
Submitted:
15 May 2023
Posted:
16 May 2023
You are already at the latest version
Abstract
Antibody-Drug Conjugates (ADCs) are a powerful therapeutic modality for cancer treatment. ADCs are multi-functional biologics in which a disease-targeting antibody is conjugated to an effector payload molecule through a linker. The success of currently used ADCs has been largely attributed to the development of linker systems which allow targeted release of cytocidal payload drugs inside cancer cells. Many lysosomal proteases are over-expressed in human cancers. They can effectively cleave a variety of peptide sequences, which can be exploited in the design of ADC linker systems. As a well-established linker, valine-citrulline-p-aminobenzyl carbamate (ValCitPABC) is used in many ADCs that are already approved or under preclinical and clinical development. Although ValCitPABC and related linkers are readily cleaved by cathepsins in the lysosome while remaining reasonably stable in human plasma, many studies have shown that they are susceptible to carboxylesterase 1C (Ces1C) in mouse and rat plasma, which hinders preclinical evaluation of the ADCs. Furthermore, neutropenia and thrombocytopenia, two of the most commonly observed dose-limiting adverse effects of ADCs, are believed to result from the premature hydrolysis of ValCitPABC by human neutrophil elastase. In addition to cathepsin-cleavable linkers, there is also growing interest in legumain-sensitive linkers for ADC development. Increasing plasma stability while maintaining lysosomal cleavability of ADC linkers is an objective of intensive current research. This review reports recent advances in the design and structure-activity relationship studies of various peptide/peptidomimetic linkers in this field.
Keywords:
antibody-drug conjugate
; lysosome
; cathepsin
; legumain
; self-immolation
; payload release
; peptidomimetic
; linker
1. Introduction
Clinical approvals of Antibody-Drug Conjugates (ADCs) have seen a big leap in the past several years. In fact, 12 out of 15 ADCs on the market were approved from 2017 to 2022 [1,2]. More than 100 new ADCs are currently at various stages of clinical development, which reflects the huge potential of this class of medicines in cancer treatment. The mechanism of action of ADCs involves antibody attachment to a specific antigen on cancerous cells and subsequent internalization through receptor-mediated endocytosis. Once inside the cancer cell, degradation of the antibody and/or cleavage of the linker in the endosomal-lysosomal compartments would release the drug payload which then exerts its cytocidal effects in the cytoplasm or nucleus. Researchers have taken advantage of two important endosomal-lysosomal features in designing linker systems for drug release: i) the acidic environment inside lysosomes for the design of acid-labile linkers, and ii) over-expression of specific lysosomal proteases for the design of protease-cleavable linkers (Figure 1A) [3,4,5,6,7,8,9]. Seven out of the fifteen approved ADCs have protease-recognizable peptide sequences in the linkers (Table 1) which are further attached to a self-immolated moiety. Upon cleavage of the peptide sequence by the protease, the self-immolated moiety readily undergoes elimination to release the free drug which then defuses out of the endosomal-lysosomal compartment. Linker-payload optimization is one of the most critical tasks in ADC development. The lysosomal protease-cleavable Valine-Citrulline-PABC (ValCitPABC) linker system is used in many of the approved ADCs [10,11], which is readily cleaved at the amide bond linking Cit and PABC, leading to self-immolate payload release. Initially, it was thought that only cathepsin B was responsible for the cleavage of ValCitPABC; however, later gene knockout studies have shown that other cathepsins, like cathepsin S, cathepsin L and cathepsin F, are also involved in the cleavage mechanism. It has also been revealed that a variety of dipeptide sequences can act as substrates for these lysosomal enzymes. After the initial development of the ValCitPABC linker, researchers have tested a plethora of peptide/peptidomimetic sequences to further improve the linker system [2,3,4,5,7,12] for faster release of payloads and to discover more enzyme-specific peptide sequences, which led to the identification of new cathepsin-sensitive dipeptide sequences such as ValAla, AlaAla, and cBuCit (cBu: cyclobutane-1,1-dicarboxamide).
The ValCitPABC linker system has proven to have good stability in human serum which is the utmost criterion for ADCs to sustain enzymatic degradation in systemic circulation. However, many studies have shown that ValCitPABC is susceptible in mouse plasma and that the degradation is caused by a protease called Carboxylesterase C1 (Ces1C) [13]. This instability in mouse plasma hampers preclinical evaluation of ADCs. Therefore, the preclinical studies have to be conducted in transgenic mice with knock-out Ces1C. Target-independent uptake toxicity [14], dose-limiting neutropenia and thrombocytopenia [3,4,5,6,8,15] are major causes of concern in ADC development. It has been shown that ADC treatment often leads to neutropenia in cancer patients [16], especially when employing ValCitPABC-MMAE. Zhao et al. [16] employed purified neutrophil elastase, a serine protease, to evaluate the in vitro stability of ADCs with a cleavable valine-citrulline linker (vc-MMAE) or a non-cleavable maleimidocaproyl linker (mc-MMAF) and showed that the ValCit linker was readily cleaved by elastase to release free MMAE, whereas the MC linker was not.
A number of recent review articles have covered ADC linkers designed on cleavage mechanisms by lysosomal enzymes like cathepsins, phosphatases, pyrophosphatases, sulfatases, β-galactosidase and β-glucuronidase as well as through reduction of disulfide linkages by glutathione [17,18,19,20,21]. In this mini review, we attempt to capture the most recent developments in the design of linkers with improved selectivity towards specific cleavage enzymes and increased stabilities in plasma.
