
Submitted:
16 May 2023
Posted:
17 May 2023
You are already at the latest version
Abstract
Parathyroid-hormone-related protein (PTHrP) is encoded by PTHLH gene which, by alternative promoter usage and splicing mechanisms, can give rise to at least three isoforms of 139, 141 and 173 amino acids with distinct C-terminals. PTHrP is subjected to different post-translational processing that generates smaller bioactive forms, comprising amino terminus, midregion (containing a nuclear/nucleolar targeting signal) and carboxy terminus peptides. Both the full-length protein and the discrete peptides are key controllers of viability, proliferation, differentiation and apoptosis in diverse normal and pathological biological systems via the reprogramming of gene expression and remodulation of PKA or PKC-mediated signalization mechanisms. The aim of this review is to pick up selected studies on PTHrP-associated signatures as revealed by molecular profiling assays, focusing on the available data about exemplary differentiating, differentiated or non-tumoral cell and tissue models. In particular, the data presented relate to adipose, bone, dental, cartilaginous and skin tissues, and also intestinal, renal, hepatic, pulmonary and pancreatic epithelia, with a focus on hepatic fibrosis-, pancreatitis- and diabetes-related changes as diseased states. Whether reported, the biochemical and/or physiological aspects associated with the specific molecular modulation of gene expression and signal transduction pathways in the target model systems under examination will be also briefly commented.
Keywords:
cell biology
; gene expression
; adipose tissue
; bone
; cartilage
; intestine
; kidney
; liver
; lung
; pancreas
; skin
1. A Briefing Note about Parathyroid Hormone-Related Protein (PTHrP) Stucture and Function
PTHrP is the product of PTHLH gene which extends more than 15 kb of genomic DNA being located on chromosome 12. The PTHLH gene exhibits a complex organization with three different promoters and alternative splicing mechanisms producing multiple mRNA variants which differentiate for their 3′ ends encompassing both coding and untranslated regions. Three isoforms of 139, 141, and 173 amino acids with distinct C-terminals, are the protein product of the different translation patterns (Table 1). The extreme N-terminus displays sequence homology with PTH, thus binding with equal affinity to the shared G protein-linked PTH/PTHrP receptor PTH1R.
PTHrP isoforms are polyhormones subjected to different post-translational processing that generates smaller secretory forms of the peptide. These include PTHrP (1-36), which contains homology with PTH and activates PTH1R. Other peptides consist of the midregion fragments, such as (38–94) and (67–86), which have been shown to influence transplacental calcium transport and the growth and invasive behavior of breast epithelial cells, and the C-terminal fragment comprised of PTHrP (107-139), a.k.a. osteostatin, which has been shown to act on skin, heart, and bone cells [1].
The presence of a lysine/arginine-rich bipartite sequence in midregion PTHrP fragment, which is homologous to the nuclear/nucleolar targeting signal (NTS) present in SV40 large tumor antigen (able to direct importin β/Ran GTPase-mediated import), may allow an “intracrine” route supplementing the autocrine/paracrine counterpart (Figure 1) [3].
Over the past 30 years, a great deal of research has demonstrated that PTHrP participates in various complex signaling pathways through its membrane and nuclear effects. It has been shown that the full-length protein and its discrete fragments are multifaceted critical regulators of proliferation, differentiation, and apoptosis acting on PKA or PKC-mediated signalization, whose main, but not exclusive, targets are P21, Akt, and NFκB. The detailed molecular dissection of the involvement of PTHrP in signal transduction mechanisms has been the object of an extensive review literature [4,5]. On the other hand, PTHrP or its discrete domains have been proven to affect gene expression in a direct and substantial way in both normal, disease-affected, and neoplastic cells. A comprehensive recapitulation of PTHrP-dependent modulation of gene signatures in cancer cells has already appeared [6].
The aim of this review is to pick up selected studies on PTHRP-associated signatures as revealed by gene expression profiling assays, focusing on the available data about exemplary differentiating, differentiated or non-tumoral cell and tissue models. Whether reported, the biochemical and/or physiological aspects associated with the specific molecular modulation in the target model systems under examination will be also briefly commented.
2. Adipose Tissue: PTHrP-Related Signatures in Adipogenesis and Transdifferentiation
Adipogenesis regulates adipose tissue expansion and function [7]. Despite the well-known role of PTHrP in the differentiation programming of stem cells into different cellular linages, its role in the regulation of adipogenic differentiation by human fat tissue–derived stem cells has been only partially elucidated at gene expression level. Roca-Rodriguez et al. [8] showed that PTHrP was expressed in both human visceral and subcutaneous adipose tissue. Moreover, PTHRP expression progressively decreased during adipogenesis from undifferentiated mesenchymal cells. As a confirmation, data collected after PTHRP silencing in adipogenesis-committed stem cells demonstrated the down-regulation of the adipogenic markers PPARG2, FABP4, ADRP and CEBPA, coding for peroxisome proliferator activated receptor γ, fatty acid-binding protein 4, lipid droplet-associated adipose differentiation-related protein and transcription factor CCAAT enhancer-binding protein α, respectively. Furthermore, they showed that PTHRP expression correlated with obesity-related morbidities, such as the setting of insulin resistance and the increase of body mass index and hip circumference in patients affected by type 2 diabetes, thereby defining PTHrP as a key regulator of their development. The mechanism by which PTHrP may switch the differentiation of stem cells from adipo- to osteogenesis, thereby inhibiting fat tissue formation, was studied in the rodent pluripotent mesenchymal cell line C3H10T1⁄2. Co-exposure of cells to bone morphogenetic protein-2 (BMP2) and PTHrP determined the down-regulation of PPARγ and aP2 (adipocyte fatty acid-binding protein) and the concurrent up-regulation of alkaline phosphatase, type I collagen, and osteocalcin mRNA levels. The PKC-mediated signalization was found to be at least in part implicated in this activity [9].
Dealing with bone-adipose tissue endocrine interplay, Zhang et al. [10] demonstrated that in Ptch1c/c;HOC-Cre mutant mice, characterized by the perturbation of energy metabolism, the up-regulated Hedgehog signalization increased bone-derived PTHrP release. This, in turn, triggered the modulation of the PKA/cAMP and Akt/Foxo pathways leading to the over-expression of UCP1, coding for the uncoupling protein-1 that mediates energy expenditure by thermogenesis, and subsequent white adipose tissue (WAT) browning and enhancement of heat production. Moreover, PTHrP determined the increase of adiponectin mRNA and protein levels [10]. Adiponectin is involved in glucose homeostasis, because of its insulin-sensitizing activity [11,12], thereby indicating that PTHrP is also involved in the regulation of energy metabolism at this level. Since skeletal muscles also contribute to energy metabolism, it was also proven that in the mutant mice they underwent atrophy and adiponectin-triggered increase of fatty acid via significant up-regulation of the genes whose products are involved in fatty acid oxidation (ACO, CPT1, FABP3) and glucose uptake (GLUT1, GLUT4). The action of adiponectin was mediated by the activation of 5′-AMP-activated protein kinase (AMPK); moreover, in the mutant mice AMPK was also activated in the liver in which the expression of GLUT1 was markedly increased. Therefore, in this experimental model PTHrP along with adiponectin contribution was responsible of hypoglycemia due to glucose uptake and systemic fatty acid oxidation. As a further support to this evidence, PTHrP was also found implicated in the higher rate of oxygen consumption and waste of fat and muscle tissues occurring in the Lewis lung carcinoma model of cancer cachexia developed in syngeneic C57BL/6 mice [13]. In particular, PTHrP (1-34), released among the tumor-derived factors, was proven to stimulate thermogenic gene expression, specifically up-regulating UCP1 and DIO2, the latter coding for type 2 iodothyronine deiodinase, a selenoenzyme which increases during cold stress only in brown adipose tissue, ultimately resulting in the onset of hypermetabolism [14].
A recent study by Qin and colleagues [15] highlighted the role of PTHrP in both opposing brown adipose tissue (BAT) whitening and promoting WAT browning in mice transduced with adeno-associated PTHrP-encoding virus vector, submitted to high fat diet (HFD) for some weeks. PTHrP was found to protect the animals from the diet–induced onset of obesity stimulating WAT transdifferentiation and maintenance of BAT via up-regulation of UCP1, UCP2 and PGC1α, the latter coding for PPARγ coactivator 1α. Also VEGFA, coding for vascular endothelial growth factor A, was up-regulated and considered responsible of the concomitant state of inflammation in BAT. In parallel, in the liver of HFD-submitted mice over-expression of PTHrP was proven not only to attenuate the transcription level of the genes coding for enzymes and receptors responsible of fatty acid synthesis (ACSL1, FASN, PPARG) but also to enhance that of lipolysis-related enzymes and receptors (ATGL, ACOX1, CPT1A and PPARA). In addition, FGF21, coding for fibroblast growth factor 21, atypical member of FGF family active on glucose and lipid metabolism [16] and on adiponectin production, was prominently up-regulated in the liver of PTHrP-overexpressing mice, thereby resulting beneficial for the mitigation of the obesity-linked metabolic diseases such as hepatic steatosis, insulin resistance and glucose intolerance.
3. Bone and Dental Tissues: PTHrP-Related Signatures in Osteoblatogenesis, Osteoclastogenesis and Ossification
The present paragraph reports selected examples referred to bone and dental tissues, since PTHrP-associated gene signatures in bone remodeling process have been the object of extensive investigation since the late 90s. One of the first evidence was provided by De Miguel et al. [17] who reported the PKC-mediated up-regulation of IL-6 mRNA, a putative osteoblast differentiation factor also involved in bone resorption in various disorders like malignant hypercalcemia and Paget’s disease, operated by PTHrP (1–34) and also (107–139) on osteoblasts from human trabecular bone (hOB). Both N- and C-terminal PTHrP were also proven to intervene in the production of VEGF by hOB cells and MG-63 osteosarcoma cells. Esbrit et al. [18] examined the underlying intracellular mechanism and demonstrated its induction at the transcriptional level and the implication of PKC signalization, thereby suggesting that the peptides may take part to the onset of vascularization in vivo during the endochondral ossification process. Additionally, Alonso et al. [19] produced interesting data on the potential anabolic action of the sole PTHrP (107-139) on bone mediated by its interplay with the VEGF system. In fact, the peptide was found to promote hOB and MG-63 cell survival by directly interacting with and transactivating VEGF receptor-2, and stimulating extracellular signal-regulated kinase (ERK) 1/2 and Akt signalization and RUNX2 activation. The anabolic action of the C-terminal peptide also aimed to tissue engineering applications was further described by Lozano et al. [20] who studied the effect of its introduction in gelatin–glutaraldehyde biopolymer-coated hydroxyapatite scaffolds aimed to improve the osteoinductive capability of this biomaterial for orthopedic implants. Using rat bone defect models and osteoblastic cell cultures, PTHrP (107-111) was found to down-regulate SOST, DKK1 and RANKL, coding for the WNT-pathway inhibitors sclerostin and Dickkopf-1 [21,22] and the osteoclastogenesis-inducer receptor activator of NF-κB ligand (RANKL), respectively, whereas up-regulating VEGF, OC and OPG, the latter two coding for the osteoblast differentiation factors osteocalcin and osteoprotegerin [23], respectively. The formation of osseous trabeculae in the cavitary bone defects with osteoblasts adhering to the trabecular surface was observed, strongly suggesting that C-terminal PTHrP may restrain osteoclastogenesis during bone regeneration.
Apart from the paper commented above, other articles highlighted the effect of PTHrP on the modulation of RANKL and OPG mRNA transcription. The periodontal ligament (PDL) plays an important role in root resorption of human deciduous teeth by odontoclasts. To assess how PDL cells are involved in osteoclastogenesis regulation, Fukushima et al. [24] examined the effects exerted by factors secreted by the tooth germ, including PTHrP. N-terminal PTHrP was found to induce osteoclast differentiation via the induction of RANKL and the reduction of OPG expression in PDL cells with the partial contribution of PKC, but not PKA, pathway. Also Sun et al. [25] reported the ability of PTHrP to up-regulate RANKL and down-regulate OPG in human dental follicle cells, thereby influencing ostoclastogenesis. Data from Mak et al. [26] demonstrated that PTHRP expression in mature osteoblasts was under the control of Hedgehog signalling. Ricarte et al. [27] analyzed the effects of PTH (1-34), PTHrP (1-36) and N-terminal PTHrP-analog abaloparatide, i.e., [Glu22,25, Leu23,28,31, Aib29, Lys26,30] PTHrP(1–34)–NH2, on RANKL expression by osteoblasts and unveiled some molecular aspects of the regulation, being based upon the induction of a particular arm of the cAMP/PKA/salt-inducible kinase (SIK) signaling axis and involving the nuclear localization of CREB-regulated transcription coactivator (CRTC)-2 and -3 mediated by the Ser/Thr phosphatases PP1 and PP2A (Figure 2).
Interestingly, in the study by Elango et al. [28] on the osteo-differentiation of mesenchymal stem cells (MSCs), the cooperative inhibitory effect of PTHrP and soluble RANKL (sRANKL), a circulating form released in vivo by disintegrin metalloproteinase-mediated cleavage of the membrane-bound component, was demonstrated. The molecular signatures associated to the coexposure-mediated osteogenesis inhibition included the enhanced down-regulation of OPG, OC, collagen, and cellular alkaline phosphatase mRNA transcription coupled with the decreased levels of mineral deposition conceivably due to the inhibition of the progressive ankylosis protein (ANK) signaling pathway, whose deficiency is known to impair osteoblastogenesis and bone formation [29]. The mineralization inhibition of bone nodules, coupled with the down-regulation of bone sialoprotein mRNA and protein levels, was also found by Kamel and Yee [30] after continuous and intermittent exposure of primary rat calvarial cells to N-terminal PTHrP. Wang et al. [31] demonstrated that administration of PTHrP was able to redress impaired bone fracture healings in PTHrP-deficient osteoporotic mice as also revealed by the increase of bone formation–related gene and protein expression levels, that is, those of alkaline phosphatase, type I collagen, RUNX2 and insulin-like growth factor (IGF)-1.
