
Submitted:
21 April 2023
Posted:
23 April 2023
You are already at the latest version
Abstract
Probabilistic and parsimony-based arguments regarding available genetics data are used to propose that Hardy and Higgin’s amyloid cascade hypothesis is valid but is commonly interpreted too narrowly to support, incorrectly, the primacy of the amyloid beta peptide (Aβ) in driving Alzheimer’s disease pathogenesis. Instead, increased activity of the βCTF (C99) fragment of APP is likely the critical pathogenic determinant altered by mutations in the APP gene. This model is consistent with the regulation of APP mRNA translation via its 5’ iron responsive element (IRE). Similar arguments support that the pathological effects of familial Alzheimer’s disease mutations in the genes PSEN1 and PSEN2 are not exerted directly via changes in APP cleavage to produce different ratios of Aβ length. Rather, these mutations likely affect the stability of presenilin holoprotein and/or γ-secretase multimers with consequences for γ-secretase activity and other important cellular functions. All fAD mutations in APP, PSEN1, and PSEN2 likely find unity of pathological mechanism in their actions on endolysosomal acidification and mitochondrial function, with detrimental effects on iron homeostasis and promotion of “pseudo-hypoxia” being of central importance. Aβ production is enhanced and distorted by oxidative stress and accumulates due to decreased lysosomal function. It may act as a disease-associated molecular pattern (DAMP) enhancing oxidative stress-driven neuroinflammation during the cognitive phase of the disease. We also discuss fascinating, but largely ignored, data on presenilin biology that may be important in understanding presenilins’ central role in familial Alzheimer’s disease.
Keywords:
Alzheimer's Disease
; Familial Alzheimer Disease (FAD)
; presenilins
; Amyloid beta Precursor Protein
; mutation
30 years of the amyloid cascade hypothesis
In 1992 Hardy and Higgins published their famous paper, “Alzheimer's disease: the amyloid cascade hypothesis” [9]. The paper captured the excitement of the Alzheimer’s disease (AD) research field over the emerging genetic evidence implicating the AMYLOID BETA A4 PRECURSOR PROTEIN (APP) gene in the inherited, early onset form of the disease (familial AD, fAD). In the following decades, this paper has been regarded as the keystone supporting the idea that AD pathogenesis is driven by accumulation of the Aβ peptide, which is derived from APP after this protein is cleaved by β- and γ-secretases (Figure 1). Any theory of AD pathogenesis seeking to challenge the central role of Aβ in AD pathogenesis must provide a more parsimonious explanation of the genetic data.
In this paper we will use arguments based in probability and parsimony to assert that the “amyloid cascade hypothesis” as originally presented by Hardy and Higgins is correct but has subsequently been interpreted too narrowly. Similarly, we will also examine the fAD-promoting mutations in the genes PRESENILIN 1 (PSEN1) and PRESENILIN 2 (PSEN2) to argue that their immediate pathogenic effects do not occur via accumulation of Aβ.
Mutations are more likely to destroy function than improve it
The amazing complexity of cellular structures and of living multicellular organisms today only exists because past changes in genetic information (mutations) have, fortuitously, resulted in improved reproductive competitiveness. However, such beneficial mutations are relatively uncommon (improbable). The great majority of mutations result either in no substantial change in cellular function or are detrimental and inhibit reproductive success. The “screwdriver analogy” illustrates this. If a photocopier (a machine for reproducing images on paper) has its screws tightened or loosened at random, most of these changes will likely have little effect, some changes will degrade photocopy quality, while a very few improbable adjustments may actually improve it.
The phenomenon that mutations with no effect are far more likely than mutations with negative effects, that themselves are far more likely than mutations with positive effects, can be referred to as the “mutation effect probability hierarchy” (MEPH). It is well illustrated by examination of mutations in the gene APP (Figure 2). The vast majority of genetic variations (mutations) in APP in the human population have no known relevance to AD – this is easily seen by inspecting the data collated in the gnomAD database. When Hardy and Higgins published their hypothesis paper in 1992, it was based on identification of three AD-causative mutations in one codon (V717) of the APP open reading frame and one mutation (E693Q) causing a related disease now known as cerebral amyloid angiopathy, APP-related, Dutch variant. Today, we know of at least 27 such deleterious mutations in APP (see Figure 2 and the mutations collated by Alzforum.org). However, only one, uncommon mutation (A673T) found mainly in the small population of Iceland is known to promote mental health by reducing AD risk [10,11,12,13,14,15].
The current “understanding” of fAD genetics
Of the ~20,000 protein-coding genes in humans, only three are known definitively to host mutations causing AD: APP, PSEN1, and PSEN2 [16,17]. A fourth gene, SORTILIN-RELATED RECEPTOR 1 (SORL1) may also cause fAD before the age of 65 years but its relationship to the other fAD genes is relatively poorly understood [17,18,19,20].
APP
Decisive for our understanding of how mutation of APP drives AD is the fact that simple increased expression of the APP coding sequence is sufficient to cause disease. Three doses of APP, whether through trisomy of chromosome 21 [21], or duplication of APP within chromosome 21 [22], causes early onset of AD. This means that increased expression of the APP protein or one of its fragments is highly likely to be driving the pathogenic changes of AD.
Within the APP gene, mutations causing AD are clustered within, and flanking, the sequence coding for Aβ (Figure 2). This fact is seen as support for the prime role of Aβ in driving AD pathology. According to the MEPH, we would expect most of these mutations to degrade/inhibit particular APP functions/abilities. Consistent with the MEPH, we find evidence that many of the mutations in the region coding for the γ-secretase cleavage site decrease the ability of the γ-secretase enzyme to cleave APP [23]. Other mutations occur in codons for the α- and β’-cleavage sites of APP respectively, and decrease the proclivity of these sites for cleavage by their relevant enzymes [24,25], thereby increasing the likelihood of cleavage at the β-site. Interestingly, one unique mutation, the “Swedish mutation” of APP (KM670/671NL) drives development of AD by improving the proclivity of the β-site for cleavage by the β-secretase enzyme BACE1 [26,27] (while the aforementioned A673T mutation near this site uniquely improves survival by reducing the ability of β-secretase to cleave at the β-site [10]).
The experimental data regarding whether fAD mutations in APP increase or decrease Aβ production is remarkably variable. The comprehensive analysis published by Xu et al. [23], that saw nearly all fAD mutations of APP decrease γ-secretase cleavage, monitored the γ-secretase cleavage needed to produce Aβ using cells transfected to express a modified βCTF. (This is also known as “C99” and is the transmembrane fragment of APP produced when APP is cleaved by β-secretase but before cleavage by γ-secretase.) The APP intracellular domain (AICD) of βCTF was fused to a transcription factor to allow detection of cleavage at the ε-site (Figure 3) through activation of luciferase expression. By reporting on γ-secretase at the ε-sites rather than after the subsequent, variable “product line” γ-secretase cleavages (see Figure 3) the rates of γ-secretase cleavage should be consistently comparable between different fAD mutant forms of βCTF. When, instead, levels of the Aβ product are used to compare γ-secretase cleavage of βCTF mutants in transfected cells, the differing levels of βCTF could have differential effects on lysosomal acidification [28], thereby affecting Aβ degradation and accumulation. This might partially explain, for example, why Xu et al. observed decreased γ-secretase cleavage of APP due to the fAD mutation D678H whereas Chen et al. [29] saw a relative increase in Aβ levels. However, Chen et al. also observed that the D678H mutation shifted the initial cleavage of APP from the α-site to the β-site which would increase βCTF levels and also promote Aβ production. Numerous mutations in the Aβ region of APP also affect its tendency to aggregate and form fibrils. However, in many cases these mutations are associated with accumulation of Aβ in brain vasculature in the disease cerebral amyloid angiopathy (CAA) that is distinct from AD e.g. APP mutations E693K [30,31] and L705V [32].
More than 100 transmembrane proteins are subject to cleavage by γ-secretase [33,34]. However, there is no clear consensus sequence determining proclivity for cleavage by γ-secretase [35] and the mechanism of operation of this enzyme causes variability in the cleavage products [36,37,38] (Figure 3). For this reason, Aβ is not one peptide but a family of related sequences of 37-49 amino acid residues (aa) in length depending on the positions of the final γ-secretase cleavages that create their carboxy termini, Figure 3. The inhibition of γ-secretase cleavage by mutations in/near the region coding for the γ-cleavage site of APP [23,39] is not obviously consistent with a primary role for Aβ in AD as some of these mutations drastically inhibit the production of Aβ [23,40]. However, rather than excluding an AD-causative role for Aβ, an Aβ-centric hypothesis has evolved positing that the ratio of different Aβ fragment lengths, and the greater tendency to aggregate of longer Aβ fragments, are critical to development of AD e.g. [41,42,43,44]. This idea is inspired by the observation of differences in the ratios of the aggregation-prone 42 aa fragment (Aβ42) relative to the most common, 40 aa Aβ fragment (Aβ40) dependent on brain disease state [42,45]. Changes in Aβ42 /Aβ40 ratios have also been observed in in vitro analyses of fAD mutations of PSEN1 [42,43,44] and PSEN2 [46,47,48] (discussed later herein). However, other general explanations for increased Aβ42 /Aβ40 ratios in AD brains are that these are caused by the influence of oxidative stress on γ-secretase [49] (since increased levels of oxidative stress are a consistent phenomenon in AD brains [50,51]) and/or that changes in the composition of the membranes in which APP is embedded also affect Aβ42 /Aβ40 ratios [52,53] since APP’s β-C-terminal fragment, βCTF, regulates the distribution of cholesterol between the plasma and endoplasmic reticulum (ER) membranes [54] and γ-secretase cleaves βCTF preferentially in cholesterol-rich lipid rafts [55,56,57].
A difficulty with much of the work analysing fAD mutations of APP, including their cleavage by γ-secretase, is that much of the research is conducted using transfected cells that significantly over-express APP. Also, the fAD genes are very responsive to stress (e.g. [58,59,60]) and cells in tissue culture (such as the transfected cells used in much fAD gene analysis) are frequently stressed by their unusual environmental conditions [61]. In contrast, fAD mutation-carrying humans are almost always heterozygous, raising the possibility of interactions between mutant and non-mutant forms of APP or its fragments. Indeed, dimerization of APP is known to affect its γ-secretase cleavage to form Aβ [62,63,64]. Studies using editing of endogenous APP genes in iPSCs to compare heterozygous mutant and non-mutant cells (e.g. [65] below) should produce information that is interpretable with greater certainty (fewer possible variables involved).
In accordance with the “Law of Parsimony” [66] it is highly likely that all the various mutations in APP cause AD through a single mechanism in common. Based on the information above, this is likely to involve an increase in production of the fragment of APP produced by cleavage at the β-site, i.e., βCTF. Increased expression of the APP gene [21,22,28], or increased proclivity for cleavage of APP by β-secretase [26,27], or decreased cleavage at APP’s α- or β’-cleavage sites that shifts cleavage towards the β-cleavage site [24,25], or decreased cleavage by γ-secretase [23], should all increase the levels of βCTF. Consistent with this, the most pathogenic mutations in APP causing the earliest onset of AD tend to be those affecting the site of γ-secretase cleavage (Figure 4) since inhibition of this cleavage would likely be very effective in increasing βCTF levels (as this inhibits the destructive cleavage of βCTF that forms the Aβ and AICD fragments.
The explanatory power of increased βCTF levels as the AD-causative effect of mutations in APP
Three decades of research focussed on Aβ has not yet established convincingly the toxic nature of this peptide under normal, physiological conditions, e.g. [67,68,69,70,71]. The apparent recent successes of anti-Aβ antibody therapies [72] supports a pathological role for this peptide in the clinical phase of the disease, possibly as a disease-associated molecular pattern (DAMP) contributing to the inflammation that is characteristic of AD brains [73,74]. However, the anti-Aβ antibody therapies are apparently unable to prevent onset of the dominantly inherited disease [75] and only slow the progression of the sporadic, late onset disease rather than stopping it [72]. This supports that something else drives the pre-clinical phase of AD. In interpreting the results of much Aβ-based AD research, it is remarkable how seldom the presence and levels of βCTF are assessed. And yet βCTF levels are likely very significantly altered in transgenic mouse models of AD that include APP transgenes (including the models 3xTg, 5xFAD, A7, APP23, APPswe/PSEN1dE9, PDAPP, J20, PDAPP, PS2APP, Tg2576, Tg-ArcSwe, TgCRND8, [76]), while βCTF has been seen to accumulate in AD brains [77,78,79]. Much of the evidence supporting a role for βCTF in both fAD and sporadic AD was reviewed in a paper by Checler and colleagues [80].
A number of papers below give very strong support for increased βCTF levels as the pathological effect of fAD mutations in APP. Note that the first two papers, and our later discussion of the effects of fAD mutations in PSEN1 and PSEN2, link these genes to normal functioning of the cellular endolysosomal system. Many of the late onset AD risk loci discovered by genome-wide association studies (GWAS) are also important for endolysosomal system function [81,82]. Indeed, the strongest genetic risk factor for sporadic (non-familial) AD, the ε4 allele of APOE (APOE4), reduces lysosomal acidification relative to the ε3 allele [83] (in common with fAD mutations in APP [28] and the presenilins [84]).
