Submitted:
05 May 2023
Posted:
08 May 2023
You are already at the latest version
Abstract
Molecular modeling studies were carried out at the DFT level of the adsorption of propane and propene on Ag surfaces as a model of the interaction of light hydrocarbons with Ag/ZrO2 catalysts for the catalytic combustion reaction. It was found that the most stable mode of adsorption of propene through its π system on Ag atom has energies consistent with chemisorption and generates an elongation of the C1=C2 bond, which would explain the increase in the activity of the catalysts as function of its metallic charge. The results obtained from the DFT calculations explain the different types of interaction between propene and propane with the metallic surface. The propene is chemisorbed on the Ag surface, distorting its bonds and generating its activation. This would imply that a higher metallic charge in the catalyst would increase the number of active sites in which this activation occurs, generating a higher activity. Besides, with the addition of O, the binding energy between the propene and the metal surface increased. On the other hand, the presence of a metallic surface is not enough for the activation of the propane molecule. This would explain why by increasing the amount of metal in the catalyst, the activity for the combustion of propane is practically not affected
Keywords:
propane
; propene
; combustion
; DFT
; silver
1. Introduction
The increasingly strict legislation regarding the control of contaminants present in sources of gaseous emissions generates a challenge for the development of clean technologies and environmental catalysis [1,2] . These emissions can come from stationary sources, mainly from the petrochemical industry, or from mobile sources, mainly from diesel engine exhausts. The pollutants usually present in gaseous emissions are nitrogen oxides (NOx), carbon oxides (COx), sulphur oxides (SOx), particulate matter, and volatile organic compounds (VOCs), among others. In particular, within the volatile organic compounds, hydrocarbons can be found in the C2-C10 range and those with the shortest chain, C2-C3, are the most difficult to remove [3].
One of the most widely used technologies to eliminate hydrocarbon molecules present in emissions is catalytic oxidation, also called catalytic combustion. To carry out this process, filters or converters are needed, which contain catalytic phases, capable of lowering the combustion temperatures to temperatures below ~400°C [4,5]. As is widely reported in the literature, several types of materials catalyse the combustion reactions of alkanes, alkenes, and polyaromatics, including those whose active phases are noble metals such as Pt, Pd, Au, Rh, Ag, etc. [6,7,8,9]. These metallic catalysts exhibit good catalytic properties due to their high activity at low temperatures and resistance to poisoning. However, the high costs and low availability of these noble metals mean their optimisation needs to be maximised. For this, much research has been conducted on the choice of the active-phase support system, the optimisation of metallic content, and in particular, the elucidation of the site where the reaction proceeds, to improve their performance.
Although it is a highly studied topic, which has given rise to numerous bibliographical reviews, the question of whether to optimise a catalytic formulation using one or a certain group of molecules as a model remains. In many cases, similar molecules share similar reaction mechanisms. However, there are catalytic phases that are very active for the combustion of some hydrocarbons that present low activity others, or they act with different efficiencies or reaction mechanisms depending on the hydrocarbons’ nature or chain. For example, for the combustion of alkanes and alkenes over Pt/Al2O3 and Pt/TiO2 catalysts, the reaction orders are positive and negative, respectively [10]. This fact suggests a different coverage of the hydrocarbon on the surface, a different rate-limiting step of the reaction, or the distinctive roles of other reactants, such as oxygen or concomitants, in the mechanism. In this context, the Langmuir–Hinshelwood and the Eley–Rideal mechanisms are two of the three proposed for the total oxidation of hydrocarbons [11,12,13]. In the first one, a bimolecular mechanism, the molecules of hydrocarbon and dioxygen adsorb onto the catalytic surface in the first reaction step [14]. In the second one, a molecule of hydrocarbon adsorbs on the surface and then reacts with oxygen from gas phase [12]. Both mechanisms propose that the first step in the reaction mechanism is the adsorption of the hydrocarbon molecule onto the catalytic surface. However, the ability of an active phase to adsorb a reactant depends on the nature of the active phase, and the adsorbate, especially, depends on its molecular structure.
For the more studied metals, for example Pt, it has been proposed that the reason for their different behaviour in catalytic combustion between alkanes and alkenes is the presence of a C=C double bond in alkenes, which can facilitate the strong adsorption of hydrocarbon molecules on the surface of noble metal through the interaction between the π orbitals of the alkene and the d orbitals of the transition metals [6,15]. On the other hand, alkanes interact with the metal surface through van der Waals forces, which are much weaker, causing the reaction mechanisms to be different [16,17]. In most of the proposed reaction mechanisms, the rate-determining step of the reaction is estimated from the reaction order data. There are few works in the literature that show the theoretical results of what occurs at the molecular level on the catalyst surface. For predictive purposes or with the purpose of being able to explain experimental results, it is very interesting to understand in depth how a different type of interaction between the substrate/hydrocarbon and the metal surface can condition the activity of a species and/or modify the step that determines the rate of a reaction or even the mechanism of it. Currently, surface science, through the application of theoretical calculations, makes it possible to study and understand fundamental aspects of the operation/behaviour of heterogeneous catalysts. For example, this method allows analysing the adsorption phenomenon of the species that are part of the reaction. In recent decades, from the implementation of DFT (density functional theory) to periodic systems, it has been possible to model with greater precision what occurs at the atomic level on the surface of catalysts. Several authors have studied the adsorption of C1, C2, and C3 species on the surfaces of different metals by applying DFT calculations. Arya et al. [18] reported the energies of the transition states for the rupture of the C-H model bond on different metals, as a model of activation energies for the alkanes’ combustion reaction. These authors found that the activation energy decreased in the following order Ag ˃ Au ˃ Al ˃ Cu ˃ Pt ˃ Pd ˃ Ni > Co > Rh > Fe. Wang et al. [19] studied the combustion of formaldehyde in a Ag cluster. The results showed that in the oxidation of formaldehyde in a Ag cluster, where there are also chemisorbed O atoms (active species), the energy of the transition state was lower than that calculated for the Ag cluster in the absence of O. A similar result was found by Porter et al. [20] who studied different reaction pathways for the epoxidation of propene on different metals from DFT calculations.
