
Submitted:
21 April 2023
Posted:
25 April 2023
You are already at the latest version
Abstract
The oncologic disease is a big global health issue that causes thousands of deaths annually, and it has a significant impact in the life quality of patients. Prostate cancer (PCa) is the second most diagnosed cancer and the fourth leading cause of cancer-related death in men in the western world. Delineation of pathogenetic pathways and key driver molecular alterations involved in PCa development has provided a roadmap for the evaluation of biomarkers in predicting disease outcome and to identify potential therapeutic targets. Chemotherapeutic agents introduced from the 1990s include the taxanes (paclitaxel, docetaxel and cabazitaxel), which are the most anticancer drugs used for PCa treatment. This review presents the current knowledge about the onset and development of PCa, state-of-art on the use of taxane-based therapy, and their combination with targeting different transmembrane oncoproteins in PCa. The silencing of some transmembrane proteins can improve taxane sensitivity, and therefore, may be a mechanism to improve the ef-fectiveness of these drugs in PCa treatment. This combined therapy needs to be explored as po-tential therapeutic agent for reducing cell proliferation, migration, and invasiveness in PCa.
Keywords:
PCa
; taxanes-based drugs
; combination therapy
; transmembrane proteins
1. Introduction
The burden of cancer incidence and mortality is rapidly growing worldwide, and expectations for 2020 pointed to, approximately, 19.3 million new cancer cases and 10.0 million cancer deaths [GLOBOCAN, https://gco.iarc.fr/, accessed on 9th March 2023]. Prostate Cancer (PCa) is currently the second most common cancer in men and represents the fourth leading cause of cancer-related mortality. In 2020, 1.4 million new cases of PCa were diagnosed worldwide and, approximately 375,000 associated deaths were reported by World Health Organization [1]. The increased number of PCa can be explained by the lack of comprehensive national control programs that contributes to substantial disparities in early detection of cancer and management of these patients, with a 3-fold higher incidence rates in countries with high human development when compared to countries with low human development (37.5 and 11.3 per 100,000 habitants, respectively), although mortality rates are less variable (8.1 and 5.9 per 100,000 habitants, respectively) [2,3]. Moreover, the aetiology of PCa is multifactorial and remain largely unknown, when compared to other types of cancer. Epidemiologic evidence has identified several biological and genetic factors, but also environmental and lifestyle factors have been shown to contribute to the appearance and progression of PCa, namely advanced age, family history and genetic predisposition, ethnicity, smoking and alcohol consumption, obesity and metabolic syndrome, physical inactivity, diet and nutrition, medications, sexual activity and vasectomy, hormones, infection, inflammation, and chemokines [4,5]. However, age is considered the highest risk factor for the development of PCa. The peak of incidence is found in older men with approximately 70-74 years old [6].
Currently, several agents received FDA approval and have been associated with beneficial effects in improving survival and life quality in patients with this pathology, including abiraterone, enzalutamide, apalutamide, and darolutamide (inhibitors of the androgen axis); paclitaxel, docetaxel and cabazitaxel (target microtubules by inhibiting depolymerization or promoting polymerization); radium-223 (radioactive agents as target bone metastases); and sipuleucel-T (trigger cellular immune mechanisms) [7]. From those agents, an appropriate drug selection is done according to clinical usage for the treatment of PCa. Several cancers are treated with drug combination, but PCa has remained an exception [8]. Transmembrane proteins are involved in many crucial cell processes, including signaling transduction pathways, transport of ions and molecules, protein targeting and intracellular transport, as well as membrane trafficking [9]. Moreover, since membrane proteins are involved in essential cellular pathways, they are often recognized in the pathophysiology of many diseases and are major targets for pharmaceutical agents, with more than 60% of drug targets being transmembrane proteins [10]. Hence, developing the effective combination of drugs and targeting some transmembrane proteins can provide insights concerning new therapeutic strategies for advanced stages of PCa. This review provides an overview of the development of PCa, and it is focused on the taxanes-based therapy currently used. Therefore, it was analyzed the scientific literature concerning the combined action of taxanes based-chemotherapeutic drugs with inhibition of transmembrane oncoproteins within the paradigm of PCa.
2. Onset and development of PCa
The human prostate gland is the major accessory gland of the male reproductive system, located frontal to the rectum and immediately below the urinary bladder, surrounding prostatic urethra and the ejaculatory ducts [11,12]. Normal prostate tissue consists of prostatic ducts lined with epithelial cells surrounded by fibromuscular stroma [13,14]. Homeostasis of normal prostate tissue is maintained by the crosstalk between epithelial cells and the surrounding stromal components [15,16]. The glandular prostatic epithelium is a well-organized tissue composed of acini and ducts constituted by three types of cells, luminal, basal and neuroendrocrine cells (Figure 1). Luminal cells are columnar epithelial cells specialized in the production of prostatic secretions, including prostate specific antigen (PSA), and responsible for the main prostate function [17]. Basal cells adhere to the basement membrane and have the ability to produce several components essential in the maintenance of cell-growth [18,19]. Neuroendocrine cells comprise less than 1% of the prostatic epithelium and express chromogranin A, synaptophysin, enolase 2, and CD56, which promote the growth of prostate [20]. Interactions between the epithelium and basement membrane are fundamental to maintain epithelial cell polarity involving apical and basal surfaces, which represent the well-differentiated cell state [13]. The non-epithelial tissue of the prostate, referred to as stroma, is composed essentially, by fibroblasts, smooth muscle cells and extracellular matrix (ECM) proteins (Figure 1) [15]. The ECM forms a dynamic and structured mixture of collagens, proteoglycans, thrombospondin, and hyaluronic acid, that respond to tissue injuries and allow its regeneration [16].
Considering the onset of PCa, there is a good agreement that this cancer develops from prostate epithelial cells [14]. However, conflicting evidence exists regarding if the oncogenic transformation in PCa arises from basal [19,21] or luminal epithelial cells [22,23]. In addition, it also has been hypothesized that PCa arising from luminal cells are more aggressive than those arising from basal cells [21]. The prostatic epithelium can be damaged and driven the carcinogenesis of prostate due to several factors, such as, inflammation, infections, genetic/epigenetic changes, persistent activation by androgens, exposure to carcinogens and/or genetic factors [14,24]. The first identifiable histologic alteration in prostate malignant transformation is so-called prostatic intraepithelial neoplasia (PIN) (Figure 1) [25]. PIN lesions can be divided into two grades, low-grade PIN (LGPIN) and high-grade PIN (HGPIN), being that HGPIN lesions are considered the most likely precursors of PCa [26,27], but they do not appear to raise serum PSA concentration [28]. Characteristically, HGPIN lesion contain basal cell layer around their periphery, although it is thin and often discontinuous. This is an important diagnostic feature because preservation of the basal cell layer can help to differentiate PIN from prostatic adenocarcinoma in which the basal cells are absent [24,29].
Prostatic adenocarcinoma mostly arises in the peripheral zone of the prostate and initially is represented as a small foci of intraductal dysplasia, that with time differentiates and progresses into an invasive adenocarcinoma (Figure 1) [30]. The tumor foci lead to a disruption of prostate tissue and a decrease on glandular activity and prostatic fluid production [31]. Histologically, PCa is characterized by the destruction of the basal cell layer, derangement of the basement membrane, decreased epithelial cell polarity, and lack of connection of the glandular acini formed by the prostate epithelial cells [32]. As the tumor progresses, neoplastic cells increase the production of proteolytic enzymes, which cause degradation of the basement membrane, allowing the spread to adjacent tissues and the development of a metastatic disease [33]. Firstly, to lymph nodes and then to distant organs, including the bones, liver, and lungs, with bone as the most common site of metastasis [34]. In fact, in the context of epithelial neoplasia, the prostate stroma induces alterations in the tumor microenvironment, it is the so-called the reactive stroma. This phenotypic histological change leads to a loss of well-differentiated smooth muscle cells, increase of fibroblast population, and increase of secretion and deposition of ECM components, such as matrix metalloproteinase (MMP). All these changes can lead to epithelial cell depolarization and formation of conduits favoring neoplastic cell migration [16,35]. All these histological changes cause a thousand-fold increased release of PSA from prostate neoplastic cells into the blood [32].
Androgens play a central role in the control of normal prostate as well as PCa cell growth and proliferation [14]. Androgens are the primary regulators of the proliferation/apoptosis ratio, stimulating proliferation and inhibiting apoptosis of prostate cells, and, thus, inducing the development of PCa [14,36]. The major circulating androgen, testosterone, can be converted into DHT by the activity of 5α-reductase enzyme. Both testosterone and DHT exert their actions through binding to the AR. PCa growth and disease progression is initially dependent on AR activation. The main mechanism of action leads to the nuclear translocation of the ligand-receptor complex and subsequent binding to the androgen response elements (AREs), which initiates the transcription of genes that regulate cellular differentiation, proliferation and apoptosis (Figure 2) [27,36,37].
In primary PCa, the action of AR keeps the same role as in normal prostate, for example, synthesis of PSA and modulating lipid metabolism [22]. However, it also triggers other events that promote epithelial cell growth, as the induction of the type II transmembrane serine protease (TMPRSS2):ETS fusion [26,38]. The TMPRSS2 is an androgen-regulated gene overexpressed in PCa, which encodes a protein belonging to the serine protease family that functions in prostate carcinogenesis and relies on gene fusion with ETS transcription factors, such as ETS related gene (ERG) and ETV1. The TMPRSS2:ETS fusion is considered the most common chromosomal rearrangement in PCa and drives the overexpression of ETS oncogenes, previously identified as the most expressed proto-oncogenes present on malignant epithelial prostate cells [38,39,40]. ARs also have two active functional domains (AFs) that initiate transcription when activated. AF-1 is present in the NTD and its activation is androgen-independent. AF-2 is located in the LBD and is ligand-dependent [41]. AF-1 may enable cross-coupling between androgenic and growth factor signaling pathways [36,42]. Therefore, these AFs are deemed clinically important as they could provide the key to understand the development of castration-resistant PCa (CRPC). At early stages of disease, PCa growth is androgen-dependent, the so-called androgen-sensitive PCa. However, with the continuous tumor development, PCa cells became androgen-insensitive, and the disease progresses to the so-called CRPC [36].
Patients that acquire resistance to the use of androgen-deprivation therapy (ADT) inevitably develop CRPC, a more lethal form of PCa. The role of AR in PCa progression and development of CRPC has been attributed to several factors, such as AR gene amplification, activating mutations and aberrant expression of co-activators [37,43,44]. These alterations lead to an increased AR expression, activation of AR by non-androgenic ligands, broadened ligand specificity and sensitivity and increased AR transactivation, which ultimately contribute to tumor cell growth in low androgen environment [36,44,45]. AR mutations in primary PCa are rare, but these mutations are prevalent in about 50% of CRPC [46,47]. These mutations lead to alterations that improve the functional activity of the receptor, such as increased AR sensitivity to low levels of ligand, non-androgen ligand binding, ligand-independent activation as well as AR-independent pathways [41,46,47]. Furthermore, recent data indicate that an increased expression of constitutively active AR splice variants follows castration and are associated with poor prognostic and a rapid recurrence of PCa [48,49]. The reduction in AR activation by endogenous androgen ligands leads to hypersensitization of other pathways of AR activation through ligand-independent mechanisms [44,50].
