
Submitted:
29 March 2023
Posted:
30 March 2023
You are already at the latest version
Abstract
Polycystic ovary syndrome (PCOS) is increasingly being characterized as an evolutionary mismatch disorder that presents with a complex mixture of metabolic and endocrine symptoms. The Evolutionary Model proposes that PCOS arises from a collection of inherited polymorphisms that have been consistently demonstrated in a variety of ethnic groups and races. In-utero developmental programming of susceptible genomic variants are thought to predispose the offspring to develop PCOS. Postnatal exposure to lifestyle and environmental risk factors results in epigenetic activation of developmentally programmed genes and disturbance of the hallmarks of health. The resulting pathophysiological changes represent the consequences of poor-quality diet, sedentary behaviour, endocrine disrupting chemicals, stress, circadian disruption, and other lifestyle factors. Emerging evidence suggests that lifestyle-induced gastrointestinal dysbiosis plays a central role in the pathogenesis of PCOS. Lifestyle and environmental exposures initiate changes that result in disturbance of the gastrointestinal microbiome (dysbiosis), immune dysregulation (chronic inflammation), altered metabolism (insulin resistance), endocrine and reproductive imbalance (hyperandrogenism), and central nervous system dysfunction (neuroendocrine, autonomic nervous system). PCOS can be a progressive metabolic condition that leads to obesity, gestational diabetes, type 2 diabetes, metabolic-associated fatty liver disease, metabolic syndrome, cardiovascular disease, and cancer. This review explores the mechanisms that underpin the evolutionary mismatch between ancient survival pathways and contemporary lifestyle factors involved in the pathogenesis and pathophysiology of PCOS.
Keywords:
polycystic ovary syndrome
; evolution
; inflammation
; insulin resistance
; hyperinsulinemia
; immune
; infertility
; endocrine disrupting chemicals
; environment
; lifestyle
; diet
1. Introduction
There is general agreement that PCOS is a polygenic multisystem disorder arising from an interaction between genetic and environmental factors (1). Comprehensive International Guidelines recommend a range of lifestyle-based interventions as first-line management for all women diagnosed with PCOS (2). These recommendations are based on evidence that lifestyle therapies, such as diet and exercise, can control and reverse many of the biochemical and endocrine features of PCOS (2,3). It has been hypothesized that contemporary lifestyle and environmental exposures are instrumental in the pathogenesis of PCOS due to a mismatch between our ancient and modern lifestyle and environment (1,4–6).
PCOS affects 8-13% of reproductive aged women, is thought to be increasing in prevalence globally, and is estimated to affect up to 200 million women world-wide (6,7). Women affected with PCOS present with a wide variety of symptoms (menstrual disturbance, acne, hirsutism, alopecia, subfertility, anxiety and depression) that reflect the underlying multisystem pathophysiology (8–10). Women with PCOS have an increased risk of pregnancy complications (deep venous thrombosis, pre-eclampsia, macrosomia, growth restriction, miscarriage, stillbirth and preterm labor) (11), psychological problems (anxiety, depression) (12), and can progress to a range of other metabolic-related conditions (obesity, gestational diabetes, type 2 diabetes (T2DM), metabolic-associated fatty liver disease, chronic kidney disease, metabolic syndrome, cardiovascular disease, and cancer) (13–16). The population attributable risk of PCOS to T2DM alone has been estimated at 19-28% of women of reproductive age (17). PCOS can be a progressive metabolic condition and therefore makes a significant contribution to the chronic disease epidemic (18).
Genome-wide association studies (GWAS) have identified common PCOS risk alleles in women from Chinese and European populations, suggesting PCOS is an ancient inherited disorder that was present before humans migrated out of Africa (19,20). Familial and twin studies (21,22) and more recent Mendelian randomization (23,24) and transcriptome-wide association studies (TWAS) (25) also support a genetic basis to PCOS. From an evolutionary perspective, decades of research has therefore characterized PCOS as an inherited polygenic trait that manifests after exposure to lifestyle and environmental risk factors (1,26,27). Nevertheless, it is thought that genetic factors contribute less than 10% to disease susceptibility, as has been found with other lifestyle-related chronic diseases such as obesity and T2DM (28). Evolutionary medicine provides a framework for understanding the pathogenesis of complex chronic diseases, including PCOS (1,29,30).
Evolutionary medicine is a discipline that involves the study of evolutionary processes as they relate to human traits and diseases (31). This involves translating research related to the origins of complex chronic diseases into the practice of clinical medicine. Evolutionary medicine incorporates considerations of the principles of natural selection (29), metabolic and reproductive trade-offs (30,32), relative reproductive fitness (30), mismatch between our evolutionary past and contemporary environment (33), and other evolutionary conflicts (7,34). Many of these evolutionary mechanisms have been associated with the pathophysiological processes identified in women with PCOS and will be discussed in the following sections of this review.
A number of evolutionary hypotheses have been developed to try to explain the pathogenesis of PCOS (1,5,26,27,35). These hypotheses refer to the relative importance of ancient metabolic survival adaptations versus improved reproductive success, which become maladaptive in modern human populations. These survival adaptations usually involve alterations to physiological, immune, metabolic, and endocrine pathways (36). Metabolism can be defined as the sum total of biochemical reactions that organisms employ for bioenergetics, creation of biomass, and for cell fate regulation (37,38). It has been hypothesized that PCOS may represent an evolutionary metabolic adaptation to balance energy substrate availability (glucose and fatty acid) to optimize reproduction (5). This shift in focus to the importance of metabolic adaptation and dysregulation has also been emphasized in the International Guidelines (2,39).
The prime directive of all life is to optimize reproduction and species survival (40). Reproduction and metabolism are intimately linked so that optimal reproductive fitness requires optimal metabolism (31,41). There is always an evolutionary trade-off to optimize metabolism and/or reproduction, depending on the species and prevailing environmental conditions (32). This is achieved by a complex network of hormonal and signaling molecules that link metabolism to reproductive cycles, via hormonal regulatory processes, post-translational modification of enzymes, substrate level inhibition of metabolic pathways, and epigenetic regulation of gene expression (42–44).
Attention has therefore been directed at identifying the mechanisms by which lifestyle and environmental exposures alter this regulatory framework (45–47). A root-cause analysis of the proximate causes of PCOS has identified a wide variety of lifestyle and environmental exposures that are likely to contribute to the pathogenesis of PCOS (1,2,5,48). These include, diet and nutritional factors, exercise and sedentary behavior, sleep and circadian disruption, endocrine disrupting chemicals, stress, direct and indirect effects of climate change, and community support systems (1). Contemporary lifestyle exposures are significantly different to the environmental conditions that existed throughout most of human evolution. Namely, starvation, predation, fear, increased maternal mortality and exposure to different climatic conditions (4,27,49). The reduction of chronic disease following lifestyle interventions such as diet, exercise and smoking cessation, has provided strong evidence for contemporary lifestyle as the primary “cause” of many diseases, including PCOS (50).
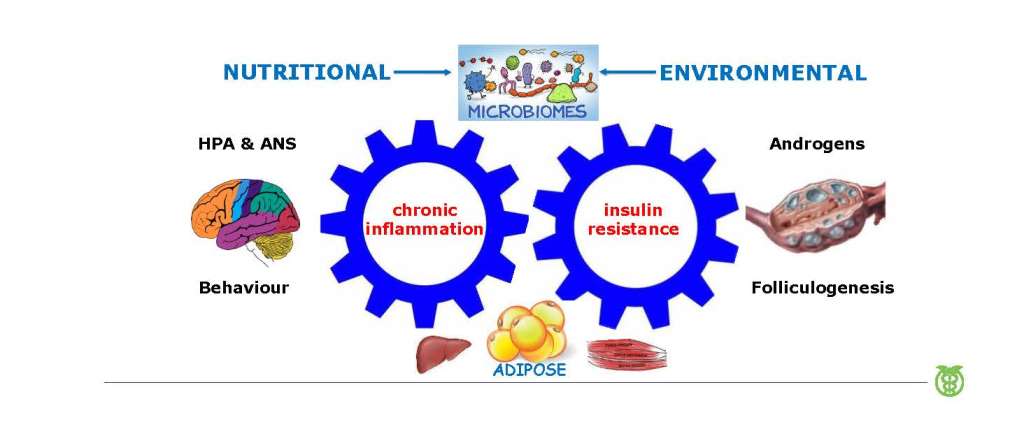
Epigenetic modifications such as DNA methylation, histone modification and noncoding RNA, control reversible gene expression and reprogramming (51,52). Control of gene expression, and resulting protein synthesis, is a dynamic process that depends on hormonal and cell signaling events that are influenced by lifestyle and environmental factors (52). Optimal health and well-being is achieved when metabolic, cellular and whole-body homeostasis is synchronized with the prevailing environmental conditions (42). Multiple adaptive survival mechanisms (immune, metabolic, neurological, hormonal) have evolved to ensure re-establishment of homeostasis during periods of environmental and personal stress, and work synergistically to maintain health (4,5,27,53,54). Epigenetic modifications provide a mechanism for organisms to rapidly respond to changing environmental conditions to ensure survival and restore homeostasis (52). Rapid social evolution has created a contemporary lifestyle and environment that is out of step with our evolutionary past (1,4). Chronic disturbance to adaptive cellular signaling systems disrupts the epigenetic machinery and other regulatory processes, resulting in maladaptive metabolic, immune and physiological responses, that manifest as complex chronic diseases such as obesity, T2DM and PCOS (Figure 1) (55,56).
It is now appreciated that more than one pathophysiological mechanism is involved in the development of PCOS (57,58). When viewed from an evolutionary perspective, PCOS represents dysregulation of the hallmarks of health (Table 2) (59). This narrative review outlines the relationship between lifestyle and environmental risk factors and the underlying pathophysiological mechanisms identified in women with PCOS. These include immune dysregulation (chronic systemic inflammation, oxidative stress), metabolic dysfunction (insulin resistance (IR), hyperglycaemia, hyperinsulinemia), hormonal dysregulation (hyperandrogenism, estrogen, follicle stimulating hormone, luteinizing hormone) and gastrointestinal dysbiosis (decreased alpha diversity, increased gastrointestinal mucosal permeability) (Figure 1). The pathological processes are discussed in the context of the Evolutionary Model of PCOS (1).
2. Materials and Methods
The literature search focused on research publications related to the pathophysiology and pathogenesis of PCOS using the keywords listed above and related mesh terms for data on the evolutionary aspects of PCOS, chronic systemic inflammation, in-utero developmental epigenetic programming, insulin resistance, hyperinsulinemia, hyperandrogenism, reproductive changes, infertility, microbiome, dysbiosis, endocrine disrupting chemicals, lifestyle, diet, and physical activity. The databases searched included PubMed, Scopus, Cochrane, and Google Scholar. The literature has been searched repeatedly over the past 10 years. A glossary of abbreviations is included.
The present manuscript provides a summary of the pathogenesis and pathophysiology of PCOS in the context of an Integrated Evolutionary Model (1).
3. Chronic Systemic Inflammation
3.1. Evolution and the advantages of a proinflammatory design
Inflammation is a normal physiological process that is an evolutionary conserved homeostatic mechanism in cells and tissues throughout the body (60). Optimal health is achieved when a balance between pro- and anti-inflammatory processes removes aging, damaged or infected cells, and restores normal cellular function (61). Inflammation is a protective mechanism in response to specific environmental conditions and occurs at a cost to normal tissue function (62,63). Acute inflammation is a physiological process that is spatially and temporally limited by multiple mechanisms (59). Local anti-inflammatory mediators attempt to limit the systemic spread of inflammation and contain the inflammatory response. Temporal limitation, or resolution of inflammation, is facilitated by removal of the primary cause, local negative feedback loops, systemic regulation by the autonomic nervous system (ANS) (64), and glucocorticoids (59). Chronic low-grade systemic inflammation can occur as a result of failure of any of these homeostatic mechanisms and is a cornerstone of PCOS pathophysiology (65). Large systematic reviews confirm an important role for chronic systemic inflammation in the pathogenesis of PCOS (8,56).
Rapid changes in the contemporary human environment have outpaced genetic adaptation, leading to a mismatch between our modern exposures and selected metabolic and reproductive traits. This mismatch has resulted in a dysregulated inflammatory response that has increased susceptibility to many common chronic diseases including obesity, type 2 diabetes, metabolic syndrome, cardiovascular disease, neuroinflammatory diseases, and PCOS (61). Chronic low-grade inflammation, or metaflammation, caused by poor-quality diet, nutritional excess, and other environmental factors, is maintained at a subacute level over long periods of time, enhancing inflammatory and metabolic signal transduction pathways that lead to the symptoms and diseases associated with PCOS (66). The concept of metaflammation refers to the pathophysiological association between metabolic disorders and the immune system and is proposed to originate from the evolutionary crosstalk between immune and metabolic pathways (63).
The central role of chronic systemic inflammation and metabolic dysregulation and their relationship to evolution, obesity, the microbiota, endocrine disrupting chemicals, and the recently described “Hallmarks of Health” are described in the following sections of this review. There is a particular focus on the molecular details and mechanisms involved in the pathophysiology.
3.2. Overview of the inflammatory response
The human body has two parallel systems of cellular defense (innate and adaptive immunity) that work co-operatively to protect cells, individuals, and ultimately the species (67). Billions of years of evolution has equipped unicellular organisms with a range of stress responses to abiotic environmental threats such as temperature, salinity, sunlight exposure, heavy metals and oxygen (68). In addition, multicellular organisms possess elaborate innate and adaptive immune responses to defend against exposure to noninfectious (reactive oxygen species, uric acid, cholesterol, microparticles, and exosomes), and infectious agents (bacteria, viruses, protozoa, parasites, fungi) (69). These responses are activated by a range of biological mechanisms including oxidative stress and reactive oxygen species (ROS), advanced glycation end-products (AGE), and via pattern recognition receptors (PRR) (70).
3.2.1. Oxidative stress in PCOS
The evolution of life chemistry and metabolism dates back 3.5 billion years (71). The first cellular life-forms arose in an anaerobic environment and most of the pathways of intermediate metabolism (glycolysis, fatty acid synthesis and oxidation, pentose phosphate pathway, Krebs cycle, electron transport, and many more), developed in an environment where oxygen was toxic (72). As a consequence, mammalian cells can revert to these “primitive” metabolic pathways to survive in hypoxic environments such as cancer, (known as the Warburg effect) and during times of cellular stress (73). Approximately 2.4 billion years ago cyanobacteria started producing oxygen from photosynthesis, raising the atmospheric oxygen to 2-4%. A billion years later, during the Pre-Cambrian period, oxygen levels rose, and multicellular organisms flourished. This resulted in the ability of cells to use oxygen to make adenosine triphosphate (ATP), produce ROS (superoxide, hydroxyl radical) as a metabolic byproduct, and for the purposes of cell signaling and defense, and to detoxify oxygen with a variety of antioxidant systems (74). ROS contain an unpaired electron that makes them extremely unstable and reactive. ROS attempt to stabilize themselves by scavenging electrons from healthy cells and cause oxidative damage.
Living cells can be differentiated from dead cells because of cessation of the coordinated flow of energy, that occurs due to electron transfer from one molecule to another during metabolism, following failure of adaptive responses to restore cellular homeostasis (75). Oxygen acts as the terminal electron acceptor from the cytochrome c oxidase enzyme in the mitochondria, at an extremely low concentration of 0.2 Torr. There is therefore an 800-fold oxygen gradient from atmospheric air (160 Torr) to interstitial tissue (30 Torr), to the inner membrane of the mitochondria (0.2 Torr). When the flow of electrons to the mitochondrial electron transport chain is disrupted by environmental factors such as microbial infection, chemical toxins, accumulation of metabolic intermediates due to nutritional excess, and other cellular stressors, metabolic mismatch occurs (74). Electrons are diverted away from mitochondria, mitochondrial oxygen consumption falls, and cytoplasmic oxygen rises. This redox imbalance creates ROS and reactive nitrogen species (RNS) that initiate innate immune responses designed to defend and protect the cell (76). Mitochondrial structure, dynamics, biogenesis, and membrane potential, are altered in women with PCOS (77).
The body has an in-built system of antioxidants to stabilize and neutralize ROS and protect the cell. Antioxidants are highly stable molecules that have the unique ability to serve as electron donors to help stabilize free radicals, without becoming reactive themselves. Oxidative stress refers to the imbalance between the production of oxidant species and antioxidant defenses, and generation of excessive amounts of ROS that underlie the various forms of cell death (78). Oxidative stress can arise from endogenous (leakage of ROS from mitochondrial oxidative phosphorylation, cytochrome P-450 detoxification enzyme systems, peroxisomal oxidases, nicotinamide dinucleotide adenine phosphate oxidases) or exogenous sources (environmental chemicals, cigarette smoke, alcohol, ionizing radiation, microbial infection, stress, sleep deprivation) (78,79), and is a potent stimulator of inflammation (80,81). Oxidative stress has been found to play a central role in the pathogenesis of PCOS (78). Oxidative stress can impair insulin signaling and cause IR (82), dysregulate follicular calcium which results in reproductive and menstrual dysfunction (83), oxidize plasma proteins that may act as pro-inflammatory mediators (84), cause lipid peroxidation (85), and induce DNA damage (86), in women with PCOS.
Cumulative studies show an association between oxidative stress and PCOS (76,78). In addition, oxidant and antioxidant status has been found to vary between individuals because of differences in diet, lifestyle, and enzymatic and dietary antioxidants (76,80). A recent case-control study showed that plant-based dietary pattern is associated with lower odds ratio of PCOS and suggested that antioxidant-rich foods may protect the body against oxidative damage (87). Dietary total antioxidant capacity was subsequently assessed using the Nutrient Data Laboratory of United States Department of Agriculture reference values. The investigators reported a significant reduction in the odds of PCOS in women that consumed a high total antioxidant containing plant-based diet (88). These findings support the existing body of dietary pattern research that recommend healthy diets for the management of PCOS (2,89).
3.2.2. Advanced glycation end products and PCOS
Advanced glycation end-products are reactive molecules that are formed by non-enzymatic reactions of carbohydrates with proteins, lipids or nucleic acids (90). Advanced glycation end-products result in irreversible cross-linking of proteins and loss of protein structure and function, and can initiate apoptosis (91). Advanced glycation end-products can be generated endogenously under normal conditions, can be ingested in food, particularly a cooked fast-food diet, and with cigarette smoking (92). Advanced glycation end-products can cause oxidative stress and inflammation resulting in cellular and tissue damage, when produced or ingested in excessive amounts (93). Protective circulating anti-inflammatory receptors called soluble receptor for advanced glycation end products and membrane-bound receptor of advanced glycation end-products (RAGE) are associated with protection against AGE (94).
The interaction of AGE with their membrane receptors activates intracellular signaling pathways that lead to increased oxidative stress, inflammation, IR, diabetes, hyperandrogenism, obesity, and ovulatory dysfunction, all of which have been associated with PCOS (95,96). Recent data have shown elevated circulating levels of AGE and increased expression of RAGE receptors in ovarian tissue (97,98). Proinflammatory AGE-RAGE signaling has been found to cause altered steroidogenesis and follicle development in ovarian granulosa cells in PCOS (99,100). In addition, hyperandrogenism induces endoplasmic reticulum stress in granulosa cells, resulting in increased accumulation of AGE in the ovary (98).
Modern western diets are rich in AGE which are absorbed through the intestine (96). A high-glycemic diet and excessive glucose ingestion results in elevated blood glucose levels and the generation of AGE that bind with cell membrane-bound RAGE and activate inflammation (101,102). Ligand binding by glycated proteins and lipids to RAGE stimulates intracellular signaling events that activate nuclear factor kappa-light-chain-enhancer of activated B (NF-κB). Nuclear factor kappa B controls several genes involved in inflammation, and RAGE itself is upregulated by NF-κB, establishing a positive feedback cycle that leads to chronic inflammation (102).