In a previous work reported in our work group, it was found that catalysts based on metallic silver supported on zirconia presented different behaviours when they were used in the combustion reactions of propane and propene [9]. No computational chemistry results that explain this behaviour have been reported in the literature; therefore, it is interesting to model the silver surface and its interaction with short-chain hydrocarbon molecules such as propane and propene. In this work, structure–activity relationships were studied from the theoretical calculations of the molecular modelling of a Ag surface. In addition, the effect of the presence of oxygen surface atoms on the hydrocarbon–catalytic surface interaction wase analysed, in order to explain the difference in activity previously found.
2. Results and Discussion
Figure 1a,b show the results of the activity, in terms of the Tini and T50 (Tini = temperature at which 5% of the hydrocarbon was converted, and T50 = temperature at which 50% of the hydrocarbon was converted), of the Ag catalysts supported on zirconia for the combustion of propane and propene. Part of these results were reported in a previous work carried out in our group [9] using catalysts with a Ag content of 1, 5, and 10 wt.% denominated Ag1/Z, Ag5/Z, and Ag10/Z, respectively. In the catalyst’s absence, propane and propene were oxidised in an O2/He atmosphere, and this homogeneous reaction started at higher temperatures (Tini propane= 510°C and Tini propene= 570°C) and reached 50 % conversion at 600°C and 630°C, respectively. The bare support exhibited activity for the combustion of both hydrocarbons, and the T50 obtained for both hydrocarbons decreased 110°C compared to that found for the reaction in the absence of a catalyst. Instead, the influence of Ag and its metallic load on the activity depended strongly on the nature of the molecule to be oxidised. For the combustion of propane, the Agx/Z catalysts presented a Tini similar to that found with the support, and a slight decrease in T50 was observed with the increase in the silver content, while for the combustion of propene, the presence of silver strongly influenced the activity. The silver’s impact on the propene conversion and the catalytic activity increased with the increase in the metallic charge. The Tini and T50 obtained with the Agx/Z catalysts decreased appreciably compared to those found with the support, indicating a notable contribution of the supported metal phase.
2.1. Adsorption on Ag (111)
Considering that the main objective of this work is to explain the difference in the behaviour of the Agx/Z catalysts facing the propane and propene combustion reactions, molecular modelling studies using DFT calculations were performed with a simplified model based on a flat surface of Ag(111). Although there is general agreement that the rate-determining step for propane combustion is the first C-H bond cleavage [21,22] in the case of propene, the elucidation of the mechanism is still under debate.
The choice of the surface was based on the following: in the Ag/support catalysts, the Ag nanoparticles are supported on zirconia (ZrO2), and the contribution of the support to the activity is similar for both reactants (Figure 1); hence, in the model proposed in this work, only the metallic surface was considered. According to the experimental data obtained in previous works, metallic and oxidic silver species coexist on the catalytic surface, but the Ag(0) species predominates [9]. With this objective, the propane and propene adsorption energies were calculated and compared as a first approach to understand which is the fundamental step in the catalytic combustion reaction of both substances on a silver metallic surface.
Figure 2 shows the optimised structures for the adsorption studies of both hydrocarbons on the Ag surface and for propene in presence of an adsorbed O atom. One stable structure was found (Figure 2a) for propane, while two stable structures were found (Figure 2b,c) for propene. In addition, only one stable structure was also found for the propene adsorption in the presence of oxygen surface atoms in the vicinity of the adsorption site (Figure 2d). Figure 2b shows the optimised structure for the adsorption mode that can be called the “bridge”, in which the C1=C2 double bond is located in parallel above the bond between two vicinal silver surface atoms. In Figure 2c, can be observed the vertical view of one of the propene adsorption modes that can be called the "top", in which the C1=C2 double bond is located just above a Ag surface atom (for more details see Figure 3, Computational details). In the top adsorption mode, propene was adsorbed onto a single Ag surface atom through a π bond, while in the bridge adsorption mode, propene interacted with two Ag atoms through two σ bonds.
Table 1 shows the results obtained for the distances between the C1 and C2 carbon atoms and the Ag surface atom(s) closest to them. In addition, the C1-C2 bond length for propane and propene molecules before and after adsorption are shown. The same table also shows the adsorption energies for the different cases calculated with Eq. (1) (please see Computational details).