Various growth factors, cytokines, kinases and other proteins have been shown to interact with and activate AR in a ligand-independent manner, including insulin-like growth factor (IGF1), fibroblast growth factor (FGF) and epidermal growth factor (EGF) [51,52]. These growth factors activate tyrosine receptor kinases, which results in the activation of phosphatidylinositol 3-kinase (PI3K) and subsequently the PI3K/AKT pathway (Figure 2) [53]. The serine/threonine protein kinase (AKT), also known as protein kinase B (PKB), is one of the major downstream effectors of PI3K. Binding of ligands to the membrane growth factor receptors initiates a cascade of events that activate PI3K, which converts phosphatidylinositol 4,5-bisphosphonate (PIP2) to phosphatidylinositol 3–5-triphosphate (PIP3). PI3K activation stimulates AKT, which recruits proteins to the luminal cell cytoplasm [53,54]. Downstream targets of AKT, namely, the mammalian target of rapamycin complex 1 (mTORC1), forkhead box protein O1 and the mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) cascade, activate several transcription factors, such as c-myc, which induces the expression of proteins associated with cell survival and proliferation, cell cycle progression, migration and angiogenesis, and, thus, contributing to the progression of PCa [44,53,55].
3. Current use of chemotherapy in PCa
Treatment approaches for PCa differ depending on the stage of the disease. Several types of therapeutic options are available such as surgery, cryosurgery, radiation therapy, hormone therapy, chemotherapy, vaccine treatment, immunotherapy and bone-directed treatment [56]. Active surveillance is the recommended treatment option for low-risk PCa, monitoring its progression while not undergoing definitive therapy [57]. Therapeutic approaches based on surgery often are used in combination with therapeutic approaches based on drugs, namely hormone therapy and chemotherapy. Similarly to the non-neoplastic prostate cells, PCa cells need androgens to growth and survive, making the ADT an effective first-line therapy. This therapy can involve two approaches: surgical castration (i.e., orchiectomy) or, more commonly, chemical castration with drugs targeting AR signaling regulated by the hypothalamic pituitary gonadal axis (e.g., GnRH agonists, AR antagonists, and CYP17A1 inhibitors). This castration reduces tissue androgens levels and also reduce the expression of several androgen-regulated genes [34]. However, several adverse effects of ADT are known, such as decreased bone mineral density, metabolic changes, hot flashes, and sexual dysfunction [58]. Although most men show positive outcomes for 1 to 2 years with ADT, clinical progression occurs with the disease entering the stage of CRPC [36]. When PCa is considered castrate resistant different treatments options are needed, which includes chemotherapy [57]. This aggressive and lethal form of PCa progresses and metastasizes, not existing currently an effective therapy, being done only palliative care [59].
As the disease progresses to CRPC stage, treatment involves the use of chemotherapeutic drugs. Mitoxantrone was the first cytotoxic chemotherapy approved by FDA for metastatic PCa [60]. Next, other therapeutic agents for the treatment of CRPC were included, such as, the chemotherapeutic taxanes paclitaxel and docetaxel. After the discovery of the mechanism of action of paclitaxel, which is tubulin binding and enhanced microtubule polymerization resulting in mitotic arrest [61], other taxanes were explored and their synthetic and semisynthetic analogues with best properties and improved water solubility were produced [62]. The most successful semisynthetic analogue of paclitaxel is docetaxel, which is a taxane derivative that induces microtubules stabilization, arresting cells in the G2/M phase of the cell cycle, and it induces bcl-2 phosphorylation promoting a cascade of events that leads to apoptotic cell death (Figure 3) [63].
Some studies using docetaxel as a single agent or in combination with other drugs showed objective response rates in up to 38% of patients, PSA declines in more than 50% of patients with hormone refractory PCa, and increased overall survival in metastatic PCa patients in approximately 24 months [60,64,65]. However, both paclitaxel and docetaxel drugs have a high affinity for multidrug resistance proteins [66]. Cabazitaxel is a novel third-generation semisynthetic analogue of docetaxel, and it is a promising treatment for docetaxel-resistant CRPC [67]. Like paclitaxel and docetaxel, cabazitaxel binds to tubulin and promotes its assembly into microtubules, while simultaneously inhibiting disassembly. This leads to the stabilization of microtubules, which results in the interference of mitotic and interphase cellular functions. The cell is then unable to progress further into the cell cycle, being stalled at metaphase, thus triggering apoptosis of the cancer cell [62]. In the last years, several studies have shown cabazitaxel as more effective in improving the life-quality of metastatic CRPC patients. Cabazitaxel induced molecular changes in favor of killing PCa cells when compared with other taxanes [68], showing a reduction of 30% of PSA levels in PCa patients [69], and cabazitaxel markedly improved the prognostic outcomes of metastatic CRPC patients [69,70].
Multiple prospective randomized clinical trials have been designed to evaluate the efficacy and toxicity of therapies and diverse combinations have been attempted [71,72,73]. The CHAARTED (Chemohormonal Therapy versus Androgen Ablation Randomized Trial for Extensive Disease in PCa) and STAMPEDE (Systemic Therapy in Advancing or Metastatic PCa: Evaluation of Drug Efficacy) trials showed a remarkable overall survival benefit when combining ADT with docetaxel, as well as increased time to progression to castration resistant status [74,75]. In the FIRSTANA (Cabazitaxel Versus Docetaxel Both With Prednisone in Patients With Metastatic CRPC) trial, cabazitaxel showed no superiority versus docetaxel for overall survival of PCa patients as first-line treatment [76]. Although the docetaxel and cabazitaxel have similar efficacy, they have different safety profiles, favoring the lower dose tested of cabazitaxel [77]. However, the CARD trial showed that high dose of cabazitaxel significantly improved a number of clinical outcomes, comparatively with the androgen-signaling-targeted inhibitor (abiraterone or enzalutamide), in patients with metastatic CRPC who had been previously treated with docetaxel and the alternative androgen-signaling-targeted agent (abiraterone or enzalutamide) [78]. These results provide the evidence of a survival benefit with taxanes treatment in CRPC patients. Furthermore, patient preference studies have increased in significance in recent years for evidence-based medicine [79]. Therefore, the most recent clinical trial aimed to evaluate patient preference between docetaxel and cabazitaxel, the CABADOC trial [80]. This study showed a significantly higher proportion of chemotherapy-naïve men with metastatic CRPC who received both taxanes preferred cabazitaxel over docetaxel. Less fatigue and better quality of life were the two main reasons driving patient choice [80].
It is evident that the taxanes are constantly in upgrade both in terms of mechanistic and clinical aspects, and their success in treatment of PCa (castrate-sensitive and castrate-resistant settings) continued development of rational combination therapy strategies with the explicit goal to improve overall survival [73]. However, a persisting obstacle in taxanes administration is the ability of tumors to acquire resistance. This further opens the way for the exploration of new combinations to improve the efficacy and anticancer activity.
4. Transmembrane proteins as a potential therapeutic target in combination with taxanes
A transmembrane protein is a type of protein located either in the lipid bilayer of the plasma membrane or in the membrane of organelles [81]. Different from monotopic proteins, transmembrane proteins structure completely crosses the membrane [82]. Representing approximately 30% of the genome, transmembrane proteins are essential for many cellular processes [83]. These proteins are responsible for cell-cell and cell-environment communication, through signal transduction, the binding of receptors to hormones and neurotransmitters, and the transport of substances across the membrane [82,83]. There are two types of transmembrane proteins regarding their structure, they are either alpha-helical proteins or beta-barrel proteins. They can also be categorized according to the protein topology, referring to the position of the N- and C-terminal domains [81,82].
Several studies have shown a link between different transmembrane proteins and cancer, due their function related in cancer progression, metastasis, patient survival, and additionally, can also be used as therapeutic targets and/or biomarkers [81,82]. Studies supporting the potential for targeting transmembrane proteins in taxane drug resistance in PCa are summarized in Table 1.
4.1. MDR1
The efflux pump MDR1 (Multidrug Resistance Protein 1), also called p-glycoprotein, is a protein composed of 12 transmembrane domains and a single monomer of 170 kDa. This protein is part of the ABC transporter family and is encoded by the ABCB1 gene, located in the region 7q21 [84]. The overexpression of MDR1 is pointed out as partially responsible for drug resistance in PCa, due to higher drug efflux [85]. Regarding p-glycoprotein expression, Kawai et al. reported that both PCa and normal prostate epithelial cells are positive for the expression of the MDR1 gene. Using monoclonal antibodies to detect the presence of p-glycoprotein, the same study confirmed that this protein is asymmetrically expressed in the inner and outer zone of nonmalignant prostate glands. Moreover, the inner zone showed a higher level of protein expression [86].
To investigate whether the presence of p-glycoprotein in blood exosomes could be a marker to diagnose docetaxel resistance in PCa, Kato et al. tested the susceptibility to docetaxel and cabazitaxel drugs in parental and docetaxel-resistant PC3 cell lines considering p-glycoprotein expression. It was demonstrated that docetaxel-sensitive PC3 cells showed little or no expression of this protein, while docetaxel-resistant PC3 cells showed high expression of p-glycoprotein [87]. The knockdown of the ABCB1 gene was also performed in docetaxel-resistant PC3 cells. The results indicated an improvement in docetaxel sensitivity when compared with the negative control. These findings confirm the relationship between p-glycoprotein expression and docetaxel resistance [87]. Additionally, another study on PC3, after demonstrating that the ETS1 transcription factor had a role in regulating the expression of the MDR1 gene, it was assessed how the downregulation of ETS1 could impact cell sensitivity to paclitaxel. Results showed that the combined treatment of paclitaxel exposure and knockdown of ETS1 induce a decrease in cell viability in paclitaxel-resistant PC3 cell line, improving the resistance to paclitaxel [88]. A further study using the C4-2B cell line, it was tested the association between the MDR1 protein and the retinoic acid receptor-related orphan receptor g (RORg) [89]. First, they established that MDR1 is regulated upstream by RORg, since the knockdown of the retinoic receptor decreased the expression of MDR1, while ectopic RORg increased it. Also, both RORg antagonists, SR2211 and GSK805, have led to the inhibition of MDR1 expression in taxane-resistant C4-2B cells [89]. Next, this study demonstrated that the knockdown alone and the use alone of RORg antagonist have led to a significant decrease in cell viability and growth in both taxane-resistant and not resistant C4-2B cell lines [89]. Furthermore, the combination between a partial RORg and a low concentration of docetaxel (20 nmol/L) led to a reduction of cell growth from 96,1% in control to 72,2% in treated cells. Likewise, the use of 1.25 mmol/L SR2211 combined with 12.5 nmol/L docetaxel reduces the viability of taxane-resistant C4-2B to 33,2%, suggesting that the downregulation of RORg can sensitize taxane-resistant CRPC cells to taxane treatment [89].
