
Submitted:
20 April 2023
Posted:
21 April 2023
You are already at the latest version
Abstract
In recent years, the FDA has approved numerous antitumor drugs that are mutation-based for clinical use. These drugs have improved the precision of treatment and reduced adverse effects and side effects. Personalized therapy is a prominent and hot topic of current medicine and also represents the future direction of development. With the continuous advancements in gene sequencing and high-throughput screening, research and development strategies for personalized clinical drugs have developed rapidly. This review elaborated the recent personalized treatment strategies, which include artificial intelligence, multi-omics analysis, chemical proteomics, and computation aided drug design. These technologies rely on the molecular classification of diseases, the global signaling network within organisms, and new models for all targets, which significantly support the development of personalized medicine. Meanwhile, we summarized chemical drugs such as lorlatinib, osimertinib, and other natural products that deliver personalized therapeutic effects based on genetic mutations. This review also highlights potential challenges in interpreting genetic mutations and combining drugs, while providing new ideas for the development of personalized medicine and pharmacogenomics in cancer study.
Keywords:
Personalized therapy
; Pharmacogenomics
; Personalized medicine
; Antitumor
; Natural products
1. Introduction
In recent years, with the completion of the Human Genome Project, the development of genomics, proteomics, imaging technology and the advent of molecularly targeted drugs, personalized medicine has come into being[1,2,3]. According to the concept of personalized medicine, the treatment plan will be selected and determined based on information about patients' individual genetics, environment and lifestyle, so that each patient can achieve the maximum health benefits while reducing ineffective treatments and side effects[4]. Personalized medicine is at the core of personalized treatment[5]. Up to 2018, 355 pharmacogenomic biomarkers and 284 drugs have been approved by the FDA. Those pharmacogenomic biomarkers which approved for use in personalized medicines are usually specific genetic variants (i.e., gene tags) or abnormally expressed proteins[6]. It can help distinguish those who will or will not respond to a drug, prevent adverse drug reactions, and play an important role in optimizing drug dosing[7,8,9]. For those personalized medicines, anti-tumor drugs have a larger proportion which mainly resulted from the discovery of oncogenic driving genes and the development of molecular targeted drugs[10]. These personalized oncology drugs mainly include receptor tyrosine kinase inhibitors, small molecule inhibitors, vaccines, antibodies/small molecule-antibody conjugates, and monoclonal antibodies[11,12,13]. Besides of new personalized drugs, repurposing old drugs is also important approach to personalized drug discovery, which mainly involves developing and gene-tagging "complementary diagnostic" biomarkers for drugs already on the market[14,15]. In the case of the lung cancer drug ramucirumab, for example, the FDA cautions that patients treated with this drug must be those with EGFR and ALK mutations whose disease progresses after targeted therapy[16,17]. The new use of old drugs can not only save a lot of time and cost while ensure efficacy, but also reduce safety risks and speed up drug approval[18]. These drugs are mainly used in cancer, neuropsychiatric diseases, infections, cardiovascular diseases, metabolic diseases and other areas[19,20,21,22,23]. With the advent of increasingly personalized medicine, Big Data analysis is creating a model of precision medicine that enables a more detailed treatment plan according to the comprehensive situation of an individual's genes, lifestyle, environment and other factors, which can not only reduce the safety risks of drugs, but also greatly improve the effectiveness of drugs[24]. Based on the theme of personalized medicine, this review mainly discusses the strategies of discovering novel drugs, chemical antitumor agents, and natural antitumor products and so on.
2. Technical system for the development of new drugs and personalized medicine
The main goal of personalized medicine is to enable precise treatment of drugs for specific populations and types of diseases through design, screening, and optimization[25,26]. In recent years, with the continuous innovation and development of life sciences and technologies, the research and development strategies for new drugs has changed from the traditional model based on the phenotype of the disease, the local signaling pathway and a single target, into the new model based on the molecular typing of the disease, the global signaling network in the organism and all targets[27,28,29]. In addition, it has greatly supported the research and development of personalized medicine due to the rapid development of new omics technologies including genome, transcriptome, proteome, metabolome, etc., the rapid accumulation of multidimensional and large-scale omics data, the rise of molecular imaging, the advent of supercomputers, and the continuous improvement of correlation analysis and mining algorithms[30,31,32,33,34,35,36,37]. Next, we will focus on the emerging research in personalized medicine and the development of new key technologies for systematic deployment.
2.1. Research and development of personalized medicine based on artificial intelligence technology
Artificial intelligence is a branch of computer science that aims to simulate human thought processes, learning abilities, and knowledge reserves [38,39]. In recent decades, the improvement of computer speed and the rapid development of artificial intelligence (AI) have promoted the study of drug discovery and personalized medicine [40,41]. As one part of the development of personalized medicine, large-scale biomedical data was being used to identify the biological principles behind drugs to more accurately simulate and predict the complex effects of drug molecules in vivo[42]. In late 2016, Goldman Sachs Group published a comprehensive report on artificial intelligence laborated at the ecology and future of artificial intelligence. This report has pointed out that the application of AI and powerful algrithims will help to get rid of the risks in the development of new drugs[43]. In 2021, one study has reported that machine learning was able to predict the higher accuracy equal to 76% for outcome of phase III clinical trials of anti-tumor agents to treat with prostate cancer[44]. In the field of disease diagnosis, a study has shown that the melanoma diagnosis ofby using machine learning can reach the level of well-experienced dermatologists[45]. In addition, Google company has developed DeepMind's Deep Learning algorithm that can quickly and accurately detect early signs of diseases such as age-related macular degeneration and diabetic retinopathy to prevent and treat them in advance[46,47]. Artificial intelligence technology, with its powerful automatic feature extraction, complex model building and image processing, opens up new possibilities for the analysis, processing and application of biomedical Big Data[48,49,50].
2.2. Approaches with multidimensional omics data
Theoretically, the large-scale integrated analysis of multidimensional omics can insight into the global and full view of the molecular mechanism of diseases and specific drugs, which means that the genome wide data including the genome, transcriptome, proteome, metabolome and other various dimensions can help to analyze the target, regulatory mechanism and biological effect of drugs at different molecular levels[51,52]. In this way, the targeting and off-target effects of the drug can be revealed globally, and different drugs can be combined according to the different characteristics of each drug to improve the efficacy and reduce the side effects, thus achieving personalized and accurate guidance for drug treatment of diseases[53]. Meanwhile, new potential targets, and new genetic variants, and regulatory mechanisms can be identified, which will help to improve the effectiveness and successful probabilities of re of the new use of old drugs to a certain extent[54,55]. At present, the strategies personalized medicine discovery based on multi omics data is not yet mature, but it has been tentatively applied into the phase of personalized medicine discovery[56]. Recent studies have shown that by integrating multi-omics data including SNP and copy number variants, mRNA expression profiling, and protein profiling of lung adenocarcinoma , normal tissue, and tumor xenograft (PDX) models, it has been predicted that the protein changes have been presented in the lung cancer which cannot been found in the single omics data, which are strongly correlated with cell metabolism and survival in patients with lung cancer and other cancer[57,58]. In addition, the integration analysis between transcriptomic and proteomic data of 24 human tumor xenograft (PDX) models from breast cancer patients have shown that proteomics can better to detect dynamic changes in some proteins and protein phosphorylation, including AKT and ARAF, BRAF, and HSP90AB1[59]. All in all, multi-omics integration will provide strong technical support for clinical personalized treatment and personalized medicine development [60].
2.3. Study on high-throughput targets of chemical proteomics technology
The function of chemical proteomics technology is to exploit the specific interaction between the drug and the target protein, and to study the drug targets and molecular regulatory mechanism by combining various enrichment methods with high-resolution biomass spectroscopy[61]. This method can identify binding targets of small molecule compounds from complex biological samples (cells or tissues) with the advantages of high throughput and unbias. Chemical proteomics technology can provide important information for further analysis of the full target spectrum of active molecules at the cellular level and evaluation of drug activity, toxicology, and indications[62,63]. Currently, drug target identification technologies based on chemical proteomics primarily include affinity-based protein profiling, activity-based protein profiling, and thermal proteome profiling, and drug affinity responsive target stability[64]. We listed the principles, advantages, and disadvantages of those four technologies.
2.4. Computer aided drug discovery system for the development of personalized medicine
CADD technology can be used in various stages of the new personalized medicine development, such as drug target identification, lead discovery and optimization, therapeutic marker discovery and prediction model, drug combination and repurpose research, ADME and safety research, etc.[65,66,67,68]. CADD can reduce costs and shorten time of research and development, while greatly improve the successful risk[69]. We will focus on the application of computational methods in target identification and lead discovery in personalized medicine. 1) Target identification: Currently, there are still a large number of potential targets to be explored for effective drug development[70]. The potential targets of chemical compounds can be predicted with the approaches of bioinformatics and cheminformatics at multiple levels to improve the reliability of data analysis, and effectively promote the use of computers, which including chemical structure similarity search, data mining using machine learning methods, reverse molecular docking, and algorithm-based bioactivity profiling[71,72,73]. Keiser et al. (2018) develop similarity ensemble approach to enable large-scale searches for known drug targets by comparing molecular fingerprint similarities of thousands of compounds[74]. In addition, the dynamic structure and thermodynamic dynamics parameters of molecules and protein can be fully obtained by simulating molecular dynamics, which is helpful in the search for new personalized drug targets[75,76]. 2) Lead compounds discovery Molecular docking is the most commonly used method for designing drugs based on receptor structure[77]. The related software programs include Gold, Autodock, GLIDE, etc.[78]. In addition, molecular dynamics (MD) can also be used to study the dynamic structure of proteins, such as AMBER, GROMACS and NAMD[79]. The free energy of the binding of the protein to the ligand was calculated using MD, and then the binding mode and the binding activity of the two were predicted to screen the lead compounds[80]. For ligand-based drug design methods, compound similarity analysis, quantitative structure-activity relationship (QSAR), and pharmacophore model analysis have been successfully used for lead discovery[81,82]. For example, Mueller et al. (2012) used QSAR to identify 27 allosteric modulators of the mGlu5 receptor that can be used to treat anxiety disorders, Parkinson's disease, and schizophrenia[83]. In addition, Ijjaali et al. (2007) performed ligand-based virtual screening of 2 million compounds and identified 16 highly active human T-type calcium channel blockers that could be used to treat epilepsy and neuropathic pain[84]. All these approaches have helped to identify the new drug design process, and they are usually combined multiple approaches to design and optimize lead structures.
3. Anti-tumor personalized medicine
The main purpose of personalized therapy is to develop effective targeted drugs for different subphenotypic patients. The application of personlized anti-cancer drugs i has been widely and largely spread due to the discovery of oncogenic driving genetic mutionsand the development of molecular targeted drugs. These personalized anticancer drugs mainly include receptor tyrosine kinase inhibitors, small molecule inhibitors, vaccines, antibodies/small molecule antibody conjugates and monoclonal antibodies[13,85,86]. Here we focused personalized antitumor agents targeting on ALK and EFGR receptors(Table 2).