Kwart et al. [65] examined the subcellular effects of a variety of fAD mutations in APP and PSEN1 after these were edited into a single iPSC cell line (for consistency of genetic background) that was then differentiated into neurons. The mutations consistently caused accumulation of, specifically, APP’s βCTF but had inconsistent effects on Aβ levels. Rab5+ early endosome size was correlated with βCTF levels and could also be increased by chemical inhibition of γ-secretase activity (which reduced Aβ to negligible levels).
Jiang et al. [28] examined fibroblasts from trisomy 21 individuals (Down syndrome) who suffer early onset Alzheimer’s disease. They showed that the increased dosage of the APP gene (present on chromosome 21) led to increased levels of βCTF. This decreased the acidification of lysosomes (and, thereby, the activity of a marker of lysosomal function, the enzyme cathepsin D, CTSD). These phenomena could be modulated by various treatments altering the levels of βCTF in normal and Down syndrome fibroblasts. For example, chemical inhibition of β-secretase cleavage of APP (to decrease βCTF levels) decreased lysosomal pH in trisomy 21 syndrome fibroblasts but, interestingly, did not lower lysosomal pH in euploid (normal diploid) fibroblasts.
Montesinos et al. [54] found that APP’s βCTF regulates the trafficking of cholesterol from the plasma membrane (PM) to the ER. Increased levels of βCTF inhibits a cell’s own cholesterol synthesis and increase the rate of cholesterol trafficking from the PM. The increased ER cholesterol increases formation of the mitochondrial associated membranes (MAM) of the ER through which cells regulate mitochondrial energy production via Ca2+ ion release [85]. Increased MAM formation is observed in fibroblasts from both fAD and late onset AD patients compared to those from cognitively healthy controls [86].
The original amyloid cascade hypothesis included βCTF as a candidate pathogenic agent
The possibility that βCTF is a pathological agent driving AD is actually suggested by Hardy and Higgins in their 1992 paper. (In the following quotation, the authors refer to Aβ as “AβP” and I have bolded the text referring to βCTF.)
“Our cascade hypothesis states that AβP itself, or APP cleavage products containing
AβP, are neurotoxic and lead to neurofibrillary tangle formation and cell death. Thus, two successive events are needed to produce Alzheimer's pathology. First, AβP must be generated as an intact entity, either by accumulation of AβP or as an AβP-containing fragment of APP. Second, this molecule must facilitate or cause neuronal death and neurofibrillary tangle formation. Neve and her colleagues have reported that the AβP-containing COOH-terminal fragment is toxic to cultured neurons(18)…
“… The evidence we have described supports the hypothesis that the AβP molecule initiates the pathological cascade of Alzheimer's disease. AβP-containing COOH-terminal derivatives of APP seem the most likely molecular candidates for initiation of the cascade, with the process presumably taking several decades to produce the full-blown pathology of the disease.”
The likelihood that βCTF represents the toxic agent in APP mutation-driven AD means that experiments looking at Aβ accumulation in human brains or animal models should also examine βCTF levels to avoid the possibility of inappropriately attributing pathological agency to Aβ.
PSEN1 and PSEN2
The majority of mutations causing fAD occur in the gene PSEN1 both in terms of their relative frequency among fAD mutations in the human population (~63%, [17]) and relative to the number of fAD mutations known in APP and PSEN2 (~80% of the known mutations, [88], Figure 5). Collectively, fAD mutations in PSEN1 also have the earliest median onset age [89]. Relatively far fewer mutations occur in the gene PSEN2 and these also have a later median age of onset than even mutations in APP [89] (Figure 6). Importantly, both PSEN1 and PSEN2 encode proteins with similar structures and functions, implying that disruption of a function they share in common causes AD. Intriguingly, while the large number and wide distribution of fAD-causative mutations in the coding sequences of PSEN1 and PSEN2 make it extremely likely that these mutations cause loss of a function(s), two characteristics of these mutations are consistent with action via a gain-of-function mechanism:
- 1)
- The great majority of fAD mutations in PSEN1 and PSEN2 are missense mutations altering the protein coding sequence. Even those mutations causing changes in transcript splicing (e.g. PSEN1 L113_I114insT [90]) always produce at least one transcript form that preserves the open reading frame to code for a “full-length” protein. We addressed this issue in a previous review [16], describing it as the reading-frame preservation rule. Consistent with this (with the exception of one unique and important gain-of-function mutation discussed later [91]), no mutations or genetic variants affecting PSEN1 or PSEN2 transcript regulation (without changing protein-coding sequences) have been discovered that increase the risk of AD [92].
- 2)
- Loss-of-function mutations that would be expected simply to reduce γ-secretase activity, without otherwise distorting it, do not cause fAD. This is dramatically illustrated by the existence of frameshift mutations in PSEN1 that truncate the open reading frame and do not cause fAD while causing a completely unrelated disease of the skin, Acne Inversa, familial 3, (ACNINV3, also known as hidradenitis suppurativa) [93,94,95]. Unlike fAD, familial Acne Inversa can also be caused by mutations in genes encoding two other components of the γ-secretase enzyme complex, NICASTRIN (NCSTN), and PRESENILIN ENHANCER, GAMMA-SECRETASE SUBUNIT (PSENEN, formerly known as PEN2) [93] (See also Figure 7). All these mutations affecting different components of the γ-secretase enzyme complex almost certainly act through reduction of cellular γ-secretase activity but mutations causing fAD only occur in PSEN1 and PSEN2 and not in genes encoding other γ-secretase complex components. Therefore, fAD cannot be due to a simple loss of γ-secretase activity. The reading frame preservation rule implies that a gain-of-function mechanism is involved.
The role of both PSEN1 and PSEN2 in γ-secretase activity, and the fixation of the AD research field on Aβ that requires γ-secretase activity for its production from βCTF, has meant that nearly all research on PSEN1 and PSEN2 has focussed on γ-secretase. Since fAD mutations in APP that affect the protein’s γ-secretase cleavage site reduce Aβ production, while fAD mutations in PSEN1 and PSEN2 do not consistently reduce γ-secretase activity (see below), research has focussed on one possible commonality of all these mutations – their effects on Aβ peptide length ratios no matter what the total level of Aβ production may be. Some early studies appeared to find correlations between the ages of AD onset caused by different fAD mutations in PSEN1 and the Aβ42 /Aβ40 ratio caused by these same mutations in transfected cell culture-based assays [96,97]. However, the comprehensive analysis of 138 PSEN1 fAD mutations by Sun et al. in 2016 [98] using cell-based and in vitro assay systems found these apparent correlations to be statistical artefacts (see Figure 5 in [98]). That work also found that, while the majority of the 138 mutations they analysed decreased γ-secretase activity (as measured by combined Aβ40 and Aβ42 production – a loss of function), ~10% of the fAD mutations increased γ-secretase cleavage of APP (a gain of function, at least in their cell-free assay system that over-expressed mutant PSEN1 proteins in the absence of wild type PSEN1). The increased γ-secretase activity of at least one of these mutations, S365A of PSEN1, has been confirmed independently by others [99]. Sun et al. saw that some fAD mutations of PSEN1 lost all apparent γ-secretase activity while others showed decreased Aβ42 /Aβ40 ratios rather than increased ratios. In summary, Sun et al. saw no consistent changes in γ-secretase activity (when mutant proteins were assayed in homogeneous states) that would support a role for γ-secretase, or for Aβ, as pathogenic agents promoted by fAD mutations in PSEN1.
The idea that Aβ fragment length is the critical determinant of the AD pathological process was reinvigorated by a study published in 2022 (Petit et al. [2]) analysing a larger number of Aβ fragments to define a Aβ(37 + 38 + 40)/Aβ(42 + 43) ratio. This ratio reflects the “processivity” of the γ-secretase enzyme (the rate at which it successively cleaves along the transmembrane domain of APP after an initial “ε-cleavage”) by comparing the common (Aβ40) plus the “maximally complete” Aβ cleavage products (Aβ37 and Aβ38) of the two alternative γ-secretase cleavage pathways of APP’s βCTF with the less common “incomplete” cleavage products of these pathways (Aβ42 and Aβ43, Figure 3). Presumably, a slower rate of γ-secretase cleavage progression along the transmembrane domain of βCTF allows more time for longer Aβ products to escape from the lipid bilayer and the enzyme complex before further cleavage (Figure 1 and Figure 3). For the mutants analysed by this study there appears to be a strong correlation between the age of disease onset and the Aβ(37 + 38 + 40)/Aβ(42 + 43) ratio when represented as a percentage of the wild type ratio (see Figure 2 of the Petit et al. paper). In a 2021 paper from some of the same authors, Szaruga et al. [100], changes in Aβ product ratios are attributed to the tendency of the mutations to destabilise γ-secretase-substrate interactions (as measured by the change in Aβ fragment production with temperature) and so can be viewed as loss-of-function. Interestingly, neither of these papers analysed any of the 12 PSEN1 gain-of-function mutations found by Sun et al. actually to increase total Aβ production (as measured by combined Aβ40 and Aβ42 production). As might be expected for fAD mutations of APP that decrease cleavage at APP’s γ-secretase site, Szaruga et al. found that these mutations also destabilise APP’s interaction with the γ-secretase enzyme complex.
Supplementary Data File 1, Spreadsheet 1, shows a simplistic re-examination (without statistical corrections) of the data presented by Petit et al. to compare the choice of Aβ “product line” (i.e. Aβ(37 + 40 + 43) relative to Aβ(38 + 42)) with the age at onset (AAO) of disease. For linear correlation it finds, r2 = 0.38 for this compared to the superior value of r2 = 0.75 for the Aβ(37 + 38 + 40)/Aβ(42 + 43) ratio that reflects processivity. This supports that the rate of γ-secretase processivity is more closely associated with disease severity than the choice of product line.
A 2023 study (Liu et al. [101]) examined the production of Aβ fragments with lengths of 37, 38, 39, 40, 42, and 43 aas produced by HEK293 cells lacking endogenous presenilins but transfected to express wild type or one of 130 different fAD mutant forms of PSEN1 (together with APP’s βCTF). It found a best correlation with disease AAO of r = 0.5807 (p < 0.0001) for the ratio of Aβ37/Aβ42. The correlation for the Aβ40/Aβ42 ratio was also highly significant. Each mutant analysis was, apparently, performed only once and, interestingly, almost a third of these showed higher levels of total Aβ production (or of Aβ40 plus Aβ42 production as Sun et al. [98] examined) than the wild type controls (see Supplementary Data File 1, Spreadsheet 2, for an analysis of the Aβ production data in Table 1 of Liu et al. [101]).
Later in this paper we argue that the correlation between AD AAO and γ-secretase processivity is consistent with an idea that disease severity is dependent on the ability of conformational changes in presenilins to disrupt formation of presenilin holoprotein and/or γ-secretase complex multimers.
Presenilin Holoproteins
The presenilin proteins at the catalytic core of the γ-secretase enzyme complex may be auto-activating zymogens. Presenilin proteins are first translated as “holoproteins” that possibly cleave themselves endoproteolytically [102] to form catalytically active heterodimers consisting of the N- and C-terminal fragments (NTF & CTF) within the γ-secretase enzyme complex (Figure 7). Some fAD mutations strongly affect this activating cleavage [103,104,105,106,107]. Petit et al. [2] and Szaruga et al [100] noted that such mutations (e.g. PSEN1 mutations P433S, R278I and L435F) have milder clinical effects (i.e. later ages of onset) than expected from correlations between other mutants. As described below, the later-than-predicted ages of onset of these mutations strongly inhibiting γ-secretase activity are consistent with an alternative presenilin fAD mutation pathogenic mechanism we shall outline.
Compared to research on γ-secretase, very little research has examined the biological role of the presenilin holoproteins or their possible role(s) in AD pathogenesis. This is likely because the holoprotein states of presenilins are commonly regarded as short-lived and transitory [108]. However, in the early days of characterisation of presenilin biology, careful analysis by Dewji et al. [109] demonstrated that, when cells are subjected to gentle lysis, most presenilin can subsequently be detected in the holoprotein state. (This suggests the tantalising possibility that presenilin proteins may be involved in responses to mechanical stress in addition to responding to hypoxia/oxidative stress [59]).