4.2. MRP4
Similarly to the MDR1 protein, the MRP4 protein, also known as multidrug resistance protein 4, is part of the ATP-binding cassette (ABC) transporters family [90]. This transmembrane protein is present in almost all tissues in the body, such as brain, kidney, liver, erythrocytes, platelets, adrenal gland, and pancreas [91]. MRP4 is responsible for the transportation of prostaglandins E1 and E2 (PGE1 and PGE2) as well as cAMP and cGMP [92]. The MRP4 protein was reported as being highly overexpressed in docetaxel-resistant C4-2B cells, while no expression of MRP4 was detected in docetaxel-sensitive C4-2B cells [93]. To assess if the overexpression of MRP4 leads to docetaxel resistance, combined treatment of MRP4 knockdown plus docetaxel exposure were given to docetaxel-resistant C4-2B cell line. The results showed a diminished cell viability, indicating a resensitization to docetaxel treatment [93]. Furthermore, researchers assessed the hypothesis that androgens are responsible for MRP4 overexpression in docetaxel-resistant cells [93]. For this, C4-2B cells were exposed to DHT or bicalutamide followed by the quantification of MRP4 mRNA and protein levels [93]. After treatment with DHT, both mRNA and protein levels were increased, displaying a dose-dependent manner [93]. However, the exposure to bicalutamide prevented the upregulation of MRP4 [93]. This data shows that MRP4 can be upregulated by androgen and downregulated by anti-androgen treatment [93].
4.3. CD44
CD44 is a non-kinase cell surface transmembrane glycoprotein. This important hyaluronate receptor is overexpressed in cancer stem cells and is involved in cellular adhesion and communication, lymphopoiesis, myelopoiesis, and angiogenesis. In regard to cancer, CD44 is implicated in metastasis, cellular growth, proliferation, migration, and invasion [94]. There are several isoforms for the CD44 protein and some of them have been associated with PCa, namely the CD44s, CD44v6, and CD44v7-10 isoforms [94]. Furthermore, CD44 is also overexpressed in this type of cancer and is associated with aggressive biological behavior and a poor prognosis [94]. CD44 expression is upregulated by transforming growth factor-beta 1 (TGF-β1) in PCa cells [94]. CD44 is expressed in PC3 cells and was demonstrated that this receptor regulates glucose metabolism, intracellular reactive oxygen species (ROS), and cell proliferation in those cells; however, CD44 is not expressed in LNCaP cells [90]. Collected data also points to the regulation of proliferation, invasion, and migration via PDK1 and PFKFB4, which are enzymes that regulated glucose metabolism and are modulated by CD44 [95]. Li et al. reported that the use of docetaxel treatment combined with SB-3CT, a possible inhibitor of CD44 cleavage, decrease the viability of PC3 cells in comparison with the docetaxel-only treatment [95]. Researchers also assessed the combination index using CompuSyn software, showing that mild to moderate synergistic effects were observed for an SB-3CT concentration of 20μmol/L in combination with docetaxel [95]. Lai et al. also reported that docetaxel-resistant PC3 and DU145 cells have a higher migration and invasion rate than the parental cells [96]. In addition, when analyzed for the CD44+ population, both docetaxel-resistant cell lines showed higher numbers than the parental cells [96]. The knockdown of CD44 reduced cell migration in both docetaxel-resistant cell lines, while invasion has been suppressed only in docetaxel-resistant PC3 cells [96].
4.4. CD133
The pentaspan transmembrane glycoprotein CD133, also known as prominin-1, is a protein mostly found in the microvilli of different epithelial cells but is also expressed in numerous types of cancer such as breast, ovarian, and PCa and other non-epithelial cell types [97,98]. CD133 is frequently used as a biomarker for the detection of cancer stem cells [98]. The molecular function of this glycoprotein has not been yet fully clarified but there is strong evidence pointing towards a role in membrane organization, due to its preferred location on the microvilli, and a role in spermatozoa biogenesis and photoreceptor disc formation [97]. Regarding the photoreceptor disc formation, it is known that a mutation on the CD133 gene is the cause of a type of macular degeneration called Stargard disease [97]. CD133 is also important in angiogenesis through the regulation of expression of vascular endothelial growth factor (VEGF) [97]. Concerning the expression of CD133 in PCa cell lines, flow cytometric analysis performed by Wang et al., found that CD133+ cells were only present in the DU145 cell line, and indetectable in PC3 and LNCaP cell lines, when cultured in normal conditions [99]. However, when cultured in a serum-free medium, the PC3 cell line was able to present an increased proportion of CD133+ cells [99]. In LNCaP cells, the presence of CD133+ remained not observable. Nonetheless, Aghajani et al. evaluated the CD133 mRNA expression levels in the same PCa cell lines and discovered that CD133 is low expressed in all three cell lines, although with higher expression levels in the LNCaP cell line [100]. Additionally, Wang et al. assessed the possibility of enriching the proportion of CD133+ cells via chemotherapy, for which a docetaxel-containing medium was used in DU145 cell culture [99]. An increase of 9.8% in the proportion of CD133+ cells was observed after treatment, corroborating that those cells are chemo-resistant [99]. Through studying the knockdown alone of CD133 and in combination with paclitaxel, Aghajani et al. reported that, in LNCaP cells, the downregulation alone did not alter cell proliferation and viability when compared to the control group [100]. However, the combination with the paclitaxel treatment led to a decrease in survival rate compared to the LNCaP cells that were uniquely treated with paclitaxel. Regarding the migration and invasiveness, both knockdown of CD133 or paclitaxel alone treatment was able to reduce it, while the combination of treatments led to a synergistic decrease [100]. Also, the combination CD133-siRNA/paclitaxel significantly reduced the metastatic potential due to a lower expression of vimentin and MMP9 [100]. Finally, an apoptosis study using the LNCaP cells showed that the knockdown of CD133 may increase the sensitivity to paclitaxel [100].
4.5. SLCO1B3
Belonging to the Solute Carriers superfamily, SLCO1B3, also called organic anion transporting polypeptide (OATP) [101] is a sodium-independent transporter of both endogenous substrates such as bilirubin, bile salts, steroid conjugates, bromosulfophthalein (BSP), Taurocholate (TCA) [101,102] as well as exogenous substrates as antihistamines, blood-glucose-lowering drugs, statins, heart medications, and also docetaxel and paclitaxel [101,103].
Konig et al. confirmed that, under normal conditions, SLCO1B3 is exclusively expressed on hepatocytes, with its subcellular location on the basolateral plasma membrane of those cells [104]. Additionally, a preferred lobular zonation was also observed, where the hepatocytes near the central vein had a higher expression of this protein when compared to other locations within the liver [104]. Meanwhile, several studies have confirmed the abnormal expression of SLCO1B3 in tumorous tissue, including PCa [105]. Wright et al. demonstrated a significantly higher expression of the gene SLCO1B3 in CRPC metastases in comparison to untreated primary PCa [101]. In addition, a higher risk for PCa-specific mortality was connected to the SNP (Single nucleotide polymorphism) SLCO1B3 rs4149117 [106]. Moreover, SLCO1B3 mRNA levels was found in 62% of the prostate tumor samples, but no expression was detected in normal prostate [103]. The same study also indicated a clear positive association between the Gleason score and SLCO1B3 expression [103].
Regarding the effects of taxanes, a study evaluated patient-derived xenografts (PDXs) of PCa and discovered that docetaxel-resistant PDX tumors presented a significant downregulation of SLCO1B3. Along with this result, the PDXs presented reduced intratumorally docetaxel concentrations. To assess if the downregulation of SLCO1B3 was responsible for the low concentration of docetaxel, the silencing of SLCO1B3, as well as other docetaxel transporters, was performed. Only cells that were transfected with the SLCO1B3 siRNA presented a significant reduction in docetaxel uptake. To further investigate the role of SLCO1B3, SLCO1B3-negative PDXs were transfected with SLCO1B3 and later exposed to docetaxel and cabazitaxel. The outcome pointed toward a higher sensitivity to both taxanes drugs treatments among SLCO1B3-overexpressing cells [107].
4.6. EGFR
The transmembrane glycoproteins epidermal growth factor receptor (EGFR) together with HER-2/neu (erbB-2), HER-3 (erbB-3) and HER-4 (erbB-4), belongs to the HER (erbB) family of membrane receptors [108]. All these receptors are expressed in both normal and malignant cells, playing important roles in cell proliferation and differentiation [109]. All four family members have a very alike structure, consisting of three regions: an extracellular ligand-binding region, which, in the case of EGFR, is the binding region for the epidermal growth factor (EGF), transforming growth factor-a (TGF-a), amphiregulin (AR), Heparin-binding EGF-like growth factor (HB-EGF), and betacellulin (BTC) [108,109]. HER2 dimerizes with EGFR [110] and has no exclusive natural ligand [108]. The second region, a transmembrane domain, consisting of a single hydrophobic anchor sequence that crosses the cell membrane only once [109]. Lastly, acting as a binding site for intracellular substrates, and therefore, activating signaling pathways. The intracellular domain has tyrosine kinase activity [108]. Rossini et al. confirmed that DU145 and PC3 cell lines express the activated form of the EGFR and HER-2 receptors [111]. LNCaP and C4-2B cell lines also express EGFR, being higher in C4-2B cells [112]. In in vivo studies, EGFR was confirmed as overexpressed in both metastatic and CRCP, as well as moderately expressed in localized primary PCa. Furthermore, the assessment of EGFR expression on circulating tumor cells from the blood of patients with metastatic disease demonstrated that 90% of patients presented circulating tumor cells positive for EGFR [112]. Vicentini et al. studied the use of ZD1839, a selective EGFR tyrosine kinase inhibitor, in both androgen-sensitive cell lines (ND1, LNCaP and ALVA-31) as well as androgen-independent cell lines (PC3, DU145 and TSU-Pr1) [113]. First, it was reported by the authors that higher levels of EGFR and its ligands were present in the androgen-receptor-negative cell lines. However, ZD1839 treatment resulted in reduced cell proliferation in all cell lines tested [113]. Furthermore, a in vivo study assessed the tumor mass response to the blockade of EGFR and HER2 [111]. In order to do that, subcutaneous DU145 or PC-3 tumors were established on male mice, and tumor volume was quantified before, during, and after treatments [111]. It was demonstrated that Cetuximab and Trastuzumab (blockers of EGFR and HER2, respectively) in combination with docetaxel treatment induced a significant tumor regression when compared to the respective control group [111]. Furthermore, 80% of mice that were given the triple combination end up tumor-free. However, even though docetaxel alone, cetuximab alone, and in combination with Trastuzumab showed significant tumor growth inhibition, tumor regrowth was observed [111]. In another study, Monteverde et al. used the tyrosine kinase inhibitor Vandetanib to target EGFR in sensitive and docetaxel-resistant PC3 cell lines [114]. The study showed that the docetaxel-resistant PC3 cells present 3 times the amount of EGFR mRNA and 12 times the amount of EGFR protein when compared to sensitive PC3 cells. Additionally, the treatment with docetaxel alone produced an increase in the pEGFR/EFGR ratio, while the combination with Vandetanib had the opposite effect [114]. In docetaxel-resistant PC3 cells, no treatment altered the pEGFR/EFGR ratio. Regarding the effect in cell proliferation, a maximum of 90% inhibition was observed in response to docetaxel treatment alone in both sensitive and resistant PC3 cells. To attain this inhibition, it was necessary a 2 x 10-9 M concentration of docetaxel for the sensitive cell line and a 0.9x 10-7 M concentration for the resistant. Vandetanib alone also displayed inhibition effects but the strongest cytotoxic effect were observed when vandetanib was combined with low concentrations of docetaxel (0.061– 0.246 nM), for which the combination index value is 0.49–0,71. However, for resistant PC3 cells, there were different results regarding if the treatment was administrated in sequence (vandetanib followed by docetaxel) or together. In the first case, the combination index value of 0.55–0.90 indicated a synergetic effect, but for the treatment when given together a combination index of 1.22–1.73 was found, indicating a possible antagonism [114].