3.1. ALK inhibitors
Anaplastic lymphoma kinase (ALK) is an important molecular marker of non-small cell lung cancer[87,88]. In 2007, EML-ALK, the ALK fusion gene, was found to be present in 3%-7% of patients with non-small cell lung cancer, triggering the research and development boom of ALK personalized medicine[89]. 1) Crizotinib Crizotinib is an orally administered ALK inhibitor that effectively inhibits phosphorylation of the NPMALK fusion protein in human degenerative cell lymphomas Karpas-299 and SU-DHL -1 cells. Cell cycle arrest and apoptosis were significantly increased after 24- and 48-hour treatment with crizotinib. In mice transplanted with Karpas-299 tumor, oral administration of 100 mg/kg crizotinib for 15 days resulted in complete tumor regression. This is consistent with antitumor activity in vivo. Ceritinib is a second-generation ALK inhibitor synthesized by Pierre-Yves Michelly's team at Novartis based on TAE684. Cell proliferation assays showed that ceritinib significantly inhibited proliferation of Karpas-299 and BaF3 tool cells with high expression of the ALK fusion protein. 25 mg/kg ceritinib significantly inhibited the growth of subcutaneously transplanted tumors in nude mice with Karpas-299 and NCLH2228 tumor cell lines. Ceritinb has some ability to cross the blood-brain barrier, and the brain tissue to plasma exposure ratio (AUCinf) measured by isotopic labeling is approximately 15%[90,91,92]. 2) Alectimib Alectimib is an orally active and highly selective inhibitor ALK. Cell-level studies have shown that alectimib can inhibit the activation of ALK and downstream STAT3 and AKT signaling pathways in NCL-H2228 non-small cell lung cancer cells. Meanwhile, alectimib significantly inhibited the proliferation of NCI-H2228 non-small cell lung cancer cells. In nude mice, alectimib at a dose of 6 mg/kg significantly inhibited the growth of NCI-H2228 and KARPAS -299 cell transplantation tumors. In terms of the ability to overcome mutations, 100nM alectimib inhibited the phosphorylation of ALK in BaF3/EML4- ALK -L1196M cells[93,94]. 3) Brigatinib Brigatinib is a dual target inhibitor of ALK and epidermal growth factor receptor (EGFR). Subsequent pharmacodynamic evaluation studies showed that brigatinib significantly inhibited ALK and ALK kinase activity mutated by C1156Y, F1174L, L1196M, G1202R and R1275Q at the molecular level, while brigatinib inhibited EGFR and other kinase activity mutated by ROS1, FLT3, D835Y. Cell-level evidence showed that brigatinib inhibited the proliferation and intracellular ALK protein phosphorylation of AlK-driven lymphoma cells Karpas-299, SU-DHL -1, L-82, SUP -M2 and lung cancer cells H3122 and NCLH2228. The results showed that 50 mg/kg brigatinib could lead to shrinkage or even complete regression of Karpas-299 and NCI-H2228 transplanted tumors in mice. No significant reversal of tumor growth inhibition was observed 15 to 30 days after drug discontinuation. 50 mg/kg brigatinib significantly inhibited intracranial lesions and prolonged survival in mouse models of brain metastases containing transplanted NCLH2228 tumors. In mice with BaF3/EML4- ALK -G1202R grafts, 25 mg/kg and 50 mg/kg brigatinib inhibited tumors 55% and 88%, respectively[90,92,95]. 5)Lorlatinib Lorlatinib, a third-generation ALK inhibitor, showed significant inhibitory activity against ROS1 and ALK kinases. The tumor transplantation experiment in mice showed that 25 mg/kg lorlatinib could significantly inhibit tumor growth in H1322 and H3122/EM1A-ALKLSH312/EMLA-ALKG1269A mice. Lorlatinib (10 mg/kg) can significantly inhibit transplanted tumor growth in the tissue-derived mouse model of tumor transplantation, and tumor can also significantly decline when lorlatinib is replaced after crizotinib resistance. In an intracranial mouse transplant tumor model, lorlatinib significantly inhibited internal tumor growth and significantly prolonged mouse survival[96,97,98].
3.2. EGFR inhibitors
EGFR is a member of the epidermal growth factor receptor (EGFR) family of ErbB receptor tyrosine kinases, which also includes BGFR, ErbB2(HER2), ErbB3(HER3), and ErbB4(HER4)[99,100]. EGFR is a transmembrane receptor protein consisting of extracellular ligand-binding domains, transmembrane domains, and intracellular kinase-active domains[101]. Upon binding of the extracellular ligand-binding region to a ligand, homodimerization or heterodimerization occurs followed by autophosphorylation in the intracellular region to activate its kinase. Phosphorylated EGFR terminals bind to various downstream adaptor proteins and perform various physiological functions such as maintaining cell growth and inhibiting cell apoptosis through movement. EGFR is expressed in a variety of tissue cells[102]. Under normal physiological conditions, EGFR regulates a number of biological processes such as cell proliferation and differentiation, while high expression of EGFR or abnormal activation has been associated with the development and progression of various tumors such as non-small cell lung cancer (NSCLC), metastatic colorectal cancer (mCRC), head and neck cancer (HNSCC), glioblastoma (GBM), ovarian cancer, etc. Among these tumors, the occurrence and development of NSCLC is most closely associated with EGFR, and the molecular mechanism of EGFR driving the occurrence and development of NSCLC is also the most profound. Several EGFR inhibitors have been used to treat NSCLC. The discovery of EGFR-activated mutations as sensitive markers for small molecule EGFR inhibitors is not only a milestone in the history of lung cancer treatment, but also a model for personalized tumor treatment[100,101,102,103].
EGFR inhibitors include primarily gefitinib and erlotinib[104]. They bind to the EGFR kinase region in a competitive ATP-binding manner, reversibly inhibiting EGFR kinase activity and thus blocking downstream signaling. 1) Gefitinib Gefitinib was approved by the FDA in 2003 for the treatment of patients with advanced NSCLC who have failed chemotherapy[105]. Erlotinib was approved by the FDA in 2004 for the treatment of locally advanced or metastatic NSCLC and was subsequently approved in combination with gemcitabine for the treatment of locally advanced or metastatic pancreatic cancer[106]. The discovery of the deletion mutation in exon EGFR19 (exon19del) and the base substitution mutation L858R in exon 21 (L858R mutation), a sensitive marker, uncovered the reason why some people are sensitive to EGFR inhibitors. EGFR-sensitive mutations are located in the intracellular ATP-linked pocket kinase region, which increases the affinity between binding pockets and ATP, leading to destruction of the EGFR self-inhibition pathway and continuous activation of downstream signaling pathways, causing carcinogenesis. The affinity of gefitinib and erlotinib for this mutant EGFR protein is stronger than that of ATP molecules, leading to more severe clinical effects in patients with these mutations. Therefore, EGFR mutation detection has been approved for first-line clinical treatment of progressive EGFR-mutated NSCLC and has become routine clinical practice in most cancer centers worldwide. Although gefitinib and erlotinib are effective in treating NSCLC with EGFR-sensitive mutations, patients develop drug resistance within an average of 9 to 14 months after treatment, severely limiting the clinical use of first-generation inhibitors[104,107,108,109]. The emergence of resistance mutations and activation of compensatory signaling pathways are the main causes of drug resistance. EGFR T790M mutation was the most common cause of drug resistance and accounted for more than 50% of acquired drug resistance. Therefore, the research and development of second-generation EGFR inhibitors targeting EGFR wild-type and EGFRT790M-resistant mutations has attracted much attention. 2) Afatinib Afatinib, which can covalently bind the EGFR ATP binding site C797 in the pocket, has significant inhibitory activity against both EGFR WT and T79M resistant mutations. The drug was approved by US FDA in 2013 for the treatment of advanced non-small cell lung cancer and HER2-positive advanced breast cancer, and in 2016 for the treatment of patients with advanced lung cancer whose disease has progressed following platinum-based chemotherapy[110]. However, afatinib has resulted in significant side effects due to its potent inhibition of wild-type EGER activity and has been unable to achieve effective blood levels of EGFR T790M in humans. Therefore, the research and development of third-generation EGFR inhibitors that selectively inhibit resistant EGFR T7M mutations has attracted much attention[103,111]. Osimertinib can covalently bind to the cysteine site of EGFR 797 and selectively inhibit EGFR-sensitive and drug-resistant mutations, with weak inhibition of wild-type EGFR, and clinically shows good efficacy and few side effects in patients with drug-resistant mutations containing EGFR T790M. Osimertinib was approved by the US FDA in November 2015 for the second-line treatment of patients with metastatic NSCLC containing EGFR T790M. In 2018, osimertinib was approved by US FDA as a first-line treatment for patients with EGFR-sensitive, mutation-positive metastatic NSCLC. In addition, osimertinib can effectively cross the blood-brain barrier and is effective in patients with brain metastases (including meningeal metastases) from lung cancer, which is a major advantage over other small molecule EGFR inhibitors[112,113,114].
4. Pharmacogenetics of the anti-cancer natural products
The action of drugs on the body is generally divided into two phases, the pharmacokinetic phase and the pharmacodynamic phase, and the actual action of drugs in vivo begins with binding to targets, so the pharmacodynamic phase cannot be ignored[115]. Therefore, the pharmacodynamic phase cannot be ignored. It is caused by targeting specific molecular mechanisms and signaling pathways, and at the same time, drug-target interactions are influenced by genetic variations in genes[116,117]. Numerous bioactive natural products have been discovered and isolated, but the role of genetic variations in targets has not been adequately explored and few pharmacological targets have been clearly confirmed compared to those of metabolizing enzymes and transporters[118,119]. It is reported that in people with different pharmacological target genotypes, drug effects of natural products differ. Some genetic variations affect drug action in other ways, such as modulating the functions of related proteins that are not the direct target proteins, enzymes, or transporters[120]. As a result, the situation is more complex and more attention needs to be paid to drug-related pathways. For some natural products, there are defined targets, but there are still a large number of natural products that are linked not only to their direct targets but also to other indirect reactions. However, evidence of direct interactions has been found to be difficult to obtain, making the study of the molecular targets of natural products very challenging. The interactions between natural products and target sites are mainly described in the following examples using tumors.
Currently, genome-wide association studies (GWAS) and other techniques have found many compounds with anti-tumor activities, including many natural products with anti-tumor activities through various mechanisms, such as cytotoxicity[121,122]. This biological action leads to a variety of biological responses, such as inhibition of mitosis, DNA damage, DNA synthesis, and repair damage[123]. The cytotoxic pathways may be related to the efficacy and adverse effects of natural tumor products.