The most important paper demonstrating the functional uniqueness of the PSEN1 holoprotein was published in 2010 by Ralph Nixon and colleagues (Lee et al. [84]). This described an important role for the holoprotein in correct assembly of the vacuolar ATPase (v-ATPase) that pumps H+ ions into the endolysosomal system – and particularly the lysosome – for acidification. Here we see a convergence of the activities of the presenilins and the βCTF of APP – all are modulating endolysosomal pH (Figure 8). In fact, Nixon and colleagues have commented (without yet presenting experimental data) that βCTF may bind to v-ATPase to modulate v-ATPase activity [28]. These researchers have,
“…identified a direct physical interaction of lysosomal-associated APP-βCTF with the cytosol-exposed domain of the transmembrane V0a1 subunit of the vATPase. Competition by APP-βCTF with specific V1 subunits for binding to the V0a1 subunit impedes association of the V1 and V0 sectors of the complex….Impaired vATPase assembly … is rescued by specifically lowering APP-βCTF levels”
Regardless of whether or not βCTF interacts physically with v-ATPase components, the fAD mutant forms of APP, PSEN1, and PSEN2 [110], all affect the acidification of the endolysomal pathway (as, interestingly, does the late onset AD risk allele APOE4 [83]). Failure to acidify lysosomes sufficiently will inhibit their ability to degrade Aβ [111], causing Aβ to accumulate in lysosomes and leading, ultimately, to the formation of neuritic Aβ plaques [112]. In addition, Kim et al. [113] recently showed evidence that both Aβ and MAPT (Tau) can bind to v-ATPase components to inhibit their function. Furthermore, Mustaly-Kalimi et al. [114] recently observed decreased v-ATPase expression when cytosolic concentrations of Ca2+ ions are raised, while an earlier 2015 paper from Lee et al. [115] found that loss of lysosomal acidification leads to increased levels of cytosolic Ca2+. These observations imply the existence of pathological positive feedback interactions between altered cytosolic Ca2+ levels and lysosomal acidification. Overall, the function of v-ATPase has emerged as a hub for the pathological influences of many agents identified as important in the development of AD.
The 2010 Lee et al paper [84] showed evidence that, in the endoplasmic reticulum, PSEN1 holoprotein interacts with the oligosaccharyltransferase complex to promote N-glycosylation of the V0a1 subunit of v-ATPase. This is necessary for correct targeting of the V0a1 subunit to the lysosome for assembly of functional v-ATPase. This PSEN1 holoprotein-dependent function in glycosylation is deficient in mouse cells lacking Psen1, but also in human fibroblasts heterozygous for fAD mutations in PSEN1. In conclusion, the numerous (and so, presumably, loss-of-function) fAD mutations of PSEN1 appear to act through a gain-of-function mechanism (see point 2 above) to cause a dominant negative (loss-of-function) effect. How is this possible? We suggest that disruption of a multimeric holoprotein complex provides a parsimonious solution to this quandary.
Presenilin holoproteins and/or γ-secretase complexes form multimers
Biochemical evidence supports that the presenilin proteins form multimers [105,116,117]. In particular, Heilig et al. [105] and Zhou et al. [99] presented strong evidence from immunoprecipitation that fAD mutant PSEN1 holoproteins can bind (directly or indirectly) to wild type PSEN1 holoproteins. Heilig et al. saw association of differentially-tagged holoproteins in cells co-transfected with expression vectors for these proteins. In contrast, Zhou et al. saw association between pre-purified γ-secretase complexes containing different mutant forms of presenilin. This raises the question of whether the multimeric association involves only the holoproteins or occurs between holoproteins in association with some or all of the other components of γ-secretase complexes. Both Heilig et al. and Zhou et al. reported dominant negative effects of the mutant proteins on the γ-secretase activity of the wild type (non-mutant) protein. However, under both experimental regimes, the relative reduction in γ-secretase activity was small (~20%) for a 1:1 ratio of mutant to wild type protein resembling a heterozygous situation. This led Zhou et al to conclude,
“To gain a better understanding of the moderate dominant negative effect, the expression level of the WT and mutant PSEN1 alleles, as well as the oligomerization state of γ-secretases, should be carefully examined under in vivo circumstances, especially in patients’ brains.”
Importantly, Zhou et al. also noted,
“We wish to stress that our experimental data provide no supporting evidence for a potential role of γ-secretase in the development of AD. In fact, a number of the γ-secretase variants with pathogenic PS1 mutations, exemplified by S365A, have WT-level proteolytic activity in terms of Aβ42 and Aβ40 production (18). The development of AD in the patients with these PS1 mutations cannot be explained by the WT-level proteolytic activity of these γ-secretase variants in vitro. Nonetheless, the dominant negative effect of these PS1 mutations in patients with AD still applies, suggesting such an effect may not be recapitulated by the catalytic function of PS1 in γ-secretase. We speculate that, for the vast majority of patients with AD, the dominant negative effect of PS1 is perhaps effected through other mechanisms that are independent of γ-secretase.”
It is also worthwhile nothing that a super-resolution microscopy study of presenilin distribution on the surface of cells saw mainly monomers with some possible dimer formation [118]. However, this may not be true of all cellular membranes as the amounts of presenilins/γ-secretase complexes vary within cells, with particularly high levels being located in the mitochondria associated membranes (MAM) of the endoplasmic reticulum [87].
The importance of PSEN1 and APP’s βCTF for the acidification of the endolysomal pathway, and the involvement of many late onset AD risk loci in endolysosomal function [81], supports that defects in endolysosomal pathway function may be a key, unifying, driver of AD pathology (Figure 8). If all fAD mutations of the presenilins caused loss of γ-secretase activity, then this would also promote accumulation of APP’s βCTF to reduce lysosomal acidification and would be consistent with this unifying endolysomal hypothesis for AD pathogenesis. However, as described above, some fAD mutations appear to increase γ-secretase cleavage of βCTF. There is evidence that correct endolysosomal pH may be important for normal γ-secretase activity [119,120,121,122] (although, as discussed by Maesako et al. [122], this conflicts with the supposed pH optimum of 6.3–7.0 for γ-secretase activity established in biochemistry-based assays). Therefore, it is conceivable that the problems with endolysosomal acidification, caused by the effect of any fAD mutation on presenilin holoprotein function, may be sufficient to decrease overall γ-secretase activity in the endolysomal system, even for those fAD mutations that show increased γ-secretase activity in homogeneous assay systems (i.e. no presence of wild type presenilin). However, a recently discovered mutation in PSEN2 is inconsistent with this idea. Pang et al [91] found that a mutation in the 3’ untranslated region (UTR) of PSEN2 transcripts increases the expression of PSEN2, apparently by destroying the binding site of the miR-183-5p microRNA. This mutation was strongly linked to fAD in a Chinese family lacking identifiable fAD mutations in other genes. Since no missense mutations were identifiable in PSEN2, PSEN1 or APP, an implication is that increased γ-secretase activity alone can drive AD pathogenesis.
A parsimonious model to account for superficially inconsistent/conflicting presenilin mutation data
Of the ~20,000 protein-coding genes in the human genome, the overwhelming majority of fAD mutations occur in only three – APP, PSEN1, and PSEN2. This restricted number of genes, and the known enzyme/substrate relationship between their protein products, supports that a single mechanism underlies fAD pathogenesis. (Alternatively, protein substrates of γ-secretase are not uncommon – they are encoded by more than 1/200th of the set of all protein-coding genes [33,34] - so it is not extremely improbable that the fAD-promoting shared activity of mutations in APP, PSEN1, and PSEN2 may be something other than effects on γ-secretase, such as reduced lysosomal acidification, i.e. while loss of γ-secretase cleavage susceptibility may be important for the action for some fAD mutations in APP, changes in γ-secretase activity in heterozygous fAD presenilin mutants need not be related to the pathogenic activity of those presenilin mutations.)
In a 2018 review examining the role of iron in AD [1] we discussed that the disease may have two distinct phases: a preclinical phase of increasing brain hypoxia with increasing reliance on glycolysis, followed by a clinical phase where cognition falters due to “metabolic inversion” of the brain into a hypometabolic state (after breaching a brain metabolic homeostasis capacity threshold - see the “Stress Threshold Change of State” model in Figure 10B). Interestingly, γ-secretase is intimately involved in hypoxia responses. It cleaves p75NTR in a feed-forward mechanism driving stabilisation of the HIF1α subunit of the transcription factor HIF1, a master regulator of cellular responses to hypoxia [123]. Importantly, γ-secretase activity is also activated by HIF1α but not via transcriptional regulation. Instead, HIF1α binds to γ-secretase [124] (possibly directly to presenilin [125]) to increase the pool of active γ-secretase [124]. In experiments using zebrafish heterozygous for a fAD-like mutation of their PSEN1-orthologous gene (psen1Q96_K97del), under normoxia we saw increased basal expression of genes known to be responsive to hypoxia in the brains of young adult fish that were, nevertheless, still able to upregulate these genes further under acute hypoxia. However, when fish became aged, the response to hypoxia of these hypoxia-responsive genes became inverted – they were downregulated by acute hypoxia. This inverted response occurred at an earlier age in fAD-like mutant fish than in non-mutants, supporting that the fAD-like mutation drives accelerated aging in this aspect of brain metabolic control [126]. The observations above would be consistent with an increase in γ-secretase activity in the brains of young fish and a decrease in γ-secretase activity after an age-dependent “state change” in energy metabolism regulation.
Is there any additional evidence that fAD mutations might drive pathogenesis by first increasing γ-secretase activity before it decreases after a metabolic change of state? When we analysed the transcriptomes of young adult zebrafish brains heterozygous for another fAD-like mutation of the zebrafish PSEN1-orthologous gene (psen1T428del, equivalent to the T440del mutation of human PSEN1), we did see overall increased expression of genes known to be upregulated by Notch signalling that is dependent on γ-secretase activity [127].
The wide distribution and missense nature of fAD mutations in the PSEN1 and PSEN2 protein-coding sequences is not consistent with their affecting any particular sub-functional domain in these proteins. However, all such mutations might be expected to affect, to some degree, the conformation of the holoprotein, and that may destabilise holoprotein multimeric complexes. If multimer dissociation is required for conversion of the holoprotein into active γ-secretase then, in heterozygous cells, a fAD-mutant form of holoprotein with decreased intrinsic γ-secretase activity might, nevertheless, promote an overall increase in γ-secretase activity by causing increased activation of non-mutant holoproteins, in addition to displaying its own γ-secretase activity (see Figure 9). Cells heterozygous for fAD mutations encoding proteins with very little intrinsic γ-secretase activity (such as the PSEN1 P433S, R278I and L435F mutations mentioned above) should show comparatively less total cellular γ-secretase activity than other fAD mutations, and would drive AD pathogenesis more slowly, giving their relatively later ages of disease onset as observed. We suggest, therefore, that a consistent, unifying pathogenic activity of fAD mutations in PSEN1 and PSEN2 may be that these all increase γ-secretase activity in heterologous systems co-expressing both fAD mutant and wild type forms of these proteins.
This hypothesis should be consistent with the mutation fAD onset age/Aβ fragment ratio correlation of Petit et al [2] described previously since, for those mutations that were examined, the Aβ fragment ratio apparently reflects the degree of destabilisation of the presenilin-substrate interaction (Szaruga et al. [100]) which likely corresponds to the degree of distortion of the presenilin protein within the γ-secretase complex. Any such fAD mutation might be expected to distort the conformation of the holoprotein (or an assembled γ-secretase complex) to a similar relative extent, and this should be reflected in the degree of destabilisation of holoprotein multimeric complexes. We note that, for this holoprotein multimer destabilisation model to function, the mutant and non-mutant forms of presenilin holoprotein must retain some ability to interact/bind each other. The N-terminal sequences of PSEN1 are known to be important for multimer formation [116,128,129] and, interestingly, there is no strong evidence for fAD mutations occurring in the PSEN1 coding sequence upstream of codon 79 in the N-terminal region or downstream of codon T440 at the C-terminal end (i.e. conservation of structure in these areas may be critical for the protein interactions needed for fAD mutations elsewhere in the coding sequence to exert a pathogenic effect – see Figure 5).
Additional evidence consistent with increased γ-secretase activity due to fAD mutations
There are a number of curious observations that appear consistent with the idea that fAD mutations of the presenilin genes may increase γ-secretase levels. These involve the gene PSEN2.
A naturally occurring transcriptional isoform of PSEN2 exists named PS2V [130]. This name is derived from the fact that PS2V transcripts exclude sequences from what was once denoted as exon 5 (i.e. “V”) of PSEN2 (but is now denoted as exon 6). PS2V levels are increased in AD brains due to an oxidative stress-sensing and -regulatory mechanism involving binding of HMGA1 protein to PSEN2 transcripts during splicing [130,131]. Interestingly, the importance of this regulatory mechanism is demonstrated by its conservation during approximately half a billion years of vertebrate evolution – except, oddly, in mice and rats [132]. PS2V mRNAs encode a truncated form of PSEN2 protein that cannot have any γ-secretase activity itself, but that nevertheless increases γ-secretase activity in cells [133]. This increase appears to be caused by suppression of the unfolded protein response – presumably in the ER [133,134,135]. (This illustrates once again the involvement of the presenilins in cellular stress responses). The existence of PS2V is relevant to early onset AD due to the existence of the fAD mutation K115Efs of PSEN2 [48]. This deletion of two nucleotides truncates the PSEN2 open reading frame so that it would encode a protein similar to that of the truncated PS2V isoform. Like PS2V, the K115Efs mutant protein can also increase γ-secretase activity [60]. Note however, that, while the K115Efs mutation was initially thought to be a possible exception to the reading frame preservation rule of presenilin fAD mutations, Braggin et al. [136] reported that this mutation also induces forms of alternative splicing that generate mRNAs with restored open reading frames but with significant coding changes. Therefore, currently, we cannot resolve whether the K115Efs mutation causes fAD through increased γ-secretase activity due to constitutive PS2V-like activity, or by encoding distorted full-length holoproteins, or both.