Studies have demonstrated that the intake of low-AGE containing diet is associated with favourable metabolic and hormonal profile, as well as less oxidative stress biomarkers in patients with PCOS (103). One study employed a low-AGE diet consisting of Mediterranean-style foods cooked at temperatures below 180 degrees by boiling, poaching, stewing or steaming (103). High temperature cooking above 220 degrees Celsius by roasting, grilling, and baking was avoided. Dietary recommendations for minimising ingestion of AGE includes increasing consumption of a whole-foods that includes vegetables, fruits, seafood, whole-grains, while reducing the consumption of high AGE containing foods. These include, highly processed foods (packaged meats, cheese and snack-foods), excessive sugar in sweets and beverages, and fried foods (89,96). Adoption of other healthy lifestyle behaviors such as exercise, maintaining normal body weight, and cessation of tobacco consumption are also important for reducing AGE (96,104).
3.2.3. Pattern recognition receptors and the innate immune system
The existence of receptors expressed by innate immune cells that were responsible for detecting microbial products was first proposed by Charles Janeway in 1989 (105). Polly Matzinger subsequently proposed the “Danger Theory” suggesting that the immune system produced molecules that initiate and propagate inflammation, in response to tissue stress, damage or infection (106). In 2013 Robert Naviaux further expanded this concept to the “Cell Danger Response”(CDR) (107). Naviaux proposed that evolutionary selection has preserved a similar response to a variety of threats, as cells have a limited number of ways they can mobilize existing cellular machinery and energy. The CDR is therefore an evolutionary-conserved cellular protective response that is activated when a cell encounters a chemical, physical, or microbial threat that could injure or kill the cell (107). More recent research has characterized the molecular details of an elaborate danger detection system involving PRR, damage-associated molecular patterns (DAMPS), pathogen-associated molecular patterns (PAMPS), inflammasomes, and an intricate system of signalling molecules that activate a system-wide network of innate and adaptive immune responses (70). This has been called the “Integrated Stress Response” and is reviewed in detail in previous reports (108,109).
There are five types of PRR that can be classified in two main groups based on their cellular localization (110). Toll-like receptors (TLR) and C-type lectin receptors (CLR) are transmembrane receptors that allow sensing of host-derived DAMPS and PAMPS, at the cell surface or within membrane-bound intracellular compartments. Nucleotide oligomerization domain-like receptors, retinoic acid- inducible gene-1-like receptors and absent in melanoma-2-like receptors are cytoplasmic-based and provide an intracellular recognition system for sensing DAMPS and PAMPS. Pattern recognition receptors are also excreted extracellularly and can be found in interstitial fluid and the blood stream, where they play an important role in pathogen recognition (70). Pattern recognition receptors also activate multiple types of cell death pathways, such as apoptosis and pyroptosis (a rapid pro-inflammatory form of cell death), if cellular defense against PAMPS and DAMPS is unsuccessful (63,70). Individual cells are the ultimate socialists and can sacrifice themselves for the good of the organism.
There are three main types of molecules involved in signal transduction following infectious or danger-related ligand binding to PRR: protein kinases, adaptor proteins, and transcription factors (110). Signal transduction occurs via several common pathways including NF-κB, mitogen-activated protein kinase (MAPK), and inflammasomes (63,110). The signals they generate can crosstalk with each other and can converge into several common pathways. Each of the PRR initiate signaling cascades that cause epigenetic modulation of gene expression and posttranslational modification of cytokine precursors. This results in activation of the innate immune response and leads to degradation of microbes, production of inflammatory cytokines, and recruitment of the adaptive immune response (70,111). Once PAMPS and DAMPS bind with TLR and nod-like receptors they activate the formation of inflammasome complexes that form an essential element of the innate immune response (63,112).
Unlike adaptive immunity, innate immunity does not recognize every possible antigen. Innate immunity recognizes PAMPS shared by related groups of microbes that are not found in mammalian cells, such as lipopolysaccharide (LPS) from gram-negative bacterial cell walls. This early induced innate immunity (4-96 hours) involves the formation of inflammasomes that lead to the release of chemokines and recruitment of defense cells. The recruited defense cells include phagocytic leukocytes such as neutrophils, eosinophils and monocytes, tissue phagocytic cells such as macrophages, macrophages and mast cells that release inflammatory mediators, and basophils, eosinophils and natural killer cells (70).
Pattern recognition receptors represent evolutionary conserved pathogen and damage recognition mechanisms that constitute the starting point for the inflammatory response. These cell-autonomous stress responses have evolved to form the basis of the non-antigen dependent defense mechanisms that characterize innate immunity (106,113). Lipopolysaccharide released by gram negative bacteria in the dysbiotic gastrointestinal microbiome, binds with TLR on sub-mucosal macrophages resulting in activation of NF-κB and inflammatory cytokine production and secretion (114). This mechanism is thought to be a major contributor to chronic inflammation in women with PCOS, and plays a significant role in the pathogenesis (discussed in section 6) (65,115).
3.2.4. Inflammasomes in PCOS
Inflammasomes are multiprotein self-assembling complexes in the cytoplasm that form an integral part of the innate immune response (116). They are produced in response to a variety of danger signals and are also involved in cellular apoptosis and pyroptosis (63). Inflammasomes act as finely tuned alarm systems that trigger and amplify innate defense mechanisms in response to cellular stresses and infection (112). The inflammasome complex contains a sensor molecule, and adaptor protein, and a pro-inflammatory caspase-1 enzyme (116). Once activated by DAMPS (like LPS) and PAMPS, the inflammasome complex converts procaspase-1 to the active caspase-1 enzyme, which subsequently activates pro-inflammatory cytokines (IL-1B, IL-18) that are released into tissues and circulation (117).
Inflammasomes form an integral part of the common CDR pathway for cellular protection from multiple types of threatening stimuli (62,106). Interruption to the flow of electrons in metabolism, generation of ROS, and other mechanisms discussed above, activate innate intracellular defense mechanisms in an attempt to contain and eliminate cellular threats (74). The CDR also includes the release of purinergic signaling to neighboring cells and immune cells, that activate inflammation (118). If the threat is contained, the CDR resolves and normal cellular function is restored (62). If the CDR is unsuccessful, pyroptosis pathways are activated and the cell is sacrificed in a further attempt to contain the threat and protect the organism (63). If the inciting stimulus persists, activation of chronic inflammation can result in tissue damage and disease, such as occurs in PCOS.
Inflammasomes and their pro-inflammatory cytokines and chemokines have been investigated for their role in inflammation, oxidative stress, ovulation, fertilization, steroidogenesis, glucose metabolism, IR, and adipogenesis, and may be involved in the pathogenesis of PCOS (112,119,120). These and other inflammatory mediators such as adipokines (leptin, adiponectin, vaspin, resistin, visfatin, omentin-1), cyclophilin A, vascular dysfunction mediators (endothelin-1, vascular cell adhesive molecule-1), NF-κB, and epigenetic regulators (microRNAs), are thought to have a role in the pathogenesis of PCOS, and have recently been reviewed (117).
3.2.5. Adaptive immune response in PCOS
The adaptive immune system involves antigen-specific defense mechanisms that are designed to react to and remove specific antigens (121). The adaptive system involves humeral and cell-mediated immunity and may take several days to become effective. The body recognizes an antigen as foreign when epitopes (fragments of an antigen that react with antibodies or lymphocyte receptors) bind to specific receptors on the surface of B-lymphocytes and/or T-lymphocytes (122). It is estimated that the human body can recognize 107 epitopes and make up to 109 different antibodies (123). Nevertheless, activation of both the innate and adaptive immune systems was primarily designed to be acute and short-lived, in order to contain and eliminate a multitude of environmental threats.
The majority of the research on the role of adaptive immunity in PCOS has been conducted on T-cells and their subpopulations (121). T-cells play a crucial role in mediating inflammation and IR by secreting proinflammatory cytokines (124,125). T-cells promote follicle development and selection by releasing specific chemokines and growth factors and produce cytotoxic signals to induce apoptosis of granulosa cells (126). Available evidence suggests that there may be a general decline in adaptive immunity and regulatory T-cell function in PCOS (121). Recent studies have suggested that immune system dysregulation, including T-cell dysfunction (127,128), may play a role in the pathogenesis of PCOS (129).
The ultimate goal of the innate and adaptive immune response, is to provide a balance between pro- and anti-inflammatory tissue turnover and protective measures, in order to achieve whole-body inflammatory homeostasis (130). As is the case with metabolism, the internal inflammatory response needs to be matched to prevailing external environmental conditions (42). The evolutionary theory of PCOS proposes that there is a mismatch between our evolutionary conserved metabolic and immune signalling systems and our modern environment (1,36,131). Accumulating evidence over the past 25 years suggests that systemic inflammation is modulated by neural and humoral communication pathways that connect the brain to the immune system (132,133). Neuroimmunomodulation is a way for organisms to regulate whole-body inflammatory homeostasis and optimize survival (130). This interconnected regulatory system has evolved over millions of years of evolutionary pressure from exposure to pathogens and other environmental threats.
3.3. Neuroimmunomodulation and the link between the nervous system and PCOS
Both the immune and nervous systems share many similarities and have unique qualities that allow them to sense changes in the internal and external environments and counteract deviations in homeostasis (130). Communication between the nervous, endocrine and immune systems involves evolutionary-conserved mechanisms that are essential for host defense and survival (132). The immune system and ANS can respond to numerous common regulatory molecules including cytokines, neurotransmitters, and glucocorticoids (134). The brain is the ultimate regulator of whole-body homeostasis of all physiological parameters. This includes blood glucose, thermoregulation, hydration, electrolyte levels, blood pressure, stress responses, feeding, behavior, reproduction, body weight, and whole-body inflammatory balance (42,58).
Regulation of the inflammatory response has previously been thought to be autonomous (135). Substantial evidence now suggests that the nervous system exerts an active role in maintaining inflammatory homeostasis (58,136,137). The brain participates in a bidirectional network of mediators including hormones, cytokines, and neurotransmitters, that monitor, co-ordinate and regulate the systemic inflammatory response (132,137). The magnitude of the inflammatory response is crucial to adaptation and survival. An inefficient response can result in immunodeficiency, infection and cancer, and an excessive response can lead to morbidity and mortality (138). Abnormalities in the neuroendocrine-immune response are implicated in the pathogenesis of many chronic diseases including obesity, atherosclerosis, autoimmune disease, depression, and PCOS (58,121,133,134).
3.3.1. Anatomy of neuroendocrine-immune connections
Animal and human research over the past six decades have investigated the extensive network of afferent and efferent communication mechanisms that co-ordinate the inflammatory response (132,133,139). There is now consensus that “the inflammatory reflex” is composed of [1] a system of sensors (that identify PAMPS and DAMPS), [2] an afferent arm, which conveys information about systemic inflammatory status to the central nervous system (CNS), [3] processing centres in the brain that integrate and interpret incoming signals (hypothalamic nuclei, brain stem autonomic neurons), [4] and an efferent arm which exerts immunomodulatory functions (Hypothalamic-Pituitary-Adrenal (HPA)-axis, ANS) (136). The brain exerts strong immunomodulatory effects on a variety of components of the immune system, by activation of the HPA-axis and ANS (Figure 2).
The hypothalamus plays a central role in sensing and coordinating neural and humoral factors and can modulate inflammatory pathways (58). Circulating cytokines, such as IL-1ẞ and Tumour Necrosis Factor-alpha (TNF-α), can cross the blood-brain barrier by a carrier-mediated mechanism (140) or via the circumventricular organs (141). The hypothalamus can also receive input from visceral vagus afferent fibres, after they synapse with the dorsal motor nucleus of the vagus nerve in the brainstem. This has been termed the “cholinergic anti-inflammatory pathway” (132). Ascending connections reach the hypothalamus via the nucleus tractus solitarius (142). The effector arm or efferent system is modulated by the HPA-axis and the sympathetic (SNS) and parasympathetic (PNS) components of the ANS (132,143).
The HPA-axis is a neurohormonal pathway that has classically been studied for its role in regulating the immune system (144) (Figure 2). Specialized neurons in the paraventricular nuclei of the hypothalamus, release corticotropin releasing hormone into the portal blood stream and stimulate production of adrenocorticotropic hormone (ACTH) from the anterior pituitary. ACTH stimulates the synthesis of immunosuppressive glucocorticoids (cortisol) from the adrenal cortex (144). Pro-inflammatory cytokines and neural inputs activate the HPA-axis to release ACTH, and the HPA-axis is subject to a classic negative feedback loop by cortisol that inhibits both corticotropin releasing hormone and ACTH (145). The immunosuppressive effects of cortisol are mainly linked to suppression of NF-κB activity and reduced cytokine synthesis (146). Glucocorticoids are also potent anti-inflammatory agents that are used in multiple clinical situations (145).
The SNS modulates both pro- and anti-inflammatory activities (132). The SNS innovates primary (thymus, bone marrow) and secondary (spleen, lymph nodes, tissues) lymphoid organs. Sympathetic neurons release noradrenalin and adrenalin that interact with adrenoreceptors on lymphocytes and macrophages, and stimulate the production of cytokines that result in anti-inflammatory effects (147). Sympathetic neural activation of chromaffin cells in the adrenal medulla leads to an increased release of catecholamines into the circulation. Sympathetic innervation of cortical cells leads to release of glucocorticoids. CNS-controlled SNS output is therefore converted to hormonal immunoregulation in peripheral tissues. In addition, recent studies have shown that the carotid body can sense proinflammatory cytokines (TNF-α) and relay information to the brainstem that activate the efferent SNS output to splanchnic nerves and suppress further TNF-α synthesis (64).
The PNS also plays a significant role in modulating immune cells and inflammatory activity (132). Evidence that vagal afferent fibres relay messages to the CNS that inflammation is present in other body sites has been demonstrated in animal models, although evidence for the mechanisms of activation is still not clear (133). The parasympathetic nervous system has an anti-inflammatory effect through release of acetylcholine that interacts directly with nicotinic receptors on macrophages (148), and also indirectly with the spleen (133). Overall, the SNS and PNS appear to act synergistically to down-regulate inflammation.
3.3.2. Neuroimmunomodulation in PCOS
Several studies have examined the bi-direction connections between the nervous and immune systems in PCOS. Women with PCOS have been found to have increased SNS activity by muscle and skin microneurography, heart rate variability, measurement of nerve growth factor, and catecholamine metabolites (137,149). Insulin has been found to stimulate SNS output, and there is a complex bidirectional relationship between IR and sympathetic activity, in both the hypothalamus, and ovary (149). Although most of the research has focused on SNS overactivity, reduced PNS activity has also been demonstrated (143). Impaired ANS function has been proposed as a contributor to hyperandrogenemia, via mechanisms in the hypothalamus, adipose tissue, and ovary (150,151). Nerve growth factor is a marker for SNS activity in women with PCOS (137).
Nerve growth factor is a signalling molecule involved in modulation of the neuro-endocrine-immune system (137). Nerve growth factor has an important role in maintaining homeostasis and may be involved in normal follicle development and ovulation. It has been proposed that excess nerve growth factor may have a role in PCOS, as women with PCOS have sympathetic hyper-innovation and enhanced production of nerve growth factor in the ovaries (152,153). Nerve growth factor may provide a link between the SNS, ovarian inflammation, hyperandrogenism, and ovulatory dysfunction, in women with PCOS (137).
SNS activity can be reduced in women with PCOS by electroacupuncture, treatment of obstructive sleep apnoea with positive airways pressure, renal denervation in refractory hypertension, pharmacotherapy, exercise training, and weight loss (149,154). Taken together, these data suggest that an imbalance of the ANS is likely to play a reversible role in the pathophysiology of PCOS.
3.4. Hyperandrogenism and chronic inflammation
Chronic systemic inflammation can cause hyperandrogenism (56), and elevated androgens can affect immune cells, resulting in predominately anti-inflammatory effects on the immune system (155). There is debate in the literature regarding the direction of causation, physiological importance, and evolutionary significance of these processes. The preponderance of evidence, and the most widely accepted current view, is that hyperandrogenism is secondary to the synergistic actions of chronic systemic inflammation and IR, through upregulation of ovarian theca cell androgen synthesis (discussed in section 4.5) (156). The evidence presented in the current review also supports this paradigm.
Serum androgens can be derived from multiple tissues including the adrenal gland, adipose tissue, and ovaries (157). Most serum androgens in PCOS are thought to be produced by the ovaries. Nevertheless, it is likely that multiple mechanisms are involved in excessive androgen production in different tissues in women with PCOS (157,158). Low-grade chronic systemic inflammation is commonly found in patients with PCOS exhibiting hyperandrogenism, and ovarian inflammation and fibrosis are part of the histopathology (159). Ovarian inflammatory mechanisms have been linked to systemic markers of inflammation (CRP, white cell count) (158–160) independent of body mass index (BMI) (161). An extensive range of inflammatory cytokines have been associated with impaired folliculogenesis and androgen synthesis in women with PCOS (158). Cytokines can influence the inflammatory response by epigenic mechanisms that alter gene expression or by posttranscriptional regulatory processes (56). Oxidative stress has been reported to enhance ovarian steroidogenic enzymes and increase androgen levels in women with PCOS (162). Endoplasmic reticular stress pathways are activated in the ovaries in mouse models and in humans, and may contribute to the pathophysiology of PCOS through multiple effects in granulosa cells (163,164). As discussed previously, AGE and RAGE have been found to cause altered steroidogenesis in granulosa cells in PCOS (99). Dehydroepiandrosterone is a circulating pre-androgen produced in the adrenal cortex that has been found to stimulate inflammation and impair ovarian function in PCOS (165). Increased luteinizing hormone (LH) levels, commonly found in PCOS, can amplify the abnormalities described in theca cell steroidogenesis (117). The combined effects of these processes result in excess follicular androgens, which combine with increased levels of insulin to downregulate aromatase levels in granulosa cells. This creates a continuous feedback loop between inflammation, IR and hyperandrogenism, with no apparent beginning or end. Despite these uncertainties, diet-induced inflammation can invoke hyperandrogenism (156), and effective treatment of inflammation can normalize androgen levels and restore fertility, in women with PCOS (166). In summary, accumulating evidence suggests that the ovaries are not the primary cause of hyperandrogenism in most women with PCOS (see section 4.5).
The influence of androgens in the pathogenesis and pathophysiology of PCOS is undisputable, despite the debate regarding the primary mechanism of causation. Data derived mostly from animal models of PCOS clearly indicate that intrauterine exposure to elevated androgen levels induces the development of PCOS traits in adult females. PCOS can also develop in women with other hyperandrogenic syndromes such as congenital adrenal hyperplasia, lipodystrophy, and ovarian or adrenal tumors. In addition, there has also been debate regarding the evolutionary “advantage” of hyperandrogenism in PCOS. Inflammation-induced hyperandrogenism has been proposed as a possible compensatory mechanism to restore homeostasis within the immune system, given the anti-inflammatory effects of androgens in women (56). It is also possible that hyperandrogenism (due to inflammation and/or hyperinsulinemia) may represent an adaptive physiological mechanism to down-regulate reproduction during periods of environmental (infection, starvation, climatic) and personal stress. In addition, there may be other individual advantages such as increased strength and fitness.
3.5. Summary of the role of inflammation in PCOS from an evolutionary perspective
A significant body of literature supports the role of chronic inflammation in the pathogenesis of PCOS. Billions of years of evolution have constructed a co-operative system of sensors, receptors, hormones, cytokines, chemokines, and other signalling molecules that invoke intracrine, autocrine, paracrine, and neuroendocrine mechanisms designed to regulate cellular, tissue and whole-body inflammatory responses. This process forms part of the repertoire of adaptive survival responses that are intimately connected with total body metabolic homeostasis to ensure individual survival. Inflammation invokes a series of adaptive metabolic survival mechanisms, such as IR, to ensure adequate energy supply to immune cells. Neuroendocrine mechanisms act as a counter-regulatory anti-inflammatory mechanism to control inflammation and restore homeostasis. In addition, immune and metabolic physiology are intimately linked to reproduction, to optimize fertility and ensure species survival. As a result, both chronic inflammation and IR can contribute to hyperandrogenemia, which also has adaptive survival advantages that restore immune homeostasis and temporarily down-regulate reproduction.