Trabectedin is a marine-derived natural product originally isolated from the marine ascidian Ecteinascidia turbinate. Trabectedin has a complex mechanism of action that affects important cell biological processes in tumor cells and is the first marine anticancer agent approved for patients with soft tissue sarcoma (STS) at European Union[124]. The DNA damage induced by the drug is largely caused by the NER protein, and an arginine residue in Rad13 (Arg961), trabectedin can kill cells by poisoning the DNA nucleotide excision repair (NER) machinery and the DNA repair pathways of homologous recombination[124]. Trabectedin has a unique multifaceted mechanism involving transcriptional regulation and DNA repair systems, and transcription-coupled nucleotide excision repair and homologous recombination repair (HRR) are the main features of its antiproliferative activity. It also has the ability to modulate the tumor microenvironment which can alter the function and expression of DNA repair genes, such as BRCA1 and BRCA2[125,126]. BRCA proteins play a critical role in DNA repair, as they are essential for the repair of double-strand breaks (HR). Cancers that have a mutation in the BRCA1 or BRCA2 genes that reduces protein activity, as in ovarian and breast cancers, may increase the activity of drugs that exert their cytotoxicity via DNA double-strand breaks[127,128]. As for BRCA2 mutations, trabectedin showed higher antitumor activity in relapsed metastatic breast cancer patients with germline BRCA2 mutations than in those with BRCA1 mutations. Loss of the wild-type BRCA2 allele in the tumor results in an excellent early complete metabolic response due to a somatic aberration that likely leads to deregulation of cellular HR function responsible for increased sensitivity to trabectedin [126]. These reports demonstrate the importance of BRCA1/2 mutations in the administration of trabectedin for the treatment of tumors with defective DNA damage repair.
Vincristine is isolated from the plant Catharanthus roseus and is a drug widely used in cancer treatment. It can be used to treat leukemias, lymphomas, brain tumors and also solid tumors[129]. The mechanism of tumor restriction is due to its interference with microtubules in the mitotic spindle[130]. In the pharmacokinetics of vincristine, CYP3A enzymes and ABC transporters may play an important role. As with other substrates of CYP3A enzymes, genetic variants of CYP3A also lead to its adverse effects, such as vincristine-induced peripheral neuropathy[131]. There are studies confirming that active CYP3A5 expressors have a lower risk of VIPN than nonexpressors[132,133]. However, a lower risk of VIPN has been observed in some children with the CYP3A5*3 genotype. This is an inverse conclusion when toxicity is accumulated to the highest concentration without CYP3A5 enzyme activity[134]. We need to pay more attention to the neurotoxicity caused by genetic variations of vincristine and perform clinical optimization its metabolism.
Gigantol, a bibenzyl phenolic compound derived from several medicinal orchids, has been shown to inhibit proliferation, migration, EMT and cancer stem cell (CSC) phenotype in lung cancer cells [135,136,137]. At non-toxic doses (below 20 μM), gigantol isolated from Dendrobium draconis could suppress tumor spheroid formation and decrease lung CSC marker proteins, including CD133 and ALDH1A1, in non-small-cell lung cancer NCI-H460 cells. Additionally, gigantol inhibited cancer stem cell-like phenotypes through down-regulation of AKT signaling pathway which leads to reduced levels of Oct4 and Nanog [135].
Paclitaxel (PTX) is one of the natural broad-spectrum antitumor drugs used as first-line chemotherapy in ovarian cancer therapy[138]. The efficacy of paclitaxel is associated with ABCB1 G2677T/A mutation. ABCB1, also known as MDR1, is the efflux pump of cells. After paclitaxel enters human tumor cells, it is pumped out of the cells by ABCB1[139]. The mutation at position 2677 reduces the transport capacity of ABCB1, allowing the drug to accumulate in tumor cells and achieve good therapeutic effect. Studies have shown that in patients with ovarian cancer, the G2677T/A mutation has a good effect on paclitaxel[140].
Chrysotoxine, a bibenzyl compound isolated from stems of Dendrobium pulchellum, has been reported to sensitize anoikis and inhibit metastasis of lung cancer cells in an anchorage-independent fashion. Bhummaphan et al. (2019) investigated the suppressive effects of chrysotoxine on CSC-rich populations of H460 and H23 cells and primary CSCs in three-dimensional (3D) culture and showed that non-toxic doses (≤20 μM) of chrysotoxine inhibited CSC-like phenotypes and decreased CSC markers CD133, CD44, ABCG2 and ALDH1A1 which were mediated through a Src-AKT-Sox2-dependent mechanism (Table 2)[141].
5. The future challenges of personalized therapy
Although a significant number of gene variants specific anti-tumor drugs have been approved by the FDA, the percentage of personalized medicines is less than 10% of all the FDA approved drugs [142]. The development of personalized medicines still confronts some challenges: 1) Interpretation of genetic variations thousands of mutant genes are scattered throughout the genome of cancer cells, and there are hundreds of different mutant genes associated with cancer[143]. According to current clinical statistical data of personalized tumor treatment, only 30-50% of patients can link the tumor to the corresponding mutations, and only 3%-13% of patients can choose personalized therapy through individual genomic analysis[144]. At present, genome sequencing technology, especially single-cell sequencing and omics analysis has been rapidly developed. We are able to obtain whole genome data in a relatively short time and low cost[145,146,147]. Therefore, people need to not only interpret gene function, but also decipher the functional effects of gene mutations. 2) Dynamic molecular changes of cancer the pathogenic genes in the cancer patients may usually evolve and escape the therapeutic effect of drugs on the lesion through genetic mutation, which is referred to as secondary drug resistance. After secondary drug resistance , current diagnostic and treatment methods may no longer be applicable in the original disease stateDue to our limited understanding of complex signal transduction pathways, the development of personalized treatment options based on basic research cannot resolve the new disease molecular state with some variants mutant [148]. Therefore, we have to confront significant challenges in monitoring the molecular typing of patients, identifying and defining the occurrence of drug resistance, and finding new treatment options for the protective mechanism of tumors. 3) Drug combination: In the development of personalized treatment, it is rare for a single drug to act on all of the gene mutations causing a patient's disease, and therefore it is more advantageous to use a combination regimen to treat diseases with multiple genes[149]. Currently, drug combinations are commonly used for cancer, infectious diseases, cardiovascular diseases, and other areas. To obtain reasonable strategies for the development of drug combinations and prediction methods for different molecular disease types to enhance the synergistic effect between drugs and reduce the occurrence of side effects[150,151,152].
6. Conclusions and future perspectives
Personalized medicine is changing the way we diagnose and treat diseases[153]. Advances in genomics have propelled the development of this field, enabling us to better understand the genetic basis of diseases and identify individuals who are most likely to benefit from certain treatments. Pharmacogenomics has been shown to be crucial for developing personalized treatment plans for patients[7]. By identifying genetic variations that affect drug metabolism, efficacy and toxicity, clinicians can tailor drug therapy according to each patient's unique genetic makeup. This approach can improve clinical care, reduce adverse drug reactions, optimize dosing, and drug selection[56]. The development of new technologies lays the groundwork for a deeper assessment of genomic data and identification of new therapeutic targets[154]. The development of new technologies and a better understanding of the underlying biology of diseases make it possible to develop more effective and targeted therapies[155]. Currently, some chemical drugs such as methotrexate and 5-fluorouracil are used for personalized therapy to treat metabolic tumors. However, studies have shown that in addition to synthetic drugs, natural products such as camptothecin also have anti-tumor effects[156]. It has been confirmed that some natural products are related to pharmacogenomics, which can affect drug-metabolizing enzymes and transport proteins. Natural products and traditional Chinese medicine are potential sources for personalized therapy[157].
Overall, the integration of pharmacogenomics and other cutting-edge technologies provides new insights into disease mechanisms and leads to the development of more personalized treatment strategies. As our understanding of the genetic and molecular mechanisms of diseases continues to grow, we can expect further breakthroughs in personalized medicine, including the identification of new drug targets and the development of more effective therapies based on natural products and traditional medicine.
Author Contributions
QZ W and XY C designed and revised the manuscript. YL Z,SQ P and HZ W reviewed the literature and drafted the original manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
We acknowledge the financial support from the National Natural Science Foundation of China (31871281), Scientific Research Foundation for Advanced Talents of Shanghai University of Traditional Chinese Medicine, Shanghai Municipal Health Commission (2020XGKY12).
Data Availability Statement
The authors declare that all data supporting the findings of this study are available within the paper and its supplementary information files.
Acknowledgments
We would like to thank all the students and staff, especially Q22 GZ, QW, and CD. They provided lots of useful suggestions for this study.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Su, Miao, Zhe Zhang, Li Zhou, Chao Han, Canhua Huang, and Edouard C Nice. "Proteomics, Personalized Medicine and Cancer." Cancers 13, no. 11 (2021): 2512. [CrossRef]
- Chen, Rui, and Michael Snyder. "Promise of Personalized Omics to Precision Medicine." Wiley Interdisciplinary Reviews: Systems Biology and Medicine 5, no. 1 (2013): 73-82. [CrossRef]
- Carrasco-Ramiro, Fernando, Ramón Peiró-Pastor, and Begoña Aguado. "Human Genomics Projects and Precision Medicine." Gene therapy 24, no. 9 (2017): 551-61. [CrossRef]
- Jain, Kewal K. "Personalized Medicine." Current opinion in molecular therapeutics 4, no. 6 (2002): 548-58.
