
Submitted:
26 April 2023
Posted:
27 April 2023
You are already at the latest version
Abstract
Leishmaniasis is a disease caused by parasites of Leishmania genus and transmitted by sand fly vectors. Tegumentary leishmaniasis is the most prevalent clinical outcome in Latin America afflicting people from 18 countries. In Panama, leishmaniasis has annual incidence rates as high as 3000 cases, representing therefore one of the major public health problems. In endemic regions, L. panamensis is responsible for almost eighty percent of the human cases that present different clinical outcomes. This differences in disease outcomes could be result of the local interplay between L. panamensis variants and human hosts with different genetic backgrounds. The genetic diversity of L. panamensis in Panama has only been partially explored, and the variability reported for this species is based on few studies restricted to small populations and/or with poor resolutive markers at low taxonomic levels. Accordingly, in this study we explored the genetic diversity of sixty-nine L. panamensis isolates from different endemic regions of Panama using a MLST approach based on four housekeeping genes (Aconitase, ALAT, GPI and HSP70). Two to seven haplotypes per locus were identified, and regional differences in L. panamensis genetic diversity were observed. Genotype analysis evidenced the circulation of thirteen L. panamensis genotypes, a fact that might have important implications for the local control of the disease.
Keywords:
tegumentary leishmaniasis
; Leishmania panamensis
; multilocus typing
; diploid sequence type
; Panama
1. Introduction
Leishmaniasis is a group of diseases caused by protozoa parasites of Leishmania genus and transmitted by sand fly vectors with an eminent neglected status. The main clinical forms of the disease are visceral (VL) and tegumentary leishmaniasis (TL) that manifest different clinical expression depending on the causative species and its genetic background, together with the immunological status of the host, and factors in sand fly’s saliva [1,2]. This disease has been considered by the World Health Organization (WHO) as an emerging/remerging disease because of the high and increasing number of cases and its geographical expansion [3]. Leishmaniasis is endemic in 98 countries worldwide having an estimated annual incidence of 2.0 million cases and an approximated prevalence of 12,000,000 cases [4]. Additionally, this disease causes around 20,000 to 40,000 deaths in endemic areas all year round [3,4].
In the Americas, TL is endemic in 18 countries where this clinical form is 15 times more frequent than VL [3]. In Panama, leishmaniasis is the most prevalent vector-borne parasitic disease, reaching incidence rates as high as 3000 annual cases and afflicting people of all ages. The local transmission of leishmaniasis occurs mainly in rural and sylvatic environments affecting economically marginated people with TL [3].
At least twenty species are included in the Leishmania genus which is divided into the sections Euleishmania and Paraleishmania which are constantly under taxonomical review [5]. In Panama, L. panamensis is the principal etiological agent of TL [6,7]. However, other Viannia species such as L. braziliensis, L. guyanensis, and L. naiffi have been found circulating sympatrically at minor frequencies also causing TL [8]. Additionally, a few cases of TL caused by L. mexicana have been reported in the country previously [6,9]. The initial reports of Leishmania species in the country were made using the gold standard approach multilocus enzyme electrophoresis (MLEE) and after the nineties by using molecular protocols based on PCR-RFLP or PCR-sequencing of specific genes such as calmodulin [10,11], HSP70 [8], cytochrome b [12], and kinetoplast minicircles [13]. Among the Leishmania species reported using these tools, L. panamensis was the most prevalent species found in human cases of TL in Panama. In Latin American countries, L. panamensis is responsible for multiple clinical forms such as localized, disseminated, mucocutaneous and diffuse forms of the tegumentary disease [14,15,16,17]. These clinical outcomes could be related to the local interplay between genetic variants of this species and the complex local population conformed by human inhabitants with different genetic backgrounds. Thus, the plethora of clinical forms caused by this species along with other medically relevant characteristics within the L. panamensis populations deserves to be addressed. To perform this task, it is initially important to assess the local genetic variability of this species using more informative molecular tools capable of finding intra-specific variation. In the case of the Leishmania genus, single locus analyses sometimes are not sufficient to find intra-specific variation, and methodologies that add more loci to the analysis should be chosen instead to increase the discriminatory power. Studies based on multilocus sequence typing (MLST) in Leishmania have demonstrated that this technique has more resolutive power than the gold standard MLEE to study intra-specific genetic diversity in Leishmania [18,19]. Furthermore, a great level of diversity has been shown when a high number of isolates from Leishmania species have been analyzed by MLST, supporting the use of MLST as a tool to study intra-specific variability within Leishmania parasites [20,21,22]. Regarding multilocus approaches, only one molecular tool based on multilocus microsatellite typing (MLMT) has been previously used to evaluate a small number of L. panamensis isolates from the Central region of Panama revealing an apparent local genetic diversity of this Leishmania species [23]. However, to elucidate the real genetic diversity of this species in Panama, it is necessary to evaluate isolates from the main endemic areas of the country applying tools with high resolutive power at lower taxonomic levels. Considering this, in this study we evaluated sixty-nine L. panamensis isolates from the main endemic areas of leishmaniasis in the country using an MLST approach based on four essential genes. Our findings suggest the circulation of different L. panamensis variants in the country and evidenced regional differences in genetic diversity, a fact that could be related to the different clinical outcomes caused by this species. The knowledge gathered herein might also provide a baseline to develop further studies on medically relevant characteristics that impact the clinical outcome of the disease as well as resistance to the first-line drugs used to treat leishmaniasis.
2. Materials and Methods
2.1. Leishmania Isolates
Sixty-nine Leishmania panamensis isolates from both the eastern border and western border of the Panama Canal were used in this study. Leishmania isolates were collected during the last five years (2015-2020) and stored in liquid nitrogen at the Dirección de Investigación en Parasitología, Instituto Conmemorativo Gorgas de Estudios de la Salud (ICGES) situated in Panama City. Parasites were isolated from patients living in Provinces located east (Panama and Darien Provinces) and west (Colon, Panama Oeste, Cocle, and Bocas del Toro) of the Panama Canal (Figure 1). These locations are recognized endemic areas of leishmaniasis in the country with a long-term average of CL incidence rates between 5 to 25 new cases per 10,000 [24].
2.1. Parasite Culture and DNA Extraction
Leishmania stocks were cultured using Schneider’s insect medium (Sigma Aldrich, Inc., St. Louis, USA) supplemented with 20% heat-inactivated fetal bovine serum (Gibco, Grand Island, USA) at 26°C. Total genomic DNA was extracted from promastigotes using a commercial kit (Wizard Genomic DNA Purification Kit, Promega, Madison, USA) according to the manufacturer’s instructions.
2.2. Molecular Identification of Leishmania panamensis Isolates
2.2.1. PCR Amplification
DNA samples from cultures parasites were used to identify L. panamensis parasites by an approach based on the PCR-amplification and sequencing of a 1364bp segment from the Leishmania HSP70 gene. To perform this PCR, we designed the set of primers PLeishF 5′-GATGGTGCTGCTGAAGATGA-3′ and PLeishR 5’-GGTCATGATCGGGTTGCATR-3′ that amplifies the HSP70 gene from the main Leishmania complexes, using the primer 3 algorithm includes in UGENE bioinformatic platform. The specificity of the primers was evaluated using the PrimerBlast software (https://www.ncbi.nlm.nih.gov/tools/primer-blast/) and the formation of inter and intra-molecular secondary structures was assessed using the OligoAnalyzer tool (OligoAnalyzer Tool - primer analysis | IDT (idtdna.com). DNA samples were amplified in a final volume of 50µl containing 10µl of 10X buffer, 200µM of DNTPs, one unit of Phusion DNA polymerase, 0.3µM of each primer, 50 nanograms of genomic DNA and nuclease-free water until reaching 50µl. The thermal conditions of the PCR reactions were an initial denaturation at 98°C for 30 seconds followed by 34 cycles of 98°C for 10 seconds, 65°C for 30 seconds, 72°C for 30 seconds, and a final extension step of 72°C for 5 minutes.
2.2.2. Sanger Sequencing and Species Discrimination by Phylogenetic Analysis
Purified products were sequenced in both senses using the BigDye™ Terminator v3.1 Cycle Sequencing Kit and analyzed in the ABI 3500XL genetic analyzer (Applied Biosystems, California, USA). The electropherograms of the HSP70 gene fragment were then assembled in both senses using the assembled-to-reference tool of the UGENE software obtaining in this way HSP70 consensus sequences in fasta format. All the HSP70 sequences obtained in this study were submitted to the GenBank database where they can be retrieved using the accession numbers OQ408828 to OQ408896. Afterward, consensus sequences were aligned using the MAFFT algorithm included in the UGENE tool and this alignment was used to find the best substitution model by JModelTest [25]. The GTR + G model then was used in Mr. Bayes v.6.12.03 software to perform a phylogenetic construction that helped to allocate isolates within specific Leishmania species clusters. To accomplish this task, 20 Markov chains were used for six million generations and trees were sampled every 10,000 generations. The HSP70 sequence CP015689.1 extracted from chromosome 39 of the Trypanosoma cruzi, Sylvio strain, was used as an outgroup to construct the phylogenetic tree. Twenty-five percent of the sampled trees were discarded, and the remaining were used to build up a consensus tree and to calculate posterior probabilities of clades. The result of Bayesian analyses was visualized using Figtree v1.4.2 (FigTree (ed.ac.uk)). Transformation of the leaves and a schematic representation of the root were applied for visualization purposes.