2. Polar acidic residues at P3 position increases plasma stability
The ValCitPABC linker is designed to be cleaved by cathepsin B at the amide bond between the P1 residue citrulline and P1′ PABC (Figure 1B) [10]. However, it has been shown that ValCitPABC can be cleaved by Ces1C in mouse plasma, as verified with purified Ces1C enzyme and Ces1C knockout mouse as positive and negative controls [12]. In the same study, the ValCit linker was found to be stable when co-incubated with Ces1C inhibitors. After clearly establishing Ces1C-mediated cleavage mechanism, the authors synthesized several novel linker-payload molecules with a substitution at P3 position and tested their stability (Figure 1B). Interestingly, they observed increased mouse plasma stability when certain hydrophilic groups were introduced at the P3 position; in particular, a 2-hydroxy acetamide group greatly increased the plasma stability. Inspired by these results, Anami et al. introduced hydrophilic amino acids at the P3 position [22] and found that, while SerValCit showed little improvement in stability, linkers with an acidic amino acid at the P3 position, GluValCit and AspValCit, showed excellent stability in mouse plasma. In contrast, having a basic amino acid Lys at P3 made the LysValCit linker more labile than the parent ValCit linker. This indicates that an acidic amino acid at the P3 position effectively blocks the access of Ces1C whereas a basic amino acid enhances the interaction between Ces1C and the linker. Importantly, all the linker-payload designs discussed above were very stable in human serum and they remained susceptible to the lysosomal enzyme cathepsin B, which is crucial for drug release inside the cancer cell, suggesting that lysosomal cleavage enzymes do not have stringent sequence specificity. It is hypothesized that the human homolog of Ces1C enzyme in human liver has the active-site serine residue deep inside the substrate binding cleft, whereas the active site of human cathepsin B is shallow and thus substituents at the P3 site are well tolerable.
3. Polar basic residues at P1 improves lysosomal cleavage
Since its initial discovery, the ValCit-PABC linker system has been used in many ADC constructs with a variety of antibodies and payloads. Researchers have designed a plethora of P2-P1 dipeptides by substitution of P2-Val and P1-Cit, aiming to increase the linker’s lysosomal cleavability and to improve its stability in mouse plasma. Poudel et al. synthesized various linkers with uncialamycin as the payload [23]. When the polar citrulline was replaced with alanine, the stability in plasma was further decreased although payload release by cathepsin B was not affected. When the polar negatively charged aspartic acid was introduced at P1 position, there was no significant change in mouse plasma stability; however, it showed decreased release of the payload by cathepsin B. On the contrary, another study on the effect of P1 amino acid on cathepsin-mediated cleavage showed that an arginine residue at P1 position improved the cleavage by nine fold [24]. These results suggest that a polar or basic P1 amino acid is preferable for efficient payload release, whereas an acidic residue decreases the cleavage efficiency.
4. The effect of substitution on the PABC benzene ring
Directly attaching Val-Cit to the payload leads to less efficient payload release due to steric factors of the payload which inhibits cathepsin binding to the ValCit peptide. This problem is partially solved when a spacer is added between the payload and ValCit. PABC (para-aminobenzyl carbamate) not only improves cathepsin binding, it also undergoes self-immolative 1,6-elimination to release the payload in the unmodified form. Currently, PABC is used with a variety of payloads and peptide linker systems. To improve stability of the ValCit-PABC linker system toward Ces1C in mouse plasma, Podule et al. synthesized several uncialamycin-linker conjugates [23,25] with substitutions on or replacement of PABC, including replacement by heterocycles like thiazole. These conjugates were prepared from the reaction of MC-peptide-PABC-uncialamycin linkers and N-acetyl cysteine. They determined the percentage of drug released after 24 h incubation with cathepsin B, human serum, and mouse serum. Introducing a thiazole amide group in the place of PABC decreased the cleavage in mouse serum; however, cleavage in human serum was also observed. When an electron-withdrawing CF3 group was introduced on the thiazole ring, the cleavage in mouse serum was further decreased, so was the cleavage in human serum. Combining CF3-substituted thiazole with an aspartic acid at P3 position generated a linker with only 6% and 4% cleavage in mouse and human serum after 24 h incubation. It is noteworthy that all these PABC linker analogues were cleaved 100% by cathepsin B, which indicates that the modifications are well tolerated by cathepsin B. Next, they tried to see the impact of substitutions on PABC benzene ring. When ValCit was attached ortho to benzyl alcohol, another possible position for self-immolation, cathepsin activity was completely lost due to high steric hindrance of the ortho substitution. Furthermore, the linker did not survive in mouse serum. Encouraging results were obtained when an N-methyl carboxyamide group was introduced at the meta position. The resultant MA-PABC was only 3% cleaved in mouse serum after 24 h incubation and its cathepsin-mediated release was not affected. When a glutamic acid was added at the P3 position to the same MA-PABC, mouse serum cleavage was further reduced to 7% over 24 h. Adding a glutamic acid not only improves the stability towards Ces1C but also the solubility which is especially important when hydrophobic payloads are used. The authors went further to increase the hydrophilicity of linkers by replacing the methyl group with 2-aminoethyl and its amino-PEGylated form (Figure 2A). These new modifications provided the linkers with excellent mouse and human serum stability without compromising cathepsin B-mediated cleavage. The ADCs generated from the above PEG-linker-payload using bacterial transglutaminase chemistry showed antigen-dependent activity without problems of linker hydrolysis in mouse serum. The above study clearly demonstrates that combining various attributes from structure-activity relationship studies can yield ideal linker-payload systems with high stability in plasma while maintaining cathepsin susceptibility.
5. Tandem cleavable linkers
A common drawback of ValCitPABC-linked ADCs is myelosuppression, which is most likely caused by premature linker cleavage and payload release. In an effort to improve in vivo stability of ValCitPABC-MMAE conjugated ADCs, Chuprakov et al. developed an interesting tandem cleavable linker [26]. They incorporated a β-glucuronide moiety onto PABC which would act as a steric blocker to protect the ValCitPABC linker from serine proteases in circulation [Figure 2B]. After internalization, β-glucuronidase-mediated cleavage of β-glucuronide would first unmask ValCitPABC which would then be readily cleaved by cathepsins to release MMAE inside the cancer cells. An anti-CD79b antibody was used for conjugation with the tandem linker-MMAE payload using Hydrazino-iso-Pictet-Spengler (HIPS) chemistry between an aldehyde and a hydrazino-indole [27] to afford an ADC with DAR1.7 [28]. An ADC with an average DAR of 3.2 was also prepared by conventional MC-ValCitPABC-MMAE using cysteine conjugation for direct comparison. They synthesized several ADCs with the β-glucuronide moiety incorporated at the P1′ PABC position (Figure 2B). All the ADCs were equally potent when tested in vitro against B-cell non-Hodgkin lymphoma derived CD79b+ JeKo-1 cells. As expected, the conventional ValCitPABC-MMAE ADC lost 20% of payload when incubated in rat plasma at 37 °C for one week, whereas no payload loss was observed in ADCs with the tandem cleavable linker. When tested in mice carrying Granta 519 xenografts, ADC prepared from cysteine-conjugated MC-ValCit-PABC-MMAE with β-glucuronide moiety on P1′ PABC showed almost equal potency in reducing the tumor volume. Interesting results were observed from in vivo tolerability and toxicokinetic studies in which the ADCs were dosed to Sprague−Dawley rats. Clinical chemistry and hematology were assessed on day 5 and day 7. Key indicators of myelosuppression, namely the levels of monocytes, neutrophils and eosinophils, were studied. Rats treated with the conventional mono-cleavage-linker ADCs had marked reductions in the levels of circulating monocytes, neutrophils and eosinophils, whereas rats treated with tandem-cleavage-linker ADCs did not show any evidence of myelosuppression. In fact, the levels of circulating monocytes, neutrophils and eosinophils of the latter group were similar to those in vehicle control groups, indicating clearly that masking with β-glucuronide moiety at P1′ greatly increased linker stability in circulation. To further confirm it, plasma samples were taken at various time points and analyzed with ELISA methods and found that there was rapid payload loss in the mono-cleavage ADCs compared to tandem-cleavage ADCs. The released payload would be the likely reason for observed myelosuppression.