4.7. STEAP1
STEAP1, together with STEAP2-4, is part of the six-transmembrane epithelial antigen of prostate (STEAP) family of proteins [115]. The STEAP1 protein is overexpressed in several human cancers, including prostate, bladder, colon ovary, breast, and cervical cancer [116]. Although its function remains unclear, some studies have pointed out that STEAP1 is involved in metal reductase activity, and also in transport of ions such as Na+, Ca2+, and K+ [117]. STEAP1 is highly expressed in LNCaP cells and also at significant levels in C4-2B cell line [118]. Regarding the effect of STEAP1 knockdown in LNCaP cells, reduced cell viability was observed in comparison to the control group. This result was supported by the cell proliferation index, showing a 0.3-fold decrease in LNCaP cells knocked down for STEAP1. Besides the effect in inhibition of cell proliferation, the STEAP1 knockdown increased the number of apoptotic cells [119]. The same study also evaluated the behavior of LNCaP cells knocked down for STEAP1 in response to DHT, and the result was that the effect of STEAP1 gene silencing was not reversed after exposure to DHT [119]. Recently, another study reported the effect of paclitaxel, docetaxel and cabazitaxel on STEAP1 expression in LNCaP and C4-2B cells [118]. It was observed that paclitaxel or cabazitaxel treatment increased the STEAP1 protein expression when compared with the control group, but no differences were observed in C4-2B cells [118]. Furthermore, it was reported that STEAP1 knockdown alone decreased the cell viability in both cell lines, as well as all taxane-based treatments when administered alone. However, the combination between STEAP1 knockdown and exposure to taxane-based therapy led to an increase in cell viability/proliferation and diminished levels of apoptosis [118]. Altough more studies are required, these data suggest that the combination of taxane-based drugs with STEAP1 knockdown may lead to PCa progression.
5. Conclusions
Taxanes based-chemotherapeutic drugs are currently the main approach when it comes to PCa treatment. Even though this type of therapy has good results in improving patient survival, the development of resistance to chemotherapeutic drugs remains a great obstacle. In this review, we have covered state-of-the-art on the use of taxane-based therapy combined with targeting different transmembrane oncoproteins in PCa. The knockdown of transmembrane oncoproteins can improve, in some cases, taxane sensitivity, and therefore, might be a mechanism to improve the efficacy of taxanes drugs. However, it should be taken into account that some combinations may even trigger harmful effects, such as the knockdown of STEAP1. Besides the proteins described in this article, there are much more transmembrane oncoproteins whose specific role in PCa and association with taxane resistance requires further elucidation.
Despite some studies have been shown a promising use of taxane treatment in combination with inhibitors of transmembrane oncoproteins, additional studies are still needed to support a translation for clinical practice. Most of the scientific studies are focused in cell lines, which present several limitations. Therefore, it is required to perform studies using animal models in order to find good combinations to evaluate in clinical trials.
Funding
This research received no external funding.
Acknowledgments
In this section, you can acknowledge any support given which is not covered by the author contribution or funding sections. This may include administrative and technical support, or donations in kind (e.g., materials used for experiments).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018 Nov;68(6):394–424.
- Rawla, P. Epidemiology of Prostate Cancer. 2019.
- Oar, A.; Moraes, F.Y.; Romero, Y.; Ilbawi, A.; Yap, M.L. Core elements of national cancer control plans: a tool to support plan development and review. Lancet Oncol. 2019, 20, e645–e652. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.L. The Etiology of Prostate Cancer. Prostate Cancer. 2021 ;17–28. 27 May.
- Malik SS, MPhil, Batool R, Honors BS, Masood N, Yasmin A. Risk factors for prostate cancer: A multifactorial case-control study. Curr Probl Cancer 2018 May 1;42(3):337–43.
- Leitzmann, M.F.; Rohrmann, S. Risk factors for the onset of prostatic cancer: age, location, and behavioral correlates. Clin. Epidemiology 2012, 4, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Swami, U.; McFarland, T.R.; Nussenzveig, R.; Agarwal, N. Advanced Prostate Cancer: Treatment Advances and Future Directions. Trends Cancer 2020, 6, 702–715. [Google Scholar] [CrossRef] [PubMed]
- Corn, P.G.; Agarwal, N.; Araujo, J.C.; Sonpavde, G. Taxane-based Combination Therapies for Metastatic Prostate Cancer. Eur. Urol. Focus 2019, 5, 369–380. [Google Scholar] [CrossRef]
- Cournia Z, Allen TW, Andricioaei I, Antonny B, Baum D, Brannigan G, et al. Membrane Protein Structure, Function and Dynamics: A Perspective from Experiments and Theory. J Membr Biol. 2015 Aug; 248(4): 611–640.
- Overington, J.P.; Al-Lazikani, B.; Hopkins, A.L. How many drug targets are there? Nat. Rev. Drug. Discov. 2006;5, 993–996.
- Lee, C.H.; Akin-Olugbade, O.; Kirschenbaum, A. Overview of prostate anatomy, histology, and pathology. Endocrinol Metab Clin North Am. 2011 Sep;40(3):565–75.
- Ittmann, M. Anatomy and Histology of the Human and Murine Prostate. Cold Spring Harb. Perspect. Med. 2017, 8, a030346. [Google Scholar] [CrossRef]
- Leong, K.G.; Wang, B.-E.; Johnson, L.; Gao, W.-Q. Generation of a prostate from a single adult stem cell. Nature 2008, 456, 804–808. [Google Scholar] [CrossRef] [PubMed]
- Packer, J.R.; Maitland, N.J. The molecular and cellular origin of human prostate cancer. Biochim Biophys Acta. 2016 Jun 1;1863(6 Pt A):1238–60.
- Corn, P.G. The tumor microenvironment in prostate cancer: elucidating molecular pathways for therapy development. Cancer Manag. Res. 2012, 4, 183–193. [Google Scholar] [CrossRef]
- Levesque, C.; Nelson, P.S. Cellular Constituents of the Prostate Stroma: Key Contributors to Prostate Cancer Progression and Therapy Resistance. Cold Spring Harb. Perspect. Med. 2018, 8, a030510. [Google Scholar] [CrossRef]
- Yadav, N.; Heemers, H.V. Androgen action in the prostate gland. 2012, 64, 35–49.
- Long, R.M.; Morrissey, C.; Fitzpatrick, J.M.; Watson, R.W.G. Prostate epithelial cell differentiation and its relevance to the understanding of prostate cancer therapies. Clin. Sci. 2005, 108, 1–11. [Google Scholar] [CrossRef]
- Lawson, D.A.; Zong, Y.; Memarzadeh, S.; Xin, L.; Huang, J.; Witte, O.N. Basal epithelial stem cells are efficient targets for prostate cancer initiation. Proc. Natl. Acad. Sci. 2010, 107, 2610–2615. [Google Scholar] [CrossRef]
- Parimi, V.; Goyal, R.; Poropatich, K.; Yang, X.J. Neuroendocrine differentiation of prostate cancer: a review. . 2014, 2, 273–85. [Google Scholar]
- Wang, Z.A.; Mitrofanova, A.; Bergren, S.K.; Abate-Shen, C.; Cardiff, R.D.; Califano, A.; Shen, M.M. Lineage analysis of basal epithelial cells reveals their unexpected plasticity and supports a cell-of-origin model for prostate cancer heterogeneity. Nature 2013, 15, 274–283. [Google Scholar] [CrossRef]
- Wang, Z.A.; Toivanen, R.; Bergren, S.K.; Chambon, P.; Shen, M.M. Luminal Cells Are Favored as the Cell of Origin for Prostate Cancer. Cell Rep. 2014, 8, 1339–1346. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Kruithof-de Julio, M.; Economides, K.D.; Walker, D.; Yu, H.; Halili, M.V.; Hu, Y.-P.; Price, S.M.; Abate-Shen, C.; Shen, M.M. A luminal epithelial stem cell that is a cell of origin for prostate cancer. Nature 2009, 461, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.M.; Abate-Shen, C. Molecular genetics of prostate cancer: new prospects for old challenges. Minerva Anestesiol. 2010, 24, 1967–2000. [Google Scholar] [CrossRef] [PubMed]
- Ayala, A.G.; Ro, J.Y. Prostatic intraepithelial neoplasia: Recent advances. Arch Pathol Lab Med. 2007 Aug;131(8):1257–66.
- Joshua, A.; Evans, A.; Van der Kwast, T.; Zielenska, M.; Meeker, A.; Chinnaiyan, A.; Squire, J. Prostatic preneoplasia and beyond. Biochim. et Biophys. Acta (BBA) - Rev. Cancer 2008, 1785, 156–181. [Google Scholar] [CrossRef] [PubMed]
- Murray, T.B.J. The Pathogenesis of Prostate Cancer. Prostate Cancer. 2021 May 27;29–42.
- Alexander, E.E.; Qian, J.; Wollan, P.C.; Myers, R.P.; Bostwick, D.G. Prostatic intraepithelial neoplasia does not appear to raise serum prostate-specific antigen concentration. Urology 1996, 47, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Bostwick, D.G.; Brawer, M.K. Prostatic Intra-Epithelial Neoplasia and Early Invasion in Prostate Cancer. Cancer 1987, 59, 788–794. [Google Scholar] [CrossRef] [PubMed]
- Schiebler, M.L.; E Tomaszewski, J.; Bezzi, M.; Pollack, H.M.; Kressel, H.Y.; Cohen, E.K.; Altman, H.G.; Gefter, W.B.; Wein, A.J.; Axel, L. Prostatic carcinoma and benign prostatic hyperplasia: correlation of high-resolution MR and histopathologic findings. . 1989, 172, 131–137. [Google Scholar] [CrossRef]
- Schiebler, M.L.; Schnall, M.D.; Pollack, H.M.; E Lenkinski, R.; E Tomaszewski, J.; Wein, A.J.; Whittington, R.; Rauschning, W.; Kressel, H.Y. Current role of MR imaging in the staging of adenocarcinoma of the prostate. . 1993, 189, 339–352. [Google Scholar] [CrossRef]
- Ulmert, D.; O'Brien, M.F.; Bjartell, A.S.; Lilja, H. Prostate kallikrein markers in diagnosis, risk stratification and prognosis. Nat. Rev. Urol. 2009, 6, 384–391. [Google Scholar] [CrossRef]
- Duffy, M.J. The role of proteolytic enzymes in cancer invasion and metastasis. Clin. Exp. Metastasis 1992, 10, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhao, D.; Spring, D.J.; DePinho, R.A. Genetics and biology of prostate cancer. Minerva Anestesiol. 2018, 32, 1105–1140. [Google Scholar] [CrossRef] [PubMed]
- Tuxhorn, J.A.; Ayala, G.E.; Smith, M.J.; Smith, V.C.; Dang, T.D.; Rowley, D.R. Reactive stroma in human prostate cancer: induction of myofibroblast phenotype and extracellular matrix remodeling. Clin. Cancer Res. 2002, 8, 2912–2923. [Google Scholar]
- Shafi, A.A.; Yen, A.E.; Weigel, N.L. Androgen receptors in hormone-dependent and castration-resistant prostate cancer. Pharmacol. Ther. 2013, 140, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Culig, Z.; Klocker, H.; Bartsch, G.; Hobisch, A. Androgen receptors in prostate cancer. Endocr Relat Cancer. 2002 Sep;9(3):155–70.