- Goetz, L. H., and N. J. Schork. "Personalized Medicine: Motivation, Challenges, and Progress." Fertil Steril 109, no. 6 (2018): 952-63. [CrossRef]
- Najjar, S., and K. H. Allison. "Updates on Breast Biomarkers." Virchows Arch 480, no. 1 (2022): 163-76. [CrossRef]
- Cecchin, E., and G. Stocco. "Pharmacogenomics and Personalized Medicine." Genes (Basel) 11, no. 6 (2020). [CrossRef]
- Chang, C. J., C. B. Chen, S. I. Hung, C. Ji, and W. H. Chung. "Pharmacogenetic Testing for Prevention of Severe Cutaneous Adverse Drug Reactions." Front Pharmacol 11 (2020): 969. [CrossRef]
- Zastrozhin, M. S., A. S. Sorokin, T. V. Agibalova, E. A. Grishina, AР Antonenko, I. N. Rozochkin, D. V. Duzhev, V. Y. Skryabin, T. E. Galaktionova, I. V. Barna, A. V. Orlova, A. D. Aguzarov, L. M. Savchenko, E. A. Bryun, and D. A. Sychev. "Using a Personalized Clinical Decision Support System for Bromdihydrochlorphenylbenzodiazepine Dosing in Patients with Anxiety Disorders Based on the Pharmacogenomic Markers." Hum Psychopharmacol 33, no. 6 (2018): e2677. [CrossRef]
- Workman, P. "Pharmacogenomics in Cancer Drug Discovery and Development: Inhibitors of the Hsp90 Molecular Chaperone." Cancer Detect Prev 26, no. 6 (2002): 405-10. [CrossRef]
- Letai, A., P. Bhola, and A. L. Welm. "Functional Precision Oncology: Testing Tumors with Drugs to Identify Vulnerabilities and Novel Combinations." Cancer Cell 40, no. 1 (2022): 26-35. [CrossRef]
- Shirley, M. "Bruton Tyrosine Kinase Inhibitors in B-Cell Malignancies: Their Use and Differential Features." Target Oncol 17, no. 1 (2022): 69-84. [CrossRef]
- Zhong, L., Y. Li, L. Xiong, W. Wang, M. Wu, T. Yuan, W. Yang, C. Tian, Z. Miao, T. Wang, and S. Yang. "Small Molecules in Targeted Cancer Therapy: Advances, Challenges, and Future Perspectives." Signal Transduct Target Ther 6, no. 1 (2021): 201. [CrossRef]
- Pushpakom, S., F. Iorio, P. A. Eyers, K. J. Escott, S. Hopper, A. Wells, A. Doig, T. Guilliams, J. Latimer, C. McNamee, A. Norris, P. Sanseau, D. Cavalla, and M. Pirmohamed. "Drug Repurposing: Progress, Challenges and Recommendations." Nat Rev Drug Discov 18, no. 1 (2019): 41-58. [CrossRef]
- Grover, A., and P. C. Sharma. "Development and Use of Molecular Markers: Past and Present." Crit Rev Biotechnol 36, no. 2 (2016): 290-302. [CrossRef]
- Nakagawa, K., E. B. Garon, T. Seto, M. Nishio, S. Ponce Aix, L. Paz-Ares, C. H. Chiu, K. Park, S. Novello, E. Nadal, F. Imamura, K. Yoh, J. Y. Shih, K. H. Au, D. Moro-Sibilot, S. Enatsu, A. Zimmermann, B. Frimodt-Moller, C. Visseren-Grul, and M. Reck. "Ramucirumab Plus Erlotinib in Patients with Untreated, Egfr-Mutated, Advanced Non-Small-Cell Lung Cancer (Relay): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial." Lancet Oncol 20, no. 12 (2019): 1655-69. [CrossRef]
- Casak, S. J., I. Fashoyin-Aje, S. J. Lemery, L. Zhang, R. Jin, H. Li, L. Zhao, H. Zhao, H. Zhang, H. Chen, K. He, M. Dougherty, R. Novak, S. Kennett, S. Khasar, W. Helms, P. Keegan, and R. Pazdur. "Fda Approval Summary: Ramucirumab for Gastric Cancer." Clin Cancer Res 21, no. 15 (2015): 3372-6. [CrossRef]
- Domingos, S., V. André, S. Quaresma, I. C. Martins, M. F. Minas da Piedade, and M. T. Duarte. "New Forms of Old Drugs: Improving without Changing." J Pharm Pharmacol 67, no. 6 (2015): 830-46. [CrossRef]
- Vlahopoulos, S., E. Critselis, I. F. Voutsas, S. A. Perez, M. Moschovi, C. N. Baxevanis, and G. P. Chrousos. "New Use for Old Drugs? Prospective Targets of Chloroquines in Cancer Therapy." Curr Drug Targets 15, no. 9 (2014): 843-51. [CrossRef]
- Joyce, C. R. "New Drugs for Mental Diseases? New Diseases for Old Drugs?" J R Soc Med 80, no. 7 (1987): 406-9. [CrossRef]
- Cortez-Maya, S., A. Moreno-Herrera, I. Palos, and G. Rivera. "Old Antiprotozoal Drugs: Are They Still Viable Options for Parasitic Infections or New Options for Other Diseases?" Curr Med Chem 27, no. 32 (2020): 5403-28. [CrossRef]
- Arbel, Y., W. Abuzeid, R. S. Rosenson, A. Weisman, and M. E. Farkouh. "Old Drugs for New Indications in Cardiovascular Medicine." Cardiovasc Drugs Ther 32, no. 2 (2018): 223-32. [CrossRef]
- Kaiser, D., and E. Oetjen. "Something Old, Something New and Something Very Old: Drugs for Treating Type 2 Diabetes." Br J Pharmacol 171, no. 12 (2014): 2940-50. [CrossRef]
- Janiaud, P., S. Serghiou, and J. P. A. Ioannidis. "New Clinical Trial Designs in the Era of Precision Medicine: An Overview of Definitions, Strengths, Weaknesses, and Current Use in Oncology." Cancer Treat Rev 73 (2019): 20-30. [CrossRef]
- Di Sanzo, M., L. Cipolloni, M. Borro, R. La Russa, A. Santurro, M. Scopetti, M. Simmaco, and P. Frati. "Clinical Applications of Personalized Medicine: A New Paradigm and Challenge." Curr Pharm Biotechnol 18, no. 3 (2017): 194-203. [CrossRef]
- Jørgensen, J. T. "Oncology Drug-Companion Diagnostic Combinations." Cancer Treat Res Commun 29 (2021): 100492. [CrossRef]
- Zwart, H., L. Landeweerd, and P. Lemmens. "Continental Philosophical Perspectives on Life Sciences and Emerging Technologies." Life Sci Soc Policy 12, no. 1 (2016): 8. [CrossRef]
- Hartl, D., V. de Luca, A. Kostikova, J. Laramie, S. Kennedy, E. Ferrero, R. Siegel, M. Fink, S. Ahmed, J. Millholland, A. Schuhmacher, M. Hinder, L. Piali, and A. Roth. "Translational Precision Medicine: An Industry Perspective." J Transl Med 19, no. 1 (2021): 245. [CrossRef]
- Lian, M. A., W. Ke-Bin, and W. U. Sheng-Xian. "[Research and Development of New Traditional Chinese Medicine Drugs for Certain Syndromes Based on "Theoretical Innovation"]." Zhongguo Zhong Yao Za Zhi 45, no. 20 (2020): 5048-56. [CrossRef]
- Jiang, Z., X. Zhou, R. Li, J. J. Michal, S. Zhang, M. V. Dodson, Z. Zhang, and R. M. Harland. "Whole Transcriptome Analysis with Sequencing: Methods, Challenges and Potential Solutions." Cell Mol Life Sci 72, no. 18 (2015): 3425-39. [CrossRef]
- Sherman, R. M., and S. L. Salzberg. "Pan-Genomics in the Human Genome Era." Nat Rev Genet 21, no. 4 (2020): 243-54. [CrossRef]
- Wang, Y., and W. Yang. "Proteome-Scale Analysis of Protein S-Acylation Comes of Age." J Proteome Res 20, no. 1 (2021): 14-26. [CrossRef]
- Balashova, E. E., O. P. Trifonova, D. L. Maslov, S. R. Lichtenberg, P. G. Lokhov, and A. I. Archakov. "[Metabolome Profiling in the Study of Aging Processes]." Biomed Khim 68, no. 5 (2022): 321-38. [CrossRef]
- Li, X., J. Ma, L. Leng, M. Han, M. Li, F. He, and Y. Zhu. "Mogcn: A Multi-Omics Integration Method Based on Graph Convolutional Network for Cancer Subtype Analysis." Front Genet 13 (2022): 806842. [CrossRef]
- Farber, G., K. E. Boczar, C. C. Wiefels, J. G. E. Zelt, E. C. Guler, R. A. deKemp, R. S. Beanlands, and B. H. Rotstein. "The Future of Cardiac Molecular Imaging." Semin Nucl Med 50, no. 4 (2020): 367-85. [CrossRef]
- Bagchi, A. "Latest Trends in Structure Based Drug Design with Protein Targets." Adv Protein Chem Struct Biol 121 (2020): 1-23. [CrossRef]
- Joshi, R. R., and V. V. Samant. "Bayesian Data Mining of Protein Domains Gives an Efficient Predictive Algorithm and New Insight." J Mol Model 13, no. 1 (2007): 275-82. [CrossRef]
- Ramesh, A. N., C. Kambhampati, J. R. Monson, and P. J. Drew. "Artificial Intelligence in Medicine." Ann R Coll Surg Engl 86, no. 5 (2004): 334-8.