2.3. PCR Amplification and Sequencing of Gene Loci used in The MLST Approach
To infer the genetic diversity of L. panamensis isolates we used an MLST approach based upon the amplification and sequencing of specific gene fragments from the metabolic enzymes GPI, aconitase, and ALAT using gene-specific primers depicted in Table 1. In this table also are depicted the gene length, chromosomal location, and product size of each gene locus used in this study. Besides, the HSP70 sequences obtained during the Leishmania species discrimination approach were used in the MLST scheme. These four markers present orthologous throughout the Leishmania genus and three of them codify for metabolic enzymes generally used to type Leishmania by Multilocus Enzyme Electrophoresis (MLEE). Additionally, aconitase, ALAT, and GPI loci have shown several polymorphisms that aided to explore the intraspecific and/or interspecific genetic variability of Leishmania species at haplotype and genotype levels when using reference sequences and/or sequences of Leishmania isolates from some Latin American countries [21,26]. As for the HSP70 gene, it has been added successfully to MLST schemes to study the genetic diversity of L. braziliensis and L. panamensis in Colombia [26] and to study L. braziliensis outbreaks in Brazil [21]. In these studies, all four markers identified several haplotypes and showed high discriminatory power in determining diploid sequence types (DSTs) of Leishmania Viannia species. Considering the facts exposed above we decided to explore the genetic diversity of L. panamensis in Panama using the abovementioned loci.
All PCR reactions were performed on a final volume of 50µl including 5µl of 10X buffer, 2.5µl of MgSO4, 200µM of dNTPs, one unit of Taq polymerase, and 50 nanograms of genomic DNA. The primer concentrations for each set were 0.4µM for ALAT and aconitase primers and 0.3µM for the GPI set of primers. Target DNA was amplified after a primary denaturation of 95ºC for 5 minutes followed by 35 cycles consisting of 95°C for 30 seconds, an aligning temperature of 55°C for aconitase/ALAT genes and 53°C for GPI fragment, and a target extension of 68°C for 90 seconds. PCR products were then extended at 68°C for 10 minutes as a final PCR step. Five microliters of all amplicons were analyzed in 1.2% agarose gel stained with GelRed for 45 minutes at 85 volts. The rest of the amplicons were purified using the commercial kit Wizard SV gel and PCR Clean-Up System following manufacturer instructions (Promega, Madison, USA). Amplification products then were sequenced in both senses using the BigDye™ Terminator v3.1 Cycle Sequencing Kit and analyzed in the ABI 3500XL genetic analyzer (Applied Biosystems, California, USA).
2.4. Sequence Assembling, Haplotype Construction, and Determination of Diversity Indexes
We used the map reads to reference tool included in the Sanger data analysis option of the bioinformatic software UGENE v.39 [27] to perform electropherograms assembling and edition using both senses in order to obtain consensus sequences from each molecular marker. The consensus sequences obtained from aconitase, ALAT, and GPI loci were placed into the GenBank on-line-servers under de accession numbers OQ408621 to OQ408027. The DNAsp software v.6.12.03 [28] was utilized subsequently to construct L. panamensis haplotypes by the algorithm PHASE setting the MCMC options of the coalescent-based Bayesian method to use 1000 interactions, one thinning interval, and 100 burn-in interactions. After constructing haplotypes, we used their sequences to infer the genetic diversity indexes including the total number of mutations (ETA), haplotype diversity (Hd), nucleotide diversity (π), and diversity index ϴ using the same bioinformatic software. To determine the diversity indexes, the number of segregation sites, and haplotype number by geographical area evaluated, we assessed independently sequences of all four loci from 34 and 35 L. panamensis isolates belonging to the western and eastern regions of the country using DNAsp v6.12.03.
2.5. Determination of L. panamensis DSTs and Their Geographical Distribution
The bioinformatics software MLSTest [29] was used to 1) determine the best number and combinations of loci that guarantee to find the highest diversity indexes of local isolates of L. panamensis, 2) Analyze incongruence among loci chosen for the MLST approach 3) calculate the efficiency and discriminatory power of the MLST scheme. To select the number of loci that offered the highest diversity index we used the scheme optimization options of the software that reveals whether the genotypic diversity increase as more loci are added to the MLST scheme. Regarding the congruency analyses, we search incongruency by determining the p-value for the incongruence length differences parameter (ILD) of all selected loci. Furthermore, we applied the Templeton test to evaluate incongruency between all loci and the topology of the concatenated tree setting the algorithm to make an overall analysis of incongruency. To obtain the allelic profile and the different types of L. panamensis DSTs, we used the average state option of the MLSTest program to handle heterozygote sites and calculate genetic distances. Also, we obtained the typing efficiency and the discriminatory power for each locus after using the option make/view allelic profile of the same program.
A map of the local distribution of L. panamensis DSTs was constructed based on the geographical coordinates of each place where DSTs were found using the software QGIZ 3.16 Hannover (https://qgis.org).
2.6. Phylogenetic Analyzes of L. panamensis DSTs
To corroborate L. panamensis genotypes by phylogenetic analysis we selected representative sequences from each DST found herein. We phased the sequences from each representative DST by DNAsp software v.6.12.03 and the alleles obtained were used to perform a Bayesian-based phylogenetic analysis. Allelic sequences from each locus used were concatenated using the software SeaView version 1:4.5.4.8-2 [30]. Sequences were then multiple aligned using the MAFFT algorithm also included in UGENE with a maximum number of iterative refinements of 3 and a gap penalty of 1.53. The HKY (G + I) model was found as the best DNA evolution model by the software JModelTest [25]. A phylogenetic tree reconstruction of Leishmania was implemented by applying Bayesian inference (BI) with Mr. Bayes v.3.2 software [31]. Ten Markov chains were proceeded for ten million generations, and trees were sampled for every 7000 generations setting the program to run the substitution model with the option invgamma and permit different rates of transition and transversion (K80 or HKY85 model). Twenty-five percent of the sampled trees were discarded, and the remaining were used to build up a consensus tree and to calculate posterior probabilities of clades. The result of Bayesian analyses was visualized using Figtree v1.4.2. Transformation of the leaves and a schematic representation of the root were applied for visualization purposes.
2.7. Clonal Complex Determination
To infer patterns of evolutive descendants for the L. panamensis DSTs we used the goeBURST algorithm included in the bioinformatic package PHYLOVIZ v.2.0 [32]. The goeBURST algorithm predicts the offprint of a founder genotype establishing groups of related strains that share a certain number of identical alleles with other members (Clonal complex).
2.8. Global Ancestry of Local Leishmania panamensis Isolates
We arrange in a matrix the haplotype information from each locus in a single text file to estimate the ancestral population proportion for each L. panamensis isolate using the software STRUCTURE V2.3.4 [33]. Structure group individuals with a similar pattern of variation into populations of genetic groups and estimates global ancestry by applying different models of population structure to the data. To estimate ancestry, we first estimated the most likely value of k (genetics groups) by running the software twenty times for values of k between 1 to 15. Software parameters were set to run the simulation for a burning period of 20000 iterations and 60,000 Markov Chain Monte Carlo repetitions using the admixture model of ancestry. The mean value and standard deviation calculated from the natural logarithm of the prior probability from each k repeat were used to assess the ∆K value (rate of variation of the log-likelihood of data). This parameter was then applied to estimate the k value that best fits our data. Also, we employed the median value of Q (estimated membership) from each cluster of the most likely k obtained (K=3) to summarize in a plot estimation of this parameter for each local L. panamensis isolate. The percentage of membership for each isolate was represented in a stacked plot constructed in LibreOffice Calc and edited in LibreOffice Draw (https://www.libreoffice.org).
3. Results
3.1. Identification of Leishmania panamensis Isolates
Leishmania species were identified by Bayesian phylogenetic inference using a set of HSP70 sequences publicly available on GenBank and TritrypDB databases (Table S1). The isolates clustered into a clade conformed by L. panamensis reference sequences used in the phylogenetic approach. Therefore, this result indicates that all local Leishmania isolates belong to this Leishmania species (Figure S1).
3.2. Genetic Diversity of L. panamensis Haplotypes
We amplified successfully the molecular markers included in the MLST approach in all local isolates of L. panamensis. Table 2 shows the diversity indexes from each molecular marker analyzed after constructing all L. panamensis haplotypes. The diversity index obtained suggest the presence of intraspecific variation of L. panamensis in Panama. The locus GPI and aconitase showed the highest diversity indexes, number of polymorphic sites, and haplotype diversity in this study. Consequently, we were able to determine six haplotypes by GPI and five haplotypes using aconitase. Conversely, the HSP70 and ALAT loci showed the lowest theta, Pi value, and haplotype diversity. However, we were able to distinguish three L. panamensis haplotypes with these loci.
