neuroprotective targets, with one study revealing an improvement in motor function and reduced brain atrophy in an Huntington's disease mouse model by overexpressing Sirt1, an NAD-dependent protein deacetylase. Notably, Sirt1 also restored brain-derived neurotrophic factor (BDNF) [121].
The following actions that happen after the translation, are represented by a condensation and oligomerization of the protein fragments in cytoplasm, which will lead to a dysfunctional proteostasis of the cell. Moreover, these fragments will go through an aggregation process inside the nucleus of the cell, binding to the DNA as inclusions and therefore altering the entire process of transcription. All of these pathological processes will alter both the axonal transport and inter-synaptic transmission, as well as the mitochondria of the cell, causing a decreased energy output [120].
According to a study conducted by G. Vonsatellel et al. 1998 [122], the mutated huntingtin protein can be found in both the dystrophic neurites and in nuclear inclusions of the neuron, with a higher prevalence in cortex and neostriatum in comparison with the globus pallidus and cerebellum where this type of mutated protein cannot be found. Moreover, this study attested that mHTT protein was found in 38% to 52% of the neurons of the patients with juvenile HD (with an age of under 20 years) and in 3% to 6% of the neurons regarding the adult on-set of the HD.
A study conducted by the GWAS (Genome-Wide Association Study) in 2017, discovered the existence of a correlation between the HD onset and a gene called MSH3, that together with the MSH2 gene, will lead to a heterodimer called MutSβ, which main goal is to repair the possible mismatches of the DNA after replication. However, a variant of the MSH3 seems to be involved in the somatic expansion of the CAG repetitive sequence, leading to an increased risk of developing the HD, affecting mostly the brain striatum, the are most affected by the HD [123] .
However, even if the today we understand the pathogeny of the HD better than anytime, the therapeutic directions are quite limited, having a hard time coming to a conclusive result. According to a study conducted by Travessa et al. [124] showed that out of 99 trials that proposed to study 41 compounds to treat the HD, only 2 trials made it to the phase 4 (2%), with a success rate of only 3.5%.
10. Treatment
Tetrabenazine is the only drug approved to treat Huntington's chorea in North America and some European countries; however, this could increase depression as a potential side effect. Post hoc analysis showed that advanced Huntington's disease patients who already took antidepressant drugs did not experience worsening depression after starting treatment with tetrabenazine. Antipsychotic drugs are most often employed for chorea treatment in Europe, while both tetrabenazine and antipsychotics are applied at an equal level in North America and Australia.
Amantadine's efficacy for chorea treatment remains inconclusive, while pridopidine, a dopaminergic stabilizer, has been assessed as symptomatic therapy in Huntington's disease; trials showed mild stimulatory and inhibitory effects depending on dopaminergic tone levels respectively.
However, a recent phase III trial involving 437 patients failed to demonstrate significant improvements in modified motor score after six months of treatment with doses up to 90 mg/day; however, further analysis suggested potential benefits in UHDRS total motor score as an endpoint. Huntington's disease offers few treatment options for cognitive dysfunction and behavioral abnormalities, and trials of cholinesterase inhibitors have proven unsuccessful in improving cognitive dysfunction. Moreover, addressing the behavioral abnormalities remains challenging.
Therapies designed to lower levels of mutant huntingtin (mHTT) represent one of the most promising approaches for disease modification. These emerging therapies target either DNA or RNA of the mHTT gene; targeting can take the form of antisense oligonucleotides (ASOs), RNA interference (RNAi) or small molecule splicing inhibitors; currently ASOs are being investigated in a human phase 1b/2a study delivered intrathecally that catalyze degradation of HTT mRNA via RNAse H; in animal models this approach resulted in up to an 80% decrease in HTT mRNA levels over time [125].
Attaining lower levels of mHTT may prove particularly effective for Huntington's disease disease modification. One approach aimed at this goal involves targeting either DNA or RNA of the mHTT gene; targeting its expression using ASOs, RNAi or small molecule splicing inhibitors are possible solutions.
RNAi-based approaches use RNA molecules that bind to mRNA in the cytoplasm and prompt its removal by Argonaute 2, an RNAse enzyme found within an RNA-induced silencing complex. While therapeutic strategies using this approach are still in their preclinical stage, one treatment could potentially provide permanent HTT reduction through intracranial injection into the striatum; small molecule splicing modifiers have shown promise in animal models of small muscular atrophy; screening is underway to identify small molecule modulators of mHTT.
Targeting the DNA of mHTT can be done using two approaches: zinc finger proteins and the CRISPR/Cas9 system. Zinc finger proteins are structural motifs that bind directly to DNA; synthetic zinc finger transcription factors targeting CAG have been used successfully to lower levels of mHTT protein in animal models; however, as they create non-native proteins, they could potentially trigger immune reactions and further research is required before conclusively targeting them.
CRISPR/Cas9 is an ingenious immune mechanism employed by bacteria to cleave foreign DNA molecules from their bodies; more recently it has also become a genome editing tool with various applications in human disease treatment. Huntington's disease patients were treated using this technology to excise promoter regions, transcription start site and CAG mutation expansion of mHTT gene found in their fibroblasts - leading to permanent allele-specific inactivation of this gene. This approach has also been successfully tested in an HD rodent model, providing proof of concept. Further preclinical work needs to be completed before CRISPR/Cas9 gene editing technology can reach clinical application. Recently raised concerns over unexpected off-target mutations have necessitated more study [126].
No treatment has yet been shown to stop or slow Huntington's disease from progressing, although various clinical trials have investigated its potential effectiveness; including minocycline, riluzole and remacemide; however none has shown significant effects.
Coenzyme Q10 (CoQ10) at 600 mg/day was observed to slow functional decline, although this did not reach statistical significance. Additional research is evaluating effects of doses up to 2400 mg/day as well as testing on preHD gene carriers. Safety and tolerability data showed that up to 3600 mg/day doses were well tolerated by most study participants without experiencing serious adverse events [127].
Creatine is widely acknowledged for its antioxidant properties and potential to improve mitochondrial function and cell bioenergetics. Unfortunately, an earlier study conducted using 10 g/day did not demonstrate significant improvements in total motor score, functional capacity, neuropsychological testing scores compared with controls. Therefore, phase III studies are now taking place with higher dosages up to 40 g/day as part of CREST-E (Creatine Safety Tolerability Efficacy in Huntington's Disease) [128].
11. Amyotrophic lateral sclerosis (ALS)
Amyotrophic lateral sclerosis is a neurodegenerative disease usually manifesting with an onset of focal muscular weakness and fatigue, having the tendency to spread selectively among upper and lower motor neurons, in some cases dysarthria, dysphonia and dysphagia can also occur [129]. It affects the health state of the patient as it spreads typically from distal muscles to more proximal ones, survival rate being about 2 to 5 years from the date of diagnosis, the cause usually being respiratory failure. ALS can be classified as being familial (10% of the cases) and sporadic (90% of the cases) both of them showing similarities between symptomatology, this shows how important epigenetic studies are in discovering new treatments for ND and how both epigenetics and environmental factors can determine the apparition of ALS and neurodegenerative disorders in general [130]. Various researches have shown that ALS has a slightly higher incidence in the female population and at the same time great geographical discrepancies shows that there is a prevalence in the European region while in Asia and Middle East far less ALS cases have been reported [131,132,133]. In addition it has been proven that environmental exposure to different agents such as infectious agents or heavy metals can lead to genetic alterations and can also facilitate the development of degenerative diseases. The most common genetic genes that cause ALS are C9orf72, SOD1, TARDBP, FUS,TBK1 [129,130]. (Figure 3)
Numerous studies have confirmed to this date that C9orf72 mutations are the most common in familial ALS disease, which only represents 10% of the ALS cases,([134]) compared to other gene mutations that lead to the apparition of this pathology. C9orf72 is a gene that is formed out of 11 exons and it is implicated in splicing mechanisms that produce transcripts and 2 isoforms. The mutation that is widely spread throughout ALS cases especially in Europe and North America is the GGGGCC hexanucleotide expansion, located in the first intron of variants 1 and 3 and in the promoter region of variant 2 [131,133]. More than 22 repeats were found in neurodegenerative disorders and it has been stated that the patients that present the GGGGCC hexanucleotide repeat might share the Finnish founder risk haplotype, statement that can be supported by the fact that numerous studies have shown that the expansion is most common in the Finnish population compared to other regions [135,136]. The mechanisms behind this expansion can be explained by its capacity to be transcribed into repetitive RNA which then forms both sense and antisense ARN foci and 5 dipeptide repeat proteins (DPR) that can explain possible mechanisms of pathology represented by dysfunction of C9orf72 protein or toxic accumulations of either ARN foci, which can sequester RNA binding proteins creating inclusions or DPRs inside the nucleus of the nervous cells [134,137].
Mutations of the SOD1 gene represents one of the most common factors of apparition for familial ALS and represents about 10-20% of FALS (familial ALS) cases, being far less common in sporadic ALS cases. At the moment there are over 155 gene mutations of the SOD1 that can have implications in ND diseases including ALS that can result in different modifications to the SOD1 protein regarding its level, structure or enzymatic activity that can eventually lead to toxic protein accumulations due to misfolding of the SOD1 proteins [138]. It was firstly stated that the SOD1 mutations have implication in the apparition of ALS by a mechanism based on the loss of function of this protein but this hypothesis was soon abandoned after some experimental studies [138,139,140]. The SOD1 protein also known as superoxide dismutase 1, is a very well conserved gene located inside the nucleus, cytoplasm and mitochondria’s membrane and is formed by 5 exons which encode 153 amino acid metalloenzyme, binding Cu and Zn ions, forming a dismutase that removes radicals from the cells and metabolize them into oxygen and hydrogen peroxide.([138]) The most common mutations of SOD1 associated with ALS are usually based on changes of structure of the protein, more exactly the amino acid positions and might include A4V [141], G93A [142], L84F [143,144].
A4V mutation changes the alanine from codon 4 to valine in exon 1 and it is associated with aggressive forms of ALS representing 50% of the SOD1 mutation cases reported in North America [138,145]. G93A is a heavily studied gene mutation of the SOD1 protein that represents a substitution of glycine with alanine from the codon 93 of the SOD1 protein, changing its conformation and also is responsible for approximately 20% of the familial ALS cases. According to a study conducted by the Chinese Pharmaceutical Association in 2023 [146] oxidative stress has a major impact on ALS pathology, patients showing different oxidative markers such as glutamate excitotoxicity and dysfunctions at several levels such as mitochondria, due to calcium influx, and axon as well as protein oxidation, modifications observed at SOD1-G93A mice. This mutation is relatively rare in general population but it is very common in familial ALS and also multiple studies on animal models have shown that having the SOD1-G93A mutation is enough to cause motor-neuron degeneration [146,147,148].Understanding the mechanisms as well as the specific effects of SOD1 mutations on protein structure and functions is an important area of research for developing effective treatments for ALS and neurodegenerative diseases in general.
The L84F mutation changes the amino acid from position 84 from leucine to phenylalanine, being associated with a slightly mild form of ALS and compared to other mutations such as G93A, resulting in both protein instability and misfolding that can lead to forming protein accumulations [143]. According to a study on the population of central Italy [144], the L84F mutation is a rare mutation that has been identified in several families as a result of a common ancestor that has been carrying the mutation. In addition, the article has reported several pathological characteristics of the patients with ALS that had this mutation including young age of onset, a relatively slow progression of the disease as well as the predominance of upper limbs motor neuron involvement, suggesting that the clinical features might be caused by the specific mutation.
Table 6.
Important SOD1 mutations associated with ALS onset.
| Mutation | Pathogenicity | Type of Mutation | Biological Effect | Citation |
| A4V | ALS Pathogenic | Substitution | This mutation is responsible for a rapidly progressive dominant form of amyotrophic lateral sclerosis (ALS) that exclusively affects lower motor neurons, and it accounts for 50% of SOD1 mutations associated with familial ALS in North America. However, it is a rare mutation in Europe. | [141,145] |
| G93A | ALS Pathogenic | Substitution | Patients showing different oxidative markers such as glutamate excitotoxicity and dysfunctions at several levels such as mitochondria, due to calcium influx, and axon as well as protein oxidation, modifications observed at SOD1-G93A mice. This mutation is relatively rare in general population but it is very common in familial ALS and also multiple studies on animal models have shown that having the SOD1-G93A mutation is enough to cause motor-neuron degeneration | [146,147,148] |
| L84F | ALS Pathogenic | Substitution | Protein instability and misfolding that can lead to forming protein accumulations. | [143,144] |
Mutations of the TARDBP gene are associated with neurodegenerative diseases, especially with amyotrophic lateral sclerosis, the gene is encoding TDP-43 protein which is implicated in the apparition of this ND. The exact mechanisms of how this mutations lead to motor neuron degeneration are not fully understood, but one of the reasons might be represented by affecting the structure of the function of TDP-43 that lead to toxic protein aggregates to form [149,150]. According to an article published in nature genetics [151], which investigated the frequency of TARDBP mutations sampling 93 familial ALS cases and 109 sporadic ALS cases, the mutations that were identified were mostly missense mutations, A382T being the most common one found. In addition, regarding the clinical symptoms, the patients found with TARDBP mutation had an early onset age, usually bulbar onset, as well as a shorter survival rate compared to other ALS cases. The TARDBP gene was present in 5% of the familial ALS cases and 1.9% of the sporadic cases. TARDBP mutations interfere with the normal function of TDP-43, leading to abnormal protein clumps in motor neurons that contribute to death and degeneration of these motor neurons. While its exact mechanisms for inducing ALS development remain enigmatic, speculation points toward changes in RNA splicing, protein translation, or degradation processes being involved. TARDBP mutations are an important genetic risk factor for ALS and studying them can provide insights into its mechanisms of development, potentially providing potential therapies targeted specifically towards these mutations [152]. TARDBP gene mutations have long been linked with amyotrophic lateral sclerosis. One of the more often-seen TARDBP mutations linked to ALS is M337V; this mutation can be found in both familial and sporadic cases of the condition. Other TARDBP variants that have also been frequently connected with this form of neurological disease include A315T, G348C and A382T mutations [149,153,154].
The TDP-43 M337V mutation has been associated with familial amyotrophic lateral sclerosis (ALS) in Japan. A study conducted on 41 families living with familial ALS revealed that 11 of those families contained TDP-43 M337V mutations. The TDP-43 M337V mutation results in production of an abnormal TDP-43 protein that aggregates abnormally and accumulates in motor neurons, ultimately leading to their degeneration and death and ultimately contributing to ALS development. It is thought that this accumulation may play a key role in its manifestation [153,154].
The TDP-43 A315T mutation has been linked with familial motor neuron disease. This genetic alteration occurs within the TARDBP gene, which contains instructions for producing TDP-43 protein; mutations disrupting this production can lead to abnormal protein accumulation within motor neurons and eventually death. According to [154] attaining abnormal levels of TDP-43 through mutations like A315T has been linked with degeneration and death of motor neurons, eventually leading to motor neuron diseases such as ALS. Although this mutation only appears in a small percentage of familial motor neuron disease cases, it is a pathogenic one responsible for inducing symptoms in those who carry it. Undergoing research into the TDP-43 A315T mutation and its impact on motor neuron disease could provide key insights into its underlying mechanisms, possible therapeutic targets, and genetic testing may play a pivotal role in identifying individuals carrying this mutation who could be at an increased risk of familial motor neuron disease. Another study shows that the TDP-43 (A315T) transgenic mouse model provides an opportunity to investigate how mutations of TDP-43 gene A315T affect amyotrophic lateral sclerosis and other motor neuron diseases, leading to abnormal accumulations of protein aggregates which eventually cause degeneration and death of motor neurons. It reveals that transgenic mice carrying the A315T mutation of TDP-43 may succumb to early death due to digestive complications before fully manifesting neurological signs associated with ALS, suggesting it also influences their digestive systems and may contribute to their early demise. Although the exact mechanisms underlying gastrointestinal complications remain poorly understood, experts speculate that abnormal TDP-43 protein build-up in intestinal cells may lead to dysfunction and damage. The early death of these transgenic mice highlights the necessity of conducting further studies into the effects of A315T mutation on non-neuronal systems within the body, and possible therapeutic approaches for managing them. TDP-43 (A315T) transgenic mouse model provides an essential way of studying its underlying mechanisms as well as devising potential treatments against it [155].
The TARDBP gene mutation A382T has been associated with an increased susceptibility for motor neuron diseases such as amyotrophic lateral sclerosis. Studies have demonstrated that this mutation results in the production of an abnormal TDP-43 protein that accumulates and damages motor neurons, ultimately leading to their decline and eventual demise. The A382T mutation is considered an uncommon pathogenic mutation found only in a small proportion of familial ALS cases. Further research is necessary to fully understand how this mutation leads to disease development and potential therapeutic approaches that target it. Genetic testing could prove invaluable in identifying individuals carrying this variant who may be at greater risk of ALS and related motor neuron diseases [156,157].
Table 7.
Important TARDBP mutations associated with ALS onset.
| Mutation | Pathogenicity | Type of Mutation | Biological Effect | Citation |
| M337V | ALS Pathogenic | Substitution | production of an abnormal TDP-43 protein that aggregates abnormally and accumulates in motor neurons, ultimately leading to their degeneration and death and ultimately contributing to ALS development | [149,153,154] |
| A315T | ALS Pathogenic | Substitution | Transgenic mice carrying the A315T mutation of TDP-43 may succumb to early death due to digestive complications before fully manifesting neurological signs associated with ALS, suggesting it also influences their digestive systems and may contribute to their early demise. Although the exact mechanisms underlying gastrointestinal complications remain poorly understood, experts speculate that abnormal TDP-43 protein build-up in intestinal cells may lead to dysfunction and damage | [155] |
| A382T | Possible ALS Pathogenic | Substitution | Unknown Mechanism | [156,157] |
The Fused in Sarcoma (FUS) gene represents another common gene that can cause ALS apparition and encodes a protein involved in controlling RNA processing and transport within cells, with mutations of this gene having been linked with neurodegenerative diseases. Mutations that alter FUS protein accumulation in neurons may result in their dysfunction and apoptosis. Both missense and truncating mutations of FUS may alter protein function and disease development. Some mutations are inherited in an autosomal dominant manner while others may occur spontaneously. Further research must be conducted in order to understand how FUS mutations work and develop therapeutic approaches; genetic testing could help identify individuals at increased risk of FUS-related diseases [158,159].
At amyotrophic lateral sclerosis (ALS), researchers have discovered several mutations of the FUS gene that may contribute to its progression. R521C, R521H and P525L mutations are among the most frequently encountered, occurring in approximately 5-10% of familial cases and smaller proportions of sporadic cases of ALS respectively.