Another curious experimental result suggests that the most intensively investigated fAD mutation of PSEN2, N141I, shows greater than normal γ-secretase activity. Mastrangelo et al. [137] attempted to rescue embryo development in mice lacking Psen1 activity by inserting transgenes expressing either wild type human PSEN2 or N141I mutant PSEN2. Only the N141I mutant form could compensate for the absence of Psen1 activity and so allow embryo survival. Whether this superior ability of the N141I mutant is due to overall increased γ-secretase activity, or increased γ-secretase activity in a particular area of cells (e.g. due to fAD mutations changing presenilin proteins’ intracellular distributions as described by [138]), or some other mechanism is not known.
Finally, most invertebrates have only one presenilin gene but a duplication event early in the evolution of the vertebrates gave rise to PSEN1 and PSEN2. As these genes have similar structures (and, therefore, similar functions) it is unsurprising that they are both loci for fAD mutations. However, PSEN2 hosts only ~10% as many fAD mutations as PSEN1, despite that these two genes represent almost identically sized “targets” for mutagenesis. Also, fAD mutations of PSEN2 tend to show less severity (later age of onset) than those of PSEN1 [89]. Keeping in mind that ~10% of the fAD mutations in PSEN1 apparently increase the intrinsic γ-secretase activity of the mutant protein [98], one possibility is that only mutations in PSEN2 that increase the intrinsic γ-secretase activity of its encoded protein are sufficiently severe to cause disease. (Alternatively, since only PSEN2 protein appears to bind directly to MFN2 protein to promote MAM formation, and since the N141I fAD mutant form of PSEN2 was shown to bind MFN2 more avidly than wild type PSEN2 [139], it may be that, in contrast to fAD mutations of PSEN1, mutations of PSEN2 must affect both endolysosomal acidification and MAM formation if they are to cause AD – possibly a less likely event than fAD mutation of PSEN1 affecting endolysosomal acidification alone.)
Notes on the importance of iron and hypoxia in fAD pathogenesis
Our transcriptome analyses of young adult brains from zebrafish heterozygous for fAD-like mutations in the psen1, psen2, or sorl1 genes identified oxidative phosphorylation in mitochondria as the cellular process disrupted in common between these brains. Some fAD mutations may alter mitochondrial activity by affecting MAM formation or function [139]. However, Yambire et al. [140] demonstrated that disturbance of lysosomal acidification has profound effects on mitochondrial activity by affecting the availability of ferrous iron ions (Fe2+) in cells required by mitochondria to form the iron-sulphur clusters in their electron transport chain (ETC) that performs oxidative phosphorylation. When lysosomal acidification is reduced, (i.e. pH is increased), lysosomes may accumulate poorly soluble ferric iron (Fe3+) since the conversion of Fe3+ to the more soluble Fe2+ form (that can be exported for uptake by mitochondria) is inhibited. Interestingly, neuritic Aβ plaques are complexed with iron [141] and, since Lee et al. [112] have explained how the plaques arise through accumulation of Aβ in distorted lysosomes, it is now clear how this association between Aβ and iron may occur.
Most ATP is produced through oxidative phosphorylation. Production of ATP is the most essential cellular function since most other cellular activities are dependent on the energy it provides. For example, ATP is consumed by v-ATPase to drive lysosomal acidification. However, an additional, underappreciated, but essential role of ATP is as a “hydrotrope” inhibiting aggregation of the proteins in cells that are, collectively, close to solution saturation concentration in the cytosol [142]. The generalised aggregation of proteins observed as cells age [143] probably reflects falling ATP levels together with molecular crosslinking due to rising oxidative stress (particularly if the proteins are associated with iron [141]).
Disturbance of mitochondria’s ETC, whether through deficiency of either oxygen or Fe2+, commonly generates reactive oxygen species causing oxidative stress [144]. The co-dependence of cells on oxygen and Fe2+ for ETC function is so tight that transcriptional responses to deficiencies of oxygen and Fe2+ are both regulated by the HIF1 transcription factor [145]. In fact, the effects of Fe2+ deficiency are sometimes described as “pseudo-hypoxia”. Hypoxia is thought to play a major role in the development of AD [146,147]. The oxidative stress caused by hypoxia greatly increases the activity of the molecular machinery producing Aβ [59,148,149,150] that may then be protective through its activity as an antioxidant [151,152,153,154,155].
It is clear that the fAD mutations in APP, PSEN1, and PSEN2 likely all generate pseudo-hypoxic effects by inhibiting correct lysosomal acidification, thereby limiting cellular Fe2+ maintenance and disturbing oxidative phosphorylation (Figure 10A). In this sense, the fAD mutations accelerate the increase in hypoxia and oxidative stress that brains normally experience during aging [156] due to the gradual degradation of vascular function [157,158,159]. At some point, the brain “shifts” into a hypometabolic state, possibly as a final effort to avoid the rising ravages of oxidative stress that also drive neuroinflammation [160] (Figure 10B). There are many observations consistent with an overarching role for hypoxia in AD, such as the increased risk of AD due to sleep apnoea [161], decreased bloodflow in AD brains [162], lower levels of HIF1α in postmortem AD brains [163] and data from model organisms studied by us [126] and others (e.g. Qian et al.’s [164] demonstration that cholinergic basal forebrain neurons – that express p75NTR [165] and are selectively lost in AD [166] - are particularly sensitive to degeneration in their mouse model of sleep apnoea, and that this is mediated by Hif1α). An additional intriguing connection between AD and hypoxia that we have not seen emphasised elsewhere relates to the “Two-Hit” hypothesis of Alzheimer’s disease described in 2004 by Mark Smith and colleagues [50,167]. They postulated that, “…although either oxidative stress or abnormalities in mitotic signalling [aka cell cycle events, CCEs] can independently serve as initiators, both processes are necessary to propagate disease pathogenesis…and [are] invariant features of disease.” CCEs are seen when neurons inappropriately enter the cell cycle but do not undergo cell death. Both oxidative stress and CCEs result from hypoxia [168] with sufficient levels of HIF1α being essential for preventing inappropriate entry of cells into the cell cycle during hypoxia [169].
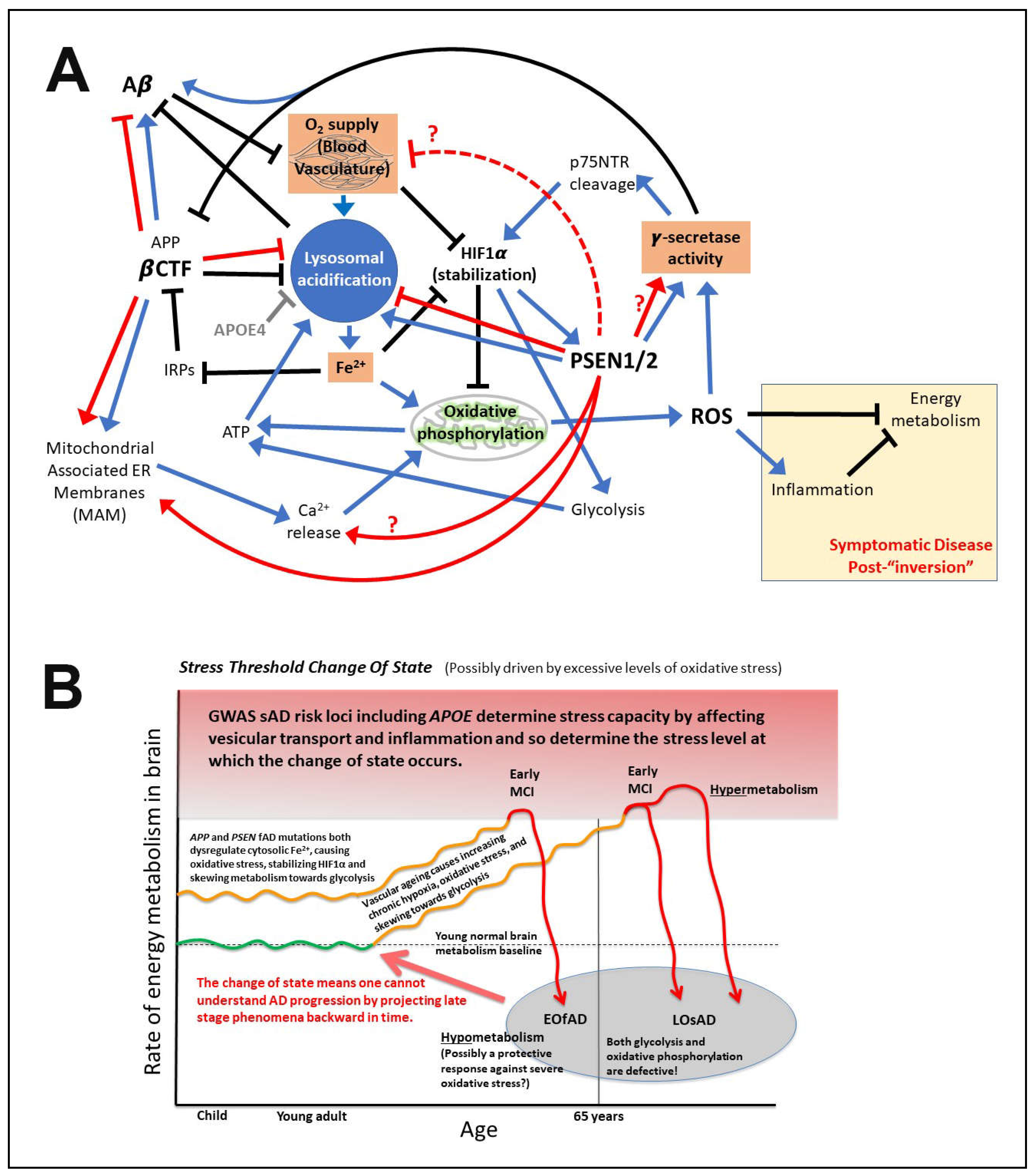
Figure 10.
Factors influencing development of Alzheimer’s disease. A, Many interconnected cellular factors place brains under stress before AD cognitive change become evident. Blue arrows indicate stimulation, black bars indicate inhibition, red arrows are the effects of fAD mutations in APP or PSEN1/2. Red dashed inhibition line refers to reported non-cell autonomous deleterious effects on vasculature of transgenic human PSEN1 fAD mutant genes when expressed solely in mouse neurons [3]. fAD mutations in APP may increase or decrease Aβ production. Components important in hypoxia/pseudo-hypoxia are indicated by brown boxes. B, Stress threshold change of state hypothesis of AD reprinted from [1]. If fAD mutations of the PSEN1/2 genes drive increased γ-secretase activity this might also drive early raised hypoxia responses. “Inversions” of gene [4,5], protein [6], and biomarker [7] expression and in other phenomena [8] have previously been observed between pre- and post-clinical disease states.
Figure 10.
Factors influencing development of Alzheimer’s disease. A, Many interconnected cellular factors place brains under stress before AD cognitive change become evident. Blue arrows indicate stimulation, black bars indicate inhibition, red arrows are the effects of fAD mutations in APP or PSEN1/2. Red dashed inhibition line refers to reported non-cell autonomous deleterious effects on vasculature of transgenic human PSEN1 fAD mutant genes when expressed solely in mouse neurons [3]. fAD mutations in APP may increase or decrease Aβ production. Components important in hypoxia/pseudo-hypoxia are indicated by brown boxes. B, Stress threshold change of state hypothesis of AD reprinted from [1]. If fAD mutations of the PSEN1/2 genes drive increased γ-secretase activity this might also drive early raised hypoxia responses. “Inversions” of gene [4,5], protein [6], and biomarker [7] expression and in other phenomena [8] have previously been observed between pre- and post-clinical disease states.
What is APP’s role in iron homeostasis?
The Iron Regulatory Proteins, IRP1 and IRP2, act to maintain cellular iron homeostasis by binding to Iron Responsive Elements (IREs) in the transcripts of genes to regulate their translation and/or stability (reviewed by [170]). The importance of iron homeostasis to cell function and survival is indicated by the fact that more than 10% of protein-coding genes may include IREs in their transcripts [171]. Considerable research has focussed on the existence of an IRE in the 5’ untranslated region (UTR) of APP mRNA that inhibits translation of APP when cells are deficient in Fe2+. A number of papers have suggested that APP acts to stabilise the cellular iron exporting protein ferroportin while others show data in conflict with this (discussed by us previously in [171]). The ability of β-secretase inhibitors to enhance ferroportin stability is regarded as evidence of a role for APP in stabilisation of ferroportin [172]. However, this stabilisation by β-secretase inhibition would also be expected if the common partner of ferroportin, the ferroxidatase hephaestin, was a β-secretase substrate. There is evidence that this is the case [173].