Zhu et al. [32] elucidated the mechanism by which PTHrP NTS and C-terminal fragment promote bone formation using mutant mice expressing PTHrP (1–84), a truncated form lacking both regions, and homozygous for p27KIP1 deletion, in particular examining whether the latter protein might function downstream of the PTHrP domains. Using the same animal model, Zhang et al. [33] examined whether the growth arrest and senescence generated in the mutant mice might be associated with oxidative stress and DNA damage response by assessing the effects of deletion of checkpoint kinase-2 (CHK2) regulator. As shown in Figure 3, their cumulative results indicated that PTHrP NTS and C-terminus inhibited p27KIP1, thus stimulating Bmi-1 protein which, in turn, inhibited p16 and p53. This inhibitory circuit allowed the cyclin D/CDK4/6 and cyclin E/CDK2 complexes to trigger the progression of bone marrow MSCs along the osteoblastic lineage via Rb phosphorylation. Moreover, activated Bmi-1 down-regulated ROS levels and suppressed the activation of the DNA damage response pathway that plays a possible role as a downstream target in the action of PTHrP to regulate skeletal development and growth.
Noteworthy, Martin-Guerrero et al. [34] reported for the first time the localization of PTH1R in bone cells’ primary cilia and proposed that PTHrP pro-survival action, likely following the mechanical stress-promoted transport of PTH1R to the appendages’ surface, may be mediated by the up-regulation of the Hedgehog effector GLI family zinc finger 1 (Gli1) protein, and the subsequent overexpression of Hedgehog transcription factor and activation of the related signaling pathway. On the other hand, PTHrP osteogenic activities, such as RUNX2, OC and OPG up-regulation, appeared to be mediated by primary cilia-dependent, but Gli1-independent, mechanisms (Figure 4).
Regarding the influence of PTHrP on senescence features led by inflammatory diseases, such as osteoarthritis (OA), in osteoblasts, the study by Platas et al. [35] showed that the C-terminal peptides PTHrP (107–111) and (107–139), but not the N-terminal domain, were able to reduce remarkably the expression of senescence markers induced in vitro by treatment of OA osteoblasts with IL-1β. In particular, the molecular signatures associated with the exposure to the peptides were the down-regulation of p53-, p21-, p16-, COX2-, caveolin 1- and AP-1 transcription factor -coding genes. In addition, the decreases in the activation of NF-κB, accumulation of γH2AX (histone marker of inflammation-induced DNA damage and aging), and production of PGE2 and IL-6, and the promotion of matrix mineralization were also reported, thereby supporting the hypothesis of the beneficial anti-senescence and anti-inflammatory action of the C-terminal moiety of PTHrP.
PTHrP protects also osteoblasts from oxidative stress, one of the several factors that prompt their apoptosis. In particular, Ardura et al. [36] demonstrated that PTHrP (1-37) could prevent the H2O2-induced p38 and ERK phosphorylation in osteoblastic MC3T3-E1 and MG-63 cells and also the high oxidative stress in an animal model, by increasing the mRNA expression of catalase and MAPK phosphatase-1. This suggested that the anti-apoptotic role of N-terminal peptide was accomplished via both reversing MAPK phosphorylation triggered by ROS and decreasing ROS levels.
In bone physiology, the cysteine-X-cysteine (CXC) family chemokine ligand 1 (CXCL1) is an important neutrophil chemoattractant acting during angiogenesis and inflammation. Interestingly, Onan et al. [37] identified its up-regulation as a PTHrP (1-141)-associated signature in osteoblastogenesis and suggested that the chemokine may play a fundamental role in attracting the osteoclast precursors, that expose its receptor CXCR2 on their surface, to the bone environment at the modeling sites.
Another example of molecular interactors associated to PTHrP, which does not exhaust the lenghty list present in the literature, is represented by the components of the WNT pathway. In this regard, a number of data were obtained in type I diabetes mellitus (TIDB) mouse models in which the inactivation of the WNT pathway and a decrease of bone mass occur, markedly augmenting fracture risk. Portal-Núñez et al. [38] reported that administration of PTHrP (1–36) and (107–139) resulted in the stabilization of β-catenin mediated by the down-regulation of CSNK1A1, coding for casein kinase I isoform α which is involved in the phosphorylation and degradation of catenin together with GSK-3β. In addition, the sole N-terminal peptide was able to reverse the decrease of Wisp-1, a final component of the pathway acting on growth promotion, whereas the sole C-terminal peptide increased the expression of WNT11, a non-canonical WNT pathway activator. Moreover, as also demonstrated by Maycas et al. [39], in the diabetic mice the peptides reversed the up-regulation of SOST, coding for the WNT-β catenin inhibitor sclerostin, thus contributing also in this way to bone formation. Noteworthy, the N-terminal PTHrP analog abaloparatide was proven to suppress, besides SOST expression, also that of DKK1, coding for the WNT signalling antagonist dickkopf-related protein 1 [40].
4. Cartilaginous tissue: PTHrP-related signatures in the repression of chondrocyte differentiation
Several early lines of evidence have revealed that PTHrP opposes chondrogenic differentiation both in vitro and in vivo [41,42,43]. Ito et al. [44] reported that in mouse chondrogenic embryonal carcinoma ATDC5 cells PTHrP negatively modulated the mRNA levels of bone morphogenetic protein-4 (BMP4), thus decreasing the amplitude of the chondrogenic signal. Moreover, in chick upper sternal chondrocytes co-treated with BMP2, PTHrP abrogated the expression of RUNX2, which in the cartilage acts as an inducer of chondrocyte maturation both in vitro and in vivo [45]. Interestingly, subsequent data indicated that PTHrP repressed chondrocyte hypertrophy via the PKA-activated dephosphorylation of histone deacetylase 4 (HDAC4) [46], thus adding the epigenetic regulation as a possible parallel further mechanism of action in PTHrP-blocked chondrogenesis.
In antler chondrocytes, PTHrP increased the expression of BCL2, whose product is involved in the suppression of chondrocytes’ apoptosis, and CCND1, coding for cyclin D1, while down-regulating RUNX2, thereby promoting cell proliferation while preventing differentiation [47]. In addition, in the same cell model system PTHrP was found able to down-regulate MMP13 and MMP9, whose products are metalloprotease 13 (collagenase-3) and -9 (92 kDa type IV collagenase), thereby restraining the proteolysis and remodeling of cartilage extracellular matrix (ECM), and blocking its maturation [48].
Particularly peculiar are the data obtained in normal articular human chondrocytes led to hypoxia. In fact, PTHRP was reported to be up-regulated by oxygen deprivation, acting, in turn, as a positive regulator of the key cartilage transcription factor SOX9 [SRY (sex determining region on the Y chromosome)-box 9], which led to the increased expression of COL2A1, coding for the α1 chain of type II collagen [49]. Hypoxia-exposed MSCs showed the PTHrP-stimulated down-regulation of MEF2C, coding for the transcriptional factor myocyte enhancement factor 2C. This suppressed chondrocytes’ hypertrophy by reducing the expression of COL10A1, coding for the α1 chain of type X collagen which is released by hypertrophic chondrocytes during endochondral osteogenesis [50].
Chondrogenesis was also studied in the case of a pulse, intermittent PTHrP treatment on MSCs. Noteworthy, the results obtained demonstrated that whether the constant application of PTHrP (1–34) suppressed chondrogenesis, conversely, pulsed exposure acted as a stimulator, as shown by the up-regulation of COL2A1 and reduced undesired hypertrophy, thus representing a best novelty in clinical treatments of cartilage defect regeneration [51].
Also, animal models in vivo showed the crucial importance of PTHrP and associated pathways in modulating chondrogenesis. From the early work of Karaplis and colleagues [52] it was known that knock-out mice for PTHRP die at birth with extensive chondrocyte hypertrophy. More than twenty years after, using multiple mouse genetic models Nishmori et al. [53] demonstrated that class I and II histone deacetylases (HDACs) are necessary for PTHrP action on chondrocyte differentiation. In particular, HDAC4 as the first protagonist and HDAC5 as an additional mediator were both involved in PTHrP signalling in chondrocytes. PTHrP action on HDAC4 appeared to suppress myocyte enhancer factor 2 (Mef2) function, thereby allowing the expression of RUNX2, necessary for chondrocyte hypertrophy. In consideration of the epigenetic role of the HDACs, these features open new scenarios of second level for the PTHrP-mediated gene expression regulation in chondrocyte differentiation.
5. Intestinal epithelium: PTHrP-related signatures in calcium uptake by enterocytes
Since PTHrP was initially identified as a tumor-derived PTH-resembling hypercalcemic factor, early studies were focused on elucidating its potential role in calcium transport using normal chick intestine as a model system. Indeed, the N-terminal PTHrP (1-40) domain was proven to stimulate the rapid Ca++ uptake in the perfused chick duodenum via a signal transduction pathway in which Ca++ channels are activated [54]. In mammals, a complex homeostatic system regulates the extracellular calcium concentrations and in particular calcium uptake in the small intestine, conceivably through paracellular or transcellular transport. Paracellular calcium transport is concentration gradient-dependent, thereby being a passive transport. On the other hand, transcellular transport is an active, energy-dependent transport, and consequently it is highly regulated. The three steps of the transcellular calcium transport include: calcium entry at the apical membranes through the epithelial calcium channel, the transport from the apical membrane to the basolateral membranes mediated by calcium-binding proteins, and the release of calcium from basolateral membrane via calcium transporters [55,56]. Liu et al. [57] investigated the molecular mechanism by which PTHrP induced calcium uptake in rat enterocytes and demonstrated that only PTHrP (1-40), but not (67-86) and (134-143), was able to induce a rapid calcium internalization. At gene expression level, this was paralleled by the up-regulation of TRPV6, CALB1, NCX1 and PMCA1 coding for potential vanilloid member 6 (a transcellular calcium transporter protein), calbindin, sodium-calcium exchanger 1 and plasma membrane calcium ATPase 1, respectively. Noteworthy, the first two proteins are known to be necessary for calcium uptake, whereas the other two ones are responsible for calcium extrusion from the basolateral membrane and therefore not involved in calcium internalization procedures. Furthermore, PTHrP (1-40) treatment increased PTHR1 mRNA level, and PKCα/β and PKA protein levels. Calcium uptake was inhibited when rat enterocytes were pre-treated with antibodies or inhibitors of either the calcium transmembrane transporters, PTHR1 receptor or PKCα/β, thus demonstrating that the intracellular pathway mediated by the last signal transducer was that involved in PTHrP-induced calcium resorption.
Furthermore, the PKA-RUNX2 pathway appeared to be involved in PTHrP-triggered epithelial-mesenchymal transition (EMT) in intestinal epithelial cells. In particular, the PTHrP/PTHR1 system was found to play an important role in the onset of EMT that promotes intestinal epithelial cell transition into fibroblasts, thus inducing intestinal fibrosis. He et al. [58] demonstrated that the PTHrP/PTHR1 system induced RUNX2 expression and subsequent deposition of type I collagen via PKA pathway activation in an animal model of intestinal fibrosis and in the intestine of Crohn disease patients. Indeed, the PTHrP-triggered stimulation of EMT-related markers and type I collagen accumulation was blocked when RUNX2 was genetically silenced with siRNA demonstrating that the PKA-RUNX2 axis was implicated in the process. Taken together, these data show that PTHrP plays a pivotal role in the establishment of the fibrotic disease.
6. Liver parenchyma: PTHrP-related signatures in the fibrotic reaction of hepatic stellate cells
Hepatic stellate cells, which reside in the subendothelial area of the Disse space, are triggered to myofibroblast-like cell differentiation by factors released by the damaged hepatocytes and are responsible of the remodelling of the ECM and the fibrotic reaction associated with the onset of chronic liver injury [59]. Liang et al. [60] reported that administration of PTHrP (1-36) to human normal hepatic stellate cells and the LX-2 cell line in vitro increased the mRNA and protein levels of α smooth muscle actin (α-SMA; myofibroblast marker), collagen I (principal constituent of the ECM), MMP-2 (contributing to the alteration of normal liver ECM [61]) and TGF-β1 (main cytokine involved in fibrosis). Additional experiments on CCl4-induced mice model of hepatic fibrosis demonstrated that the PTHrP mRNA levels increased in a time-dependent manner, and also in this case they were related with the up-regulation of the genes coding for TGF-β1, collagen I and α-SMA. The same result was achieved by overexpressing PTHrP via administration of a liver-targeted PTHrP recombinant adeno-associated vector through the mice’s tail vein and the animals developed spontaneously liver fibrosis within 6 months. Moreover, it was observed that in PTHrP-induced fibrosis, the expressions levels of PTCH, SHH and GLI2 genes, coding for Patched-1, Sonic Hedgehog and GLI Family Zinc Finger-2 proteins, respectively, were also increased indicating that PTHrP effect on the activation of stellate cells was mediated through Hedgehog signalization by the PTH1R-PKCθ-Hedgehog axis [62]. These cumulative results strongly suggested that PTHrP might be considered as one of the cytokines playing a central role in the onset of liver fibrotic disease
7. Lung epithelium: PTHrP-related signatures in pulmonary surfactant production.
In the lung, the PTHrP-associated gene regulatory network is involved in the control of fundamental aspects of the development and homeostasis of the organ, such as the epithelial-mesenchymal paracrine cross-talk and the production of surfactant [63]. In their study aimed to understand the mechanism of these events during the differentiation process of epithelial type II (TII) cells, Torday and Rehan [64] showed that PTHrP acted as a “stretch-sensitive” product of such cells. In particular, stretching stimuli induced pulmonary surfactant production through a paracrine epithelial-mesenchymal-epithelial loop also mediated by leptin, a soluble product of the mature lipofibroblast (LFs). Their data demonstrated that stretching of LFs and TIIs in co-culture resulted in the enhancement of PTHrP signalling from the epithelium to the mesenchyme through the coordinated up-regulation of both PTHrP itself and PTH1R expression by TFII cells and LFs, respectively. In turn, stretching increased the signalling from the mesenchyme to the epithelium by augmenting leptin stimulation of surfactant’s phospholipid synthesis by TII cells, which could be blocked by inhibitors of PTHrP or leptin, thus supporting the existence of the paracrine feedback loop. A PTHrP-related signature identified in this cAMP/protein kinase A-dependent mechanism was the up-regulation of the mRNA of adipocyte differentiation-related protein (ADRP), involved in the mechanism of lipid uptake by LFs [65]. As a further confirmation of the leptin-PTHrP axis, Oruqaj et al. [66] showed that elevated plasma leptin levels induced in high fat diet-fed mice up-regulated the pulmonary mRNA expression of PTHrP, PTH1R and other classical PTHrP-dowstream target genes such as ADRP or the key lipogenic marker PPARG; conversely, genetically leptin-deficient mice displayed reduced steady-state levels of PTHrP. Interestingly, PTHrP was also proven to exert an inhibitory effect on the trans-differentiation of LFs into myofibroblasts, commonly associated to nicotine exposure, via PPARγ up-regulation [67]. Thus, the sustained activation of the PTHrP-driven epithelial-mesenchymal paracrine cross-talk may also act as a protective mechanism for prevention of the nicotine-induced lung injury.