Mutations result in production of mutated FUS protein that accumulates in motor neuron cytoplasm, eventually leading to their degeneration and death. While the exact mechanisms underlying ALS development due to such mutations remain enigmatic, experts speculate that they disrupt RNA metabolism and transport processes that are vitally important in maintaining proper motor nerve functionality [158].
According to a study which investigates the prevalence and clinical features of FUS mutations among Italian patients with familial ALS. From 54 cases studied, 2 individuals (3.7%) carried FUS R521C mutations that showed early disease onset as well as more severe disease progression than patients without this mutation; suggesting FUS mutations may play an integral role in familial ALS prevalence among Italian populations. This suggests FUS mutations may play a vital part in creating familial ALS within Italy's population [160,161].
The TBK1 gene is also implicated in ALS apparition and plays an essential role in regulating various cellular processes, such as autophagy, inflammation and immune response. Mutations have been identified as one cause of ALS and FTD; such mutations result in abnormal protein aggregate accumulations within motor neurons causing degeneration and death; interference with regular cellular processes may play a part in contributing to this condition and contributing to its progression. Mutations in TBK1 have been identified as an uncommon cause of ALS and FTD. These mutations cause abnormal protein aggregates to build up in motor neurons, eventually leading to their degeneration and death - possibly interfering with normal cellular processes that contribute to disease development in this way, contributing further to ALS or FTD development [162,163]. Mutations in the TBK1 gene have been identified in patients suffering from amyotrophic lateral sclerosis (ALS), including missense, frameshift and truncating mutations. Some of the more prevalent mutations include the frameshift mutation TBK1 p.Ile383Thrfs17 which results in truncated protein expression; missense mutations such as p.Arg357Ser and p.Gly290Val have also been found; these latter two also appear familial cases as these affect highly conserved amino acids found within its protein. Additionally, missense mutations impairing its ability to bind its target proteins allowing greater protein expression from within.
These mutations are thought to disrupt TBK1's role in regulating various cellular processes, such as autophagy and immune responses, potentially contributing to motor neuron degeneration in ALS patients [163,164].
According to [163] various studies have demonstrated TBK1 gene mutations among familial and sporadic cases of ALS, suggesting TBK1 dysfunction may play an integral part in its progression. Furthermore, this piece delves into potential pathways through which these mutations could contribute to ALS progression, such as disrupted autophagy, neuroinflammation and mitochondrial malfunction.
This article highlights the various functions of TBK1 in terms of its involvement with innate immunity, autophagy, protein aggregate clearance and neuroinflammation - processes which have been implicated as causal in ALS. Studies have identified mutations of the TBK1 gene among ALS patients indicating its involvement. Furthermore, dysfunction caused by mutations may contribute to neuroinflammation which results in protein aggregate accumulation as well as motor neuron death resulting in the progression of the condition.
This article suggests that TBK1 could be an effective therapeutic target for treating ALS, specifically by increasing autophagy and decreasing neuroinflammation. Furthermore, further research needs to be conducted in order to fully comprehend how this gene contributes to ALS as well as creating effective treatments that target it [163,164,165].
In recent years, epigenetic studies have led to major discoveries regarding neurodegenerative diseases and appears to be the key element in finding a treatment that can cure the condition rather than just alleviate it. The term epigenetic is referred to as any genetic alterations occurred at any molecular level associated with several factors such as environmental agents or stress, that do not implicate the modification of the DNA directly [166]. The most common mechanisms regarding epigenetics are DNA methylation, miRNA’s and PTM (post-translational modification) of histones [167].
ALS is a disease characterized by the loss of neuromuscular junctions also being associated with apoptosis of UMN (upper motor neuron) and LMN (lower motor neuron) cells, as well as the surrounding astrocytes and microglial cells and with inclusions in which the main protein involved is TDP-43, a protein that can cause protein aggregation by its mislocalization between the nucleus and the cytoplasm [168]. In addition there are multiple pathological causes of ALS, the most relevant ones being failure of protein degradation, changes in RNA metabolism and axonal transport.
The failure of protein degradation can be the leading cause implicated in the formation of protein aggregates that can disturb the homeostasis of the cells and it is also one of the similarities between neurodegenerative diseases. Protein chaperones can aid the cells to the point of saturation by refolding the misfolded protein that can explain the formation of protein aggregates, but if the chaperones are overloaded, ubiquitin inclusions are formed inside the nucleus. The genes that lead to protein accumulation relating to ALS pathologies are UBQLN2 which is associated with the formation of the inclusions, SQSTM1, TBK1, VCP and C9orf72 protein which interact with the inclusions previously formed [130,169,170,171].
Another mechanism that contributes to the pathogenesis of ALS is abnormal RNA metabolism which includes abnormalities in RNA processing and miRNA expression that lead to misfolding proteins and formation of aggregates. RNA processing includes many mechanisms such as transcription, splicing, editing, transport, and degradation. The key elements implicated in the deviations are RNA-binding proteins such as TDP-43 and FUS, but there are additional targeted proteins that can present mutations such as ANG, STX, ATXN2, MATR3, hnRNPA1, hnRNPA2B1 which demonstrate that any mutations occurred at this level have an important role in ALS but we cannot state that these mutations lead to the apparition of ALS [172].
Specific gene mutations that may contribute to ALS are PFN1, SOD1 and TUBA4A and by the mutations of C-terminus of kinesin-1,the mechanism implicated being related to affecting the integrity of the cytoarchitecture and the transport throughout the axon by destabilizing the tubulin networks which may also affect the molecules carried inside the axon. The binding of the molecules transported inside are stabilized by DCTN1, with the mention that mutations at this level are not very common and do not play an important role in familial ALS apparition but they rather contribute to diagnosing several ND diseases especially in late-onset Parkinson, ALS and FTD [130,173,174].
The different motor phenotypes of ALS can be classified based on whether upper or lower motor neurons are affected, and which regions they impact. Each subtype may present with differing life expectancies; some may even present with cognitive and behavioral deficits. Classic ALS is the most prevalent subtype, marked by signs of both upper and lower motor neuron loss in various body areas. By contrast, primary lateral sclerosis (PLS) typically presents as progressive spasticity with slowing movements as well as isolated upper motor neuron signs. PLS patients exhibit no muscle atrophy, visible fasciculations, or denervation on EMG four years post symptom onset. Their median survival is over 20 years while PLS can transition into ALS three to four years after disease onset. A subtype called UMN-predominant ALS shows some LMN involvement but less prominently than classic ALS; patients in this subtype progress more slowly but live shorter lives than PLS patients [175,176].
There are multiple motor phenotypes of ALS, defined by their degree of involvement with upper versus lower motor neurons and regional distribution of symptoms. Understanding each subtype's life expectancy and degree of cognitive and behavioral impairment is imperative.
One form of ALS, called LMN predominant ALS, involves limited UMN involvement with variable rates of progression. PMA (Progressive muscular atrophy), on the other hand, involves progressive isolated LMN signs without evidence of UMN dysfunction; up to 30% of PMA patients may develop these signs during follow-up.
Bulbar ALS is a devastating form of the disease characterized by rapid decline and an average lifespan of only two years from disease onset. It manifests as spastic dysarthria due to bulbar UMN dysfunction and tongue wasting and fasciculation due to bulbar LMN dysfunction; while only 30% initially exhibit bulbar symptoms initially, most ultimately experience difficulty speaking or swallowing due to this form of the condition.
Pseudobulbar Palsy (PBP) is another subtype, distinguished by absent facial expressions, spastic dysarthria, difficulty chewing, dysphagia, tongue protrusion due to spasticity but no fasciculation or wasting. This disorder originates in the upper motor neuron (UMN), distinguishing it from progressive bulbar Palsy which only affects LMN but may not be universally recognized [177,178].
12. Perspectives for Treatment
The complexity of ALS has been demonstrated by its failure in over 40 randomized-controlled trials that attempted to find effective disease-modifying medications . Riluzole is currently approved in most European countries as the sole disease-modifying medication; administered twice daily at 50 mg dose, its antiglutamatergic properties increase mean patient survival by approximately six months. The most frequently experienced side effects include nausea, diarrhea, fatigue dizziness and liver issues.
More recently, edaravone (a free radical scavenger) has been studied in ALS patients. A Phase III, double-blind study administering 60 mg/day intravenously over 2 weeks per month of intravenous edaravone treatment resulted in significantly less decline in scores on the revised ALS Functional Rating Scale (ALSFRS-R). After six months, significantly fewer scores had decreased [179,180,181].
Masitinib, an oral tyrosine kinase inhibitor, is currently under study as a possible treatment for ALS. A randomized controlled trial administered masitinib as add-on therapy with riluzole at 4.5 mg/kg/day showed positive effects in decreasing ALSFRS-R scores - especially among those experiencing typical disease progression . Its effectiveness will further be examined in a confirmatory study [182].
Honokiol was found to provide neuroprotective benefits by mitigating oxidative stress and improving mitochondrial function in affected neurons, while researchers also observed increased expression of key antioxidant enzymes while reduced expression of pro-inflammatory cytokines in these neurons treated with honokiol.
Overall, studies suggest that honokiol could serve as an effective therapy to treat ALS by decreasing oxidative stress and improving mitochondrial function in affected neurons. More research should be conducted in order to confirm these results as well as determine optimal dosing and treatment regimens for honokiol in ALS patients [146].
13. Discussions
Over the last five years, tremendous advances have been made in understanding both the pathophysiology and genetic basis of Alzheimer's disease. Revamp of amyloid b cascade hypothesis and greater insight into Alzheimer's disease preclinical phases has enabled improved comprehension. Genetic investigations have progressed from identifying three causal and one risk gene to discovering multiple genetic markers that can be used to create a polygenic risk score for Alzheimer's disease. Biomarker diagnosis has fundamentally transformed how Alzheimer's disease is classified, making it possible to enroll patients earlier into studies when blood biomarkers become accessible. Molecular imaging will enhance diagnostic categorization and pathophysiology of any disease by providing visual evidence of copathology or regional protein aggregated deposits. At this rate, insights into risk reduction, primary and secondary prevention, non-pharmacological and pharmacological treatments could become available earlier and in parallel than ever before; the early identification and multimodal treatment of patients could soon become a reality [183].
Parkinson's disease has been recognized for over 200 years and presents serious healthcare challenges worldwide. However, Parkinson's is treatable when interventions are tailored specifically for each patient by trained healthcare providers. Here we highlight several promising advancements in Parkinson's research and treatments which offer hope that services will continue to evolve to make an impactful difference to those living with the disorder worldwide [184].
Understanding of HD has advanced substantially due to advances in genetic technology and large cohorts of individuals living with HD. Thus, new genetic modifiers of the disease have been identified. Somatic instability of CAG repeats is most prevalent in tissues most susceptible to HD pathology, and its degree is negatively correlated with age at disease onset. DNA repair components involved with mismatch repair may act to control somatic instability and disease course. MutSb's attempts at repair may induce loop-outs within CAG tracts targeted by MutSb, leading to potentially significant expansion. Reducing MSH3, PMS2, and LIG1 pro-instability factors or hindering their function is thought to reduce somatic instability while being well-tolerated. FAN1 expression decreases somatic instability while postponing disease onset; its upregulation could serve as protection from HD. Modulating these DNA repair components may also reduce instability in other pathogenic repeat sequences, suggesting these potential therapeutic options could also prove effective against repeat expansion diseases. Additionally, mHTT sequesters components of the NPC in aggregates, disrupting nucleocytoplasmic transport. Modulating nuclear transport pathways has proven protective in cell models of HD, opening new avenues for therapeutic intervention. Cerebrospinal Fluid (CSF) offers an ideal source of CNS proteins for clinical trials due to its accessibility throughout. Notably, neurofilament light chain (NfL) is one of the proteins released into CSF and plasma after neuronal damage to be used as biomarkers or surrogate endpoints for clinical trials; its levels seem to correlate strongly with disease progression; thus its levels could serve both functions simultaneously. Furthermore, mHTT could also be released by damaged neurons, with CSF concentration changes representing early changes to be found premanifest HD [111,185].