The fact that excessive expression of APP can cause fAD supports that iron-dependent regulation of APP mRNA translation may be a significant influence in fAD pathogenesis. We note that the IRE-dependent regulation of APP mRNA translation is consistent with a role for APP’s βCTF in modulating pH in the endolysomal system. When cells are replete in Fe2+, βCTF levels would be higher since APP translation would not be repressed by the binding of Iron Responsive Proteins (IRPs) to APP’s 5’ IRE. The consequent increase in endolysomal pH would reduce importation of Fe3+ and its conversion to Fe2+.
Iron is seen to accumulate in AD brains leading to the common speculation that this excessive iron load drives oxidative stress via the Fenton reaction [174,175]. However, the mechanism described in this paper, whereby insufficient endolysosomal acidification contributes to fAD, supports an alternative idea – AD brains suffer from a functional ferrous iron deficiency in a background of ferric iron accumulation. This is supported by transcriptome analysis of postmortem AD brains indicting increased expression of iron importation protein TFRC [171], the transcripts of which are stabilised during Fe2+ deficiency through binding of IRPs to multiple IREs in their 3’ UTR [176,177].
Predictions arising from these proposed mechanisms
If fAD mutations in APP exert their effect by increasing βCTF levels, then we expect that ongoing research from the AD research community will continue to find a strong association between these levels and disease pathologies – when researchers take the care to examine βCTF levels in addition to their usual examination of Aβ levels.
If fAD mutations in APP, PSEN1, and PSEN2 all cause AD though their action on endolysomal system acidification, then a prediction of this model would be existence of an inverse correlation between mutations’ effects on lysosomal pH and age of disease onset. The most severe mutations with the earliest ages of disease onset should show the relatively highest lysosomal pH (when comparing heterozygous mutations under uniform conditions in cellular systems with otherwise identical genetic backgrounds). Unfortunately, it is currently challenging to make precise measurements of lysosomal pH.
Testing of the hypothesis that all fAD mutations in the presenilins lead to increased γ-secretase activity could be performed by restoring the wild type sequence in iPSCs derived from an individual heterozygous for a fAD mutation in PSEN1 or PSEN2. Comparison of the γ-secretase activity between the mutant and the wild type cells (possessing otherwise identical genetic backgrounds) could then be performed after their differentiation to neurons or other cell types.
To test the idea that brains “invert” into a hypometabolic state upon exceeding a particular oxidative stress threshold, a model organism such as the zebrafish could be exposed to varying levels of reactive oxygen species (e.g. by placement under varying degrees of hypoxia) and then metabolic states assessed via metabolomics and transcriptomics strategies.
Conclusions
The most parsimonious explanation for the many types of fAD mutation affecting APP is that these increase the expression of APP’s βCTF. This alters cellular cholesterol distribution and reduces lysosomal acidification (likely affecting Fe2+ ion homeostasis thus driving pseudo-hypoxia) to impact mitochondrial function. The identification of excessive βCTF as the pathogenic agent promoted by fAD mutations in APP is consistent with Hardy and Higgins’ amyloid cascade hypothesis as originally published.
A unifying mechanism for fAD promotion by mutations in PSEN1 and PSEN2 is that these drive pseudo-hypoxia by reducing lysosomal acidification thereby disrupting Fe2+ ion homeostasis. The possibility exists that these mutations also promote a pseudo-hypoxic state by increasing γ-secretase activity. The formation of multimeric complexes by presenilin holoproteins and/or γ-secretase complexes may be fundamental to understanding how mutations in the presenilin genes cause fAD. Nevertheless, this phenomenon remains almost completely uninvestigated.
Hypoxia and pseudo-hypoxia cause increased production of Aβ as brains age. Failure of lysosomal acidification results in accumulation of undegraded Aβ, likely with co-accumulation of Fe3+, in the lysosomes of neurons. Upon neuronal death these are observed as neuritic plaques.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Supplementary Data File 1: Spreadsheet 1, Data copied from Petit et al. [2] supplementary Table S2 with SDs removed and AAOs added from Table 1 of the paper (with ranges removed). Spreadsheet 2, Data from Liu et al 2022 [101], "TABLE 1 Aβs secretion from PS dKO293 cells co-transfected with WT or mutant PSEN1 and APP-C99 (pg/mL)"
Funding
The author is an academic employee of the University of Adelaide.
Data Availability Statement
The data supporting the findings of this study are available within the article and/or its supplementary material.
Acknowledgments
The author wishes to thank Eric Schon, Greg Sutherland, and Giuseppe Verdile for their feedback after critical reading of the manuscript. All opinions and discussion in this work are the responsibility of the author alone.
Conflict of Interest Statement
The author declares no conflict of interest.
References
- Lumsden, A.L.; Rogers, J.T.; Majd, S.; Newman, M.; Sutherland, G.T.; Verdile, G.; Lardelli, M. Dysregulation of Neuronal Iron Homeostasis as an Alternative Unifying Effect of Mutations Causing Familial Alzheimer's Disease. Frontiers in neuroscience 2018, 12, 533. [Google Scholar] [CrossRef] [PubMed]
- Petit, D.; Fernandez, S.G.; Zoltowska, K.M.; Enzlein, T.; Ryan, N.S.; O'Connor, A.; Szaruga, M.; Hill, E.; Vandenberghe, R.; Fox, N.C.; et al. Abeta profiles generated by Alzheimer's disease causing PSEN1 variants determine the pathogenicity of the mutation and predict age at disease onset. Molecular psychiatry 2022, 27, 2821–2832. [Google Scholar] [CrossRef] [PubMed]
- Gama Sosa, M.A.; Gasperi, R.D.; Rocher, A.B.; Wang, A.C.; Janssen, W.G.; Flores, T.; Perez, G.M.; Schmeidler, J.; Dickstein, D.L.; Hof, P.R.; et al. Age-related vascular pathology in transgenic mice expressing presenilin 1-associated familial Alzheimer's disease mutations. The American journal of pathology 2010, 176, 353–368. [Google Scholar] [CrossRef] [PubMed]
- Berchtold, N.C.; Sabbagh, M.N.; Beach, T.G.; Kim, R.C.; Cribbs, D.H.; Cotman, C.W. Brain gene expression patterns differentiate mild cognitive impairment from normal aged and Alzheimer's disease. Neurobiology of aging 2014, 35, 1961–1972. [Google Scholar] [CrossRef] [PubMed]
- Donertas, H.M.; Izgi, H.; Kamacioglu, A.; He, Z.; Khaitovich, P.; Somel, M. Gene expression reversal toward pre-adult levels in the aging human brain and age-related loss of cellular identity. Sci Rep 2017, 7, 5894. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.A.; Varma, V.R.; An, Y.; Varma, S.; Candia, J.; Fantoni, G.; Tiwari, V.; Anerillas, C.; Williamson, A.; Saito, A.; et al. A brain proteomic signature of incipient Alzheimer's disease in young APOE epsilon4 carriers identifies novel drug targets. Sci Adv 2021, 7, eabi8178. [Google Scholar] [CrossRef]
- Sutphen, C.L.; McCue, L.; Herries, E.M.; Xiong, C.; Ladenson, J.H.; Holtzman, D.M.; Fagan, A.M.; Adni. Longitudinal decreases in multiple cerebrospinal fluid biomarkers of neuronal injury in symptomatic late onset Alzheimer's disease. Alzheimer's & dementia : the journal of the Alzheimer's Association 2018, 14, 869–879. [Google Scholar] [CrossRef]
- Merlo, S.; Spampinato, S.F.; Sortino, M.A. Early compensatory responses against neuronal injury: A new therapeutic window of opportunity for Alzheimer's Disease? CNS neuroscience & therapeutics 2019, 25, 5–13. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer's disease: the amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Maloney, J.A.; Bainbridge, T.; Gustafson, A.; Zhang, S.; Kyauk, R.; Steiner, P.; van der Brug, M.; Liu, Y.; Ernst, J.A.; Watts, R.J.; et al. Molecular mechanisms of Alzheimer disease protection by the A673T allele of amyloid precursor protein. The Journal of biological chemistry 2014, 289, 30990–31000. [Google Scholar] [CrossRef]
- Wang, L.S.; Naj, A.C.; Graham, R.R.; Crane, P.K.; Kunkle, B.W.; Cruchaga, C.; Murcia, J.D.; Cannon-Albright, L.; Baldwin, C.T.; Zetterberg, H.; et al. Rarity of the Alzheimer disease-protective APP A673T variant in the United States. JAMA Neurol 2015, 72, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Ting, S.K.; Chong, M.S.; Kandiah, N.; Hameed, S.; Tan, L.; Au, W.L.; Prakash, K.M.; Pavanni, R.; Lee, T.S.; Foo, J.N.; et al. Absence of A673T amyloid-beta precursor protein variant in Alzheimer's disease and other neurological diseases. Neurobiology of aging 2013, 34, 2441–e2447. [Google Scholar] [CrossRef] [PubMed]
- Mengel-From, J.; Jeune, B.; Pentti, T.; McGue, M.; Christensen, K.; Christiansen, L. The APP A673T frequency differs between Nordic countries. Neurobiology of aging 2015, 36, 2909–e2901. [Google Scholar] [CrossRef] [PubMed]
- Kero, M.; Paetau, A.; Polvikoski, T.; Tanskanen, M.; Sulkava, R.; Jansson, L.; Myllykangas, L.; Tienari, P.J. Amyloid precursor protein (APP) A673T mutation in the elderly Finnish population. Neurobiology of aging 2013, 34, 1518. [Google Scholar] [CrossRef]
- Bamne, M.N.; Demirci, F.Y.; Berman, S.; Snitz, B.E.; Rosenthal, S.L.; Wang, X.; Lopez, O.L.; Kamboh, M.I. Investigation of an amyloid precursor protein protective mutation (A673T) in a North American case-control sample of late-onset Alzheimer's disease. Neurobiology of aging 2014, 35, 1779. [Google Scholar] [CrossRef] [PubMed]
- Jayne, T.; Newman, M.; Verdile, G.; Sutherland, G.; Munch, G.; Musgrave, I.; Moussavi Nik, S.H.; Lardelli, M. Evidence For and Against a Pathogenic Role of Reduced gamma-Secretase Activity in Familial Alzheimer's Disease. Journal of Alzheimer's disease : JAD 2016, 52, 781–799. [Google Scholar] [CrossRef] [PubMed]
- Pottier, C.; Hannequin, D.; Coutant, S.; Rovelet-Lecrux, A.; Wallon, D.; Rousseau, S.; Legallic, S.; Paquet, C.; Bombois, S.; Pariente, J.; et al. High frequency of potentially pathogenic SORL1 mutations in autosomal dominant early-onset Alzheimer disease. Molecular psychiatry 2012, 17, 875–879. [Google Scholar] [CrossRef]
- Verheijen, J.; Van den Bossche, T.; van der Zee, J.; Engelborghs, S.; Sanchez-Valle, R.; Llado, A.; Graff, C.; Thonberg, H.; Pastor, P.; Ortega-Cubero, S.; et al. A comprehensive study of the genetic impact of rare variants in SORL1 in European early-onset Alzheimer's disease. Acta Neuropathol 2016, 132, 213–224. [Google Scholar] [CrossRef]
- Bellenguez, C.; Charbonnier, C.; Grenier-Boley, B.; Quenez, O.; Le Guennec, K.; Nicolas, G.; Chauhan, G.; Wallon, D.; Rousseau, S.; Richard, A.C.; et al. Contribution to Alzheimer's disease risk of rare variants in TREM2, SORL1, and ABCA7 in 1779 cases and 1273 controls. Neurobiology of aging 2017, 59, 220. [Google Scholar] [CrossRef]