8. Exocrine and endocrine pancreas: PTHrP-related signatures in β cell biology and in the onset of pancreatitis
Sawada et al. [68] examined the effect of the differentiation status of pancreatic insulin-producing cells on PTHrP synthesis using different passages of the MIN6 cell line as an in vitro model system of well-differentiated and less-differentiated β cells. PTHrP was found expressed and secreted in both conditions, but increased with increasing passage, i.e., moving toward a less-differentiated phenotype. Also the expression and secretion of furin, the PTHrP precursor-processing enzyme [69], followed the same trend whereas PTH1R expression was similar in all the experimental conditions tested. Furthermore, PTHrP increased the insulin mRNA content through the cAMP pathway in highly-differentiated MIN6 cells and also in primary cultured islets; on the other hand, it stimulated DNA synthesis, a marker of cell proliferation, in the less-differentiated MIN61 cultures. These data strongly suggested that PTHrP might be a regulator of the balance between growing and differentiated cells in the heterogenous β cell population of pancreatic islets. Within this context, PTHrP responded to an insulin shortage by inducing cell growth in the proliferation-prone cell fraction in an autocrine manner and induced insulin up-regulation in highly differentiated cells, thus compensating for a loss in insulin-producing capacity in the whole cell mass in a paracrine manner.
Studies by Guthalu Kondegowda et al. [70] showed that PTHrP (1-36) was sufficient to exert biological effects on β cell proliferation and function in vitro. The molecular signatures associated to cell growth following peptide administration were the late G1/S cell cycle activators cyclin E and cdk2, over-expressed in treated human islets, which caused an increase in the percentage of cells in the S-phase of the cell cycle acting in a synergistic way. This was coupled with a significant enhancement of insulin secretion at both physiological and increased glucose concentrations, thus confirming that exposure to PTHrP did not lead to cell de-differentiation. Noteworthy, different results were obtained by analyzing the islets from rat insulin II promoter (RIP)-PTHRP transgenic mice [71] since the difference in cell cycle-related gene expression levels lied in the up-regulation of the G1/S activator cyclin D2 and the down-regulation of the inhibitor p16Ink4a, thereby suggesting that the systemic and acute administration of the sole N-terminal peptide to adult mice may stimulate β cell replication via different mechanisms.
Apart from the effect exerted by PTHrP in normal physiology, its involvement in pancreatic diseases was also studied. Within this context, the studies by Bhatia at al. [72] focused onto the molecular framework associated to the onset of acute pancreatitis, a common and potentially lethal necro-inflammatory disease of the exocrine pancreas that can degenerate into a chronic form with an elevated risk of cancer development [73]. Indeed, treatment of acinar and stellate cells, both from primary and stabilized (AR42J and irPSCc3) cultures with cerulein or ethanol induced an increase of PTHrP at mRNA and protein level. The molecular signatures associated to acinar cell exposure were the up-regulation of the mRNA levels for IL-6 and ICAM-1, both factors involved in the control of the inflammatory response, whereas those associated to stellate cell exposure were the up-regulation of the mRNA levels for fibronectin and procollagen type I, which are key mediators for the development of fibrosis. In addition, the treatment with PTHrP addressed acinar cells to apoptosis. Conceivably, the increase in PTHrP levels following alcohol abuse may be the starting point of a cascade of events that ultimately leads to the inflammatory and fibrotic response and to the loss of acinar cells due to apoptotic promotion. The pathway proposed by the Authors for the contribution of PTHrP to the development of acute pancreatitis is shown in Figure 5.
The complex PTHrP-involving molecular network occurring in chronic pancreatitis was investigated by Rastellini et al. [74]. It is known that BMP2, apelin, and PTHrP signalization systems are expressed in the rodent and human pancreas, and that TGF-β is a major effector in the genesis and progression of the disease [75,76,77]. They found a bidirectional, negative feedback loop that targeted the transcriptional regulation of apelin and PTHrP; in particular, the anti-inflammatory and anti-fibrotic role of BMP2 appeared to be mediated by the concurrent activation of apelin- and inhibition of PTHrP-signaling during pancreatitis. In addition, TGF-β up-regulated PTHrP expression, and, in turn, both PTHrP and TGF-β stimulated the expression of gremlin10, an inhibitor of the BMP2-apelin axis that regulates the downstream inflammatory response and fibrosis. On the other hand, BMP2 and apelin inhibited PTHrP expression, thereby quenching the inducing effects of PTHrP, and possibly TGF-β, on inflammation and fibrosis. Therefore, these cumulative results substantiated the occurrence of a composite interplay underlying the response to the chronic injury that can either block or amplify organ deterioration associated to the disease, and that may offer insights for pharmacologic interventions for the treatment of this pathology.
9. Renal parenchyma: PTHrP-related signatures in the promotion of kidney cell survival and in diabetes-related changes
Within the context of the investigation on the pro-survival role of PTHrP in cells subjected to apoptotic stimuli, studies by Okoumsassoun et al. [78] on human Hek293 embryonic kidney cells addressed to apoptosis during treatment with TNFα showed that the expression of full-length PTHrP, or the pre-exposure to its NTS peptide, blocked the onset of programmed cell death as evidenced by the lack of caspase activation and the up-regulation of anti-apoptotic proteins at the mitochondrial membrane. This was accomplished through the increase in the mRNA expression of casein kinase 2 (CK2), a known potent suppressor of apoptosis [79], followed by its sustained accumulation and rapid co-translocation with PTHrP to the nuclear compartment. In addition, albeit homogeneously present in the mitochondria regardless of the presence of PTHrP, CK2 activity was significantly increased in cells expressing PTHrP or pre-treated with the NTS peptide. A link between PTHrP and CK2 in mediating nuclear and mitochondrial events related to cell survival was postulated, based upon the possible phosphorylation activity of the kinase on PTHrP which may modify its RNA-binding activity, and CK2 targeting of the caspase 8 repressor protein ARC and the mitochondria-associated proteins Bid and Bad. Moreover, NTS exposure reduced the expression level of BAK, BAD and BID genes, thereby suggesting also an intervention at the transcription level.
It is known that after renal injury, the EMT process is committed to the rescue of the damaged tubules. In fact, the surviving tubular epithelial cells de-differentiate into mesenchymal cells that migrate toward the damaged areas and subsequently re-differentiate into the original phenotype to restore tubular integrity. TGF-β is a key regulator of EMT and also PTHrP (1-36) was proven to intervene in modulating the process. In particular, Ardura et al. [80] showed that the peptide up-regulated TGF-β mRNA and protein in mouse tubular cells and that, in turn, antagonizing TGF-β strongly diminished EMT induction by N-terminal PTHrP. This suggested that TGF-β was a downstream mediator of PTHrP action, which occurs via EGFR activation of ERK1/2 pathway known to interfere with the integrity of intercellular junctions and to induce the accumulation of cytoplasmic β-catenin, both events linked to EMT onset [81]. These first data were subsequently supplemented by evidence demonstrating that the pro-survival action of PTHrP (1-36) was also mediated by the over-expression and increased nuclear translocation of RUNX2, instrumental for the up-regulation of the anti-apoptotic proteins Bcl-2 and osteopontin in mouse and human tubuloepithelial cell lines and in PTHrP-overexpressing transgenic mice in vivo [82]. A schematic model for the mechanism of N-terminal PTHrP pro-survival and anti-apoptotic activities in tubuloepithelial cells has been proposed by Kramann and Schneider [83] (Figure 6).
The viability-promoting role of PTHrP was confirmed by Hochane et al. [84] using primary cultures of murine mesangial cells (MC) in an in vitro model of inflammatory response mimicking the onset of mesangial proliferative glomerulonephritis. Exposure to IL1-β and TNFα was proven to up-regulate PTHRP expression and also to extend the half-life of its mRNA through binding with HuR, an RNA-stabilizing factor known to interact with the 3′-UTR of the PTHrP-141 mRNA isoform, and delaying its degradation [85]. In addition, PTHrP treatment on MC cells induced an up-regulation of a panel of cytokines and chemokines and also of IL1-β itself suggesting a positive feedback loop on this gene expression. Furthermore, PTHrP treatment up-regulated COX2, a target gene of NF-κB [86], and this over-expression was blocked by an inhibitor of IκB kinase indicating the PTHrP-mediated activation of NF-kB pathway in MC. Interestingly, the up-regulation of COX2 was able to prevent apoptosis through the cAMP/PKA and PI3K/Akt pathways. Thus, Hochane and coworkers proposed a mechanism in which PTHrP is overexpressed after the exposition of MC to IL1-β and TNFα proinflammatory cytochines and acts itself as an inflammatory cytokine, increasing the expression levels of interleukines, chemokines and COX2 through the NF-kB pathways, while acting also as a survival factor through the reduction of the apoptotic effect of IL1-β and TNFα via COX-2 metabolites.
In early diabetes, renal hyperfiltration occurs due to an increase in proximal tubule reabsorption. The hyperfiltration state may contribute to structural renal lesion which are preceeded by the hypertrophy of the organ. A number of cytokines and molecules are involved in renal changes occurring in diabetes [87,88]. Both PTHrP and PTH1R are abundantly expressed throughout the kidney’s parenchyma and PTHrP was proven to modulate renal plasma flow and glomerular filtration rate, as well as renal cell proliferation [89]. Experimental evidence suggests that it is involved in the pathophysiology of the diabetic kidney, being up-regulated during renal injury [90]. Romero et al. [91] and Ortega et al. [92] investigated the mechanism of high glucose-induced hypertrophy in cultured mouse podocytes, renal tissue explants and animal models of diabetes, and found that angiotensin II-induced PTHrP (1-36) overproduction resulted in the increased expression of TGFβ. This, in turn, mediated an early increase of both cyclins D1 and E and cdk2 activity followed by inactivation of cyclinE/cdk which is an acknowledged molecular signal directing the growth pattern towards hypertrophy. In addition, they also found that p27Kip1, inhibitor of the cyclin E/cdk2 complex, was up-regulated thus contributing to the hypertrophy response. The N-terminal PTHrP-stimulated overproduction of p47phox, involved in ROS production, was also demonstrated by Chen et al. [93] in rat mesangial cells and linked to the activation of EGFR, Akt and ERK1/2 signaling, and to the resulting ECM accumulation associated to the fibrotic reaction occurring in the diabetic rat kidney (Figure 7).
10. Skin: PTHrP-related signatures in keratinocyte development and anti-aging effect
Epidermal keratinocytes are PTH1R-lacking and PTHrP-expressing cells which do not produce but can respond to IGF-1 binding to their surface receptors able to promote their proliferation as well as ECM protein production [94]. Shin et al. [95] demonstrated the possible paracrine interaction between dermal fibroblasts and keratinocytes in which keratinocyte-derived PTHrP activated cAMP production in PTH1R-exposing dermal fibroblasts and enhanced the PKA-dependent steady state mRNA levels of IGF-1 and its secretion by the latters that, in turn, enhanced keratinocyte development and matrix synthesis, instrumental for skin remodeling and repair (Figure 8).
Keratinocyte growth factor (KGF) is highly expressed by stromal cells from different tissues, including the skin. In contrast, it acts exclusively on epithelial cell types through the KGF receptor (KGFR), stimulating DNA synthesis and supporting the growth of various types of epithelial cells including keratinocytes. To shed further light on the complex epidermis/dermis interplay, Blomme et al. [96] studied the effects of KGF on PTHrP expression and secretion by normal human foreskin keratinocytes (NHFK) and the effects of PTHrP on KGF expression and secretion by normal human dermal fibroblasts (NHDF). Their studies demonstrated that N-terminal PTHrP stimulated KGF mRNA and protein expression and secretion by NHDF in a dose-dependent manner, whereas KGF did not regulate PTHrP expression and secretion by NHFK, thereby qualifying the keratinocyte-derived PTHrP as a potential paracrine regulator of KGF expression by dermal fibroblasts in vivo.
Miao et al. [97] generated PTHrP knock-in (PTHrP KI) mice expressing PTHrP (1–84), i.e., a form lacking the NTS and the C-terminus, and characterized by the premature aging of the skin. Further, in PTHrP KI mice they found high levels of the cdk inhibitor P27, whose involvenment in skin differentiation process has long been known [98]. To determine the effects of p27 deficiency on premature skin aging of PTHrP KI mice, Jiang et al. [99] compared the skin phenotypes of PTHrP KI mice to those of p27 knockout mice (p27-/-) and of double homozygous p27-deficient PTHrP KI mice (p27 -/- /PTHrP KI). P27 shortfall in PTHrP KI mice partially corrected skin premature impairment through a reduction of cellular senescence, being the down-regulation of the expression of cdk inhibitors (including p19, p27, and p53) and the up-regulation of the expression of cyclin E and CDK2 the associated molecular signatures evidenced. In addition, ROS accumulation was reduced significantly in p27 -/- PTHrP KI mice compared with PTHrP KI mice and therefore the expression levels of the genes coding for antioxidant enzymes were examined in the skin of the animal models by real-time RT-PCR. Unlike wild-type mice, SOD1, SOD2, GPX1, GPX4, CAT and GSR, coding for superoxide dismutase-1 and -2, glutathione peroxidase-1 and -4, catalase and glutathione-disulfide reductase, respectively, resulted up-regulated in the skin of p27 knockout mice and down-regulated significantly in PTHrP KI and p27 -/- PTHrP KI mice, thus suggesting that the rescue of the skin-aging phenotype in the latter animals occurred via some degree of recovery of the redox balance.