4. Insulin Resistance and reduced insulin sensitivity
4.1. Physiological actions of insulin
Insulin is a peptide hormone (51 amino acids) produced by the beta cells in the islets of Langerhans in the pancreas, where it is stored in cytoplasmic granules as an inactive hexamer with zinc and calcium (167). Glucose diffuses into the beta cell via insulin-independent GLUT-2 transporters, is converted to glucose-6-phosphate by glucokinase at normal serum concentrations (4-6mmol/L), and is then metabolized via glycolysis to produce diacylglycerol or ATP (168). Diacylglycerol and ATP are the main signal molecules that trigger insulin release from beta-cell granules. Insulin release is characterized by low level basal secretion in the fasting state and an oscillating pulsatile ultradian pattern (1-2-hour cycle), when stimulated. In summary, glucose metabolism by beta-cells is the main mechanism that triggers insulin release.
Insulin binds with its receptor which is a large (320 kDa) transmembrane protein with an intracellular domain that acts as a tyrosine kinase. The insulin receptor is highly conserved from an evolutionary perspective, and is present on all mammalian cells, although the distribution is unequal (40 per red blood cell, 300,000 per adipocyte) (169). Insulin binds to the alpha-subunit and induces autophosphorylation of specific tyrosine residues on the cytoplasmic side of the membrane (54,170). The activated insulin receptor initiates signal transduction via the phosphatidylinositol-3 kinase (PI-3K) metabolic pathway and the mitogen-activated protein kinase pathway (MAPK) which is involved in cell growth and proliferation (170) . There are multiple physiological stimulators (vagal PNS, growth hormone, chronic cortisol, prolactin, gonadotropins) and inhibitors of insulin (adrenaline, noradrenaline, SNS, parathyroid hormone, somatostatin, acute cortisol, pancreatic polypeptide) (171). Glucose is by far the most potent stimulator of insulin and all the others are regulatory and much less effective.
Insulin has pleiotropic metabolic and growth-promoting anabolic effects throughout the body (171). Importantly, the cellular effects of insulin are tissue specific, and not all cells require insulin to transport glucose into cells. Insulin signalling increases the availability of GLUT-4 glucose transport proteins to the surface of insulin-dependent cells (skeletal and cardiac muscle, adipose tissue, vascular endothelium). Liver and brain are insulin-independent and use other GLUT transporters, in addition to using insulin-responsive GLUT-4 transporters. Insulin modulates a wide range of tissue specific physiological processes, in addition to facilitating glucose removal from the blood (42). Insulin enhances fat storage and inhibits lipolysis in adipose tissue, stimulates glycogen synthesis in muscle and liver, inhibits hepatic glucose output (gluconeogenesis), causes vasodilation in vascular endothelium and the heart (via production of nitric oxide), and enhances sodium reabsorption in the kidney (171,172). Insulin has widely different dose-response curves in different tissues and organs. Myocardium has the highest rate of glucose uptake (approximately 1000 mmol/g/minute), but skeletal muscle accounts for most of the uptake quantitatively (estimated at 75-80% of total glucose movement), as these cells constitute 40-50% of the body mass. Appropriate lean body mass, age-related sarcopaenia, and physical activity, therefore play a significant role in health and disease, due to the metabolic impact of total body muscle.
Insulin also has a direct anti-inflammatory role by preventing hyperglycaemia-related generation of ROS and AGE, inhibiting NF-kB (by reducing the production of inflammatory cytokines), inducing vasodilation (via nitric oxide release), reducing leukocyte adhesion to the endothelium (173), and by inhibiting formation of the NLR inflammasome complex (174). As a result, hyperglycaemia and IR are pro-inflammatory states. Insulin resistance can cause inflammation, and inflammation can cause IR, in women with PCOS (173,175). Most of the literature has focused on the evolutionary benefit of the pro-inflammatory effects of IR in starvation, trauma, and infection (see section 4.4) (175). In contrast, the anti-inflammatory effects of insulin may have an evolutionary protective effect against excessive activation of the systemic immune response following exposure to small amounts of ingested antigens, in a similar manner to the gastrointestinal mucosal immune system (176,177).
In summary, insulin’s overall role is to control energy conservation and utilization during feeding and fasting states (167). Insulin acts as a metabolic switch between anabolic and catabolic processes to regulate blood glucose levels and has a significant anti-inflammatory effect. Insulin provides a direct link between metabolism and immune regulation, and is paramount in activating numerous adaptive survival mechanisms (178). Dysregulation of the protective physiological functions of insulin are instrumental in the pathogenesis and progression of many chronic diseases, including PCOS (167,179,180). The physiological actions of insulin are summarized in table 1.

Table 1.
Physiological actions of insulin.
| Functions of Insulin | Mechanism |
|---|---|
| Energy storage |
lipolysisglycogen synthesis gluconeogenesis |
| Anti-inflammatory |
AGE) Inhibits NF-κB activated cytokine production Inflammasome formation reduced Leukocyte adhesion to endothelium reduced |
| Volume expansion | sodium reabsorption |
| Vasodilation | endothelial nitric oxide |
| Tissue perfusion | Volume expansion and vasodilation |
| Evolutionary role of insulin | Tissue-specific energy storage and anabolic growth effects. Anti-inflammatory protective effect against excessive immune activation |
| Evolutionary role of insulin resistance | Maintain blood glucose to supply brain, immune cells, and fetus, during starvation, infection, stress and trauma. Proinflammatory |
BSL, blood sugar levels; ROS, reactive oxygen species; AGE, advanced glycation end-products; NF-κB, nuclear factor kappa-B.
4.2. Reduced insulin sensitivity versus insulin resistance in PCOS
Diminished tissue sensitivity to insulin has become characterized as a pathological condition known as IR, as a result of the association between IR, metabolic conditions, and chronic disease (17,180). Being able to vary the sensitivity of the cellular and tissue response to insulin is an evolutionarily-conserved protective mechanism used by many species (insects, worms, vertebrates, humans), to enhance survival (181). Insulin resistance is a categorical variable that has an arbitrary definition in research studies, but has no agreed definition or normal range in clinical practice (2,182). Reduced insulin sensitivity is a continuous variable that is considered a quantitative trait (interaction of multiple genes with the environment that results in a continuous distribution of phenotypes), in evolutionary medicine (183).
The gold standard for assessing IR is the hyperinsulinemic-euglycemic clamp (clamp) test. The clamp test is performed using insulin and glucose infusions and is time-consuming, labor-intensive, expensive, and has associated risks (184). A high-dose insulin infusion (80mU/m2/min) is used to suppress hepatic gluconeogenesis and create a steady-state blood glucose concentration. The rate of glucose infused, in order to maintain a steady state, is equal to whole-body glucose disposal. Decreased insulin sensitivity is defined as IR when the glucose disposal rate is less than an arbitrary cut-off of 4.45 mg/kg/min (184).
It is important to distinguish between reduced insulin sensitivity and IR, as most women with PCOS have reduced insulin sensitivity, but not all women with PCOS meet the experimental criteria for insulin resistance. The reported prevalence of IR in PCOS has varied widely due to the heterogeneity of diagnostic criteria for PCOS, variety of assessment methods, and the arbitrary definition of IR selected for different studies (185). Nevertheless, IR has been considered a central feature in the majority of women with PCOS (186). A systematic review of hyperinsulinemic-euglycemic clamp studies found that women with PCOS have a 27% reduction in insulin sensitivity compared to matched controls (179).
When considered from an evolutionary perspective, women with a PCOS phenotype would have improved survival chances during times of increased physiological demand or imposed environmental stress, but be more vulnerable to the pathological effects of IR when exposed to contemporary lifestyle factors (1,5). When viewed as a continuous variable, it is likely that all women with PCOS, whether obese or lean, have reduced insulin sensitivity (179,187,188).
4.3. Mechanisms of insulin resistance in PCOS
Insulin resistance can be caused by numerous mechanisms including hyperinsulinemia, insulin receptor variants, receptor antagonists and agonists, autoantibodies, oxidative stress, advanced glycation end-products, hormones, nutrient sensors, inflammatory cytokines, and metabolic intermediates (171,189). It is likely that more than one mechanism is involved in any individual, given the interactive nature of many of the pathophysiological processes and signalling pathways.
Insulin resistance has been related to the western diet via a variety of mechanisms (190). These include high-glycemic diet-related dysbiosis (65,115), chronic inflammation, and intracellular accumulation of metabolic intermediates (171). These mechanisms may act individually, or together, to produce the observed features of IR. A graphical description is given in Figure 3.
4.4. Evolutionary adaptive role of insulin resistance in PCOS
Human survival has relied on the ability to alter our physiology according to the changing demands of the environment, or to different internal states during various life stages (191). In evolutionary terms, physiological IR is an adaptive survival mechanism that allows organisms to selectively modulate cellular and tissue responses to a variety of environmental challenges (infection, starvation, dehydration, psychological stress, physical stress from injury) (172,192,193), and internal states (pregnancy, puberty, adolescence) (194,195). Pathological IR is a detrimental condition associated with metabolic syndrome, other chronic diseases, and PCOS (189). The suggested mechanisms and evolutionary adaptive roles of insulin resistance are discussed in the following sections.
4.4.1. Insulin resistance and infection
Bidirectional immune-endocrine interactions regulate metabolism in the context of infection, to ensure pathogen clearance, provide the appropriate metabolic requirements, and restore homeostasis (191,192). The immune system uses PRR-activated cytokines (see section 3.2.3) to communicate with the endocrine system and modify the responsiveness of peripheral tissues to endocrine signals. Immune cells are highly dependent on glucose for metabolism and are responsible for nearly 20% of total body energy consumption (196). This can rise to 30% during infection and needs an effective and rapid response system to facilitate the switch from oxidative phosphorylation to more nutrient intensive glycolytic metabolism (192). In addition, inflammatory cytokines can influence processes normally regulated by the endocrine system such as hunger, temperature, insulin sensitivity and glucose uptake. Similarly, the endocrine system can regulate immune cells via a complex system of hormones (leptin, adiponectin, insulin), receptors, nutrient sensors, and metabolic signalling pathways (192,196).
When an infectious threat is detected by the immune system, the body activates neuroendocrine components of the HPA-axis and SNS (see section 3.3) to increase glucocorticoid and catecholamine release (196). The HPA-axis and SNS act together with a state of cytokine-mediated IR, to mobilize free fatty acids from adipose tissue and glucose from hepatic glycogen, to ensure appropriate supply of energy to immune cells. The ability to regulate insulin sensitivity and selectively redistribute glucose and energy, is an adaptive survival mechanism conserved in many species throughout evolutionary history (191).
In contemporary society, stress is more likely to be psychological rather than physical or infectious, but the systemic stress response is similar (191). The HPA-axis and SNS are activated, IR is implemented, but the metabolic demand is much less, and the mobilized energy is re-stored in adipose tissue. The stress is often protracted resulting in anxiety, increased appetite from cortisol excess and leptin-resistance, stress-eating, and central obesity (197). In addition, sustained adverse nutritional and environmental exposures can result in “trained immunity” (adaptive changes to innate immune responses), that may partly explain IR-related pathologies (198). In evolutionary terms, chronic IR represents an evolutionary mismatch between ancestral survival responses and modern cultural demands and lifestyle. Women with PCOS experience increased levels of anxiety, depression and stress, and the International Guidelines recommend screening for emotional well-being (199,200).
4.4.2. Insulin resistance, starvation and dehydration
Insulin is an anabolic hormone that promotes cellular growth and energy storage in adipose tissue, liver and muscle (171). Prolonged calorie restriction during starvation activates adaptive survival mechanisms that reduce energy expenditure and prevent muscle loss (201). Clamp studies have demonstrated that starvation-related IR is caused by decreased insulin signalling and reduced glucose uptake (202). The implementation of IR in starvation states diminishes oxidation of glucose, provides energy by mobilizing fatty acids from adipose tissue, limits protein loss, and redirects energy to in-demand organs (201).
Hyperinsulinemia may have played a role in preventing dehydration in ancestral populations (172). Insulin regulates sodium channels and increases the reabsorption of sodium and water in the renal tubules (203). Hyperinsulinemia may increase blood volume and blood pressure by this mechanism, or via activation of the renin-angiotensin-aldosterone system (204). Insulin also stimulates nitric oxide resulting in vasodilation and decreased blood pressure. Insulin resistance results in selective impairment of the nitric oxide pathway, and hyperinsulinemia may activate the MAPK signalling pathway and cause vasoconstriction and elevated blood pressure (205). The combined effects of these mechanisms may contribute to maintenance of blood volume and increased cerebral perfusion during starvation. Pathological activation of these mechanisms could contribute to the increased risk of hypertension and metabolic syndrome observed in women with PCOS (206).
Women with a PCOS phenotype may have had an evolutionary advantage from having a better response to infection, dehydration and starvation, but are now more susceptible to stress, hypertension, metabolic syndrome, and PCOS, as a result of contemporary lifestyle and environmental exposures (1,5).
4.4.3. Insulin resistance and pregnancy
Insulin sensitivity decreases throughout pregnancy and is an evolutionary-conserved mechanism to limit maternal glucose use and shunt energy to the foetus, particularly during the second half of gestation (194). Insulin sensitivity gradually decreases up to 20 weeks gestation, followed by a more rapid decrease to 50% of the non-pregnant values by 40 weeks, in normal pregnancy (207). The pancreatic beta-cells respond by increasing insulin secretion by up to 250% to maintain euglycaemia (208). An inability to secrete adequate amounts of insulin results in elevated blood sugar levels and Gestational Diabetes Mellitis (GDM).
Maternal IR results in the use of more lipids for energy by the mother and spares carbohydrates for the foetus. The decreased maternal response to insulin is mediated by oestrogen, progesterone, human placental lactogen, human placental growth hormone, cortisol, prolactin, inflammatory cytokines, adipokines, exosomes, and the microbiome (194,209,210). Clamp studies in late pregnancy show that hepatic gluconeogenesis is only 80% suppressed in late pregnancy, in women with GDM, compared to non-diabetic pregnant women (208). Insulin sensitivity is also affected by obesity, and fat mass increases in both lean and obese women in pregnancy. Women with PCOS have higher gestational weight gain and proportionately more visceral adiposity, which are associated with greater increases in IR and GDM (194).
Women with PCOS have a 25-50% chance of developing GDM in pregnancy (211,212). Women with GDM have up to 50% risk of developing T2DM in the 5-10 years following pregnancy (213,214). The population attributable risk of PCOS to T2DM has been estimated at 19-28% (17). Therefore, up to 28% of adult women with T2DM have pre-existing PCOS that progresses to T2DM. Up to 50% of individuals diagnosed with T2DM have complications (retinopathy, nephropathy, neuropathy, vascular), at the time of diagnosis (215). The International Diabetes Federation estimates that there are approximately 537 million diabetics in the world, and women with PCOS therefore make a significant contribution to this global epidemic (216). These data support the characterization of PCOS is a progressive metabolic disease. Recent studies have demonstrated that the risk of progression of GDM to T2DM can be reduced by greater than 90% by dietary and lifestyle interventions (217). PCOS therefore appears to be a progressive metabolic disease that is preventable.
In summary, the downregulation of insulin signalling and implementation of IR can be viewed as a physiological adaptive mechanism that facilitates the switch from an anabolic, to a catabolic state. This results in mobilization of fatty acids from adipose tissue, increased gluconeogenesis, release of glucose from the liver, and redistribution of energy to the immune system and brain. This process is mediated by the co-operative interaction of the immune, nervous, and endocrine systems. Chronic overactivation of any of these mechanisms creates a disturbance to whole-body homeostasis that results in chronic inflammation and IR. Women with PCOS appear to have a genetically determined proinflammatory design and increased insulin sensitivity, that would be protective in an ancestral environment, but becomes maladaptive in modern society.
4.5. Insulin resistance and hyperandrogenism
Androgens play an essential role in many aspects of male and female physiology, metabolism, sex differentiation and reproductive biology (218). Optimal health is achieved when bioactive metabolites (testosterone, dihydrotestosterone) are maintained in a defined homeostatic range. In women, low levels of androgens have been associated with endometriosis and high levels with PCOS (219). As with all other pathophysiological processes in PCOS, there appears to be a bidirectional relationship between hyperinsulinemia and hyperandrogenism. Hyperinsulinemia can cause hyperandrogenism via mechanisms discussed below, and hyperinsulinemia has been shown to occur after administration of androgens in clamp studies (220,221). This may represent a self-reinforcing positive feed-forward mechanism to augment the effects of IR.
Ovarian androgen synthesis occurs in a co-operative two-step process involving luteinizing hormone (LH) stimulation of steroidogenesis in the theca cell, transfer of androstenedione to the granulosa cells, and follicle stimulating hormone (FSH) induced production of testosterone (222). Androgens are irreversibly converted to oestrogen in granulosa cells by the rate-limiting aromatase enzyme. Aromatase belongs to the cytochrome P450 superfamily and is the product of the CYP19A1 gene. The principal role of aromatase is to convert androgens to estrogen. Disruption of aromatase function can lead to elevated or reduced levels of androgens or estrogen, which are associated with a wide variety of common pathologies (endometriosis, osteoporosis, hypogonadism, Alzheimer’s disease, cancer, PCOS) (218). Aromatase is crucial for many metabolic functions due to its role as an estrogen biosynthetic enzyme (glucose and lipid homeostasis, bone mineralization, brain function) (223). Ovarian aromatase activity is an important regulator of ovulation and sequential endometrial changes during the normal menstrual cycle. Dysregulation of aromatase has been linked to altered steroidogenesis, hyperandrogenism, impaired folliculogenesis, and anovulation, and is thought to play a central role in the pathogenesis of PCOS (224,225).
Structural models used to investigate the molecular evolution of aromatase have characterized the amino-acid sequences and configurations, substrate recognition sites, catalytic mechanisms and inhibitor specificities (226–228). It is clear that there is a complex molecular network that regulates the normal physiology of aromatase activity, and subsequent androgen and estrogen production, to maintain tissue-specific homeostasis and function (228). The CYP19A1 gene and aromatase enzyme are present in virtually all vertebrates and have been identified in many invertebrates. The core structure, active site amino acid sequences, and substrate recognition sites have been highly conserved throughout evolutionary history (226). In humans there are eleven promoters that regulate tissue-specific aromatase gene expression. Post-translational modification by phosphorylation allows rapid modulation of aromatase activity, compared to gene regulation. While FSH has been identified as the most important factor that regulates the expression of aromatase (224), other hormones such as melatonin and leptin also play an important stimulatory role (229,230). In addition, hormonal contraceptives and endocrine disrupting chemicals (Bisphenol A) have been shown to increase aromatase activity (231,232). Multiple endogenous (insulin, leptin, adiponectin, cortisol, vitamin D, AGE, inflammatory cytokines) (218) and exogenous molecules (glyphosate, phytoestrogens, resveratrol, curcumin, nicotine, alcohol, azole agricultural antifungals) (233–238), can act as aromatase inhibitors and potentially contribute to hyperandrogenism in PCOS. In summary, aromatase appears to be a significant physiological intersection point for regulatory molecules involved in immune, metabolic, hormonal, and reproductive function. In evolutionary terms, varying aromatase activity adjusts the homeostatic balance between oestrogen and androgens, modulating adaptive survival pathways in multiple tissues throughout the body.
As previously discussed, IR is thought to have evolved as an adaptation to environmental stressors such as starvation, infection and fear (1). Varying the levels of insulin sensitivity permits the redistribution of total body energy to organs of greater need, such as the brain, immune system, and fetus (191). In addition, it has been proposed that the development of selective insulin resistance is a mechanism that activates specific behavioural and reproductive survival strategies (54). Specific cells and tissues are protected from developing IR, including the brain, immune cells, placenta and ovaries (239,240). Areas of the brain do not develop IR and benefit from the redistribution of glucose from muscle and fat tissue. In pregnancy, the placenta is an insulin independent organ, and the development of maternal IR is expected to divert more nutrients through the placenta. The ovary does not develop IR and remains sensitive to the high levels of insulin that occur in IR and hyperinsulinemia (241,242). This may be a mechanism to downregulate fertility at times of physiological or psychological stress. In summary, both the physiological actions of insulin and the development of insulin resistance, are tissue specific and facilitate a variety of adaptive survival responses.