The distances between the carbon atoms of the propane molecule and the Ag metal surface C1-Ag and C2-Ag were 3.57 Å and 3.87 Å, respectively. These distances were greater than those found between the surface and the C1 and C2 atoms of propene in its two adsorption modes (bridge and top). On the other hand, the results showed that in the top adsorption mode, the substrate was closer to the surface than in the bridge adsorption mode. The calculated distances between C1 and C2 and the Ag atom for the top adsorption mode were 2.58 Å and 2.74, respectively, while the values obtained for the bridge adsorption mode were 3.10 Å and 3.25 Å, respectively.
The C1-C2 bond length of a free hydrocarbon molecule in the vacuum can be different from that adsorbed onto a catalytic surface. Changes in this bond length can provide information about the interactions between the C1-C2 bond and the metallic surface. As shown in Table 1, the C1-C2 bond length calculated for propane was constant, at a value of 1.52 Å, before and after its adsorption over the surface. From these results, a stretching of the C-C bond, which allowed supposing its activation, was not observed.
The calculations indicated that propene exhibits a different behaviour than that observed with propane. The C1-C2 bond length of the propene, adsorbed in the top mode, was slightly greater (1.35 Å) than that found for the same bond in the vacuum. These results show that there may be an interaction between the alkene molecule and the catalytic surface. The silver surface has the capacity to activate the double bond and lead to a rupture of the C=C bond.
Finally, the last column of Table 1 shows the adsorption energies calculated from Eq. 1 (Section 4. Experimental). The adsorption energies obtained corroborated the analysis carried out from the results of the bond lengths. The most stable structure was found for propene adsorption. An adsorption energy of -0.77 eV was calculated for the top mode, and -0.21 eV was found for the adsorption in the bridge mode. These values showed a strong interaction, chemisorption, between the alkene and the Ag surface. On the other hand, the propane adsorption on the metallic surface presented an energy of -0.14 eV. This low adsorption energy was consistent with physical adsorption or physisorption.
The results obtained from the DFT calculations explain the different types of interaction between propene and propane with the metallic surface. The propene is chemisorbed on the Ag surface, distorting its bonds and generating its activation. This would imply that a higher metallic charge in the catalyst would increase the number of active sites in which this activation occurs, generating a higher activity. On the other hand, the presence of a metallic surface is not enough for the activation of the propane molecule. This would explain why by increasing the amount of metal in the catalyst, the activity for the combustion of propane is practically not affected.
The interaction energy of the propane with the metallic surface calculated through the DFT was -0.14 eV; this energy suggests a weaker interaction than the one found for propene in the two adsorption modes, and it is also of the order of van der Waals-type interactions. These results agree with what was reported by Lian et al. [15], who studied propane adsorption on Pt catalysts. The interaction energies between propane and the platinum metal surface were between -0.01 and -0.35 eV, depending on the functional used. These values are typical for physisorption energies. In this work, by DFT calculations, it was found that for the optimised structure of the physiosorbed propane molecule on the Ag(111) surface, the C1-C2 bond length was the same as for free propane. This indicated that this physisorption did not generate any distortion in the C1-C2 bond and, consequently, did not induce the activation of hydrocarbon. These results explain the low dependence of the catalyst activity on the silver content for the propane combustion reaction. Since metallic silver does not intervene in a decisive way in the reaction mechanism, the increase in the metallic charge of this metal in the catalyst does not lead to a substantial increase in its activity.
On the other hand, the adsorption energies calculated for the propene adsorption on the metal surface in the bridge mode and the top mode were -0.21 and -0.77 eV, respectively. The propene adsorption on a silver surface atom, through the π system, was more stable than their adsorption on two silver surface atoms, through two σ type bonds of C1 and C2, in the bridge mode. In the top mode, the propene molecule adsorbed was closer to the metallic surface than in the bridge mode. In addition, in the top mode, a slight stretching of the C1=C2 bond was observed. This stretching was not observed in the bridge mode.
These results suggest the presence of a chemisorption phenomenon, preferentially through the propene π system on a Ag atom, which generates a slight stretching of the C1=C2 bond, activating it for a break. These results agree with those reported by other authors, who propose that this rupture is the determining step of the reaction rate for the combustion of propene [2,16,17].
2.2. Adsorption on Ag (111) in the Presence of Oxygen
As suggested by the XPS results (Table 2), metallic and oxidised silver species coexist on the catalytic surface [9]. As was previously reported, all three silver catalysts, Ag1/Z, Ag5/Z, and Ag10/Z, contained silver on their catalytic surface. From the quantification of the Ag3d and Zr3d signals, it was determined that the Ag/Zr atomic ratio increased with the increase of the silver concentration. Furthermore, from the analysis of the silver signals in the Auger region of the spectra, it was possible to propose the coexistence of metallic and oxidic silver species on the catalytic surface. The signal representing the kinetic energy of the AgM5N45N45 Auger transition presented a shoulder at ca. 349.5 eV associated with the presence of oxidic species [9]. This shoulder represents 3% and 6% of the signal for the Ag5/Z and Ag10/Z catalysts, respectively. These results suggest the presence of O atoms associated with surface silver species. Therefore, an O atom was added to the surface to model/simulate a slightly oxidised surface for the propene adsorption. The influence of a superficial O adatom was studied only for the case of propene adsorption in top mode because it is the one that presented the highest adsorption energy.