Recent years have witnessed an increasing focus on developing precision medicine approaches for ALS subtypes with known genetic causes. One promising therapeutic avenue involves antisense oligonucleotides (ASOs), short sequences of nucleotides that can modulate gene expression and splicing. ASOs have proven effective against preclinical models of ALS characterized by SOD1 mutations and C9orf72 repeat expansions; clinical studies utilizing intrathecal administration of ASOs targeting these genes are currently ongoing. Stem cell therapies such as Granulocyte Colony Stimulating Factor (G-CSF) are being explored as possible therapies for ALS. Peripheral blood stem cells, bone marrow mesenchymal stem cells and non-neural progenitor cells induced by granulocyte-colony stimulating factor have all shown safety and tolerability when administered to ALS patients; however, their efficacy against disease progression remains undetermined. Current Phase II and III clinical trials offer hope that more precise classification of ALS cases based on pathogenic mechanisms will lead to targeted therapies with favorable results in particular ALS subgroups, eventually making ALS manageable disease [186,187].
Disclaimer
The authors declare no conflict of interests.
Abbreviations
| ND | Neurodegenerative Disease |
| AD | Alzheimer’s disease |
| APP | Amyloid precursor protein |
| Aβ | Amyloid beta peptide |
| HCHWA-D | Hereditary cerebral hemorrhage with amyloidosis |
| PiD | Pick's disease |
| GFAP | Glial fibrillary acidic protein |
| SPECT | Single-photon emission computed tomography |
| PET | Positron emission tomography |
| APOE | Apolipoprotein E |
| BBB | Brain Blood Barrier |
| HDL | High density lipoprotein |
| CNS | Central Nervous System |
| NFTs | Neural Focal Thresholds |
| GWAS | Genome-wide association study |
| PP2A | Protein phosphatase 2A |
| FTD | Frontotemporal Dementia |
| CTD | Chronic traumatic encephalopathy |
| CBD | Corticobasal degeneration |
| DAM | Disease-associated microglia |
| MGnD | Microglial neurodegenerative |
| TREM2 | Triggering receptor expressed on myeloid cells 2 |
| CRP | C-reactive protein |
| MCI | Mild cognitive impairment |
| MAPT | Microtubule-associated protein tau |
| PSP | Progressive supranuclear palsy |
| PHF | Abnormally hyperphosphorylated taus |
| CL | Centiloids |
| SUVR | Standardized Uptake Ration |
| PD | Parkinson's disease |
| SNpc | Substantia nigra pars compacta |
| DA | Dopamine |
| DMV | Dorsal motor nucleus of vagus |
| OB | Olfactory bulb |
| LC | Locus coeruleus |
| IML | Intermediolateral nucleus in spinal cord |
| ENS | Enteric nervous system |
| EOPD | Early-onset Parkinson's disease |
| mitoQC | Mitochondrial quality control |
| DFO | Deferrioxamine |
| DFP | Deferiprone |
| HD | Huntington’s disease |
| MSNs | Medium spiny neurons |
| mHTT | Mutation of the huntingtin gene |
| HTT | Huntingtin gene |
| CBP | cAMP response element-binding protein |
| MSK1 | Mitogen-activated and stress-activated protein kinase 1 |
| PGC-1a | Proliferator-activated receptor gamma coactivator alpha |
| HDAC | Histone deacetylase |
| BDNF | Brain-derived neurotrophic factor |
| ASOs | Antisense oligonucleotides |
| RNAi | RNA interference |
| CoQ10 | Coenzyme Q10 |
| CREST-E | Creatine safety tolerability efficacy in Huntington's disease |
| ALS | Amyotrophic lateral sclerosis |
| FALS | Familial amyotrophic lateral sclerosis |
| DPR | Dipeptide repeat proteins |
| FUS | Fused in Sarcoma |
| miRNA | Micro RNA |
| PTM | Post-translational modification |
| UMN | Upper motor neuron |
| LMN | Lower motor neuron |
| PMA | Progressive muscular atrophy |
| PBP | Pseudobulbar Palsy |
References
- Berry, R. M. The Genetic Revolution and the Physician’s Duty of Confidentiality: The Role of the Old Hippocratic Virtues in the Regulation of the New Genetic Intimacy. J. Leg. Med. 1997, 18 (4), 401–441. [CrossRef]
- De Castro, M. Johann Gregor Mendel: Paragon of Experimental Science. Mol. Genet. Genomic Med. 2016, 4 (1), 3–8. [CrossRef]
- Saceleanu, V. M.; Mohan, A. G.; Covache-Busuioc, R. A.; Costin, H. P.; Ciurea, A. V. Wilhelm von Waldeyer: Important Steps in Neural Theory, Anatomy and Citology. Brain Sci. 2022, 12 (2), 224. [CrossRef]
- Kovacs, G. G. Concepts and Classification of Neurodegenerative Diseases. In Handbook of Clinical Neurology; Elsevier, 2018; Vol. 145, pp 301–307. [CrossRef]
- Brettschneider, J.; Tredici, K. D.; Lee, V. M.-Y.; Trojanowski, J. Q. Spreading of Pathology in Neurodegenerative Diseases: A Focus on Human Studies. Nat. Rev. Neurosci. 2015, 16 (2), 109–120. [CrossRef]
- Guo, J. L.; Lee, V. M. Y. Cell-to-Cell Transmission of Pathogenic Proteins in Neurodegenerative Diseases. Nat. Med. 2014, 20 (2), 130–138. [CrossRef]
- Castellani, R. J.; Rolston, R. K.; Smith, M. A. Alzheimer Disease. Dis. Mon. 2010, 56 (9), 484–546. [CrossRef]
- Ciurea, V. A.; Neurosurgery Department, Carol Davila University of Medicine and Pharmacy, Bucharest, Romania; Neurosurgery Department, Sanador Clinical Hospital, Bucharest, Romania; Covache-Busuioc, R.-A.; Carol Davila University of Medicine and Pharmacy, Bucharest, Romania; Mohan, A. G.; Department of Neurosurgery, Bihor County Emergency Clinical Hospital, Oradea, Romania; Neurosurgery Department, Faculty of Medicine, Oradea University, Oradea, Romania; Costin, H. P.; Carol Davila University of Medicine and Pharmacy, Bucharest, Romania; Voicu, V.; Pharmacology, Toxicology and Clinical Psychopharmacology Department, Carol Davila University of Medicine and Pharmacy, Bucharest, Romania; Romanian Academy, Bucharest, Romania. Alzheimer’s Disease: 120 Years of Research and Progress. J. Med. Life 2023, 16 (2), 173–177. [CrossRef]
- Gatz, M.; Reynolds, C. A.; Fratiglioni, L.; Johansson, B.; Mortimer, J. A.; Berg, S.; Fiske, A.; Pedersen, N. L. Role of Genes and Environments for Explaining Alzheimer Disease. Arch. Gen. Psychiatry 2006, 63 (2), 168. [CrossRef]
- An, F.; Zhao, R.; Xuan, X.; Xuan, T.; Zhang, G.; Wei, C. Calycosin Ameliorates Advanced Glycation End Product-Induced Neurodegenerative Changes in Cellular and Rat Models of Diabetes-Related Alzheimer’s Disease. Chem. Biol. Interact. 2022, 368, 110206. [CrossRef]
- Van Cauwenberghe, C.; Van Broeckhoven, C.; Sleegers, K. The Genetic Landscape of Alzheimer Disease: Clinical Implications and Perspectives. Genet. Med. 2016, 18 (5), 421–430. [CrossRef]
- Roberts, S. B.; Ripellino, J. A.; Ingalls, K. M.; Robakis, N. K.; Felsenstein, K. M. Non-Amyloidogenic Cleavage of the Beta-Amyloid Precursor Protein by an Integral Membrane Metalloendopeptidase. J. Biol. Chem. 1994, 269 (4), 3111–3116. [CrossRef]
- Wu, Q.; Sun, J.-X.; Song, X.-H.; Wang, J.; Xiong, C.-Q.; Teng, F.-X.; Gao, C.-X. Blocking Beta 2-Adrenergic Receptor Inhibits Dendrite Ramification in a Mouse Model of Alzheimer’s Disease. Neural Regen. Res. 2017, 12 (9), 1499–1506. [CrossRef]
- Okochi, M.; Tagami, S.; Yanagida, K.; Takami, M.; Kodama, T. S.; Mori, K.; Nakayama, T.; Ihara, Y.; Takeda, M. γ-Secretase Modulators and Presenilin 1 Mutants Act Differently on Presenilin/γ-Secretase Function to Cleave Aβ42 and Aβ43. Cell Rep. 2013, 3 (1), 42–51. [CrossRef]
- Hampel, H.; Hu, Y.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S. H.; Villemagne, V. L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; Masters, C. L.; Cho, M.; Lannfelt, L.; Cummings, J. L.; Vergallo, A. The Amyloid-β Pathway in Alzheimer’s Disease: A Plain Language Summary. Neurodegener. Dis. Manag. 2023, nmt-2022-0037. [CrossRef]
- Tosh, J. L.; Rhymes, E. R.; Mumford, P.; Whittaker, H. T.; Pulford, L. J.; Noy, S. J.; Cleverley, K.; LonDownS Consortium; Strydom, A.; Fisher, E.; Wiseman, F.; Nizetic, D.; Hardy, J.; Tybulewicz, V.; Karmiloff-Smith, A.; Walker, M. C.; Tybulewicz, V. L. J.; Wykes, R. C.; Fisher, E. M. C.; Wiseman, F. K. Genetic Dissection of down Syndrome-Associated Alterations in APP/Amyloid-β Biology Using Mouse Models. Sci. Rep. 2021, 11 (1), 5736. [CrossRef]
- Jonsson, T.; Atwal, J. K.; Steinberg, S.; Snaedal, J.; Jonsson, P. V.; Bjornsson, S.; Stefansson, H.; Sulem, P.; Gudbjartsson, D.; Maloney, J.; Hoyte, K.; Gustafson, A.; Liu, Y.; Lu, Y.; Bhangale, T.; Graham, R. R.; Huttenlocher, J.; Bjornsdottir, G.; Andreassen, O. A.; Jönsson, E. G.; Palotie, A.; Behrens, T. W.; Magnusson, O. T.; Kong, A.; Thorsteinsdottir, U.; Watts, R. J.; Stefansson, K. A Mutation in APP Protects against Alzheimer’s Disease and Age-Related Cognitive Decline. Nature 2012, 488 (7409), 96–99. [CrossRef]
- Benilova, I.; Gallardo, R.; Ungureanu, A.-A.; Castillo Cano, V.; Snellinx, A.; Ramakers, M.; Bartic, C.; Rousseau, F.; Schymkowitz, J.; De Strooper, B. The Alzheimer Disease Protective Mutation A2T Modulates Kinetic and Thermodynamic Properties of Amyloid-β (Aβ) Aggregation. J. Biol. Chem. 2014, 289 (45), 30977–30989. [CrossRef]
- Liu, Y.-W.; He, Y.-H.; Zhang, Y.-X.; Cai, W.-W.; Yang, L.-Q.; Xu, L.-Y.; Kong, Q.-P. Absence of A673T Variant in APP Gene Indicates an Alternative Protective Mechanism Contributing to Longevity in Chinese Individuals. Neurobiol. Aging 2014, 35 (4), 935.e11-935.e12. [CrossRef]
- Mengel-From, J.; Jeune, B.; Pentti, T.; McGue, M.; Christensen, K.; Christiansen, L. The APP A673T Frequency Differs between Nordic Countries. Neurobiol. Aging 2015, 36 (10), 2909.e1-2909.e4. [CrossRef]
- Giaccone, G.; Morbin, M.; Moda, F.; Botta, M.; Mazzoleni, G.; Uggetti, A.; Catania, M.; Moro, M. L.; Redaelli, V.; Spagnoli, A.; Rossi, R. S.; Salmona, M.; Di Fede, G.; Tagliavini, F. Neuropathology of the Recessive A673V APP Mutation: Alzheimer Disease with Distinctive Features. Acta Neuropathol. (Berl.) 2010, 120 (6), 803–812. [CrossRef]
- Villemagne, V. L.; Ataka, S.; Mizuno, T.; Brooks, W. S.; Wada, Y.; Kondo, M.; Jones, G.; Watanabe, Y.; Mulligan, R.; Nakagawa, M.; Miki, T.; Shimada, H.; O’Keefe, G. J.; Masters, C. L.; Mori, H.; Rowe, C. C. High Striatal Amyloid β-Peptide Deposition Across Different Autosomal Alzheimer Disease Mutation Types. Arch. Neurol. 2009, 66 (12). [CrossRef]
- Readhead, B.; Haure-Mirande, J.-V.; Zhang, B.; Haroutunian, V.; Gandy, S.; Schadt, E. E.; Dudley, J. T.; Ehrlich, M. E. Molecular Systems Evaluation of Oligomerogenic APPE693Q and Fibrillogenic APPKM670/671NL/PSEN1Δexon9 Mouse Models Identifies Shared Features with Human Alzheimer’s Brain Molecular Pathology. Mol. Psychiatry 2016, 21 (8), 1099–1111. [CrossRef]
- Natté, R.; Maat-Schieman, M. L. C.; Haan, J.; Bornebroek, M.; Roos, R. A. C.; Van Duinen, S. G. Dementia in Hereditary Cerebral Hemorrhage with Amyloidosis-Dutch Type Is Associated with Cerebral Amyloid Angiopathy but Is Independent of Plaques and Neurofibrillary Tangles: Dementia in HCHWA-D Is Associated with CAA. Ann. Neurol. 2001, 50 (6), 765–772. [CrossRef]
- Nishitsuji, K.; Tomiyama, T.; Ishibashi, K.; Kametani, F.; Ozawa, K.; Okada, R.; Maat-Schieman, M. L.; Roos, R. A. C.; Iwai, K.; Mori, H. Cerebral Vascular Accumulation of Dutch-Type Aβ42, but Not Wild-Type Aβ42, in Hereditary Cerebral Hemorrhage with Amyloidosis, Dutch Type. J. Neurosci. Res. 2007, 85 (13), 2917–2923. [CrossRef]
- Uddin, Md. S.; Tewari, D.; Sharma, G.; Kabir, Md. T.; Barreto, G. E.; Bin-Jumah, M. N.; Perveen, A.; Abdel-Daim, M. M.; Ashraf, G. M. Molecular Mechanisms of ER Stress and UPR in the Pathogenesis of Alzheimer’s Disease. Mol. Neurobiol. 2020, 57 (7), 2902–2919. [CrossRef]
- Alberdi, E.; Wyssenbach, A.; Alberdi, M.; Sánchez-Gómez, M. V.; Cavaliere, F.; Rodríguez, J. J.; Verkhratsky, A.; Matute, C. Ca 2+ -Dependent Endoplasmic Reticulum Stress Correlates with Astrogliosis in Oligomeric Amyloid β-Treated Astrocytes and in a Model of Alzheimer’s Disease. Aging Cell 2013, 12 (2), 292–302. [CrossRef]
- Tomiyama, T.; Nagata, T.; Shimada, H.; Teraoka, R.; Fukushima, A.; Kanemitsu, H.; Takuma, H.; Kuwano, R.; Imagawa, M.; Ataka, S.; Wada, Y.; Yoshioka, E.; Nishizaki, T.; Watanabe, Y.; Mori, H. A New Amyloid β Variant Favoring Oligomerization in Alzheimer’s-Type Dementia. Ann. Neurol. 2008, 63 (3), 377–387. [CrossRef]
- McKnelly, K. J.; Kreutzer, A. G.; Howitz, W. J.; Haduong, K.; Yoo, S.; Hart, C.; Nowick, J. S. Effects of Familial Alzheimer’s Disease Mutations on the Assembly of a β-Hairpin Peptide Derived from Aβ 16–36. Biochemistry 2022, 61 (6), 446–454. [CrossRef]
- Paik, Y. K.; Chang, D. J.; Reardon, C. A.; Walker, M. D.; Taxman, E.; Taylor, J. M. Identification and Characterization of Transcriptional Regulatory Regions Associated with Expression of the Human Apolipoprotein E Gene. J. Biol. Chem. 1988, 263 (26), 13340–13349.
- Maloney, B.; Ge, Y.-W.; Alley, G. M.; Lahiri, D. K. Important Differences between Human and Mouse APOE Gene Promoters: Limitation of Mouse APOE Model in Studying Alzheimer’s Disease. J. Neurochem. 2007, 103 (3), 1237–1257. [CrossRef]
- Yee, A.; Tsui, N. B. Y.; Kwan, R. Y. C.; Leung, A. Y. M.; Lai, C. K. Y.; Chung, T.; Lau, J. Y. N.; Fok, M.; Dai, D. L. K.; Lau, L.-T. Apolipoprotein E Gene Revisited: Contribution of Rare Variants to Alzheimer’s Disease Susceptibility in Southern Chinese. Curr. Alzheimer Res. 2021, 18 (1), 67–79. [CrossRef]
- Zhou, Y.; Mägi, R.; Milani, L.; Lauschke, V. M. Global Genetic Diversity of Human Apolipoproteins and Effects on Cardiovascular Disease Risk. J. Lipid Res. 2018, 59 (10), 1987–2000. [CrossRef]
- Saunders, A. M. Gene Identification in Alzheimer’s Disease. Pharmacogenomics 2001, 2 (3), 239–249. [CrossRef]
- Theuns, J. Genetic Variability in the Regulatory Region of Presenilin 1 Associated with Risk for Alzheimer’s Disease and Variable Expression. Hum. Mol. Genet. 2000, 9 (3), 325–331. [CrossRef]
- Cruts, M.; Theuns, J.; Van Broeckhoven, C. Locus-specific Mutation Databases for Neurodegenerative Brain Diseases. Hum. Mutat. 2012, 33 (9), 1340–1344. [CrossRef]
- Sun, L.; Zhou, R.; Yang, G.; Shi, Y. Analysis of 138 Pathogenic Mutations in Presenilin-1 on the in Vitro Production of Aβ42 and Aβ40 Peptides by γ-Secretase. Proc. Natl. Acad. Sci. 2017, 114 (4). [CrossRef]
- Wines-Samuelson, M.; Schulte, E. C.; Smith, M. J.; Aoki, C.; Liu, X.; Kelleher, R. J.; Shen, J. Characterization of Age-Dependent and Progressive Cortical Neuronal Degeneration in Presenilin Conditional Mutant Mice. PLoS ONE 2010, 5 (4), e10195. [CrossRef]
- Kauwe, J. S. K.; Jacquart, S.; Chakraverty, S.; Wang, J.; Mayo, K.; Fagan, A. M.; Holtzman, D. M.; Morris, J. C.; Goate, A. M. Extreme Cerebrospinal Fluid Amyloid β Levels Identify Family with Late-Onset Alzheimer’s Disease Presenilin 1 Mutation. Ann. Neurol. 2007, 61 (5), 446–453. [CrossRef]
- Kumar-Singh, S.; Theuns, J.; Van Broeck, B.; Pirici, D.; Vennekens, K.; Corsmit, E.; Cruts, M.; Dermaut, B.; Wang, R.; Van Broeckhoven, C. Mean Age-of-Onset of Familial Alzheimer Disease Caused by Presenilin Mutations Correlates with Both Increased Aβ42 and Decreased Aβ40. Hum. Mutat. 2006, 27 (7), 686–695. [CrossRef]