In our modern environment, insulin resistance and hyperinsulinemia are thought to be primary factors in the development of hyperandrogenism in PCOS, in addition to chronic inflammation (discussed in section 3.4) (242). Insulin has a number of known mechanisms that can increase androgen levels in the serum, liver, and ovaries (162,242). Insulin is reported to stimulate ovarian androgen production directly (via the PI3K and MAPK pathways), or indirectly by augmenting LH-stimulated androgen synthesis (162). Insulin increases the availability of insulin-like growth factor and decreases its binding protein, resulting in increased androgen stimulation (243). Insulin can increase the amplitude of gonadotropin releasing hormone stimulated LH pulses (244) that are known to occur in PCOS. Both insulin and testosterone decrease hepatic production of sex hormone binding globulin, resulting in increased free testosterone (245). In addition, hyperinsulinemia stimulates the HPA-axis leading to increased adrenal androgen production (246).
Accumulating evidence suggests that the ovaries are not the primary abnormality in PCOS (242,247,248). Women with PCOS often respond promptly to gonadotropin stimulation with clomiphene and gonadotropin releasing hormone agonists, or antiandrogenic compounds (249). Furthermore, lifestyle interventions such as weight loss and exercise, reduce IR and insulin levels, normalize gonadotropin secretion, and regulate menstrual cycles in women with PCOS (250). Therapy with metformin results in reduced androgens and restores ovulation by reducing insulin levels, and altering the effect of insulin on androgen biosynthesis and theca cell proliferation (251). As discussed previously, inflammation also contributes to the pathophysiology of hyperandrogenism. Lifestyle interventions reduce both insulin levels and inflammation, highlighting the likelihood that hyperinsulinemia and chronic inflammation act together to alter ovarian steroidogenesis, increase androgen levels, impair follicular development, and reduce ovulation (10,252). From an evolutionary perspective, inhibition of aromatase may act as another physiological mechanism (in addition to hypothalamic effects discussed in section 3.3), to downregulate reproduction until inflammatory and metabolic stressors are contained.
5. Evolutionary significance of adipose tissue in PCOS
Hundreds of millions of years of evolution have shaped adipose tissue (AT) into its current form (253–256). Taking an evolutionary perspective provides insight into the complex range of AT-related adaptive survival functions that form part of the network of interdependent homeostatic systems previously discussed. Adipose tissue is involved in a variety of functions including immune responses (innate, adaptive, inflammatory), metabolism (glucose and lipid metabolism, appetite regulation, maintenance of body weight, insulin resistance), and reproduction (pregnancy, lactation, hyperandrogenism) (253,257). Adipose tissue has a bidirectional relationship with the neuroendocrine, immune, metabolic, and reproductive systems, that facilitate these functions. This communication is achieved via a variety of cellular receptors and secretion of a large number of signalling molecules. These include adipokines (adiponectin, leptin, resistin, visfatin, retinol-binding protein 4, pigment epithelium-derived factor, endocannabinoids, and many more), cytokines (50 cytokines have been identified), metabolites, lipids, non-coding RNA’s or extracellular vesicles, and chemokines (256,258).
Adipose tissue contains a diverse range of cell types such as immune cells (macrophages, monocytes, granulocytes, T-cells), stromal cells, fibroblasts, preadipocytes, and adipocytes (257,258) and is divided into brown AT (BAT) and white AT (WAT). WAT is classified by its anatomical location as subcutaneous AT (SAT), or visceral AT (VAT) (259,260). Brown AT is primarily involved in thermoregulation and there are distinct differences between the location, structure, and function of VAT and SAT.
Subcutaneous AT functions as an endocrine organ and energy storage depot, and represents a normal physiological buffer to store excess consumed energy. This energy can be released during periods of limited food availability, and is an important evolutionary-conserved adaptive survival mechanism (5). Carbohydrates (glucose, fructose) that are not converted to glycogen are metabolized to triglycerides, and along with dietary lipids, are transported to AT and stored in lipid droplets (254). Subcutaneous AT is widely distributed, but predominantly located in the abdominal wall (males) and femurogluteal region (females). The lower body distribution of SAT in women is thought to have evolved to provide insulation in the cooler temperatures of the Pleistocene open Savana (261). This fat distribution is unique to human females and may have provided extra energy storage during lactation and pregnancy (191).
Genetic differences in the ability to store lipids in SAT, may partly explain the apparent lean paradox in 20% of women with PCOS (see reference 1 for detailed discussion) (1). According to the “adipose expandability hypothesis,” when the genetically-determined storage capacity is exceeded, or the ability to generate new adipocytes is impaired (genetic, inflammation, hyperandrogenism, stress-related elevated cortisol), fat begins to accumulate in areas outside SAT (262). In addition, excess lipid accumulation in SAT, leads to adipocyte hypertrophy, hypoxia, insulin resistance, inflammation, and release of inflammatory cytokines into the circulation (263). Chronic stress, hyperandrogenism, chronic inflammation, and insulin resistance, found in women with PCOS, may contribute to accumulation of additional VAT (258,264,265).
Up to 80% of women with PCOS are overweight or obese (266). Both lean and obese women with PCOS have been found to have an increased proportion of VAT using a variety of body composition assessment methods (267). In addition, there are many anatomical and physiological differences between VAT and SAT. Visceral AT contains larger adipocytes, is more vascular, has more glucocorticoid, androgen, ẞ-adrenoreceptors receptors, angiotensinogen, adiponectin and leptin, has less oestrogen receptors, and reduced fatty acid uptake (256–258). Visceral AT also has increased catecholamine-induced lipolysis, greater insulin sensitivity, and produces more inflammatory cytokines (253,257). It has been proposed that having an increased proportion of VAT provides an adaptive survival advantage against infection (253). This has been called the “VAT prioritization hypothesis,” and is supported by multiple lines of evidence.
Most VAT is in the omentum and mesentery (253). Mesenteric VAT surrounds the small intestine and is the next line of defense against pathogens and endotoxins (LPS) translocated from the intestine, following passage through the submucosal immune system (268). The mesenteric VAT contains large numbers of immune cells that can mount a rapid energy intensive immune response in case of infection. Visceral adipocytes, like lymph nodes, are specialized to store diet-derived polyunsaturated fatty acids that are required for an immune response. Mesenteric VAT contains lymphoid clusters that expand in response to inflammation and infection (268). Blood from the mesenteric and omental VAT drains into the portal vein and is moved directly to the liver. The junction of the portal vein and liver contains another cluster of immunologically important cells, stellate cells (specialized fat cells) and Kupffer cells (macrophages), that provide a further layer of immunological protection (269). The VAT prioritization hypothesis proposes that these evolutionary adaptive changes are developmentally programmed in a similar way to the proposed developmental programming of PCOS. Readers are referred to articles by West-Eberhard (253) and Parker (270) for detailed discussion.
In summary, adipose tissue and obesity have a significant bidirectional role in the pathophysiology of chronic systemic inflammation and insulin resistance, in women with PCOS. The anatomical and functional redistribution of adipose tissue, in women with PCOS, may have adaptive survival benefits in an ancestral environment (improved energy storage capacity, greater protection from infection) that become maladaptive in response to contemporary lifestyle and environmental exposures (diet, inactivity, stress). In addition, adipose tissue is closely linked to the reproductive system and clearly plays a significant role in the pathophysiology of PCOS.
6. Central role of the microbiome in the pathogenesis of PCOS
The microbiota is the sum of microbial organisms (bacteria, virus, archaea, fungi) that inhabit the interface between the external environment or habitat and the internal environment of the human body (271). This interface mainly occurs at mucosal surfaces (nasal, oral, respiratory, gastrointestinal, genitourinary), eyes, and skin. The gastrointestinal (GI) microbiome (microbial organisms and their genetic material), makes up 80% of the microbiota and is now considered to have a central role in human health and disease, including PCOS (115,272,273). The functions of the microbiome include inhibition of pathogen colonization, regulation of the mucosal and systemic immune systems, alteration of metabolism, energy balance, hormonal action, maintenance of the integrity of the GI barrier, and bidirectional signalling with most organs and tissues throughout the body (gut-brain, gut-bone, gut-immune, gut-liver etc). The GI microbiome therefore forms part of the whole-body homeostatic regulatory framework that maintains health, and is now appreciated to be part of the human-microbe meta-organism (59).
Microbiome eubiosis (a balanced microbial ecosystem) can be disrupted by external factors (lifestyle, diet, environmental chemicals, pathogenic microbes, medications), or endogenous factors (hormones, circadian, exercise, stress). Many of these factors can cause dysbiosis (an imbalance of the composition, metabolism or distribution of the microbiome associated with an unhealthy outcome) and have been investigated for their role in a wide range of human diseases (274). These include obesity, diabetes, gestational diabetes, metabolic syndrome, cardiovascular disease, inflammatory bowel disease, multiple sclerosis, dementia, PCOS, and many others (275,276). Lifestyle-related disturbance to the balanced microbial ecosystem contributes to a variety of diseases, depending on the level of exposure, length of exposure, combination of exposures, and individual disease susceptibility (274). Accumulating evidence suggests that dysbiosis plays a fundamental role in the pathogenesis of PCOS (65,115).
The dysbiosis of gut microbiota theory of PCOS proposes that poor-quality Western-style diets (high-glycemic, high fat, high calorie, highly processed, low nutrient, low fibre), result in an imbalanced microbiome which induces increased GI permeability and results in endotoxin-mediated chronic inflammation (65). Dysbiosis results in breakdown of the GI barrier function (loss of protective mucous, activation of the zonulin pathway and breakdown of intercellular tight junctions), and release of lipopolysaccharide from the cell walls of gram-negative bacteria, which can traverse the “leaky gut.” Lipopolysaccharide binds with lipopolysaccharide binding protein which together bind with Toll-like receptor 4 on the surface of submucosal macrophages. This activates the NF-κB inflammatory pathway resulting in release of inflammatory cytokines. Continuing ingestion of a poor-quality diet results in chronic inflammation, insulin resistance, hyperandrogenism, ovulatory dysfunction, and the clinical features of PCOS (1,65).
A recent review of 31 proof-of-concept studies that specifically investigated this mechanism, concluded that preliminary evidence supports this theory (115). Most studies showed reduced alpha-diversity (lower numbers of bacterial taxa) of the microbiome, in women with PCOS, compared to healthy individuals. Despite this finding, no specific microbial signature has been found in women with PCOS (272). This is likely due to immense individual variability in microbiomes, the existence of functional redundancy (microbiotas contain many species and strains that can perform the same function) (277), as well as the fact that PCOS is a quantitative trait (interaction between environment and individual). Individual variation in the composition of the microbiome has likely contributed to the ability of humans to adapt to so many varied environments and types of diets, throughout evolutionary history (271).
Many other dysbiosis-related pathophysiological mechanisms have been proposed and investigated since the dysbiosis model was proposed in 2012. These include altered choline pathways, changes in bile acid metabolites that affect inflammation, glucose and lipid metabolism, processes involved in energy absorption, short-chain fatty acids, effects on gastrointestinal hormones, and other host metabolites (trimethylamine N-oxide, lactate, primary bile acids) (272,278). Many food components are not digested in the small intestine and transit to the colon, where they are fermented by the colonic microbiota. Bacterial conversion of these compounds results in a wide variety of metabolites that enter the host, alter immunity, metabolism, hormones and reproduction, and can influence the risk of disease (89). Fermentation of undigested carbohydrates (soluble fibre), proteins and plant-derived phytochemicals (polyphenols), results in a range of bioactive metabolites such as short-chain fatty acids, amines, secondary metabolites of polyphenols, and gases (hydrogen, hydrogen sulphide, methane). It is likely that future research will identify a range of pathophysiological mechanisms related to the role of dysbiosis in the pathogenesis of PCOS.
In summary, as with the rest of the biological components involved in PCOS, the microbiome appears to have a significant bidirectional role in the pathophysiology of chronic systemic inflammation, insulin resistance, hyperandrogenism, and ovulatory dysfunction. Dysbiosis is an important part of the evolutionary model and represents a maladaptive response of the microbiome to contemporary lifestyle and the environment. Dysbiosis is a modifiable change that can be reversed with lifestyle, diet, prebiotics and probiotics (115).
7. Environmental and endocrine disrupting chemicals in PCOS
Endocrine disrupting chemicals are a global problem for human health and the environment (279). There is no doubt that anthropomorphic chemicals interfere with human physiology and have adverse health effects (280–283). Human exposure mainly occurs through mucosal surfaces (oral, gastrointestinal, respiratory, genitourinary), or via dermal absorption. EDC exposure is ubiquitous and numerous international organizations have issued warnings to doctors and patients, regarding the possible dangers to human health and pregnant women. These include The Royal College of Obstetricians and Gynaecologists (281), the International Federation of Gynecology and Obstetrics (282), and the Endocrine Society (283). They have recommended that all pregnant women be advised of the possible risks of EDC, and that education programmes be developed to inform health professionals of the risks.
Endocrine disrupting chemicals have a significant role in chronic systemic inflammation (284), insulin resistance (285), hyperandrogenism (286), microbiome disruption (287) neuroendocrine imbalance (288), obesity (289), oxidative stress (290), AGE (291), and inflammasomes (292), all of which are involved in the pathophysiology of PCOS. Intervention trials, randomized-controlled trials (RCT), and systematic reviews of RCT, will never be possible or ethical in humans. Hypotheses about mechanisms of causation need to be based on observational studies in humans, human bio-monitoring studies, intervention studies in animals, and disasters from inadvertent human exposure (279). Large amounts of data are already available that provide “evidence” of the potential harmful effects of EDC in humans (284–293). The implementation of the “Precautionary Principle” (294), is supported by a long list of previous known disasters that have occurred due to delayed action, or negligent inaction (diethylstilbestrol, thalidomide, nicotine, dioxin, asbestos, mesh) (295).
Many EDC have been investigated and implicated in the pathogenesis and pathophysiology of PCOS. These include heavy metals, persistent organic pollutants, polychlorinated biphenyls, organochloride pesticides, air pollutants, pesticides, herbicides, nanomaterials, and plastics (288,296–300). EDC can act individually, but humans are usually exposed to mixtures of chemicals on a regular basis (301). EDC have been identified in most body fluids in women with PCOS (302,303). Six classes of EDC have been shown to cross the placenta (302,304), and EDC may be involved in transgenerational transmission of PCOS (115,305). Anthropometric production of EDC is intimately connected to climate change and related adverse health outcomes, and climate change can exacerbate the effects of EDC (301). Improved regulatory processes, bio-detection methods, climate change mitigation strategies, and dietary interventions, can result in reduced exposure to EDC and other environmental chemicals (306).
Women with a genetic susceptibility to PCOS may be at increased risk of adverse effects of EDC due to having a heightened proinflammatory design (section 3.1) and increased metabolic sensitivity (section 4.2). In addition, the in-utero developmental effects of EDC can be inherited by transgenerational transmission (270,295). Every effort should be made to inform women with PCOS about the potential risks of environmental chemicals and discuss ways to avoid or minimize exposure. Detailed discussion of the mechanisms of action of EDC, animal, and epidemiological studies related to PCOS, is beyond the scope of this review. Readers are referred to published reports for further information (297–299,307).
8. Evolutionary Model of PCOS and the Hallmarks of Health
There has been a gradual paradigm shift in conceptualizing the causes of health and disease over the past 20 years. Seminal publications on the “Hallmarks of Cancer” summarize the properties of malignant cells and their interaction with their non-malignant environment (308). The “Hallmarks of Aging” focus on the interactions of molecular, cellular, and systemic processes that explain the deterioration of organisms over time (309). This is a paradigm shift that signals a move away from conventional anatomical and physiological-based conceptions of disease (individual cells, tissues, organs, and systems) to a more “organizational” structure focused on factors that are causatively involved in maintaining homeostasis and equilibrium.
The “Hallmarks of Health” has endeavored to define health as a “compendium of organizational and dynamic features that maintain physiology (59).” This contrasts with the usual definition of health as the “absence of disease.” The current conception of PCOS as a polygenic disorder (collection of normal alleles), that is programmed in-utero and then manifests in adolescence following exposure to lifestyle, nutritional and environmental influences, seems to be an excellent example of this new paradigm (1). Many of the inter-related components of the hallmarks of health are dysregulated in women with PCOS. There are disturbances at the molecular (ROS, AGE, metabolites), organelle (mitochondria, endoplasmic reticulum), cellular (immune, endocrine, neural), supracellular (gastrointestinal mucosa, mucosal immunity), organ (ovary, pancreas), systemic (endocrine, reproductive, immune), and meta-organism levels (host-microbiota interaction) (table 2). The previous sections of this manuscript have attempted to describe some of the detailed interactions that disrupt the organizational dynamics of the hallmarks of health in women with PCOS.
The Evolutionary Model of PCOS is consistent with the hallmarks of health paradigm (1,5). Seen from this new perspective, PCOS is a progressive disturbance of the overall organism involving multiple levels that usually operate to maintain internal homeostasis and equilibrium with the environment. Multiple contemporary lifestyle factors can derail the overall organization of the system resulting in complex pathophysiological interactions and feedback mechanisms, that have no apparent beginning or end. All the components are inter-related and inter-dependent, in a system where multiple lifestyle and environmental “causative” factors operate simultaneously and synergistically. Defining PCOS in a positive way, with an evolutionary and hallmarks of health perspective, opens new opportunities for understanding this complex condition, improving patient communication and compliance, and informing future research, prevention, and intervention strategies.
Table 2.
Dysregulation of the Hallmarks of Health in Polycystic Ovary Syndrome.
| Hallmark | Polycystic Ovary Syndrome |
|---|---|
| Barrier integrity | Gastrointestinal permeability Respiratory mucosa |
| Containment of local perturbations | Mucosal and adaptive immunity ROS, AGE, DAMPS, PAMPS |
| Recycling and turnover | Autophagy, apoptosis, pyroptosis GI stem cell turnover |
| Integration of circuitries | Metabolic, reproductive neurological, circadian |
| Rhythmic oscillations | Menstrual cycle, circadian GI peristalsis |
| Homeostatic resilience | Endocrine, ANS metabolic, immune, microbiome |
| Hormetic regulation | oxidative stress, inactivity xenohormesis# |
| Repair and regeneration | Endoplasmic reticular stress mitochondrial stress |
# xenohormesis=dietary phytochemical-induced; ANS=autonomic nervous system; DAMPS=danger-associated molecular patterns; ROS=reactive oxygen species; PAMPS=pathogen-associated molecular patterns; GI=Gastrointestinal; AGE=advanced glycation end-products
9. Conclusions
Polycystic ovary syndrome is a common condition that affects women at all stages of the life cycle. The pathogenesis is related to a combination of nutritional and environmental exposures, in a genetically susceptible individual. Polycystic ovary syndrome can be characterized as an evolutionary mismatch disorder resulting from a disturbance to the hallmarks of health. Chronic systemic inflammation and insulin resistance are core mechanisms that operate at an organismal level in the physiology of survival, and play a central role in the pathophysiology of PCOS. Polycystic ovary syndrome is usually diagnosed in adolescence by the combination of oligomenorrhoea and hyperandrogenism and is a progressive condition that is associated with significant metabolic, hormonal, reproductive, and psychological problems. The symptoms and chronic sequelae can be controlled and prevented by lifestyle interventions, and the diagnosis of PCOS provides the ideal opportunity for prevention of chronic disease. This narrative review has outlined some components of the complex web of biological and pathophysiological changes that contribute to the pathogenesis of PCOS.
Author Contributions
JP conceptualized and wrote the manuscript.
Funding
Not applicable.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
I would like to thank Geoffrey Reid, Helen Rienits, Eugene Cheah, and Claire O’Brien and for their helpful comments and suggestions in reviewing this manuscript. I would also like to thank Hannah Toogood for her assistance with the graphics.
Conflicts of Interest
No conflicts of interest to declare.