- Carrera-Rivera, A., W. Ochoa, F. Larrinaga, and G. Lasa. "How-to Conduct a Systematic Literature Review: A Quick Guide for Computer Science Research." MethodsX 9 (2022): 101895. [CrossRef]
- Rasteau, S., D. Ernenwein, C. Savoldelli, and P. Bouletreau. "Artificial Intelligence for Oral and Maxillo-Facial Surgery: A Narrative Review." J Stomatol Oral Maxillofac Surg 123, no. 3 (2022): 276-82. [CrossRef]
- Wang, A., X. Xiu, S. Liu, Q. Qian, and S. Wu. "Characteristics of Artificial Intelligence Clinical Trials in the Field of Healthcare: A Cross-Sectional Study on Clinicaltrials.Gov." Int J Environ Res Public Health 19, no. 20 (2022). [CrossRef]
- Wang, L., and C. A. Alexander. "Big Data Analytics in Medical Engineering and Healthcare: Methods, Advances and Challenges." J Med Eng Technol 44, no. 6 (2020): 267-83. [CrossRef]
- Dallas, S., C. Sensenhauser, A. Batheja, M. Singer, M. Markowska, C. Zakszewski, R. N. Mamidi, M. McMillia, C. Han, H. Zhou, and J. Silva. "De-Risking Bio-Therapeutics for Possible Drug Interactions Using Cryopreserved Human Hepatocytes." Curr Drug Metab 13, no. 7 (2012): 923-9. [CrossRef]
- Lee, Y. W., J. W. Choi, and E. H. Shin. "Machine Learning Model for Predicting Malaria Using Clinical Information." Comput Biol Med 129 (2021): 104151. [CrossRef]
- Chan, S., V. Reddy, B. Myers, Q. Thibodeaux, N. Brownstone, and W. Liao. "Machine Learning in Dermatology: Current Applications, Opportunities, and Limitations." Dermatol Ther (Heidelb) 10, no. 3 (2020): 365-86. [CrossRef]
- Jiang, Y., M. Yang, S. Wang, X. Li, and Y. Sun. "Emerging Role of Deep Learning-Based Artificial Intelligence in Tumor Pathology." Cancer Commun (Lond) 40, no. 4 (2020): 154-66. [CrossRef]
- Dong, L., W. He, R. Zhang, Z. Ge, Y. X. Wang, J. Zhou, J. Xu, L. Shao, Q. Wang, Y. Yan, Y. Xie, L. Fang, H. Wang, Y. Wang, X. Zhu, J. Wang, C. Zhang, H. Wang, Y. Wang, R. Chen, Q. Wan, J. Yang, W. Zhou, H. Li, X. Yao, Z. Yang, J. Xiong, X. Wang, Y. Huang, Y. Chen, Z. Wang, C. Rong, J. Gao, H. Zhang, S. Wu, J. B. Jonas, and W. B. Wei. "Artificial Intelligence for Screening of Multiple Retinal and Optic Nerve Diseases." JAMA Netw Open 5, no. 5 (2022): e229960. [CrossRef]
- Yu, K. H., A. L. Beam, and I. S. Kohane. "Artificial Intelligence in Healthcare." Nat Biomed Eng 2, no. 10 (2018): 719-31. [CrossRef]
- Ji, Y., S. Liu, X. Hong, Y. Lu, X. Wu, K. Li, K. Li, and Y. Liu. "Advances in Artificial Intelligence Applications for Ocular Surface Diseases Diagnosis." Front Cell Dev Biol 10 (2022): 1107689. [CrossRef]
- Song, Q., and X. Li. "[Application and Development of Voice Analysis and Endoscopic Technology Combined with Artificial Intelligence in the Diagnosis and Treatment of Throat Disease]." Lin Chung Er Bi Yan Hou Tou Jing Wai Ke Za Zhi 36, no. 8 (2022): 647-50. [CrossRef]
- Ye, Y., Z. Zhang, Y. Liu, L. Diao, and L. Han. "A Multi-Omics Perspective of Quantitative Trait Loci in Precision Medicine." Trends Genet 36, no. 5 (2020): 318-36. [CrossRef]
- Chong, J., D. S. Wishart, and J. Xia. "Using Metaboanalyst 4.0 for Comprehensive and Integrative Metabolomics Data Analysis." Curr Protoc Bioinformatics 68, no. 1 (2019): e86. [CrossRef]
- Nygaard, H. B. "Targeting Fyn Kinase in Alzheimer's Disease." Biol Psychiatry 83, no. 4 (2018): 369-76. [CrossRef]
- Wang, M., A. Li, M. Sekiya, N. D. Beckmann, X. Quan, N. Schrode, M. B. Fernando, A. Yu, L. Zhu, J. Cao, L. Lyu, E. Horgusluoglu, Q. Wang, L. Guo, Y. S. Wang, R. Neff, W. M. Song, E. Wang, Q. Shen, X. Zhou, C. Ming, S. M. Ho, S. Vatansever, HÜ Kaniskan, J. Jin, M. M. Zhou, K. Ando, L. Ho, P. A. Slesinger, Z. Yue, J. Zhu, P. Katsel, S. Gandy, M. E. Ehrlich, V. Fossati, S. Noggle, D. Cai, V. Haroutunian, K. M. Iijima, E. Schadt, K. J. Brennand, and B. Zhang. "Transformative Network Modeling of Multi-Omics Data Reveals Detailed Circuits, Key Regulators, and Potential Therapeutics for Alzheimer's Disease." Neuron 109, no. 2 (2021): 257-72.e14. [CrossRef]
- Parker, G., and H. Brotchie. "Do the Old Psychostimulant Drugs Have a Role in Managing Treatment-Resistant Depression?" Acta Psychiatr Scand 121, no. 4 (2010): 308-14. [CrossRef]
- Whirl-Carrillo, M., E. M. McDonagh, J. M. Hebert, L. Gong, K. Sangkuhl, C. F. Thorn, R. B. Altman, and T. E. Klein. "Pharmacogenomics Knowledge for Personalized Medicine." Clin Pharmacol Ther 92, no. 4 (2012): 414-7. [CrossRef]
- Qu, J., F. S. Kalyani, L. Liu, T. Cheng, and L. Chen. "Tumor Organoids: Synergistic Applications, Current Challenges, and Future Prospects in Cancer Therapy." Cancer Commun (Lond) 41, no. 12 (2021): 1331-53. [CrossRef]
- Park, M., D. Kim, K. Moon, and T. Park. "Integrative Analysis of Multi-Omics Data Based on Blockwise Sparse Principal Components." Int J Mol Sci 21, no. 21 (2020). [CrossRef]
- Ding, Z., N. Wang, N. Ji, and Z. S. Chen. "Proteomics Technologies for Cancer Liquid Biopsies." Mol Cancer 21, no. 1 (2022): 53. [CrossRef]
- Battaglini, D., L. Al-Husinat, A. G. Normando, A. P. Leme, K. Franchini, M. Morales, P. Pelosi, and P. R. Rocco. "Personalized Medicine Using Omics Approaches in Acute Respiratory Distress Syndrome to Identify Biological Phenotypes." Respir Res 23, no. 1 (2022): 318. [CrossRef]
- Spradlin, J. N., E. Zhang, and D. K. Nomura. "Reimagining Druggability Using Chemoproteomic Platforms." Acc Chem Res 54, no. 7 (2021): 1801-13. [CrossRef]
- Kanduc, D. "The Role of Proteomics in Defining Autoimmunity." Expert Rev Proteomics 18, no. 3 (2021): 177-84. [CrossRef]
- Elyada, E., M. Bolisetty, P. Laise, W. F. Flynn, E. T. Courtois, R. A. Burkhart, J. A. Teinor, P. Belleau, G. Biffi, M. S. Lucito, S. Sivajothi, T. D. Armstrong, D. D. Engle, K. H. Yu, Y. Hao, C. L. Wolfgang, Y. Park, J. Preall, E. M. Jaffee, A. Califano, P. Robson, and D. A. Tuveson. "Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts." Cancer Discov 9, no. 8 (2019): 1102-23. [CrossRef]
- Pang, Z., M. A. Schafroth, D. Ogasawara, Y. Wang, V. Nudell, N. K. Lal, D. Yang, K. Wang, D. M. Herbst, J. Ha, C. Guijas, J. L. Blankman, B. F. Cravatt, and L. Ye. "In Situ Identification of Cellular Drug Targets in Mammalian Tissue." Cell 185, no. 10 (2022): 1793-805.e17. [CrossRef]
- Ambure, P., and K. Roy. "Cadd Modeling of Multi-Target Drugs against Alzheimer'S Disease." Curr Drug Targets 18, no. 5 (2017): 522-33. [CrossRef]
- Lomenick, B., R. Hao, N. Jonai, R. M. Chin, M. Aghajan, S. Warburton, J. Wang, R. P. Wu, F. Gomez, J. A. Loo, J. A. Wohlschlegel, T. M. Vondriska, J. Pelletier, H. R. Herschman, J. Clardy, C. F. Clarke, and J. Huang. "Target Identification Using Drug Affinity Responsive Target Stability (Darts)." Proc Natl Acad Sci U S A 106, no. 51 (2009): 21984-9. [CrossRef]
- Li, G., P. Lin, K. Wang, C. C. Gu, and S. Kusari. "Artificial Intelligence-Guided Discovery of Anticancer Lead Compounds from Plants and Associated Microorganisms." Trends Cancer 8, no. 1 (2022): 65-80. [CrossRef]
- Wong, M., A. Badri, C. Gasparis, G. Belfort, and M. Koffas. "Modular Optimization in Metabolic Engineering." Crit Rev Biochem Mol Biol 56, no. 6 (2021): 587-602. [CrossRef]
- Gomeni, R., M. Bani, C. D'Angeli, M. Corsi, and A. Bye. "Computer-Assisted Drug Development (Cadd): An Emerging Technology for Designing First-Time-in-Man and Proof-of-Concept Studies from Preclinical Experiments." Eur J Pharm Sci 13, no. 3 (2001): 261-70. [CrossRef]
- Vintonyak, V. V., A. P. Antonchick, D. Rauh, and H. Waldmann. "The Therapeutic Potential of Phosphatase Inhibitors." Curr Opin Chem Biol 13, no. 3 (2009): 272-83. [CrossRef]
- Avram, S., M. Mernea, C. Limban, F. Borcan, and C. Chifiriuc. "Potential Therapeutic Approaches to Alzheimer's Disease by Bioinformatics, Cheminformatics and Predicted Adme-Tox Tools." Curr Neuropharmacol 18, no. 8 (2020): 696-719. [CrossRef]
- Akalin, P. K. "Introduction to Bioinformatics." Mol Nutr Food Res 50, no. 7 (2006): 610-9. [CrossRef]
- Wishart, D. S. "Introduction to Cheminformatics." Curr Protoc Bioinformatics Chapter 14 (2007): Unit 14.1. [CrossRef]
- Irwin, J. J., G. Gaskins, T. Sterling, M. M. Mysinger, and M. J. Keiser. "Predicted Biological Activity of Purchasable Chemical Space." J Chem Inf Model 58, no. 1 (2018): 148-64. [CrossRef]
- Mitusińska, K., A. Raczyńska, M. Bzówka, W. Bagrowska, and A. Góra. "Applications of Water Molecules for Analysis of Macromolecule Properties." Comput Struct Biotechnol J 18 (2020): 355-65. [CrossRef]
- Zobdeh, F., A. Ben Kraiem, M. M. Attwood, V. N. Chubarev, V. V. Tarasov, H. B. Schiöth, and J. Mwinyi. "Pharmacological Treatment of Migraine: Drug Classes, Mechanisms of Action, Clinical Trials and New Treatments." Br J Pharmacol 178, no. 23 (2021): 4588-607. [CrossRef]
- Pinzi, L., and G. Rastelli. "Molecular Docking: Shifting Paradigms in Drug Discovery." Int J Mol Sci 20, no. 18 (2019). [CrossRef]
- Reddy, K. K., R. S. Rathore, P. Srujana, R. R. Burri, C. R. Reddy, M. Sumakanth, P. Reddanna, and M. R. Reddy. "Performance Evaluation of Docking Programs- Glide, Gold, Autodock & Surflexdock, Using Free Energy Perturbation Reference Data: A Case Study of Fructose-1, 6-Bisphosphatase-Amp Analogs." Mini Rev Med Chem 20, no. 12 (2020): 1179-87. [CrossRef]
- Collier, T. A., T. J. Piggot, and J. R. Allison. "Molecular Dynamics Simulation of Proteins." Methods Mol Biol 2073 (2020): 311-27. [CrossRef]
- Wang, L., J. Chambers, and R. Abel. "Protein-Ligand Binding Free Energy Calculations with Fep." Methods Mol Biol 2022 (2019): 201-32.