In the top adsorption mode, the presence of an O atom close to the Ag site caused a decrease in the distance between the propene carbons and the surface respect to the Ag(111) surface without oxygen. The results obtained suggest that the presence of oxygen atoms in the vicinity of the active sites lead to an increase in the interaction. Furthermore, the calculated C1-C2 bond length of propene molecule when is adsorbed on a surface containing surface O atoms increase from 1.34 Å to 1.36 Å.
With the addition of O, the binding energy between the propene and the metal surface increased 0.1 eV from -0.77 eV to -0.87 eV. The slightly increased of C1=C2 bond length, the decreased of distance between the propene molecule and the surface (0.08 Å) and the increase in the adsorption energy, are evidence of the strengthened propene–surface interaction, and, in the presence of oxygen surface atoms, the system was even more capable of activating the double bond. These results agreed with that reported by Huang and White [18] who, through O2-TPD and RAIRS studies, demonstrated that at low O coverages, propene was adsorbed onto the surface of a Ag(111) preferentially through its π system. This adsorption occurred on a Ag atom with a slight inclination of the plane of the molecule concerning the Ag surface. Probably, the presence of a surface oxygen atom generates Ag atoms deficient in electrons, and due to this, the π system of the propene interacts more strongly with the surface.
The results of the theoretical calculations indicate that the greater interaction of propene with the metallic surface, the energies of the order of chemisorption, the lower distances between the molecule and the surface, the increase in the C1-C2 bond length, and the presence of oxygen surface atoms are essential for bond activation in a possible reaction. Therefore, on a surface with higher silver content, there exists a greater possibility of interaction between the propene molecules that come from the gas phase and the catalyst. This interaction is not favoured when the reactant is propane; therefore, its poor activity does not depend on the metallic charge.
4. Experimental
4.1. Computational Details
Ab initio calculations implementing the density functional theory (DFT) with plane waves were performed using Quantum Espresso software [23,24]. An exchange and correlation functional with density gradient correction PBESOL [25] and pseudopotentials type PAW (projector augmented-wave) [26] was used. For all calculations, whether for bulk, surface (slab) or, for free molecules in a vacuum, the cutoff kinetic energy for plane waves and the charge density used in the calculations was 30 Ry and 120 Ry, respectively. The convergence criterion used for the total energy was <10-4 Ry, and for the atomic forces, it was <10-3 Ry/ua. A 4x4 Ag(111) supercell and a depth of three layers were used for the surface calculations, as can be seen in Figure 3. The spacing on the Z-axis of the supercell was around 20 Å of vacuum between one surface and the other. These dimensions of the supercell allowed us to avoid the effects of interactions with near neighbours in any dimension. During the development of the surface optimisation calculations, the positions of the Ag atoms of the two innermost layers were kept frozen, while the atoms of the most superficial layer were allowed to relax freely. In the case of the propane and propene adsorption calculations, the two internal layers were kept frozen and the relaxation of the Ag surface layer and all the atoms of the molecules and/or atoms to be adsorbed were allowed to relax. A grid of k-points-3x3x1 was used in the reciprocal space using the Monkhorst–Pack method [27]. For the adsorption studies of the molecules on the surface, two adsorption positions were analysed for the case of propylene and one for propane. The adsorption of propylene in the presence of oxygen adsorbed at a nearby site was also studied. As can be seen in Figure 4, the difference in the two propylene adsorption modes was that in the top mode, the double bond of the molecule was located above an Ag atom, while in the bridge mode, the double bond was located above a surface Ag-Ag bond.
The adsorption energy for each species was calculated using Equation 1, where Eads is the adsorption energy for each adsorbed molecule, Emolec+surf is the total electronic energy of the molecule system adsorbed onto the surface, Esurf is the total electronic energy of the metallic surface, and Emolec is the energy of each of the free molecules in a vacuum.
5. Conclusions
From the molecular modelling studies using DFT calculations, it was possible to explain the difference in the behaviour of the Ag catalysts supported on ZrO2 for both reagents in the catalytic combustion reaction.
In the case of the catalytic combustion reaction of propene, the increase in the activity of the catalysts can be explained as a function of their metal charge because the determining step of the reaction rate is the breaking of the C1=C2 bond; for this, the propene needs to be adsorbed onto the Ag nanoparticles that activate and weaken this double bond.
On the other hand, the low adsorption energy calculated for propane suggest weak interaction energies consistent with physisorption and the null deformation or activation of the C1-C2 bond. This fact explains the low dependence of the catalytic activity concerning the Ag metallic charge.
Supplementary Materials
This article does not contain supplementary files.
Author Contributions
Conceptualization, I.D.Lick. and M.L.Casella, methodology, J.F. Ruggera.; software, J.F.Ruggera.; formal analysis, J.F. Ruggera, M.A. Ocsachoque and M. Montaña.; investigation, J.F. Ruggera, M.A. Ocsachoque and M. Montaña, writing—original draft preparation, J.F. Ruggera.; writing—review and editing, J.F.Ruggera, M.L. Casella and I. D. Lick.; supervision, M.L. Casella.; project administration, M.L.Casella and I.D. Lick; funding acquisition, M.L.Casella and I.D. Lick. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by UNLP (project X903, Expte 100-1282/19), CONICET (project PUE 005/2018, EXP. 8085/17; PIP 086/2022 and PIP 0134/2021) and ANPCyT FONCyT (PICT 2019/1962 and PICT 2021/601).