Abbreviations
| ACTH | Adrenocorticotropic Hormone |
| AGE | Advanced Glycation End-Products |
| ANS | Autonomic Nervous System |
| AT | Adipose Tissue |
| BAT | Brown Adipose Tissue |
| CDR | Cell Danger Response |
| DAMPS | Danger Associated Molecular Patterns |
| DNA | Deoxyribose Nucleic Acid |
| EDC | Endocrine Disrupting Chemical |
| GDM | Gestational Diabetes |
| GI | Gastrointestinal |
| GLUT | Glucose Transporter |
| HPA | Hypothalamic-Pituitary-Adrenal |
| IR | Insulin Resistance |
| LH | Luteinizing Hormone |
| LPS | Lipopolysaccharide |
| PAMPS | Pathogen Associated Molecular Patterns |
| PCOS | Polycystic Ovary Syndrome |
| PNS | Peripheral Nervous System |
| PRR | Pattern Recognition Receptors |
| RAGE | Receptor of Advanced Glycation End-Products |
| RNA | Ribose Nucleic Acid |
| RNS | Reactive Nitrogen Species |
| ROS | Reactive Oxygen Species |
| SAT | Subcutaneous Adipose Tissue |
| SNS | Sympathetic Nervous System |
| TLR | Toll-like Receptor |
| TNF | Tumour Necrosis Factor |
| T2DM | Type 2 Diabetes Mellitis |
| WAT | White Adipose Tissue |
| VAT | Visceral Adipose Tissue |
References
- Parker J, O’brien C, Hawrelak J, Gersh FL. Polycystic Ovary Syndrome: An Evolutionary Adaptation to Lifestyle and the Environment. Int J Environ Res Public Health. 2022;19(3). [CrossRef]
- Teede HJ, Misso ML, Costello MF, Dokras A, Laven J, Moran L, et al. Recommendations from the international evidence-based guideline for the assessment and management of polycystic ovary syndrome. Fertil Steril. 2018;110(3):364–79. [CrossRef]
- Cowan S, Lim S, Alycia C, Pirotta S, Thomson R, Gibson-Helm M, et al. Lifestyle management in polycystic ovary syndrome - beyond diet and physical activity. BMC Endocr Disord [Internet]. 2023;23(1):14. Available from: http://www.ncbi.nlm.nih.gov/pubmed/36647089%0Ahttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC9841505. [CrossRef]
- Pei Z, Lu W, Feng Y, Xu C, Hsueh AJW. Out of step societal and Darwinian adaptation during evolution is the cause of multiple women’s health issues. Hum Reprod. 2022;37(9):1959–69. [CrossRef]
- Dumesic DA, Padmanabhan V, Chazenbalk GD, Abbott DH. Polycystic ovary syndrome as a plausible evolutionary outcome of metabolic adaptation. Reprod Biol Endocrinol [Internet]. 2022;20(1):1–12. Available from: https://doi.org/10.1186/s12958-021-00878-y. [CrossRef]
- Pathak G, Nichter M. Polycystic ovary syndrome in globalizing India: An ecosocial perspective on an emerging lifestyle disease. Soc Sci Med [Internet]. 2015;146(March 2017):21–8. Available from: http://dx.doi.org/10.1016/j.socscimed.2015.10.007. [CrossRef]
- Parker J, O’Brien C. Evolutionary and genetic antecedents to the pathogenesis of polycystic ovary syndrome (PCOS). J ACNEM. 2021;40(1).
- Aboeldalyl S, James C, Seyam E, Ibrahim EM, Shawki HED, Amer S. The role of chronic inflammation in polycystic ovarian syndrome—a systematic review and meta-analysis. Int J Mol Sci. 2021;22(5):1–31. [CrossRef]
- Giampaolino P, Foreste V, Di Filippo C, Gallo A, Mercorio A, Serafino P, et al. Microbiome and PCOS: State-of-art and future aspects. Int J Mol Sci. 2021;22(4):1–16. [CrossRef]
- Wang J, Wu D, Guo H, Li M. Hyperandrogenemia and insulin resistance: The chief culprit of polycystic ovary syndrome. Life Sci. 2019;236(October). [CrossRef]
- Palomba S, De Wilde MA, Falbo A, Koster MPH, La Sala GB, Fauser BCJM. Pregnancy complications in women with polycystic ovary syndrome. Hum Reprod Update. 2015;21(5):575–92. [CrossRef]
- Brutocao C, Zaiem F, Alsawas M, Morrow AS, Murad MH, Javed A. Psychiatric disorders in women with polycystic ovary syndrome: a systematic review and meta-analysis. Endocrine [Internet]. 2018;62(2):318–25. Available from: http://dx.doi.org/10.1007/s12020-018-1692-3. [CrossRef]
- Zore T, Joshi N V., Lizneva D, Azziz R. Polycystic Ovarian Syndrome: Long-Term Health Consequences. Vol. 35, Seminars in Reproductive Medicine. 2017. p. 271–81. [CrossRef]
- Wu J, Yao XY, Shi RX, Liu SF, Wang XY. A potential link between polycystic ovary syndrome and non-alcoholic fatty liver disease: An update meta-analysis. Reprod Health. 2018;15(1):1–9. [CrossRef]
- Yumiceba, V., Lopez-Cortes, A., Perez-Villa A. Oncology and Pharmacogenomics Insights in Polycystic Ovary Syndrome : An Integrative Analysis. Front Endocrinol (Lausanne). 2020;11(October):1–21. [CrossRef]
- Du Y, Li F, Li S, Ding L, Liu M. Causal relationship between polycystic ovary syndrome and chronic kidney disease : A Mendelian randomization study. Front Endocrinol 15 March 2023;(14):1–7. [CrossRef]
- Rodgers RJ, Avery JC, Moore VM, Davies MJ, Azziz R, Stener-Victorin E, et al. Complex diseases and co-morbidities: Polycystic ovary syndrome and type 2 diabetes mellitus. Endocr Connect. 2019;8(3):R71–5. [CrossRef]
- Parker J. NEM : A New Paradigm for Understanding the Common Origins of the Chronic Disease Epidemic. J ACNEM. 2018;37(3):6–11.
- Dunaif A. Perspectives in polycystic ovary syndrome: From hair to eternity. J Clin Endocrinol Metab. 2016;101(3):759–68. [CrossRef]
- Day F, Karaderi T, Jones MR, Meun C, He C, Drong A, et al. Large-scale genome-wide meta-analysis of polycystic ovary syndrome suggests shared genetic architecture for different diagnosis criteria. PLoS Genet. 2018.
- Vink JM, Sadrzadeh S, Lambalk CB, Boomsma DI. Heritability of polycystic ovary syndrome in a Dutch twin-family study. J Clin Endocrinol Metab. 2006;91(6):2100–4. [CrossRef]
- Kahsar-Miller MD, Nixon C, Boots LR, Go RC, Azziz R. Prevalence of polycystic ovary syndrome (PCOS) in first-degree relatives of patients with PCOS. Fertil Steril. 2001;75(1):53–8. [CrossRef]
- Sun Q, Gao Y, Yang J, Lu J, Feng W, Yang W. Mendelian Randomization Analysis Identified Potential Genes Pleiotropically Associated with Polycystic Ovary Syndrome. Reprod Sci [Internet]. 2021; Available from: https://doi.org/10.1007/s43032-021-00776-z. [CrossRef]
- Zhu, Tiantian. Goodarzi M. Causes and consequences of polycystic ovary syndrome: Insights from Mendelian Randomization. J Clin Endocrinol Metab. epub ahead. [CrossRef]
- Lyle SM, Ahmed S, Elliott JE, Stener-Victorin E, Nachtigal MW, Drögemöller BI. Transcriptome-wide association analyses identify an association between ARL14EP and polycystic ovary syndrome. J Hum Genet. 2023;(August 2022). [CrossRef]
- Charifson MA, Trumble BC. Evolutionary origins of polycystic ovary syndrome: An environmental mismatch disorder. Evol Med Public Heal. 2019;2019(1):50–63. [CrossRef]
- Shaw LMA, Elton S. Polycystic ovary syndrome: A transgenerational evolutionary adaptation. BJOG An Int J Obstet Gynaecol. 2008;115(2):144–8.
- Diamanti-Kandarakis E, Piperi C. Genetics of polycystic ovary syndrome: Searching for the way out of the labyrinth. Hum Reprod Update. 2005;11(6):631–43. [CrossRef]
- Futuyma DJ. Evolutionary constraint and ecological consequences. Evolution (N Y). 2010;64(7):1865–84. [CrossRef]
- Ricklefs R, Wikelski M. The physiology/lifehistory nexus. Trends Ecol Evol [Internet]. 2002;17(10):462–8. Available from: https://ac.els-cdn.com/S0169534702025788/1-s2.0-S0169534702025788-main.pdf?_tid=9d9d8202-a223-11e7-9008-00000aacb35e&acdnat=1506366131_56ecc99336113d5fe5d529b4572abb84.
- Benton ML. The influence of evolutionary history on human health and disease. Nat Rev Genet [Internet]. 2021; Available from: http://dx.doi.org/10.1038/s41576-020-00305-9. [CrossRef]
- Brady SP, Bolnick DI, Barrett RDH, Chapman L, Crispo E, Derry AM, et al. Understanding maladaptation by uniting ecological and evolutionary perspectives. Am Nat. 2019;194(4):495–515. [CrossRef]
- Gluckman PD, Hanson MA, Spencer HG. Predictive adaptive responses and human evolution. Trends Ecol Evol. 2005;20(10):527–33. [CrossRef]
- Fay JC. Disease consequences of human adaptation. Appl Transl Genomics [Internet]. 2013;2(1):42–7. Available from: http://dx.doi.org/10.1016/j.atg.2013.08.001. [CrossRef]
- Casarini L, Simoni M, Brigante G. Is polycystic ovary syndrome a sexual conflict? A review. Reprod Biomed Online [Internet]. 2016;32(4):350–61. Available from: http://dx.doi.org/10.1016/j.rbmo.2016.01.011. [CrossRef]
- Itoh H, Ueda M, Suzuki M, Kohmura-Kobayashi Y. Developmental Origins of Metaflammation; A Bridge to the Future Between the DOHaD Theory and Evolutionary Biology. Front Endocrinol (Lausanne). 2022;13(February):1–7. [CrossRef]
- Chiang J. Liver Physiology: MetaboLism and Detoxification [Internet]. Pathobiology of Human Disease: A Dynamic Encyclopedia of Disease Mechanisms. Elsevier Inc.; 2014. 1770–1782 p. Available from: http://dx.doi.org/10.1016/B978-0-12-386456-7.04202-7. [CrossRef]
- Roh E, Song DK, Kim MS. Emerging role of the brain in the homeostatic regulation of energy and glucose metabolism. Exp Mol Med [Internet]. 2016;48(3):e216-12. Available from: http://dx.doi.org/10.1038/emm.2016.4. [CrossRef]
- Norman, R. Teede H. A new evidence-based guideline for assessment and management of polycystic ovary syndrome. Med J Aust. 2018;209(7):299–300. [CrossRef]
- Chodasewicz K. Evolution, reproduction and definition of life. Theory Biosci. 2014;133(1):39–45. [CrossRef]
- Corbett S, Morin-Papunen L. The Polycystic Ovary Syndrome and recent human evolution. Mol Cell Endocrinol [Internet]. 2013;373(1–2):39–50. Available from: http://dx.doi.org/10.1016/j.mce.2013.01.001. [CrossRef]
- Parker J. Glucose metabolism , energy production and regulation of cellular and whole-body metabolism. J ACNEM. 2020;39(1):29–33.
- Chantranupong L, Wolfson RL, Sabatini DM. Nutrient-sensing mechanisms across evolution. Cell [Internet]. 2015;161(1):67–83. Available from: http://dx.doi.org/10.1016/j.cell.2015.02.041. [CrossRef]
- Holly JMP, Biernacka K, Perks CM. Systemic metabolism, its regulators, and cancer: Past mistakes and future potential. Front Endocrinol (Lausanne). 2019;10(FEB):1–17. [CrossRef]
- Eiras MC, Pinheiro DP, Romcy KAM, Ferriani RA, Reis RM Dos, Furtado CLM. Polycystic Ovary Syndrome: the Epigenetics Behind the Disease. Reprod Sci. 2022;29(3):680–94. [CrossRef]
- Dapas M, Dunaif A. Deconstructing a Syndrome: Genomic Insights Into PCOS Causal Mechanisms and Classification. Endocr Rev. 2022;(February):927–65. [CrossRef]
- Smirnov, V. Beeraka, N. Yu Butko, D. Nikolenko, V. Bondarev, S. Achkasov, E. Sinelnikov, M. Vikram P. Updates on Molecular Targets and Epigenetic-Based Therapies for PCOS. Reprod Sci. 2022.
- Parker J. Emerging Concepts in the Pathogenesis and Treatment of Polycystic Ovary Syndrome. Curr Womens Health Rev. 2016;10(2):107–12. [CrossRef]
- Azziz R, Dumesic DA, Goodarzi MO. Polycystic ovary syndrome: An ancient disorder? Fertil Steril [Internet]. 2011;95(5):1544–8. Available from: http://dx.doi.org/10.1016/j.fertnstert.2010.09.032. [CrossRef]
- Gluckman PD, Low FM, Hanson MA. Anthropocene-related disease. Evol Med Public Heal. 2020;2020(1):304–10. [CrossRef]
- Lee HJ, Hore TA, Reik W. Reprogramming the methylome: Erasing memory and creating diversity. Cell Stem Cell [Internet]. 2014;14(6):710–9. Available from: http://dx.doi.org/10.1016/j.stem.2014.05.008. [CrossRef]
- Monk D, Mackay DJG, Eggermann T, Maher ER, Riccio A. Genomic imprinting disorders: lessons on how genome, epigenome and environment interact. Nat Rev Genet [Internet]. 2019;20(4):235–48. Available from: http://dx.doi.org/10.1038/s41576-018-0092-0. [CrossRef]
- Rubio-Ruiz ME, Peredo-Escárcega AE, Cano-Martínez A, Guarner-Lans V. An Evolutionary Perspective of Nutrition and Inflammation as Mechanisms of Cardiovascular Disease. Int J Evol Biol. 2015;2015:1–10. [CrossRef]
- Watve MG, Yajnik CS. Evolutionary origins of insulin resistance: A behavioral switch hypothesis. BMC Evol Biol. 2007;7:1–13. [CrossRef]
- Ling C, Rönn T. Epigenetics in Human Obesity and Type 2 Diabetes. Cell Metab. 2019;29(5):1028–44. [CrossRef]
- Szukiewicz D, Trojanowski S, Kociszewska A, Szewczyk G. Modulation of the Inflammatory Response in Polycystic Ovary Syndrome (PCOS)—Searching for Epigenetic Factors. Int J Mol Sci. 2022;23(23). [CrossRef]
- Shorakae S, Ranasinha S, Abell S, Lambert G, Lambert E, de Courten B, et al. Inter-related effects of insulin resistance, hyperandrogenism, sympathetic dysfunction and chronic inflammation in PCOS. Clinical Endocrinology. 2018;89:628–33. [CrossRef]
- Barlampa D, Bompoula MS, Bargiota A, Kalantaridou S, Mastorakos G, Valsamakis G. Hypothalamic inflammation as a potential pathophysiologic basis for the heterogeneity of clinical, hormonal, and metabolic presentation in pcos. Nutrients. 2021;13(2):1–17. [CrossRef]
- López-Otín C, Kroemer G. Hallmarks of Health. Cell. 2021;184(1):33–63.
- Okin D, Medzhitov R. Evolution of inflammatory diseases. Curr Biol [Internet]. 2012;22(17):R733–40. Available from: http://dx.doi.org/10.1016/j.cub.2012.07.029. [CrossRef]
- Furman D, Campisi J, Verdin E, Carrera-Bastos P, Targ S, Franceschi C, et al. Chronic inflammation in the etiology of disease across the life span. Nat Med [Internet]. 2019;25(12):1822–32. Available from: http://dx.doi.org/10.1038/s41591-019-0675-0. [CrossRef]
- Naviaux RK. Metabolic features and regulation of the healing cycle—A new model for chronic disease pathogenesis and treatment. Mitochondrion [Internet]. 2019;46(April 2018):278–97. Available from: https://doi.org/10.1016/j.mito.2018.08.001. [CrossRef]
- Yu SY, Li XL. Pyroptosis and inflammasomes in obstetrical and gynecological diseases. Gynecol Endocrinol [Internet]. 2021;37(5):385–91. Available from: https://doi.org/10.1080/09513590.2021.1871893. [CrossRef]
- Katayama PL, Leirão IP, Kanashiro A, Luiz JPM, Cunha FQ, Navegantes LCC, et al. The carotid body detects circulating tumor necrosis factor-alpha to activate a sympathetic anti-inflammatory reflex. Brain Behav Immun. 2022;102(December 2021):370–86. [CrossRef]
- Tremellen K, Pearce K. Dysbiosis of Gut Microbiota (DOGMA) - A novel theory for the development of Polycystic Ovarian Syndrome. Med Hypotheses [Internet]. 2012;79(1):104–12. Available from: http://dx.doi.org/10.1016/j.mehy.2012.04.016. [CrossRef]
- Xiong P, Zhang F, Liu F, Zhao J, Huang X. Metaflammation in glucolipid metabolic disorders : Pathogenesis and treatment. Biomed Pharmacother [Internet]. 2023;161(March):114545. Available from: https://doi.org/10.1016/j.biopha.2023.114545. [CrossRef]
- Netea MG, Balkwill F, Chonchol M, Cominelli F, Donath MY, Giamarellos-Bourboulis EJ, et al. A guiding map for inflammation. Nat Immunol [Internet]. 2017;18(8):826–31. Available from: http://dx.doi.org/10.1038/ni.3790. [CrossRef]
- Zhang H, Zhu J, Gong Z, Zhu JK. Abiotic stress responses in plants. Nat Rev Genet. 2022;23(2):104–19.
- Khan RN, Hay DP. A clear and present danger: Inflammasomes DAMPing down disorders of pregnancy. Hum Reprod Update. 2015;21(3):388–405.
- Amarante-Mendes GP, Adjemian S, Branco LM, Zanetti LC, Weinlich R, Bortoluci KR. Pattern recognition receptors and the host cell death molecular machinery. Front Immunol. 2018;9(OCT):1–19. [CrossRef]
- Cavalier-Smith T. Cell evolution and Earth history: Stasis and revolution. Philos Trans R Soc B Biol Sci. 2006;361(1470):969–1006. [CrossRef]
- Tang KH, Blankenship RE. Both forward and reverse TCA cycles operate in green sulfur bacteria. J Biol Chem [Internet]. 2010;285(46):35848–54. Available from: http://dx.doi.org/10.1074/jbc.M110.157834. [CrossRef]
- Medeiros HCD, Lunt SY. The Warburg effect: Saturation of mitochondrial NADH shuttles triggers aerobic lactate fermentation. Mol Cell [Internet]. 2022;82(17):3119–21. Available from: https://doi.org/10.1016/j.molcel.2022.08.004. [CrossRef]
- Naviaux RK. Oxidative shielding or oxidative stress? J Pharmacol Exp Ther. 2012;342(3):608–18.
- Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25(3):486–541. [CrossRef]
- Dabravolski SA, Nikiforov NG, Eid AH, Nedosugova L V., Starodubova A V., Popkova T V., et al. Mitochondrial dysfunction and chronic inflammation in polycystic ovary syndrome. Int J Mol Sci. 2021;22(8):1–20. [CrossRef]
- Zhao H, Zhao Y, Li T, Li M, Li J, Li R, et al. Metabolism alteration in follicular niche: The nexus among intermediary metabolism, mitochondrial function, and classic polycystic ovary syndrome. Free Radic Biol Med [Internet]. 2015;86(49):295–307. Available from: http://dx.doi.org/10.1016/j.freeradbiomed.2015.05.013. [CrossRef]
- Mohammadi M. Oxidative Stress and Polycystic Ovary Syndrome: A Brief Review. Int J Prev Med. 2017;8:1–7. [CrossRef]
- Zuo T, Zhu M, Xu W. Roles of oxidative stress in polycystic ovary syndrome and cancers. Oxid Med Cell Longev. 2016;2016. [CrossRef]
- Sharifi-Rad M, Anil Kumar N V., Zucca P, Varoni EM, Dini L, Panzarini E, et al. Lifestyle, Oxidative Stress, and Antioxidants: Back and Forth in the Pathophysiology of Chronic Diseases. Front Physiol. 2020;11(July):1–21. [CrossRef]
- Herman R, Jensterle M, Janež A, Goričar K, Dolžan V. Genetic variability in antioxidative and inflammatory pathways modifies the risk for PCOs and influences metabolic profile of the syndrome. Metabolites. 2020;10(11):1–18. [CrossRef]
- Diamanti-Kandarakis E, Dunaif A. Insulin resistance and the polycystic ovary syndrome revisited: An update on mechanisms and implications. Endocr Rev. 2012;33(6):981–1030. [CrossRef]
- Rashidi B, Haghollahi F, Shariat M, Zayerii F. The Effects of Calcium-Vitamin D and Metformin on Polycystic Ovary Syndrome: A Pilot Study. Taiwan J Obstet Gynecol [Internet]. 2009;48(2):142–7. Available from: http://dx.doi.org/10.1016/S1028-4559(09)60275-8. [CrossRef]
- Kaya C, Erkan AF, Cengiz SD, Dünder I, Demirel ÖE, Bilgihan A. Advanced oxidation protein products are increased in women with polycystic ovary syndrome: relationship with traditional and nontraditional cardiovascular risk factors in patients with polycystic ovary syndrome. Fertil Steril. 2009;92(4):1372–7. [CrossRef]
- Enechukwu CI, Onuegbu AJ, Olisekodiaka MJ, Eleje GU, Ikechebelu JI, Ugboaja JO, et al. Oxidative stress markers and lipid profiles of patients with polycystic ovary syndrome in a Nigerian tertiary hospital. Obstet Gynecol Sci. 2019;62(5):335–43. [CrossRef]
- Dincer Y, Akcay T, Erdem T, Ilker Saygili E, Gundogdu S. DNA damage, DNA susceptibility to oxidation and glutathione level in women with polycystic ovary syndrome. Scand J Clin Lab Invest. 2005;65(8):721–8. [CrossRef]
- Noormohammadi M, Eslamian G, Malek S, Shoaibinobarian N, Mirmohammadali SN. The association between fertility diet score and polycystic ovary syndrome: A Case-Control study. Health Care Women Int [Internet]. 2022;43(1–3):70–84. Available from: https://doi.org/10.1080/07399332.2021.1886298. [CrossRef]
- Shoaibinobarian N, Eslamian G, Noormohammadi M, Malek S, Rouhani S, Mirmohammadali SN. Dietary Total Antioxidant Capacity and Risk of Polycystic Ovary Syndrome: A Case-Control Study. Int J Fertil Steril. 2022;16(3):200–5. [CrossRef]
- Parker, J. Hawrelak, J. Gersh F. Nutritional Role of Polyphenols As a Component of a Wholefood Diet in the Management of Polycystic Ovary Syndrome. J ACNEM [Internet]. 2021;40(2):6–12. Available from: https://search.ebscohost.com/login.aspx?direct=true&db=rzh&AN=154032844&lang=ja&site=ehost-live.
- Piperi C, Adamopoulos C, Dalagiorgou G, Diamanti-Kandarakis E, Papavassiliou AG. Crosstalk between advanced glycation and endoplasmic reticulum stress: Emerging therapeutic targeting for metabolic diseases. J Clin Endocrinol Metab. 2012;97(7):2231–42. [CrossRef]
- Inagi R. Inhibitors of advanced glycation and endoplasmic reticulum stress [Internet]. 1st ed. Vol. 491, Methods in Enzymology. Elsevier Inc.; 2011. 361–380 p. Available from: http://dx.doi.org/10.1016/B978-0-12-385928-0.00020-1. [CrossRef]
- Goldberg T, Cai W, Peppa M, Dardaine V, Baliga BS, Uribarri J, et al. Advanced glycoxidation end products in commonly consumed foods. J Am Diet Assoc. 2004;104(8):1287–91. [CrossRef]
- Garg D, Merhi Z. Advanced glycation end products: Link between diet and ovulatory dysfunction in PCOS? Nutrients. 2015;7(12):10129–44.
- Basta G. Receptor for advanced glycation endproducts and atherosclerosis: From basic mechanisms to clinical implications. Atherosclerosis. 2008;196(1):9–21. [CrossRef]
- Kathryn C. B. Tan; Sammy W. M. Shiu; Ying Wong; Xystus Tam. Serum advanced glycation end products (AGEs) are associated with insulin resistance. Diabetes Metab Res Rev. 2011;27:1488–92. [CrossRef]
- Palimeri S, Palioura E, Diamanti-Kandarakis E. Current perspectives on the health risks associated with the consumption of advanced glycation end products: Recommendations for dietary management. Diabetes, Metab Syndr Obes Targets Ther. 2015;8:415–26. [CrossRef]
- Diamanti-Kandarakis E, Piperi C, Patsouris E, Korkolopoulou P, Panidis D, Pawelczyk L, et al. Immunohistochemical localization of advanced glycation end-products (AGEs) and their receptor (RAGE) in polycystic and normal ovaries. Histochem Cell Biol. 2007;127(6):581–9. [CrossRef]
- Azhary JMK, Harada M, Kunitomi C, Kusamoto A, Takahashi N, Nose E, et al. Androgens Increase Accumulation of Advanced Glycation End Products in Granulosa Cells by Activating ER Stress in PCOS. Endocrinol (United States). 2020;161(2):1–13.
- Tatone C, Di Emidio G, Placidi M, Rossi G, Ruggieri S, Taccaliti C, et al. AGEs-related dysfunctions in PCOS: Evidence from animal and clinical research. J Endocrinol. 2021;251(2):R1–9. [CrossRef]
- Garg D, Merhi Z. Relationship between Advanced Glycation End Products and Steroidogenesis in PCOS. Reprod Biol Endocrinol [Internet]. 2016;14(1):1–13. Available from: http://dx.doi.org/10.1186/s12958-016-0205-6. [CrossRef]
- Van Der Lugt T, Weseler AR, Gebbink WA, Vrolijk MF, Opperhuizen A, Bast A. Dietary advanced glycation endproducts induce an inflammatory response in human macrophages in vitro. Nutrients. 2018;10(12):1–10. [CrossRef]
- Uribarri J, del Castillo MD, de la Maza MP, Filip R, Gugliucci A, Luevano-Contreras C, et al. Dietary advanced glycation end products and their role in health and disease. Adv Nutr. 2015;6(4):461–73. [CrossRef]
- Tantalaki E, Piperi C, Livadas S, Kollias A, Adamopoulos C, Koulouri A, et al. Impact of dietary modification of advanced glycation end products (AGEs) on the hormonal and metabolic profile of women with polycystic ovary syndrome (PCOS). Hormones. 2014;13(1):65–73. [CrossRef]
- Vlassara H, Striker GE. AGE restriction in diabetes mellitus: A paradigm shift. Nat Rev Endocrinol. 2011;7(9):526–39. [CrossRef]
- Janeway CA. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol. 1989;54(1):1–13.
- Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045.
- Naviaux RK. Metabolic features of the cell danger response. Mitochondrion [Internet]. 2014;16:7–17. Available from: http://dx.doi.org/10.1016/j.mito.2013.08.006. [CrossRef]
- Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. The integrated stress response. EMBO Rep. 2016;17(10):1374–95.
- Costa-Mattioli M, Walter P. The integrated stress response: From mechanism to disease. Science. 2020;368(6489). [CrossRef]
- Li D, Wu M. Pattern recognition receptors in health and diseases. Signal Transduct Target Ther [Internet]. 2021;6(1):1–24. Available from: http://dx.doi.org/10.1038/s41392-021-00687-0. [CrossRef]
- Moretti J, Blander JM. Insights into phagocytosis-coupled activation of pattern recognition receptors and inflammasomes. Curr Opin Immunol [Internet]. 2014;26(1):100–10. Available from: http://dx.doi.org/10.1016/j.coi.2013.11.003. [CrossRef]
- Zhou F, Li C, Zhang SY. NLRP3 inflammasome: a new therapeutic target for high-risk reproductive disorders? Chin Med J (Engl). 2020;134(1):20–7.
- Moretti J, Blander JM. Cell-autonomous stress responses in innate immunity. J Leukoc Biol. 2017;101(1):77–86. [CrossRef]
- Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56(7):1761–72. [CrossRef]
- Parker J, O’Brien C, Hawrelak J. A narrative review of the role of gastrointestinal dysbiosis in the pathogenesis of polycystic ovary syndrome. Obstet Gynecol Sci. 2022;65(1):14-28. [CrossRef]
- Murthi P, Pinar AA, Dimitriadis E, Samuel CS. Inflammasomes—a molecular link for altered immunoregulation and inflammation mediated vascular dysfunction in preeclampsia. Int J Mol Sci. 2020;21(4). [CrossRef]
- Rostamtabar, M. Esmaeilzadeh, M. Tourani, M. Rahmani, A. Baee, M. Shirafkan, F. Saleki, K. Mirzababayi, S. Ebrahimpour, S. Nouri H. Pathophysiological roles of chronic low-grade inflammation in polycystic ovary syndrome. J Cell Physiol. 2020;236:824–38.
- Eltzschig, HK. Sitkovsky, MV. Robson S. Purinergic Signaling during Inflammation. N Engl J Med. 2013;368(13):1260–1260.
- Rostamtabar M, Esmaeilzadeh S, Karkhah A, Amiri M, Rahmani A, Bakouei F, et al. Elevated expression of IL-18 but not IL-1β gene is associated with NALP3 and AIM2 inflammasome in Polycystic Ovary Syndrome. Gene [Internet]. 2020;731(January):144352. Available from: https://doi.org/10.1016/j.gene.2020.144352. [CrossRef]
- Guo QJ, Shan J, Xu YF, Hu YY, Huo CL, Song JY, et al. Pioglitazone Metformin Complex Improves Polycystic Ovary Syndrome Comorbid Psychological Distress via Inhibiting NLRP3 Inflammasome Activation: A Prospective Clinical Study. Mediators Inflamm. 2020;2020. [CrossRef]
- Hu C, Pang B, Ma Z, Yi H. Immunophenotypic profiles in polycystic ovary syndrome. Mediators Inflamm. 2020;2020. [CrossRef]
- Mahajan S, Vita R, Shackelford D, Lane J, Schulten V, Zarebski L, et al. Epitope specific antibodies and T cell receptors in the immune epitope database. Front Immunol. 2018;9(NOV):1–10. [CrossRef]
- Rees AR. Understanding the human antibody repertoire. MAbs [Internet]. 2020;12(1):1–16. Available from: https://doi.org/10.1080/19420862.2020.1729683. [CrossRef]
- Alberto Kölliker Frers R, Otero-Losada M, Inés Herrera M, Porta S, Cosentino V, Kerzberg E, et al. Immune-Mediated Inflammation: Human T CD4 Helper Lymphocyte Diversity and Plasticity in Health and Disease. In: Ota Fuchs and Seyyed Shamsadin Athari, editor. Cells of the Immune System [Internet]. 1st ed. Intehopen; 2020. p. 1–14. Available from: https://www.intechopen.com/chapters/69166.
- Sbierski-Kind J, Goldeck D, Buchmann N, Spranger J, Volk HD, Steinhagen-Thiessen E, et al. T cell phenotypes associated with insulin resistance: results from the Berlin Aging Study II. Immun Ageing. 2020;17(1):1–11. [CrossRef]
- Wu R, Fujii S, Ryan NK, Van der Hoek KH, Jasper MJ, Sini I, et al. Ovarian leukocyte distribution and cytokine/chemokine mRNA expression in follicular fluid cells in women with polycystic ovary syndrome. Hum Reprod. 2007;22(2):527–35. [CrossRef]
- Krishna MB, Joseph A, Subramaniam AG, Gupta A, Pillai SM, Laloraya M. Reduced tregs in peripheral blood of PCOS patients - A consequence of aberrant Il2 signaling. J Clin Endocrinol Metab. 2015;100(1):282–92. [CrossRef]
- Ma M, Wang M, Xu F, Hao S. The Imbalance in Th17 and Treg Cells in Polycystic Ovarian Syndrome Patients with Autoimmune Thyroiditis. Immunol Invest [Internet]. 2022;51(5):1170–81. Available from: https://doi.org/10.1080/08820139.2021.1915329. [CrossRef]
- Luan Y yi, Zhang L, Peng Y qiu, Li Y ying, Liu R xia, Yin C hong. Immune regulation in polycystic ovary syndrome. Clin Chim Acta [Internet]. 2022;531(January):265–72. Available from: https://doi.org/10.1016/j.cca.2022.04.234. [CrossRef]
- Salvador AF, de Lima KA, Kipnis J. Neuromodulation by the immune system: a focus on cytokines. Nat Rev Immunol [Internet]. 2021;21(8):526–41. Available from: http://dx.doi.org/10.1038/s41577-021-00508-z. [CrossRef]
- Patel S. Polycystic ovary syndrome (PCOS), an inflammatory, systemic, lifestyle endocrinopathy. Vol. 182, Journal of Steroid Biochemistry and Molecular Biology. 2018;182:27–36.
- Pavlov VA, Wang H, Czura CJ, Friedman SG, Tracey KJ. The Cholinergic Anti-inflammatory Pathway: A Missing Link in Neuroimmunomodulation. Mol Med. 2003;9(5–8):125–34. [CrossRef]
- Bellocchi C, Carandina A, Montinaro B, Targetti E, Furlan L, Rodrigues GD, et al. The Interplay between Autonomic Nervous System and Inflammation across Systemic Autoimmune Diseases. Int J Mol Sci. 2022;23(5). [CrossRef]
- Ingegnoli F, Buoli M, Antonucci F, Coletto LA, Esposito CM, Caporali R. The Link Between Autonomic Nervous System and Rheumatoid Arthritis: From Bench to Bedside. Front Med. 2020;7(December):1–11. [CrossRef]
- del Rey A, Besedovsky HO, Sorkin E, da Prada M, Arrenbrecht S. Immunoregulation mediated by the sympathetic nervous system, II. Cell Immunol. 1981;63(2):329–34. [CrossRef]
- Martelli D. The inflammatory reflex reloaded. Brain Behav Immun [Internet]. 2022;104:137–8. Available from: https://doi.org/10.1016/j.bbi.2022.06.001. [CrossRef]
- Zangeneh FZ, Bagheri M, Naghizadeh MM. Hyponeurotrophinemia in Serum of Women with Polycystic Ovary Syndrome as a Low Grade Chronic Inflammation. Open J Obstet Gynecol. 2015;05(09):459–69. [CrossRef]
- Tracey KJ. The inflammatory reflex. Nature. 2002;420(December):853–8.
- WEXLER BC, DOLGIN AE, TRYCZYNSKI EW. Effects of a bacterial polysaccharide (piromen) on the pituitary-adrenal axis: adrenal ascorbic acid, cholesterol and histologic alterations. Endocrinology. 1957;61(3):300–8.
- Banks, WA. Kastin, AJ. Broadwell R. یلیPassage of cytokines across the blood-brain-barrier. Neuroimmunomodulation. 1995;2(24):1–8.
- Buller KM. Role of circumventricular organs in pro-inflammatory cytokine-induced activation of the hypothalamic-pituitary-adrenal axis. Clin Exp Pharmacol Physiol. 2001;28(7):581–9.
- Berthoud HR, Neuhuber WL. Functional and chemical anatomy of the afferent vagal system. Auton Neurosci Basic Clin. 2000;85(1–3):1–17. [CrossRef]
- Dag ZO, Alpua M, Turkel Y, Isik Y. Autonomic dysfunction in patients with polycystic ovary syndrome. Taiwan J Obstet Gynecol [Internet]. 2015;54(4):381–4. Available from: http://dx.doi.org/10.1016/j.tjog.2015.03.002. [CrossRef]
- Webster JI, Tonelli L, Sternberg EM. Neuroendocrine regulation of immunity. Annu Rev Immunol. 2002;20:125–63. [CrossRef]
- Shaikh S, Verma H, Yadav N, Jauhari M, Bullangowda J. Applications of Steroid in Clinical Practice: A Review. ISRN Anesthesiol. 2012;2012:1–11. [CrossRef]
- McKay LI, Cidlowski JA. Molecular control of immune/inflammatory responses: Interactions between nuclear factor-κB and steroid receptor-signaling pathways. Endocr Rev. 1999;20(4):435–59.
- Kox M, Van Eijk LT, Zwaag J, Van Den Wildenberg J, Sweep FCGJ, Van Der Hoeven JG, et al. Voluntary activation of the sympathetic nervous system and attenuation of the innate immune response in humans. Proc Natl Acad Sci U S A. 2014;111(20):7379–84. [CrossRef]
- Sun P, Liu DG, Ye XM. Nicotinic Acetylcholine Receptor α7 Subunit Is an Essential Regulator of Seizure Susceptibility. Front Neurol. 2021;12(January):1–5. [CrossRef]
- Lansdown A, Rees DA. The sympathetic nervous system in polycystic ovary syndrome: A novel therapeutic target? Clin Endocrinol (Oxf). 2012;77(6):791–801.
- Chen MJ, Yang WS, Yang JH, Chen CL, Ho HN, Yang YS. Relationship between androgen levels and blood pressure in young women with polycystic ovary syndrome. Hypertension. 2007;49(6):1442–7. [CrossRef]
- Perciaccante A, Fiorentini A, Valente R, Tubani L. Polycystic ovary syndrome: Androgens, autonomic nervous system, and hypertension [7]. Hypertension. 2007;50(1):91710. [CrossRef]
- Dissen GA, Parrott JA, Skinner MK, Hill DF, Costa ME, Ojeda SR. Direct effects of nerve growth factor on thecal cells from antral ovarian follicles. Endocrinology. 2000;141(12):4736–50. [CrossRef]
- Dissen GA, Garcia-Rudaz C, Paredes A, Mayer C, Mayerhofer A, Ojeda SR. Excessive ovarian production of nerve growth factor facilitates development of cystic ovarian morphology in mice and is a feature of polycystic ovarian syndrome in humans. Endocrinology. 2009;150(6):2906–14. [CrossRef]
- Giallauria F, Palomba S, Maresca L, Vuolo L, Tafuri D, Lombardi G, et al. Exercise training improves autonomic function and inflammatory pattern in women with polycystic ovary syndrome (PCOS). Clin Endocrinol (Oxf). 2008;69(5):792–8. [CrossRef]
- 155. Trigunaite A, Dimo J, Jørgensen TN. Suppressive effects of androgens on the immune system. Cell Immunol, Available from. [CrossRef]
- González, F. Inflammation in Polycystic Ovary Syndrome: Underpinning of insulin resistance and ovarian dysfunction. Steroids.
- 157. Wagner IV, Savchuk I, Sahlin L, Kulle A, Klöting N, Dietrich A, et al. De Novo and Depot-Specific Androgen Production in Human Adipose Tissue: A Source of Hyperandrogenism in Women with Obesity. Obes Facts.
- 158. Velez LM, Seldin M, Motta AB. Inflammation and reproductive function in women with polycystic ovary syndrome. Biol Reprod.
- 159. Xiong YL, Liang XY, Yang X, Li Y, Wei LN. Low-grade chronic inflammation in the peripheral blood and ovaries of women with polycystic ovarian syndrome. Eur J Obstet Gynecol Reprod Biol.
- 160. Diamanti-Kandarakis E, Alexandraki K, Piperi C, Protogerou A, Katsikis I, Paterakis T, et al. Inflammatory and endothelial markers in women with polycystic ovary syndrome. Eur J Clin Invest.
- 161. Escobar-Morreale HF, Luque-Ramírez M, González F. Circulating inflammatory markers in polycystic ovary syndrome: A systematic review and metaanalysis. Fertil Steril.
- 162. Zuo T, Zhu M, Xu W. Roles of oxidative stress in polycystic ovary syndrome and cancers. Oxid Med Cell Longev.
- 163. Azhary JMK, Harada M, Takahashi N, Nose E, Kunitomi C, Koike H, et al. Endoplasmic reticulum stress activated by androgen enhances apoptosis of granulosa cells via induction of death receptor 5 in PCOS. Endocrinology.
- 164. Koike H, Harada M, Kusamoto A, Xu Z, Tanaka T, Sakaguchi N, et al. Roles of endoplasmic reticulum stress in the pathophysiology of polycystic ovary syndrome. Front Endocrinol, 1124.
- 165. Li Y, Zheng Q, Sun D, Cui X, Chen S, Bulbul A, et al. Dehydroepiandrosterone stimulates inflammation and impairs ovarian functions of polycystic ovary syndrome. J Cell Physiol.
- 166. Zhai Y, Pang Y. Systemic and ovarian inflammation in women with polycystic ovary syndrome. J Reprod Immunol, Available from, 1036. [CrossRef]
- 167. Rahman MS, Hossain KS, Das S, Kundu S, Adegoke EO, Rahman MA, et al. Role of insulin in health and disease: An update. Int J Mol Sci.