11. Conclusions
Molecular profiling has identified signatures associated to full-length PTHrP or its distinct domains in various differentiated and differentiating model systems in vitro and in vivo, also correlated to the onset of diverse non-tumoral and tumoral diseases. In this review, we have examined a number of distinct sets of molecular signatures which have been distinguished on the basis of the different histo- and cytotypes considered. These molecular markers are linked to relevant cellular responses and to the activation of transduction pathways, thus expanding the knowledge on the multiple roles of PTHrP in controlling the transcriptional activity and signaling mechanism in a variety of organs with respect to their functional/pathological states. Enhanced understanding of the role of PTHrP in cell fate determination at the molecular level will facilitate new approaches to improve the maintenance of organ homeostasis and assist in developing more effective therapies.
Author Contributions
Conceptualization, C.L. and F.C..; writing—original draft preparation, M.L., F.N., G.A., C.L. and F.C.; writing—review, C.L.; writing—editing, M.L., F.N., G.A., C.L. and F.C.; supervision, C.L. and F.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the University of Palermo (Italy), grant number FFR 2023 to C.L., F.C. and F.N.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Luparello, C. Parathyroid Hormone-Related Protein (PTHrP): A Key Regulator of Life/Death Decisions by Tumor Cells with Potential Clinical Applications. Cancers (Basel) 2011, 3, 396–407. [Google Scholar] [CrossRef] [PubMed]
- Longo, A.; Librizzi, M.; Naselli, F.; Caradonna, F.; Tobiasch, E.; Luparello, C. PTHrP in Differentiating Human Mesenchymal Stem Cells: Transcript Isoform Expression, Promoter Methylation, and Protein Accumulation. Biochimie 2013, 95, 1888–1896. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.J.; Johnson, R.W. Multiple actions of parathyroid hormone-related protein in breast cancer bone metastasis. Br J Pharmacol 2021, 178, 1923–1935. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.J. PTH1R Actions on Bone Using the cAMP/Protein Kinase A Pathway. Front Endocrinol (Lausanne) 2022, 12, 833221. [Google Scholar] [CrossRef] [PubMed]
- Suva, L.J.; Friedman, P.A. Structural pharmacology of PTH and PTHrP. Vitam Horm 2022, 120, 1–21. [Google Scholar] [PubMed]
- Luparello, C.; Librizzi, M. Parathyroid hormone-related protein (PTHrP)-dependent modulation of gene expression signatures in cancer cells. Vitam Horm 2022, 120, 179–214. [Google Scholar]
- Lagathu, C.; Christodoulides, C.; Tan, C.Y.; Virtue, S.; Laudes, M.; Campbell, M.; Ishikawa, K.; Ortega, F.; Tinahones, F.J.; Fernández-Real, J.-M.; Orešič, M.; Sethi, J.K.; Vidal-Puig, A. Secreted Frizzled-Related Protein 1 Regulates Adipose Tissue Expansion and Is Dysregulated in Severe Obesity. Int J Obes 2010, 34, 1695–1705. [Google Scholar] [CrossRef]
- Roca-Rodríguez, M.M.; el Bekay, R.; Garrido-Sanchez, L.; Gómez-Serrano, M.; Coin-Aragüez, L.; Oliva-Olivera, W.; Lhamyani, S.; Clemente-Postigo, M.; García-Santos, E.; de Luna Diaz, R.; Yubero-Serrano, E.M.; Fernández Real, J.M.; Peral, B.; Tinahones, F.J. Parathyroid Hormone-Related Protein, Human Adipose-Derived Stem Cells Adipogenic Capacity and Healthy Obesity. J Clin Endocrinol Metab 2015, 100, E826–E835. [Google Scholar] [CrossRef]
- Chan, G.K.; Miao, D.; Deckelbaum, R.; Bolivar, I.; Karaplis, A.; Goltzman, D. Parathyroid Hormone-Related Peptide Interacts with Bone Morphogenetic Protein 2 to Increase Osteoblastogenesis and Decrease Adipogenesis in Pluripotent C3H10T½ Mesenchymal Cells. Endocrinology 2003, 144, 5511–5520. [Google Scholar] [CrossRef]
- Zhang, X.; Cheng, Q.; Wang, Y.; Leung, P.S.; Mak, K.K. Hedgehog Signaling in Bone Regulates Whole-Body Energy Metabolism through a Bone–Adipose Endocrine Relay Mediated by PTHrP and Adiponectin. Cell Death Differ 2017, 24, 225–237. [Google Scholar] [CrossRef]
- Combs, T.P.; Berg, A.H.; Obici, S.; Scherer, P.E.; Rossetti, L. Endogenous Glucose Production Is Inhibited by the Adipose-Derived Protein Acrp30. J Clin Invest 2001, 108, 1875–1881. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Kamon, J.; Ito, Y.; Tsuchida, A.; Yokomizo, T.; Kita, S.; Sugiyama, T.; Miyagishi, M.; Hara, K.; Tsunoda, M.; Murakami, K.; Ohteki, T.; Uchida, S.; Takekawa, S.; Waki, H.; Tsuno, N.H.; Shibata, Y.; Terauchi, Y.; Froguel, P.; Tobe, K.; Koyasu, S.; Taira, K.; Kitamura, T.; Shimizu, T.; Nagai, R.; Kadowaki, T. Cloning of Adiponectin Receptors That Mediate Antidiabetic Metabolic Effects. Nature 2003, 423, 762–769. [Google Scholar] [CrossRef] [PubMed]
- Kir, S.; White, J.P.; Kleiner, S.; Kazak, L.; Cohen, P.; Baracos, V.E.; Spiegelman, B.M. Tumour-Derived PTH-Related Protein Triggers Adipose Tissue Browning and Cancer Cachexia. Nature 2014, 513, 100–104. [Google Scholar] [CrossRef] [PubMed]
- De Jesus, L.A.; Carvalho, S.D.; Ribeiro, M.O.; Schneider, M.; Kim, S.-W.; Harney, J.W.; Larsen, P.R.; Bianco, A.C. The Type 2 Iodothyronine Deiodinase Is Essential for Adaptive Thermogenesis in Brown Adipose Tissue. J Clin Invest 2001, 108, 1379–1385. [Google Scholar] [CrossRef] [PubMed]
- Qin, B.; Qincao, L.; He, S.; Liao, Y.; Shi, J.; Xie, F.; Diao, N.; Bai, L. Parathyroid Hormone-Related Protein Prevents High-Fat-Diet-Induced Obesity, Hepatic Steatosis and Insulin Resistance in Mice. Endocr J 2022, 69, EJ20–EJ0728. [Google Scholar] [CrossRef] [PubMed]
- Tezze, C.; Romanello, V.; Sandri, M. FGF21 as Modulator of Metabolism in Health and Disease. Front Physiol 2019, 10, 419. [Google Scholar] [CrossRef] [PubMed]
- De Miguel, F.; Martinez-Fernandez, P.; Guillen, C.; Valin, A.; Rodrigo, A.; Martinez, M.E.; Esbrit. P. Parathyroid hormone-related protein (107-139) stimulates interleukin-6 expression in human osteoblastic cells. J Am Soc Nephrol 1999, 10, 796–803. [Google Scholar] [CrossRef]
- Esbrit, P.; Alvarez-Arroyo, M.V.; DE Miguel, F.; Martin, O.; Martinez, M.E.; Caramelo, C. C-terminal parathyroid hormone-related protein increases vascular endothelial growth factor in human osteoblastic cells. J Am Soc Nephrol 2000, 11, 1085–1092. [Google Scholar] [CrossRef]
- Alonso, V.; de Gortázar, A.R.; Ardura, J.A.; Andrade-Zapata, I.; Alvarez-Arroyo, M.V.; Esbrit, P. Parathyroid hormone-related protein (107-139) increases human osteoblastic cell survival by activation of vascular endothelial growth factor receptor-2. J Cell Physiol 2008, 217, 717–727. [Google Scholar] [CrossRef]
- Lozano, D.; Sánchez-Salcedo, S.; Portal-Núñez, S.; Vila, M.; López-Herradón, A.; Ardura, J.A.; Mulero, F.; Gómez-Barrena, E.; Vallet-Regí, M.; Esbrit, P. Parathyroid hormone-related protein (107-111) improves the bone regeneration potential of gelatin-glutaraldehyde biopolymer-coated hydroxyapatite. Acta Biomater 2014, 10, 3307–3316. [Google Scholar] [CrossRef]
- Niida, A.; Hiroko, T.; Kasai, M.; Furukawa, Y.; Nakamura, Y.; Suzuki, Y.; Sugano, S.; Akiyama, T. DKK1, a negative regulator of Wnt signaling, is a target of the β-catenin/TCF pathway. Oncogene 2004, 23, 8520–8526. [Google Scholar] [CrossRef] [PubMed]
- Semënov, M.; Tamai, K.; He, X. SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J Biol Chem 2005, 80, 26770–26775. [Google Scholar] [CrossRef] [PubMed]
- Rutkovskiy, A.; Stensløkken, K.O.; Vaage, I.J. Osteoblast Differentiation at a Glance. Med Sci Monit Basic Res 2016, 22, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, H.; Jimi, E.; Kajiya, H.; Motokawa, W.; Okabe, K. Parathyroid-hormone-related protein induces expression of receptor activator of NF-κB ligand in human periodontal ligament cells via a cAMP/protein kinase A-independent pathway. J Dent Res 2005, 84, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Li, Q.; Zhang, Y.; Bi, Y.; Li, X.; Shu, Y.; Chen, X.; Jin, Z.; Ge, C. Regulation of OPG and RANKL expressed by human dental follicle cells in osteoclastogenesis. Cell Tissue Res 2015, 362, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Mak, K.K.; Bi, Y.; Wan, C.; Chuang, P.T.; Clemens, T.; Young, M.; Yang, Y. Hedgehog signaling in mature osteoblasts regulates bone formation and resorption by controlling PTHrP and RANKL expression. Dev Cell 2008, 14, 674–688. [Google Scholar] [CrossRef] [PubMed]
- Ricarte, F.R.; Le Henaff, C.; Kolupaeva, V.G.; Gardella, T.J.; Partridge, N.C. Parathyroid hormone (1-34) and its analogs differentially modulate osteoblastic Rankl expression via PKA/SIK2/SIK3 and PP1/PP2A-CRTC3 signaling. J Biol Chem 2018, 293, 20200–20213. [Google Scholar] [CrossRef]
- Elango, J.; Rahman, S.U.; Henrotin, Y.; de Val, J.E.M.S.; Bao, B.; Wang, S.; Li, B.; Wu, W. Parathyroid Hormone-Related Protein (PTHrP) Accelerates Soluble RANKL Signals for Downregulation of Osteogenesis of Bone Mesenchymal Stem Cells. J. Clin. Med. 2019, 8, 836. [Google Scholar] [CrossRef]
- Kim, H.J.; Minashima, T.; McCarthy, E.F.; Winkles, J.A.; Kirsch, T. Progressive ankylosis protein (ANK) in osteoblasts and osteoclasts controls bone formation and bone remodeling. J Bone Miner Res 2010, 25, 1771–1783. [Google Scholar] [CrossRef]
- Kamel, S.A.; Yee, J.A. Continuous and intermittent exposure of neonatal rat calvarial cells to PTHrP (1-36) inhibits bone nodule mineralization in vitro by downregulating bone sialoprotein expression via the cAMP signaling pathway. F1000Res. 2013, 2, 77. [Google Scholar] [CrossRef]