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
The authors acknowledge the financial support of CONICET, ANPCyT, and UNLP, and Dra. María Laura Barbelli for collaborations.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Twigg, M.V. Progress and Future Challenges in Controlling Automotive Exhaust Gas Emissions. Applied Catalysis B: Environmental 2007, 70, 2–15. [CrossRef]
- Yang, A.-C.; Streibel, V.; Choksi, T.S.; Aljama, H.; Werghi, B.; Bare, S.R.; Sánchez-Carrera, R.S.; Schäfer, A.; Li, Y.; Abild-Pedersen, F.; et al. Insights and Comparison of Structure–Property Relationships in Propane and Propene Catalytic Combustion on Pd- and Pt-Based Catalysts. Journal of Catalysis 2021, 401, 89–101. [CrossRef]
- Choudhary, T.V.; Banerjee, S.; Choudhary, V.R. Catalysts for Combustion of Methane and Lower Alkanes. Applied Catalysis A: General 2002, 234, 1–23. [CrossRef]
- Shelef, M.; McCabe, R.W. Twenty-Five Years after Introduction of Automotive Catalysts: What Next? Catalysis Today 2000, 62, 35–50. [CrossRef]
- Stratakis, G.A.; Stamatelos, A.M. Thermogravimetric Analysis of Soot Emitted by a Modern Diesel Engine Run on Catalyst-Doped Fuel. Combustion and Flame 2003, 132, 157–169. [CrossRef]
- Liotta, L.F. Catalytic Oxidation of Volatile Organic Compounds on Supported Noble Metals. Applied Catalysis B: Environmental 2010, 100, 403–412. [CrossRef]
- Cortés-Reyes, M.; Herrera, C.; Larrubia, M.Á.; Alemany, L.J. Intrinsic Reactivity Analysis of Soot Removal in LNT-Catalysts. Applied Catalysis B: Environmental 2016, 193, 110–120. [CrossRef]
- Demoulin, O.; Le Clef, B.; Navez, M.; Ruiz, P. Combustion of Methane, Ethane and Propane and of Mixtures of Methane with Ethane or Propane on Pd/γ-Al2O3 Catalysts. Applied Catalysis A: General 2008, 344, 1–9. [CrossRef]
- Montaña, M.; Leguizamón Aparicio, M.S.; Ocsachoque, M.A.; Navas, M.B.; de C. L. Barros, I.; Rodriguez-Castellón, E.; Casella, M.L.; Lick, I.D. Zirconia-Supported Silver Nanoparticles for the Catalytic Combustion of Pollutants Originating from Mobile Sources. Catalysts 2019, 9, 297. [CrossRef]
- Avila, M.S.; Vignatti, C.I.; Apesteguía, C.R.; Garetto, T.F. Effect of Support on the Deep Oxidation of Propane and Propylene on Pt-Based Catalysts. Chemical Engineering Journal 2014, 241, 52–59. [CrossRef]
- Bratan, V.; Vasile, A.; Chesler, P.; Hornoiu, C. Insights into the Redox and Structural Properties of CoOx and MnOx: Fundamental Factors Affecting the Catalytic Performance in the Oxidation Process of VOCs. Catalysts 2022, 12, 1134. [CrossRef]
- Ertl, G. Reactions at Well-Defined Surfaces. Surface Science 1994, 299–300, 742–754. [CrossRef]
- Zhang, Z.; Jiang, Z.; Shangguan, W. Low-Temperature Catalysis for VOCs Removal in Technology and Application: A State-of-the-Art Review. Catalysis Today 2016, 264, 270–278. [CrossRef]
- Langmuir, I. Part II.—“Heterogeneous Reactions”. Chemical Reactions on Surfaces. Trans. Faraday Soc. 1922, 17, 607–620. [CrossRef]
- Huang, W.; Jiang, Z.; White, J.M. Distinct Oxidation Behaviors of π-Bonded and Di-σ-Bonded Propylene on Ag(111). Catalysis Today 2008, 131, 360–366. [CrossRef]
- Sattler, J.J.H.B.; Ruiz-Martinez, J.; Santillan-Jimenez, E.; Weckhuysen, B.M. Catalytic Dehydrogenation of Light Alkanes on Metals and Metal Oxides. Chem. Rev. 2014, 114, 10613–10653. [CrossRef]
- Yang, M.-L.; Zhu, J.; Zhu, Y.-A.; Sui, Z.-J.; Yu, Y.-D.; Zhou, X.-G.; Chen, D. Tuning Selectivity and Stability in Propane Dehydrogenation by Shaping Pt Particles: A Combined Experimental and DFT Study. Journal of Molecular Catalysis A: Chemical 2014, 395, 329–336. [CrossRef]
- Arya, M.; Mirzaei, A.A.; Davarpanah, A.M.; Barakati, S.M.; Atashi, H.; Mohsenzadeh, A.; Bolton, K. DFT Studies of Hydrocarbon Combustion on Metal Surfaces. J Mol Model 2018, 24, 47. [CrossRef]
- Wang, X.; Rui, Z.; Ji, H. DFT Study of Formaldehyde Oxidation on Silver Cluster by Active Oxygen and Hydroxyl Groups: Mechanism Comparison and Synergistic Effect. Catalysis Today 2020, 347, 124–133. [CrossRef]
- Porter, W.N.; Lin, Z.; Chen, J.G. Experimental and Theoretical Studies of Reaction Pathways of Direct Propylene Epoxidation on Model Catalyst Surfaces. Surface Science Reports 2021, 76, 100524. [CrossRef]
- Burch, R.; Hayes, M.J. CH Bond Activation in Hydrocarbon Oxidation on Solid Catalysts. Journal of Molecular Catalysis A: Chemical 1995, 100, 13–33. [CrossRef]
- Tomita, A.; Miki, T.; Tai, Y. Effect of Water Treatment and Fe Doping on Pt Sintering and the Propane Oxidation Activity of Pt/Al2O3. Applied Catalysis A: General 2016, 522, 138–144. [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A Modular and Open-Source Software Project for Quantum Simulations of Materials. J. Phys.: Condens. Matter 2009, 21, 395502. [CrossRef]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Nardelli, M.B.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced Capabilities for Materials Modelling with Quantum ESPRESSO. J. Phys.: Condens. Matter 2017, 29, 465901. [CrossRef]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin, L.A.; Zhou, X.; Burke, K. Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces. Phys. Rev. Lett. 2008, 100, 136406. [CrossRef]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. [CrossRef]
Figure 1.