- Fu Z, R. Gilbert E, Liu D. Regulation of Insulin Synthesis and Secretion and Pancreatic Beta-Cell Dysfunction in Diabetes. Curr Diabetes Rev.
- 169. Watanabe M, Hayasaki H, Tamayama T, Shimada M. Histologic distribution of insulin and glucagon receptors. Brazilian J Med Biol Res.
- 170. Khalid M, Alkaabi J, Khan MAB, Adem A. Insulin signal transduction perturbations in insulin resistance. Int J Mol Sci.
- 171. Petersen MC, Shulman GI. Mechanisms of insulin action and insulin resistance. Physiol Rev.
- 172. Zhou MS, Wang A, Yu H. Link between insulin resistance and hypertension: What is the evidence from evolutionary biology? Diabetol Metab Syndr.
- 173. Sun Q, Li J, Gao F. New insights into insulin: The anti-inflammatory effect and its clinical relevance. World J Diabetes.
- 174. Chang YW, Hung LC, Chen YC, Wang WH, Lin CY, Tzeng HH, et al. Insulin Reduces Inflammation by Regulating the Activation of the NLRP3 Inflammasome. Front Immunol.
- 175. Fernández-Real JM, Ricart W. Insulin resistance and inflammation in an evolutionary perspective: The contribution of cytokine genotype/phenotype to thriftiness. Diabetologia.
- Wan, YY. Regulatory T cells: Immune suppression and beyond. Cell Mol Immunol, 2010;7(3):204–10. Available from, 2010. [Google Scholar] [CrossRef]
- 177. Jacobse J, Li J, Rings EHHM, Samsom JN, Goettel JA. Intestinal Regulatory T Cells as Specialized Tissue-Restricted Immune Cells in Intestinal Immune Homeostasis and Disease. Front Immunol.
- Makhijani, P. Basso, PJ. Chan, YT. Chen, N. Baechle, J. Khan, S. Furman, D. Tsai, S. Winer D. Regulation of the immune system by the insulin receptor in health and disease. Front Endocrinol, 1: 2023;(March), 2023. [Google Scholar]
- 179. Cassar S, Misso ML, Hopkins WG, Shaw CS, Teede HJ, Stepto NK. Insulin resistance in polycystic ovary syndrome: A systematic review and meta-analysis of euglycaemic-hyperinsulinaemic clamp studies. Hum Reprod.
- 180. Stepto NK, Cassar S, Joham AE, Hutchison SK, Harrison CL, Goldstein RF, et al. Women with polycystic ovary syndrome have intrinsic insulin resistance on euglycaemic-hyperinsulaemic clamp. Hum Reprod.
- 181. Katic M, Kahn CR. The role of insulin and IGF-1 signaling in longevity. Cell Mol Life Sci.
- Cibula, D. Is insulin resistance an essential component of PCOS? The influence of confounding factors. Hum Reprod.
- 183. Plomin R, Haworth CMA, Davis OSP. Common disorders are quantitative traits. Nat Rev Genet, Available from. [CrossRef]
- 184. Tam CS, Xie W, Johnson WD, Cefalu WT, Redman LM, Ravussin E. Defining insulin resistance from hyperinsulinemic-euglycemic clamps. Diabetes Care.
- 185. Dunaif A, Segal KR, Futterweit W, Dobrjansky A. Profound peripheral insulin resistance, independent of obesity, in polycystic ovary syndrome. Diabetes.
- 186. Teede HJ, Hutchison SK, Zoungas S. The management of insulin resistance in polycystic ovary syndrome. Trends Endocrinol Metab.
- 187. Toosy S, Sodi R, Pappachan JM. Lean polycystic ovary syndrome (PCOS): an evidence-based practical approach. J Diabetes Metab Disord.
- 188. Morciano A, Romani F, Sagnella F, Scarinci E, Palla C, Moro F, et al. Assessment of insulin resistance in lean women with polycystic ovary syndrome. Fertil Steril, Available from. [CrossRef]
- 189. Gorjão R, Takahashi HK, Pan JA, Massao Hirabara S. Molecular mechanisms involved in inflammation and insulin resistance in chronic diseases and possible interventions. J Biomed Biotechnol.
- 190. Fernandez M, Murillo A. Dietary Treatments to Reduce Insulin Resistance and Inflammation in Type-2 Diabetic Patients. Med Res Arch.
- Tsatsoulis, A. Mantzaris, MD. Sofia, B. Andrikoula M. Insulin resistance: An adaptive mechanism becomes maladaptive in the current environment - An evolutionary perspective. Metabolism.
- 192. Wensveen FM, Šestan M, Turk Wensveen T, Polić B. ‘Beauty and the beast’ in infection: How immune–endocrine interactions regulate systemic metabolism in the context of infection. Eur J Immunol.
- 193. Wang P, Mariman ECM. Insulin resistance in an energy-centered perspective. Physiol Behav.
- 194. Kampmann U, Knorr S, Fuglsang J, Ovesen P. Determinants of Maternal Insulin Resistance during Pregnancy: An Updated Overview. J Diabetes Res.
- 195. Sprague JE, Gandica R, Kelsey MM. Insulin Resistance in Puberty. Contemporary Endocrinology.
- 196. Straub RH, Cutolo M, Buttgereit F, Pongratz G. Energy regulation and neuroendocrine-immune control in chronic inflammatory diseases. J Intern Med.
- 197. Rabasa C, Dickson SL. Impact of stress on metabolism and energy balance. Curr Opin Behav Sci, Available from. [CrossRef]
- 198. Ieronymaki E, Daskalaki MG, Lyroni K, Tsatsanis C. Insulin signaling and insulin resistance facilitate trained immunity in macrophages through metabolic and epigenetic changes. Front Immunol.
- 199. Teede H, Misso M, Costello M, Dokras A, Laven J, Moran L, et al. International evidence-based guideline for the assessment and management of polycystic ovary syndrome 2018. National Health and Medical Research Council.
- Chaudhari, AP. Mazumdar K, Deepak P. Anxiety, Depression, and Quality of Life in Women with Polycystic Ovarian Syndrome. Indian J Psychol Med.
- 201. Soeters MR, Soeters PB. The evolutionary benefit of insulin resistance. Clin Nutr.
- 202. Soeters MR, Sauerwein HP, Dubbelhuis PF, Groener JE, Ackermans MT, Fliers E, et al. Muscle adaptation to short-term fasting in healthy lean humans. J Clin Endocrinol Metab.
- 203. Horita S, Seki G, Yamada H, Suzuki M, Koike K, Fujita T. Insulin resistance, obesity, hypertension, and renal sodium transport. Int J Hypertens.
- 204. Velloso LA, Folli F, Perego L, Saad MJA. The multi-faceted cross-talk between the insulin and angiotensin II signaling systems. Diabetes/Metabolism Research and Reviews.
- 205. Zhou MS, Schulman IH, Raij L. Vascular inflammation, insulin resistance, and endothelial dysfunction in salt-sensitive hypertension: Role of nuclear factor kappa B activation. J Hypertens.
- 206. Ehrmann DA, Liljenquist DR, Kasza K, Azziz R, Legro RS, Ghazzi MN, et al. Prevalence and predictors of the metabolic syndrome in women with polycystic ovary syndrome. J Clin Endocrinol Metab.
- Sonagra, AD. Normal Pregnancy- A State of Insulin Resistance. J Clin Diagnostic Res.
- 208. Catalano PM, Huston L, Amini SB, Kalhan SC. Longitudinal changes in glucose metabolism during pregnancy in obese women with normal glucose tolerance and gestational diabetes mellitus. Am J Obstet Gynecol.
- 209. Jayabalan N, Nair S, Nuzhat Z, Rice GE, Zuñiga FA, Sobrevia L, et al. Cross talk between adipose tissue and placenta in obese and gestational diabetes mellitus pregnancies via exosomes. Front Endocrinol.
- 210. Koren O, Goodrich JK, Cullender TC, Spor A, Laitinen K, Kling Bäckhed H, et al. Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell.
- 211. Li X, Liu X, Zuo Y, Gao J, Liu Y, Zheng W. The risk factors of gestational diabetes mellitus in patients with polycystic ovary syndrome: What should we care. Med, 2652.
- 212. Yan Q, Qiu D, Liu X, Xing Q, Liu R, Hu Y. The incidence of gestational diabetes mellitus among women with polycystic ovary syndrome: a meta-analysis of longitudinal studies. BMC Pregnancy Childbirth, Available from. [CrossRef]
- Noctor, E. Type 2 diabetes after gestational diabetes: The influence of changing diagnostic criteria. World J Diabetes.
- 214. Vounzoulaki E, Khunti K, Abner SC, Tan BK, Davies MJ, Gillies CL. Progression to type 2 diabetes in women with a known history of gestational diabetes: Systematic review and meta-analysis. BMJ.
- 215. Bonora E, Trombetta M, Dauriz M, Travia D, Cacciatori V, Brangani C, et al. Chronic complications in patients with newly diagnosed type 2 diabetes: Prevalence and related metabolic and clinical features: The Verona Newly Diagnosed Type 2 Diabetes Study (VNDS) 9. BMJ Open Diabetes Res Care.
- Bolton, A. International Diabetes Federation Diabetes Atlas [Internet]. 10th ed. Bolton A, editor. Vol. 102, Diabetes Research and Clinical Practice. 2021. Available from: www.diabetesatlas.
- 217. Yang J, Qian F, Chavarro JE, Ley SH, Tobias DK, Yeung E, et al. Modifiable risk factors and long term risk of type 2 diabetes among individuals with a history of gestational diabetes mellitus: prospective cohort study. BMJ.
- Patel, S. Disruption of aromatase homeostasis as the cause of a multiplicity of ailments: A comprehensive review. J Steroid Biochem Mol Biol, 2017;168:19–25. Available from, 2017. [Google Scholar] [CrossRef]
- 219. Evans SF, Hull ML, Hutchinson MR, Rolan PE. Androgens, Endometriosis and Pain. Front Reprod Heal.
- 220. Diamond MP, Grainger D, Diamond MC, Sherwin RS, Defronzo RA. Effects of methyltestosterone on insulin secretion and sensitivity in women. J Clin Endocrinol Metab.
- POLDERMAN, KH. GOOREN, LJ. ASSCHEMAN H. Induction of Insulin Resistance by Androgens and Estrogens. J Clin Endocrinol Metab, /: 1994;79(79):265–71. Available from: https, 1994. [Google Scholar]
- 222. Skinner MK, Schmidt M, Savenkova MI, Sadler-Riggleman I, Nilsson EE. Regulation of granulosa and theca cell transcriptomes during ovarian antral follicle development. Mol Reprod Dev.
- Clegg, DJ. Minireview: The year in review of estrogen regulation of metabolism. Mol Endocrinol. 1957. [Google Scholar]
- Stocco, C. Aromatase expression in the ovary: Hormonal and molecular regulation. Steroids.
- 225. Armanini D, Boscaro M, Bordin L, Sabbadin C. Controversies in the Pathogenesis, Diagnosis and Treatment of PCOS: Focus on Insulin Resistance, Inflammation, and Hyperandrogenism. Int J Mol Sci.
- 226. Di Nardo G, Zhang C, Marcelli AG, Gilardi G. Molecular and structural evolution of cytochrome p450 aromatase. Int J Mol Sci.
- 227. Simeon S, Spjuth O, Lapins M, Nabu S, Anuwongcharoen N, Prachayasittikul V, et al. Origin of aromatase inhibitory activity via proteochemometric modeling. Peer J.
- 228. Conley A, Mapes S, Jo Corbin C, Greger D, Graham S. Structural determinants of aromatase cytochrome p450 inhibition in substrate recognition site-1. Mol Endocrinol.
- 229. Jin Y, Choi YJ, Heo K, Park SJ. Melatonin as an oncostatic molecule based on its anti-aromatase role in breast cancer. Int J Mol Sci.
- 230. Cheng JC, Fang L, Li Y, Wang S, Li Y, Yan Y, et al. Melatonin stimulates aromatase expression and estradiol production in human granulosa-lutein cells: relevance for high serum estradiol levels in patients with ovarian hyperstimulation syndrome. Exp Mol Med.
- Kim JY, Han EH, Kim HG, Oh KN, Kim SK, Lee KY, et al. Bisphenol A-induced aromatase activation is mediated by cyclooxygenase-2 up-regulation in rat testicular Leydig cells. Toxicol Lett [Internet]. 2010;193(2):200–8. Available from. [CrossRef]
- 232. Maia H, Haddad C, Pinheiro N, Casoy J. The effect of oral contraceptives on aromatase and Cox-2 expression in the endometrium of patients with idiopathic menorrhagia or adenomyosis. Int J Womens Health.
- 233. Wang C, Mäkelä T, Hase T, Adlercreutz H, Kurzer MS. Lignans and flavonoids inhibit aromatase enzyme in human preadipocytes. J Steroid Biochem Mol Biol.
- 234. Singh S, Awasthi M, Pandey VP, Dwivedi UN. Plant derived anti-cancerous secondary metabolites as multipronged inhibitor of COX, Topo, and aromatase: molecular modeling and dynamics simulation analyses. J Biomol Struct Dyn, Available from, 3082. [CrossRef]
- 235. Richard S, Moslemi S, Sipahutar H, Benachour N, Seralini GE. Differential effects of glyphosate and roundup on human placental cells and aromatase. Environ Health Perspect.
- 236. Zarn JA, Brüschweiler BJ, Schlatter JR. Azole fungicides affect mammalian steroidogenesis by inhibiting sterol 14α-demethylase and aromatase. Environ Health Perspect.
- 237. Rojas J, Chávez-Castillo M, Torres W, Arraiz N, Cabrera M. Metformin in Cancer: Chemical Pathways for Tumoral Control Independent of Amp-Dependent Kinase. J Endocrinol Diabetes Obes.
- 238. Biegon A, Alia-Klein N, Fowler JS. Potential contribution of aromatase inhibition to the effects of nicotine and related compounds on the brain. Front Pharmacol.
- 239. Lee SH, Park SY, Choi CS. Insulin Resistance: From Mechanisms to Therapeutic Strategies. Diabetes Metab J.
- Baillargeon JP, Nestler JE. Commentary: Polycystic ovary syndrome: A syndrome of ovarian hypersensitivity to insulin? J Clin Endocrinol Metab. 2006;91(1):22–4.
- 241. Zhao H, Zhang J, Cheng X, Nie X, He B. Insulin resistance in polycystic ovary syndrome across various tissues: an updated review of pathogenesis, evaluation, and treatment. J Ovarian Res, Available from. [CrossRef]
- 242. Bremer AA, Miller WL. The serine phosphorylation hypothesis of polycystic ovary syndrome: a unifying mechanism for hyperandrogenemia and insulin resistance. Fertil Steril.
- Ibáñez L, Potau N, Zampolli M, Riqué S, Saenger P, Carrascosa A. Hyperinsulinemia and decreased insulin-like growth factor-binding protein-1 are common features in prepubertal and pubertal girls with a history of premature pubarche. J Clin Endocrinol Metab. 1997;82(7):2283–8.
- 244. Soldani R, Cagnacci A, Yen SSC. Insulin insulin-like growth factor I (IGF-I) and IGF-II enhance basal and gonadotrophin-releasing hormone-stimulated luteinizing hormone release from rat anterior pituitary cells in vitro. Eur J Endocrinol.
- 245. Nestler JE, Powers LP, Matt DW, Steingold KA, Plymate SR, Rittmaster RS, et al. A direct effect of hyperinsulinemia on serum sex hormone-binding globulin levels in obese women with the polycystic ovary syndrome. J Clin Endocrinol Metab.
- 246. Chan O, Inouye K, Akirav E, Park E, Riddell MC, Vranic M, et al. Insulin alone increases hypothalamo-pituitary-adrenal activity, and diabetes lowers peak stress responses. Endocrinology.
- 247. Kanbour SA, Dobs AS. Hyperandrogenism in Women with Polycystic Ovarian Syndrome: Pathophysiology and Controversies. Androgens.
- 248. Pateguana NB, Janes A. The contribution of hyperinsulinemia to the hyperandrogenism of polycystic ovary syndrome. J Insul Resist.
- 249. Vyrides AA, El Mahdi E, Giannakou K. Ovulation induction techniques in women with polycystic ovary syndrome. Front Med.
- 250. Panth N, Gavarkovs A, Tamez M, Mattei J. The Influence of Diet on Fertility and the Implications for Public Health Nutrition in the United States. Front Public Heal.
- Elnashar, AM. The role of metformin in ovulation induction: Current status. Middle East Fertil Soc J, 2011;16(3):175–81. Available from, 2011. [Google Scholar] [CrossRef]
- 252. Nehir Aytan A, Bastu E, Demiral I, Bulut H, Dogan M, Buyru F. Relationship between hyperandrogenism, obesity, inflammation and polycystic ovary syndrome. Gynecol Endocrinol.
- West-Eberhard, MJ. Nutrition, the visceral immune system, and the evolutionary origins of pathogenic obesity. Proc Natl Acad Sci U S A. 2019;116(3):723–31.
- 254. Ottaviani E, Malagoli D, Franceschi C. The evolution of the adipose tissue: A neglected enigma. Gen Comp Endocrinol, Available from. [CrossRef]
- Kuzawa, CW. Adipose Tissue in Human Infancy and Childhood: An Evolutionary Perspective. Yearb Phys Anthropol.
- 256. Parra-Peralbo E, Talamillo A, Barrio R. Origin and Development of the Adipose Tissue, a Key Organ in Physiology and Disease. Front Cell Dev Biol.
- Ibrahim, MM. Subcutaneous and visceral adipose tissue: Structural and functional differences. Obes Rev. 2010;11(1):11–8.
- 258. Spritzer PM, Lecke SB, Satler F, Morsch DM. Adipose tissue dysfunction, adipokines, and low-grade chronic inflammation in polycystic ovary syndrome. Reproduction.
- 259. Scheja L, Heeren J. The endocrine function of adipose tissues in health and cardiometabolic disease. Nat Rev Endocrinol, Available from. [CrossRef]
- 260. Choe SS, Huh JY, Hwang IJ, Kim JI, Kim JB. Adipose tissue remodeling: Its role in energy metabolism and metabolic disorders. Front Endocrinol.
- 261. Pawłowski B, Żelaźniewicz A. The evolution of perennially enlarged breasts in women: a critical review and a novel hypothesis. Biol Rev.
- 262. Tan CY, Vidal-Puig A. Adipose tissue expandability: The metabolic problems of obesity may arise from the inability to become more obese. Biochem Soc Trans.
- 263. Villa J, Pratley RE. Adipose tissue dysfunction in polycystic ovary syndrome. Curr Diab Rep.
- 264. Echiburú B, Pérez-Bravo F, Galgani JE, Sandoval D, Saldías C, Crisosto N, et al. Enlarged adipocytes in subcutaneous adipose tissue associated to hyperandrogenism and visceral adipose tissue volume in women with polycystic ovary syndrome. Steroids, Available from, 20 November. [CrossRef]
- 265. Arpaci D, Gurkan Tocoglu A, Yilmaz S, Ergenc H, Tamer A, Keser N, et al. The relationship between epicardial fat tissue thickness and visceral adipose tissue in lean patients with polycystic ovary syndrome. J Ovarian Res, Available from. [CrossRef]
- 266. Glueck CJ, Goldenberg N. Characteristics of obesity in polycystic ovary syndrome: Etiology, treatment, and genetics. Metabolism, Available from. [CrossRef]
- 267. Kałużna M, Czlapka-Matyasik M, Bykowska-Derda A, Moczko J, Ruchala M, Ziemnicka K. Indirect predictors of visceral adipose tissue in women with polycystic ovary syndrome: A comparison of methods. Nutrients.
- 268. Jones GW, Hill DG, Jones SA. Understanding immune cells in tertiary lymphoid organ development: It is all starting to come together. Front Immunol.
- 269. Dixon LJ, Barnes M, Tang H, Pritchard MT, Nagy LE. Kupffer cells in the liver. Compr Physiol.
- 270. Parker J, O’Brien C, Gersh FL. Developmental origins and transgenerational inheritance of polycystic ovary syndrome. Aust New Zeal J Obstet Gynaecol.
- 271. Davenport ER, Sanders JG, Song SJ, Amato KR, Clark AG, Knight R. The human microbiome in evolution. BMC Biol.