- Koriath, C.; Kenny, J.; Adamson, G.; Druyeh, R.; Taylor, W.; Beck, J.; Quinn, L.; Mok, T. H.; Dimitriadis, A.; Norsworthy, P.; Bass, N.; Carter, J.; Walker, Z.; Kipps, C.; Coulthard, E.; Polke, J. M.; Bernal-Quiros, M.; Denning, N.; Thomas, R.; Raybould, R.; Williams, J.; Mummery, C. J.; Wild, E. J.; Houlden, H.; Tabrizi, S. J.; Rossor, M. N.; Hummerich, H.; Warren, J. D.; Rowe, J. B.; Rohrer, J. D.; Schott, J. M.; Fox, N. C.; Collinge, J.; Mead, S. Predictors for a Dementia Gene Mutation Based on Gene-Panel next-Generation Sequencing of a Large Dementia Referral Series. Mol. Psychiatry 2020, 25 (12), 3399–3412. [CrossRef]
- Gallo, M.; Frangipane, F.; Cupidi, C.; De Bartolo, M.; Turone, S.; Ferrari, C.; Nacmias, B.; Grimaldi, G.; Laganà, V.; Colao, R.; Bernardi, L.; Anfossi, M.; Conidi, M. E.; Vasso, F.; Curcio, S. A. M.; Mirabelli, M.; Smirne, N.; Torchia, G.; Muraca, M. G.; Puccio, G.; Di Lorenzo, R.; Piccininni, M.; Tedde, A.; Maletta, R. G.; Sorbi, S.; Bruni, A. C. The Novel PSEN1 M84V Mutation Associated to Frontal Dysexecutive Syndrome, Spastic Paraparesis, and Cerebellar Atrophy in a Dominant Alzheimer’s Disease Family. Neurobiol. Aging 2017, 56, 213.e7-213.e12. [CrossRef]
- Dominantly Inherited Alzheimer Network (DIAN); Hsu, S.; Gordon, B. A.; Hornbeck, R.; Norton, J. B.; Levitch, D.; Louden, A.; Ziegemeier, E.; Laforce, R.; Chhatwal, J.; Day, G. S.; McDade, E.; Morris, J. C.; Fagan, A. M.; Benzinger, T. L. S.; Goate, A. M.; Cruchaga, C.; Bateman, R. J.; Karch, C. M. Discovery and Validation of Autosomal Dominant Alzheimer’s Disease Mutations. Alzheimers Res. Ther. 2018, 10 (1), 67. [CrossRef]
- Kakuda, N.; Takami, M.; Okochi, M.; Kasuga, K.; Ihara, Y.; Ikeuchi, T. Switched Aβ43 Generation in Familial Alzheimer’s Disease with Presenilin 1 Mutation. Transl. Psychiatry 2021, 11 (1), 558. [CrossRef]
- López-García, S.; Jiménez-Bonilla, J.; López Delgado, A.; Orizaola Balaguer, P.; Infante Ceberio, J.; Banzo Marraco, I.; Rodríguez Rodríguez, E.; Sánchez-Juan, P. A Rare PSEN1 (Leu85Pro) Mutation Causing Alzheimer’s Disease in a 29-Year-Old Woman Presenting as Corticobasal Syndrome. J. Alzheimers Dis. JAD 2019, 70 (3), 655–658. [CrossRef]
- Arber, C.; Villegas-Llerena, C.; Toombs, J.; Pocock, J. M.; Ryan, N. S.; Fox, N. C.; Zetterberg, H.; Hardy, J.; Wray, S. Amyloid Precursor Protein Processing in Human Neurons with an Allelic Series of the PSEN1 Intron 4 Deletion Mutation and Total Presenilin-1 Knockout. Brain Commun. 2019, 1 (1), fcz024. [CrossRef]
- De Jonghe, C.; MarcCruts; Rogaeva, E. A.; Tysoe, C.; Singleton, A.; Vanderstichele, H.; Meschino, W.; Dermaut, B.; Vanderhoeven, I.; Backhovens, H.; Vanmechelen, E.; Morris, C. M.; Hardy, J.; Rubinsztein, D. C.; St George-Hyslop, P. H.; Van Broeckhoven, C. Aberrant Splicing in the Presenilin-1 Intron 4 Mutation Causes Presenile Alzheimer’s Disease by Increased A 42 Secretion. Hum. Mol. Genet. 1999, 8 (8), 1529–1540. [CrossRef]
- Szaruga, M.; Veugelen, S.; Benurwar, M.; Lismont, S.; Sepulveda-Falla, D.; Lleo, A.; Ryan, N. S.; Lashley, T.; Fox, N. C.; Murayama, S.; Gijsen, H.; De Strooper, B.; Chávez-Gutiérrez, L. Qualitative Changes in Human γ-Secretase Underlie Familial Alzheimer’s Disease. J. Exp. Med. 2015, 212 (12), 2003–2013. [CrossRef]
- Fox, N. Clinicopathological Features of Familial Alzheimer’s Disease Associated with the M139V Mutation in the Presenilin 1 Gene. Pedigree but Not Mutation Specific Age at Onset Provides Evidence for a Further Genetic Factor. Brain 1997, 120 (3), 491–501. [CrossRef]
- Chávez-Gutiérrez, L.; Bammens, L.; Benilova, I.; Vandersteen, A.; Benurwar, M.; Borgers, M.; Lismont, S.; Zhou, L.; Van Cleynenbreugel, S.; Esselmann, H.; Wiltfang, J.; Serneels, L.; Karran, E.; Gijsen, H.; Schymkowitz, J.; Rousseau, F.; Broersen, K.; De Strooper, B. The Mechanism of γ-Secretase Dysfunction in Familial Alzheimer Disease: Mechanisms of Alzheimer Disease-Causing Mutations. EMBO J. 2012, 31 (10), 2261–2274. [CrossRef]
- Ringman, J. M.; Gylys, K. H.; Medina, L. D.; Fox, M.; Kepe, V.; Flores, D. L.; Apostolova, L. G.; Barrio, J. R.; Small, G.; Silverman, D. H.; Siu, E.; Cederbaum, S.; Hecimovic, S.; Malnar, M.; Chakraverty, S.; Goate, A. M.; Bird, T. D.; Leverenz, J. B. Biochemical, Neuropathological, and Neuroimaging Characteristics of Early-Onset Alzheimer’s Disease Due to a Novel PSEN1 Mutation. Neurosci. Lett. 2011, 487 (3), 287–292. [CrossRef]
- Devi, G.; Fotiou, A.; Jyrinji, D.; Tycko, B.; DeArmand, S.; Rogaeva, E.; Song, Y.-Q.; Medieros, H.; Liang, Y.; Orlacchio, A.; Williamson, J.; St George-Hyslop, P.; Mayeux, R. Novel Presenilin 1 Mutations Associated With Early Onset of Dementia in a Family With Both Early-Onset and Late-Onset Alzheimer Disease. Arch. Neurol. 2000, 57 (10). [CrossRef]
- Levy-Lahad, E.; Wasco, W.; Poorkaj, P.; Romano, D. M.; Oshima, J.; Pettingell, W. H.; Yu, C.; Jondro, P. D.; Schmidt, S. D.; Wang, K.; Crowley, A. C.; Fu, Y.-H.; Guenette, S. Y.; Galas, D.; Nemens, E.; Wijsman, E. M.; Bird, T. D.; Schellenberg, G. D.; Tanzi, R. E. Candidate Gene for the Chromosome 1 Familial Alzheimer’s Disease Locus. Science 1995, 269 (5226), 973–977. [CrossRef]
- Perrone, F.; Cacace, R.; Van Mossevelde, S.; Van Den Bossche, T.; De Deyn, P. P.; Cras, P.; Engelborghs, S.; Van Der Zee, J.; Van Broeckhoven, C. Genetic Screening in Early-Onset Dementia Patients with Unclear Phenotype: Relevance for Clinical Diagnosis. Neurobiol. Aging 2018, 69, 292.e7-292.e14. [CrossRef]
- Jia, L.; Fu, Y.; Shen, L.; Zhang, H.; Zhu, M.; Qiu, Q.; Wang, Q.; Yan, X.; Kong, C.; Hao, J.; Wei, C.; Tang, Y.; Qin, W.; Li, Y.; Wang, F.; Guo, D.; Zhou, A.; Zuo, X.; Yu, Y.; Li, D.; Zhao, L.; Jin, H.; Jia, J. PSEN1, PSEN2, and APP Mutations in 404 Chinese Pedigrees with Familial Alzheimer’s Disease. Alzheimers Dement. J. Alzheimers Assoc. 2020, 16 (1), 178–191. [CrossRef]
- Pang, Y.; Li, T.; Wang, Q.; Qin, W.; Li, Y.; Wei, Y.; Jia, L. A Rare Variation in the 3’ Untranslated Region of the Presenilin 2 Gene Is Linked to Alzheimer’s Disease. Mol. Neurobiol. 2021, 58 (9), 4337–4347. [CrossRef]
- The collaborators of the GMAJ project; Wallon, D.; Rousseau, S.; Rovelet-Lecrux, A.; Quillard-Muraine, M.; Guyant-Maréchal, L.; Martinaud, O.; Pariente, J.; Puel, M.; Rollin-Sillaire, A.; Pasquier, F.; Le Ber, I.; Sarazin, M.; Croisile, B.; Boutoleau-Bretonnière, C.; Thomas-Antérion, C.; Paquet, C.; Moreaud, O.; Gabelle, A.; Sellal, F.; Sauvée, M.; Laquerrière, A.; Duyckaerts, C.; Delisle, M.-B.; Streichenberger, N.; Lannes, B.; Frebourg, T.; Hannequin, D.; Campion, D. The French Series of Autosomal Dominant Early Onset Alzheimer’s Disease Cases: Mutation Spectrum and Cerebrospinal Fluid Biomarkers. J. Alzheimers Dis. 2012, 30 (4), 847–856. [CrossRef]
- Marcon, G.; Giaccone, G.; Cupidi, C.; Balestrieri, M.; Beltrami, C. A.; Finato, N.; Bergonzi, P.; Sorbi, S.; Bugiani, O.; Tagliavini, F. Neuropathological and Clinical Phenotype of an Italian Alzheimer Family with M239V Mutation of Presenilin 2 Gene. J. Neuropathol. Exp. Neurol. 2004, 63 (3), 199–209. [CrossRef]
- Huang, Y.; Weisgraber, K. H.; Mucke, L.; Mahley, R. W. Apolipoprotein E: Diversity of Cellular Origins, Structural and Biophysical Properties, and Effects in Alzheimer’s Disease. J. Mol. Neurosci. 2004, 23 (3), 189–204. [CrossRef]
- Liu, C.-C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer Disease: Risk, Mechanisms and Therapy. Nat. Rev. Neurol. 2013, 9 (2), 106–118. [CrossRef]
- Kara, E.; Marks, J. D.; Fan, Z.; Klickstein, J. A.; Roe, A. D.; Krogh, K. A.; Wegmann, S.; Maesako, M.; Luo, C. C.; Mylvaganam, R.; Berezovska, O.; Hudry, E.; Hyman, B. T. Isoform- and Cell Type-Specific Structure of Apolipoprotein E Lipoparticles as Revealed by a Novel Forster Resonance Energy Transfer Assay. J. Biol. Chem. 2017, 292 (36), 14720–14729. [CrossRef]
- Lin, Y.-T.; Seo, J.; Gao, F.; Feldman, H. M.; Wen, H.-L.; Penney, J.; Cam, H. P.; Gjoneska, E.; Raja, W. K.; Cheng, J.; Rueda, R.; Kritskiy, O.; Abdurrob, F.; Peng, Z.; Milo, B.; Yu, C. J.; Elmsaouri, S.; Dey, D.; Ko, T.; Yankner, B. A.; Tsai, L.-H. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human IPSC-Derived Brain Cell Types. Neuron 2018, 98 (6), 1141-1154.e7. [CrossRef]
- Kang, S. S.; Ebbert, M. T. W.; Baker, K. E.; Cook, C.; Wang, X.; Sens, J. P.; Kocher, J.-P.; Petrucelli, L.; Fryer, J. D. Microglial Translational Profiling Reveals a Convergent APOE Pathway from Aging, Amyloid, and Tau. J. Exp. Med. 2018, 215 (9), 2235–2245. [CrossRef]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J. Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; Martorell, A. J.; Ransohoff, R. M.; Hafler, B. P.; Bennett, D. A.; Kellis, M.; Tsai, L.-H. Single-Cell Transcriptomic Analysis of Alzheimer’s Disease. Nature 2019, 570 (7761), 332–337. [CrossRef]
- Therriault, J.; Benedet, A. L.; Pascoal, T. A.; Mathotaarachchi, S.; Chamoun, M.; Savard, M.; Thomas, E.; Kang, M. S.; Lussier, F.; Tissot, C.; Parsons, M.; Qureshi, M. N. I.; Vitali, P.; Massarweh, G.; Soucy, J.-P.; Rej, S.; Saha-Chaudhuri, P.; Gauthier, S.; Rosa-Neto, P. Association of Apolipoprotein E Ε4 With Medial Temporal Tau Independent of Amyloid-β. JAMA Neurol. 2020, 77 (4), 470. [CrossRef]
- Alzheimer’s Disease Neuroimaging Initiative; Shi, Y.; Yamada, K.; Liddelow, S. A.; Smith, S. T.; Zhao, L.; Luo, W.; Tsai, R. M.; Spina, S.; Grinberg, L. T.; Rojas, J. C.; Gallardo, G.; Wang, K.; Roh, J.; Robinson, G.; Finn, M. B.; Jiang, H.; Sullivan, P. M.; Baufeld, C.; Wood, M. W.; Sutphen, C.; McCue, L.; Xiong, C.; Del-Aguila, J. L.; Morris, J. C.; Cruchaga, C.; Fagan, A. M.; Miller, B. L.; Boxer, A. L.; Seeley, W. W.; Butovsky, O.; Barres, B. A.; Paul, S. M.; Holtzman, D. M. ApoE4 Markedly Exacerbates Tau-Mediated Neurodegeneration in a Mouse Model of Tauopathy. Nature 2017, 549 (7673), 523–527. [CrossRef]
- Harris, F. M.; Brecht, W. J.; Xu, Q.; Mahley, R. W.; Huang, Y. Increased Tau Phosphorylation in Apolipoprotein E4 Transgenic Mice Is Associated with Activation of Extracellular Signal-Regulated Kinase. J. Biol. Chem. 2004, 279 (43), 44795–44801. [CrossRef]
- Wadhwani, A. R.; Affaneh, A.; Van Gulden, S.; Kessler, J. A. Neuronal Apolipoprotein E4 Increases Cell Death and Phosphorylated Tau Release in Alzheimer Disease. Ann. Neurol. 2019, 85 (5), 726–739. [CrossRef]
- Gibb, G. M.; Pearce, J.; Betts, J. C.; Lovestone, S.; Hoffmann, M. M.; Maerz, W.; Blackstock, W. P.; Anderton, B. H. Differential Effects of Apolipoprotein E Isoforms on Phosphorylation at Specific Sites on Tau by Glycogen Synthase Kinase-3β Identified by Nano-Electrospray Mass Spectrometry. FEBS Lett. 2000, 485 (2–3), 99–103. [CrossRef]
- Park, J.-S.; Ji, I. J.; An, H. J.; Kang, M.-J.; Kang, S.-W.; Kim, D.-H.; Yoon, S.-Y. Disease-Associated Mutations of TREM2 Alter the Processing of N-Linked Oligosaccharides in the Golgi Apparatus: Impaired Trafficking of TREM2. Traffic 2015, 16 (5), 510–518. [CrossRef]
- Saceleanu, V. M.; Covache-Busuioc, R.-A.; Costin, H.-P.; Glavan, L.-A.; Ciurea, A. V. An Important Step in Neuroscience: Camillo Golgi and His Discoveries. Cells 2022, 11 (24), 4112. [CrossRef]
- Ulrich, J. D.; Ulland, T. K.; Mahan, T. E.; Nyström, S.; Nilsson, K. P.; Song, W. M.; Zhou, Y.; Reinartz, M.; Choi, S.; Jiang, H.; Stewart, F. R.; Anderson, E.; Wang, Y.; Colonna, M.; Holtzman, D. M. ApoE Facilitates the Microglial Response to Amyloid Plaque Pathology. J. Exp. Med. 2018, 215 (4), 1047–1058. [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T. K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; Itzkovitz, S.; Colonna, M.; Schwartz, M.; Amit, I. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169 (7), 1276-1290.e17. [CrossRef]
- Hutton, M.; Lendon, C. L.; Rizzu, P.; Baker, M.; Froelich, S.; Houlden, H.; Pickering-Brown, S.; Chakraverty, S.; Isaacs, A.; Grover, A.; Hackett, J.; Adamson, J.; Lincoln, S.; Dickson, D.; Davies, P.; Petersen, R. C.; Stevens, M.; De Graaff, E.; Wauters, E.; Van Baren, J.; Hillebrand, M.; Joosse, M.; Kwon, J. M.; Nowotny, P.; Che, L. K.; Norton, J.; Morris, J. C.; Reed, L. A.; Trojanowski, J.; Basun, H.; Lannfelt, L.; Neystat, M.; Fahn, S.; Dark, F.; Tannenberg, T.; Dodd, P. R.; Hayward, N.; Kwok, J. B. J.; Schofield, P. R.; Andreadis, A.; Snowden, J.; Craufurd, D.; Neary, D.; Owen, F.; Oostra, B. A.; Hardy, J.; Goate, A.; Van Swieten, J.; Mann, D.; Lynch, T.; Heutink, P. Association of Missense and 5′-Splice-Site Mutations in Tau with the Inherited Dementia FTDP-17. Nature 1998, 393 (6686), 702–705. [CrossRef]
- Spillantini, M. G.; Bird, T. D.; Ghetti, B. Frontotemporal Dementia and Parkinsonism Linked to Chromosome 17: A New Group of Tauopathies. Brain Pathol. 2006, 8 (2), 387–402. [CrossRef]
- Poorkaj, P.; Bird, T. D.; Wijsman, E.; Nemens, E.; Garruto, R. M.; Anderson, L.; Andreadis, A.; Wiederholt, W. C.; Raskind, M.; Schellenberg, G. D. Tau Is a Candidate Gene for Chromosome 17 Frontotemporal Dementia. Ann. Neurol. 1998, 43 (6), 815–825. [CrossRef]
- Goedert, M.; Spillantini, M. G.; Jakes, R.; Rutherford, D.; Crowther, R. A. Multiple Isoforms of Human Microtubule-Associated Protein Tau: Sequences and Localization in Neurofibrillary Tangles of Alzheimer’s Disease. Neuron 1989, 3 (4), 519–526. [CrossRef]
- Baudier, J.; Cole, R. D. Interactions between the Microtubule-Associated Tau Proteins and S100b Regulate Tau Phosphorylation by the Ca2+/Calmodulin-Dependent Protein Kinase II. J. Biol. Chem. 1988, 263 (12), 5876–5883.
- Drewes, G.; Ebneth, A.; Preuss, U.; Mandelkow, E.-M.; Mandelkow, E. MARK, a Novel Family of Protein Kinases That Phosphorylate Microtubule-Associated Proteins and Trigger Microtubule Disruption. Cell 1997, 89 (2), 297–308. [CrossRef]
- Drewes, G.; Lichtenberg-Kraag, B.; Döring, F.; Mandelkow, E. M.; Biernat, J.; Goris, J.; Dorée, M.; Mandelkow, E. Mitogen Activated Protein (MAP) Kinase Transforms Tau Protein into an Alzheimer-like State. EMBO J. 1992, 11 (6), 2131–2138. [CrossRef]
- Bancher, C.; Brunner, C.; Lassmann, H.; Budka, H.; Jellinger, K.; Wiche, G.; Seitelberger, F.; Grundke-Iqbal, I.; Iqbal, K.; Wisniewski, H. M. Accumulation of Abnormally Phosphorylated τ Precedes the Formation of Neurofibrillary Tangles in Alzheimer’s Disease. Brain Res. 1989, 477 (1–2), 90–99. [CrossRef]
- Wang, J.; Wu, Q.; Smith, A.; Grundke-Iqbal, I.; Iqbal, K. τ Is Phosphorylated by GSK-3 at Several Sites Found in Alzheimer Disease and Its Biological Activity Markedly Inhibited Only after It Is Prephosphorylated by A-Kinase. FEBS Lett. 1998, 436 (1), 28–34. [CrossRef]
- Yasuda, M.; Takamatsu, J.; D’Souza, I.; Crowther, R. A.; Kawamata, T.; Hasegawa, M.; Hasegawa, H.; Spillantini, M. G.; Tanimukai, S.; Poorkaj, P.; Varani, L.; Varani, G.; Iwatsubo, T.; Goedert, M.; Schellenberg, D. G.; Tanaka, C. A Novel Mutation at Position +12 in the Intron Following Exon 10 of the Tau Gene in Familial Frontotemporal Dementia (FTD-Kumamoto). Ann. Neurol. 2000, 47 (4), 422–429.