- Gupta, R., D. Srivastava, M. Sahu, S. Tiwari, R. K. Ambasta, and P. Kumar. "Artificial Intelligence to Deep Learning: Machine Intelligence Approach for Drug Discovery." Mol Divers 25, no. 3 (2021): 1315-60. [CrossRef]
- Hessler, G., and K. H. Baringhaus. "Artificial Intelligence in Drug Design." Molecules 23, no. 10 (2018). [CrossRef]
- Mueller, R., E. S. Dawson, J. Meiler, A. L. Rodriguez, B. A. Chauder, B. S. Bates, A. S. Felts, J. P. Lamb, U. N. Menon, S. B. Jadhav, A. S. Kane, C. K. Jones, K. J. Gregory, C. M. Niswender, P. J. Conn, C. M. Olsen, D. G. Winder, K. A. Emmitte, and C. W. Lindsley. "Discovery of 2-(2-Benzoxazoyl Amino)-4-Aryl-5-Cyanopyrimidine as Negative Allosteric Modulators (Nams) of Metabotropic Glutamate Receptor 5 (Mglu₅): From an Artificial Neural Network Virtual Screen to an in Vivo Tool Compound." ChemMedChem 7, no. 3 (2012): 406-14. [CrossRef]
- Ijjaali, I., C. Barrere, J. Nargeot, F. Petitet, and E. Bourinet. "Ligand-Based Virtual Screening to Identify New T-Type Calcium Channel Blockers." Channels (Austin) 1, no. 4 (2007): 300-4. [CrossRef]
- Dembic, Z. "Antitumor Drugs and Their Targets." Molecules 25, no. 23 (2020). [CrossRef]
- Jain, K. K. "Personalized Immuno-Oncology." Med Princ Pract 30, no. 1 (2021): 1-16. [CrossRef]
- Du, X., Y. Shao, H. F. Qin, Y. H. Tai, and H. J. Gao. "Alk-Rearrangement in Non-Small-Cell Lung Cancer (Nsclc)." Thorac Cancer 9, no. 4 (2018): 423-30. [CrossRef]
- Golding, B., A. Luu, R. Jones, and A. M. Viloria-Petit. "The Function and Therapeutic Targeting of Anaplastic Lymphoma Kinase (Alk) in Non-Small Cell Lung Cancer (Nsclc)." Mol Cancer 17, no. 1 (2018): 52. [CrossRef]
- Lin, J. J., G. J. Riely, and A. T. Shaw. "Targeting Alk: Precision Medicine Takes on Drug Resistance." Cancer Discov 7, no. 2 (2017): 137-55. [CrossRef]
- Camidge, D. R., H. R. Kim, M. J. Ahn, J. C. Yang, J. Y. Han, J. S. Lee, M. J. Hochmair, J. Y. Li, G. C. Chang, K. H. Lee, C. Gridelli, A. Delmonte, R. Garcia Campelo, D. W. Kim, A. Bearz, F. Griesinger, A. Morabito, E. Felip, R. Califano, S. Ghosh, A. Spira, S. N. Gettinger, M. Tiseo, N. Gupta, J. Haney, D. Kerstein, and S. Popat. "Brigatinib Versus Crizotinib in Alk-Positive Non-Small-Cell Lung Cancer." N Engl J Med 379, no. 21 (2018): 2027-39. [CrossRef]
- Horn, L., Z. Wang, G. Wu, E. Poddubskaya, T. Mok, M. Reck, H. Wakelee, A. A. Chiappori, D. H. Lee, V. Breder, S. Orlov, I. Cicin, Y. Cheng, Y. Liu, Y. Fan, J. G. Whisenant, Y. Zhou, V. Oertel, K. Harrow, C. Liang, L. Mao, G. Selvaggi, and Y. L. Wu. "Ensartinib Vs Crizotinib for Patients with Anaplastic Lymphoma Kinase-Positive Non-Small Cell Lung Cancer: A Randomized Clinical Trial." JAMA Oncol 7, no. 11 (2021): 1617-25. [CrossRef]
- Camidge, D. R., H. R. Kim, M. J. Ahn, J. C. H. Yang, J. Y. Han, M. J. Hochmair, K. H. Lee, A. Delmonte, M. R. García Campelo, D. W. Kim, F. Griesinger, E. Felip, R. Califano, A. Spira, S. N. Gettinger, M. Tiseo, H. M. Lin, N. Gupta, M. J. Hanley, Q. Ni, P. Zhang, and S. Popat. "Brigatinib Versus Crizotinib in Advanced Alk Inhibitor-Naive Alk-Positive Non-Small Cell Lung Cancer: Second Interim Analysis of the Phase Iii Alta-1l Trial." J Clin Oncol 38, no. 31 (2020): 3592-603. [CrossRef]
- Popat, S., G. Liu, S. Lu, G. Song, X. Ma, and J. C. Yang. "Brigatinib Vs Alectinib in Crizotinib-Resistant Advanced Anaplastic Lymphoma Kinase-Positive Non-Small-Cell Lung Cancer (Alta-3)." Future Oncol 17, no. 32 (2021): 4237-47. [CrossRef]
- Taniguchi, K., H. Konishi, A. Yoshinaga, M. Tsugane, H. Takahashi, F. Nishisaka, Y. Shishido, and A. Asai. "Efficacy of Combination Treatment Using Yho-1701, an Orally Active Stat3 Inhibitor, with Molecular-Targeted Agents on Cancer Cell Lines." Sci Rep 11, no. 1 (2021): 6685. [CrossRef]
- Camidge, D. R., H. R. Kim, M. J. Ahn, J. C. H. Yang, J. Y. Han, M. J. Hochmair, K. H. Lee, A. Delmonte, M. R. Garcia Campelo, D. W. Kim, F. Griesinger, E. Felip, R. Califano, A. I. Spira, S. N. Gettinger, M. Tiseo, H. M. Lin, Y. Liu, F. Vranceanu, H. Niu, P. Zhang, and S. Popat. "Brigatinib Versus Crizotinib in Alk Inhibitor-Naive Advanced Alk-Positive Nsclc: Final Results of Phase 3 Alta-1l Trial." J Thorac Oncol 16, no. 12 (2021): 2091-108. [CrossRef]
- Cooper, A. J., L. V. Sequist, and J. J. Lin. "Third-Generation Egfr and Alk Inhibitors: Mechanisms of Resistance and Management." Nat Rev Clin Oncol 19, no. 8 (2022): 499-514. [CrossRef]
- Shaw, A. T., B. J. Solomon, B. Besse, T. M. Bauer, C. C. Lin, R. A. Soo, G. J. Riely, S. I. Ou, J. S. Clancy, S. Li, A. Abbattista, H. Thurm, M. Satouchi, D. R. Camidge, S. Kao, R. Chiari, S. M. Gadgeel, E. Felip, and J. F. Martini. "Alk Resistance Mutations and Efficacy of Lorlatinib in Advanced Anaplastic Lymphoma Kinase-Positive Non-Small-Cell Lung Cancer." J Clin Oncol 37, no. 16 (2019): 1370-79. [CrossRef]
- Baba, K., and Y. Goto. "Lorlatinib as a Treatment for Alk-Positive Lung Cancer." Future Oncol 18, no. 24 (2022): 2745-66. [CrossRef]
- Hyman, D. M., S. A. Piha-Paul, H. Won, J. Rodon, C. Saura, G. I. Shapiro, D. Juric, D. I. Quinn, V. Moreno, B. Doger, I. A. Mayer, V. Boni, E. Calvo, S. Loi, A. C. Lockhart, J. P. Erinjeri, M. Scaltriti, G. A. Ulaner, J. Patel, J. Tang, H. Beer, S. D. Selcuklu, A. J. Hanrahan, N. Bouvier, M. Melcer, R. Murali, A. M. Schram, L. M. Smyth, K. Jhaveri, B. T. Li, A. Drilon, J. J. Harding, G. Iyer, B. S. Taylor, M. F. Berger, R. E. Cutler, Jr., F. Xu, A. Butturini, L. D. Eli, G. Mann, C. Farrell, A. S. Lalani, R. P. Bryce, C. L. Arteaga, F. Meric-Bernstam, J. Baselga, and D. B. Solit. "Her Kinase Inhibition in Patients with Her2- and Her3-Mutant Cancers." Nature 554, no. 7691 (2018): 189-94. [CrossRef]
- Harrison, P. T., S. Vyse, and P. H. Huang. "Rare Epidermal Growth Factor Receptor (Egfr) Mutations in Non-Small Cell Lung Cancer." Semin Cancer Biol 61 (2020): 167-79. [CrossRef]
- Sigismund, S., D. Avanzato, and L. Lanzetti. "Emerging Functions of the Egfr in Cancer." Mol Oncol 12, no. 1 (2018): 3-20. [CrossRef]
- Voldborg, B. R., L. Damstrup, M. Spang-Thomsen, and H. S. Poulsen. "Epidermal Growth Factor Receptor (Egfr) and Egfr Mutations, Function and Possible Role in Clinical Trials." Ann Oncol 8, no. 12 (1997): 1197-206. [CrossRef]
- Wu, S. G., and J. Y. Shih. "Management of Acquired Resistance to Egfr Tki-Targeted Therapy in Advanced Non-Small Cell Lung Cancer." Mol Cancer 17, no. 1 (2018): 38. [CrossRef]
- Yang, Z., A. Hackshaw, Q. Feng, X. Fu, Y. Zhang, C. Mao, and J. Tang. "Comparison of Gefitinib, Erlotinib and Afatinib in Non-Small Cell Lung Cancer: A Meta-Analysis." Int J Cancer 140, no. 12 (2017): 2805-19. [CrossRef]