- Knuuttila, M.; Mehmood, A.; Mäki-Jouppila, J.; Ryberg, H.; Taimen, P.; Knaapila, J.; Ettala, O.; Boström, P.J.; Ohlsson, C.; Venäläinen, M.S.; et al. Intratumoral androgen levels are linked to TMPRSS2-ERG fusion in prostate cancer. Endocrine-Related Cancer 2018, 25, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, T.R.; Strittmatter, B.G.; Hollenhorst, P.C. Oncogenic ETS Factors in Prostate Cancer. Adv Exp Med Biol. 2019; 1210:409–36.
- Wei, T.; Lu, J.; Ma, T.; Huang, H.; Kocher, J.-P.; Wang, L. Re-Evaluate Fusion Genes in Prostate Cancer. Cancer Informatics 2021, 20. [Google Scholar] [CrossRef] [PubMed]
- Davey, R.A.; Grossmann, M. Androgen Receptor Structure, Function and Biology: From Bench to Bedside. Clin. Biochem. Rev. 2016, 37, 3–15. [Google Scholar]
- Bevan, C.; Parker, M. The Role of Coactivators in Steroid Hormone Action. Exp. Cell Res. 1999, 253, 349–356. [Google Scholar] [CrossRef]
- Tan, M.H.E.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E.-L. Androgen receptor: structure, role in prostate cancer and drug discovery. Acta Pharmacol. Sin. 2015, 36, 3–23. [Google Scholar] [CrossRef]
- Saraon, P.; Drabovich, A.P.; Jarvi, K.A.; Diamandis, E.P. Mechanisms of Androgen-Independent Prostate Cancer. 2014, 25, 42–54.
- Devlin, H.-L.; Mudryj, M. Progression of prostate cancer: Multiple pathways to androgen independence. Cancer Lett. 2009, 274, 177–186. [Google Scholar] [CrossRef]
- Gottlieb, B.; Beitel, L.K.; Nadarajah, A.; Paliouras, M.; Trifiro, M. The androgen receptor gene mutations database: 2012 update. Hum Mutat. 2012 May;33(5):887–94.
- Beltran, H.; Yelensky, R.; Frampton, G.M.; Park, K.; Downing, S.R.; MacDonald, T.Y.; Jarosz, M.; Lipson, D.; Tagawa, S.T.; Nanus, D.M.; et al. Targeted Next-generation Sequencing of Advanced Prostate Cancer Identifies Potential Therapeutic Targets and Disease Heterogeneity. Eur. Urol. 2013, 63, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.C.; Li, Y.; Dehm, S.M. Androgen Receptor Splice Variants Activate Androgen Receptor Target Genes and Support Aberrant Prostate Cancer Cell Growth Independent of Canonical Androgen Receptor Nuclear Localization Signal. J. Biol. Chem. 2012, 287, 19736–19749. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Sprenger, C.C.; Vessella, R.L.; Haugk, K.; Soriano, K.; Mostaghel, E.A.; Page, S.T.; Coleman, I.M.; Nguyen, H.M.; Sun, H.; et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J. Clin. Investig. 2010, 120, 2715–2730. [Google Scholar] [CrossRef] [PubMed]
- Jenster, G. Ligand-independent activation of the androgen receptor in prostate cancer by growth factors and cytokines. J Pathol. 2000;191(3):227–8.
- Culig, Z.; Hobisch, A.; Cronauer, M.V.; Radmayr, C.; Trapman, J.; Hittmair, A.; Bartsch, G.; Klocker, H. Androgen receptor activation in prostatic tumor cell lines by insulin-like growth factor-I, keratinocyte growth factor, and epidermal growth factor. . 1994, 54, 5474–8. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.-L.; Kyprianou, N. Androgen receptor and growth factor signaling cross-talk in prostate cancer cells. Endocrine-Related Cancer 2008, 15, 841–849. [Google Scholar] [CrossRef]
- Shorning BY, Dass MS, Smalley MJ, HB, Pearson. The PI3K-AKT-mTOR Pathway and Prostate Cancer: At the Crossroads of AR, MAPK, and WNT Signaling. Int J Mol Sci. 2020 Jun 2;21(12):1–47.
- Martini, M.; De Santis, M.C.; Braccini, L.; Gulluni, F.; Hirsch, E. PI3K/AKT signaling pathway and cancer: an updated review. Ann. Med. 2014, 46, 372–383. [Google Scholar] [CrossRef]
- Labbé, D.P.; Brown, M. Transcriptional Regulation in Prostate Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a030437. [Google Scholar] [CrossRef]
- Schatten, H. Brief Overview of Prostate Cancer Statistics, Grading, Diagnosis and Treatment Strategies. Adv. Exp. Med. Biol. 2018, 1095, 1–14. [Google Scholar] [CrossRef]
- Litwin, M.S.; Tan, H.J. The diagnosis and treatment of prostate cancer: A review. JAMA - J Am Med Assoc. 2017;317(24):2532–42.
- Liao, C.H.; Li, H.Y.; Chung, S.D.; Chiang, H.S.; Yu, H.J. Significant association between serum dihydrotestosterone level and prostate volume among Taiwanese men aged 40–79 years. Aging Male 2012, 15, 28–33. [Google Scholar] [CrossRef]
- Catt, S.; Matthews, L.; May, S.; Payne, H.; Mason, M.; et al, P.a.t.i.e.n.t.s.; et al. ’.; partners’ views of care treatment provided for metastatic castrate-resistant prostate cancer in the, U.K. Eur J Cancer Care (Engl). 2019 Nov 1;28(6).
- Tannock, I.F.; Osoba, D.; Stockler, M.R.; Ernst, D.S.; Neville, A.J.; Moore, M.J.; Armitage, G.R.; Wilson, J.J.; Venner, P.M.; Coppin, C.M.; et al. Chemotherapy with mitoxantrone plus prednisone or prednisone alone for symptomatic hormone-resistant prostate cancer: a Canadian randomized trial with palliative end points. J. Clin. Oncol. 1996, 14, 1756–1764. [Google Scholar] [CrossRef]
- Long, H.J. Paclitaxel (Taxol): A Novel Anticancer Chemotherapeutic Drug. 69. [CrossRef]
- kubník J, Pavlíčková V, Ruml T, Rimpelová S. Current Perspectives on Taxanes: Focus on Their Bioactivity, Delivery and Combination Therapy. Plants. 2021 Mar 1;10(3):1–35.
- Pienta, K.J. Preclinical mechanisms of action of docetaxel and docetaxel combinations in prostate cancer. Semin Oncol. 2001 Aug;28(4 Suppl 15):3–7.
- Roumiguié, M.; Paoletti, X.; Neuzillet, Y.; Mathieu, R.; Vincendeau, S.; Kleinclauss, F.; Mejean, A.; Guy, L.; Timsit, M.O.; Lebret, T. Apalutamide, darolutamide and enzalutamide in nonmetastatic castration-resistant prostate cancer: a meta-analysis. Futur. Oncol. 2021, 17, 1811–1823. [Google Scholar] [CrossRef] [PubMed]
- Rathkopf DE, Morris MJ, Fox JJ, Danila DC, Slovin SF, et al. Phase I study of ARN-509, a novel antiandrogen, in the treatment of castration-resistant prostate cancer. J Clin Oncol. 2013 Oct 1;31(28):3525–30.
- Madan, R.A.; Pal, S.K.; Sartor, O.; Dahut, W.L. Overcoming Chemotherapy Resistance in Prostate Cancer. Clin. Cancer Res. 2011, 17, 3892–3902. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Kawakami, A.; Sato, R.; Watanabe, K.; Matsushita, Y.; Miyake, H. Molecular Mechanism Mediating Cytotoxic Activity of Cabazitaxel in Docetaxel-resistant Human Prostate Cancer Cells. Anticancer. Res. 2021, 41, 3753–3758. [Google Scholar] [CrossRef] [PubMed]
- Cevik, O.; Acidereli, H.; Turut, F.A.; Yildirim, S.; Acilan, C. Cabazitaxel exhibits more favorable molecular changes compared to other taxanes in androgen-independent prostate cancer cells. J. Biochem. Mol. Toxicol. 2020, 34, e22542. [Google Scholar] [CrossRef] [PubMed]
- Takai, M.; Kato, S.; Nakano, M.; Fujimoto, S.; Iinuma, K.; Ishida, T.; Taniguchi, M.; Tamaki, M.; Uno, M.; Takahashi, Y.; et al. Efficacy of cabazitaxel and the influence of clinical factors on the overall survival of patients with castration-resistant prostate cancer: A local experience of a multicenter retrospective study. Asia-Pacific J. Clin. Oncol. 2021, 17, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Miyake, H.; Sato, R.; Watanabe, K.; Matsushita, Y.; Watanabe, H.; Motoyama, D.; Ito, T.; Sugiyama, T.; Otsuka, A. Prognostic significance of third-line treatment for patients with metastatic castration-resistant prostate cancer: comparative assessments between cabazitaxel and other agents. Int. J. Clin. Oncol. 2021, 26, 1745–1751. [Google Scholar] [CrossRef] [PubMed]
- Van Soest, R.J.; De Wit, R. Irrefutable evidence for the use of docetaxel in newly diagnosed metastatic prostate cancer: results from the STAMPEDE and CHAARTED trials. BMC Med. 2015, 13, 1–3. [Google Scholar] [CrossRef]
- Damodaran, S.; Lang, J.M.; Jarrard, D.F. Targeting Metastatic Hormone Sensitive Prostate Cancer: Chemohormonal Therapy and New Combinatorial Approaches. J. Urol. 2019, 201, 876–885. [Google Scholar] [CrossRef]
- Huebner, N.A.; Shariat, S.F.; Resch, I.; Gust, K.; Kramer, G. The role of taxane-based chemotherapy in the treatment of prostate cancer. Curr. Opin. Urol. 2020, 30, 527–533. [Google Scholar] [CrossRef]
- James, N.D.; Sydes, M.R.; Clarke, N.W.; Mason, M.D.; Dearnaley, D.P.; Spears, M.R.; Ritchie, A.W.S.; Parker, C.C.; Russell, J.M.; Attard, G.; et al. Addition of docetaxel, zoledronic acid, or both to first-line long-term hormone therapy in prostate cancer (STAMPEDE): Survival results from an adaptive, multiarm, multistage, platform randomised controlled trial. Lancet 2016, 387, 1163–1177. [Google Scholar] [CrossRef] [PubMed]
- Clarke NW, Ali A, Ingleby FC, Hoyle A, Amos CL, et al. Addition of docetaxel to hormonal therapy in low- and high-burden metastatic hormone sensitive prostate cancer: long-term survival results from the STAMPEDE trial. Ann Oncol Off J Eur Soc Med Oncol. 2019 Dec 1;30(12):1992–2003.