Besides plasma instability, the dipeptide-based linkers are often hydrophobic in nature, which may cause ADC aggregation. To address these issues, Bargh et al. reported a dual enzyme-cleavable 3-O-sulfo-β-galactose linker [29] (Figure 2C), which can be cleaved in a cascade reaction by the consecutive actions of arylsulfatase A (ARSA) and β-galactsedase (β-gal). To validate this concept, they synthesized the 3-O-sulfo-β-galactose linker with 7-amino-4-methyl coumarin (AMC) as a model payload, and incubated it with ARSA, β-gal and mixture of both ARSA and β-gal and measured the fluorescence signal of released AMC. As anticipated, either ARSA or β-gal alone did not give any fluorescence signal, but incubation with both enzymes gave dramatic fluorescence, indicating that both enzymes were needed for the payload release. This ARSA/β-gal combination greatly improved the lysosome-selective cleavage. Further, the presence of anionic sulfate group and galactose moiety greatly improved the solubility of the linker in aqueous medium. Having encouraging results in fluorescence studies, they synthesized 3-O-sulfo-β-galactose-MMAE linker payloads with bis-thiol reactive divinylprimidine (DVP), which reacted with reduced α-HER2-trastuzumab to afford ADCs with DAR4. The generated ADCs were tested against HER2-positive SKBR3 and HER2-negative MFC7 cells. Excellent potency (IC50 = 49 pM), which is comparable to Val-Ala-PABC-MMAE (IC50 = 41 pM), and selectivity were observed. The 3-O-sulfo-β-galactose linker system would be really useful in generating ADCs having highly hydrophobic payloads, which are otherwise known to aggregate easily in aqueous medium.
6. Legumain cleavable linkers
Legumain, an asparaginyl endopeptidase [30,31,32], is known to have high expression levels in cancer cells. Like cathepsins, it is also present in lysosomes. Legumain is first synthesized as a pro-enzyme and an acidic pH (pH < 4-5) is needed for its activation. It has increased expression in solid tumors where it plays a key role in tumor invasion and metastasis [33], which makes it an attractive alternative to cathepsins for the development of protease-cleavable linkers [34] [33]. Bajjuri et al. found that the tripeptide-conjugated MMAE, AlaAlaAsn-PABC-MMAE, was a prodrug to MMAE, which is otherwise too toxic to use as a free drug [35]. It was shown that AlaAlaAsn-PABC-MMAE was effectively cleaved by legumain and was less toxic than free MMAE in mouse models. Cheng et al. used the cysteine conjugation approach to generate several ADCs with different cleavable linkers with varying length of PEG as the spacer and microtubule inhibitors as the payloads. They showed that an ADC prepared using the polyethylene glycol-extended legumain-cleavable tripeptide linker payload system, PEG2-AlaAlaAsn-PABC-eribulin, showed similar cytotoxicity when compared to other ADCs prepared from other linkers [36]. However, they did not disclose in vivo efficacy data in mouse models. Lerchen et al. reported rather interesting results on legumain-cleavable linkers [37]. They prepared TWEAKR-targeting ADCs with a kinesin spindle protein inhibitor (KSPi) as the payload [38,39]. The KSPi has a primary amine group which was directly attached to the peptide linker without a self-immolative system. The alpha carbon of the KSPi primary amine was functionalized with a propionyl-Glu moiety (Figure 3A) to modulate physiochemical properties of the active metabolite as the polar glutamate reduced the permeability, thus enabled longer retention inside the tumor cells. They synthesized constructs with varying linker sequences, such as L-Ala-L-Ala-L-Asn (Lm-1), L-Ala-D-Ala-L-Asn (Lm-2), L-Ala-L-Ala(NMe)-L-Asn (Lm-3), L-Ala-L-Ala(NMe)-D-Asn (lm-4), L-Asn (Lm-5), D-Asn (Lm-6), L-Leu (Lm-7), and L-Gln (Lm-8) (Figure 3A) and evaluated in vitro protease cleavage activity with legumain, cathepsin B and neutrophil elastase. As anticipated, D-Asn-containing constructs and constructs with a single Asn showed very poor hydrolysis activity by legumain. All the constructs showed no cleavage with cathepsin B and neutrophil elastase. Surprisingly, when the corresponding ADCs were incubated with NCI-H292 cells, for ADCs with linkers Lm-2 and Lm-5, similar amounts of active metabolite was observed in cell lysate indicating expression of high levels of legumain in tumor cells. In in vitro efficacy studies against TWEAKR-expressing NCI-H292 lung cancer, BxPC3 pancreatic cancer and LoVo colon cancer cell lines, the Lm-1, Lm-2 and Lm-3 ADCs showed excellent potency. Replacing the central L-Ala with D-Ala did not affect the activity; however, when the L-Asn was replaced with unnatural D-Asn, the activity was completely lost. The most intriguing results were that the ADC with a single L-Asn in the linker retained the activity. Similar activity trends were observed with Her-2-targeting ADCs. Liver toxicity of the ADCs were evaluated by incubating in the lysosomal preparation of rat liver for 48 h using the classic ValCit-PABC ADC as a positive control which showed 85% cleavage; however, the new ADCs with legumain-cleavable linkers were found to be much more stable with little hydrolysis, 18% for Lm-1, 5% for Lm-3 and less than 1% for Lm-2 and Lm-5. Excellent in vivo anti-tumor activity was observed with these ADCs when treated with mice bearing NCI-H292 and Ku-19-19 tumors.