- Takamatsu, J.; Kondo, A.; Ikegami, K.; Kimura, T.; Fujii, H.; Mitsuyama, Y.; Hashizume, Y. Selective Expression of Ser 199/202 Phosphorylated Tau in a Case of Frontotemporal Dementia. Dement. Geriatr. Cogn. Disord. 1998, 9 (2), 82–89. [CrossRef]
- Labbé, C.; Ogaki, K.; Lorenzo-Betancor, O.; Soto-Ortolaza, A. I.; Walton, R. L.; Rayaprolu, S.; Fujioka, S.; Murray, M. E.; Heckman, M. G.; Puschmann, A.; McCarthy, A.; Lynch, T.; Siuda, J.; Opala, G.; Rudzinska, M.; Krygowska-Wajs, A.; Barcikowska, M.; Czyzewski, K.; Sanotsky, Y.; Rektorová, I.; McLean, P. J.; Rademakers, R.; Ertekin-Taner, N.; Hassan, A.; Ahlskog, J. E.; Boeve, B. F.; Petersen, R. C.; Maraganore, D. M.; Adler, C. H.; Ferman, T. J.; Parisi, J. E.; Graff-Radford, N. R.; Uitti, R. J.; Wszolek, Z. K.; Dickson, D. W.; Ross, O. A. Role for the Microtubule-Associated Protein Tau Variant p.A152T in Risk of α-Synucleinopathies. Neurology 2015, 85 (19), 1680–1686. [CrossRef]
- Jin, S.; Pastor, P.; Cooper, B.; Cervantes, S.; Benitez, B. A.; Razquin, C.; Goate, A.; Ibero-American Alzheimer Disease Genetics Group Researchers; Cruchaga, C. Pooled-DNA Sequencing Identifies Novel Causative Variants in PSEN1, GRN and MAPT in a Clinical Early-Onset and Familial Alzheimer’s Disease Ibero-American Cohort. Alzheimers Res. Ther. 2012, 4 (4), 34. [CrossRef]
- Coppola, G.; Chinnathambi, S.; Lee, J. J.; Dombroski, B. A.; Baker, M. C.; Soto-Ortolaza, A. I.; Lee, S. E.; Klein, E.; Huang, A. Y.; Sears, R.; Lane, J. R.; Karydas, A. M.; Kenet, R. O.; Biernat, J.; Wang, L.-S.; Cotman, C. W.; DeCarli, C. S.; Levey, A. I.; Ringman, J. M.; Mendez, M. F.; Chui, H. C.; Le Ber, I.; Brice, A.; Lupton, M. K.; Preza, E.; Lovestone, S.; Powell, J.; Graff-Radford, N.; Petersen, R. C.; Boeve, B. F.; Lippa, C. F.; Bigio, E. H.; Mackenzie, I.; Finger, E.; Kertesz, A.; Caselli, R. J.; Gearing, M.; Juncos, J. L.; Ghetti, B.; Spina, S.; Bordelon, Y. M.; Tourtellotte, W. W.; Frosch, M. P.; Vonsattel, J. P. G.; Zarow, C.; Beach, T. G.; Albin, R. L.; Lieberman, A. P.; Lee, V. M.; Trojanowski, J. Q.; Van Deerlin, V. M.; Bird, T. D.; Galasko, D. R.; Masliah, E.; White, C. L.; Troncoso, J. C.; Hannequin, D.; Boxer, A. L.; Geschwind, M. D.; Kumar, S.; Mandelkow, E.-M.; Wszolek, Z. K.; Uitti, R. J.; Dickson, D. W.; Haines, J. L.; Mayeux, R.; Pericak-Vance, M. A.; Farrer, L. A.; Ross, O. A.; Rademakers, R.; Schellenberg, G. D.; Miller, B. L.; Mandelkow, E.; Geschwind, D. H. Evidence for a Role of the Rare p.A152T Variant in MAPT in Increasing the Risk for FTD-Spectrum and Alzheimer’s Diseases. Hum. Mol. Genet. 2012, 21 (15), 3500–3512. [CrossRef]
- Rizzini, C.; Goedert, M.; Hodges, J. R.; Smith, M. J.; Jakes, R.; Hills, R.; Xuereb, J. H.; Crowther, R. A.; Spillantini, M. G. Tau Gene Mutation K257T Causes a Tauopathy Similar to Pick’s Disease. J. Neuropathol. Exp. Neurol. 2000, 59 (11), 990–1001. [CrossRef]
- Kobayashi, T.; Ota, S.; Tanaka, K.; Ito, Y.; Hasegawa, M.; Umeda, Y.; Motoi, Y.; Takanashi, M.; Yasuhara, M.; Anno, M.; Mizuno, Y.; Mori, H. A Novel L266V Mutation of the Tau Gene Causes Frontotemporal Dementia with a Unique Tau Pathology. Ann. Neurol. 2003, 53 (1), 133–137. [CrossRef]
- Hogg, M.; Grujic, Z. M.; Baker, M.; Demirci, S.; Guillozet, A. L.; Sweet, A. P.; Herzog, L. L.; Weintraub, S.; Mesulam, M.-M.; LaPointe, N. E.; Gamblin, T. C.; Berry, R. W.; Binder, L. I.; De Silva, R.; Lees, A.; Espinoza, M.; Davies, P.; Grover, A.; Sahara, N.; Ishizawa, T.; Dickson, D.; Yen, S.-H.; Hutton, M.; Bigio, E. H. The L266V Tau Mutation Is Associated with Frontotemporal Dementia and Pick-like 3R and 4R Tauopathy. Acta Neuropathol. (Berl.) 2003, 106 (4), 323–336. [CrossRef]
- Schilling, S.; Zeitschel, U.; Hoffmann, T.; Heiser, U.; Francke, M.; Kehlen, A.; Holzer, M.; Hutter-Paier, B.; Prokesch, M.; Windisch, M.; Jagla, W.; Schlenzig, D.; Lindner, C.; Rudolph, T.; Reuter, G.; Cynis, H.; Montag, D.; Demuth, H.-U.; Rossner, S. Glutaminyl Cyclase Inhibition Attenuates Pyroglutamate Aβ and Alzheimer’s Disease–like Pathology. Nat. Med. 2008, 14 (10), 1106–1111. [CrossRef]
- Frost, J. L.; Liu, B.; Kleinschmidt, M.; Schilling, S.; Demuth, H.-U.; Lemere, C. A. Passive Immunization against Pyroglutamate-3 Amyloid-β Reduces Plaque Burden in Alzheimer-Like Transgenic Mice: A Pilot Study. Neurodegener. Dis. 2012, 10 (1–4), 265–270. [CrossRef]
- Frost, J. L.; Liu, B.; Rahfeld, J.-U.; Kleinschmidt, M.; O’Nuallain, B.; Le, K. X.; Lues, I.; Caldarone, B. J.; Schilling, S.; Demuth, H.-U.; Lemere, C. A. An Anti-Pyroglutamate-3 Aβ Vaccine Reduces Plaques and Improves Cognition in APPswe/PS1ΔE9 Mice. Neurobiol. Aging 2015, 36 (12), 3187–3199. [CrossRef]
- Hoffmann, T.; Meyer, A.; Heiser, U.; Kurat, S.; Böhme, L.; Kleinschmidt, M.; Bühring, K.-U.; Hutter-Paier, B.; Farcher, M.; Demuth, H.-U.; Lues, I.; Schilling, S. Glutaminyl Cyclase Inhibitor PQ912 Improves Cognition in Mouse Models of Alzheimer’s Disease—Studies on Relation to Effective Target Occupancy. J. Pharmacol. Exp. Ther. 2017, 362 (1), 119–130. [CrossRef]
- DeMattos, R. B.; Lu, J.; Tang, Y.; Racke, M. M.; DeLong, C. A.; Tzaferis, J. A.; Hole, J. T.; Forster, B. M.; McDonnell, P. C.; Liu, F.; Kinley, R. D.; Jordan, W. H.; Hutton, M. L. A Plaque-Specific Antibody Clears Existing β-Amyloid Plaques in Alzheimer’s Disease Mice. Neuron 2012, 76 (5), 908–920. [CrossRef]
- Lowe, S. L.; Willis, B. A.; Hawdon, A.; Natanegara, F.; Chua, L.; Foster, J.; Shcherbinin, S.; Ardayfio, P.; Sims, J. R. Donanemab (LY3002813) Dose-escalation Study in Alzheimer’s Disease. Alzheimers Dement. Transl. Res. Clin. Interv. 2021, 7 (1). [CrossRef]
- Lowe, S. L.; Duggan Evans, C.; Shcherbinin, S.; Cheng, Y.-J.; Willis, B. A.; Gueorguieva, I.; Lo, A. C.; Fleisher, A. S.; Dage, J. L.; Ardayfio, P.; Aguiar, G.; Ishibai, M.; Takaichi, G.; Chua, L.; Mullins, G.; Sims, J. R. Donanemab (LY3002813) Phase 1b Study in Alzheimer’s Disease: Rapid and Sustained Reduction of Brain Amyloid Measured by Florbetapir F18 Imaging. J. Prev. Alzheimers Dis. 2021, 1–11. [CrossRef]
- Shcherbinin, S.; Evans, C. D.; Lu, M.; Andersen, S. W.; Pontecorvo, M. J.; Willis, B. A.; Gueorguieva, I.; Hauck, P. M.; Brooks, D. A.; Mintun, M. A.; Sims, J. R. Association of Amyloid Reduction After Donanemab Treatment With Tau Pathology and Clinical Outcomes: The TRAILBLAZER-ALZ Randomized Clinical Trial. JAMA Neurol. 2022, 79 (10), 1015. [CrossRef]
- Raza, C.; Anjum, R.; Shakeel, N. U. A. Parkinson’s Disease: Mechanisms, Translational Models and Management Strategies. Life Sci. 2019, 226, 77–90. [CrossRef]
- Beitz, J. M. Parkinson s Disease a Review. Front. Biosci. 2014, S6 (1), 65–74. [CrossRef]
- Day, J. O.; Mullin, S. The Genetics of Parkinson’s Disease and Implications for Clinical Practice. Genes 2021, 12 (7), 1006. [CrossRef]
- Ye, H.; Robak, L. A.; Yu, M.; Cykowski, M.; Shulman, J. M. Genetics and Pathogenesis of Parkinson’s Syndrome. Annu. Rev. Pathol. Mech. Dis. 2023, 18 (1), 95–121. [CrossRef]
- Siddiqui, I. J.; Pervaiz, N.; Abbasi, A. A. The Parkinson Disease Gene SNCA: Evolutionary and Structural Insights with Pathological Implication. Sci. Rep. 2016, 6 (1), 24475. [CrossRef]
- Rui, Q.; Ni, H.; Li, D.; Gao, R.; Chen, G. The Role of LRRK2 in Neurodegeneration of Parkinson Disease. Curr. Neuropharmacol. 2018, 16 (9), 1348–1357. [CrossRef]
- Vizziello, M.; Borellini, L.; Franco, G.; Ardolino, G. Disruption of Mitochondrial Homeostasis: The Role of PINK1 in Parkinson’s Disease. Cells 2021, 10 (11), 3022. [CrossRef]
- Crichton, R. R.; Dexter, D. T.; Ward, R. J. Brain Iron Metabolism and Its Perturbation in Neurological Diseases. In Metal Ions in Neurological Systems; Linert, W., Kozlowski, H., Eds.; Springer Vienna: Vienna, 2012; pp 1–15. [CrossRef]
- Chandra, G.; Shenoi, R. A.; Anand, R.; Rajamma, U.; Mohanakumar, K. P. Reinforcing Mitochondrial Functions in Aging Brain: An Insight into Parkinson’s Disease Therapeutics. J. Chem. Neuroanat. 2019, 95, 29–42. [CrossRef]
- C Borgna-Pignatti; S Rugolotto; P De Stefano; H Zhao; MD Cappellini; GC Del Vecchio; MA Romeo; GL Forni; MR Gamberini; R Ghilardi; A Piga; A Cnaan. Survival and Complications in Patients with Thalassemia Major Treated with Transfusion and Deferoxamine. Haematologica 2004, 89 (10), 1187–1193.
- Boddaert, N.; Le Quan Sang, K. H.; Rötig, A.; Leroy-Willig, A.; Gallet, S.; Brunelle, F.; Sidi, D.; Thalabard, J.-C.; Munnich, A.; Cabantchik, Z. I. Selective Iron Chelation in Friedreich Ataxia: Biologic and Clinical Implications. Blood 2007, 110 (1), 401–408. [CrossRef]
- Devos, D.; Moreau, C.; Devedjian, J. C.; Kluza, J.; Petrault, M.; Laloux, C.; Jonneaux, A.; Ryckewaert, G.; Garçon, G.; Rouaix, N.; Duhamel, A.; Jissendi, P.; Dujardin, K.; Auger, F.; Ravasi, L.; Hopes, L.; Grolez, G.; Firdaus, W.; Sablonnière, B.; Strubi-Vuillaume, I.; Zahr, N.; Destée, A.; Corvol, J.-C.; Pöltl, D.; Leist, M.; Rose, C.; Defebvre, L.; Marchetti, P.; Cabantchik, Z. I.; Bordet, R. Targeting Chelatable Iron as a Therapeutic Modality in Parkinson’s Disease. Antioxid. Redox Signal. 2014, 21 (2), 195–210. [CrossRef]
- McColgan, P.; Tabrizi, S. J. Huntington’s Disease: A Clinical Review. Eur. J. Neurol. 2018, 25 (1), 24–34. [CrossRef]
- Wexler, A.; Wild, E. J.; Tabrizi, S. J. George Huntington: A Legacy of Inquiry, Empathy and Hope. Brain 2016, 139 (8), 2326–2333. [CrossRef]
- Bates, G. P.; Dorsey, R.; Gusella, J. F.; Hayden, M. R.; Kay, C.; Leavitt, B. R.; Nance, M.; Ross, C. A.; Scahill, R. I.; Wetzel, R.; Wild, E. J.; Tabrizi, S. J. Huntington Disease. Nat. Rev. Dis. Primer 2015, 1 (1), 15005. [CrossRef]
- Macdonald, M. A Novel Gene Containing a Trinucleotide Repeat That Is Expanded and Unstable on Huntington’s Disease Chromosomes. Cell 1993, 72 (6), 971–983. [CrossRef]
- Squitieri, F.; Andrew, S. E.; Goldberg, Y. P.; Kremer, B.; Spence, N.; Zelsler, J.; Nichol, K.; Theilmann, J.; Greenberg, J.; Goto, J.; Kanazawa, I.; Vesa, J.; Peltonen, L.; Almqvist, E.; Anvret, M.; Telenius, H.; Lin, B.; Napolitano, G.; Morgan, K.; Hayden, M. R. DNA Haplotype Analysis of Huntington Disease Reveals Clues to the Origins and Mechanisms of CAG Expansion and Reasons for Geographic Variations of Prevalence. Hum. Mol. Genet. 1994, 3 (12), 2103–2114. [CrossRef]
- Persichetti, F.; Srinidhi, J.; Kanaley, L.; Ge, P.; Myers, R. H.; D’Arrigo, K.; Barnes, G. T.; MacDonald, M. E.; Vonsattel, J.-P.; Gusella, J. F.; Bird, E. D. Huntington’s Disease CAG Trinucleotide Repeats in Pathologically Confirmed Post-Mortem Brains. Neurobiol. Dis. 1994, 1 (3), 159–166. [CrossRef]
- Gusella, J. F.; MacDonald, M. E.; Lee, J.-M. Genetic Modifiers of Huntington’s Disease: GENETIC MODIFIERS OF HD. Mov. Disord. 2014, 29 (11), 1359–1365. [CrossRef]
- Lee, J.-M.; Gillis, T.; Mysore, J. S.; Ramos, E. M.; Myers, R. H.; Hayden, M. R.; Morrison, P. J.; Nance, M.; Ross, C. A.; Margolis, R. L.; Squitieri, F.; Griguoli, A.; Di Donato, S.; Gomez-Tortosa, E.; Ayuso, C.; Suchowersky, O.; Trent, R. J.; McCusker, E.; Novelletto, A.; Frontali, M.; Jones, R.; Ashizawa, T.; Frank, S.; Saint-Hilaire, M.-H.; Hersch, S. M.; Rosas, H. D.; Lucente, D.; Harrison, M. B.; Zanko, A.; Abramson, R. K.; Marder, K.; Sequeiros, J.; MacDonald, M. E.; Gusella, J. F. Common SNP-Based Haplotype Analysis of the 4p16.3 Huntington Disease Gene Region. Am. J. Hum. Genet. 2012, 90 (3), 434–444. [CrossRef]
- Landles, C.; Sathasivam, K.; Weiss, A.; Woodman, B.; Moffitt, H.; Finkbeiner, S.; Sun, B.; Gafni, J.; Ellerby, L. M.; Trottier, Y.; Richards, W. G.; Osmand, A.; Paganetti, P.; Bates, G. P. Proteolysis of Mutant Huntingtin Produces an Exon 1 Fragment That Accumulates as an Aggregated Protein in Neuronal Nuclei in Huntington Disease. J. Biol. Chem. 2010, 285 (12), 8808–8823. [CrossRef]
- Ha, A. D.; Fung, V. S. C. Huntingtonʼs Disease: Curr. Opin. Neurol. 2012, 25 (4), 491–498. [CrossRef]
- Ross, C. A.; Tabrizi, S. J. Huntington’s Disease: From Molecular Pathogenesis to Clinical Treatment. Lancet Neurol. 2011, 10 (1), 83–98. [CrossRef]
- G. Vonsattel, J. P.; DiFiglia, M. Huntington Disease: J. Neuropathol. Exp. Neurol. 1998, 57 (5), 369–384. [CrossRef]
- Moss, D. J. H.; Pardiñas, A. F.; Langbehn, D.; Lo, K.; Leavitt, B. R.; Roos, R.; Durr, A.; Mead, S.; Holmans, P.; Jones, L.; Tabrizi, S. J.; Coleman, A.; Santos, R. D.; Decolongon, J.; Sturrock, A.; Bardinet, E.; Ret, C. J.; Justo, D.; Lehericy, S.; Marelli, C.; Nigaud, K.; Valabrègue, R.; Van Den Bogaard, S.; Dumas, E. M.; Van Der Grond, J.; t’Hart, E.; Jurgens, C.; Witjes-Ane, M.-N.; Arran, N.; Callaghan, J.; Stopford, C.; Frost, C.; Jones, R.; Hobbs, N.; Lahiri, N.; Ordidge, R.; Owen, G.; Pepple, T.; Read, J.; Say, M.; Wild, E.; Patel, A.; Fox, N. C.; Gibbard, C.; Malone, I.; Crawford, H.; Whitehead, D.; Keenan, S.; Cash, D. M.; Berna, C.; Bechtel, N.; Bohlen, S.; Man, A. H.; Kraus, P.; Axelson, E.; Wang, C.; Acharya, T.; Lee, S.; Monaco, W.; Campbell, C.; Queller, S.; Whitlock, K.; Campbell, C.; Campbell, M.; Frajman, E.; Milchman, C.; O’Regan, A.; Labuschagne, I.; Stout, J.; Landwehrmeyer, B.; Craufurd, D.; Scahill, R.; Hicks, S.; Kennard, C.; Johnson, H.; Tobin, A.; Rosas, H.; Reilmann, R.; Borowsky, B.; Pourchot, C.; Andrews, S. C.; Bachoud-Lévi, A.-C.; Bentivoglio, A. R.; Biunno, I.; Bonelli, R.; Burgunder, J.-M.; Dunnett, S.; Ferreira, J.; Handley, O.; Heiberg, A.; Illmann, T.; Landwehrmeyer, G. B.; Levey, J.; Ramos-Arroyo, M. A.; Nielsen, J.; Koivisto, S. P.; Päivärinta, M.; Roos, R. A. C.; Sebastián, A. R.; Tabrizi, S.; Vandenberghe, W.; Verellen-Dumoulin, C.; Uhrova, T.; Wahlström, J.; Zaremba, J.; Baake, V.; Barth, K.; Garde, M. B.; Betz, S.; Bos, R.; Callaghan, J.; Come, A.; Guedes, L. C.; Ecker, D.; Finisterra, A. M.; Fullam, R.; Gilling, M.; Gustafsson, L.; Handley, O. J.; Hvalstedt, C.; Held, C.; Koppers, K.; Lamanna, C.; Laurà, M.; Descals, A. M.; Martinez-Horta, S.; Mestre, T.; Minster, S.; Monza, D.; Mütze, L.; Oehmen, M.; Orth, M.; Padieu, H.; Paterski, L.; Peppa, N.; Koivisto, S. P.; Di Renzo, M.; Rialland, A.; Røren, N.; Šašinková, P.; Timewell, E.; Townhill, J.; Cubillo, P. T.; Da Silva, W. V.; Van Walsem, M. R.; Whalstedt, C.; Witjes-Ané, M.-N.; Witkowski, G.; Wright, A.; Zielonka, D.; Zielonka, E.; Zinzi, P.; Bonelli, R. M.; Lilek, S.; Hecht, K.; Herranhof, B.; Holl, A.; Kapfhammer, H.-P.; Koppitz, M.; Magnet, M.; Müller, N.; Otti, D.; Painold, A.; Reisinger, K.; Scheibl, M.; Schöggl, H.; Ullah, J.; Braunwarth, E.