3.3. Diversity Indexes by Locus and Geographical Region
Panama presents ecological differences between the western and eastern sites of the country, particularly in the distribution of seasonal precipitation, annual average, and diary fluctuations of temperature and vegetal coverage that might influence the local transmission of leishmaniasis and consequently the populational structure of the parasites circulating in both regions. Therefore, we evaluated diversity indexes in the western and eastern regions of the country. After this evaluation, two loci indicated clear differences between genetic diversity indexes found in both regions (Figure 2, Table 3). The differences were more evident when we applied aconitase and GPI approaches to assess L. panamensis haplotype diversity. We found a higher diversity index in the L. panamensis population from the western region using these two markers. With the ALAT approach, slight differences between the diversity index from both areas were observed, while with the HSP70 analysis, we detect no diversity in the east region as only one haplotype was found circulating in this area. The number of polymorphic sites and haplotypes also supports the diversity differences between local L. panamensis populations from both regions (Table 3). All diversity indexes evaluated in this study showed to be higher when GPI and aconitase were used as markers and demonstrated the highest values in the western region where the L. panamensis populations seem to be more diverse.
Conversely, the ALAT approach found the same number of polymorphic sites and haplotypes in both regions with two haplotypes defined by one single nucleotide polymorphism (SNP). On the other hand, the HSP70 selected fragment demonstrated the presence of only one haplotype in the east region and three haplotypes circulating in the west region due to the presence of two more SNPs in the HSP70 locus of this population.
3.4. Genotyping Approach by MLST
To optimize the MLST approach we first determined the best number and combination of loci that produce the highest diversity values. We found that as the number of loci used in the MLST scheme was increased, there was an increment in the minimum, maximum, and mean number of DSTs found by the algorithm of the MLSTest software. As a result, we decided to use the MLST scheme containing all four loci to find the highest intraspecific diversity in the L. panamensis isolates evaluated in this study. As a part of the optimization process, we also performed a congruency analysis for the four gene selections. After evaluating incongruency length differences and comparing the topology of the concatenated locus with the loci selected for the MLST approach, we found no incongruency as the p-value for the ILDP parameter and Templeton test was 1. Moreover, our MLST approach was able to determine thirteen L. panamensis DSTs because of a collective typing efficiency of 0.85, a combined discriminatory power of 0.491, and the presence of 20 SNPs considering all loci (Table S2).
3.5. Geographic Distribution of DSTs
The analysis of the allele combination from selected loci obtained in this study evidenced the circulation of thirteen L. panamensis genotypes that were named DST1 to DST13. Figure 1 depicts the geographical distribution of the thirteen DSTs found in the studied areas. The L. panamensis genotype DST1 was the most prevalent in all studied provinces from the western and eastern regions of the country. The second most frequent genotype was DST6 which was found in the provinces of Panama, Panama Oeste, and Cocle. The rest of the DSTs seem to have a restricted geographical distribution as they were found only in particular provinces. However, because a low number of isolates were evaluated in some provinces, we cannot rule out the presence of these DSTs or further ones in other regions of the country. Genotypes DST2 and DST4 seem to be restricted to the northern region of Panama province. Also, in the eastern region of Panama province, we found the DST6, DST7, DST8, DST9, and DST13 and nearby, in Darien province, we found circulating the genotype DST3. On the other hand, in the western region of the country, we found circulating the genotypes DST7 and DST6 in Panama Oeste, DST6, DST11, and DST10 in Cocle province and genotypes DST5 and DST12 in Bocas del Toro province (Figure 1).
3.6. Haplotype Resolution of local L. panamensis Isolates
To establish the haplotype compositions of local L. panamensis isolates, we resolve the ambiguities found in all loci using the PHASE algorithm of the DNAsp software (Table S). The isolates grouped into DST1 showed to be homologous genotypes comprised of the same haplotype in all studied loci. The remaining genotypes were found to be heterologous isolates bearing one to three ambiguities in one locus to three loci evaluated. The genotypes defined by ambiguities located in the aconitase loci were DST5, DST6, DST7, DST9, and DST13. Only one ambiguity located in the ALAT locus discriminates DST3 from the rest of the L. panamensis genotypes. Regarding the GPI locus, SNPs found in this locus made it easy to distinguish DST2, DST4, and DST8 from the other L. panamensis genotypes. In the case of the isolate FID16203-674 which belongs to DST12, it was the only genotype defined by an ambiguity located in the HSP70 locus. On the other hand, the heterologous isolates FID16203-453(DST10) and FDI16203-454(DST11) presented ambiguities in two and three loci, respectively. The isolate FID16203-453 showed ambiguities in the aconitase and ALAT loci and the isolate FID16203-454 in the aconitase, GPI, and HSP70 loci.
The evidence found in this study suggests that most of the DSTs found herein are heterologous genotypes circulating at low frequencies in most of the endemic areas evaluated. Additionally, the clonal complex analysis indicates that these heterologous genotypes arose from a homologous genotype (DST1) that diversified by genetic exchange and its variant nowadays might participate in different transmission cycles. The results also suggest that exist local areas such as Cocle province (Table S) where the intense genetic exchange between L. panamensis genotypes is giving rise to new variants of this Leishmania species. This could be the case of the heterologous genotypes bearing ambiguities in two or more loci found in this study.
3.7. Phylogenetic Analysis and Clonal Complex Determination of L. panamensis DSTs
The phylogenetic analysis by Bayesian inference using the concatenated sequences from the loci analyzed in this study supported the existence of 13 L. panamensis DSTs that are depicted branching with high credibility values in Figure 3. The alleles from L. panamensis DSTs grouped together in a major clade with a credibility value of 0.98 corroborating that they are phylogenetically related, and the DSTs obtained represent different genotypes of L. panamensis. Also, the phylogenetic analysis revealed a close association among the L. panamensis genotypes DST6, DST7, DST10, and DST11 that cluster together in the same clade. The clonal complex analysis supports this fact as indicates that the genotype DST11 arose from DST6 and the genotype DST10 from DST7 (Figure 3). These genotypes overlap in distribution and some of them seem to be circumscribed to endemic areas from Cocle province indicating this geographical point as a possible hotspot where inbreeding events and genetic exchange are contributing to the high genetical variability found in the L. panamensis population from this area.
Additionally, the clonal complex test performed on the sequences of representative alleles from L. panamensis DSTs suggests the presence of one L. panamensis clonal complex conformed by DST1 as a founder genotype and other 12 L. panamensis DSTs that differ in one or two loci from DST1 (Figure 3).
3.8. Global Ancestry of Local L. panamensis Isolates
To better characterized the population structure of L. panamensis in the studied areas we assessed the median value of the ancestral population proportion for each local isolate (Figure 4). Global ancestry estimates the proportion of ancestry from each contributing population, and each proportion could be considered as an average over the individual´s entire genome [34]. Taking this fact into consideration, we estimated the contribution of each ancestral population to the genetic constitution of local L. panamensis isolates. Interestingly, we found that the genetic make-up of all isolates regardless of their geographic origin is a product of the combination of different proportions of genetic segments from three ancestral populations. Furthermore, the local L. panamensis genotypes inferred by the program MLSTest showed unique patterns of the ancestral contribution that mirror the specific contribution of the three ancestral populations to each L. panamensis genotype studied herein. This fact also points out that in the case of admixture DSTs, genotype-specific patterns of the ancestral contribution could be used to identify different genotypes in endemic areas.
4. Discussion
Leishmania species are comprised of different populations bearing medically important characteristics that make their identification relevant in all epidemiological landscapes. Indeed, the genetic variability at the species level greatly impacts the biological diversity of its populations, influencing important properties such as virulency and tolerance to certain drugs. In this sense, it is highly recommended to type Leishmania parasites beyond the species level to properly understand their population structure, population dynamics, geographical distribution, and transmission cycles of medically relevant genotypes.
In this study, we used an MLST approach based on four housekeeping genes segments to infer the genetic diversity of L. panamensis at haplotype and genotype levels and to determine the distribution of genetic variants of this species in Panama. We based the molecular characterization of L. panamensis isolates on an MLST approach as it will probably become the recommended gold standard for Leishmania genotyping, including phylogenetic and epidemiological studies in different geographical areas [22]. In fact, this tool has been already used with Leishmania strain/isolates of subgenus Viannia to study phylogeny and population genetics [20], discriminate Viannia species [35] investigate leishmaniasis outbreaks [36], and study parasite variability and phylodynamics of this disease [21,36].