- 272. Rizk MG, Thackray VG. Intersection of Polycystic Ovary Syndrome and the Gut Microbiome. J Endocr Soc, /: 2021;5(2):bvaa177. Available from: https, 2021.
- 273. Valdes AM, Walter J, Segal E, Spector TD. Role of the gut microbiota in nutrition and health. BMJ.
- 274. Martinez JE, Kahana DD, Ghuman S, Wilson HP, Wilson J, Kim SCJ, et al. Unhealthy Lifestyle and Gut Dysbiosis: A Better Understanding of the Effects of Poor Diet and Nicotine on the Intestinal Microbiome. Front Endocrinol.
- 275. Degruttola AK, Low D, Mizoguchi A, Mizoguchi E. Current understanding of dysbiosis in disease in human and animal models. Inflamm Bowel Dis.
- 276. Shahi SK, Ghimire S, Lehman P, Mangalam AK. Obesity induced gut dysbiosis contributes to disease severity in an animal model of multiple sclerosis. Front Immunol.
- 277. Moya A, Ferrer M. Functional Redundancy-Induced Stability of Gut Microbiota Subjected to Disturbance. Trends Microbiol, Available from. [CrossRef]
- 278. Zhao X, Jiang Y, Xi H, Chen L, Feng X. Exploration of the Relationship between Gut Microbiota and Polycystic Ovary Syndrome (PCOS): A Review. Geburtshilfe Frauenheilkd.
- 279. Yilmaz B, Terekeci H, Sandal S, Kelestimur F. Endocrine disrupting chemicals: exposure, effects on human health, mechanism of action, models for testing and strategies for prevention. Rev Endocr Metab Disord.
- 280. Fabozzi G, Rebuzzini P, Cimadomo D, Allori M, Franzago M, Stuppia L, et al. Endocrine-Disrupting Chemicals, Gut Microbiota, and Human (In) Fertility — It Is Time to Consider the Triad. Cells.
- 281. Bellingham M, Sharpe R. Chemical Exposures During Pregnancy: Dealing with Potential, but Unproven, Risks to Child Health. R Coll Obstet Gynaecol, /: Available from: http.
- 282. Di Renzo GC, Conry JA, Blake J, Defrancesco MS, Denicola N, Martin JN, et al. International Federation of Gynecology and Obstetrics opinion on reproductive health impacts of exposure to toxic environmental chemicals. Int J Gynecol Obstet.
- 283. Gore AC, Chappell VA, Fenton SE, Flaws JA, Nadal A, Prins GS, et al. EDC-2: The Endocrine Society’s Second Scientific Statement on Endocrine-Disrupting Chemicals. Endocr Rev.
- 284. Liu Z, Lu Y, Zhong K, Wang C, Xu X. The associations between endocrine disrupting chemicals and markers of inflammation and immune responses: A systematic review and meta-analysis. Ecotoxicol Environ Saf.
- 285. Alonso-Magdalena P, Morimoto S, Ripoll C, Fuentes E, Nadal A. The estrogenic effect of bisphenol a disrupts pancreatic β-cell function in vivo and induces insulin resistance. Environ Health Perspect.
- 286. Wang Y, Zhu Q, Dang X, He Y, Li X, Sun Y. Local effect of bisphenol A on the estradiol synthesis of ovarian granulosa cells from PCOS. Gynecol Endocrinol.
- 287. Sun Y, Gao S, Ye C, Zhao W. Gut microbiota dysbiosis in polycystic ovary syndrome : Mechanisms of progression and clinical applications. Frontiers in Cellular and Infection Microbiology, 3389.
- 288. Ananthasubramanian P, Ananth S, Kumaraguru S, Barathi S, Santosh W, Vasantharekha R. Associated Effects of Endocrine Disrupting Chemicals (EDCs) on Neuroendocrine Axes and Neurotransmitter Profile in Polycystic Ovarian Syndrome Condition. Proc Zool Soc, Available from. [CrossRef]
- 289. Gupta R, Kumar P, Fahmi N, Garg B, Dutta S, Sachar S, et al. Endocrine disruption and obesity: A current review on environmental obesogens. Curr Res Green Sustain Chem, Available from, 1000. [CrossRef]
- 290. Aydemir D, Ulusu NN. The possible role of the endocrine disrupting chemicals on the premature and early menopause associated with the altered oxidative stress metabolism. Front Endocrinol.
- 291. Ravichandran G, Lakshmanan DK, Raju K, Elangovan A, Nambirajan G, Devanesan AA, et al. Food advanced glycation end products as potential endocrine disruptors: An emerging threat to contemporary and future generation. Environ Int, Available from, 20 December. [CrossRef]
- 292. Bansal A, Henao-Mejia J, Simmons RA. Immune system: An emerging player in mediating effects of endocrine disruptors on metabolic health. Endocrinology.
- 293. Tudurí E, Marroqui L, Dos Santos RS, Quesada I, Fuentes E, Alonso-Magdalena P. Timing of exposure and Bisphenol-A: Implications for diabetes development. Front Endocrinol.
- Resnik, DB. The precautionary principle and medical decision making. J Med Philos.
- 295. Schug TT, Johnson AF, Birnbaum LS, Colborn T, Guillette LJ, Crews DP, et al. Minireview: Endocrine disruptors: Past lessons and future directions. Mol Endocrinol.
- 296. More S, Bampidis V, Benford D, Bragard C, Halldorsson T, Hernández-Jerez A, et al. Guidance on risk assessment of nanomaterials to be applied in the food and feed chain: human and animal health. EFSA J.
- 297. Rutkowska AZ, Diamanti-Kandarakis E. Polycystic ovary syndrome and environmental toxins. Fertil Steril.
- 298. Jala A, Varghese B, Kaur G, Rajendiran K, Dutta R, Adela R, et al. Implications of endocrine-disrupting chemicals on polycystic ovarian syndrome: A comprehensive review. Environ Sci Pollut Res, Available from, 5848. [CrossRef]
- 299. Jozkowiak M, Piotrowska-Kempisty H, Kobylarek D, Gorska N, Mozdziak P, Kempisty B, et al. Endocrine Disrupting Chemicals in Polycystic Ovary Syndrome: The Relevant Role of the Theca and Granulosa Cells in the Pathogenesis of the Ovarian Dysfunction. Cells.
- Parker, J. A new hypothesis for the mechanism of glyphosate induced intestinal permeability in the pathogenesis of polycystic ovary syndrome. J Australas Coll Nutr Environ Med.
- 301. Kumar M, Sarma DK, Shubham S, Kumawat M, Verma V, Prakash A, et al. Environmental Endocrine-Disrupting Chemical Exposure: Role in Non-Communicable Diseases. Front Public Heal.
- 302. Mitro SD, Johnson T, Zota AR. Cumulative Chemical Exposures During Pregnancy and Early Development. Curr Environ Heal reports.
- 303. Starling AP, Adgate JL, Hamman RF, Kechris K, Calafat AM, Ye X, et al. Perfluoroalkyl substances during pregnancy and offspring weight and adiposity at birth: Examining mediation by maternal fasting glucose in the healthy start study. Environ Health Perspect.
- Rosenfeld, CS. Transcriptomics and Other Omics Approaches to Investigate Effects of Xenobiotics on the Placenta. Front Cell Dev Biol.
- 305. Hewlett M, Chow E, Aschengrau A, Mahalingaiah S. Prenatal Exposure to Endocrine Disruptors: A Developmental Etiology for Polycystic Ovary Syndrome. Reprod Sci.
- 306. Corbett GA, Lee S, Woodruff TJ, Hanson M, Hod M, Charlesworth AM, et al. Nutritional interventions to ameliorate the effect of endocrine disruptors on human reproductive health: A semi-structured review from FIGO. Int J Gynecol Obstet.
- 307. Piazza MJ, Urbanetz AA. Environmental toxins and the impact of other endocrine disrupting chemicals in women’s reproductive health. J Bras Reprod Assist.
- 308. Hanahan D, Weinberg RA. The Hallmarks of Cancer. Cell.
- 309. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell.
Figure 1.
Pathophysiology of Polycystic Ovary Syndrome. Depicts the multi-directional interactions between nutritional and environmental lifestyle-related risk factors and the identified pathophysiological processes and symptoms in PCOS. Health is a result of the successful integration of multidimensional subcellular, cellular, and systemic, integrated circuits and networks. Disturbances in these networks due to dysbiosis, chronic inflammation, insulin resistance and neuroendocrine deregulation, in isolation or in combination, can lead to loss of homeostatic resilience in the system. Combinations of adverse nutritional and environmental factors can disturb this network in a myriad of ways, and at multiple different sites, and are responsible for the pathogenesis of PCOS. The influence of the exposome on developmentally programmed susceptibility genes programs the embryo/fetus to express the phenotypic manifestations of PCOS during childhood, adolescence, and adulthood, following exposure to lifestyle, dietary, and environmental factors. QOL, quality of life.
Figure 1.
Pathophysiology of Polycystic Ovary Syndrome. Depicts the multi-directional interactions between nutritional and environmental lifestyle-related risk factors and the identified pathophysiological processes and symptoms in PCOS. Health is a result of the successful integration of multidimensional subcellular, cellular, and systemic, integrated circuits and networks. Disturbances in these networks due to dysbiosis, chronic inflammation, insulin resistance and neuroendocrine deregulation, in isolation or in combination, can lead to loss of homeostatic resilience in the system. Combinations of adverse nutritional and environmental factors can disturb this network in a myriad of ways, and at multiple different sites, and are responsible for the pathogenesis of PCOS. The influence of the exposome on developmentally programmed susceptibility genes programs the embryo/fetus to express the phenotypic manifestations of PCOS during childhood, adolescence, and adulthood, following exposure to lifestyle, dietary, and environmental factors. QOL, quality of life.
Figure 2.
Hypothalamic-Pituitary-Adrenal-Immune Axis (HPA). ANS, Autonomic Nervous System; Parasympathetic Nervous System (PNS); Sympathetic Nervous System (SNS); CRH, Corticotropin Releasing Hormone; Adrenocorticotropic Hormone (ACTH); IL-1ẞ, interleukin-1ẞ; TNF-α, tumour necrosis factor-α.
Figure 2.
Hypothalamic-Pituitary-Adrenal-Immune Axis (HPA). ANS, Autonomic Nervous System; Parasympathetic Nervous System (PNS); Sympathetic Nervous System (SNS); CRH, Corticotropin Releasing Hormone; Adrenocorticotropic Hormone (ACTH); IL-1ẞ, interleukin-1ẞ; TNF-α, tumour necrosis factor-α.
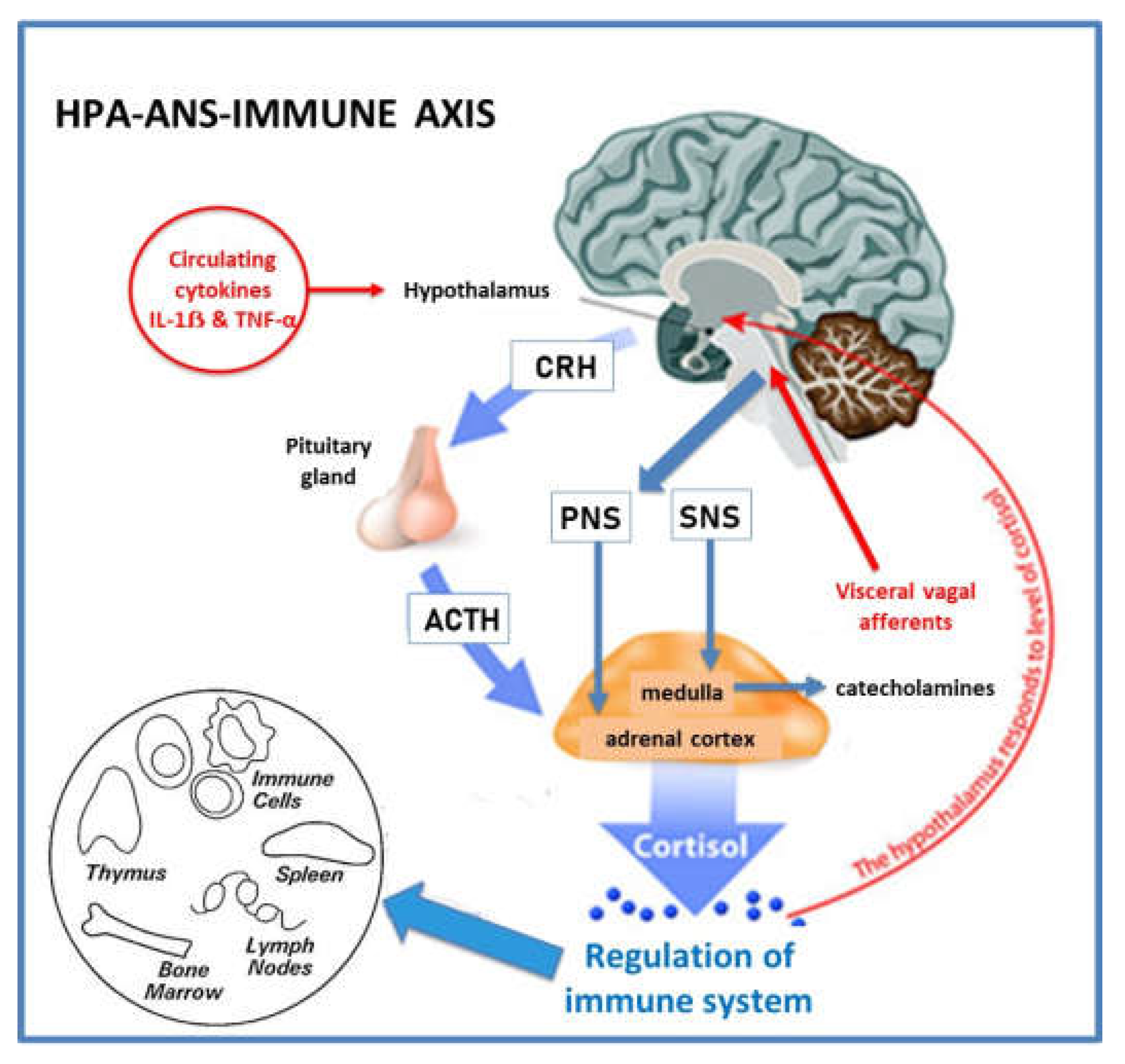
Figure 3.
Pathophysiology of insulin resistance. Any of the causes listed above can impact insulin signaling pathways and lead to tissue-selective impairment of insulin action. Once insulin resistance occurs in muscle, glycogen synthesis and glucose uptake from the circulation are reduced. Since muscle constitutes approximately 50% of body mass, insulin resistance in muscle makes a significant contribution to hyperglycaemia. Insulin resistance in adipose tissue leads to impaired lipogenesis and continued lipolysis, with release of glycerol and free fatty acids into the circulation. Hepatic insulin resistance impairs glycogen synthesis and prevents insulin from inhibiting gluconeogenesis. Adipose lipolysis supplies substrates for continued hepatic gluconeogenesis. Together, the effects of decreased muscle glucose uptake, adipose lipolysis, and hepatic gluconeogenesis, result in hyper-glycaemia. This causes compensatory release of insulin from the pancreas and hyperinsulinemia. DNL, de-novo lipogenesis; DAG, diacylglycerol; FFA, free fatty acid; IR, insulin resistance; VLDL, very low-density lipoprotein.
Figure 3.
Pathophysiology of insulin resistance. Any of the causes listed above can impact insulin signaling pathways and lead to tissue-selective impairment of insulin action. Once insulin resistance occurs in muscle, glycogen synthesis and glucose uptake from the circulation are reduced. Since muscle constitutes approximately 50% of body mass, insulin resistance in muscle makes a significant contribution to hyperglycaemia. Insulin resistance in adipose tissue leads to impaired lipogenesis and continued lipolysis, with release of glycerol and free fatty acids into the circulation. Hepatic insulin resistance impairs glycogen synthesis and prevents insulin from inhibiting gluconeogenesis. Adipose lipolysis supplies substrates for continued hepatic gluconeogenesis. Together, the effects of decreased muscle glucose uptake, adipose lipolysis, and hepatic gluconeogenesis, result in hyper-glycaemia. This causes compensatory release of insulin from the pancreas and hyperinsulinemia. DNL, de-novo lipogenesis; DAG, diacylglycerol; FFA, free fatty acid; IR, insulin resistance; VLDL, very low-density lipoprotein.
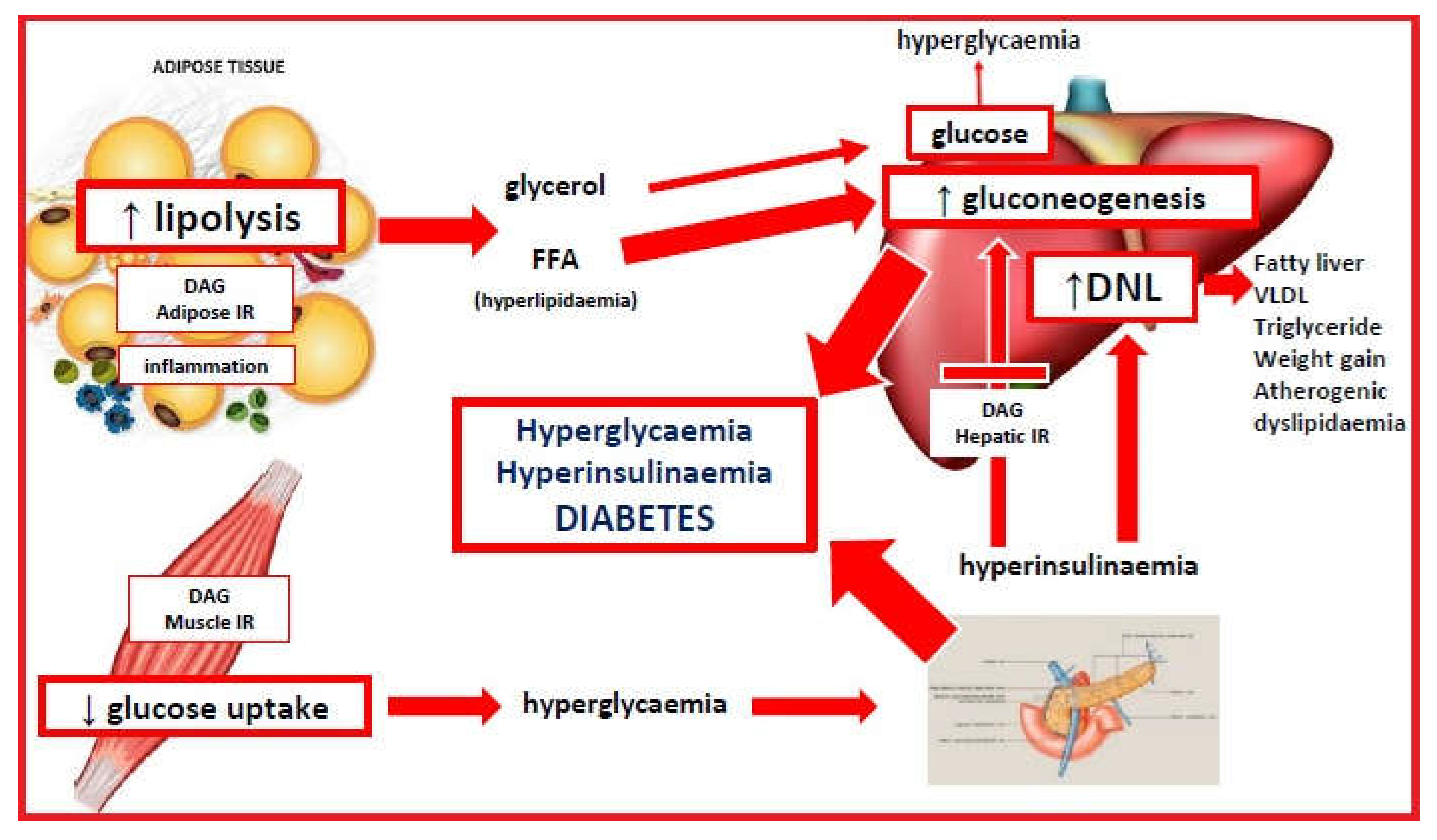
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