- Barthelson, K.; Newman, M.; Lardelli, M. Sorting Out the Role of the Sortilin-Related Receptor 1 in Alzheimer's Disease. J Alzheimers Dis Rep 2020, 4, 123–140. [Google Scholar] [CrossRef]
- Fortea, J.; Zaman, S.H.; Hartley, S.; Rafii, M.S.; Head, E.; Carmona-Iragui, M. Alzheimer's disease associated with Down syndrome: a genetic form of dementia. The Lancet. Neurology 2021, 20, 930–942. [Google Scholar] [CrossRef] [PubMed]
- Rovelet-Lecrux, A.; Hannequin, D.; Raux, G.; Le Meur, N.; Laquerrière, A.; Vital, A.; Dumanchin, C.; Feuillette, S.; Brice, A.; Vercelletto, M.; et al. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nature genetics 2006, 38, 24–26. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.H.; Yan, Y.; Kang, Y.; Jiang, Y.; Melcher, K.; Xu, H.E. Alzheimer's disease-associated mutations increase amyloid precursor protein resistance to gamma-secretase cleavage and the Abeta42/Abeta40 ratio. Cell Discov 2016, 2, 16026. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Hung, A.Y.; Selkoe, D.J.; Teplow, D.B. Mutations associated with a locus for familial Alzheimer's disease result in alternative processing of amyloid beta-protein precursor. The Journal of biological chemistry 1994, 269, 17741–17748. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Brouwers, N.; Benilova, I.; Vandersteen, A.; Mercken, M.; Van Laere, K.; Van Damme, P.; Demedts, D.; Van Leuven, F.; Sleegers, K.; et al. Amyloid precursor protein mutation E682K at the alternative beta-secretase cleavage beta'-site increases Abeta generation. EMBO Mol Med 2011, 3, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Mullan, M.; Crawford, F.; Axelman, K.; Houlden, H.; Lilius, L.; Winblad, B.; Lannfelt, L. A pathogenic mutation for probable Alzheimer's disease in the APP gene at the N-terminus of beta-amyloid. Nature genetics 1992, 1, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.D.; Golde, T.E.; Younkin, S.G. Release of excess amyloid beta protein from a mutant amyloid beta protein precursor. Science 1993, 259, 514–516. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Sato, Y.; Im, E.; Berg, M.; Bordi, M.; Darji, S.; Kumar, A.; Mohan, P.S.; Bandyopadhyay, U.; Diaz, A.; et al. Lysosomal Dysfunction in Down Syndrome Is APP-Dependent and Mediated by APP-betaCTF (C99). The Journal of neuroscience : the official journal of the Society for Neuroscience 2019, 39, 5255–5268. [Google Scholar] [CrossRef]
- Chen, W.T.; Hong, C.J.; Lin, Y.T.; Chang, W.H.; Huang, H.T.; Liao, J.Y.; Chang, Y.J.; Hsieh, Y.F.; Cheng, C.Y.; Liu, H.C.; et al. Amyloid-beta (Abeta) D7H mutation increases oligomeric Abeta42 and alters properties of Abeta-zinc/copper assemblies. PloS one 2012, 7, e35807. [Google Scholar] [CrossRef]
- Bugiani, O.; Giaccone, G.; Rossi, G.; Mangieri, M.; Capobianco, R.; Morbin, M.; Mazzoleni, G.; Cupidi, C.; Marcon, G.; Giovagnoli, A.; et al. Hereditary cerebral hemorrhage with amyloidosis associated with the E693K mutation of APP. Archives of neurology 2010, 67, 987–995. [Google Scholar] [CrossRef]
- Tagliavini, J.; Williot, P.; Congiu, L.; Chicca, M.; Lanfredi, M.; Rossi, R.; Fontana, F. Molecular cytogenetic analysis of the karyotype of the European Atlantic sturgeon, Acipenser sturio. Heredity (Edinb) 1999, 83 ( Pt 5) Pt 5, 520–525. [Google Scholar] [CrossRef]
- Obici, L.; Demarchi, A.; de Rosa, G.; Bellotti, V.; Marciano, S.; Donadei, S.; Arbustini, E.; Palladini, G.; Diegoli, M.; Genovese, E.; et al. A novel AbetaPP mutation exclusively associated with cerebral amyloid angiopathy. Annals of neurology 2005, 58, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Haapasalo, A.; Kovacs, D.M. The many substrates of presenilin/gamma-secretase. Journal of Alzheimer's disease : JAD 2011, 25, 3–28. [Google Scholar] [CrossRef] [PubMed]
- Guner, G.; Lichtenthaler, S.F. The substrate repertoire of gamma-secretase/presenilin. Semin Cell Dev Biol 2020, 105, 27–42. [Google Scholar] [CrossRef]
- Yan, Y.; Xu, T.H.; Melcher, K.; Xu, H.E. Defining the minimum substrate and charge recognition model of gamma-secretase. Acta Pharmacol Sin 2017, 38, 1412–1424. [Google Scholar] [CrossRef] [PubMed]
- Takami, M.; Nagashima, Y.; Sano, Y.; Ishihara, S.; Morishima-Kawashima, M.; Funamoto, S.; Ihara, Y. gamma-Secretase: successive tripeptide and tetrapeptide release from the transmembrane domain of beta-carboxyl terminal fragment. The Journal of neuroscience : the official journal of the Society for Neuroscience 2009, 29, 13042–13052. [Google Scholar] [CrossRef] [PubMed]
- Qi-Takahara, Y.; Morishima-Kawashima, M.; Tanimura, Y.; Dolios, G.; Hirotani, N.; Horikoshi, Y.; Kametani, F.; Maeda, M.; Saido, T.C.; Wang, R.; et al. Longer forms of amyloid beta protein: implications for the mechanism of intramembrane cleavage by gamma-secretase. The Journal of neuroscience : the official journal of the Society for Neuroscience 2005, 25, 436–445. [Google Scholar] [CrossRef]
- Chavez-Gutierrez, L.; Bammens, L.; Benilova, I.; Vandersteen, A.; Benurwar, M.; Borgers, M.; Lismont, S.; Zhou, L.; Van Cleynenbreugel, S.; Esselmann, H.; et al. The mechanism of gamma-Secretase dysfunction in familial Alzheimer disease. The EMBO journal 2012, 31, 2261–2274. [Google Scholar] [CrossRef]
- Tan, J.; Mao, G.; Cui, M.Z.; Kang, S.C.; Lamb, B.; Wong, B.S.; Sy, M.S.; Xu, X. Effects of gamma-secretase cleavage-region mutations on APP processing and Abeta formation: interpretation with sequential cleavage and alpha-helical model. Journal of neurochemistry 2008, 107, 722–733. [Google Scholar] [CrossRef]
- Suarez-Calvet, M.; Belbin, O.; Pera, M.; Badiola, N.; Magrane, J.; Guardia-Laguarta, C.; Munoz, L.; Colom-Cadena, M.; Clarimon, J.; Lleo, A. Autosomal-dominant Alzheimer's disease mutations at the same codon of amyloid precursor protein differentially alter Abeta production. Journal of neurochemistry 2014, 128, 330–339. [Google Scholar] [CrossRef]
- Jarrett, J.T.; Lansbury, P.T., Jr. Seeding "one-dimensional crystallization" of amyloid: a pathogenic mechanism in Alzheimer's disease and scrapie? Cell 1993, 73, 1055–1058. [Google Scholar] [CrossRef] [PubMed]
- Tamaoka, A.; Odaka, A.; Ishibashi, Y.; Usami, M.; Sahara, N.; Suzuki, N.; Nukina, N.; Mizusawa, H.; Shoji, S.; Kanazawa, I.; et al. APP717 missense mutation affects the ratio of amyloid beta protein species (A beta 1-42/43 and a beta 1-40) in familial Alzheimer's disease brain. The Journal of biological chemistry 1994, 269, 32721–32724. [Google Scholar] [CrossRef]
- Borchelt, D.R.; Thinakaran, G.; Eckman, C.B.; Lee, M.K.; Davenport, F.; Ratovitsky, T.; Prada, C.M.; Kim, G.; Seekins, S.; Yager, D.; et al. Familial Alzheimer's disease-linked presenilin 1 variants elevate Abeta1-42/1-40 ratio in vitro and in vivo. Neuron 1996, 17, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Lichtenthaler, S.F.; Ida, N.; Multhaup, G.; Masters, C.L.; Beyreuther, K. Mutations in the transmembrane domain of APP altering gamma-secretase specificity. Biochemistry 1997, 36, 15396–15403. [Google Scholar] [CrossRef] [PubMed]
- Naslund, J.; Schierhorn, A.; Hellman, U.; Lannfelt, L.; Roses, A.D.; Tjernberg, L.O.; Silberring, J.; Gandy, S.E.; Winblad, B.; Greengard, P.; et al. Relative abundance of Alzheimer A beta amyloid peptide variants in Alzheimer disease and normal aging. Proceedings of the National Academy of Sciences of the United States of America 1994, 91, 8378–8382. [Google Scholar] [CrossRef] [PubMed]
- Scheuner, D.; Eckman, C.; Jensen, M.; Song, X.; Citron, M.; Suzuki, N.; Bird, T.D.; Hardy, J.; Hutton, M.; Kukull, W.; et al. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nature medicine 1996, 2, 864–870. [Google Scholar] [CrossRef]
- Citron, M.; Westaway, D.; Xia, W.; Carlson, G.; Diehl, T.; Levesque, G.; Johnson-Wood, K.; Lee, M.; Seubert, P.; Davis, A.; et al. Mutant presenilins of Alzheimer's disease increase production of 42-residue amyloid beta-protein in both transfected cells and transgenic mice. Nature medicine 1997, 3, 67–72. [Google Scholar] [CrossRef]
- Jayadev, S.; Leverenz, J.B.; Steinbart, E.; Stahl, J.; Klunk, W.; Yu, C.E.; Bird, T.D. Alzheimer's disease phenotypes and genotypes associated with mutations in presenilin 2. Brain : a journal of neurology 2010, 133, 1143–1154. [Google Scholar] [CrossRef]
- Arimon, M.; Takeda, S.; Post, K.L.; Svirsky, S.; Hyman, B.T.; Berezovska, O. Oxidative stress and lipid peroxidation are upstream of amyloid pathology. Neurobiology of disease 2015, 84, 109–119. [Google Scholar] [CrossRef]
- Zhu, X.; Raina, A.K.; Perry, G.; Smith, M.A. Alzheimer's disease: the two-hit hypothesis. The Lancet. Neurology 2004, 3, 219–226. [Google Scholar] [CrossRef]
- Martins, R.N.; Harper, C.G.; Stokes, G.B.; Masters, C.L. Increased cerebral glucose-6-phosphate dehydrogenase activity in Alzheimer's disease may reflect oxidative stress. Journal of neurochemistry 1986, 46, 1042–1045. [Google Scholar] [CrossRef] [PubMed]
- Holmes, O.; Paturi, S.; Ye, W.; Wolfe, M.S.; Selkoe, D.J. Effects of membrane lipids on the activity and processivity of purified gamma-secretase. Biochemistry 2012, 51, 3565–3575. [Google Scholar] [CrossRef]
- Song, Y.; Mittendorf, K.F.; Lu, Z.; Sanders, C.R. Impact of bilayer lipid composition on the structure and topology of the transmembrane amyloid precursor C99 protein. J Am Chem Soc 2014, 136, 4093–4096. [Google Scholar] [CrossRef] [PubMed]
- Montesinos, J.; Pera, M.; Larrea, D.; Guardia-Laguarta, C.; Agrawal, R.R.; Velasco, K.R.; Yun, T.D.; Stavrovskaya, I.G.; Xu, Y.; Koo, S.Y.; et al. The Alzheimer's disease-associated C99 fragment of APP regulates cellular cholesterol trafficking. The EMBO journal 2020, 39, e103791. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.Y.; Kwon, O.H.; Chung, S. Preferred Endocytosis of Amyloid Precursor Protein from Cholesterol-Enriched Lipid Raft Microdomains. Molecules 2020, 25. [Google Scholar] [CrossRef] [PubMed]
- Vetrivel, K.S.; Cheng, H.; Lin, W.; Sakurai, T.; Li, T.; Nukina, N.; Wong, P.C.; Xu, H.; Thinakaran, G. Association of gamma-secretase with lipid rafts in post-Golgi and endosome membranes. The Journal of biological chemistry 2004, 279, 44945–44954. [Google Scholar] [CrossRef] [PubMed]
- Pera, M.; Larrea, D.; Guardia-Laguarta, C.; Montesinos, J.; Velasco, K.R.; Agrawal, R.R.; Xu, Y.; Chan, R.B.; Di Paolo, G.; Mehler, M.F.; et al. Increased localization of APP-C99 in mitochondria-associated ER membranes causes mitochondrial dysfunction in Alzheimer disease. The EMBO journal 2017, 36, 3356–3371. [Google Scholar] [CrossRef]
- Cui, J.G.; Fraser, P.E.; St George-Hyslop, P.; Westaway, D.; Lukiw, W.J. Potential roles for presenilin-1 in oxygen sensing and in glial-specific gene expression. Neuroreport 2004, 15, 2025–2028. [Google Scholar] [CrossRef]