The present study represents a pioneering initiative aiming to assess the local genetic variability of L. panamensis by MLST in the country. For this purpose, we first evaluated the genetic variability of this species at the haplotype level. The combination of haplotype reconstruction by locus, along with the diversity index evaluation, allows us to assess local L. panamensis variation. All evaluated markers presented different degrees of polymorphisms mirrored by the differences in diversity indexes and the number of haplotypes reported by locus in this study. This finding gave us a preliminary glance at the local variation of L. panamensis and suggested a certain degree of intra-specific variability of this Leishmania species at the haplotype level. In fact, our results suggest differences between the haplotype diversity found in the local L. panamensis isolates from the eastern and western regions of the country. The western region showed higher haplotype diversity indexes and outnumbered the eastern region in the number of haplotypes. Differences in genetic diversity at the haplotype level between these two regions have also been reported for Plasmodium vivax [36]. In the case of P. vivax, it was suggested that different ecological and climate conditions as well as vector diversity prevalent in both areas have influenced local malaria transmission and therefore Plasmodium genetic structure. Indeed, western, and eastern regions have different ecological conditions, land covering, and weather conditions, such as seasonal distribution of rainfall, mean annual temperatures, and daily fluctuations in temperatures [37]. These different ecoclimatic conditions between areas might be shaping the genetic make-up of L. panamensis in Panama as macroecological and climate conditions have been shown to influence the patterns of transmission and seasonality of this disease in the country [24,38]. A study carried out in Colombia using an MLST approach also found geographical differences in the haplotype diversity of L. panamensis in regions with different ecoclimatic profiles [26]. Changes in climatic conditions and ambient temperature can affect the distribution of leishmaniasis through sand fly abundance or through the effect of temperature on parasite development in vectors [39]. In this regard, it has been demonstrated the capability of L. braziliensis to produce heavy late infections in sand flies at the range of temperature between 20 to 26°C [40]. This plasticity of Viannia species might be one of the factors that drive parasite dissemination to different ecoclimatic areas and further divergency to produce different L. panamensis haplotypes in the Americas.
After combining the haplotype reconstruction algorithm of DNAsp and the capacity of MLSTest software to determine DSTs, we found more diversity in the local population of L. panamensis by inferring the haplotype resolution at the diploid state of the parasite. Applying this approach, we found thirteen L. panamensis genotypes causing TL in most of the endemic areas of the country. This finding and the one obtained by haplotype analysis, confirm the circulation of genetic variants of L. panamensis in Panama. The phylogenetic analysis supports this result by grouping all DSTs alleles with L. panamensis reference sequences and confirming them as specific genotypes of this species that come from the same population. Few studies in Panama have described the genetic diversity of L. panamensis. A preliminary study using a microsatellite panel carried out in a small geographic region from Central Panama revealed extensive genetic diversity in 27 L. panamensis isolates [23]. However, no repeated genotypes were detected in that study revealing the high resolution of this set of microsatellites in such a small group of isolates. This fact might hinder a further association between local L. panamensis variants and the clinical expression of TL in the country, validating the probable need for less resolutive tools such as multilocus typing to address this question. A recent study that evaluated a set of SNPs from 24 L. panamensis genomes in Panamá found a genetically divergent group of isolates circulating in some endemic areas of the country corroborating therefore the presence of genetic variants of this parasite in the country [41]. Recently, our research group reported three haplotypes of L. panamensis circulating in the country [12]. In that study, the typing approach was based on one molecular marker, a fact that might explain the lack of resolution in finding more L. panamensis haplotypes using a one-market strategy. As mentioned, when evaluating levels of characterization under the sub-species category, it is recommended to add more locus to typing schemes in order to increase the discriminatory power [22]. In this regard, our MLST scheme based on four housekeeping genes support the previous evidence on the circulation of L. panamensis variants in Panama and showed a great resolution, identifying several genotypes of this species. Additionally, each local L. panamensis genotype presented a specific profile in the summary plot of estimates of Q, indicating the inheritance of different allelic proportions by the local L. panamensis genotypes. As these proportions are obtained as an average over organisms’ genomes, this finding also supports the circulation of L. panamensis genotypes with different genetic make-up in the local epidemiological scenario. These genetic differences might be further mirrored in different medically important characteristics such as the clinical expression of the disease, parasite internalization into macrophages, immune evasion, virulence, pathogenicity, and drug resistance. In this sense, the medical staff from the clinical branch of the Instituto Conmemorativo Gorgas de Estudios de la Salud located in Panama City has reported in its clinical practice human cases of localized, disseminated, diffuse and mucocutaneous forms of TL associated with L. panamensis [16]. Such variability in clinical forms in its broadest sense could be the result of the local interplay between variants of this species and inhabitants with different genetic profiles. Therefore, it is important to carry out further studies using high-resolutive techniques such as the one developed herein to unveil the association between L. panamensis make-up and the clinical expression of leishmaniasis in the country. In this regard, we are gathering all the necessary clinical information to evaluate the association between these two variables using the molecular tools developed herein.
Regarding the distribution of L. panamensis in the studied areas, no specific pattern was found. It seems that DST1 is widely spread in the country as it was found in all studied areas. Unlike this genotype, the rest of L. panamensis DTSs were less frequently found and most of them circumscribed to specific geographical points where DST1 was also reported. This association between DST1 and other L. panamensis genotypes in specific points of the country might indicate that DST1 diversified to produce new genotypes allied to specific transmission foci. The clonal complex analysis supports this assumption indicating DST1 as a founder genotype that differentiates from the rest of the DSTs by one to three allele differences. This result suggests that DST1 might represent the ancestral genotype from which the rest of L. panamensis DSTs emerged in the country. This genotype could have arrived in the country after the formation of the isthmus that prompted the settlement of vectors and reservoirs of Leishmania parasites. In fact, Valderrama and collaborators [42] proposed that Lutzomyia gomezi, one of the most important local vectors, was established and disseminated in the country after the closure of the Isthmus. Also, the same authors stated that several mammals’ reservoirs of Leishmania parasites settled in the country after this event. After that, the spreading of DST1 to areas presenting different ecoclimatic conditions with particular vectors and reservoirs host composition might have contributed to the emergence of new local L. panamensis genotypes. Furthermore, geographical barriers in Panama as the Central Mountain range, possibly restricted Lutzomyia populations to specific demographic areas where different Leishmania cycles were established. This circumstance might have driven the local L. panamensis diversification and expansion as it was suggested elsewhere for the Lu gomezi vector [42].
As for the low frequency and geographical constraint of most of the L. panamensis genotypes, this fact could be the result of the geographical isolation followed by divergence from DST1. Additionally, these genotypes might be participating in specific transmission cycles along with particular vectors and reservoirs circumscribed to specific transmission focus. Leishmania strains structure at a small geographic scale and patterns of structuration have been reported for L. guyanensis in French Guyana [43]. and L. braziliensis in Peru and Bolivia [44]. In such transmission focus, Leishmania vectors and reservoirs might have an important role in shaping the genetic make-up of Leishmania species. Sexual events happening in vectors represent a great source of Leishmania diversity and the clonal expansion and dissemination of parasite populations are ensured by mammal hosts. In Panama, Choloepus hoffmani is the main reservoir host of L. panamensis [45,46]. and its presence might be associated with high human population densities facilitating the local transmission of TL [47]. Additionally, secondary mammal reservoirs responsible for parasite population maintenance in enzootics cycles have also been described in the country [48,49]. Anthropophilic vectors participating in both enzootic and domestic/peridomestic cycles because of their adaptation to disrupted environments might be responsible for bringing up new genotypes to local endemic areas [50]. This could be the case of the L. panamensis genotypes found in this study at low frequencies across the country.
Our study presented a few important limitations that need to be stated. First, the study sample size was rather small, and, therefore, our results do not necessarily represent the entire genetic diversity of L. panamensis that could be seen in the country nor in the endemic regions evaluated. For this reason, we cannot rule out the presence of other genotypes in regions where they were not identified, although overall patterns of increased diversity in western Panama when compared with western Panama are likely robust. Second, we did not address the temporal distribution of DSTs thus it is unknown whether these genotypes are totally fixed in the local L. panamensis populations and have enough continuity throughout time to be responsible for the local cases of TL during all seasons. Indeed, the temporal presence of the L. panamensis DSTs found herein needs to be evaluated before considering them in any local programs of leishmaniasis control and surveillance.
The global ancestry result inferred by STRUCTURE showed that all L. panamensis genotypes found in this study are admixture individuals that originated from three ancestral populations. A recent analysis of medically relevant SNPs from a small sample of 24 L. panamensis isolates from Panama also found admixture individuals [41]. Events of endogamy are common in Leishmania parasites recurrently happening in species from the same L. panamensis subgenus such as L. guyanensis [43] and L. braziliensis [44] Inbreeding also was suggested for L. panamensis during a study that analyzed a set of microsatellites in 27 isolates of this species from regions located in Central Panama [23].
Despite L. panamensis being an important etiological agent of TL in Mesoamerica and some South American countries [51], few studies have been conducted to explore the genetic diversity of this Leishmania species. A study carried out in Colombia by MLST obtained a greater diversity index using the ALAT locus to evaluate the genetic diversity of local populations of L. panamensis than the one inferred for us using the same marker. These variations in diversity index could be explained by either difference in sample size between studies or geographical isolation and independent evolution of both L. panamensis populations. As a matter of fact, a recent study using 43 isolates of Colombian and Panamanian origin found that the Panamanian population of L. panamensis is genetically divergent from the Colombian counterpart [41]. As parasites from the Viannia subgenus structure in small geographical areas [43], it is not rare to find divergent groups of L. panamensis separated by distance and geographical barriers as the ones existing between Panama and Colombia. Furthermore, geographic isolation and restricted genetic flow of parasites due to the low dispersion of infected vectors and mammals might be favoring the structuration of Leishmania populations at a small scale and the emergency of genetic variants constrained to specific endemic areas. This could be the case of the genetic variants of L. panamensis found herein and in other studies carried out in Panama [12,23,41] and Colombia [26] and probably in other Latin American countries where this species is circulating. This phenomenon might be potentiated by the presence of highlands and mountain ranges along the endemic territories that have facilitated isolation, genetic divergence, and speciation events. For instance, the Mesoamerican region is considered the second most threatened biodiversity hotspot of the Americas [52], is highly fragmented, like an archipelago, and taxa are thus frequently represented as sets of isolated populations, each restricted to particular mountain ranges and often showing a high degree of divergence, both morphologically and genetically [53].