(a). Tini obtained with Agx/Z catalysts (x = 1, 5 and 10). (b). T50 obtained with Agx/Z catalysts (x= 1, 5 and 10).
Figure 1.
(a). Tini obtained with Agx/Z catalysts (x = 1, 5 and 10). (b). T50 obtained with Agx/Z catalysts (x= 1, 5 and 10).
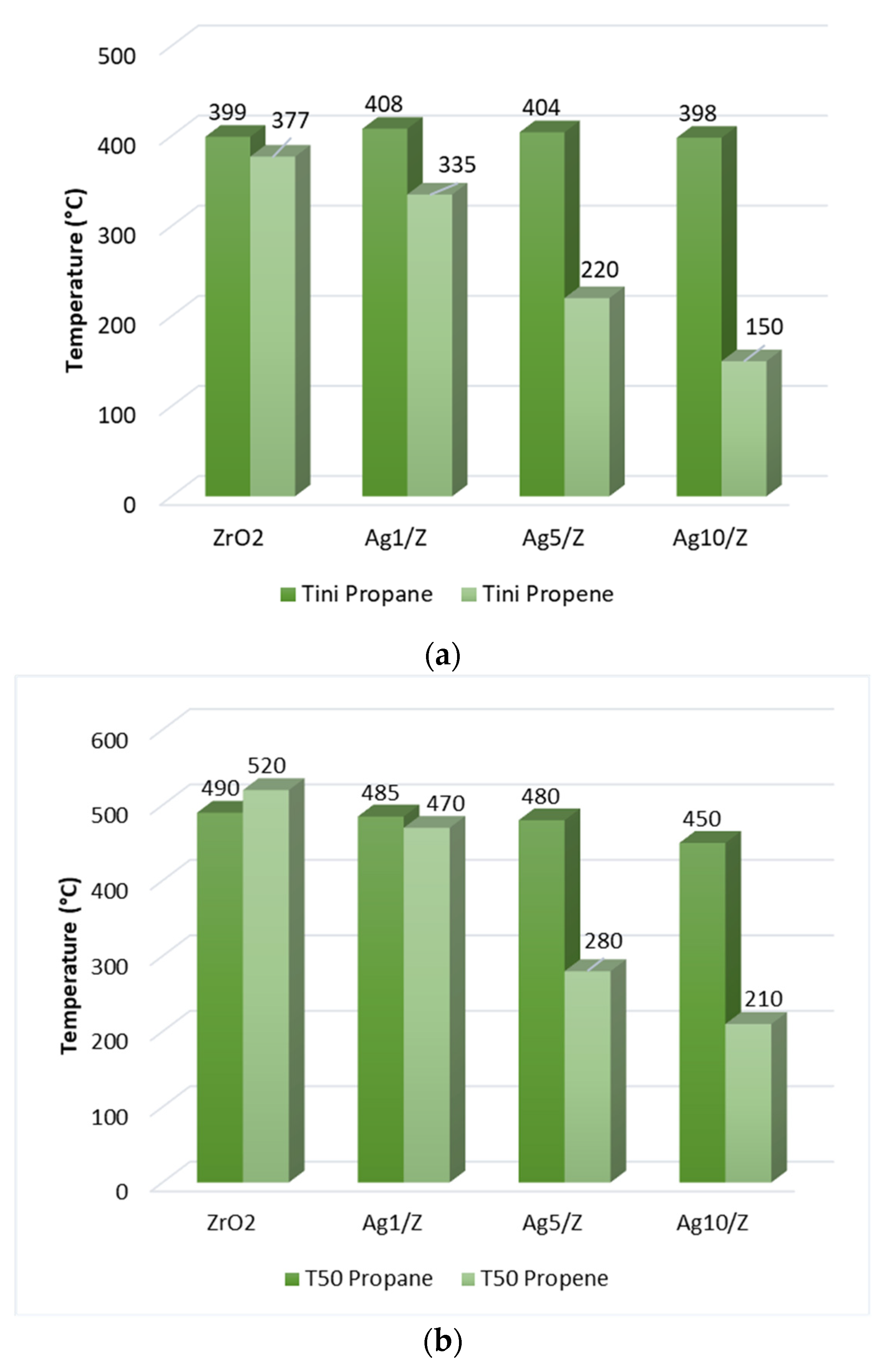
Figure 2.