- Moussavi Nik, S.H.; Wilson, L.; Newman, M.; Croft, K.; Mori, T.A.; Musgrave, I.; Lardelli, M. The BACE1-PSEN-AbetaPP regulatory axis has an ancient role in response to low oxygen/oxidative stress. Journal of Alzheimer's disease : JAD 2012, 28, 515–530. [Google Scholar] [CrossRef]
- Moussavi Nik, S.H.; Newman, M.; Wilson, L.; Ebrahimie, E.; Wells, S.; Musgrave, I.; Verdile, G.; Martins, R.N.; Lardelli, M. Alzheimer's disease-related peptide PS2V plays ancient, conserved roles in suppression of the unfolded protein response under hypoxia and stimulation of gamma-secretase activity. Human molecular genetics 2015, 24, 3662–3678. [Google Scholar] [CrossRef]
- Halliwell, B. Oxidative stress in cell culture: an under-appreciated problem? FEBS Lett 2003, 540, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Eggert, S.; Gonzalez, A.C.; Thomas, C.; Schilling, S.; Schwarz, S.M.; Tischer, C.; Adam, V.; Strecker, P.; Schmidt, V.; Willnow, T.E.; et al. Dimerization leads to changes in APP (amyloid precursor protein) trafficking mediated by LRP1 and SorLA. Cellular and molecular life sciences : CMLS 2018, 75, 301–322. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.I.; Premraj, S.; Cruz, P.E.; Ladd, T.B.; Kwak, Y.; Koo, E.H.; Felsenstein, K.M.; Golde, T.E.; Ran, Y. Independent Relationship between Amyloid Precursor Protein (APP) Dimerization and γ-Secretase Processivity. PloS one 2014, 9, e111553. [Google Scholar] [CrossRef]
- Scheuermann, S.; Hambsch, B.; Hesse, L.; Stumm, J.; Schmidt, C.; Beher, D.; Bayer, T.A.; Beyreuther, K.; Multhaup, G. Homodimerization of amyloid precursor protein and its implication in the amyloidogenic pathway of Alzheimer's disease. The Journal of biological chemistry 2001, 276, 33923–33929. [Google Scholar] [CrossRef] [PubMed]
- Kwart, D.; Gregg, A.; Scheckel, C.; Murphy, E.A.; Paquet, D.; Duffield, M.; Fak, J.; Olsen, O.; Darnell, R.B.; Tessier-Lavigne, M. A Large Panel of Isogenic APP and PSEN1 Mutant Human iPSC Neurons Reveals Shared Endosomal Abnormalities Mediated by APP beta-CTFs, Not Abeta. Neuron 2019, 104, 1022. [Google Scholar] [CrossRef] [PubMed]
- Laird, J. The Law of Parsimony. The Monist 1919, 29, 321–344. [Google Scholar] [CrossRef]
- Morris, G.P.; Clark, I.A.; Vissel, B. Questions concerning the role of amyloid-beta in the definition, aetiology and diagnosis of Alzheimer's disease. Acta Neuropathol 2018, 136, 663–689. [Google Scholar] [CrossRef]
- Castellani, R.J.; Lee, H.G.; Siedlak, S.L.; Nunomura, A.; Hayashi, T.; Nakamura, M.; Zhu, X.; Perry, G.; Smith, M.A. Reexamining Alzheimer's disease: evidence for a protective role for amyloid-beta protein precursor and amyloid-beta. Journal of Alzheimer's disease : JAD 2009, 18, 447–452. [Google Scholar] [CrossRef]
- Castellani, R.J.; Smith, M.A. Compounding artefacts with uncertainty, and an amyloid cascade hypothesis that is 'too big to fail'. The Journal of pathology 2011, 224, 147–152. [Google Scholar] [CrossRef]
- Mondragon-Rodriguez, S.; Basurto-Islas, G.; Lee, H.G.; Perry, G.; Zhu, X.; Castellani, R.J.; Smith, M.A. Causes versus effects: the increasing complexities of Alzheimer's disease pathogenesis. Expert Rev Neurother 2010, 10, 683–691. [Google Scholar] [CrossRef]
- Kepp, K.P. Alzheimer's disease due to loss of function: A new synthesis of the available data. Progress in neurobiology 2016, 143, 36–60. [Google Scholar] [CrossRef] [PubMed]
- Perneczky, R.; Jessen, F.; Grimmer, T.; Levin, J.; Floel, A.; Peters, O.; Froelich, L. Anti-amyloid antibody therapies in Alzheimer's disease. Brain : a journal of neurology, 1093. [Google Scholar] [CrossRef]
- Richards, R.I.; Robertson, S.A.; O'Keefe, L.V.; Fornarino, D.; Scott, A.; Lardelli, M.; Baune, B.T. The Enemy within: Innate Surveillance-Mediated Cell Death, the Common Mechanism of Neurodegenerative Disease. Frontiers in neuroscience 2016, 10, 193. [Google Scholar] [CrossRef] [PubMed]
- Ihnatovych, I.; Birkaya, B.; Notari, E.; Szigeti, K. iPSC-Derived Microglia for Modeling Human-Specific DAMP and PAMP Responses in the Context of Alzheimer's Disease. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Kolata, G. An Alzheimer’s Treatment Fails: ‘We Don’t Have Anything Now’. The New York Times 2020. [Google Scholar]
- Yokoyama, M.; Kobayashi, H.; Tatsumi, L.; Tomita, T. Mouse Models of Alzheimer's Disease. Front Mol Neurosci 2022, 15, 912995. [Google Scholar] [CrossRef]
- Pulina, M.V.; Hopkins, M.; Haroutunian, V.; Greengard, P.; Bustos, V. C99 selectively accumulates in vulnerable neurons in Alzheimer's disease. Alzheimer's & dementia : the journal of the Alzheimer's Association 2020, 16, 273–282. [Google Scholar] [CrossRef]
- Vaillant-Beuchot, L.; Mary, A.; Pardossi-Piquard, R.; Bourgeois, A.; Lauritzen, I.; Eysert, F.; Kinoshita, P.F.; Cazareth, J.; Badot, C.; Fragaki, K.; et al. Accumulation of amyloid precursor protein C-terminal fragments triggers mitochondrial structure, function, and mitophagy defects in Alzheimer's disease models and human brains. Acta Neuropathol 2021, 141, 39–65. [Google Scholar] [CrossRef]
- Hebert, S.S.; Horre, K.; Nicolai, L.; Papadopoulou, A.S.; Mandemakers, W.; Silahtaroglu, A.N.; Kauppinen, S.; Delacourte, A.; De Strooper, B. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer's disease correlates with increased BACE1/beta-secretase expression. Proceedings of the National Academy of Sciences of the United States of America 2008, 105, 6415–6420. [Google Scholar] [CrossRef]
- Checler, F.; Afram, E.; Pardossi-Piquard, R.; Lauritzen, I. Is gamma-secretase a beneficial inactivating enzyme of the toxic APP C-terminal fragment C99? The Journal of biological chemistry 2021, 296, 100489. [Google Scholar] [CrossRef]
- Gao, S.; Casey, A.E.; Sargeant, T.J.; Mäkinen, V.P. Genetic variation within endolysosomal system is associated with late-onset Alzheimer's disease. Brain : a journal of neurology 2018, 141, 2711–2720. [Google Scholar] [CrossRef]
- Szabo, M.P.; Mishra, S.; Knupp, A.; Young, J.E. The role of Alzheimer's disease risk genes in endolysosomal pathways. Neurobiology of disease 2022, 162, 105576. [Google Scholar] [CrossRef]
- Prasad, H.; Rao, R. Amyloid clearance defect in ApoE4 astrocytes is reversed by epigenetic correction of endosomal pH. Proceedings of the National Academy of Sciences of the United States of America 2018, 115, E6640–E6649. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 2010, 141, 1146–1158. [Google Scholar] [CrossRef] [PubMed]
- Csordas, G.; Hajnoczky, G. SR/ER-mitochondrial local communication: calcium and ROS. Biochimica et biophysica acta 2009, 1787, 1352–1362. [Google Scholar] [CrossRef] [PubMed]
- Area-Gomez, E.; Del Carmen Lara Castillo, M.; Tambini, M.D.; Guardia-Laguarta, C.; de Groof, A.J.; Madra, M.; Ikenouchi, J.; Umeda, M.; Bird, T.D.; Sturley, S.L.; et al. Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. The EMBO journal 2012, 31, 4106–4123. [Google Scholar] [CrossRef] [PubMed]
- Area-Gomez, E.; de Groof, A.J.C.; Boldogh, I.; Bird, T.D.; Gibson, G.E.; Koehler, C.M.; Yu, W.H.; Duff, K.E.; Yaffe, M.P.; Pon, L.A.; et al. Presenilins Are Enriched in Endoplasmic Reticulum Membranes Associated with Mitochondria. The American journal of pathology 2009, 175, 1810–1816. [Google Scholar] [CrossRef]
- Lanoiselee, H.M.; Nicolas, G.; Wallon, D.; Rovelet-Lecrux, A.; Lacour, M.; Rousseau, S.; Richard, A.C.; Pasquier, F.; Rollin-Sillaire, A.; Martinaud, O.; et al. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med 2017, 14, e1002270. [Google Scholar] [CrossRef]
- Ryman, D.C.; Acosta-Baena, N.; Aisen, P.S.; Bird, T.; Danek, A.; Fox, N.C.; Goate, A.; Frommelt, P.; Ghetti, B.; Langbaum, J.B.; et al. Symptom onset in autosomal dominant Alzheimer disease: a systematic review and meta-analysis. Neurology 2014, 83, 253–260. [Google Scholar] [CrossRef]
- De Jonghe, C.; Cruts, M.; Rogaeva, E.A.; Tysoe, C.; Singleton, A.; Vanderstichele, H.; Meschino, W.; Dermaut, B.; Vanderhoeven, I.; Backhovens, H.; et al. Aberrant splicing in the presenilin-1 intron 4 mutation causes presenile Alzheimer's disease by increased Abeta42 secretion. Human molecular genetics 1999, 8, 1529–1540. [Google Scholar] [CrossRef]
- Pang, Y.; Li, T.; Wang, Q.; Qin, W.; Li, Y.; Wei, Y.; Jia, L. A Rare Variation in the 3' Untranslated Region of the Presenilin 2 Gene Is Linked to Alzheimer's Disease. Molecular neurobiology 2021, 58, 4337–4347. [Google Scholar] [CrossRef]
- Brenowitz, W.D.; Fornage, M.; Launer, L.J.; Habes, M.; Davatzikos, C.; Yaffe, K. Alzheimer's Disease Genetic Risk, Cognition, and Brain Aging in Midlife. Annals of neurology, 1002. [Google Scholar] [CrossRef]
- Wang, Z.; Yan, Y.; Wang, B. gamma-Secretase Genetics of Hidradenitis Suppurativa: A Systematic Literature Review. Dermatology 2021, 237, 698–704. [Google Scholar] [CrossRef] [PubMed]
- Duchatelet, S.; Miskinyte, S.; Delage, M.; Ungeheuer, M.N.; Lam, T.; Benhadou, F.; Del Marmol, V.; Vossen, A.; Prens, E.P.; Cogrel, O.; et al. Low Prevalence of GSC Gene Mutations in a Large Cohort of Predominantly Caucasian Patients with Hidradenitis Suppurativa. J Invest Dermatol 2020, 140, 2085–2088. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Yang, W.; Wen, W.; Sun, J.; Su, B.; Liu, B.; Ma, D.; Lv, D.; Wen, Y.; Qu, T.; et al. Gamma-secretase gene mutations in familial acne inversa. Science 2010, 330, 1065. [Google Scholar] [CrossRef] [PubMed]
- Duering, M.; Grimm, M.O.; Grimm, H.S.; Schroder, J.; Hartmann, T. Mean age of onset in familial Alzheimer's disease is determined by amyloid beta 42. Neurobiology of aging 2005, 26, 785–788. [Google Scholar] [CrossRef] [PubMed]
- Kumar-Singh, S.; Theuns, J.; Van Broeck, B.; Pirici, D.; Vennekens, K.; Corsmit, E.; Cruts, M.; Dermaut, B.; Wang, R.; Van Broeckhoven, C. Mean age-of-onset of familial alzheimer disease caused by presenilin mutations correlates with both increased Abeta42 and decreased Abeta40. Hum Mutat 2006, 27, 686–695. [Google Scholar] [CrossRef]
- Sun, L.; Zhou, R.; Yang, G.; Shi, Y. Analysis of 138 pathogenic mutations in presenilin-1 on the in vitro production of Abeta42 and Abeta40 peptides by gamma-secretase. Proceedings of the National Academy of Sciences of the United States of America 2017, 114, E476–E485. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yang, G.; Shi, Y. Dominant negative effect of the loss-of-function gamma-secretase mutants on the wild-type enzyme through heterooligomerization. Proceedings of the National Academy of Sciences of the United States of America 2017, 114, 12731–12736. [Google Scholar] [CrossRef] [PubMed]
- Szaruga, M.; Munteanu, B.; Lismont, S.; Veugelen, S.; Horre, K.; Mercken, M.; Saido, T.C.; Ryan, N.S.; De Vos, T.; Savvides, S.N.; et al. Alzheimer's-causing mutations shift Abeta length by destabilizing gamma-secretase-Abetan interactions. Cell 2021, 184, 2257–2258. [Google Scholar] [CrossRef]