-M.; Brugger, F.; Buratti, L.; Hametner, E.-M.; Hepperger, C.; Holas, C.; Hotter, A.; Hussl, A.; Müller, C.; Poewe, W.; Seppi, K.; Sprenger, F.; Wenning, G.; Boogaerts, A.; Calmeyn, G.; Delvaux, I.; Liessens, D.; Somers, N.; Dupuit, M.; Minet, C.; Van Paemel, D.; Ribaï, P.; Verellen-Dumoulin, C.; Boogaerts, A.; Vandenberghe, W.; Van Reijen, D.; Klempír, J.; Majerová, V.; Roth, J.; Stárková, I.; Hjermind, L. E.; Jacobsen, O.; Nielsen, J. E.; Larsen, I. U.; Vinther-Jensen, T.; Hiivola, H.; Hyppönen, H.; Martikainen, K.; Tuuha, K.; Allain, P.; Bonneau, D.; Bost, M.; Gohier, B.; Guérid, M.-A.; Olivier, A.; Prundean, A.; Scherer-Gagou, C.; Verny, C.; Babiloni, B.; Debruxelles, S.; Duché, C.; Goizet, C.; Jameau, L.; Lafoucrière, D.; Spampinato, U.; Barthélémy, R.; De Bruycker, C.; Carette, M. C. A.-S.; Defebvre, E. D. L.; Delliaux, M.; Delval, A.; Destee, A.; Dujardin, K.; Lemaire, M.-H.; Manouvrier, S.; Peter, M.; Plomhouse, L.; Sablonnière, B.; Simonin, C.; Thibault-Tanchou, S.; Vuillaume, I.; Bellonet, M.; Berrissoul, H.; Blin, S.; Courtin, F.; Duru, C.; Fasquel, V.; Godefroy, O.; Krystkowiak, P.; Mantaux, B.; Roussel, M.; Wannepain, S.; Azulay, J.-P.; Delfini, M.; Eusebio, A.; Fluchere, F.; Mundler, L.; Anheim, M.; Julié, C.; Boukbiza, O. L.; Longato, N.; Rudolf, G.; Tranchant, C.; Zimmermann, M.-A.; Kosinski, C. M.; Milkereit, E.; Probst, D.; Reetz, K.; Sass, C.; Schiefer, J.; Schlangen, C.; Werner, C. J.; Gelderblom, H.; Priller, J.; Prüß, H.; Spruth, E. J.; Ellrichmann, G.; Herrmann, L.; Hoffmann, R.; Kaminski, B.; Kotz, P.; Prehn, C.; Saft, C.; Lange, H.; Maiwald, R.; Löhle, M.; Maass, A.; Schmidt, S.; Bosredon, C.; Storch, A.; Wolz, A.; Wolz, M.; Capetian, P.; Lambeck, J.; Zucker, B.; Boelmans, K.; Ganos, C.; Heinicke, W.; Hidding, U.; Lewerenz, J.; Münchau, A.; Orth, M.; Schmalfeld, J.; Stubbe, L.; Zittel, S.; Diercks, G.; Dressler, D.; Gorzolla, H.; Schrader, C.; Tacik, P.; Ribbat, M.; Longinus, B.; Bürk, K.; Möller, J. C.; Rissling, I.; Mühlau, M.; Peinemann, A.; Städtler, M.; Weindl, A.; Winkelmann, J.; Ziegler, C.; Bechtel, N.; Beckmann, H.; Bohlen, S.; Hölzner, E.; Lange, H.; Reilmann, R.; Rohm, S.; Rumpf, S.; Schepers, S.; Weber, N.; Dose, M.; Leythäuser, G.; Marquard, R.; Raab, T.; Wiedemann, A.; Barth, K.; Buck, A.; Connemann, J.; Ecker, D.; Geitner, C.; Held, C.; Kesse, A.; Landwehrmeyer, B.; Lang, C.; Lewerenz, J.; Lezius, F.; Nepper, S.; Niess, A.; Orth, M.; Schneider, A.; Schwenk, D.; Süßmuth, S.; Trautmann, S.; Weydt, P.; Cormio, C.; Sciruicchio, V.; Serpino, C.; De Tommaso, M.; Capellari, S.; Cortelli, P.; Galassi, R.; Rizzo, G.; Poda, R.; Scaglione, C.; Bertini, E.; Ghelli, E.; Ginestroni, A.; Massaro, F.; Mechi, C.; Paganini, M.; Piacentini, S.; Pradella, S.; Romoli, A. M.; Sorbi, S.; Abbruzzese, G.; Di Poggio, M. B.; Ferrandes, G.; Mandich, P.; Marchese, R.; Albanese, A.; Di Bella, D.; Castaldo, A.; Di Donato, S.; Gellera, C.; Genitrini, S.; Mariotti, C.; Monza, D.; Nanetti, L.; Paridi, D.; Soliveri, P.; Tomasello, C.; De Michele, G.; Di Maio, L.; Massarelli, M.; Peluso, S.; Roca, A.; Russo, C. V.; Salvatore, E.; Sorrentino, P.; Amico, E.; Favellato, M.; Griguoli, A.; Mazzante, I.; Petrollini, M.; Squitieri, F.; D’Alessio, B.; Esposito, C.; Bentivoglio, R.; Frontali, M.; Guidubaldi, A.; Ialongo, T.; Jacopini, G.; Piano, C.; Romano, S.; Soleti, F.; Spadaro, M.; Zinzi, P.; Van Hout, M. S. E.; Verhoeven, M. E.; Van Vugt, J. P. P.; De Weert, A. M.; Bolwijn, J. J. W.; Dekker, M.; Kremer, B.; Leenders, K. L.; Van Oostrom, J. C. H.; Van Den Bogaard, S. J. A.; Bos, R.; Dumas, E. M.; ’T Hart, E. P.; Roos, R. A. C.; Kremer, B.; Verstappen, C. C. P.; Aaserud, O.; C, J. F.; Heiberg, A.; Van Walsem, M. R.; Wehus, R.; Bjørgo, K.; Fannemel, M.; Gørvell, P. F.; Lorentzen, E.; Koivisto, S. P.; Retterstøl, L.; Stokke, B.; Bjørnevoll, I.; Sando, S. B.; Dziadkiewicz, A.; Nowak, M.; Robowski, P.; Sitek, E.; Slawek, J.; Soltan, W.; Szinwelski, M.; Blaszcyk, M.; Boczarska-Jedynak, M.; Ciach-Wysocka, E.; Gorzkowska, A.; Jasinska-Myga, B.; Klodowska-Duda, G.; Opala, G.; Stompel, D.; Banaszkiewicz, K.; Bocwinska, D.; Bojakowska-Jaremek, K.; Dec, M.; Krawczyk, M.; Rudzinska, M.; Szczygiel, E.; Szczudlik, A.; Wasielewska, A.; Wójcik, M.; Bryl, A.; Ciesielska, A.; Klimberg, A.; Marcinkowski, J.; Samara, H.; Sempolowicz, J.; Zielonka, D.; Gogol, A.; Janik, P.; Kwiecinski, H.; Jamrozik, Z.; Antczak, J.; Jachinska, K.; Krysa, W.; Rakowicz, M.; Richter, P.; Rola, R.; Ryglewicz, D.; Sienkiewicz-Jarosz, H.; Stepniak, I.; Sulek, A.; Witkowski, G.; Zaremba, J.; Zdzienicka, E.; Zieora-Jakutowicz, K.; Ferreira, J. J.; Coelho, M.; Guedes, L. C.; Mendes, T.; Mestre, T.; Valadas, A.; Andrade, C.; Gago, M.; Garrett, C.; Guerra, M. R.; Herrera, C. D.; Garcia, P. M.; Barbera, M. A.; Guia, D. B.; Hernanz, L. C.; Catena, J. L.; Ferrer, P. Q.; Sebastián, A. R.; Carruesco, G. T.; Bas, J.; Busquets, N.; Calopa, M.; Robert, M. F.; Viladrich, C. M.; Idiago, J. M. R.; Riballo, A. V.; Cubo, E.; Polo, C. G.; Mariscal, N.; Rivadeneyra, P. J.; Barrero, F.; Morales, B.; Fenollar, M.; García, R. G.-R.; Ortega, P.; Villanueva, C.; Alegre, J.; Bascuñana, M.; Caldentey, J. G.; Ventura, M. F.; Ribas, G. G.; De Yébenes, J. G.; Moreno, J. L. L.-S.; Cubillo, P. T.; Alegre, J.; Frech, F. A.; De Yébenes, J. G.; Ruíz, P. J. G.; Martínez-Descals, A.; Guerrero, R.; Artiga, M. J. S.; Sánchez, V.; Perea, M. F. N.; Fortuna, L.; Manzanares, S.; Reinante, G.; Torres, M. M. A.; Moreau, L. V.; González González, S.; Guisasola, L. M.; Salvador, C.; Martín, E. S. S.; Ramirez, I. L.; Gorospe, A.; Lopera, M. R.; Arques, P. N.; Rodríguez, M. J. T.; Pastor, B. V.; Gaston, I.; Martinez-Jaurrieta, M. D.; Ramos-Arroyo, M. A.; Moreno, J. M. G.; Lucena, C. M.; Damas, F.; Cortegana, H. E. P.; Peña, J. C.; Redondo, L.; Carrillo, F.; Teresa Cáceres, M.; Mir, P.; Suarez, M. J. L.; Vargas-González, L.; Bosca, M. E.; Brugada, F. C.; Burguera, J. A.; Campos, A.; Vilaplana, G. C. P.; Berglund, P.; Constantinescu, R.; Fredlund, G.; Høsterey-Ugander, U.; Linnsand, P.; Neleborn-Lingefjärd, L.; Wahlström, J.; Wentzel, M.; Loutfi, G.; Olofsson, C.; Stattin, E.-L.; Westman, L.; Wikström, B.; Burgunder, J.-M.; Stebler, Y.; Kaelin, A.; Romero, I.; Schüpbach, M.; Weber Zaugg, S.; Hauer, M.; Gonzenbach, R.; Jung, H. H.; Mihaylova, V.; Petersen, J.; Jack, R.; Matheson, K.; Miedzybrodzka, Z.; Rae, D.; Simpson, S. A.; Summers, F.; Ure, A.; Vaughan, V.; Akhtar, S.; Crooks, J.; Curtis, A.; De Souza, J.; Piedad, J.; Rickards, H.; Wright, J.; Coulthard, E.; Gethin, L.; Hayward, B.; Sieradzan, K.; Wright, A.; Armstrong, M.; Barker, R. A.; O’Keefe, D.; Di Pietro, A.; Fisher, K.; Goodman, A.; Hill, S.; Kershaw, A.; Mason, S.; Paterson, N.; Raymond, L.; Swain, R.; Guzman, N. V.; Busse, M.; Butcher, C.; Callaghan, J.; Dunnett, S.; Clenaghan, C.; Fullam, R.; Handley, O.; Hunt, S.; Jones, L.; Jones, U.; Khalil, H.; Minster, S.; Owen, M.; Price, K.; Rosser, A.; Townhill, J.; Edwards, M.; Ho, C.; Hughes, T.; McGill, M.; Pearson, P.; Porteous, M.; Smith, P.; Brockie, P.; Foster, J.; Johns, N.; McKenzie, S.; Rothery, J.; Thomas, G.; Yates, S.; Burrows, L.; Chu, C.; Fletcher, A.; Gallantrae, D.; Hamer, S.; Harding, A.; Klöppel, S.; Kraus, A.; Laver, F.; Lewis, M.; Longthorpe, M.; Markova, I.; Raman, A.; Robertson, N.; Silva, M.; Thomson, A.; Wild, S.; Yardumian, P.; Chu, C.; Evans, C.; Gallentrae, D.; Hamer, S.; Kraus, A.; Markova, I.; Raman, A.; Chu, C.; Hamer, S.; Hobson, E.; Jamieson, S.; Kraus, A.; Markova, I.; Raman, A.; Musgrave, H.; Rowett, L.; Toscano, J.; Wild, S.; Yardumian, P.; Bourne, C.; Clapton, J.; Clayton, C.; Dipple, H.; Freire-Patino, D.; Grant, J.; Gross, D.; Hallam, C.; Middleton, J.; Murch, A.; Thompson, C.; Alusi, S.; Davies, R.; Foy, K.; Gerrans, E.; Pate, L.; Andrews, T.; Dougherty, A.; Golding, C.; Kavalier, F.; Laing, H.; Lashwood, A.; Robertson, D.; Ruddy, D.; Santhouse, A.; Whaite, A.; Andrews, T.; Bruno, S.; Doherty, K.; Golding, C.; Haider, S.; Hensman, D.; Lahiri, N.; Lewis, M.; Novak, M.; Patel, A.; Robertson, N.; Rosser, E.; Tabrizi, S.; Taylor, R.; Warner, T.; Wild, E.; Arran, N.; Bek, J.; Callaghan, J.; Craufurd, D.; Fullam, R.; Hare, M.; Howard, L.; Huson, S.; Johnson, L.; Jones, M.; Murphy, H.; Oughton, E.; Partington-Jones, L.; Rogers, D.; Sollom, A.; Snowden, J.; Stopford, C.; Thompson, J.; Trender-Gerhard, I.; Verstraelen, N.; Westmoreland, L.; Armstrong, R.; Dixon, K.; Nemeth, A. H.; Siuda, G.; Valentine, R.; Harrison, D.; Hughes, M.; Parkinson, A.; Soltysiak, B.; Bandmann, O.; Bradbury, A.; Gill, P.; Fairtlough, H.; Fillingham, K.; Foustanos, I.; Kazoka, M.; O’Donovan, K.; Peppa, N.; Taylor, C.; Tidswell, K.; Quarrell, O.; Burgunder, J.-M.; Lau, P. N.; Pica, E.; Tan, L. Identification of Genetic Variants Associated with Huntington’s Disease Progression: A Genome-Wide Association Study. Lancet Neurol. 2017, 16 (9), 701–711. [CrossRef]
- Travessa, A. M.; Rodrigues, F. B.; Mestre, T. A.; Ferreira, J. J. Fifteen Years of Clinical Trials in Huntington’s Disease: A Very Low Clinical Drug Development Success Rate. J. Huntingt. Dis. 2017, 6 (2), 157–163. [CrossRef]
- Kordasiewicz, H. B.; Stanek, L. M.; Wancewicz, E. V.; Mazur, C.; McAlonis, M. M.; Pytel, K. A.; Artates, J. W.; Weiss, A.; Cheng, S. H.; Shihabuddin, L. S.; Hung, G.; Bennett, C. F.; Cleveland, D. W. Sustained Therapeutic Reversal of Huntington’s Disease by Transient Repression of Huntingtin Synthesis. Neuron 2012, 74 (6), 1031–1044. [CrossRef]
- Agustín-Pavón, C.; Mielcarek, M.; Garriga-Canut, M.; Isalan, M. Deimmunization for Gene Therapy: Host Matching of Synthetic Zinc Finger Constructs Enables Long-Term Mutant Huntingtin Repression in Mice. Mol. Neurodegener. 2016, 11 (1), 64. [CrossRef]
- Yang, L.; Calingasan, N. Y.; Wille, E. J.; Cormier, K.; Smith, K.; Ferrante, R. J.; Flint Beal, M. Combination Therapy with Coenzyme Q 10 and Creatine Produces Additive Neuroprotective Effects in Models of Parkinson’s and Huntington’s Diseases. J. Neurochem. 2009, 109 (5), 1427–1439. [CrossRef]
- McGarry, A.; Auinger, P.; Kieburtz, K. D.; Bredlau, A.-L.; Hersch, S. M.; Rosas, H. D. Suicidality Risk Factors Across the CARE-HD, 2CARE, and CREST-E Clinical Trials in Huntington Disease. Neurol. Clin. Pract. 2022, 12 (2), 131–138. [CrossRef]
- Basavarajappa, B. S.; Subbanna, S. Histone Methylation Regulation in Neurodegenerative Disorders. Int. J. Mol. Sci. 2021, 22 (9), 4654. [CrossRef]
- Masrori, P.; Van Damme, P. Amyotrophic Lateral Sclerosis: A Clinical Review. Eur. J. Neurol. 2020, 27 (10), 1918–1929. [CrossRef]
- Renton, A. E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J. R.; Schymick, J. C.; Laaksovirta, H.; van Swieten, J. C.; Myllykangas, L.; Kalimo, H.; Paetau, A.; Abramzon, Y.; Remes, A. M.; Kaganovich, A.; Scholz, S. W.; Duckworth, J.; Ding, J.; Harmer, D. W.; Hernandez, D. G.; Johnson, J. O.; Mok, K.; Ryten, M.; Trabzuni, D.; Guerreiro, R. J.; Orrell, R. W.; Neal, J.; Murray, A.; Pearson, J.; Jansen, I. E.; Sondervan, D.; Seelaar, H.; Blake, D.; Young, K.; Halliwell, N.; Callister, J. B.; Toulson, G.; Richardson, A.; Gerhard, A.; Snowden, J.; Mann, D.; Neary, D.; Nalls, M. A.; Peuralinna, T.; Jansson, L.; Isoviita, V.-M.; Kaivorinne, A.-L.; Hölttä-Vuori, M.; Ikonen, E.; Sulkava, R.; Benatar, M.; Wuu, J.; Chiò, A.; Restagno, G.; Borghero, G.; Sabatelli, M.; Heckerman, D.; Rogaeva, E.; Zinman, L.; Rothstein, J. D.; Sendtner, M.; Drepper, C.; Eichler, E. E.; Alkan, C.; Abdullaev, Z.; Pack, S. D.; Dutra, A.; Pak, E.; Hardy, J.; Singleton, A.; Williams, N. M.; Heutink, P.; Pickering-Brown, S.; Morris, H. R.; Tienari, P. J.; Traynor, B. J. A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron 2011, 72 (2), 257–268. [CrossRef]
- Woollacott, I. O. C.; Mead, S. The C9ORF72 Expansion Mutation: Gene Structure, Phenotypic and Diagnostic Issues. Acta Neuropathol. (Berl.) 2014, 127 (3), 319–332. [CrossRef]
- Balendra, R.; Isaacs, A. M. C9orf72-Mediated ALS and FTD: Multiple Pathways to Disease. Nat. Rev. Neurol. 2018, 14 (9), 544–558. [CrossRef]
- Herdewyn, S.; Zhao, H.; Moisse, M.; Race, V.; Matthijs, G.; Reumers, J.; Kusters, B.; Schelhaas, H. J.; van den Berg, L. H.; Goris, A.; Robberecht, W.; Lambrechts, D.; Van Damme, P. Whole-Genome Sequencing Reveals a Coding Non-Pathogenic Variant Tagging a Non-Coding Pathogenic Hexanucleotide Repeat Expansion in C9orf72 as Cause of Amyotrophic Lateral Sclerosis. Hum. Mol. Genet. 2012, 21 (11), 2412–2419. [CrossRef]
- Majounie, E.; Renton, A. E.; Mok, K.; Dopper, E. G.; Waite, A.; Rollinson, S.; Chiò, A.; Restagno, G.; Nicolaou, N.; Simon-Sanchez, J.; Van Swieten, J. C.; Abramzon, Y.; Johnson, J. O.; Sendtner, M.; Pamphlett, R.; Orrell, R. W.; Mead, S.; Sidle, K. C.; Houlden, H.; Rohrer, J. D.; Morrison, K. E.; Pall, H.; Talbot, K.; Ansorge, O.; Hernandez, D. G.; Arepalli, S.; Sabatelli, M.; Mora, G.; Corbo, M.; Giannini, F.; Calvo, A.; Englund, E.; Borghero, G.; Floris, G. L.; Remes, A. M.; Laaksovirta, H.; McCluskey, L.; Trojanowski, J. Q.; Van Deerlin, V. M.; Schellenberg, G. D.; Nalls, M. A.; Drory, V. E.; Lu, C.-S.; Yeh, T.-H.; Ishiura, H.; Takahashi, Y.; Tsuji, S.; Le Ber, I.; Brice, A.; Drepper, C.; Williams, N.; Kirby, J.; Shaw, P.; Hardy, J.; Tienari, P. J.; Heutink, P.; Morris, H. R.; Pickering-Brown, S.; Traynor, B. J. Frequency of the C9orf72 Hexanucleotide Repeat Expansion in Patients with Amyotrophic Lateral Sclerosis and Frontotemporal Dementia: A Cross-Sectional Study. Lancet Neurol. 2012, 11 (4), 323–330. [CrossRef]
- Xi, Z.; Zinman, L.; Grinberg, Y.; Moreno, D.; Sato, C.; Bilbao, J. M.; Ghani, M.; Hernández, I.; Ruiz, A.; Boada, M.; Morón, F. J.; Lang, A. E.; Marras, C.; Bruni, A.; Colao, R.; Maletta, R. G.; Puccio, G.; Rainero, I.; Pinessi, L.; Galimberti, D.; Morrison, K. E.; Moorby, C.; Stockton, J. D.; Masellis, M.; Black, S. E.; Hazrati, L.-N.; Liang, Y.; Van Haersma De With, J.; Fornazzari, L.; Villagra, R.; Rojas-Garcia, R.; Clarimón, J.; Mayeux, R.; Robertson, J.; St George-Hyslop, P.; Rogaeva, E. Investigation of C9orf72 in 4 Neurodegenerative Disorders. Arch. Neurol. 2012, 69 (12), 1583. [CrossRef]
- Česnik, A. B.; Darovic, S.; Mihevc, S. P.; Štalekar, M.; Malnar, M.; Motaln, H.; Lee, Y.-B.; Mazej, J.; Pohleven, J.; Grosch, M.; Modic, M.; Fonovič, M.; Turk, B.; Drukker, M.; Shaw, C. E.; Rogelj, B. Nuclear RNA Foci from C9ORF72 Expansion Mutation Form Paraspeckle-like Bodies. J. Cell Sci. 2019, jcs.224303. [CrossRef]