- Cheng, H., H. Liu, Q. Du, H. Zhang, X. Zhang, Y. Wang, J. Shao, F. Yang, B. Zhang, J. Shi, Y. Liu, N. Wu, S. Xu, Q. Wei, Y. Sun, Q. Zhai, and B. Yu. "Efficacy and Safety of Domestic and Imported Gefitinib in Patients with Advanced Non-Small Cell Lung Cancer." Ann Palliat Med 10, no. 1 (2021): 10-15. [CrossRef]
- Çoban, Ö, and Z. Değim. "Development of Nanocochleates Containing Erlotinib Hcl and Dexketoprofen Trometamol and Evaluation of in Vitro Characteristic Properties." Turk J Pharm Sci 15, no. 1 (2018): 16-21. [CrossRef]
- Lynch, T. J., D. W. Bell, R. Sordella, S. Gurubhagavatula, R. A. Okimoto, B. W. Brannigan, P. L. Harris, S. M. Haserlat, J. G. Supko, F. G. Haluska, D. N. Louis, D. C. Christiani, J. Settleman, and D. A. Haber. "Activating Mutations in the Epidermal Growth Factor Receptor Underlying Responsiveness of Non-Small-Cell Lung Cancer to Gefitinib." N Engl J Med 350, no. 21 (2004): 2129-39. [CrossRef]
- Zhong, W. Z., Q. Wang, W. M. Mao, S. T. Xu, L. Wu, Y. C. Wei, Y. Y. Liu, C. Chen, Y. Cheng, R. Yin, F. Yang, S. X. Ren, X. F. Li, J. Li, C. Huang, Z. D. Liu, S. Xu, K. N. Chen, S. D. Xu, L. X. Liu, P. Yu, B. H. Wang, H. T. Ma, J. J. Yang, H. H. Yan, X. N. Yang, S. Y. Liu, Q. Zhou, and Y. L. Wu. "Gefitinib Versus Vinorelbine Plus Cisplatin as Adjuvant Treatment for Stage Ii-Iiia (N1-N2) Egfr-Mutant Nsclc: Final Overall Survival Analysis of Ctong1104 Phase Iii Trial." J Clin Oncol 39, no. 7 (2021): 713-22. [CrossRef]
- Fu, Y., Y. Zhang, Z. Lei, T. Liu, T. Cai, A. Wang, W. Du, Y. Zeng, J. Zhu, Z. Liu, and J. A. Huang. "Abnormally Activated Opn/Integrin Avβ3/Fak Signalling Is Responsible for Egfr-Tki Resistance in Egfr Mutant Non-Small-Cell Lung Cancer." J Hematol Oncol 13, no. 1 (2020): 169. [CrossRef]
- Harvey, R. D., V. R. Adams, T. Beardslee, and P. Medina. "Afatinib for the Treatment of Egfr Mutation-Positive Nsclc: A Review of Clinical Findings." J Oncol Pharm Pract 26, no. 6 (2020): 1461-74. [CrossRef]
- Passaro, A., T. Mok, S. Peters, S. Popat, M. J. Ahn, and F. de Marinis. "Recent Advances on the Role of Egfr Tyrosine Kinase Inhibitors in the Management of Nsclc with Uncommon, Non Exon 20 Insertions, Egfr Mutations." J Thorac Oncol 16, no. 5 (2021): 764-73. [CrossRef]
- Leonetti, A., S. Sharma, R. Minari, P. Perego, E. Giovannetti, and M. Tiseo. "Resistance Mechanisms to Osimertinib in Egfr-Mutated Non-Small Cell Lung Cancer." Br J Cancer 121, no. 9 (2019): 725-37. [CrossRef]
- Ramalingam, S. S., J. Vansteenkiste, D. Planchard, B. C. Cho, J. E. Gray, Y. Ohe, C. Zhou, T. Reungwetwattana, Y. Cheng, B. Chewaskulyong, R. Shah, M. Cobo, K. H. Lee, P. Cheema, M. Tiseo, T. John, M. C. Lin, F. Imamura, T. Kurata, A. Todd, R. Hodge, M. Saggese, Y. Rukazenkov, and J. C. Soria. "Overall Survival with Osimertinib in Untreated, Egfr-Mutated Advanced Nsclc." N Engl J Med 382, no. 1 (2020): 41-50. [CrossRef]
- Remon, J., C. E. Steuer, S. S. Ramalingam, and E. Felip. "Osimertinib and Other Third-Generation Egfr Tki in Egfr-Mutant Nsclc Patients." Ann Oncol 29, no. suppl_1 (2018): i20-i27. [CrossRef]
- Fan, J., and I. A. de Lannoy. "Pharmacokinetics." Biochem Pharmacol 87, no. 1 (2014): 93-120. [CrossRef]
- Evans, W. E., and H. L. McLeod. "Pharmacogenomics--Drug Disposition, Drug Targets, and Side Effects." N Engl J Med 348, no. 6 (2003): 538-49. [CrossRef]
- Mougey, E. B., J. E. Lang, X. Wen, and J. J. Lima. "Effect of Citrus Juice and Slco2b1 Genotype on the Pharmacokinetics of Montelukast." J Clin Pharmacol 51, no. 5 (2011): 751-60. [CrossRef]
- Harvey, A. L., R. Edrada-Ebel, and R. J. Quinn. "The Re-Emergence of Natural Products for Drug Discovery in the Genomics Era." Nat Rev Drug Discov 14, no. 2 (2015): 111-29. [CrossRef]
- Xu, L., Y. Li, Y. Dai, and J. Peng. "Natural Products for the Treatment of Type 2 Diabetes Mellitus: Pharmacology and Mechanisms." Pharmacol Res 130 (2018): 451-65. [CrossRef]
- Rodrigues, T., D. Reker, P. Schneider, and G. Schneider. "Counting on Natural Products for Drug Design." Nat Chem 8, no. 6 (2016): 531-41. [CrossRef]
- Liu, X., J. M. Simon, H. Xie, L. Hu, J. Wang, G. Zurlo, C. Fan, T. S. Ptacek, L. Herring, X. Tan, M. Li, A. S. Baldwin, W. Y. Kim, T. Wu, M. W. Kirschner, K. Gong, and Q. Zhang. "Genome-Wide Screening Identifies Sfmbt1 as an Oncogenic Driver in Cancer with Vhl Loss." Mol Cell 77, no. 6 (2020): 1294-306.e5. [CrossRef]
- Pan, H., C. Xue, B. J. Auerbach, J. Fan, A. C. Bashore, J. Cui, D. Y. Yang, S. B. Trignano, W. Liu, J. Shi, C. O. Ihuegbu, E. C. Bush, J. Worley, L. Vlahos, P. Laise, R. A. Solomon, E. S. Connolly, A. Califano, P. A. Sims, H. Zhang, M. Li, and M. P. Reilly. "Single-Cell Genomics Reveals a Novel Cell State During Smooth Muscle Cell Phenotypic Switching and Potential Therapeutic Targets for Atherosclerosis in Mouse and Human." Circulation 142, no. 21 (2020): 2060-75. [CrossRef]
- Warren, N. J. H., and A. Eastman. "Comparison of the Different Mechanisms of Cytotoxicity Induced by Checkpoint Kinase I Inhibitors When Used as Single Agents or in Combination with DNA Damage." Oncogene 39, no. 7 (2020): 1389-401. [CrossRef]
- Cuevas, C., and A. Francesch. "Development of Yondelis (Trabectedin, Et-743). A Semisynthetic Process Solves the Supply Problem." Nat Prod Rep 26, no. 3 (2009): 322-37. [CrossRef]
- Italiano, A., N. Touati, S. Litière, F. Collin, P. Pourquier, and A. Gronchi. "Prospective Assessment of the Predictive Value of the Brca1 Gene Status in Sarcoma Patients Treated with Trabectedin: An Updated Analysis of the Eortc 62091 Trial." Cancer Med 7, no. 5 (2018): 1575-77. [CrossRef]
- Monk, B. J., D. Lorusso, A. Italiano, S. B. Kaye, M. Aracil, A. Tanović, and M. D'Incalci. "Trabectedin as a Chemotherapy Option for Patients with Brca Deficiency." Cancer Treat Rev 50 (2016): 175-82. [CrossRef]
- Risdon, E. N., C. H. Chau, D. K. Price, O. Sartor, and W. D. Figg. "Parp Inhibitors and Prostate Cancer: To Infinity and Beyond Brca." Oncologist 26, no. 1 (2021): e115-e29. [CrossRef]
- Yoshida, K., and Y. Miki. "Role of Brca1 and Brca2 as Regulators of DNA Repair, Transcription, and Cell Cycle in Response to DNA Damage." Cancer Sci 95, no. 11 (2004): 866-71. [CrossRef]
- Evans, D. G. "Neurofibromatosis 2." In Genereviews(®), edited by M. P. Adam, G. M. Mirzaa, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. W. Gripp and A. Amemiya. Seattle (WA): University of Washington, Seattle Copyright © 1993-2023, University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved., 1993.