Even though L. panamensis has shown lower genomic variability than members of the Viannia subgenus such as L. braziliensis [54], the polymorphisms found in a selected group of loci by MLST and the evaluation of selected sets of SNPs at the genomic level in Latin America [26,41,54] indicate that exist intra-specific genetic variability in L. panamensis. Other members of the L. guyanensis complex such as L. guyanensis and L. naiffi have shown low structural variations and a high degree of structural homogeneity among Leishmania strains of the same species regardless of the heterogeneity observed in selected sets of SNPs/Indels analyzed [55]. It has been hypothesized that such SNPs heterogeneity might be attributed to the parasite’s adaptation to new human hosts and different ecological niches [55]. Consequently, the diversity in human population groups and ecological niches present along Latin American countries where L. panamensis is circulating might be shaping the genetic structure of this parasite causing somehow the emergency of genotypes restricted to epidemiological sceneries characteristic of each country. The structuration of L. panamensis into subgroups, variants, or lineages at a small geographical scale highlights the necessity of developing regional initiatives aiming at revealing how the genetic composition of this species impacts medically relevant characteristics of the parasite that might be driving the disease outcome in Latin America.
5. Conclusion
As far as we are concerned, this is the first study describing the genetic diversity of L. panamensis in most of the endemic regions of the country. Our results clearly indicate the circulation of thirteen L. panamensis genotypes belonging to the same population associated with TL. Moreover, these results support the high intra-specific genetic diversity found in L. panamensis using SNPs as a marker by either MLST or NGS approaches in other Latin American countries such as Colombia. Our findings pave the way for developing further studies aiming at shedding light on the relationship between the genetic composition of L. panamensis and important medically relevant characteristics of this parasite. Furthermore, the MLST approach developed in this study could be used as a feasible option for typing and inferring the genetic structure of L. panamensis in developing countries of Latin America where obtaining complete genomes of this species involves higher costs.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: title HSP70-based Bayesian phylogenetic tree of Leishmania panamensis isolates evaluated in this study; Table S1: References sequences used in this study to determine Leishmania species by an approach based on HSP70 gene; Table S2: Determination of the local Leishmania panamensis diploid sequences by the MLSTest software; Table S3: Haplotype resolution of the Leishmania panamensis isolates found in this study.
Author Contributions
D.M. performed the molecular characterization of Leishmania parasites using the GPI gene and aided in interpreting the main results; V.V. carried out the molecular characterization of Leishmania isolates using aconitase and alanine transferase genes as molecular markers; L.J. standardized a methodology to perform the molecular characterization of Leishmania isolates using the HSP70 gene; V.P. carried out Leishmania parasites thawing and in vitro culture expansion; J.C. contributed to the design and implementation of the research and to the analysis of the results; A.Z. contributed to design the study and to the interpretation of the results; F.S. devised the project, the main conceptual ideas, proof outline and supervised the project activities. Also, this author performed all the computations related to the phylogenetic analysis and haplotype descriptions. All authors have read and agreed to the published version of the manuscript.
Funding
This study was sponsored by the National Secretary of Sciences and Technology (SENACYT), grant No. FID16-203 as a part of the research for development initiative (I + D) in Panama.
Institutional Review Board Statement
This research was considered by the Comité de Bioética de la Investigación del Instituto Conmemorativo Gorgas de Estudios de la Salud (CBI-ICGES) and deemed exempt (Note No. 1084/CBI/ICGES/16).
Informed Consent Statement
Not applicable
Data Availability Statement
Individual alignments of each locus used in this study as well as the concatenated alignments containing sequences from aconitase, ALAT, GPI and HSP70 loci are available on request. The sequences obtained in this study are publicly available in GenBank database and can be retrieved using accession numbers described in the materials and method section of this study.
Acknowledgments
We thank the technical staff of the Sanger sequence facility from the Instituto Conmemorativo Gorgas de Estudios de la Salud for its support in performing capillary electrophoresis of the molecular targets employed in this study. We acknowledge the National Research System (SNI-SENACYT-PANAMA) for supporting Franklyn Samudio, José E. Calzada, and Azael Saldaña.
Conflicts of Interest
The authors declare no conflict of interest.
References
- de Moura, T.R.; Oliveira, F.; Novais, F.O.; Miranda, J.C.; Clarêncio, J.; Follador, I.; Carvalho, E.M.; Valenzuela, J.G.; Barral-Netto, M.; Barral, A.; et al. Enhanced Leishmania braziliensis infection following pre-exposure to sandfly saliva. PLoS Negl. Trop. Dis. 2007, 1, 1–10. [CrossRef]
- Vanloubbeeck, Y.; Jones, D.E. The immunology of Leishmania infection and the implications for vaccine development. In Proceedings of the Annals of the New York Academy of Sciences; New York Academy of Sciences, 2004; Vol. 1026, pp. 267–272. [CrossRef]
- Pan American Health Organization. Leishmaniasis: Epidemiological Report in the Americas.; Washington, D.C, 2019; pp 2-3.
- Alvar, J.; Vélez, I.D.; Bern, C.; Herrero, M.; Desjeux, P.; Cano, J.; Jannin, J.; de Boer, M. Leishmaniasis worldwide and global estimates of its incidence. PLoS ONE 2012, 7, e35671. [CrossRef]
- Schönian, G.; Mauricio, I.; Cupolillo, E. Is it time to revise the nomenclature of Leishmania? Trends Parasitol. 2010, 26, 466–469. [CrossRef]
- Christensen, H.A.; De Vasquez, A.M.; Petersen, J.L. Short report: Epidemiologic studies on cutaneous leishmaniasis in eastern Panama. Am. J. Trop. Med. Hyg. 1999, 60, 54–57. [CrossRef]
- Miranda, A.; Carrasco, R.; Paz, H.; Pascale, J.M.; Samudio, F.; Saldaña, A.; Santamaría, G.; Mendoza, Y.; Calzada, J.E. Molecular epidemiology of American tegumentary leishmaniasis in Panama. Am. J. Trop. Med. Hyg. 2009, 81, 565–571. [CrossRef]
- Del Miranda, A.C.; González, K.A.; Samudio, F.; Pineda, V.J.; Calzada, J.E.; Capitan-Barrios, Z.; Jiménez, A.; Castillo, J.; Mendoza, Y.; Suárez, J.A.; et al. Molecular identification of parasites causing cutaneous leishmaniasis in Panama. Am. J. Trop. Med. Hyg. 2021, 104, 1326–1334. [CrossRef]
- Vasquez, A.N.A.M.D.E.; Saenz, R.E.; Petersen, J.L.; Johnson, C.M. Leishmania Mexicana complex: Human infections in the Republic of Panama. Am. J. Trop. Med. Hyg. 1990, 43, 619–622. [CrossRef]
- Miranda, A.; Samudio, F.; Saldaña, A.; Castillo, J.; Brandão, A.; Calzada, J.E. The calmodulin intergenic spacer as molecular target for characterization of Leishmania species. Parasites and Vectors 2014, 7, 1–9. [CrossRef]
- Miranda, A.; Samudio, F.; González, K.; Saldaña, A.; Brandão, A.; Calzada, J.E. Calmodulin polymerase chain reaction-restriction fragment length polymorphism for leishmania identification and typing. Am. J. Trop. Med. Hyg. 2016, 95, 383–387. [CrossRef]
- Davila, M.; Pineda, V.; Calzada, J.E.; Saldaña, A.; Samudio, F. Evaluation of cytochrome b sequence to identify Leishmania species and variants: The case of Panama. Mem. Inst. Oswaldo Cruz 2021, 116, 1–10. [CrossRef]
- Miranda, A.; Carrasco, R.; Paz, H.; Pascale, J.M.; Samudio, F.; Saldaña, A.; Santamaría, G.; Mendoza, Y.; Calzada, J.E. Molecular epidemiology of American tegumentary leishmaniasis in Panama. Am. J. Trop. Med. Hyg. 2009, 81, 565–571. [CrossRef]
- Hashiguchi, Y.; Gomez, E.L.; Kato, H.; Martini, L.R.; Velez, L.N.; Uezato, H. Diffuse and disseminated cutaneous leishmaniasis: Clinical cases experienced in Ecuador and a brief review. Trop. Med. Health 2016, 44, 1–9. [CrossRef]
- Osorio, L.E.; Castillo, C.M.; Ochoa, M.T. Mucosal Leishmaniasis due to Leishmania (Viannia) panamensis in Colombia: Clinical characteristics. Am. J. Trop. Med. Hyg. 1998, 59, 49–52. [CrossRef]