- Saccon, R. A.; Bunton-Stasyshyn, R. K. A.; Fisher, E. M. C.; Fratta, P. Is SOD1 Loss of Function Involved in Amyotrophic Lateral Sclerosis? Brain 2013, 136 (8), 2342–2358. [CrossRef]
- Bosco, D. A.; Morfini, G.; Karabacak, N. M.; Song, Y.; Gros-Louis, F.; Pasinelli, P.; Goolsby, H.; Fontaine, B. A.; Lemay, N.; McKenna-Yasek, D.; Frosch, M. P.; Agar, J. N.; Julien, J.-P.; Brady, S. T.; Brown, R. H. Wild-Type and Mutant SOD1 Share an Aberrant Conformation and a Common Pathogenic Pathway in ALS. Nat. Neurosci. 2010, 13 (11), 1396–1403. [CrossRef]
- Bruijn, L. I.; Houseweart, M. K.; Kato, S.; Anderson, K. L.; Anderson, S. D.; Ohama, E.; Reaume, A. G.; Scott, R. W.; Cleveland, D. W. Aggregation and Motor Neuron Toxicity of an ALS-Linked SOD1 Mutant Independent from Wild-Type SOD1. Science 1998, 281 (5384), 1851–1854. [CrossRef]
- Saeed, M.; Yang, Y.; Deng, H.-X.; Hung, W.-Y.; Siddique, N.; Dellefave, L.; Gellera, C.; Andersen, P. M.; Siddique, T. Age and Founder Effect of SOD1 A4V Mutation Causing ALS. Neurology 2009, 72 (19), 1634–1639. [CrossRef]
- Hensley, K.; Mhatre, M.; Mou, S.; Pye, Q. N.; Stewart, C.; West, M.; Williamson, K. S. On the Relation of Oxidative Stress to Neuroinflammation: Lessons Learned from the G93A-SOD1 Mouse Model of Amyotrophic Lateral Sclerosis. Antioxid. Redox Signal. 2006, 8 (11–12), 2075–2087. [CrossRef]
- Curti, D.; Rognoni, F.; Alimonti, D.; Malaspina, A.; Feletti, F.; Tessera, S.; Finotti, N.; Rehak, L.; Mazzini, L.; Zerbi, F.; Poloni, T.; Ceroni, M. SOD1 Activity and Protective Factors in Familial ALS Patients with L84F SOD1 Mutation. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2002, 3 (3), 115–122. [CrossRef]
- Ceroni, M.; Malaspina, A.; Poloni, T. E.; Alimonti, D.; Rognoni, F.; Habgood, J.; Imbesi, F.; Antonelli, P.; Alfonsi, E.; Curti, D.; deBelleroche, J. Clustering of ALS Patients in Central Italy Due to the Occurrence of the L84F SOD1 Gene Mutation. Neurology 1999, 53 (5), 1064–1064. [CrossRef]
- Juneja, T.; Pericak-Vance, M. A.; Laing, N. G.; Dave, S.; Siddique, T. Prognosis in Familial Amyotrophic Lateral Sclerosis: Progression and Survival in Patients with Glu100gly and Ala4val Mutations in Cu,Zn Superoxide Dismutase. Neurology 1997, 48 (1), 55–57. [CrossRef]
- Zhou, Y.; Tang, J.; Lan, J.; Zhang, Y.; Wang, H.; Chen, Q.; Kang, Y.; Sun, Y.; Feng, X.; Wu, L.; Jin, H.; Chen, S.; Peng, Y. Honokiol Alleviated Neurodegeneration by Reducing Oxidative Stress and Improving Mitochondrial Function in Mutant SOD1 Cellular and Mouse Models of Amyotrophic Lateral Sclerosis. Acta Pharm. Sin. B 2023, 13 (2), 577–597. [CrossRef]
- Cassina, P.; Cassina, A.; Pehar, M.; Castellanos, R.; Gandelman, M.; De León, A.; Robinson, K. M.; Mason, R. P.; Beckman, J. S.; Barbeito, L.; Radi, R. Mitochondrial Dysfunction in SOD1 G93A -Bearing Astrocytes Promotes Motor Neuron Degeneration: Prevention by Mitochondrial-Targeted Antioxidants. J. Neurosci. 2008, 28 (16), 4115–4122. [CrossRef]
- Zhang, X.; Li, L.; Chen, S.; Yang, D.; Wang, Y.; Zhang, X.; Wang, Z.; Le, W. Rapamycin Treatment Augments Motor Neuron Degeneration in SOD1 G93A Mouse Model of Amyotrophic Lateral Sclerosis. Autophagy 2011, 7 (4), 412–425. [CrossRef]
- Daoud, H.; Valdmanis, P. N.; Kabashi, E.; Dion, P.; Dupre, N.; Camu, W.; Meininger, V.; Rouleau, G. A. Contribution of TARDBP Mutations to Sporadic Amyotrophic Lateral Sclerosis. J. Med. Genet. 2008, 46 (2), 112–114. [CrossRef]
- Lattante, S.; Rouleau, G. A.; Kabashi, E. TARDBP and FUS Mutations Associated with Amyotrophic Lateral Sclerosis: Summary and Update. Hum. Mutat. 2013, 34 (6), 812–826. [CrossRef]
- Kabashi, E.; Valdmanis, P. N.; Dion, P.; Spiegelman, D.; McConkey, B. J.; Velde, C. V.; Bouchard, J.-P.; Lacomblez, L.; Pochigaeva, K.; Salachas, F.; Pradat, P.-F.; Camu, W.; Meininger, V.; Dupre, N.; Rouleau, G. A. TARDBP Mutations in Individuals with Sporadic and Familial Amyotrophic Lateral Sclerosis. Nat. Genet. 2008, 40 (5), 572–574. [CrossRef]
- Corcia, P.; Valdmanis, P.; Millecamps, S.; Lionnet, C.; Blasco, H.; Mouzat, K.; Daoud, H.; Belzil, V.; Morales, R.; Pageot, N.; Danel-Brunaud, V.; Vandenberghe, N.; Pradat, P. F.; Couratier, P.; Salachas, F.; Lumbroso, S.; Rouleau, G. A.; Meininger, V.; Camu, W. Phenotype and Genotype Analysis in Amyotrophic Lateral Sclerosis with TARDBP Gene Mutations. Neurology 2012, 78 (19), 1519–1526. [CrossRef]
- Tamaoka, A.; Arai, M.; Itokawa, M.; Arai, T.; Hasegawa, M.; Tsuchiya, K.; Takuma, H.; Tsuji, H.; Ishii, A.; Watanabe, M.; Takahashi, Y.; Goto, J.; Tsuji, S.; Akiyama, H. TDP-43 M337V Mutation in Familial Amyotrophic Lateral Sclerosis in Japan. Intern. Med. 2010, 49 (4), 331–334. [CrossRef]
- Watanabe, S.; Oiwa, K.; Murata, Y.; Komine, O.; Sobue, A.; Endo, F.; Takahashi, E.; Yamanaka, K. ALS-Linked TDP-43M337V Knock-in Mice Exhibit Splicing Deregulation without Neurodegeneration. Mol. Brain 2020, 13 (1), 8. [CrossRef]
- Esmaeili, M. A.; Panahi, M.; Yadav, S.; Hennings, L.; Kiaei, M. Premature Death of TDP-43 (A315T) Transgenic Mice Due to Gastrointestinal Complications Prior to Development of Full Neurological Symptoms of Amyotrophic Lateral Sclerosis. Int. J. Exp. Pathol. 2013, 94 (1), 56–64. [CrossRef]
- Orrù, S.; Coni, P.; Floris, A.; Littera, R.; Carcassi, C.; Sogos, V.; Brancia, C. Reduced Stress Granule Formation and Cell Death in Fibroblasts with the A382T Mutation of TARDBP Gene: Evidence for Loss of TDP-43 Nuclear Function. Hum. Mol. Genet. 2016, ddw276. [CrossRef]
- Zanini, G.; Selleri, V.; Nasi, M.; De Gaetano, A.; Martinelli, I.; Gianferrari, G.; Lofaro, F. D.; Boraldi, F.; Mandrioli, J.; Pinti, M. Mitochondrial and Endoplasmic Reticulum Alterations in a Case of Amyotrophic Lateral Sclerosis Caused by TDP-43 A382T Mutation. Int. J. Mol. Sci. 2022, 23 (19), 11881. [CrossRef]
- Deng, H.; Gao, K.; Jankovic, J. The Role of FUS Gene Variants in Neurodegenerative Diseases. Nat. Rev. Neurol. 2014, 10 (6), 337–348. [CrossRef]
- Corrado, L.; Del Bo, R.; Castellotti, B.; Ratti, A.; Cereda, C.; Penco, S.; Soraru, G.; Carlomagno, Y.; Ghezzi, S.; Pensato, V.; Colombrita, C.; Gagliardi, S.; Cozzi, L.; Orsetti, V.; Mancuso, M.; Siciliano, G.; Mazzini, L.; Comi, G. P.; Gellera, C.; Ceroni, M.; D’Alfonso, S.; Silani, V. Mutations of FUS Gene in Sporadic Amyotrophic Lateral Sclerosis. J. Med. Genet. 2010, 47 (3), 190–194. [CrossRef]
- Rademakers, R.; Stewart, H.; Dejesus-Hernandez, M.; Krieger, C.; Graff-Radford, N.; Fabros, M.; Briemberg, H.; Cashman, N.; Eisen, A.; Mackenzie, I. R. A. Fus Gene Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Muscle Nerve 2010, 42 (2), 170–176. [CrossRef]
- Ticozzi, N.; Silani, V.; LeClerc, A. L.; Keagle, P.; Gellera, C.; Ratti, A.; Taroni, F.; Kwiatkowski, T. J.; McKenna-Yasek, D. M.; Sapp, P. C.; Brown, R. H.; Landers, J. E. Analysis of FUS Gene Mutation in Familial Amyotrophic Lateral Sclerosis within an Italian Cohort. Neurology 2009, 73 (15), 1180–1185. [CrossRef]
- Weinreich, M.; Shepheard, S. R.; Verber, N.; Wyles, M.; Heath, P. R.; Highley, J. R.; Kirby, J.; Shaw, P. J. Neuropathological Characterization of a Novel TANK Binding Kinase ( TBK1 ) Gene Loss of Function Mutation Associated with Amyotrophic Lateral Sclerosis. Neuropathol. Appl. Neurobiol. 2020, 46 (3), 279–291. [CrossRef]
- Oakes, J. A.; Davies, M. C.; Collins, M. O. TBK1: A New Player in ALS Linking Autophagy and Neuroinflammation. Mol. Brain 2017, 10 (1), 5. [CrossRef]
- Gurfinkel, Y.; Polain, N.; Sonar, K.; Nice, P.; Mancera, R. L.; Rea, S. L. Functional and Structural Consequences of TBK1 Missense Variants in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Neurobiol. Dis. 2022, 174, 105859. [CrossRef]
- Zarei, S.; Carr, K.; Reiley, L.; Diaz, K.; Guerra, O.; Altamirano, P.; Pagani, W.; Lodin, D.; Orozco, G.; Chinea, A. A Comprehensive Review of Amyotrophic Lateral Sclerosis. Surg. Neurol. Int. 2015, 6 (1), 171. [CrossRef]
- Deans, C.; Maggert, K. A. What Do You Mean, “Epigenetic”? Genetics 2015, 199 (4), 887–896. [CrossRef]
- Burggren, W. Epigenetic Inheritance and Its Role in Evolutionary Biology: Re-Evaluation and New Perspectives. Biology 2016, 5 (2), 24. [CrossRef]
- Neumann, M.; Sampathu, D. M.; Kwong, L. K.; Truax, A. C.; Micsenyi, M. C.; Chou, T. T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C. M.; McCluskey, L. F.; Miller, B. L.; Masliah, E.; Mackenzie, I. R.; Feldman, H.; Feiden, W.; Kretzschmar, H. A.; Trojanowski, J. Q.; Lee, V. M.-Y. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314 (5796), 130–133. [CrossRef]
- Fecto, F. SQSTM1 Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Arch. Neurol. 2011, 68 (11), 1440. [CrossRef]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; Komure, O.; Matsuura, S.; Kobatake, K.; Morimoto, N.; Abe, K.; Suzuki, N.; Aoki, M.; Kawata, A.; Hirai, T.; Kato, T.; Ogasawara, K.; Hirano, A.; Takumi, T.; Kusaka, H.; Hagiwara, K.; Kaji, R.; Kawakami, H. Mutations of Optineurin in Amyotrophic Lateral Sclerosis. Nature 2010, 465 (7295), 223–226. [CrossRef]
- Johnson, J. O.; Mandrioli, J.; Benatar, M.; Abramzon, Y.; Van Deerlin, V. M.; Trojanowski, J. Q.; Gibbs, J. R.; Brunetti, M.; Gronka, S.; Wuu, J.; Ding, J.; McCluskey, L.; Martinez-Lage, M.; Falcone, D.; Hernandez, D. G.; Arepalli, S.; Chong, S.; Schymick, J. C.; Rothstein, J.; Landi, F.; Wang, Y.-D.; Calvo, A.; Mora, G.; Sabatelli, M.; Monsurrò, M. R.; Battistini, S.; Salvi, F.; Spataro, R.; Sola, P.; Borghero, G.; Galassi, G.; Scholz, S. W.; Taylor, J. P.; Restagno, G.; Chiò, A.; Traynor, B. J. Exome Sequencing Reveals VCP Mutations as a Cause of Familial ALS. Neuron 2010, 68 (5), 857–864. [CrossRef]
- Boeynaems, S.; Bogaert, E.; Van Damme, P.; Van Den Bosch, L. Inside out: The Role of Nucleocytoplasmic Transport in ALS and FTLD. Acta Neuropathol. (Berl.) 2016, 132 (2), 159–173. [CrossRef]
- Vilarino-Guell, C.; Wider, C.; Soto-Ortolaza, A. I.; Cobb, S. A.; Kachergus, J. M.; Keeling, B. H.; Dachsel, J. C.; Hulihan, M. M.; Dickson, D. W.; Wszolek, Z. K.; Uitti, R. J.; Graff-Radford, N. R.; Boeve, B. F.; Josephs, K. A.; Miller, B.; Boylan, K. B.; Gwinn, K.; Adler, C. H.; Aasly, J. O.; Hentati, F.; Destee, A.; Krygowska-Wajs, A.; Chartier-Harlin, M.-C.; Ross, O. A.; Rademakers, R.; Farrer, M. J. Characterization of DCTN1 Genetic Variability in Neurodegeneration. Neurology 2009, 72 (23), 2024–2028. [CrossRef]
- Araki, E.; Tsuboi, Y.; Daechsel, J.; Milnerwood, A.; Vilarino-Guell, C.; Fujii, N.; Mishima, T.; Oka, T.; Hara, H.; Fukae, J.; Farrer, M. J. A Novel DCTN1 Mutation with Late-Onset Parkinsonism and Frontotemporal Atrophy: A Novel DCTN1 Mutation. Mov. Disord. 2014, 29 (9), 1201–1204. [CrossRef]
- Al-Chalabi, A.; Hardiman, O.; Kiernan, M. C.; Chiò, A.; Rix-Brooks, B.; Van Den Berg, L. H. Amyotrophic Lateral Sclerosis: Moving towards a New Classification System. Lancet Neurol. 2016, 15 (11), 1182–1194. [CrossRef]
- Chio, A.; Calvo, A.; Moglia, C.; Mazzini, L.; Mora, G.; PARALS study group. Phenotypic Heterogeneity of Amyotrophic Lateral Sclerosis: A Population Based Study. J. Neurol. Neurosurg. Psychiatry 2011, 82 (7), 740–746. [CrossRef]
- Finegan, E.; Chipika, R. H.; Li Hi Shing, S.; Hardiman, O.; Bede, P. Pathological Crying and Laughing in Motor Neuron Disease: Pathobiology, Screening, Intervention. Front. Neurol. 2019, 10, 260. [CrossRef]
- Wijesekera, L. C.; Mathers, S.; Talman, P.; Galtrey, C.; Parkinson, M. H.; Ganesalingam, J.; Willey, E.; Ampong, M. A.; Ellis, C. M.; Shaw, C. E.; Al-Chalabi, A.; Leigh, P. N. Natural History and Clinical Features of the Flail Arm and Flail Leg ALS Variants. Neurology 2009, 72 (12), 1087–1094. [CrossRef]
- Doble, A. The Pharmacology and Mechanism of Action of Riluzole. Neurology 1996, 47 (Issue 6, Supplement 4), 233S-241S. [CrossRef]
- Amyotrophic Lateral Sclerosis/Riluzole Study Group II; Lacomblez, L.; Bensimon, G.; Meininger, V.; Leigh, P. N.; Guillet, P. Dose-Ranging Study of Riluzole in Amyotrophic Lateral Sclerosis. The Lancet 1996, 347 (9013), 1425–1431. [CrossRef]
- Hinchcliffe, M.; Smith, A. Riluzole: Real-World Evidence Supports Significant Extension of Median Survival Times in Patients with Amyotrophic Lateral Sclerosis. Degener. Neurol. Neuromuscul. Dis. 2017, Volume 7, 61–70. [CrossRef]
- Mora, J. S.; Genge, A.; Chio, A.; Estol, C. J.; Chaverri, D.; Hernández, M.; Marín, S.; Mascias, J.; Rodriguez, G. E.; Povedano, M.; Paipa, A.; Dominguez, R.; Gamez, J.; Salvado, M.; Lunetta, C.; Ballario, C.; Riva, N.; Mandrioli, J.; Moussy, A.; Kinet, J.-P.; Auclair, C.; Dubreuil, P.; Arnold, V.; Mansfield, C. D.; Hermine, O.; on behalf of the AB10015 STUDY GROUP. Masitinib as an Add-on Therapy to Riluzole in Patients with Amyotrophic Lateral Sclerosis: A Randomized Clinical Trial. Amyotroph. Lateral Scler. Front. Degener. 2020, 21 (1–2), 5–14. [CrossRef]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C. E.; Cummings, J.; Van Der Flier, W. M. Alzheimer’s Disease. The Lancet 2021, 397 (10284), 1577–1590. [CrossRef]
- Bloem, B. R.; Henderson, E. J.; Dorsey, E. R.; Okun, M. S.; Okubadejo, N.; Chan, P.; Andrejack, J.; Darweesh, S. K. L.; Munneke, M. Integrated and Patient-Centred Management of Parkinson’s Disease: A Network Model for Reshaping Chronic Neurological Care. Lancet Neurol. 2020, 19 (7), 623–634. [CrossRef]
- Tabrizi, S. J.; Flower, M. D.; Ross, C. A.; Wild, E. J. Huntington Disease: New Insights into Molecular Pathogenesis and Therapeutic Opportunities. Nat. Rev. Neurol. 2020, 16 (10), 529–546. [CrossRef]
- Deda, H.; Inci, M.; Kürekçi, A.; Sav, A.; Kayıhan, K.; Özgün, E.; Üstünsoy, G.; Kocabay, S. Treatment of Amyotrophic Lateral Sclerosis Patients by Autologous Bone Marrow-Derived Hematopoietic Stem Cell Transplantation: A 1-Year Follow-Up. Cytotherapy 2009, 11 (1), 18–25. [CrossRef]
- Moviglia, G. A.; Moviglia-Brandolino, M. T.; Varela, G. S.; Albanese, G.; Piccone, S.; Echegaray, G.; Martinez, G.; Blasseti, N.; Farias, J.; Farina, P.; Perusso, A.; Gaeta, C. A. Feasibility, Safety, and Preliminary Proof of Principles of Autologous Neural Stem Cell Treatment Combined with T-Cell Vaccination for ALS Patients. Cell Transplant. 2012, 21 (1_suppl), 57–63. [CrossRef]
Figure 1.