In a high throughput enzyme agnostic screening, Miller et al. used Förster resonance energy transfer (FRET) assay to study cleavage efficiency in lysosomal extracts and stability in human and mouse plasma of 75 peptide FERT pairs (Figure 3A) [40]. They found that various Asn-containing dipeptide sequences were readily cleaved by legumain. In enzyme-based assays, peptide sequences like AsnAsn, AsnAla were selectively cleaved by legumain and very stable towards cathepsin B. The newly identified Asn peptide linkers were found to be very stable in mouse plasma and resistant to Ces1c-mediated cleavage. They synthesized anti-Her2 and anti-CD20 ADCs with the AsnAsn, GlnAsn, AlaAsn, ValCit dipeptide-PABC-MMAE linker-payload system and using cysteine-maleimide chemistry for conjugation (DAR ~ 6). In vitro cytotoxicity of the ADCs was evaluated using Her-2 expressing SKBR3 (+++) cells and CD20 expressing Ramos cells. Compared with the cathepsin-cleavable ValCit ADCs, the legumain cleavable ADCs showed similar activities; however, Asn-containing ADCs showed 2-3-fold higher off-target activity than the ValCit ADC. The reduced selectivity of legumain-cleavable ADCs is likely because that legumain is also secreted in tumor microenvironment and thus might have caused extracellular linker cleavage.
7. Peptidomimetic substitution at P2 position
In a structure-based study of novel peptidomimetic substitutions at the P2 position, Wei et al. took the key assumption that reducing the number of hydrolyzable peptide bonds would yield cathepsin B-specific linkers with improved extracellular stability [24]. In this study, they used simplified constructs where linkers were attached to norfloxacin through PABC and the N-terminal was protected as benzyl carbamate. Cleavage efficiency was evaluated using Michaelis-Menten steady state Vmax and Km data by keeping cathepsin B concentration constant. When they replaced the P2-P1 peptide bond with fluoroolefin and triazole, the cleavage activity by cathespsin B was greatly reduced, which suggests hydrogen bond interactions are crucial for enzyme binding. Based on computational shape similarity search, they identified cyclobutane-1,1-dicarboxamide as a suitable P2 residue. This novel structural unit is able to provide three hydrogen bonding interactions and the cyclobutyl group has optimum size to fit in the S2 binding pocket. They synthesized various linkers with varying size of cycloalkyl rings and side chains at the P1 site; however, none of these showed increased cleavage activity compared to the ValCit counterpart. To address the discrepancies between computational model and observed cleavage results, they obtained crystal structure of human cathepsin B complexed with thiophen-cBuCit-CN (Figure 3B, insert) where the electrophilic CN group was introduced to capture the Cys thiol group at the active site. Indeed, the crystal structure showed expected hydrogen bonding interactions and the key thioimidate bond formed between the cysteine and C≡N and the cyclobutyl group perfectly sitting in the S2 binding pocket. Encouraged from the crystal structure results, they went on to synthesize the ADCs, anti-Her2-mc-cBuCitPABC-MMAE and anti-Her2-mc-ValCitPABC-MMAE (Figure 3B), using engineered-cysteine strategy with DAR between 1.8 and 2.0. When these ADCs were evaluated against Her-2 expressing SKBR-3 cells, both ADCs showed almost similar antigen dependent anti-cancer activity, and both ADCs were equally stable in in vivo mouse models. When incubating the ValCit and cBuCit ADCs with inhibitors of cathepsin B, they found that cBuCit cleavage activity was reduced by 90% vs 50% for ValCit, demonstrating the high specificity of the cBuCit linker towards cathepsin B.
8. Conclusion and outlook
The immense research effort put into ADC linker-payload design during the past decade has contributed to a sharp increase in regulatory approvals. However, ADC development still faces dose-limiting toxicity issues during clinical trials, which could be partly attributed to extracellular release of the payload and poor PK properties. The development of linker systems more stable in mouse and rat plasma would accelerate preclinical evaluation of ADC drug candidates. In particular, the identification of new peptide/peptidomimetic sequences will allow highly specific cleavage which can greatly reduce extracellular cleavage of the linker, hence increasing the safety profiles of the ADCs. Site-specific conjugation using engineered antibodies and enzyme systems now enables the development of homogeneous ADCs with superior PK properties. The use of improved linker-payload designs and highly active payloads in combination with site-specific conjugation chemistry will lead to more successes in the ADC field in the future.
Author Contributions
Seetharamsing Balamkundu wrote a draft and Chuan-Fa Liu edited and finalized the manuscript.