- Oudard, S.; Fizazi, K.; Sengeløv, L.; Daugaard, G.; Saad, F.; Hansen, S.; Hjälm-Eriksson, M.; Jassem, J.; Thiery-Vuillemin, A.; Caffo, O.; et al. Cabazitaxel Versus Docetaxel As First-Line Therapy for Patients With Metastatic Castration-Resistant Prostate Cancer: A Randomized Phase III Trial—FIRSTANA. J. Clin. Oncol. 2017, 35, 3189–3197. [Google Scholar] [CrossRef] [PubMed]
- Eisenberger, M.; Hardy-Bessard, A.-C.; Kim, C.S.; Géczi, L.; Ford, D.; Mourey, L.; Carles, J.; Parente, P.; Font, A.; Kacso, G.; et al. Phase III Study Comparing a Reduced Dose of Cabazitaxel (20 mg/m2) and the Currently Approved Dose (25 mg/m2) in Postdocetaxel Patients With Metastatic Castration-Resistant Prostate Cancer—PROSELICA. J. Clin. Oncol. 2017, 35, 3198–3206. [Google Scholar] [CrossRef] [PubMed]
- de Wit, R.; de Bono, J.; Sternberg, C.N.; Fizazi, K.; Tombal, B.; Wülfing, C.; Kramer, G.; Eymard, J.-C.; Bamias, A.; Carles, J.; et al. Cabazitaxel versus Abiraterone or Enzalutamide in Metastatic Prostate Cancer. New Engl. J. Med. 2019, 381, 2506–2518. [Google Scholar] [CrossRef]
- Menges, D.; Piatti, M.C.; Cerny, T.; A Puhan, M. Patient Preference Studies for Advanced Prostate Cancer Treatment Along the Medical Product Life Cycle: Systematic Literature Review. Patient Preference Adherence 2022, ume 16, 1539–1557. [Google Scholar] [CrossRef]
- Baciarello, G.; Delva, R.; Gravis, G.; Tazi, Y.; Beuzeboc, P.; Gross-Goupil, M.; Bompas, E.; Joly, F.; Greilsamer, C.; Hon, T.N.T.; et al. Patient Preference Between Cabazitaxel and Docetaxel for First-line Chemotherapy in Metastatic Castration-resistant Prostate Cancer: The CABADOC Trial. Eur. Urol. 2021, 81, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Schmit, K.; Michiels, C. TMEM Proteins in Cancer: A Review. Front. Pharmacol. 2018, 9, 1345. [Google Scholar] [CrossRef]
- Ryu, H.; Fuwad, A.; Yoon, S.; Jang, H.; Lee, J.C.; Kim, S.M.; Jeon, T.-J. Biomimetic Membranes with Transmembrane Proteins: State-of-the-Art in Transmembrane Protein Applications. Int. J. Mol. Sci. 2019, 20, 1437. [Google Scholar] [CrossRef]
- Marx, S.; Maso, T.D.; Chen, J.-W.; Bury, M.; Wouters, J.; Michiels, C.; Le Calvé, B. Transmembrane (TMEM) protein family members: Poorly characterized even if essential for the metastatic process. Semin. Cancer Biol. 2020, 60, 96–106. [Google Scholar] [CrossRef]
- Bossennec M, Di Roio A, Caux C, Ménétrier-Caux C. MDR1 in immunity: friend or foe? Oncoimmunology. 2018 Dec 2;7(12).
- Ganju, A.; Yallapu, M.M.; Khan, S.; Behrman, S.W.; Chauhan, S.C.; Jaggi, M. Nanoways to overcome docetaxel resistance in prostate cancer. Drug Resist. Updat. 2014, 17, 13–23. [Google Scholar] [CrossRef]
- Kawai, K.; Sakurai, M.; Sakai, T.; Misaki, M.; Kusano, I.; Shiraishi, T.; Yatani, R. Demonstration of MDR1 P-glycoprotein isoform expression in benign and malignant human prostate cells by isoform-specific monoclonal antibodies. Cancer Lett. 2000, 150, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Mizutani, K.; Kameyama, K.; Kawakami, K.; Fujita, Y.; Nakane, K.; Kanimoto, Y.; Ehara, H.; Ito, H.; Seishima, M.; et al. Serum exosomal P-glycoprotein is a potential marker to diagnose docetaxel resistance and select a taxoid for patients with prostate cancer. Urol. Oncol. Semin. Orig. Investig. 2015, 33, 385–e15. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Fujita, Y.; Nakane, K.; Kojima, T.; Nozawa, Y.; Deguchi, T.; Ito, M. ETS1 promotes chemoresistance and invasion of paclitaxel-resistant, hormone-refractory PC3 prostate cancer cells by up-regulating MDR1 and MMP9 expression. Biochem. Biophys. Res. Commun. 2012, 417, 966–971. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, Z.; Chen, C.Z.; Liu, C.; Evans, C.P.; Gao, A.C.; Zhou, F.; Chen, H.-W. Therapeutic Targeting of MDR1 Expression by RORγ Antagonists Resensitizes Cross-Resistant CRPC to Taxane via Coordinated Induction of Cell Death Programs. Mol. Cancer Ther. 2020, 19, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Hardy, D.; Bill, R.M.; Jawhari, A.; Rothnie, A.J. Functional Expression of Multidrug Resistance Protein 4 MRP4/ABCC4. SLAS Discov. 2019 Dec;24(10):1000-1008.
- Ravna, A.W.; Sager, G. Molecular model of the outward facing state of the human multidrug resistance protein 4 (MRP4/ABCC4). Bioorg Med Chem Lett. 2008 Jun 15;18(12):3481-3.
- Sauna, Z.E.; Nandigama, K.; Ambudkar, S.V. Multidrug resistance protein 4 (ABCC4)-mediated ATP hydrolysis: effect of transport substrates and characterization of the post-hydrolysis transition state. J Biol Chem. 2004 Nov 19;279(47):48855-64.
- Li, Y.-F.; Ji, H.-H.; Zhang, Z.-L.; Zhang, T.-T.; Gan, W.; Zhang, S.-F. Targeting MRP4 expression by anti-androgen treatment reverses MRP4-mediated docetaxel resistance in castration-resistant prostate cancer. Oncol. Lett. 2017, 14, 1748–1756. [Google Scholar] [CrossRef] [PubMed]
- Mesrati, M.H.; Syafruddin, S.E.; Mohtar, M.A.; Syahir, A. CD44: A Multifunctional Mediator of Cancer Progression. Biomolecules 2021, 11, 1850. [Google Scholar] [CrossRef]
- Li, W.; Qian, L.; Lin, J.; Huang, G.; Hao, N.; Wei, X.; Wang, W.; Liang, J. CD44 regulates prostate cancer proliferation, invasion and migration via PDK1 and PFKFB4. Oncotarget 2017, 8, 65143–65151. [Google Scholar] [CrossRef]
- Lai, C.-J.; Lin, C.-Y.; Liao, W.-Y.; Hour, T.-C.; Wang, H.-D.; Chuu, C.-P. CD44 Promotes Migration and Invasion of Docetaxel-Resistant Prostate Cancer Cells Likely via Induction of Hippo-Yap Signaling. Cells 2019, 8, 295. [Google Scholar] [CrossRef]
- Behrooz, A.B.; Syahir, A.; Ahmad, S. CD133: beyond a cancer stem cell biomarker. J. Drug Target. 2019, 27, 257–269. [Google Scholar] [CrossRef]
- Glumac, P.M.; LeBeau, A.M. The role of CD133 in cancer: a concise review. Clin. Transl. Med. 2018, 7, 18. [Google Scholar] [CrossRef]
- Wang, L.; Huang, X.; Zheng, X.; Wang, X.; Li, S.; Zhang, L.; Yang, Z.; Xia, Z. Enrichment of Prostate Cancer Stem-Like Cells from Human Prostate Cancer Cell Lines by Culture in Serum-Free Medium and Chemoradiotherapy. Int. J. Biol. Sci. 2013, 9, 472–479. [Google Scholar] [CrossRef]
- Aghajani, M.; Mokhtarzadeh, A.; Aghebati-Maleki, L.; Mansoori, B.; Mohammadi, A.; Safaei, S.; Asadzadeh, Z.; Hajiasgharzadeh, K.; Shahgoli, V.K.; Baradaran, B. CD133 suppression increases the sensitivity of prostate cancer cells to paclitaxel. Mol. Biol. Rep. 2020, 47, 3691–3703. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Ying, Y.; Tang, Z.; Liu, T.; Shi, F.; Li, H.; Guo, T.; Huang, S.; Lai, R. The Emerging Role of the SLCO1B3 Protein in Cancer Resistance. Protein Pept. Lett. 2020, 27, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Peng, T.; Wang, Z.; Li, Y.; Zhang, H.; Gui, C. Effect of rare coding variants of charged amino acid residues on the function of human organic anion transporting polypeptide 1B3 (SLCO1B3). Biochem. Biophys. Res. Commun. 2021, 557, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Pressler, H.; Sissung, T.M.; Venzon, D.; Price, D.K.; Figg, W.D. Expression of OATP Family Members in Hormone-Related Cancers: Potential Markers of Progression. PLOS ONE 2011, 6, e20372. [Google Scholar] [CrossRef] [PubMed]
- König, J.; Cui, Y.; Nies, A.T.; Keppler, D. Localization and Genomic Organization of a New Hepatocellular Organic Anion Transporting Polypeptide. J. Biol. Chem. 2000, 275, 23161–23168. [Google Scholar] [CrossRef]
- Thakkar, N.; Kim, K.; Jang, E.R.; Han, S.; Kim, K.; Kim, D.; Merchant, N.; Lockhart, A.C.; Lee, W. A Cancer-Specific Variant of the SLCO1B3 Gene Encodes a Novel Human Organic Anion Transporting Polypeptide 1B3 (OATP1B3) Localized Mainly in the Cytoplasm of Colon and Pancreatic Cancer Cells. Mol. Pharm. 2013, 10, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Wright JL, Kwon EM, Ostrander EA, Montgomery RB, Lin DW, et al. Expression of SLCO transport genes in castration resistant prostate cancer and impact of genetic variation in SCLO1B3 and SLCO2B1 on prostate cancer outcomes. Cancer Epidemiol Biomarkers Prev. 2011 Apr; 20(4): 619–627.
- de Morrée, E.S.; Böttcher, R.; van Soest, R.J.; Aghai, A.; de Ridder, C.M.; A Gibson, A.; Mathijssen, R.H.; Burger, H.; Wiemer, E.A.; Sparreboom, A.; et al. Loss of SLCO1B3 drives taxane resistance in prostate cancer. Br. J. Cancer 2016, 115, 674–681. [Google Scholar] [CrossRef]
- Bellezza, I.; Bracarda, S.; Caserta, C.; Minelli, A. Targeting of EGFR tyrosine kinase by ZD1839 (“Iressa”) in androgen-responsive prostate cancer in vitro. Mol. Genet. Metab. 2006, 88, 114–122. [Google Scholar] [CrossRef]
- Rude Voldborg, B.; Damstrup, L.; Spang-Thomsen, M.; Skovgaard Poulsen, H. Epidermal growth factor receptor (EGFR) and EGFR mutations, function and possible role in clinical trials. Ann Oncol. 1997 Dec 1;8(12):1197–206.