Amyloid precursor protein (APP) is a protein that spans the cell membrane. Its processing can occur through two pathways: the non-amyloidogenic and amyloidogenic pathways. In the non-amyloidogenic pathway, APP is cleaved in the middle of Aβ by α-secretase, resulting in the production of soluble APPα (sAPPα) and C-terminal fragment α (CTFα), which is then hydrolyzed by γ-secretase to generate the APP intracellular domain (AICD). In the amyloidogenic pathway, APP is cleaved by β-secretase, resulting in the release of N-terminal soluble APPβ (sAPPβ) and the C-terminal fragment β (CTFβ), which is then hydrolyzed by γ-secretase to produce Aβ and AICD. γ-secretase is composed of several parts, including presenilin, nicastrin, anterior pharynx-defective 1 (APH-1), and presenilin enhancer 2 (PEN-2). Mutations in the PSEN gene may increase the activity of γ-secretase, leading to the formation of plaques.
Figure 1.
Amyloid precursor protein (APP) is a protein that spans the cell membrane. Its processing can occur through two pathways: the non-amyloidogenic and amyloidogenic pathways. In the non-amyloidogenic pathway, APP is cleaved in the middle of Aβ by α-secretase, resulting in the production of soluble APPα (sAPPα) and C-terminal fragment α (CTFα), which is then hydrolyzed by γ-secretase to generate the APP intracellular domain (AICD). In the amyloidogenic pathway, APP is cleaved by β-secretase, resulting in the release of N-terminal soluble APPβ (sAPPβ) and the C-terminal fragment β (CTFβ), which is then hydrolyzed by γ-secretase to produce Aβ and AICD. γ-secretase is composed of several parts, including presenilin, nicastrin, anterior pharynx-defective 1 (APH-1), and presenilin enhancer 2 (PEN-2). Mutations in the PSEN gene may increase the activity of γ-secretase, leading to the formation of plaques.
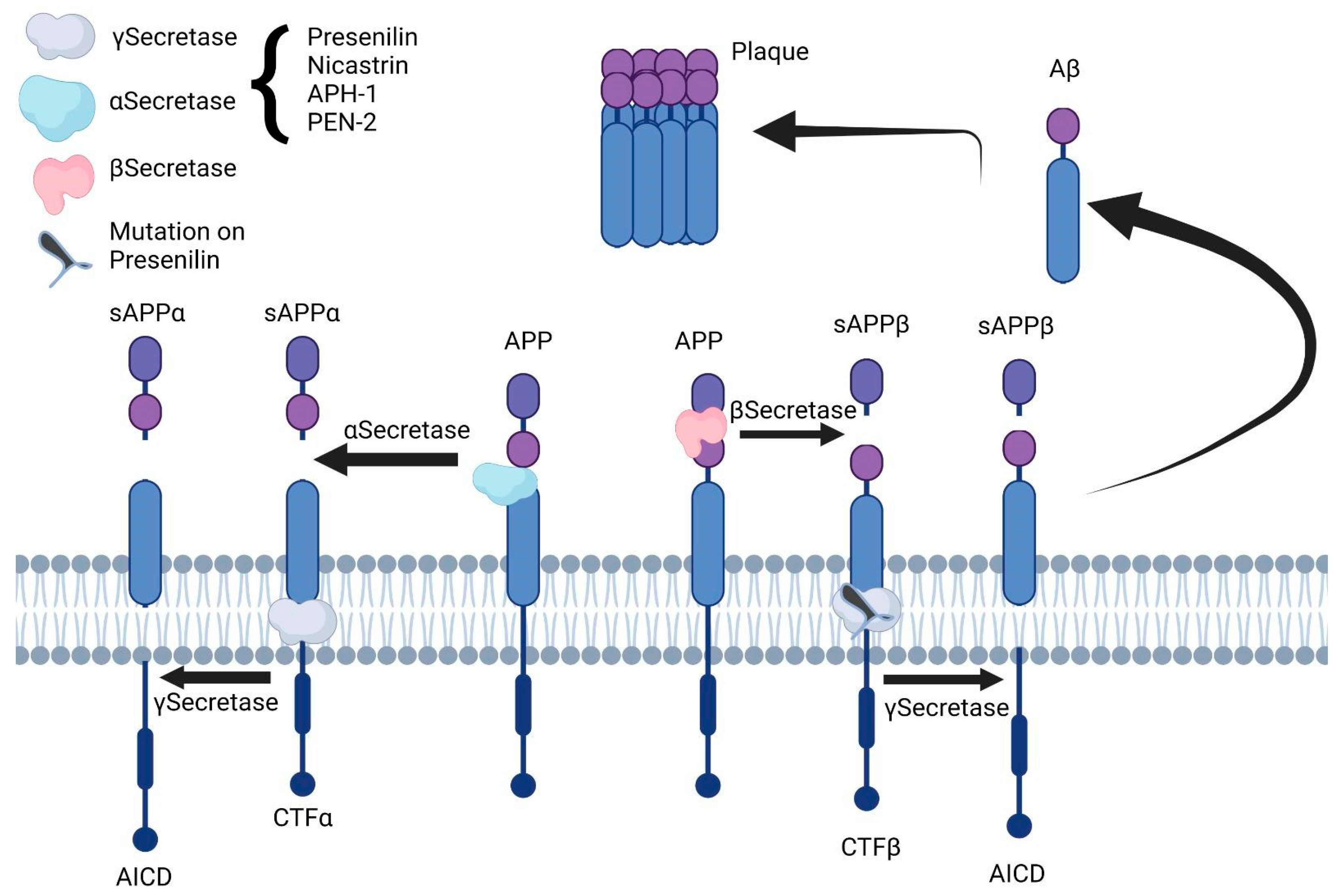
Figure 2.
The huntingtin protein can take on various forms and undergo changes depending on the expression of the HTT gene. Normally, the expression of HTT results in the production of an RNA transcript that codes for the complete huntingtin protein. However, if the gene has an expanded CAG repeat, the RNA transcript can be processed abnormally, producing an mRNA that encodes only the HTT exon1 protein.
Figure 2.
The huntingtin protein can take on various forms and undergo changes depending on the expression of the HTT gene. Normally, the expression of HTT results in the production of an RNA transcript that codes for the complete huntingtin protein. However, if the gene has an expanded CAG repeat, the RNA transcript can be processed abnormally, producing an mRNA that encodes only the HTT exon1 protein.
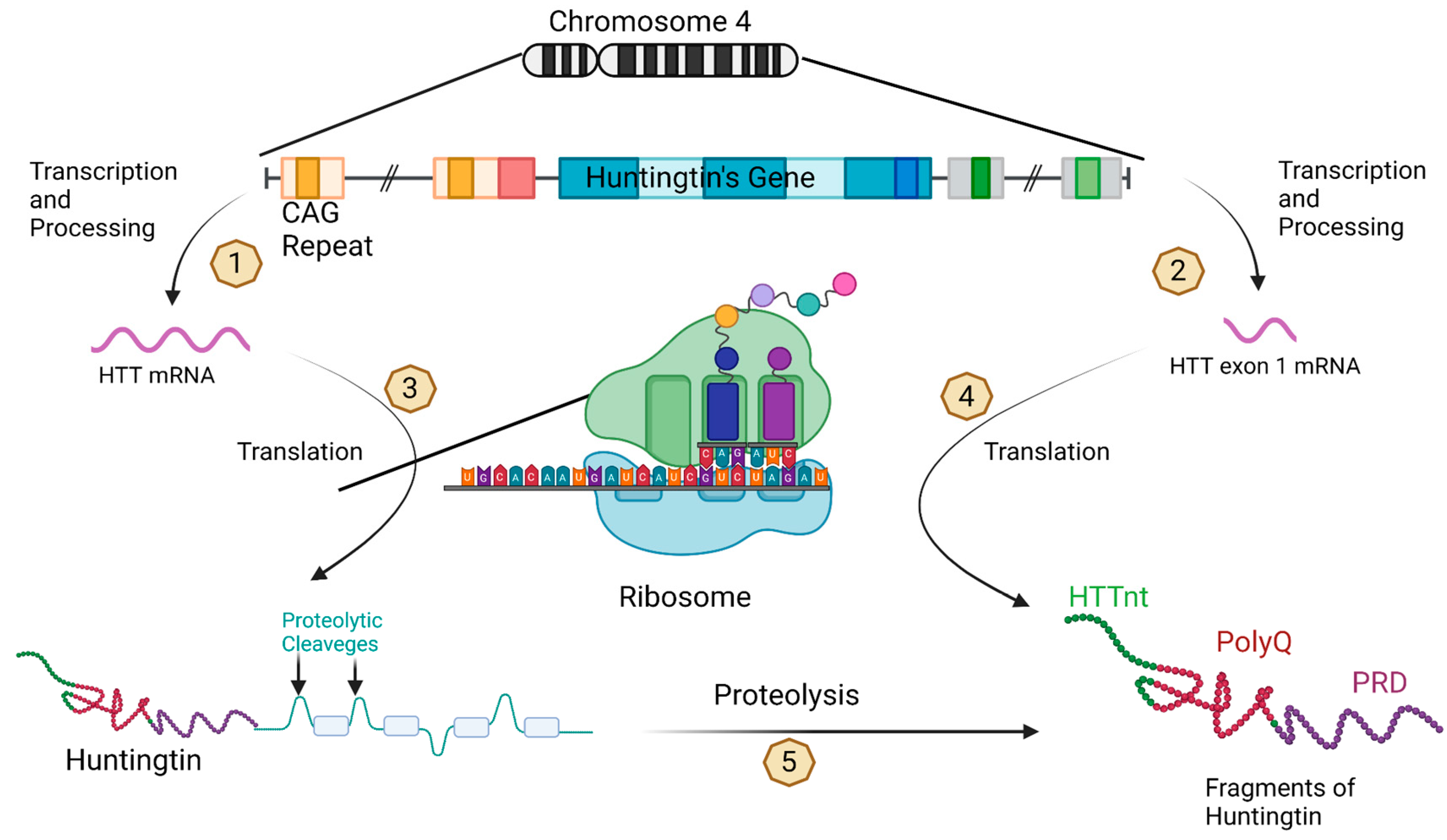
Figure 3.
Advances in large-scale genomic analysis have uncovered a variety of causative genes and risk factors for amyotrophic lateral sclerosis (ALS). These gene variants map onto key pathogenic mechanisms relevant to all motor neuron cellular compartments as well as neighboring cells such as glia and interneurons. In this way, these mechanisms are genetically validated, enabling a greater confidence in their targeting for therapeutic benefit. Some of these mechanisms have emerged only in recent years due to new genetic information, including gene changes highlighting dysregulation of RNA processing and metabolism. There is significant overlap of some genes with those found in closely related disorders such as frontotemporal dementia (for example, C9orf72, CHCHD10, SQSTM1, TBK1, CCNF, FUS, TARDBP, OPTN, UBQLN2, TUBA4A, ATAXN2, VCP and CHMP2B). This suggests a closer relationship to broader neurodegenerative disorders, and indeed many of the pathways depicted are relevant in, for example, Alzheimer disease. ER, endoplasmic reticulum.
Figure 3.
Advances in large-scale genomic analysis have uncovered a variety of causative genes and risk factors for amyotrophic lateral sclerosis (ALS). These gene variants map onto key pathogenic mechanisms relevant to all motor neuron cellular compartments as well as neighboring cells such as glia and interneurons. In this way, these mechanisms are genetically validated, enabling a greater confidence in their targeting for therapeutic benefit. Some of these mechanisms have emerged only in recent years due to new genetic information, including gene changes highlighting dysregulation of RNA processing and metabolism. There is significant overlap of some genes with those found in closely related disorders such as frontotemporal dementia (for example, C9orf72, CHCHD10, SQSTM1, TBK1, CCNF, FUS, TARDBP, OPTN, UBQLN2, TUBA4A, ATAXN2, VCP and CHMP2B). This suggests a closer relationship to broader neurodegenerative disorders, and indeed many of the pathways depicted are relevant in, for example, Alzheimer disease. ER, endoplasmic reticulum.
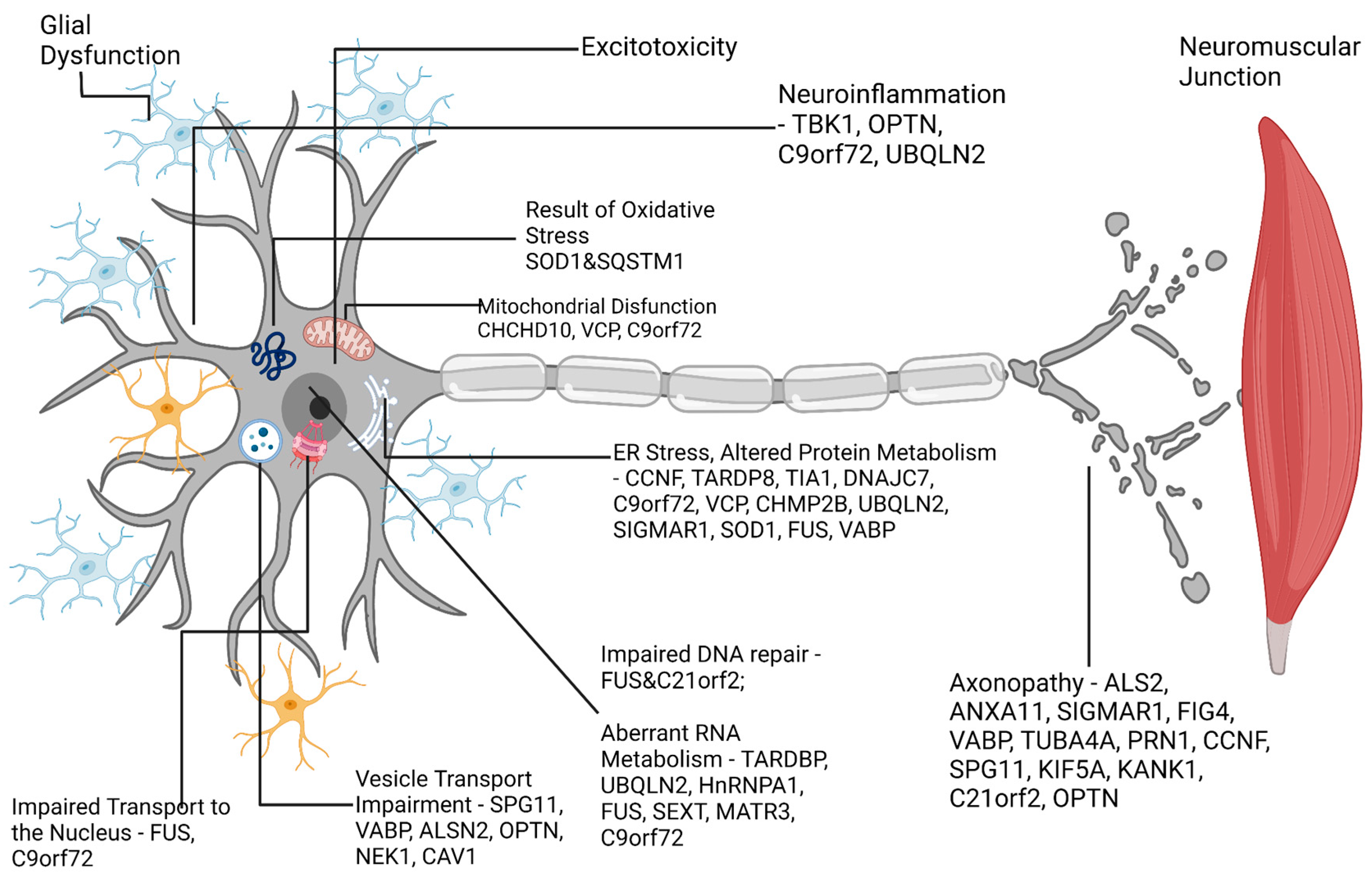
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