- Freites, C.O.; Gundacker, N.D.; Pascale, J.M.; Saldaña, A.; Diaz-Suarez, R.; Jimenez, G.; Sosa, N.; García, E.; Jimenez, A.; Suarez, J.A. First case of diffuse leishmaniasis associated with leishmania panamensis. Open Forum Infect. Dis. 2018, 5, 1–3. [CrossRef]
- Grimaldi, G.; Tesh, R.B.; McMahon-Pratt, D. A review of the geographic distribution and epidemiology of leishmaniasis in the New World. Am. J. Trop. Med. Hyg. 1989, 41, 687–725. [CrossRef]
- Mauricio, I.L.; Yeo, M.; Baghaei, M.; Doto, D.; Pratlong, F.; Zemanova, E.; Dedet, J.P.; Lukes, J.; Miles, M.A. Towards multilocus sequence typing of the Leishmania donovani complex: Resolving genotypes and haplotypes for five polymorphic metabolic enzymes (ASAT, GPI, NH1, NH2, PGD). Int. J. Parasitol. 2006, 36, 757–769. [CrossRef]
- Zemanová, E.; Jirků, M.; Mauricio, I.L.; Horák, A.; Miles, M.A.; Lukeš, J. The Leishmania donovani complex: Genotypes of five metabolic enzymes (ICD, ME, MPI, G6PDH, and FH), new targets for multilocus sequence typing. Int. J. Parasitol. 2007, 37, 149–160. [CrossRef]
- Boité, M.C.; Mauricio, I.L.; Miles, M.A.; Cupolillo, E. New Insights on Taxonomy, Phylogeny and Population Genetics of Leishmania (Viannia) Parasites Based on Multilocus Sequence Analysis. PLoS Negl. Trop. Dis. 2012, 6, 1–14. [CrossRef]
- Marco, J.D.; Barroso, P.A.; Locatelli, F.M.; Cajal, S.P.; Hoyos, C.L.; Nevot, M.C.; Lauthier, J.J.; Tomasini, N.; Juarez, M.; Estévez, J.O.; et al. Multilocus sequence typing approach for a broader range of species of Leishmania genus: Describing parasite diversity in Argentina. Infect. Genet. Evol. 2015, 30, 308–317. [CrossRef]
- Lauthier, J.J.; Ruybal, P.; Barroso, P.A.; Hashiguchi, Y.; Marco, J.D.; Korenaga, M. Development of a Multilocus sequence typing (MLST) scheme for Pan-Leishmania. Acta Trop. 2020, 201, 1–13. [CrossRef]
- Restrepo, C.M.; Llanes, A.; De La Guardia, C.; Lleonart, R. Genome-wide discovery and development of polymorphic microsatellites from Leishmania panamensis parasites circulating in central Panama. Parasit. Vectors 2015, 8, 527. [CrossRef]
- Yamada, K.; Valderrama, A.; Gottdenker, N.; Cerezo, L.; Minakawa, N.; Saldaña, A.; Calzada, J.E.; Chaves, L.F. Macroecological patterns of American Cutaneous Leishmaniasis transmission across the health areas of Panamá (1980–2012). Parasite Epidemiol. Control 2016, 1, 42–55. [CrossRef]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [CrossRef]
- Herrera, G.; Hernández, C.; Ayala, M.S.; Flórez, C.; Teherán, A.A.; Ramírez, J.D. Evaluation of a Multilocus Sequence Typing (MLST) scheme for Leishmania (Viannia) braziliensis and Leishmania (Viannia) panamensis in Colombia. Parasit. Vectors 2017, 10, 236. [CrossRef]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; Varlamov, A.; Vaskin, Y.; Efremov, I.; German Grehov, O.G.; Kandrov, D.; Rasputin, K.; Syabro, M.; et al. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [CrossRef]
- Rozas, J.; Rozas, R. DnaSP, DNA sequence polymorphism: An interactive program for estimating population genetics parameters from DNA sequence data. Comput. Appl. Biosci. 1995, 11, 621–625. [CrossRef]
- Tomasini, N.; Lauthier, J.J.; Llewellyn, M.S.; Diosque, P. MLSTest: Novel software for multi-locus sequence data analysis in eukaryotic organisms. Infect. Genet. Evol. 2013, 20, 188–196. [CrossRef]
- Gouy, M.; Guindon, S.; Gascuel, O. Sea view version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 2010, 27, 221–224. [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. Mrbayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [CrossRef]
- Nascimento, M.; Sousa, A.; Ramirez, M.; Francisco, A.P.; Carriço, J.A.; Vaz, C. PHYLOViZ 2.0: Providing scalable data integration and visualization for multiple phylogenetic inference methods. Bioinformatics 2017, 33, 128–129. [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [CrossRef]
- Veland, N.; Boggild, A.K.; Valencia, C.; Valencia, B.M.; Llanos-Cuentas, A.; Van Der Auwera, G.; Dujardin, J.C.; Arevalo, J. Leishmania (Viannia) species identification on clinical samples from cutaneous leishmaniasis patients in Peru: Assessment of a molecular stepwise approach. J. Clin. Microbiol. 2012, 50, 495–498. [CrossRef]
- Santamaría, A.M.; Vásquez, V.; Rigg, C.; Samudio, F.; Moreno, D.; Romero, L.; Saldaña, A.; Chaves, L.F.; Calzada, J.E. Plasmodium vivax genetic diversity in Panama: Challenges for malaria elimination in Mesoamerica. Pathogens 2021, 10, 1–18. [CrossRef]
- Instituto Geográfico Nacional “Tommy Guardia” Atlas Nacional de la República de Panamá; 5th edition.; Instituto Tommy Guardia: Panama, 2016; ISBN 978-9962-11-048-4.
- Chaves, L.F.; Calzada, J.E.; Valderrama, A.; Saldaña, A. Cutaneous Leishmaniasis and Sand Fly Fluctuations Are Associated with El Niño in Panamá. PLoS Negl. Trop. Dis. 2014, 8, 1–11. [CrossRef]
- Ready, P.D. Leishmaniasis emergence and climate change. OIE Rev. Sci. Tech. 2008, 27, 399–412. [CrossRef]
- Hlavacova, J.; Votypka, J.; Volf, P. The effect of temperature on Leishmania (Kinetoplastida: Trypanosomatidae) development in sand flies. J. Med. Entomol. 2013, 50, 955–958. [CrossRef]
- Llanes, A.; Cruz, G.; Morán, M.; Vega, C.; Pineda, V.J.; Ríos, M.; Penagos, H.; Suárez, J.A.; Saldaña, A.; Lleonart, R.; et al. Genomic diversity and genetic variation of Leishmania panamensis within its endemic range. Infect. Genet. Evol. 2022, 103, 1–11. [CrossRef]
- Valderrama, A.; Tavares, M.G.; Filho, J.D.A. Phylogeography of the Lutzomyia gomezi (Diptera: Phlebotominae) on the Panama Isthmus. Parasites and Vectors 2014, 7, 1–11. [CrossRef]
- Rougeron, V.; Bañuls, A.L.; Carme, B.; Simon, S.; Couppié, P.; Nacher, M.; Hide, M.; De Meeûs, T. Reproductive strategies and population structure in Leishmania: Substantial amount of sex in Leishmania Viannia guyanensis. Mol. Ecol. 2011, 20, 3116–3127. [CrossRef]
- Rougeron, V.; De Meeûs, T.; Hide, M.; Waleckx, E.; Bermudez, H.; Arevalo, J.; Llanos-Cuentas, A.; Dujardin, J.C.; De Doncker, S.; Le Ray, D.; et al. Extreme inbreeding in Leishmania braziliensis. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 10224–10229. [CrossRef]
- Herrer, A.; Christensen, H.A. Epidemiological patterns of cutaneous leishmaniasis in Panama. III. Endemic persistence of the disease. Am. J. Trop. Med. Hyg. 1976, 25, 54–58. [CrossRef]
- González, K.; Calzada, J.E.; Saldaña, A.; Rigg, C.A.; Alvarado, G.; Rodríguez-Herrera, B.; Kitron, U.D.; Adler, G.H.; Gottdenker, N.L.; Chaves, L.F.; et al. Survey of wild mammal hosts of cutaneous leishmaniasis parasites in Panamá and Costa Rica. Trop. Med. Health 2015, 43, 75–8. [CrossRef]
- Dutari, L.C.; Loaiza, J.R. American Cutaneous Leishmaniasis in Panama: A historical review of entomological studies on anthropophilic Lutzomyia sand fly species. Parasit. Vectors 2014, 7, 218. [CrossRef]
- Christensen, H.A.; De Vasquez, A.M. The tree-buttress biotope: A pathobiocenose of Leishmania braziliensis. Am. J. Trop. Med. Hyg. 1982, 31, 243–251. [CrossRef]
- Herrer, A.; Christensen, H.A.; Beumer, R.J. Reservoir hosts of cutaneous leishmaniasis among Panamanian forest mammals. Am. J. Trop. Med. Hyg. 1973, 22, 585–591. [CrossRef]
- Valderrama, A.; Tavares, M.G.; Filho, J.D.A. Anthropogenic influence on the distribution, abundance, and diversity of sandfly species (Diptera: Phlebotominae: Psychodidae), vectors of cutaneous leishmaniasis in Panama. Mem. Inst. Oswaldo Cruz 2011, 106, 1024–1031. [CrossRef]
- PAHO. Manual de Procedimientos para Vigilancia y Control de Las Leishmaniasis en las Américas; Organización Panamericana de la Salud: Washington, D.C, 2019; ISBN 978-92-75-32063-1. [CrossRef]
- Myers, N.; R. A. Mittermeier; C. G. Mittermeier; G. A. B. da Fonseca and J. Kent. Biodiversity hotspots for conservation priorities. Nature. 2000, 403:853-858. [CrossRef]
- García-Moreno, J., A. G. Navarro-Sigüenza, A. T. Peterson, and L. A. Sánchez-González. Genetic variation coincides with geographic structure in the common bush-tanager (Chlorospingus opthalmicus) complex from Mexico. Mol. Phylogenet. Evol 2004, 206, 33, 186-196. [CrossRef]
- Patino L.H.; Muñoz M.; Cruz-Saavedra L.; Muskus, C.; Ramírez J.D. Genomic Diversification, Structural Plasticity, and Hybridization in Leishmania (Viannia) braziliensis. Front. Cell. Infect. Microbiol. 2020, 10, 582196. [CrossRef]
- Patiño L.H.; Muñoz M.; Pavia P.; Muskus C.; Shaban M.; Paniz-Mondolfi A.; Ramírez J.D. Filling the gaps in Leishmania naiffi and Leishmania guyanensis genome plasticity. G3, 2022, 12, 1, jkab377. [CrossRef]
Figure 1.