Funding
This research was supported by AcRF Tier 1 grant (2019-T1-002-100) from the Singapore Ministry of Education (MOE) and a CRP grant (NRF-CRP24-2020-0005) from the National Research Foundation (NRF) under the Prime Minister's Office, Singapore.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors thank the School of Biological Sciences at Nanyang Technological University for the excellent facilities and research environment.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Fu, Z.; Li, S.; Han, S.; Shi, C.; Zhang, Y. Antibody drug conjugate: the “biological missile” for targeted cancer therapy. Signal Transduct Target Ther 2022, 7, 93. [Google Scholar] [CrossRef] [PubMed]
- Tong, J. T.; Harris, P. W.; Brimble, M. A.; Kavianinia, I. An insight into FDA approved antibody-drug conjugates for cancer therapy. Molecules 2021, 26, 5847. [Google Scholar] [CrossRef]
- Lyon, R. Drawing lessons from the clinical development of antibody-drug conjugates. Drug Discov Today Technol 2018, 30, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Chau, C. H.; Steeg, P. S.; Figg, W. D. Antibody–drug conjugates for cancer. Lancet 2019, 394, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody–drug conjugates. Nat Rev Drug Discov 2017, 16, 315–337. [Google Scholar] [CrossRef] [PubMed]
- Hughes, B. Antibody-drug conjugates for cancer: poised to deliver? Highlighted by Genentech's recent US regulatory submission for trastuzumab-DM1, antibody-drug conjugation technology could be heading for the mainstream in anticancer drug development. Nat Rev Drug Discov 2010, 9, 665–668. [Google Scholar] [CrossRef] [PubMed]
- Carter, P. J.; Senter, P. D. Antibody-drug conjugates for cancer therapy. Cancer J 2008, 14, 154–169. [Google Scholar] [CrossRef]
- Donaghy, H. Effects of antibody, drug and linker on the preclinical and clinical toxicities of antibody-drug conjugates. MAbs 2016, 8, 659–671. [Google Scholar] [CrossRef]
- Drago, J. Z.; Modi, S.; Chandarlapaty, S. Unlocking the potential of antibody–drug conjugates for cancer therapy. Nat Rev Clin Oncol 2021, 18, 327–344. [Google Scholar] [CrossRef]
- Doronina, S. O.; Toki, B. E.; Torgov, M. Y.; Mendelsohn, B. A.; Cerveny, C. G.; Chace, D. F.; DeBlanc, R. L.; Gearing, R. P. ’ Bovee, T. D.; Siegall, C. B.; Francisco, J. A.; Wahl, A. F.; Meyer D. L.; Senter P. D. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat Biotech 2003, 2003 21, 778–784. [Google Scholar] [CrossRef]
- Doronina, S. O.; Bovee, T. D.; Meyer, D. W.; Miyamoto, J. B.; Anderson, M. E.; Morris-Tilden, C. A.; Senter, P. D. Novel peptide linkers for highly potent antibody− auristatin conjugate. Bioconjug Chem 2008, 19, 1960–1963. [Google Scholar] [CrossRef] [PubMed]
- Salomon, P. L.; Reid, E. E.; Archer, K. E.; Harris, L.; Maloney, E. K.; Wilhelm, A. J.; Miller, M. L.; Chari, R. V.; Keating, T. A.; Singh, R. Optimizing Lysosomal Activation of Antibody–Drug Conjugates (ADCs) by Incorporation of Novel Cleavable Dipeptide Linkers. Mol Pharm 2019, 16, 4817–4825. [Google Scholar] [CrossRef] [PubMed]
- Dorywalska, M.; Dushin, R.; Moine, L.; Farias, S. E.; Zhou, D.; Navaratnam, T.; Lui, V.; Hasa-Moreno, A.; Casas, M. G.; Tran, T.-T. Molecular basis of valine-citrulline-pabc linker instability in site-specific ADCs and its mitigation by linker designmolecular basis of VC-PABC linker instability. Mol Cancer Ther 2016, 15, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Mahalingaiah, P. K.; Ciurlionis, R.; Durbin, K. R.; Yeager, R. L.; Philip, B. K.; Bawa, B.; Mantena, S. R.; Enright, B. P.; Liguori, M. J.; Van Vleet, T. R. Potential mechanisms of target-independent uptake and toxicity of antibody-drug conjugates. Pharmacol Ther 2019, 200, 110–125. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.; Gopal, A. K.; Smith, S. E.; Ansell, S. M.; Rosenblatt, J. D.; Savage, K. J.; Ramchandren, R.; Bartlett, N. L.; Cheson, B. D.; De Vos, S. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin's lymphoma. J Clin Oncol 2012, 30, 2183. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Gulesserian, S.; Malinao, M. C.; Ganesan, S. K.; Song, J.; Chang, M. S.; Williams, M. M.; Zeng, Z.; Mattie, M.; Mendelsohn, B. A. A potential mechanism for ADC-induced neutropenia: role of neutrophils in their own demisemechanism for ADC-induced neutropenia. Mol Cancer Ther 2017, 16, 1866–1876. [Google Scholar] [CrossRef] [PubMed]
- Bargh, J. D.; Isidro-Llobet, A.; Parker, J. S.; Spring, D. R. Cleavable linkers in antibody–drug conjugates. Chem Soc Rev 2019, 48, 4361–4374. [Google Scholar] [CrossRef]
- Poreba, M. Protease-activated prodrugs: strategies, challenges, and future directions. Febs j 2020, 287, 1936–1969. [Google Scholar] [CrossRef]
- Su, Z.; Xiao, D.; Xie, F.; Liu, L.; Wang, Y.; Fan, S.; Zhou, X.; Li, S. Antibody–drug conjugates: Recent advances in linker chemistry. Acta Pharm Sin B 2021. [Google Scholar] [CrossRef]
- Mckertish, C. M.; Kayser, V. Advances and limitations of antibody drug conjugates for cancer. Biomedicines 2021, 9, 872. [Google Scholar] [CrossRef]
- Sheyi, R.; de la Torre, B. G.; Albericio, F. Linkers: An assurance for controlled delivery of antibody-drug conjugate. Pharmaceutics 2022, 14, 396. [Google Scholar] [CrossRef] [PubMed]
- Anami, Y.; Yamazaki, C. M.; Xiong, W.; Gui, X.; Zhang, N.; An, Z.; Tsuchikama, K. Glutamic acid–valine–citrulline linkers ensure stability and efficacy of antibody–drug conjugates in mice. Nat Commun 2018, 9, 2512. [Google Scholar] [CrossRef]