- Wang, Y.; Fang, X.; Wang, C.; Ding, C.; Lin, H.; Liu, A.; Wang, L.; Cao, Y. Exogenous PTHrP Repairs the Damaged Fracture Healing of PTHrP+/− Mice and Accelerates Fracture Healing of Wild Mice. Int. J. Mol. Sci. 2017, 18, 337. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Zhang, J.; Dong, Z.; Zhang, Y.; Wang, R.; Karaplis, A.; Goltzman, D.; Miao, D. The p27 Pathway Modulates the Regulation of Skeletal Growth and Osteoblastic Bone Formation by Parathyroid Hormone-Related Peptide. J Bone Miner Res 2015, 30, 1969–1979. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, G.; Gu, Z.; Sun, H.; Karaplis, A.; Goltzman, D.; Miao, D. DNA damage checkpoint pathway modulates the regulation of skeletal growth and osteoblastic bone formation by parathyroid hormone-related peptide. Int J Biol Sci 2018, 14, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Martín-Guerrero, E.; Tirado-Cabrera, I.; Buendía, I.; Alonso, V.; Gortázar, A.R.; Ardura, J.A. Primary cilia mediate parathyroid hormone receptor type 1 osteogenic actions in osteocytes and osteoblasts via Gli activation. J Cell Physiol 2020, 235, 7356–7369. [Google Scholar] [CrossRef] [PubMed]
- Platas, J.; Guillén, M.I.; Gomar, F.; Castejón, M.A.; Esbrit, P.; Alcaraz, M.J. Anti-senescence and Anti-inflammatory Effects of the C-terminal Moiety of PTHrP Peptides in OA Osteoblasts. J Gerontol A Biol Sci Med Sci 2017, 72, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Ardura, J.A.; Portal-Núñez, S.; Castelbón-Calvo, I.; Martínez de Toda, I.; De la Fuente, M.; Esbrit, P. Parathyroid Hormone-Related Protein Protects Osteoblastic Cells From Oxidative Stress by Activation of MKP1 Phosphatase. J Cell Physiol 2017, 232, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Onan, D.; Allan, E.H.; Quinn, J.M.; Gooi, J.H.; Pompolo, S.; Sims, N.A.; Gillespie, M.T.; Martin, T.J. The chemokine Cxcl1 is a novel target gene of parathyroid hormone (PTH)/PTH-related protein in committed osteoblasts. Endocrinology 2009, 150, 2244–2253. [Google Scholar] [CrossRef]
- Portal-Núñez, S.; Lozano, D.; de Castro, L.F.; de Gortázar, A.R.; Nogués, X.; Esbrit, P. Alterations of the Wnt/beta-catenin pathway and its target genes for the N- and C-terminal domains of parathyroid hormone-related protein in bone from diabetic mice. FEBS Lett 2010, 584, 3095–3100. [Google Scholar] [CrossRef]
- Maycas, M.; McAndrews, K.A.; Sato, A.Y.; Pellegrini, G.G.; Brown, D.M.; Allen, M.R.; Plotkin, L.I.; Gortazar, A.R.; Esbrit, P.; Bellido, T. PTHrP-Derived Peptides Restore Bone Mass and Strength in Diabetic Mice: Additive Effect of Mechanical Loading. J Bone Miner Res 2017, 32, 486–497. [Google Scholar] [CrossRef]
- Makino, A.; Takagi, H.; Takahashi, Y.; Hase, N.; Sugiyama, H.; Yamana, K.; Kobayashi, T. Abaloparatide Exerts Bone Anabolic Effects with Less Stimulation of Bone Resorption-Related Factors: A Comparison with Teriparatide. Calcif Tissue Int 2018, 103, 289–297. [Google Scholar] [CrossRef]
- Amizuka, N.; Warshawsky, H.; Henderson, J.E.; Goltzman, D.; Karaplis, A.C. Parathyroid hormone-related peptide-depleted mice show abnormal epiphyseal cartilage development and altered endochondral bone formation. J Cell Biol 1994, 126, 1611–1623. [Google Scholar] [CrossRef] [PubMed]
- Carey, D.E.; Liu, X. Expression of bone morphogenetic protein-6 messenger RNA in bovine growth plate chondrocytes of different size. J Bone Miner Res 1995, 10, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Lanske, B.; Karaplis, A.C.; Lee, K.; Luz, A.; Vortkamp, A.; Pirro, A.; Karperien, M.; Defize, L.H.; Ho, C.; Mulligan, R.C.; Abou-Samra, A.B.; Jüppner, H.; Segre, G.V.; Kronenberg, H.M. PTH/PTHrP receptor in early development and Indian hedgehog-regulated bone growth. Science 1996, 273, 663–666. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Akiyama, H.; Shigeno, C.; Nakamura, T. Parathyroid hormone-related peptide inhibits the expression of bone morphogenetic protein-4 mRNA through a cyclic AMP/protein kinase A pathway in mouse clonal chondrogenic EC cells, ATDC5. Biochim Biophys Acta 2000, 1497, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Li, T.F.; Dong, Y.; Ionescu, A.M.; Rosier, R.N.; Zuscik, M.J.; Schwarz, E.M.; O’Keefe, R.J.; Drissi, H. Parathyroid hormone-related peptide (PTHrP) inhibits Runx2 expression through the PKA signaling pathway. Exp Cell Res 2004, 299, 128–136. [Google Scholar] [CrossRef]
- Kozhemyakina, E.; Cohen, T.; Yao, T.P.; Lassar, A.B. Parathyroid hormone-related peptide represses chondrocyte hypertrophy through a protein phosphatase 2A/histone deacetylase 4/MEF2 pathway. Mol Cell Biol 2009, 29, 5751–5762. [Google Scholar] [CrossRef]
- Guo, B.; Wang, S.T.; Duan, C.C.; Li, D.D.; Tian, X.C.; Wang, Q.Y.; Yue, Z.P. Effects of PTHrP on chondrocytes of sika deer antler. Cell Tissue Res 2013, 354, 451–460. [Google Scholar] [CrossRef]
- Wang, S.T.; Gao, Y.J.; Duan, C.C.; Li, D.D.; Tian, X.C.; Zhang, Q.L.; Guo, B.; Yue, Z.P. Effects of PTHrP on expression of MMP9 and MMP13 in sika deer antler chondrocytes. Cell Biol Int 2013, 37, 1300–1307. [Google Scholar] [CrossRef]
- Pelosi, M.; Lazzarano, S.; Thoms, B.L.; Murphy, C.L. Parathyroid hormone-related protein is induced by hypoxia and promotes expression of the differentiated phenotype of human articular chondrocytes. Clin Sci (Lond) 2013, 125, 461–470. [Google Scholar] [CrossRef]
- Browe, D.C.; Coleman, C.M.; Barry, F.P.; Elliman, S.J. Hypoxia Activates the PTHrP-MEF2C Pathway to Attenuate Hypertrophy in Mesenchymal Stem Cell Derived Cartilage. Sci Rep 2019, 9, 13274. [Google Scholar] [CrossRef]
- Fischer, J.; Aulmann, A.; Dexheimer, V.; Grossner, T.; Richter, W. Intermittent PTHrP(1-34) exposure augments chondrogenesis and reduces hypertrophy of mesenchymal stromal cells. Stem Cells Dev 2014, 23, 2513–2523. [Google Scholar] [CrossRef] [PubMed]
- Karaplis, A.C.; Luz, A.; Glowacki, J.; Bronson, R.T.; Tybulewicz, V.L.; Kronenberg, H.M.; Mulligan, R.C. Lethal skeletal dysplasia from targeted disruption of the parathyroid hormone-related peptide gene. Genes Dev 1994, 8, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Nishimori, S.; Lai, F.; Shiraishi, M.; Kobayashi, T.; Kozhemyakina, E.; Yao, T.P.; Lassar, A.B.; Kronenberg, H.M. PTHrP targets HDAC4 and HDAC5 to repress chondrocyte hypertrophy. JCI Insight 2019, 4, e97903. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.X.; Nemere, I.; Norman, A.W. A parathyroid-related peptide induces transcaltachia (the rapid, hormonal stimulation of intestinal Ca2+ transport). Biochem Biophys Res Commun 1992, 186, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Larsson, D.; Nemere, I. Vectorial Transcellular Calcium Transport in Intestine: Integration of Current Models. J Biomed Biotechnol 2002, 2, 117–119. [Google Scholar] [CrossRef] [PubMed]
- Beggs, M.R.; Alexander, R.T. Intestinal absorption and renal reabsorption of calcium throughout postnatal development. Exp Biol Med (Maywood) 2017, 242, 840–849. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Shao, G.; Lu, Y.; Xue, M.; Liang, F.; Zhang, Z.; Bai, L. Parathyroid Hormone-Related Protein (1-40) Enhances Calcium Uptake in Rat Enterocytes Through PTHR1 Receptor and Protein Kinase Cα/β Signaling. Cell Physiol Biochem 2018, 51, 1695–1709. [Google Scholar] [CrossRef]
- He, S.; Xue, M.; Liu, C.; Xie, F.; Bai, L. Parathyroid Hormone-Like Hormone Induces Epithelial-to-Mesenchymal Transition of Intestinal Epithelial Cells by Activating the Runt-Related Transcription Factor 2. Am J Pathol 2018, 188, 1374–1388. [Google Scholar] [CrossRef]
- Zhang, D.; Zhang, Y.; Sun, B. The Molecular Mechanisms of Liver Fibrosis and Its Potential Therapy in Application. Int J Mol Sci 2022, 23, 12572. [Google Scholar] [CrossRef]
- Liang, F.F.; Liu, C.P.; Li, L.X.; Xue, M.M.; Xie, F.; Guo, Y.; Bai, L. Activated effects of parathyroid hormone-related protein on human hepatic stellate cells. PLoS One 2013, 8, e76517. [Google Scholar] [CrossRef]
- Gardi, C.; Arezzini, B.; Fortino, V.; Comporti, M. Effect of free iron on collagen synthesis, cell proliferation and MMP-2 expression in rat hepatic stellate cells. Biochem Pharmacol 2002, 64, 1139–1145. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Tang, J.; Diao, N.; Liao, Y.; Shi, J.; Xu, X.; Xie, F.; Bai, L. Parathyroid hormone-related protein activates HSCs via hedgehog signalling during liver fibrosis development. Artif Cells Nanomed Biotechnol 2019, 47, 1984–1994. [Google Scholar] [CrossRef] [PubMed]
- Torday, J.S.; Rehan, V.K. The evolutionary continuum from lung development to homeostasis and repair. Am J Physiol Lung Cell Mol Physiol 2007, 292, L608–L611. [Google Scholar] [CrossRef] [PubMed]
- Torday, J.S.; Rehan, V.K. Stretch-stimulated surfactant synthesis is coordinated by the paracrine actions of PTHrP and leptin. Am J Physiol Lung Cell Mol Physiol 2002, 283, L130–L135. [Google Scholar] [CrossRef] [PubMed]
- Torday, J.; Rehan, V. Neutral lipid trafficking regulates alveolar type II cell surfactant phospholipid and surfactant protein expression. Exp Lung Res 2011, 37, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Oruqaj, L.; Forst, S.; Schreckenberg, R.; Inserte, J.; Poncelas, M.; Bañeras, J.; Garcia-Dorado, D.; Rohrbach, S.; Schlüter, K.D. Effect of high fat diet on pulmonary expression of parathyroid hormone-related protein and its downstream targets. Heliyon 2016, 2, e00182. [Google Scholar] [CrossRef]
- Rehan, V.K.; Sakurai, R.; Wang, Y.; Santos, J.; Huynh, K.; Torday, J.S. Reversal of nicotine-induced alveolar lipofibroblast-to-myofibroblast transdifferentiation by stimulants of parathyroid hormone-related protein signaling. Lung 2007, 185, 151–159. [Google Scholar] [CrossRef]
- Sawada, Y.; Zhang, B.; Okajima, F.; Izumi, T.; Takeuchi, T. PTHrP increases pancreatic β-cell-specific functions in well-differentiated cells. Mol Cell Endocrinol 2001, 182, 265–275. [Google Scholar] [CrossRef]