Side and top view of optimized geometries of 2.a) propane on Ag 111; 2.b) propene bridge mode on Ag 111; 2.c) propene top mode on Ag 111; 2.d) propene top mode on Ag 111 with O adatom.
Figure 2.
Side and top view of optimized geometries of 2.a) propane on Ag 111; 2.b) propene bridge mode on Ag 111; 2.c) propene top mode on Ag 111; 2.d) propene top mode on Ag 111 with O adatom.
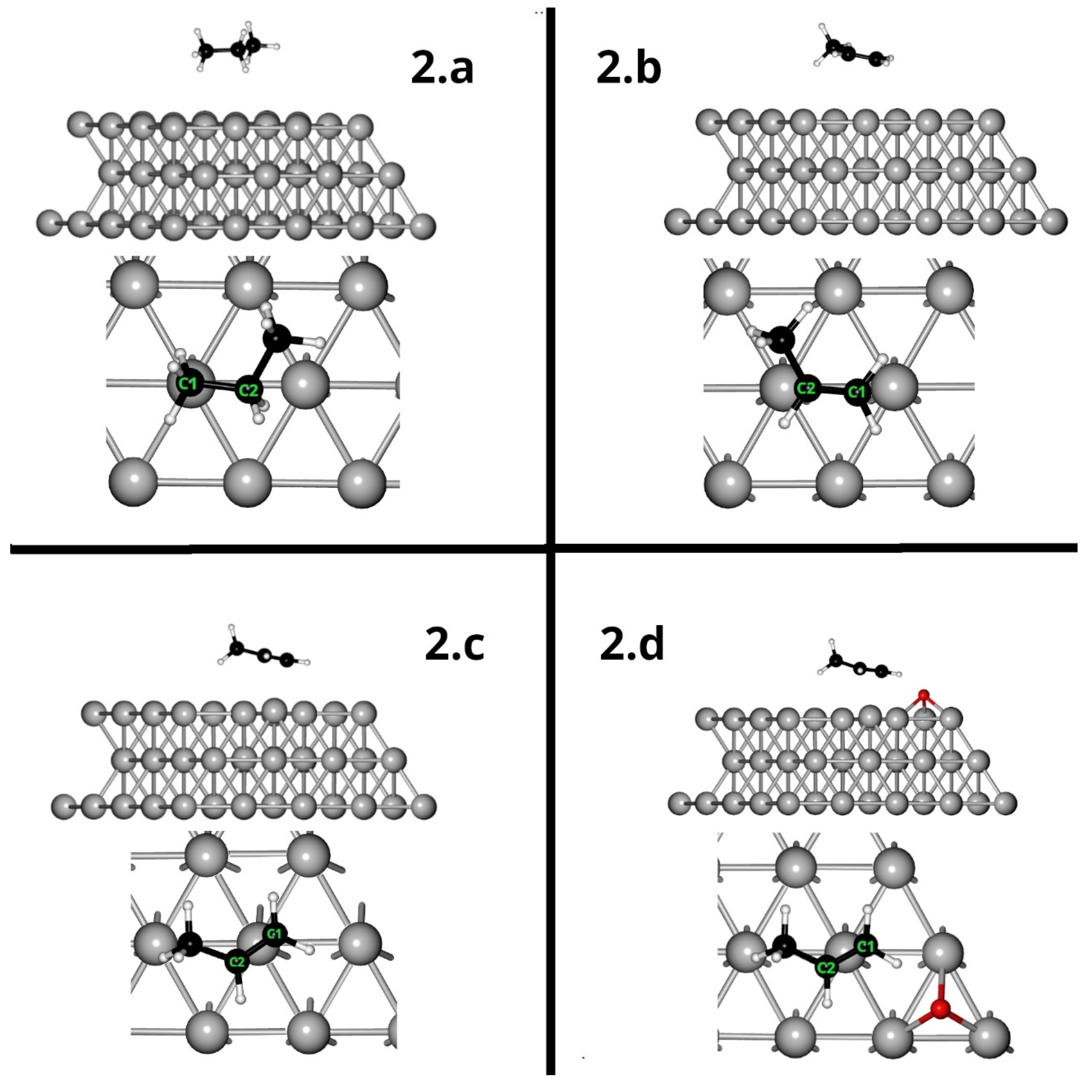
Figure 3.
Side and top view of the Ag 111 supercell used for the DFT model.