- Jathal, M.K.; Steele, T.M.; Siddiqui, S.; Mooso, B.A.; D’abronzo, L.S.; Drake, C.M.; Whang, Y.E.; Ghosh, P.M. Dacomitinib, but not lapatinib, suppressed progression in castration-resistant prostate cancer models by preventing HER2 increase. Br. J. Cancer 2019, 121, 237–248. [Google Scholar] [CrossRef]
- Rossini, A.; Giussani, M.; Ripamonti, F.; Aiello, P.; Regondi, V.; Balsari, A.; Triulzi, T.; Tagliabue, E. Combined targeting of EGFR and HER2 against prostate cancer stem cells. Cancer Biol. Ther. 2020, 21, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Day KC, Hiles GL, Kozminsky M, Dawsey SJ, Paul A, et al. HER2 and EGFR Overexpression Support Metastatic Progression of Prostate Cancer to Bone. Cancer Res. 2017 Jan 1;77(1):74–85.
- Vicentini, C.; Festuccia, C.; Gravina, G.L.; Angelucci, A.; Marronaro, A.; Bologna, M. Prostate cancer cell proliferation is strongly reduced by the epidermal growth factor receptor tyrosine kinase inhibitor ZD1839 in vitro on human cell lines and primary cultures. J. Cancer Res. Clin. Oncol. 2003, 129, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Monteverde, M.; Tonissi, F.; Fischel, J.-L.; Etienne-Grimaldi, M.-C.; Milano, G.; Merlano, M.; Nigro, C.L. Combination of docetaxel and vandetanib in docetaxel-sensitive or resistant PC3 cell line. Urol. Oncol. Semin. Orig. Investig. 2013, 31, 776–786. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Evans, L.; Bizzaro, C.L.; Quaglia, F.; Verrillo, C.E.; Li, L.; Stieglmaier, J.; Schiewer, M.J.; Languino, L.R.; Kelly, W.K. STEAP1–4 (Six-Transmembrane Epithelial Antigen of the Prostate 1–4) and Their Clinical Implications for Prostate Cancer. Cancers 2022, 14, 4034. [Google Scholar] [CrossRef] [PubMed]
- Gomes IM, Maia CJ, Santos CR. STEAP proteins: From structure to applications in cancer therapy Mol Cancer Res. 2012 May;10(5):573-87.
- Zhao, C.; Xiong, K.; Ji, Z.; Liu, F.; Li, X. The Prognostic Value and Immunological Role of STEAP1 in Pan-Cancer: A Result of Data-Based Analysis. Oxidative Med. Cell. Longev. 2022, 2022, 1–28. [Google Scholar] [CrossRef]
- Rocha, S.M.; Nascimento, D.; Coelho, R.S.; Cardoso, A.M.; Passarinha, L.A.; Socorro, S.; Maia, C.J. STEAP1 Knockdown Decreases the Sensitivity of Prostate Cancer Cells to Paclitaxel, Docetaxel and Cabazitaxel. Int. J. Mol. Sci. 2023, 24, 6643. [Google Scholar] [CrossRef]
- Gomes, I.M.; Rocha, S.M.; Gaspar, C.; Alvelos, M.I.; Santos, C.R.; Socorro, S.; Maia, C.J. Knockdown of STEAP1 inhibits cell growth and induces apoptosis in LNCaP prostate cancer cells counteracting the effect of androgens. Med Oncol. 2018, 35, 1–10. [Google Scholar] [CrossRef]
Figure 1.
Schematic representation of the proposed model of the cellular events associated with the development and progression of PCa. Prostate epithelium is composed by the luminal cells responsible for the production of prostatic secretions, basal cells that are on the base of epithelium in contact with the basement membrane. Located among the epithelial cells also exist neuroendocrine cells that are involved in the regulation of secretory activity and prostate cell growth. Prostate epithelial cells maintain contact with the stroma, including smooth muscle cells, fibroblast cells and components of the extracellular matrix (ECM). Damage in the prostate normal epithelium induces the development of pre-neoplastic lesions called prostatic intraepithelial neoplasia (PIN). This stage progresses to localized prostate adenocarcinoma where the basal cell layer is lost, which then becomes invasive adenocarcinoma when the basement membrane is degraded, and neoplastic cells can invade to lymphatic system and other organs including liver, lungs and bones.
Figure 1.
Schematic representation of the proposed model of the cellular events associated with the development and progression of PCa. Prostate epithelium is composed by the luminal cells responsible for the production of prostatic secretions, basal cells that are on the base of epithelium in contact with the basement membrane. Located among the epithelial cells also exist neuroendocrine cells that are involved in the regulation of secretory activity and prostate cell growth. Prostate epithelial cells maintain contact with the stroma, including smooth muscle cells, fibroblast cells and components of the extracellular matrix (ECM). Damage in the prostate normal epithelium induces the development of pre-neoplastic lesions called prostatic intraepithelial neoplasia (PIN). This stage progresses to localized prostate adenocarcinoma where the basal cell layer is lost, which then becomes invasive adenocarcinoma when the basement membrane is degraded, and neoplastic cells can invade to lymphatic system and other organs including liver, lungs and bones.
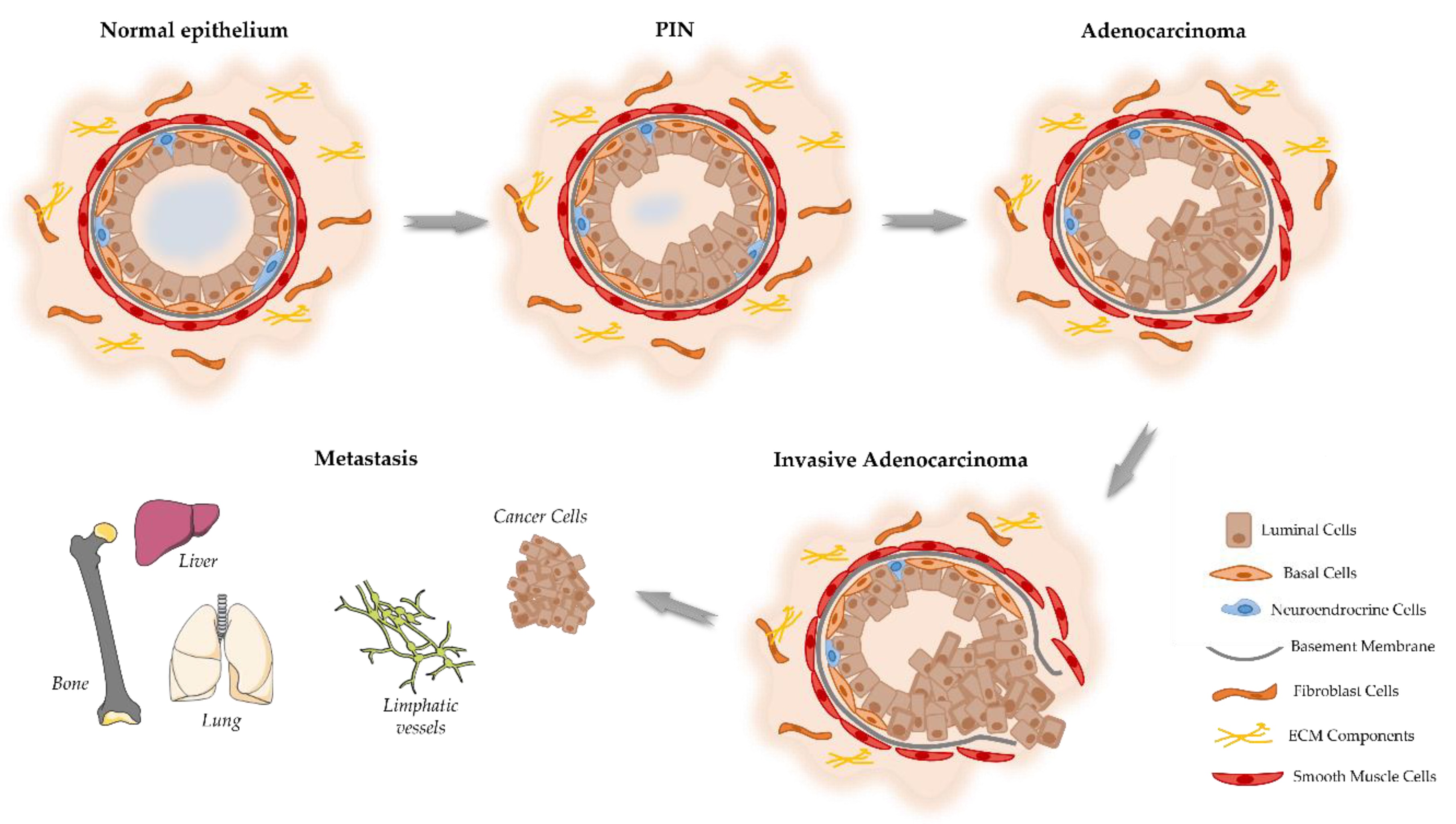
Figure 2.
Overview of the molecular pathways associated with the development of CRPC. In the cytoplasm, activity of AR is regulated by ligand-binding and heat shock proteins (HSP). Testosterone is transported into the cytoplasm of androgen-receptive cells and is converted to 5α-dihydrotestosterone (DHT) by the enzyme 5α-reductase. DHT binding leads to dissociation of AR from HSP and its phosphorylation by the mitogen-activated protein kinase (MAPK), which is followed by receptor dimerization and translocation into the nucleus where it binds to the androgen response elements (AREs) in the DNA activating transcription of genes essential for cell growth, survival and proliferation. On the other hand, PCa cell fate is controlled by receptor tyrosine kinases (RTK) activated by several growth factors, such as insulin-like growth factor (IGF1), fibroblast growth factor (FGF) and epidermal growth factor (EGF). RTK activation leads to the stimulation of phosphatidylinositol 3-kinase (PI3K) that phosphorylates phosphatidylinositol 4,5-bisphosphonate (PIP2) into phosphatidylinositol 3–5-triphosphate (PIP3). This process is inhibited by the tumor suppressor phosphatase and tensin homolog (PTEN). PIP3 activates, which subsequently removes the inhibition on the mTOR/Raptor complex (also known as mTORC1), thus leading to mTORC1 activation. mTORC1 is pivotal in the translation of proteins for protein synthesis and activation of transcription factors that translocate to the nucleus inducing the expression of pro-proliferation and anti-apoptotic genes. Other intracellular pathways also converge on the mTORC1 complex is constituted by the Ras-dependent pathway. Activated Ras (a small GTPase) phosphorylates and activates the mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) cascade, regulating the activity of several transcription factors that are important for the cell cycle and proliferation. The activation of these signaling pathways inhibits apoptosis and induce the proliferation, invasion, and migration of PCa cells, being also implicated in tumor metastization.
Figure 2.