- Sawada, Y.; Kameya, T.; Aizama, T.; Izumi, T.; Takeuchi, T. Proprotein-Processing Endoprotease Furin and its Substrate Parathyroid Hormone-Related Protein are Coexpressed in Insulinoma Cells. Endocr Pathol 2000, 11, 31–39. [Google Scholar] [CrossRef]
- Guthalu Kondegowda, N.; Joshi-Gokhale, S.; Harb, G.; Williams, K.; Zhang, X.Y.; Takane, K.K.; Zhang, P.; Scott, D.K.; Stewart, A.F.; Garcia-Ocaña, A.; Vasavada, R.C. Parathyroid hormone-related protein enhances human ß-cell proliferation and function with associated induction of cyclin-dependent kinase 2 and cyclin E expression. Diabetes 2010, 59, 3131–3138. [Google Scholar] [CrossRef]
- Williams, K.; Abanquah, D.; Joshi-Gokhale, S.; Otero, A.; Lin, H.; Guthalu, N.K.; Zhang, X.; Mozar, A.; Bisello, A.; Stewart, A.F.; Garcia-Ocaña, A.; Vasavada, R.C. Systemic and acute administration of parathyroid hormone-related peptide(1-36) stimulates endogenous beta cell proliferation while preserving function in adult mice. Diabetologia 2011, 54, 2867–2877. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, V.; Kim, S.O.; Aronson, J.F.; Chao, C.; Hellmich, M.R.; Falzon, M. Role of parathyroid hormone-related protein in the pro-inflammatory and pro-fibrogenic response associated with acute pancreatitis. Regul Pept 2012, 175, 49–60, Erratum in: Regul Pept 2014, 192-193, 59. [Google Scholar] [CrossRef] [PubMed]
- Sadr-Azodi, O.; Oskarsson, V.; Discacciati, A.; Videhult, P.; Askling, J.; Ekbom, A. Pancreatic Cancer Following Acute Pancreatitis: A Population-based Matched Cohort Study. Am J Gastroenterol 2018, 113, 1711–1719. [Google Scholar] [CrossRef] [PubMed]
- Rastellini, C.; Han, S.; Bhatia, V.; Cao, Y.; Liu, K.; Gao, X.; Ko, T.C.; Greeley, G.H. Jr.; Falzon, M. Induction of chronic pancreatitis by pancreatic duct ligation activates BMP2, apelin, and PTHrP expression in mice. Am J Physiol Gastrointest Liver Physiol 2015, 309, G554–G565. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Cao, Y.; Yang, W.; Duan, C.; Aronson, J.F.; Rastellini, C.; Chao, C.; Hellmich, M.R.; Ko, T.C. BMP2 inhibits TGF-β-induced pancreatic stellate cell activation and extracellular matrix formation. Am J Physiol Gastrointest Liver Physiol 2013, 304, G804–G813. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Englander, E.W.; Gomez, G.A.; Aronson, J.F.; Rastellini, C.; Garofalo, R.P.; Kolli, D.; Quertermous, T.; Kundu, R.; Greeley, G.H. Jr. Pancreatitis activates pancreatic apelin-APJ axis in mice. Am J Physiol Gastrointest Liver Physiol 2013, 305, G139–G150. [Google Scholar] [CrossRef] [PubMed]
- Morrell, N.W.; Archer, S.L.; Defelice, A.; Evans, S.; Fiszman, M.; Martin, T.; Saulnier, M.; Rabinovitch, M.; Schermuly, R.; Stewart, D.; Truebel, H.; Walker, G.; Stenmark, K.R. Anticipated classes of new medications and molecular targets for pulmonary arterial hypertension. Pulm Circ 2013, 3, 226–244. [Google Scholar] [CrossRef]
- Okoumassoun, L.E.; Russo, C.; Denizeau, F.; Averill-Bates, D.; Henderson, Dr.J.E. Parathyroid hormone-related protein (PTHrP) inhibits mitochondrial-dependent apoptosis through CK2. J Cell Physiol 2007, 212, 591–599. [Google Scholar] [CrossRef]
- Hanif, I.M.; Hanif, I.M.; Shazib, M.A.; Ahmad, K.A.; Pervaiz, S. Casein Kinase II: an attractive target for anti-cancer drug design. Int J Biochem Cell Biol 2010, 42, 1602–1605. [Google Scholar] [CrossRef]
- Ardura, J.A.; Rayego-Mateos, S.; Rámila, D.; Ruiz-Ortega, M.; Esbrit, P. Parathyroid Hormone–Related Protein Promotes Epithelial–Mesenchymal Transition. J Am Soc Nephrol 2010, 21, 237–248. [Google Scholar] [CrossRef]
- Hua, K.; Li, Y.; Zhou, H.; Hu, X.; Chen, Y.; He, R.; Luo, R.; Zhou, R.; Bi, D.; Jin, H. Haemophilus parasuis Infection Disrupts Adherens Junctions and Initializes EMT Dependent on Canonical Wnt/β-Catenin Signaling Pathway. Front Cell Infect Microbiol 2018, 8, 324. [Google Scholar] [CrossRef] [PubMed]
- Ardura, J.A.; Sanz, A.B.; Ortiz, A.; Esbrit, P. Parathyroid hormone–related protein protects renal tubuloepithelial cells from apoptosis by activating transcription factor Runx2. Kidney Int 2013, 83, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Kramann, R.; Schneider, R.K. Parathyroid hormone-related protein and regulation of cell survival in the kidney. Kidney Int 2013, 83, 777–779. [Google Scholar] [CrossRef] [PubMed]
- Hochane, M.; Raison, D.; Coquard, C.; Béraud, C.; Bethry, A.; Danilin, S.; Massfelder, T.; Barthelmebs, M. Parathyroid hormone-related protein modulates inflammation in mouse mesangial cells and blunts apoptosis by enhancing COX-2 expression. Am J Physiol Cell Physiol 2018, 314, C242–C253. [Google Scholar] [CrossRef] [PubMed]
- Danilin, S.; Sourbier, C.; Thomas, L.; Rothhut, S.; Lindner, V.; Helwig, J.J.; Jacqmin, D.; Lang, H.; Massfelder, T. von Hippel-Lindau tumor suppressor gene-dependent mRNA stabilization of the survival factor parathyroid hormone-related protein in human renal cell carcinoma by the RNA-binding protein HuR. Carcinogenesis 2009, 30, 387–96. [Google Scholar] [CrossRef] [PubMed]
- Kaltschmidt, B.; Linker, R.A.; Deng, J.; Kaltschmidt, C. Cyclooxygenase-2 is a neuronal target gene of NF-kappaB. BMC Mol Biol 2002, 3, 16. [Google Scholar] [CrossRef]
- Cooper, M.E.; Vranes, D.; Youssef, S.; Stacker, S.A.; Cox, A.J.; Rizkalla, B.; Casley, D.J.; Bach, L.A.; Kelly, D.J.; Gilbert, R.E. Increased renal expression of vascular endothelial growth factor (VEGF) and its receptor VEGFR-2 in experimental diabetes. Diabetes 1999, 48, 2229–2239. [Google Scholar] [CrossRef]
- Burns, K.D. Angiotensin II and its receptors in the diabetic kidney. Am J Kidney Dis 2000, 36, 449–467. [Google Scholar] [CrossRef]
- Izquierdo, A.; López-Luna, P.; Ortega, A.; Romero, M.; Guitiérrez-Tarrés, M.A.; Arribas, I.; Alvarez, M.J.; Esbrit, P.; Bosch, R.J. The parathyroid hormone-related protein system and diabetic nephropathy outcome in streptozotocin-induced diabetes. Kidney Int 2006, 69, 2171–2177. [Google Scholar] [CrossRef]
- Esbrit, P.; Santos, S.; Ortega, A.; Fernández-Agulló, T.; Vélez, E.; Troya, S.; Garrido, P.; Peña, A.; Bover, J.; Bosch, R.J. Parathyroid hormone-related protein as a renal regulating factor. From vessels to glomeruli and tubular epithelium. Am J Nephrol 2001, 21, 179–184. [Google Scholar] [CrossRef]
- Romero, M.; Ortega, A.; Izquierdo, A.; López-Luna, P.; Bosch, R.J. Parathyroid hormone-related protein induces hypertrophy in podocytes via TGF-beta(1) and p27(Kip1): implications for diabetic nephropathy. Nephrol Dial Transplant 2010, 25, 2447–2457. [Google Scholar] [CrossRef] [PubMed]
- Ortega, A.; Romero, M.; Izquierdo, A.; Troyano, N.; Arce, Y.; Ardura, J.A.; Arenas, M.I.; Bover, J.; Esbrit, P.; Bosch, R.J. Parathyroid hormone-related protein is a hypertrophy factor for human mesangial cells: Implications for diabetic nephropathy. J Cell Physiol 2012, 227, 1980–1987. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.M.; Dai, J.J.; Zhu, R.; Peng, F.F.; Wu, S.Z.; Yu, H.; Krepinsky, J.C.; Zhang, B.F. Parathyroid hormone-related protein induces fibronectin up-regulation in rat mesangial cells through reactive oxygen species/Src/EGFR signaling. Biosci Rep 2019, 39, BSR20182293. [Google Scholar] [CrossRef] [PubMed]
- Póvoa, G.; Diniz, L.M. Growth Hormone System: skin interactions. An Bras Dermatol 2011, 86, 1159–1165. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Ji, C.; Casinghino, S.; McCarthy, T.L.; Centrella, M. Parathyroid hormone-related protein enhances insulin-like growth factor-I expression by fetal rat dermal fibroblasts. J Biol Chem 1997, 272, 23498–23502. [Google Scholar] [CrossRef] [PubMed]
- Blomme, E.A.; Sugimoto, Y.; Lin, Y.C.; Capen, C.C.; Rosol, T.J. Parathyroid hormone-related protein is a positive regulator of keratinocyte growth factor expression by normal dermal fibroblasts. Mol Cell Endocrinol 1999, 152, 189–197. [Google Scholar] [CrossRef]
- Miao, D.; Su, H.; He, B.; Gao, J.; Xia, Q.; Zhu, M.; Gu, Z.; Goltzman, D.; Karaplis, A.C. Severe growth retardation and early lethality in mice lacking the nuclear localization sequence and C-terminus of PTH-related protein. Proc Natl Acad Sci USA 2008, 105, 20309–20314. [Google Scholar] [CrossRef]
- Harvat, B.L.; Wang, A.; Seth, P.; Jetten, A.M. Up-regulation of p27Kip1, p21WAF1/Cip1 and p16Ink4a is associated with, but not sufficient for, induction of squamous differentiation. J Cell Sci 1998, 111, 1185–1196. [Google Scholar] [CrossRef]
- Jiang, M.; Chen, G.; Lu, N.; Zhang, Y.; Jin, S.; Karaplis, A.; Goltzman, D.; Miao, D. Deficiency of the parathyroid hormone-related peptide nuclear localization and carboxyl terminal sequences leads to premature skin ageing partially mediated by the downregulation of p27. Exp Dermatol 2015, 24, 847–852. [Google Scholar] [CrossRef]
Figure 1.
PTHrP regulation of gene expression via paracrine/autocrine (a) and intracrine (b) actions. In (a) PTHrP-bound PTH1R stimulates cAMP/CREB signalization, whereas in (b) gene expression is activated by alternative mechanisms, such as calcium signalling. Reprinted from [3].
Figure 1.
PTHrP regulation of gene expression via paracrine/autocrine (a) and intracrine (b) actions. In (a) PTHrP-bound PTH1R stimulates cAMP/CREB signalization, whereas in (b) gene expression is activated by alternative mechanisms, such as calcium signalling. Reprinted from [3].
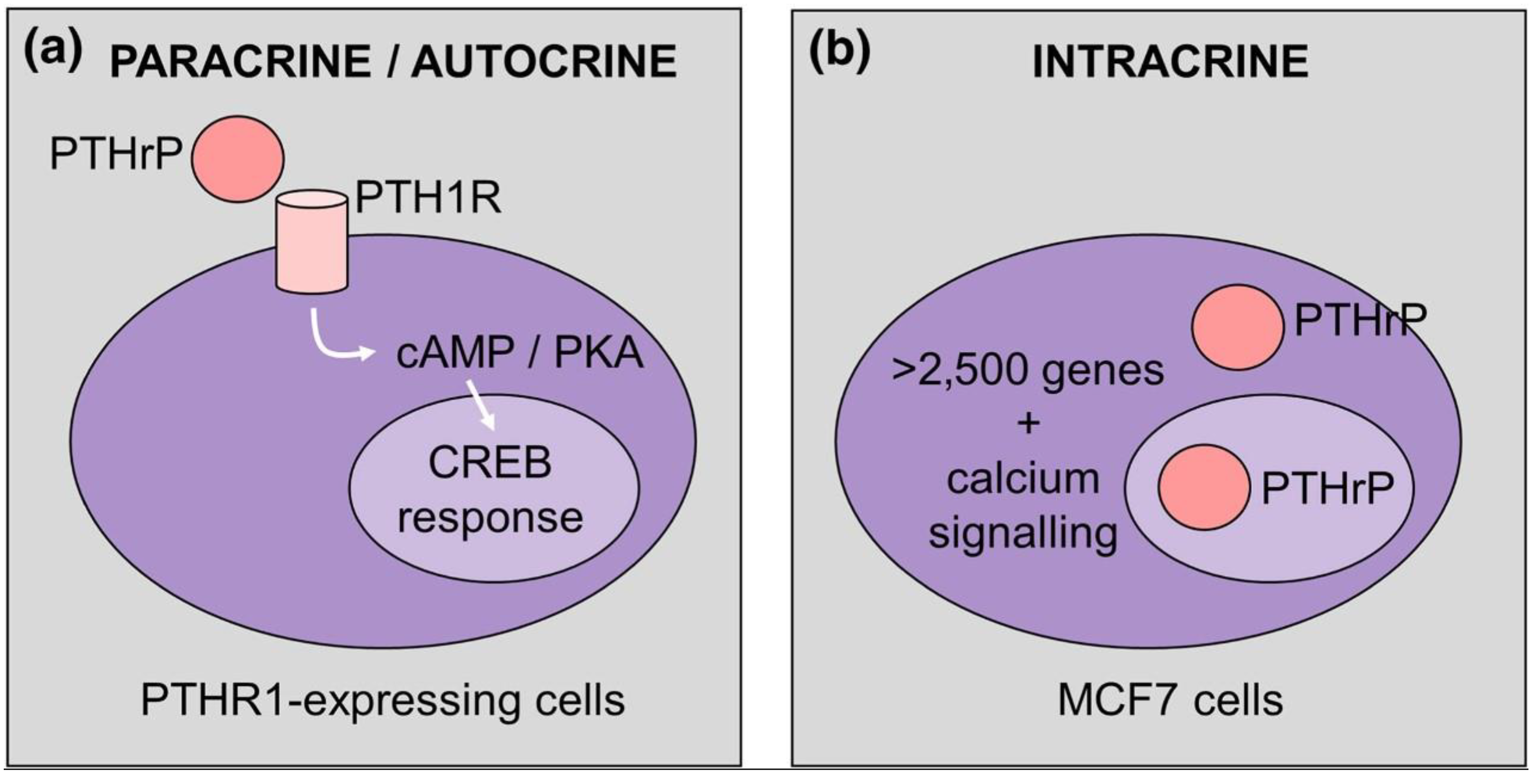
Figure 2.
Model depicting the pathway induced by PTH (1-34), PTHrP (1-36) and abaloparatide (ABL) which leads to RANKL up-regulation. Reprinted from [27]. Distributed under the terms of the Creative Commons Attribution licence (CC BY 4.0).
Figure 2.
Model depicting the pathway induced by PTH (1-34), PTHrP (1-36) and abaloparatide (ABL) which leads to RANKL up-regulation. Reprinted from [27]. Distributed under the terms of the Creative Commons Attribution licence (CC BY 4.0).