Map of Panama showing the geographical distribution of the local Leishmania panamensis diploid sequences type (DSTs) found in this study.
Figure 1.
Map of Panama showing the geographical distribution of the local Leishmania panamensis diploid sequences type (DSTs) found in this study.

Figure 2.
Leishmania panamensis diversity index by molecular marker and geographical areas in Panama evaluated in this study. A. Haplotype diversity index; B. Phi Diversity index; C. Theta diversity index.
Figure 2.
Leishmania panamensis diversity index by molecular marker and geographical areas in Panama evaluated in this study. A. Haplotype diversity index; B. Phi Diversity index; C. Theta diversity index.
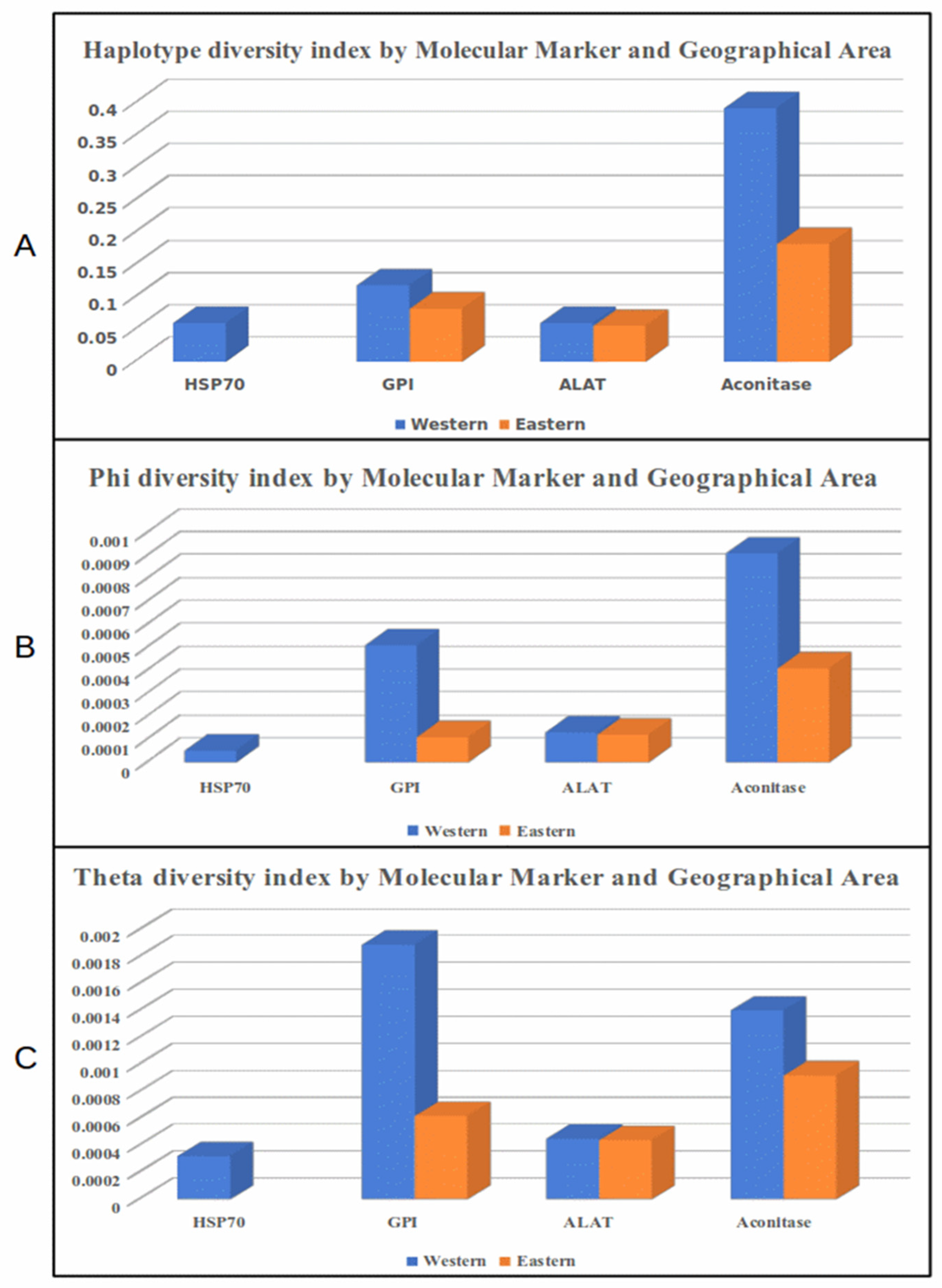
Figure 3.
A. Bayesian phylogenetic tree inferred using the concatenated sequences of each DSTs alleles from L. panamensis found in this study. The code of each DST allele is highlighted in red. B. Clonal complex analysis showing in green the founder (DST1) and its genetically related groups DST2-DST13 represented by numbers from 1-13.
Figure 3.
A. Bayesian phylogenetic tree inferred using the concatenated sequences of each DSTs alleles from L. panamensis found in this study. The code of each DST allele is highlighted in red. B. Clonal complex analysis showing in green the founder (DST1) and its genetically related groups DST2-DST13 represented by numbers from 1-13.
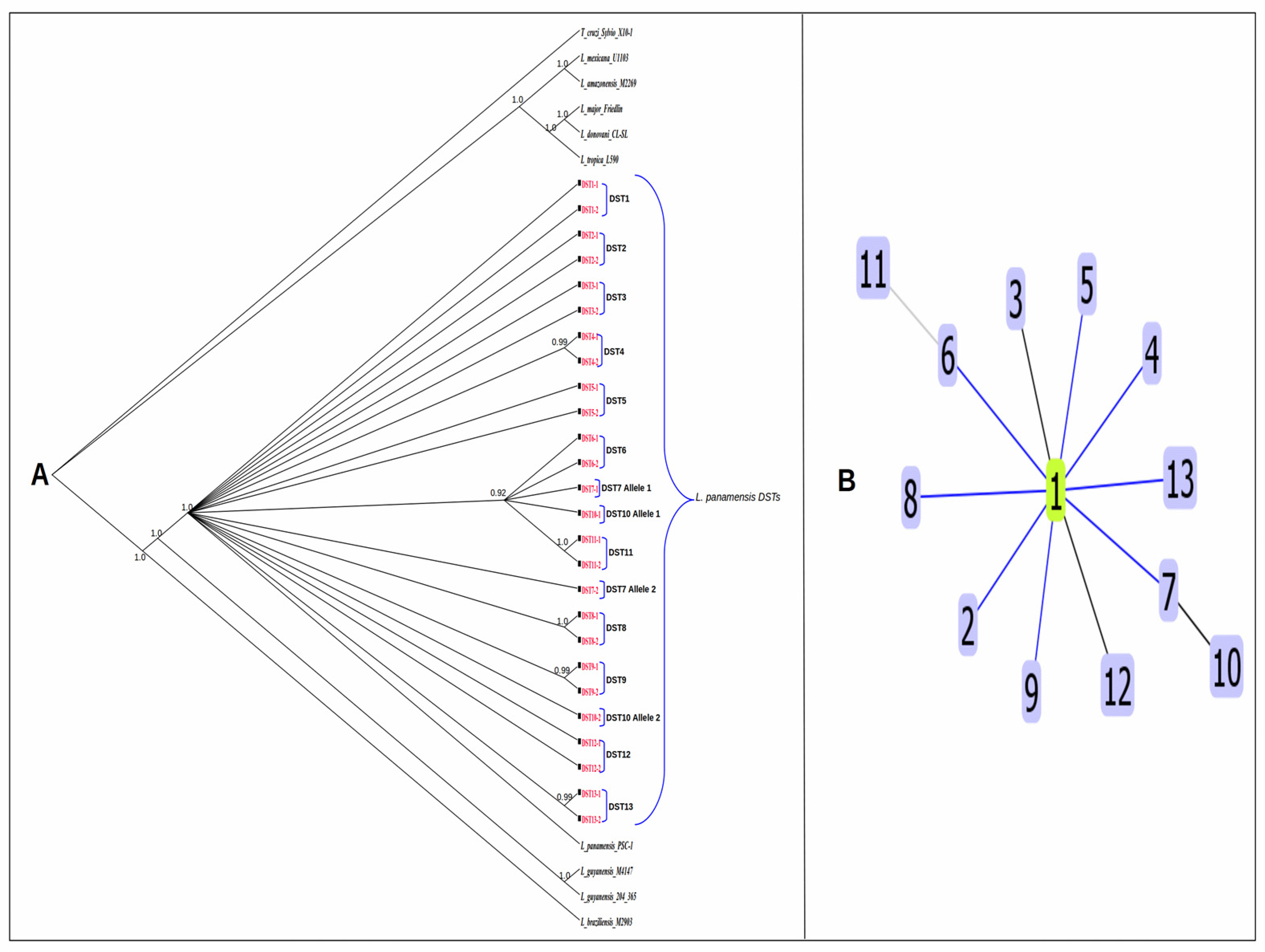
Figure 4.