- Poudel, Y. B.; Chowdari, N. S.; Cheng, H.; Iwuagwu, C. I.; King, H. D.; Kotapati, S.; Passmore, D.; Rampulla, R.; Mathur, A.; Vite, G. Chemical modification of linkers provides stable linker–payloads for the generation of antibody–drug conjugates. ACS Med Chem Lett 2020, 11, 2190–2194. [Google Scholar] [CrossRef]
- Wei, B.; Gunzner-Toste, J.; Yao, H.; Wang, T.; Wang, J.; Xu, Z.; Chen, J.; Wai, J.; Nonomiya, J.; Tsai, S. P. Discovery of peptidomimetic antibody–drug conjugate linkers with enhanced protease specificity. J Med Chem 2018, 61, 989–1000. [Google Scholar] [CrossRef] [PubMed]
- Poudel, Y. B.; Rao, C.; Kotapati, S.; Deshpande, M.; Thevanayagam, L.; Pan, C.; Cardarelli, J.; Chowdari, N.; Kaspady, M.; Samikannu, R. Design, synthesis and biological evaluation of phenol-linked uncialamycin antibody-drug conjugates. Bioorg Med Chem Lett 2020, 30, 126782. [Google Scholar] [CrossRef] [PubMed]
- Chuprakov, S.; Ogunkoya, A. O.; Barfield, R. M.; Bauzon, M.; Hickle, C.; Kim, Y. C.; Yeo, D.; Zhang, F.; Rabuka, D.; Drake, P. M. Tandem-cleavage linkers improve the in vivo stability and tolerability of antibody–drug conjugates. Bioconjug Chem 2021, 32, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Kudirka, R.; Albers, A. E.; Barfield, R. M.; de Hart, G. W.; Drake, P. M.; Jones, L. C.; Rabuka, D. Hydrazino-pictet-spengler ligation as a biocompatible method for the generation of stable protein conjugates. Bioconjug Chem 2013, 24, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Rabuka, D.; Rush, J. S.; Dehart, G. W.; Wu, P.; Bertozzi, C. R. Site-specific chemical protein conjugation using genetically encoded aldehyde tags. Nat Protoc 2012, 7, 1052–1067. [Google Scholar] [CrossRef]
- Bargh, J. D.; Walsh, S. J.; Ashman, N.; Isidro-Llobet, A.; Carroll, J. S.; Spring, D. R. A dual-enzyme cleavable linker for antibody–drug conjugates. Chem Commun 2021, 57, 3457–3460. [Google Scholar] [CrossRef]
- Ishii, S. Legumain: Asparaginyl endopeptidase. Methods Enzymol 1994, 244, 604–615. [Google Scholar]
- Chen, J. M.; Dando, P. M.; Stevens, R. A. E.; Fortunato, M.; Barrett, A. J. Cloning and expression of mouse legumain, a lysosomal endopeptidase. Biochem J 1998, 335, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-M.; Dando, P. M.; Rawlings, N. D.; Brown, M. A.; Young, N. E.; Stevens, R. A.; Hewitt, E.; Watts, C.; Barrett, A. J. Cloning, isolation, and characterization of mammalian legumain, an asparaginyl endopeptidase. J Biol Chem 1997, 272, 8090–8098. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Sun, C.; Huang, H.; Janda, K.; Edgington, T. Overexpression of legumain in tumors is significant for invasion/metastasis and a candidate enzymatic target for prodrug therapy. Cancer Res 2003, 63, 2957–2964. [Google Scholar] [PubMed]
- Wu, W.; Luo, Y.; Sun, C.; Liu, Y.; Kuo, P.; Varga, J.; Xiang, R.; Reisfeld, R.; Janda, K. D.; Edgington, T. S. Targeting cell-impermeable prodrug activation to tumor microenvironment eradicates multiple drug-resistant neoplasms. Cancer Res 2006, 66, 970–980. [Google Scholar] [CrossRef] [PubMed]
- Bajjuri, K. M.; Liu, Y.; Liu, C.; Sinha, S. C. The legumain protease-activated auristatin prodrugs suppress tumor growth and metastasis without toxicity. ChemMedChem 2011, 6, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Li, J.; Tanaka, K.; Majumder, U.; Milinichik, A. Z.; Verdi, A. C.; Maddage, C. J.; Rybinski, K. A.; Fernando, S.; Fernando, D. MORAb-202, an antibody–drug conjugate utilizing humanized anti-human frα farletuzumab and the microtubule-targeting agent eribulin, has potent antitumor activityMORAb-202, an anti-FRA ADC utilizing eribulin as payload. Mol Cancer Ther 2018, 17, 2665–2675. [Google Scholar] [CrossRef]
- Lerchen, H.-G.; Stelte-Ludwig, B.; Sommer, A.; Berndt, S.; Rebstock, A.-S.; Johannes, S.; Mahlert, C.; Greven, S.; Dietz, L.; Jörißen, H. Tailored linker chemistries for the efficient and selective activation of ADCs with KSPi payloads. Bioconjug Chem 2020, 31, 1893–1898. [Google Scholar] [CrossRef]
- Lerchen, H. G.; Wittrock, S.; Stelte-Ludwig, B.; Sommer, A.; Berndt, S.; Griebenow, N.; Rebstock, A. S.; Johannes, S.; Cancho-Grande, Y.; Mahlert, C. Antibody–drug conjugates with pyrrole-based KSP inhibitors as the payload class. Angew Chem Int Ed 2018, 57, 15243–15247. [Google Scholar] [CrossRef]
- Karpov, A. S.; Nieto-Oberhuber, C. M.; Abrams, T.; Beng-Louka, E.; Blanco, E.; Chamoin, S.; Chene, P.; Dacquignies, I.; Daniel, D.; Dillon, M. P.; Doumampouom-Metoul, L.; Drosos, N.; Fedoseev, P.; Furegati, M.; Granda, B.; Grotzfeld, R. M.; Hess Clark, S.; Joly, E.; Jones, D.; Lacaud-Baumlin, M.; Lagasse-Guerro, S.; Lorenzana, E. G.; Mallet, W.; Martyniuk, P.; Marzinzik, A. L.; Mesrouze, Y.; Nocito, S.; Oei, Y.; Perruccio, F.; Piizzi, G.; Richard, E.; Rudewicz, P. J.; Schindler, P.; Velay, M.; Venstrom, K.; Wang, P.; Zurini, M.; Lafrance, M. Discovery of potent and selective antibody–drug conjugates with Eg5 inhibitors through linker and payload optimization. ACS Med Chem Lett 2019, 10, 1674–1679. [Google Scholar] [CrossRef]
- Miller, J. T.; Vitro, C. N.; Fang, S.; Benjamin, S. R.; Tumey, L. N. Enzyme-agnostic lysosomal screen identifies new legumain-cleavable ADC linkers. Bioconjug Chem 2021, 32, 842–858. [Google Scholar] [CrossRef]
Figure 1.