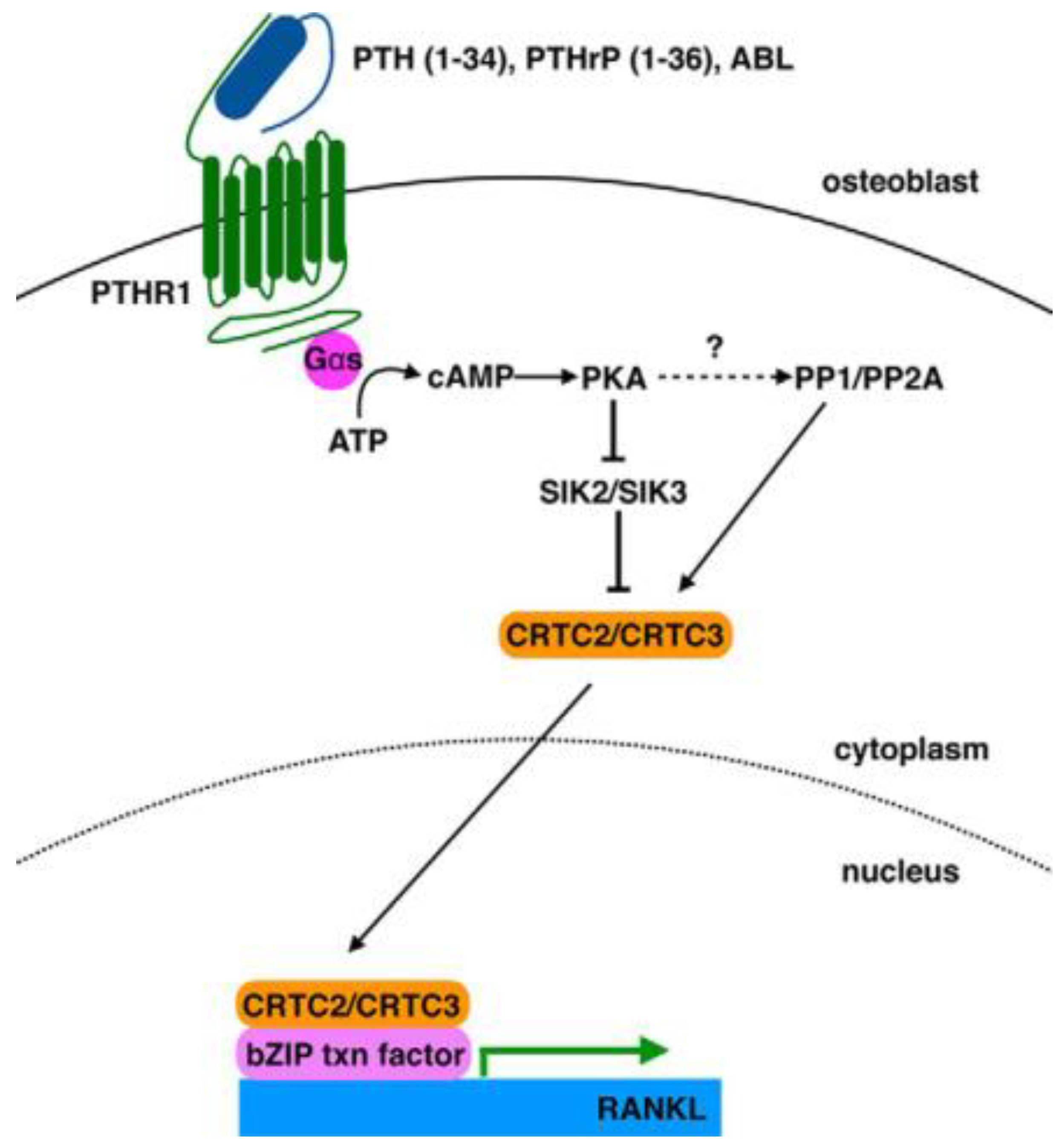
Figure 3.
Model depicting the pathways modulated by PTHrP NTS (here referred as NLS, i.e., nuclear localization sequence) and C-terminus and stimulating osteogenesis by bone marrow MSC. Reprinted from [33]. Distributed under the terms of the Creative Commons Attribution (CC BY-NC) license.
Figure 3.
Model depicting the pathways modulated by PTHrP NTS (here referred as NLS, i.e., nuclear localization sequence) and C-terminus and stimulating osteogenesis by bone marrow MSC. Reprinted from [33]. Distributed under the terms of the Creative Commons Attribution (CC BY-NC) license.
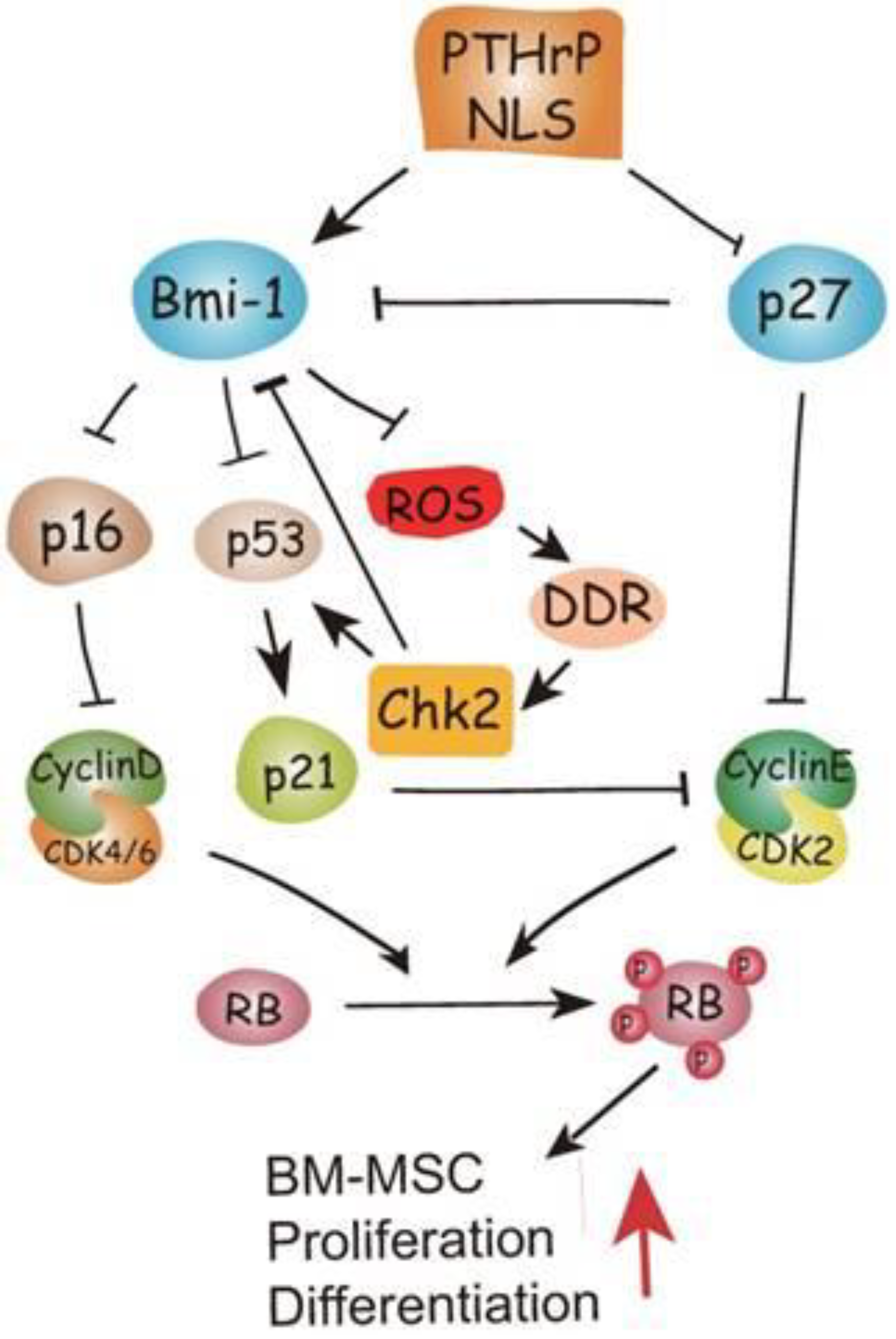
Figure 4.
Model depicting the primary cilia-dependent pathways activated by PTHrP and inducing survival and bone-formation related gene expression by mouse osteoblasts and osteocytes. Reprinted from [34].
Figure 4.
Model depicting the primary cilia-dependent pathways activated by PTHrP and inducing survival and bone-formation related gene expression by mouse osteoblasts and osteocytes. Reprinted from [34].
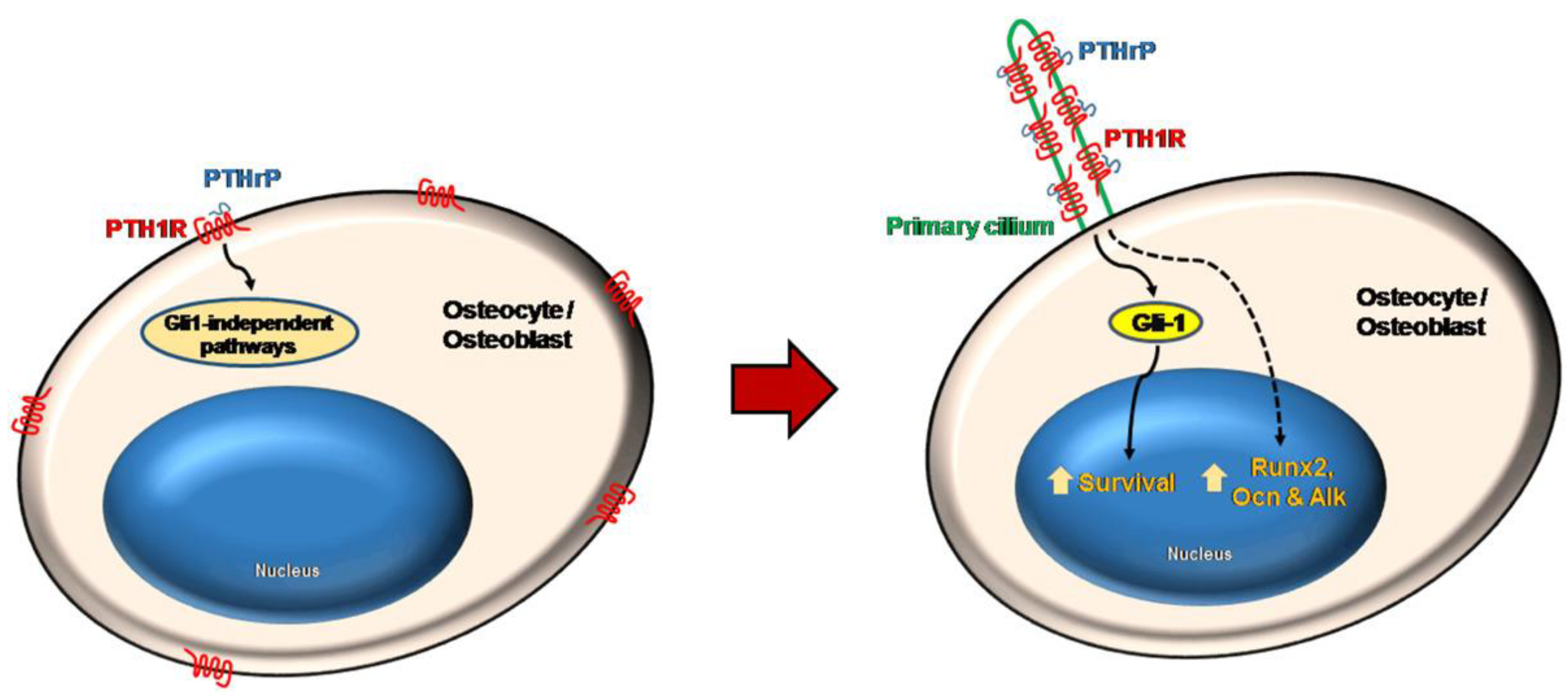
Figure 5.
Scheme depicting the pathways activated by PTHrP which induce the release of cytokines and chemokines and the deposition of the extracellular matrix (ECM). Activation of the stellate cells, exacerbates the inflammatory and fibrogenic responses that accompany acute pancreatitis (AP), whose repeated episodes (RAP) may eventually lead to chronic pancreatitis (CP). Reprinted from [72].
Figure 5.
Scheme depicting the pathways activated by PTHrP which induce the release of cytokines and chemokines and the deposition of the extracellular matrix (ECM). Activation of the stellate cells, exacerbates the inflammatory and fibrogenic responses that accompany acute pancreatitis (AP), whose repeated episodes (RAP) may eventually lead to chronic pancreatitis (CP). Reprinted from [72].
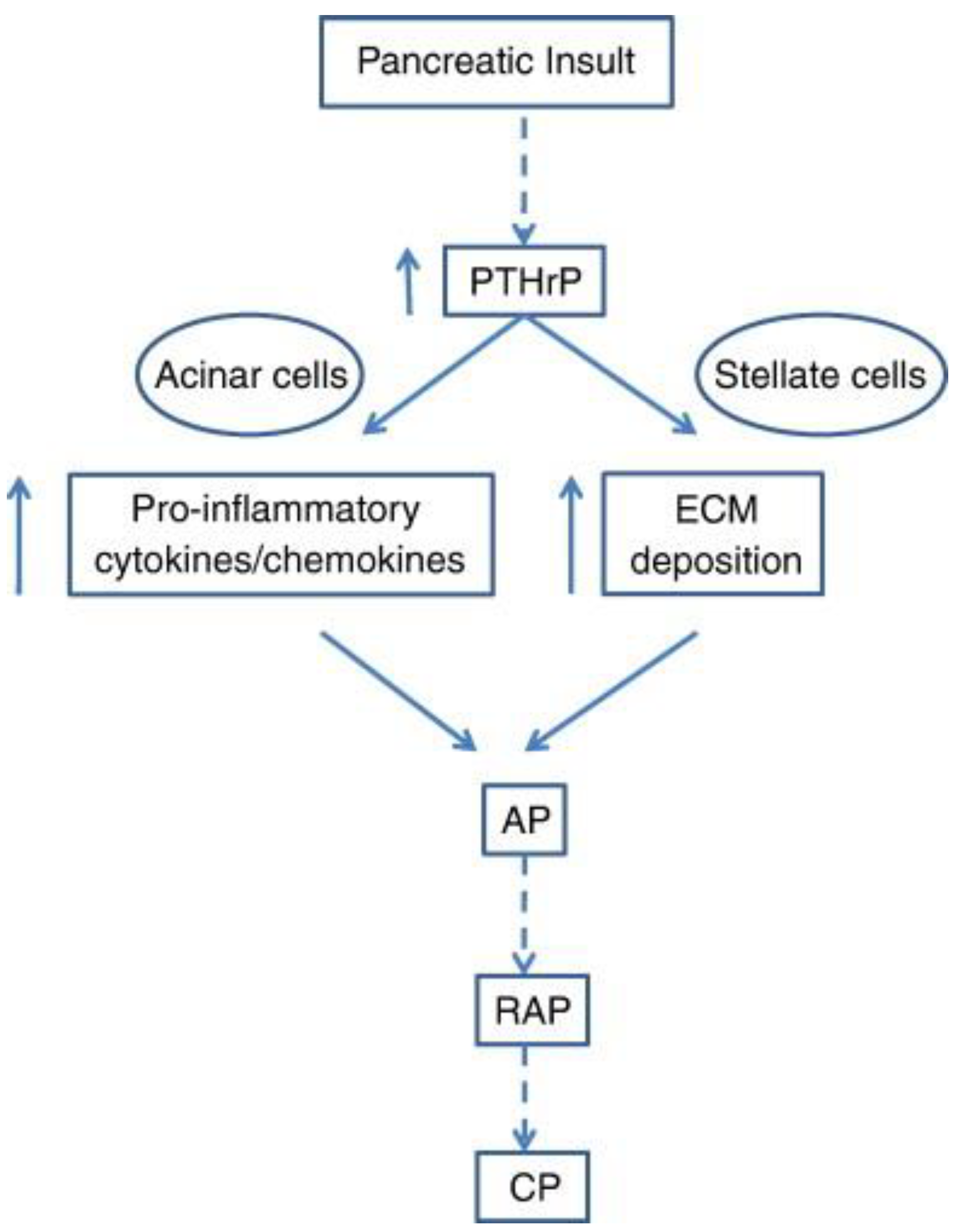
Figure 6.
a) Model summarizing the effect of PTHrP (1-36) in tubuloepitelial cell survival and anti-apoptotic mechanisms. b) Role played by the up-regulation of PTHrP following kidney injury. The complete explaination of the pathways depicted can be found in the original paper [83].