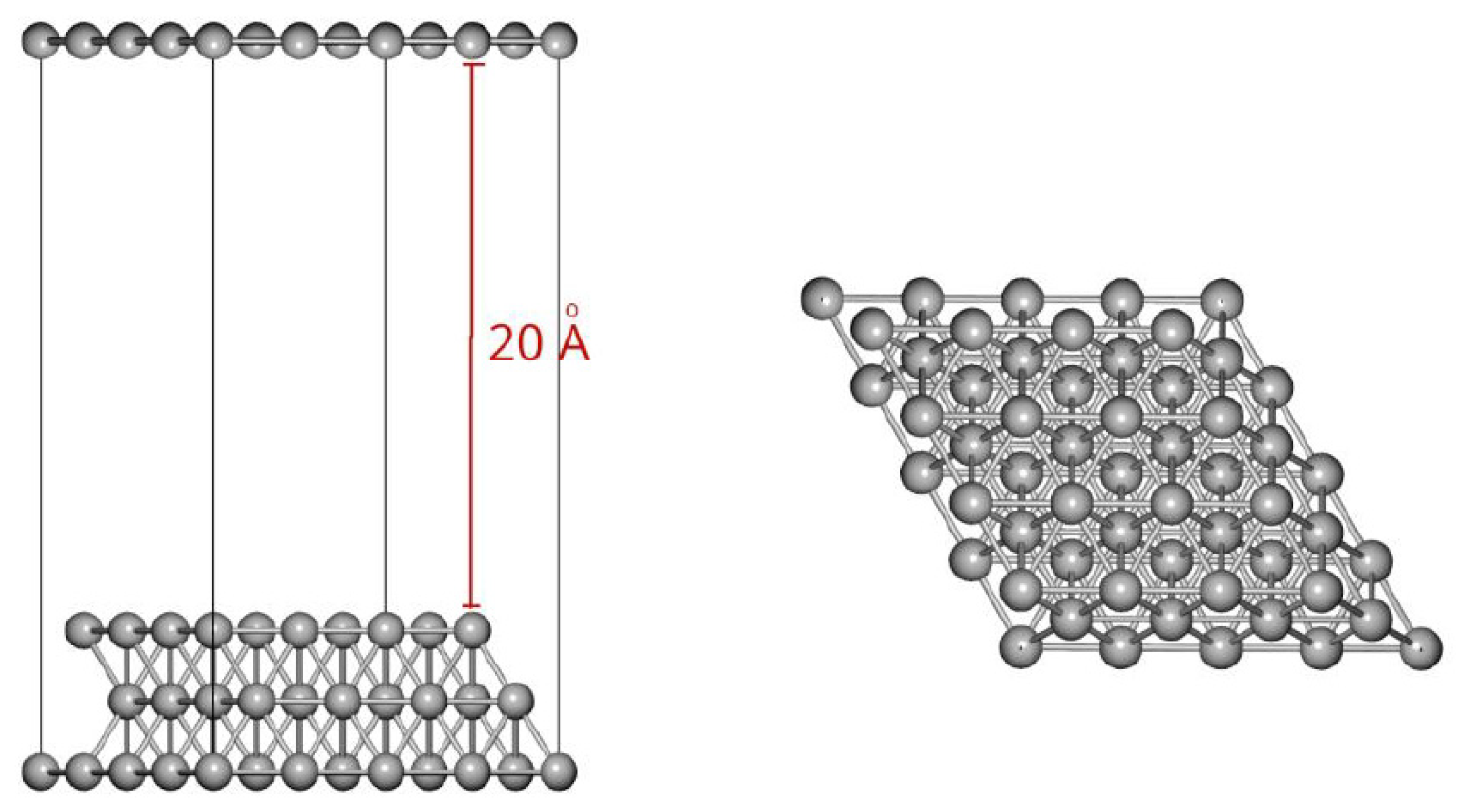
Figure 4.
Top view of first layer of the Ag(111) surface and the considered sites (shown in the colors indicated) used in the simulation.
Figure 4.
Top view of first layer of the Ag(111) surface and the considered sites (shown in the colors indicated) used in the simulation.

Table 1.
Distances C1-Ag y C2-Ag for all adsorbed species (Å), bond length C1-C2 before and after adsorbed (Å) y adsorption energies calculated with Eq. (1) (eV).
Table 1.
Distances C1-Ag y C2-Ag for all adsorbed species (Å), bond length C1-C2 before and after adsorbed (Å) y adsorption energies calculated with Eq. (1) (eV).
| Distances C1-Ag (Å) | Distances C2-Ag (Å) | Bond lentgh C1-C2 free (Å) | Bond lentgh C1-adsorbed C2 (Å) | Eads (eV) | |
|---|---|---|---|---|---|
| Propane | 3.57 | 3.87 | 1.52 | 1.52 | -0.14 |
| Propene bridge | 3.10 | 3.25 | 1.34 | 1.34 | -0.21 |
| Propene top | 2.58 | 2.74 | 1.34 | 1.35 | -0.77 |
| Propene O | 2.50 | 2.66 | 1.34 | 1.36 | -0.87 |
Table 2.
Silver XPS and Auger signals [9].
Table 2.
Silver XPS and Auger signals [9].
| Ag1/Z | Ag5/Z | Ag10/Z | |
|---|---|---|---|
| KE AgM4N45N45 | - | 357.5 | 357.9 |
| KE AgM5N45N45 | - | 352.5 (97)* 349.5 (3) |
352.8 (94) 349.4 (6) |
| Ag/Zr atomic ratio | 0.009 | 0.154 | 0.213 |
* the percentage contribution is shown in parentheses.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