Global Ancestry analyses of each L. panamensis isolate obtained in this study. The name observed on the X-axis is a composed code that stands for the L. panamensis genotypes (letters G1-G13) and the L. panamensis isolation code (numbers after the dashes).
Figure 4.
Global Ancestry analyses of each L. panamensis isolate obtained in this study. The name observed on the X-axis is a composed code that stands for the L. panamensis genotypes (letters G1-G13) and the L. panamensis isolation code (numbers after the dashes).
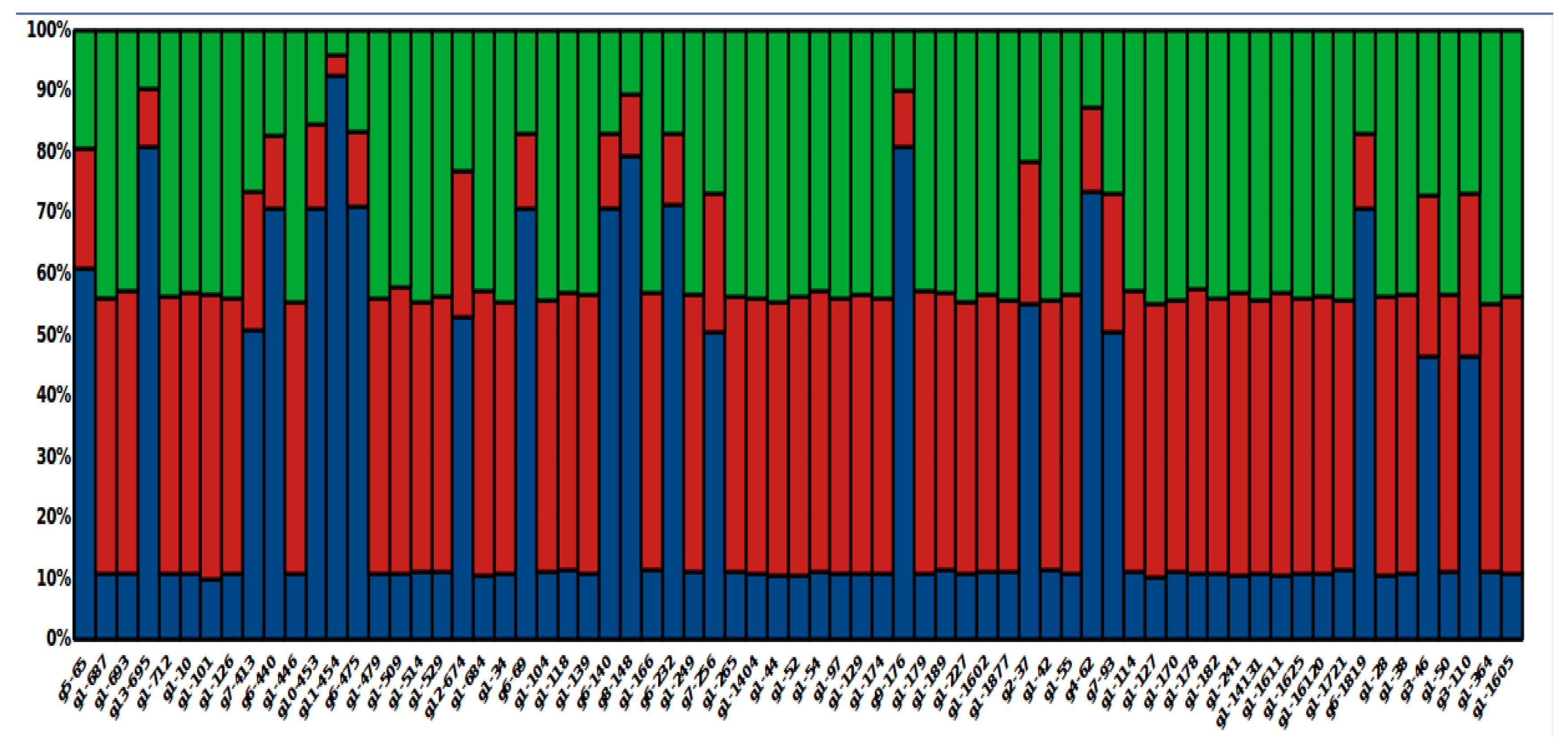

Table 1.
List of gene loci and primers used in this study.
| Target gene | Enzyme entry | Gene length (bp) | Chromosomal location | Amplicon size (bp) | Primers sequences | Reference |
|---|---|---|---|---|---|---|
| Heat Shock Protein 70 | AAG01344.1 | 2,566 | 28 | 1364 | PLeishF: GATGGTGCTGCTGAAGATGA PLeishR: GGTCATGATCGGGTTGCATR |
This study |
| Glucose-6-phosphate isomerase | EC 5.3.1.9 | 2,084 | 12 | 1,745 | GPIextF: AAT GTT CTT CAT ACC CCT CT GPIextR: TTC CGT CCG TCT CCT G GPIintF: TGG GAT TGG CGG CAG CGA CCTT GPIintR: CGC CAC AGG TAC TGG TCG T |
[37] |
| Alanine aminotransferase | EC 2.6.1.21 | 1,493 | 12 | 589 | ALAT.F: GTGTGCATCAACCCMGGGAA ALAT.R: CGTTCAGCTCCTCGTTCCGC |
[21] |
| Aconitase | EC 4.2.1.3 | 2,690 | 18 | 579 | ACO.F: CAAGTTCCTGRCGTCTCTGC ACO.R: GAGTCCGGGTATAGCAKCCC |
[21] |
Table 2.
Genetic diversity of local Leishmania panamensis population by locus evaluated.
| Loci | s | h | hd | π | ϴ |
|---|---|---|---|---|---|
| HSP70 | 2 | 3 | 0.029 | 0.00002 | 0.00028 |
| GPI | 12 | 6 | 0.099 | 0.00030 | 0.00217 |
| ALAT | 2 | 3 | 0.057 | 0.00012 | 0.00077 |
| Aconitase | 4 | 5 | 0.290 | 0.00067 | 0.00162 |
s: segregations sites; h: number of haplotypes; hd: haplotype diversity; π: pi: diversity index; ϴ: theta diversity index.
Table 3.
Genetic diversity of local Leishmania panamensis populations by molecular marker and geographical area.
Table 3.
Genetic diversity of local Leishmania panamensis populations by molecular marker and geographical area.
| Western N=34 |
Eastern N=35 |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Marker | s | h | hd | π | ϴ | s | h | hd | π | ϴ |
| HSP70 | 2 | 3 | 0.060 | 0.00005 | 0.00032 | 0 | 1 | 0 | 0 | 0 |
| GPI | 9 | 4 | 0.118 | 0.00051 | 0.00189 | 3 | 3 | 0.082 | 0.00011 | 0.00062 |
| ALAT | 1 | 2 | 0.060 | 0.00013 | 0.00045 | 1 | 2 | 0.055 | 0.00012 | 0.00044 |
| Aconitase | 3 | 4 | 0.392 | 0.00091 | 0.00140 | 2 | 3 | 0.182 | 0.00041 | 0.00092 |
S: segregations sites; h: number of haplotypes; hd: haplotype diversity; π: pi diversity index; ϴ: theta diversity index, N= number of L. panamensis isolates.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