Figure 6.
a) Model summarizing the effect of PTHrP (1-36) in tubuloepitelial cell survival and anti-apoptotic mechanisms. b) Role played by the up-regulation of PTHrP following kidney injury. The complete explaination of the pathways depicted can be found in the original paper [83].
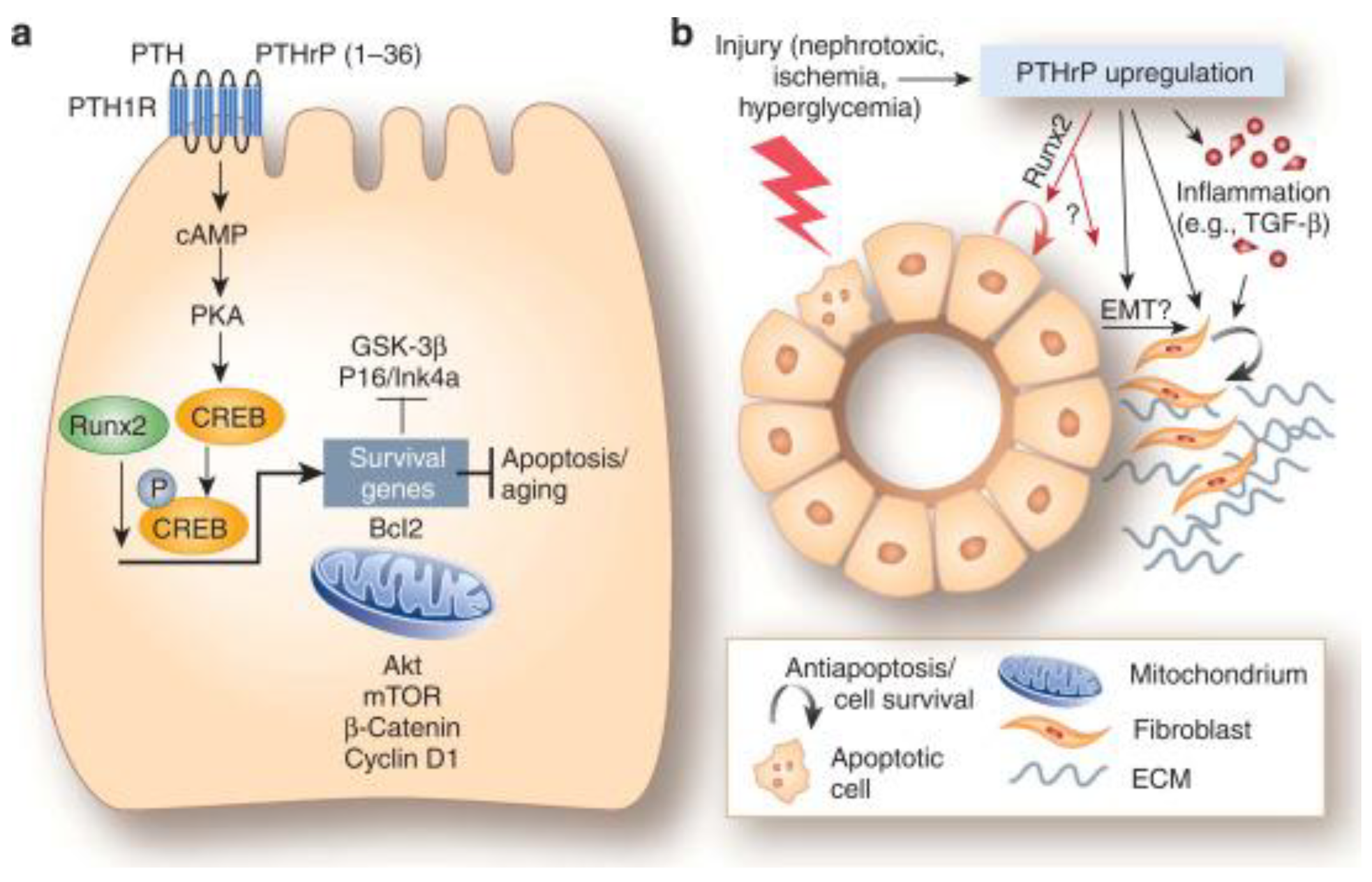
Figure 7.
Model depicting the N-terminal PTHrP-induced and ROS-triggered signalling cascade leading to ECM overdeposition in rat mesangial cells. Reprinted from [93]. Distributed under the terms of the Creative Commons Attribution 4.0 (CC BY) license.
Figure 7.
Model depicting the N-terminal PTHrP-induced and ROS-triggered signalling cascade leading to ECM overdeposition in rat mesangial cells. Reprinted from [93]. Distributed under the terms of the Creative Commons Attribution 4.0 (CC BY) license.
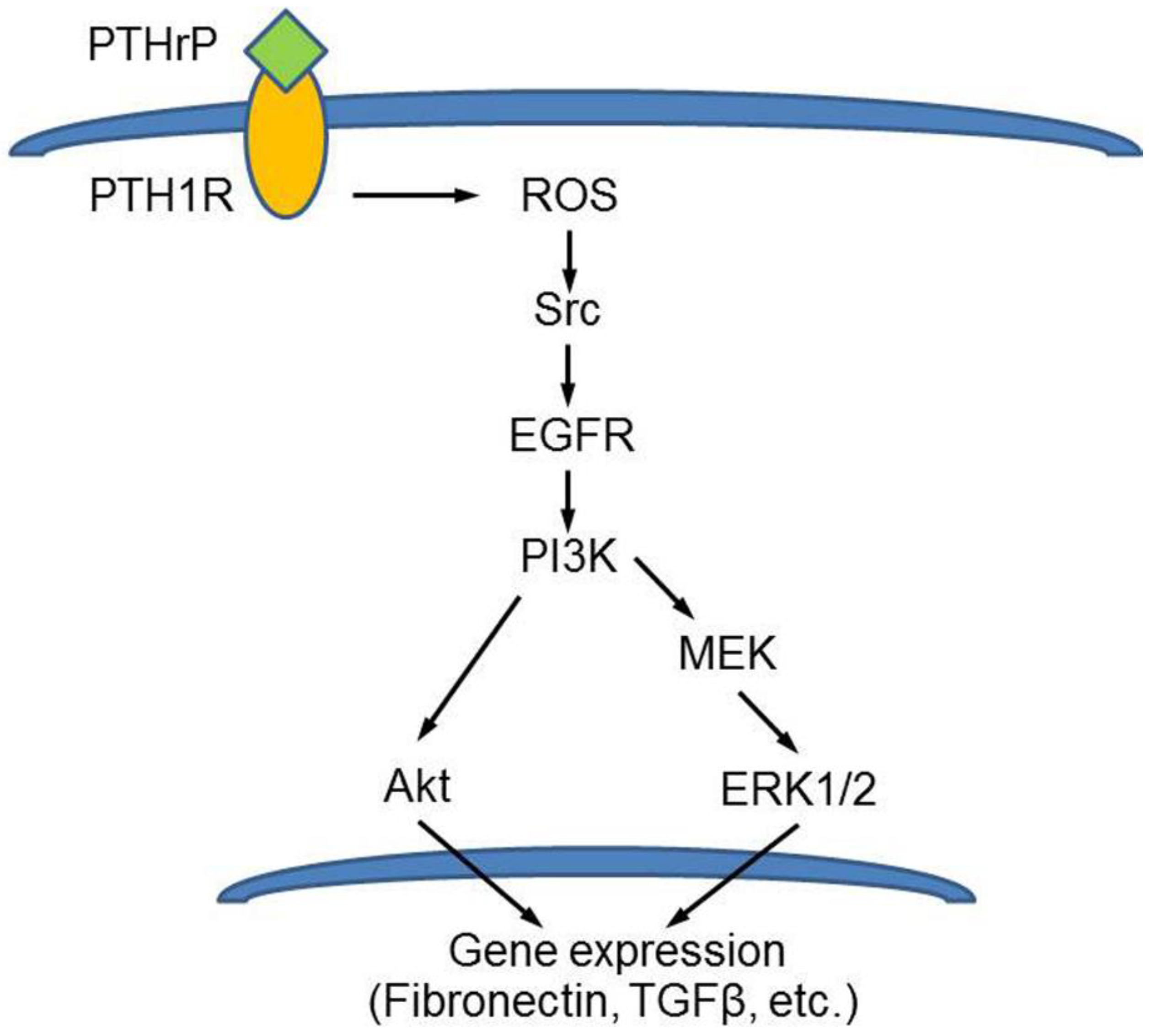
Figure 8.
Model depicting the paracrine interactions between dermal fibroblasts and keratinocytes via the PTHrP/IGF-1 circuit. Reprinted from [95]. Distributed under the terms of the Creative Commons Attribution (CC BY) license.
Figure 8.
Model depicting the paracrine interactions between dermal fibroblasts and keratinocytes via the PTHrP/IGF-1 circuit. Reprinted from [95]. Distributed under the terms of the Creative Commons Attribution (CC BY) license.
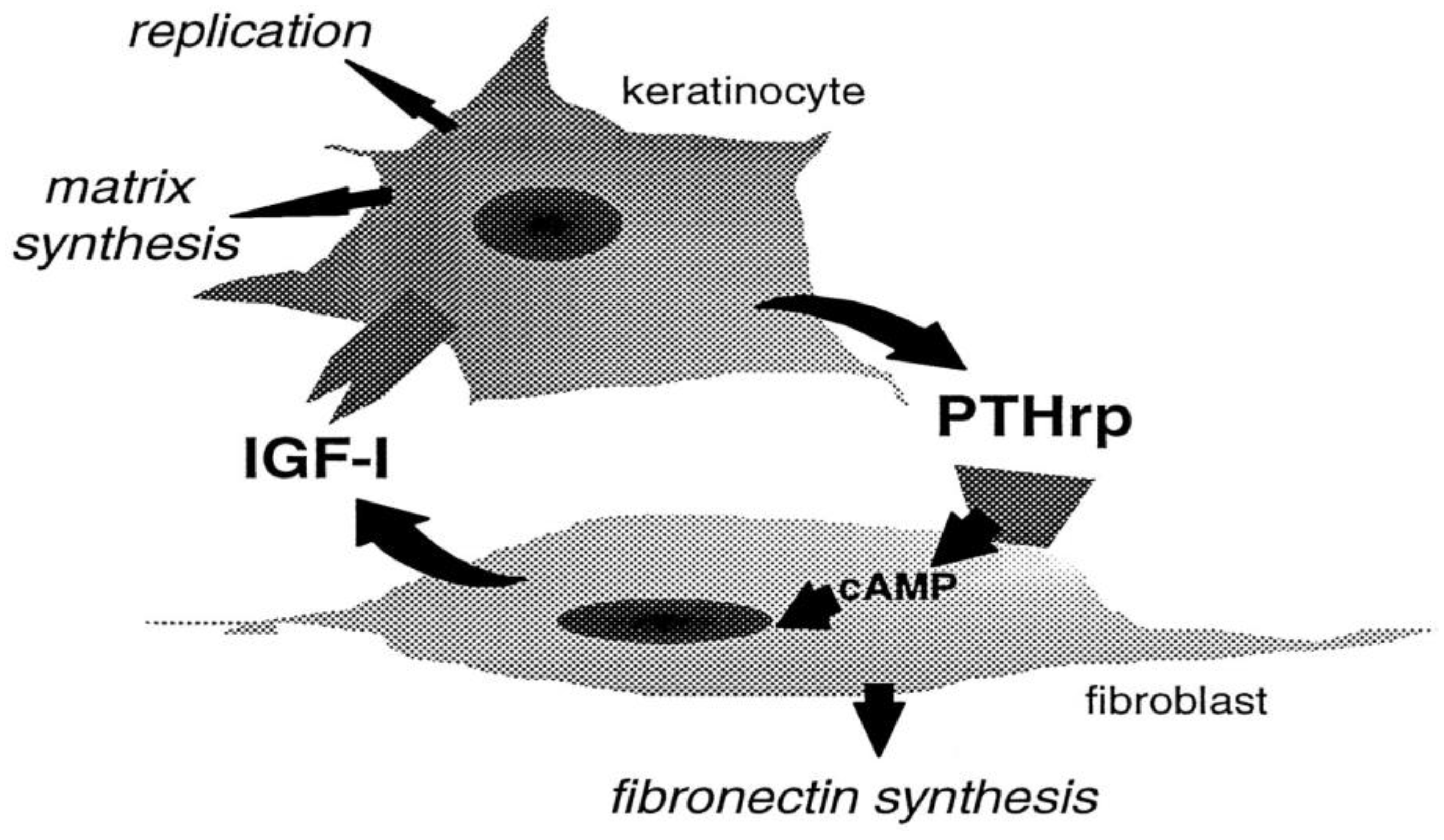

Table 1.
Variants of PTHrP mRNA produced by alternative splicing at the 5′ and 3′ ends. Reprinted from [2].
Table 1.
Variants of PTHrP mRNA produced by alternative splicing at the 5′ and 3′ ends. Reprinted from [2].

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