(A) Fate of an ADC before and after internalization. Premature cleavage of the linker in extracellular matrix is often associated with off-target toxicity. Antigen-mediated endocytosis delivers ADC in the endosomal-lysosomal system and lysosomal linker cleavage releases the drug which acts to exert its cytotoxicity. (B) Effect of amino acid composition in the linker peptide and substitution on the benzene ring of PABC on linker stability in mouse plasma. The SAR data can guide design of ideal ADC linkers with efficient payload release while maintaining good extracellular stability.
Figure 1.
(A) Fate of an ADC before and after internalization. Premature cleavage of the linker in extracellular matrix is often associated with off-target toxicity. Antigen-mediated endocytosis delivers ADC in the endosomal-lysosomal system and lysosomal linker cleavage releases the drug which acts to exert its cytotoxicity. (B) Effect of amino acid composition in the linker peptide and substitution on the benzene ring of PABC on linker stability in mouse plasma. The SAR data can guide design of ideal ADC linkers with efficient payload release while maintaining good extracellular stability.
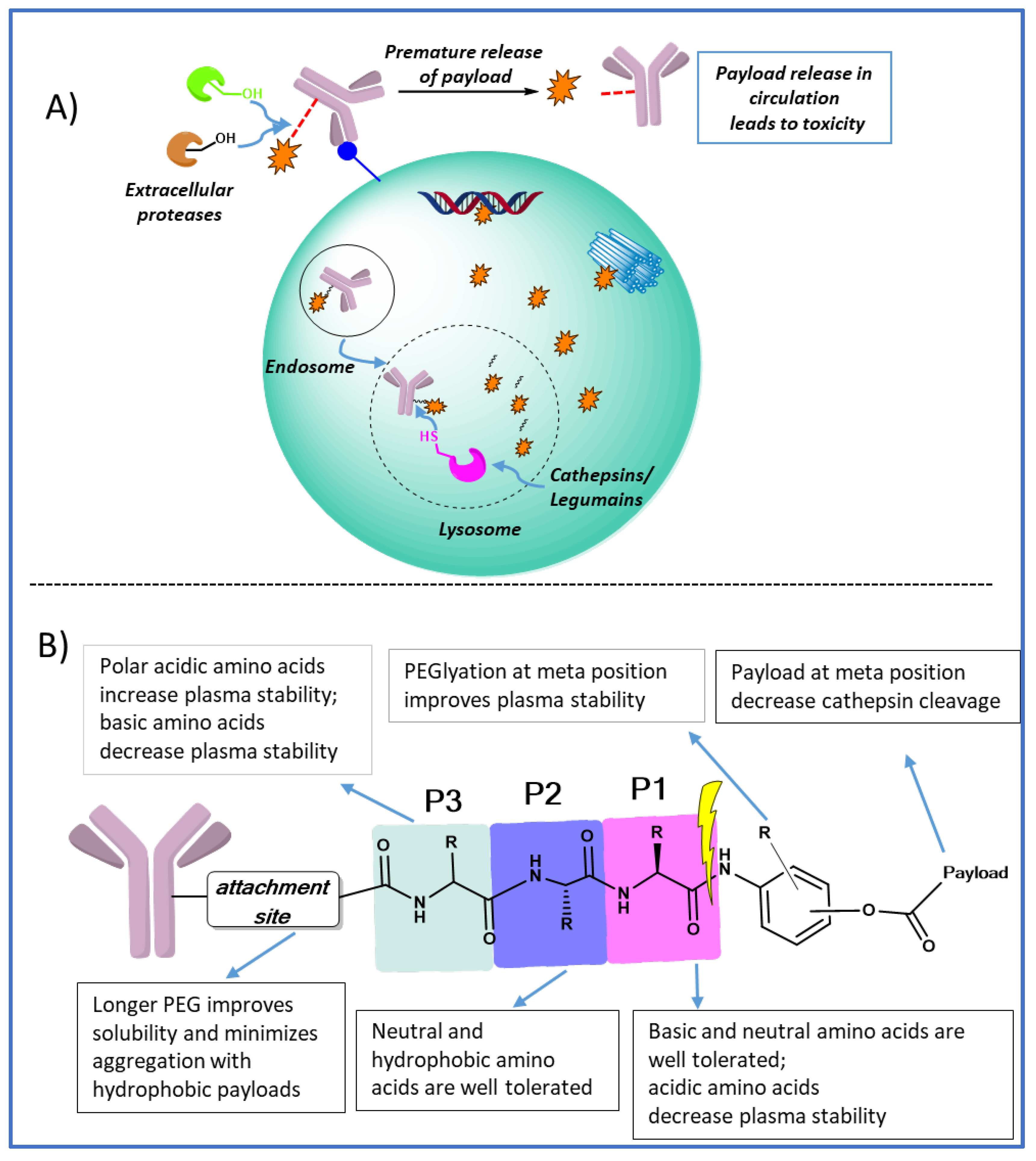
Figure 2.
Various Linker payload designs for faster lysosomal cleavage and improved plasma stability, (A) Modifications to central PABC ring, (B) Tandem cleavable linkers, glucuronide group masks the linker system to maintain stability in extracellular environment, (C) Aryalsulfatase A (ARSA) and β-galactosidase dual cleavable 3-O-sulfo-β-galactose linker.
Figure 2.
Various Linker payload designs for faster lysosomal cleavage and improved plasma stability, (A) Modifications to central PABC ring, (B) Tandem cleavable linkers, glucuronide group masks the linker system to maintain stability in extracellular environment, (C) Aryalsulfatase A (ARSA) and β-galactosidase dual cleavable 3-O-sulfo-β-galactose linker.