Overview of the molecular pathways associated with the development of CRPC. In the cytoplasm, activity of AR is regulated by ligand-binding and heat shock proteins (HSP). Testosterone is transported into the cytoplasm of androgen-receptive cells and is converted to 5α-dihydrotestosterone (DHT) by the enzyme 5α-reductase. DHT binding leads to dissociation of AR from HSP and its phosphorylation by the mitogen-activated protein kinase (MAPK), which is followed by receptor dimerization and translocation into the nucleus where it binds to the androgen response elements (AREs) in the DNA activating transcription of genes essential for cell growth, survival and proliferation. On the other hand, PCa cell fate is controlled by receptor tyrosine kinases (RTK) activated by several growth factors, such as insulin-like growth factor (IGF1), fibroblast growth factor (FGF) and epidermal growth factor (EGF). RTK activation leads to the stimulation of phosphatidylinositol 3-kinase (PI3K) that phosphorylates phosphatidylinositol 4,5-bisphosphonate (PIP2) into phosphatidylinositol 3–5-triphosphate (PIP3). This process is inhibited by the tumor suppressor phosphatase and tensin homolog (PTEN). PIP3 activates, which subsequently removes the inhibition on the mTOR/Raptor complex (also known as mTORC1), thus leading to mTORC1 activation. mTORC1 is pivotal in the translation of proteins for protein synthesis and activation of transcription factors that translocate to the nucleus inducing the expression of pro-proliferation and anti-apoptotic genes. Other intracellular pathways also converge on the mTORC1 complex is constituted by the Ras-dependent pathway. Activated Ras (a small GTPase) phosphorylates and activates the mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) cascade, regulating the activity of several transcription factors that are important for the cell cycle and proliferation. The activation of these signaling pathways inhibits apoptosis and induce the proliferation, invasion, and migration of PCa cells, being also implicated in tumor metastization.
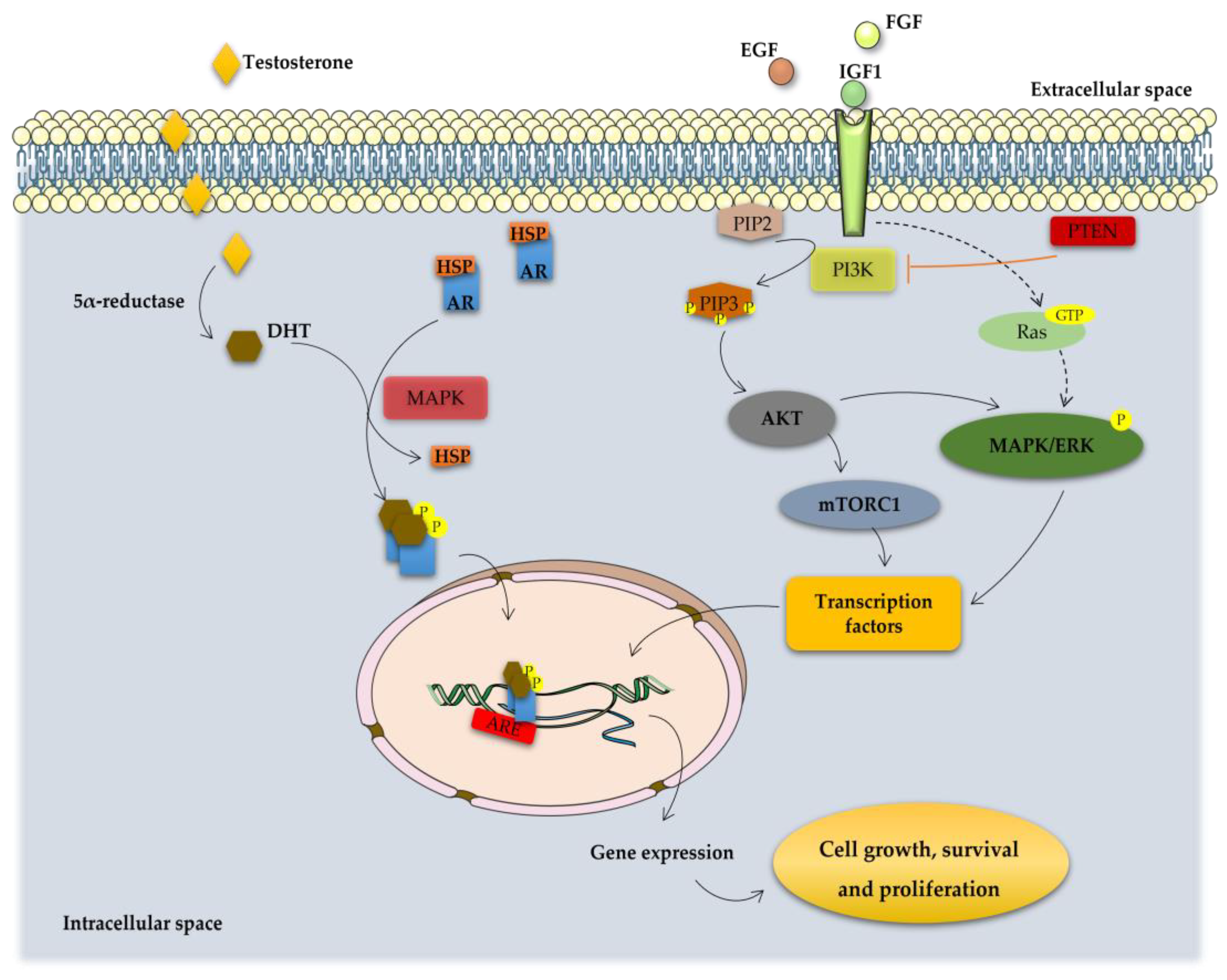
Figure 3.
Schematic representation of mode of action of taxanes on cancer cell. Taxanes have been described to exert their antitumor efficacy via distinct modes of action: mitotic and apoptotic action. Taxanes bind to microtubules and thereby prevent their disassembly, resulting in G2/M cell cycle arrest and apoptosis. Alternatively, taxanes may inhibit the expression of antiapoptotic Bcl-2, favoring apoptotic cell death through the relief of BAX-mediated cytochrome c release.
Figure 3.
Schematic representation of mode of action of taxanes on cancer cell. Taxanes have been described to exert their antitumor efficacy via distinct modes of action: mitotic and apoptotic action. Taxanes bind to microtubules and thereby prevent their disassembly, resulting in G2/M cell cycle arrest and apoptosis. Alternatively, taxanes may inhibit the expression of antiapoptotic Bcl-2, favoring apoptotic cell death through the relief of BAX-mediated cytochrome c release.
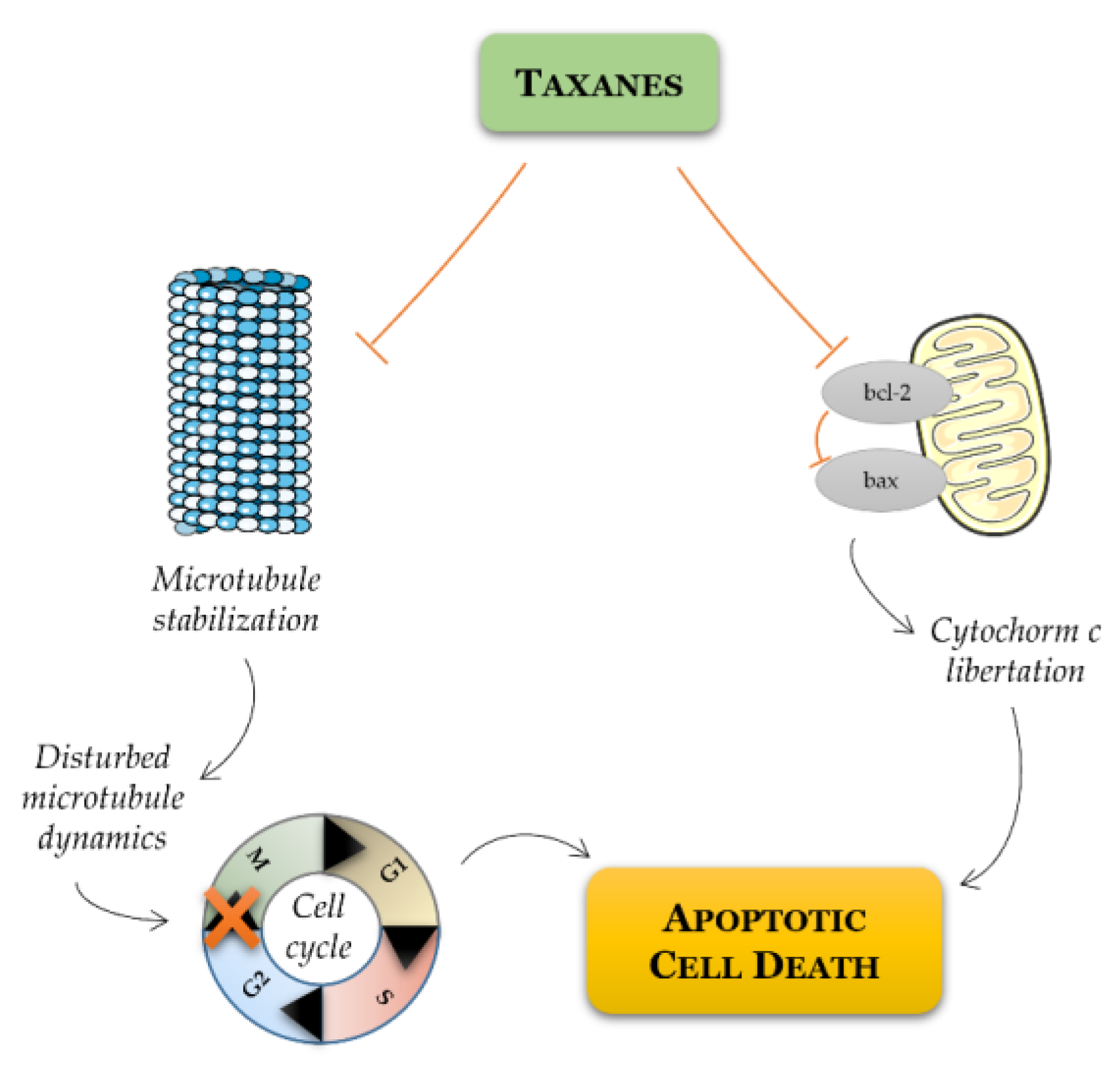

Table 1.
Identification of transmembrane proteins with combined effect of taxanes in PCa.
| Protein | Function | Effect of knockdown alone | Effect of knockdown + taxane treatment |
|---|---|---|---|
| MDR1 | Efflux pump | - | Improvement in docetaxel sensitivity |
| MRP4 | Efflux pump | - | Resensitization to docetaxel treatment |
| CD44 | Hyaluronate receptor | Reduced cell migration | Decrease viability of PC3 cells |
| CD133 | Membrane organization | No alteration in cell proliferation and viability | Decrease in survival rate of cell, reduced metastatic potential, sensibilization to paclitaxel |
| SLCO1B3 | Sodium-independent transporter | Reduction in cellular uptake of docetaxel | - |
| EGFR | Membrane receptor | Reduce cell proliferation | Tumor regression |
| STEAP1 | Metalloreductase | Reduce cell viability and proliferation | Increase cell viability |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