- van der Valk, M. J. M., D. E. Hilling, E. Bastiaannet, E. Meershoek-Klein Kranenbarg, G. L. Beets, N. L. Figueiredo, A. Habr-Gama, R. O. Perez, A. G. Renehan, and C. J. H. van de Velde. "Long-Term Outcomes of Clinical Complete Responders after Neoadjuvant Treatment for Rectal Cancer in the International Watch & Wait Database (Iwwd): An International Multicentre Registry Study." Lancet 391, no. 10139 (2018): 2537-45. [CrossRef]
- Skiles, J. L., C. Chiang, C. H. Li, S. Martin, E. L. Smith, G. Olbara, D. R. Jones, T. A. Vik, S. Mostert, F. Abbink, G. J. Kaspers, L. Li, F. Njuguna, T. J. Sajdyk, and J. L. Renbarger. "Cyp3a5 Genotype and Its Impact on Vincristine Pharmacokinetics and Development of Neuropathy in Kenyan Children with Cancer." Pediatr Blood Cancer 65, no. 3 (2018). [CrossRef]
- Maki, K., T. Harada, Y. Akiyama, H. Ogasawara, and F. Kishi. "Giant Cell Tumor of the Rib." Intern Med 46, no. 14 (2007): 1151-2. [CrossRef]
- Egbelakin, A., M. J. Ferguson, E. A. MacGill, A. S. Lehmann, A. R. Topletz, S. K. Quinney, L. Li, K. C. McCammack, S. D. Hall, and J. L. Renbarger. "Increased Risk of Vincristine Neurotoxicity Associated with Low Cyp3a5 Expression Genotype in Children with Acute Lymphoblastic Leukemia." Pediatr Blood Cancer 56, no. 3 (2011): 361-7. [CrossRef]
- Uittenboogaard, A., C. L. G. Neutel, J. C. F. Ket, F. Njuguna, A. D. R. Huitema, G. J. L. Kaspers, and M. E. van de Velde. "Pharmacogenomics of Vincristine-Induced Peripheral Neuropathy in Children with Cancer: A Systematic Review and Meta-Analysis." Cancers (Basel) 14, no. 3 (2022). [CrossRef]
- Losuwannarak, N., A. Maiuthed, N. Kitkumthorn, A. Leelahavanichkul, S. Roytrakul, and P. Chanvorachote. "Gigantol Targets Cancer Stem Cells and Destabilizes Tumors Via the Suppression of the Pi3k/Akt and Jak/Stat Pathways in Ectopic Lung Cancer Xenografts." Cancers (Basel) 11, no. 12 (2019). [CrossRef]
- Bhummaphan, N., and P. Chanvorachote. "Gigantol Suppresses Cancer Stem Cell-Like Phenotypes in Lung Cancer Cells." Evid Based Complement Alternat Med 2015 (2015): 836564. [CrossRef]
- Losuwannarak, N., S. Roytrakul, and P. Chanvorachote. "Gigantol Targets Myc for Ubiquitin-Proteasomal Degradation and Suppresses Lung Cancer Cell Growth." Cancer Genomics Proteomics 17, no. 6 (2020): 781-93. [CrossRef]
- Verweij, J., M. Clavel, and B. Chevalier. "Paclitaxel (Taxol) and Docetaxel (Taxotere): Not Simply Two of a Kind." Ann Oncol 5, no. 6 (1994): 495-505. [CrossRef]
- McCorkle, J. R., J. W. Gorski, J. Liu, M. B. Riggs, A. B. McDowell, N. Lin, C. Wang, F. R. Ueland, and J. M. Kolesar. "Lapatinib and Poziotinib Overcome Abcb1-Mediated Paclitaxel Resistance in Ovarian Cancer." PLoS One 16, no. 8 (2021): e0254205. [CrossRef]
- van Eerden, R. A. G., L. van Doorn, F. M. de Man, N. Heersche, M. Doukas, T. P. P. van den Bosch, E. Oomen-de Hoop, P. de Bruijn, S. Bins, E. Ibrahim, S. Nikkessen, L. E. Friberg, S. L. W. Koolen, M. C. W. Spaander, and R. H. J. Mathijssen. "Tissue Type Differences in Abcb1 Expression and Paclitaxel Tissue Pharmacokinetics in Patients with Esophageal Cancer." Front Pharmacol 12 (2021): 759146. [CrossRef]
- Bhummaphan, N., N. Petpiroon, O. Prakhongcheep, B. Sritularak, and P. Chanvorachote. "Lusianthridin Targeting of Lung Cancer Stem Cells Via Src-Stat3 Suppression." Phytomedicine 62 (2019): 152932. [CrossRef]
- Wu, Q., W. Qian, X. Sun, and S. Jiang. "Small-Molecule Inhibitors, Immune Checkpoint Inhibitors, and More: Fda-Approved Novel Therapeutic Drugs for Solid Tumors from 1991 to 2021." J Hematol Oncol 15, no. 1 (2022): 143. [CrossRef]
- Nam, A. S., R. Chaligne, and D. A. Landau. "Integrating Genetic and Non-Genetic Determinants of Cancer Evolution by Single-Cell Multi-Omics." Nat Rev Genet 22, no. 1 (2021): 3-18. [CrossRef]
- Jardim, D. L., A. Goodman, D. de Melo Gagliato, and R. Kurzrock. "The Challenges of Tumor Mutational Burden as an Immunotherapy Biomarker." Cancer Cell 39, no. 2 (2021): 154-73. [CrossRef]
- Pareek, C. S., R. Smoczynski, and A. Tretyn. "Sequencing Technologies and Genome Sequencing." J Appl Genet 52, no. 4 (2011): 413-35. [CrossRef]
- Reuter, J. A., D. V. Spacek, and M. P. Snyder. "High-Throughput Sequencing Technologies." Mol Cell 58, no. 4 (2015): 586-97. [CrossRef]
- Hwang, B., J. H. Lee, and D. Bang. "Single-Cell Rna Sequencing Technologies and Bioinformatics Pipelines." Exp Mol Med 50, no. 8 (2018): 1-14. [CrossRef]
- Ngamcherdtrakul, W., and W. Yantasee. "Sirna Therapeutics for Breast Cancer: Recent Efforts in Targeting Metastasis, Drug Resistance, and Immune Evasion." Transl Res 214 (2019): 105-20. [CrossRef]
- Mu, Q., J. Yu, L. A. McConnachie, J. C. Kraft, Y. Gao, G. K. Gulati, and R. J. Y. Ho. "Translation of Combination Nanodrugs into Nanomedicines: Lessons Learned and Future Outlook." J Drug Target 26, no. 5-6 (2018): 435-47. [CrossRef]
- Stuver, R., G. L. Shah, N. S. Korde, L. E. Roeker, A. R. Mato, C. L. Batlevi, D. J. Chung, S. Doddi, L. Falchi, B. Gyurkocza, A. Hamilton, Y. H. Lin, A. A. Jakubowski, E. Joffe, H. L. Landau, R. J. Lin, S. Mailankody, M. L. Palomba, J. H. Park, M. A. Perales, D. M. Ponce, L. V. Ramanathan, G. A. Salles, M. Scordo, S. K. Seo, U. A. Shah, E. M. Stein, D. Straus, S. Z. Usmani, J. W. Young, A. D. Zelenetz, A. Noy, and S. A. Vardhana. "Activity of Azd7442 (Tixagevimab-Cilgavimab) against Omicron Sars-Cov-2 in Patients with Hematologic Malignancies." Cancer Cell 40, no. 6 (2022): 590-91. [CrossRef]
- Cilento, M. E., Y. T. Ong, P. R. Tedbury, and S. G. Sarafianos. "Drug Interactions in Lenacapavir-Based Long-Acting Antiviral Combinations." Viruses 14, no. 6 (2022). [CrossRef]
- Liang, Z., Y. He, and X. Hu. "Cardio-Oncology: Mechanisms, Drug Combinations, and Reverse Cardio-Oncology." Int J Mol Sci 23, no. 18 (2022). [CrossRef]
- McDonald, E. S., A. S. Clark, J. Tchou, P. Zhang, and G. M. Freedman. "Clinical Diagnosis and Management of Breast Cancer." J Nucl Med 57 Suppl 1 (2016): 9s-16s. [CrossRef]
- Ho, D., S. R. Quake, E. R. B. McCabe, W. J. Chng, E. K. Chow, X. Ding, B. D. Gelb, G. S. Ginsburg, J. Hassenstab, C. M. Ho, W. C. Mobley, G. P. Nolan, S. T. Rosen, P. Tan, Y. Yen, and A. Zarrinpar. "Enabling Technologies for Personalized and Precision Medicine." Trends Biotechnol 38, no. 5 (2020): 497-518. [CrossRef]
- Litman, T. "Personalized Medicine-Concepts, Technologies, and Applications in Inflammatory Skin Diseases." Apmis 127, no. 5 (2019): 386-424. [CrossRef]
- Botella, P., and E. Rivero-Buceta. "Safe Approaches for Camptothecin Delivery: Structural Analogues and Nanomedicines." J Control Release 247 (2017): 28-54. [CrossRef]
- Thomford, N. E., D. A. Senthebane, A. Rowe, D. Munro, P. Seele, A. Maroyi, and K. Dzobo. "Natural Products for Drug Discovery in the 21st Century: Innovations for Novel Drug Discovery." Int J Mol Sci 19, no. 6 (2018). [CrossRef]

Table 1.
Screening and comparison of drug targets based on chemical proteomics.
| Technology | Principle | Advantage | Disadvantage |
| AfBPP | Affinity of target proteins to active small molecules on stationary phases | 1. No bias; 2. Systematic study of total protein; 3. It can enrich the target and is suitable for identification of low abundance proteins. |
1. A detailed understanding of the structure-activity relationship of active molecules is required; 2. Chemical derivatization of active molecules is required; 3. Targets with low abundance and low affinity are easy to be washed off; 4. Probes usually cannot enter cells. |
| ABPP | The target protein forms a covalent bond with a covalent small molecule. | 1. No bias; 2. Systematic study of whole protein; 3. It can enrich the target and is suitable for identification of low abundance proteins; Grasp low affinity targets; 4. Probes usually get into cells. |
1. A thorough understanding of the structure-activity relationship of active molecules is required; 2. Chemical derivatization of active molecules is required; 3. Non-specific covalent binding is easy to occur. |
| TPP | The thermal stability of the target protein increases after binding with small molecules and it is not easy to precipitate | 1. No bias; 2. Systematic study of whole protein; 3. No derivations of small active molecules are required. |
1. Limited effect on extreme conditions, such as heat insensitivity or heat unstable proteins; 2. Further measures should be taken to reduce the complexity of samples so as to realize the identification of low abundance proteins. |
| DARTS | The stability of the target protein increases after binding with small molecules and is not easily degraded by enzymes | 1. No bias; 2. Systematic study of whole protein; 3. No derivations of small active molecules are required. |
1. The protein that is not sensitive to enzyme digestion has limited effect; 2. Further measures should be taken to reduce the complexity of samples so as to realize the identification of low abundance proteins. |
Table 2.
FDA approved personalized antitumor drugs.
| Seq_ID | Medicine | Personalized tag | Approval time | Molecular formula | Mechanism of action | Disease |
| 1 | Crizotinib | ALK+ | 2014 | 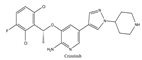 |
ALK inhibitor | Metastatic non-small cell lung cancer with ALK or ROS1 positive |
| 2 | Ceritinib | ALK+ | 2014 | 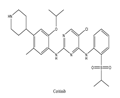 |
ALK inhibitor | Non-small-cell lung cancer |
| 3 | Alectinib | ALK+ | 2015 | 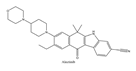 |
ALK inhibitor | Non-small-cell lung cancer |
| 4 | Brigatinib | ALK+ | 2017 | 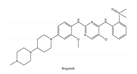 |
ALK inhibitor | Non-small-cell lung cancer |
| 5 | Lorlatinib | ALK+ is positive | 2018 | 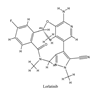 |
A dual-target inhibitoror of ALK/ROS1 | Non-small-cell lung cancer |
| 6 | Gefitinib | EGFR | 2003 | 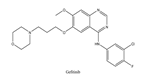 |
EGFR inhibitor | Non-small-cell lung cancer |
| 7 | Erlotinib | EGFR | 2004 | 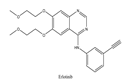 |
EGFR inhibitor | Non-small-cell lung cancer |
| 8 | Afatinib | EGFR | 2013 | 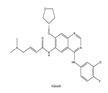 |
EGFR inhibitor | Non-small-cell lung cancer |
| 9 | Osimertinib | EGFR | 2015 | 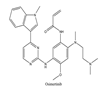 |
EGFR inhibitor | Non-small-cell lung cancer |
Table 3.
Pharmacogenetics in the pharmacological targets and pathways of natural products.
| Seq_ID | Natural products | Main Sources | Molecular formula | Related Gene | Disease |
| 1 | Trabectedin | Ecteinascidia turbinata | 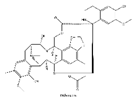 |
BRCA1, BRCA2 | Soft tissue sarcoma, Breast cancer |
| 2 | Vincristine | Catharanthus roseus | 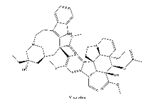 |
CYP3A enzymes, ABC transporters |
Leukemias, Lymphomas, Brain tumors, Solid tumors |
| 5 | Paclitaxel | Taxus baccata Linn | 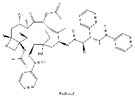 |
ABCB1 G2677T/A mutation | Ovarian cancer |
| 3 | Gigantol | Dendrobium draconis | 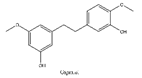 |
CD133, ALDH1A1 | Non-small-cell lung cancer |
| 6 | Chrysotoxine | Dendrobium pulchellum | 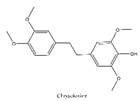 |
ABCG2 | Lung cancer |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