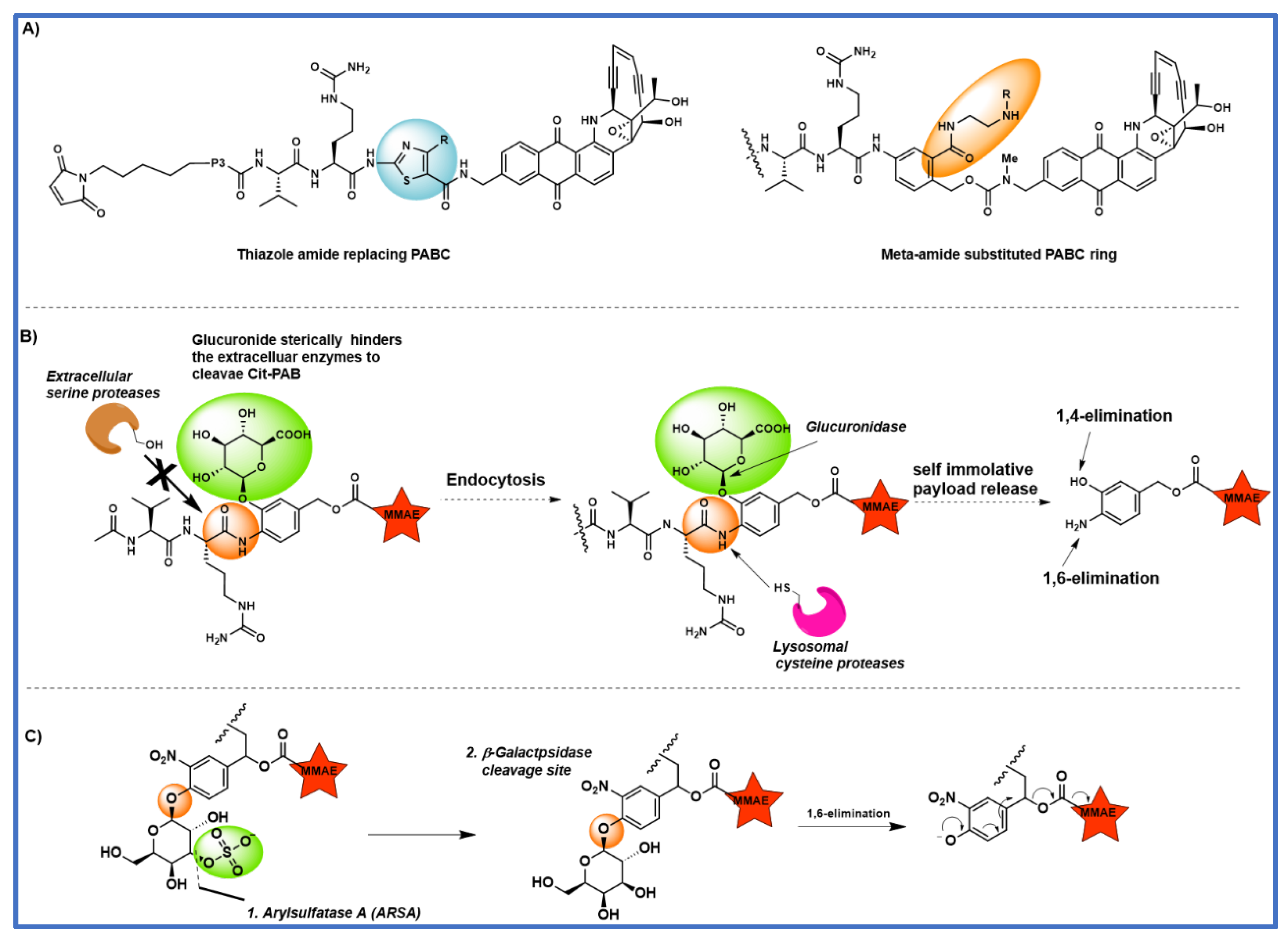
Figure 3.
(A) Asn-containing peptide linkers with improved selectivity towards legumain, (B) Peptidomimetic linkers specifically cleaved by cathepsin B.
Figure 3.
(A) Asn-containing peptide linkers with improved selectivity towards legumain, (B) Peptidomimetic linkers specifically cleaved by cathepsin B.

Table 1.
Clinically approved ADCs .
| S.No | ADC | Linker system | Cleavage mechanism | Payload | Company |
|---|---|---|---|---|---|
| 1 | Gemtuzumab ozogamicin (Mylotarg) |
4-(4-acetylphenoxy) butanoic acid |
pH sensitive | Calicheamicin | Pfizer/Wyeth |
| 2 | Brentuximab vedotin (Adcetris) |
mc- ValCitPABC |
Lysosomal | MMAE | Seattle/Takeda |
| 3 | Trastuzumab emtansine (Kadcyla) |
MCC | Non cleavable | Maytansine DM1 | Genentech Roche |
| 4 | Inotuzumab ozogamicin (Besponsa) |
Hydrazone | pH sensitive | Calicheamicin | Pfizer/Wyeth |
| 5 | Polatuzumab vedotin (Polivy) |
mc- ValCitPABC |
Lysosomal degradation | MMAE | Genentech Roche |
| 6 | Enfortumab vedotin (Padcev) |
mc- ValCitPABC |
Lysosomal degradation | MMAE | Astellas/Seattle Genetics |
| 7 | Trastuzumab deruxtecan (Enhertu) |
mc-GGFG-aminol | Lysosomal degradation | Deruxtecan, Dxd | Daiichi-Sankyo/AstraZeneca |
| 8 | Sacituzumab govitecan (Trodelvy) |
mc-PEG-carbonate | pH | SN-98 | Immunomedics |
| 9 | Belantamab mafodotin (Blenerp)* | mc-MMAF | Non cleavable | MMAF | GSK |
| 10 | Loncastuximab tesirine (Zynlonta) |
mc- ValCitPABC |
Lysosomal degradation | SG3199, PDB dimer | ADC Therapeutics |
| 11 | Tisotumab vedotin (Tivdak) |
mc- ValCitPABC | Lysosomal degradation | MMAE | Genmab and Seattle Genetics |
| 12 | Disitamab Vedotin (Aidixi) |
mc- ValCitPABC |
Lysosomal degradation | MMAE | RemeGen |
| 13 | Moxetumomab pasudotox (Lumoxiti) | mc-ValCitPABC | Lysosomal degradation | PE38 | AstraZeneca |
| 14 | Cetuximab sarotalocan (Akalux) | NA | NA | IRDye700DX | Rakuten Medical |
| 15 | Mirvetuximab Soravtansine (ELAHERE) |
Disulfide-containing cleavable linker sulfo-SPDB | Glutathione cleavable | Maytansinoid DM4 | ImmunoGen |
* Following the request of U.S. FDA, belantamab mafodotin was withdrawn from the market based on the outcome of DREAMM-3 phase-III confirmatory trials.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