- Liu, L.; Lauro, B.M.; He, A.; Lee, H.; Bhattarai, S.; Wolfe, M.S.; Bennett, D.A.; Karch, C.M.; Young-Pearse, T.; Dominantly Inherited Alzheimer, N.; et al. Identification of the Abeta37/42 peptide ratio in CSF as an improved Abeta biomarker for Alzheimer's disease. Alzheimer's & dementia : the journal of the Alzheimer's Association 2023, 19, 79–96. [Google Scholar] [CrossRef]
- Fukumori, A.; Fluhrer, R.; Steiner, H.; Haass, C. Three-amino acid spacing of presenilin endoproteolysis suggests a general stepwise cleavage of gamma-secretase-mediated intramembrane proteolysis. The Journal of neuroscience : the official journal of the Society for Neuroscience 2010, 30, 7853–7862. [Google Scholar] [CrossRef]
- Steiner, H.; Romig, H.; Grim, M.G.; Philipp, U.; Pesold, B.; Citron, M.; Baumeister, R.; Haass, C. The biological and pathological function of the presenilin-1 Deltaexon 9 mutation is independent of its defect to undergo proteolytic processing. The Journal of biological chemistry 1999, 274, 7615–7618. [Google Scholar] [CrossRef] [PubMed]
- Xia, D.; Watanabe, H.; Wu, B.; Lee, S.H.; Li, Y.; Tsvetkov, E.; Bolshakov, V.Y.; Shen, J.; Kelleher, R.J. , 3rd. Presenilin-1 knockin mice reveal loss-of-function mechanism for familial Alzheimer's disease. Neuron 2015, 85, 967–981. [Google Scholar] [CrossRef] [PubMed]
- Heilig, E.A.; Gutti, U.; Tai, T.; Shen, J.; Kelleher, R.J. , 3rd. Trans-dominant negative effects of pathogenic PSEN1 mutations on gamma-secretase activity and Abeta production. The Journal of neuroscience : the official journal of the Society for Neuroscience 2013, 33, 11606–11617. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Qin, W.; Wu, L.; Zhou, A.; Tang, Y.; Wang, Q.; Jia, L.; Jia, J. Two novel presenilin-1 mutations (I249L and P433S) in early onset Chinese Alzheimer's pedigrees and their functional characterization. Biochemical and biophysical research communications 2019, 516, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Kretner, B.; Trambauer, J.; Fukumori, A.; Mielke, J.; Kuhn, P.H.; Kremmer, E.; Giese, A.; Lichtenthaler, S.F.; Haass, C.; Arzberger, T.; et al. Generation and deposition of Abeta43 by the virtually inactive presenilin-1 L435F mutant contradicts the presenilin loss-of-function hypothesis of Alzheimer's disease. EMBO Mol Med 2016, 8, 458–465. [Google Scholar] [CrossRef]
- Steiner, H.; Capell, A.; Pesold, B.; Citron, M.; Kloetzel, P.M.; Selkoe, D.J.; Romig, H.; Mendla, K.; Haass, C. Expression of Alzheimer's disease-associated presenilin-1 is controlled by proteolytic degradation and complex formation. The Journal of biological chemistry 1998, 273, 32322–32331. [Google Scholar] [CrossRef] [PubMed]
- Dewji, N.N.; Do, C.; Singer, S.J. On the spurious endoproteolytic processing of the presenilin proteins in cultured cells and tissues. Proceedings of the National Academy of Sciences of the United States of America 1997, 94, 14031–14036. [Google Scholar] [CrossRef] [PubMed]
- Raut, S.; Patel, R.; Al-Ahmad, A.J. Presence of a mutation in PSEN1 or PSEN2 gene is associated with an impaired brain endothelial cell phenotype in vitro. Fluids Barriers CNS 2021, 18, 3. [Google Scholar] [CrossRef]
- Vidoni, C.; Follo, C.; Savino, M.; Melone, M.A.B.; Isidoro, C. The Role of Cathepsin D in the Pathogenesis of Human Neurodegenerative Disorders. Medicinal Research Reviews 2016, 36, 845–870. [Google Scholar] [CrossRef]
- Lee, J.H.; Yang, D.S.; Goulbourne, C.N.; Im, E.; Stavrides, P.; Pensalfini, A.; Chan, H.; Bouchet-Marquis, C.; Bleiwas, C.; Berg, M.J.; et al. Faulty autolysosome acidification in Alzheimer's disease mouse models induces autophagic build-up of Abeta in neurons, yielding senile plaques. Nature neuroscience 2022, 25, 688–701. [Google Scholar] [CrossRef]
- Kim, S.H.; Cho, Y.S.; Kim, Y.; Park, J.; Yoo, S.M.; Gwak, J.; Kim, Y.; Gwon, Y.; Kam, T.I.; Jung, Y.K. Endolysosomal impairment by binding of amyloid beta or MAPT/Tau to V-ATPase and rescue via the HYAL-CD44 axis in Alzheimer disease. Autophagy, 1080. [Google Scholar] [CrossRef]
- Mustaly-Kalimi, S.; Gallegos, W.; Marr, R.A.; Gilman-Sachs, A.; Peterson, D.A.; Sekler, I.; Stutzmann, G.E. Protein mishandling and impaired lysosomal proteolysis generated through calcium dysregulation in Alzheimer's disease. Proceedings of the National Academy of Sciences of the United States of America 2022, 119, e2211999119. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; McBrayer, M.K.; Wolfe, D.M.; Haslett, L.J.; Kumar, A.; Sato, Y.; Lie, P.P.; Mohan, P.; Coffey, E.E.; Kompella, U.; et al. Presenilin 1 Maintains Lysosomal Ca(2+) Homeostasis via TRPML1 by Regulating vATPase-Mediated Lysosome Acidification. Cell reports 2015, 12, 1430–1444. [Google Scholar] [CrossRef] [PubMed]
- Schroeter, E.H.; Ilagan, M.X.; Brunkan, A.L.; Hecimovic, S.; Li, Y.M.; Xu, M.; Lewis, H.D.; Saxena, M.T.; De Strooper, B.; Coonrod, A.; et al. A presenilin dimer at the core of the gamma-secretase enzyme: insights from parallel analysis of Notch 1 and APP proteolysis. Proceedings of the National Academy of Sciences of the United States of America 2003, 100, 13075–13080. [Google Scholar] [CrossRef] [PubMed]
- Brautigam, H.; Moreno, C.L.; Steele, J.W.; Bogush, A.; Dickstein, D.L.; Kwok, J.B.; Schofield, P.R.; Thinakaran, G.; Mathews, P.M.; Hof, P.R.; et al. Physiologically generated presenilin 1 lacking exon 8 fails to rescue brain PS1-/- phenotype and forms complexes with wildtype PS1 and nicastrin. Sci Rep 2015, 5, 17042. [Google Scholar] [CrossRef]
- Escamilla-Ayala, A.A.; Sannerud, R.; Mondin, M.; Poersch, K.; Vermeire, W.; Paparelli, L.; Berlage, C.; Koenig, M.; Chavez-Gutierrez, L.; Ulbrich, M.H.; et al. Super-resolution microscopy reveals majorly mono- and dimeric presenilin1/gamma-secretase at the cell surface. Elife 2020, 9. [Google Scholar] [CrossRef]
- Valapala, M.; Hose, S.; Gongora, C.; Dong, L.; Wawrousek, E.F.; Samuel Zigler, J., Jr.; Sinha, D. Impaired endolysosomal function disrupts Notch signalling in optic nerve astrocytes. Nature communications 2013, 4, 1629. [Google Scholar] [CrossRef]
- Vaccari, T.; Duchi, S.; Cortese, K.; Tacchetti, C.; Bilder, D. The vacuolar ATPase is required for physiological as well as pathological activation of the Notch receptor. Development 2010, 137, 1825–1832. [Google Scholar] [CrossRef]
- Yan, Y.; Denef, N.; Schupbach, T. The vacuolar proton pump, V-ATPase, is required for notch signaling and endosomal trafficking in Drosophila. Developmental cell 2009, 17, 387–402. [Google Scholar] [CrossRef]
- Maesako, M.; Houser, M.C.Q.; Turchyna, Y.; Wolfe, M.S.; Berezovska, O. Presenilin/gamma-Secretase Activity Is Located in Acidic Compartments of Live Neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience 2022, 42, 145–154. [Google Scholar] [CrossRef]
- Le Moan, N.; Houslay, D.M.; Christian, F.; Houslay, M.D.; Akassoglou, K. Oxygen-dependent cleavage of the p75 neurotrophin receptor triggers stabilization of HIF-1alpha. Mol Cell 2011, 44, 476–490. [Google Scholar] [CrossRef]
- Villa, J.C.; Chiu, D.; Brandes, A.H.; Escorcia, F.E.; Villa, C.H.; Maguire, W.F.; Hu, C.J.; de Stanchina, E.; Simon, M.C.; Sisodia, S.S.; et al. Nontranscriptional role of Hif-1alpha in activation of gamma-secretase and notch signaling in breast cancer. Cell reports 2014, 8, 1077–1092. [Google Scholar] [CrossRef] [PubMed]
- De Gasperi, R.; Sosa, M.A.; Dracheva, S.; Elder, G.A. Presenilin-1 regulates induction of hypoxia inducible factor-1alpha: altered activation by a mutation associated with familial Alzheimer's disease. Molecular neurodegeneration 2010, 5, 38. [Google Scholar] [CrossRef] [PubMed]
- Newman, M.; Nik, H.M.; Sutherland, G.T.; Hin, N.; Kim, W.S.; Halliday, G.M.; Jayadev, S.; Smith, C.; Laird, A.S.; Lucas, C.W.; et al. Accelerated loss of hypoxia response in zebrafish with familial Alzheimer's disease-like mutation of presenilin 1. Human molecular genetics 2020, 29, 2379–2394. [Google Scholar] [CrossRef] [PubMed]
- Barthelson, K.; Dong, Y.; Newman, M.; Lardelli, M. PRESENILIN 1 mutations causing early-onset familial Alzheimer’s disease or familial acne inversa differ in their effects on genes facilitating energy metabolism and signal transduction. Journal of Alzheimer's Disease 2021, 82, 327–347. [Google Scholar] [CrossRef] [PubMed]
- Cervantes, S.; Gonzalez-Duarte, R.; Marfany, G. Homodimerization of presenilin N-terminal fragments is affected by mutations linked to Alzheimer's disease. FEBS Lett 2001, 505, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Hebert, S.S.; Godin, C.; Tomiyama, T.; Mori, H.; Levesque, G. Dimerization of presenilin-1 in vivo: suggestion of novel regulatory mechanisms leading to higher order complexes. Biochemical and biophysical research communications 2003, 301, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Manabe, T.; Katayama, T.; Sato, N.; Gomi, F.; Hitomi, J.; Yanagita, T.; Kudo, T.; Honda, A.; Mori, Y.; Matsuzaki, S.; et al. Induced HMGA1a expression causes aberrant splicing of Presenilin-2 pre-mRNA in sporadic Alzheimer's disease. Cell Death and Differentiation 2003, 10, 698–708. [Google Scholar] [CrossRef]
- Manabe, T.; Ohe, K.; Katayama, T.; Matsuzaki, S.; Yanagita, T.; Okuda, H.; Bando, Y.; Imaizumi, K.; Reeves, R.; Tohyama, M.; et al. HMGA1a: sequence-specific RNA-binding factor causing sporadic Alzheimer's disease-linked exon skipping of presenilin-2 pre-mRNA. Genes to Cells 2007, 12, 1179–1191. [Google Scholar] [CrossRef]
- Sharman, M.J.; Moussavi Nik, S.H.; Chen, M.M.; Ong, D.; Wijaya, L.; Laws, S.M.; Taddei, K.; Newman, M.; Lardelli, M.; Martins, R.N.; et al. The Guinea Pig as a Model for Sporadic Alzheimer's Disease (AD): The Impact of Cholesterol Intake on Expression of AD-Related Genes. PloS one 2013, 8, e66235. [Google Scholar] [CrossRef]
- Sato, N.; Imaizumi, K.; Manabe, T.; Taniguchi, M.; Hitomi, J.; Katayama, T.; Yoneda, T.; Morihara, T.; Yasuda, Y.; Takagi, T.; et al. Increased production of beta-amyloid and vulnerability to endoplasmic reticulum stress by an aberrant spliced form of presenilin 2. Journal of Biological Chemistry 2001, 276, 2108–2114. [Google Scholar] [CrossRef]
- Katayama, T.; Imaizumi, K.; Sato, N.; Miyoshi, K.; Kudo, T.; Hitomi, J.; Morihara, T.; Yoneda, T.; Gomi, F.; Mori, Y.; et al. Presenilin-1 mutations downregulate the signalling pathway of the unfolded-protein response. Nature cell biology 1999, 1, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Katayama, T.; Imaizumi, K.; Manabe, T.; Hitomi, J.; Kudo, T.; Tohyama, M. Induction of neuronal death by ER stress in Alzheimer's disease. Journal of chemical neuroanatomy 2004, 28, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Braggin, J.E.; Bucks, S.A.; Course, M.M.; Smith, C.L.; Sopher, B.; Osnis, L.; Shuey, K.D.; Domoto-Reilly, K.; Caso, C.; Kinoshita, C.; et al. Alternative splicing in a presenilin 2 variant associated with Alzheimer disease. Ann Clin Transl Neurol 2019, 6, 762–777. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, P.; Mathews, P.M.; Chishti, M.A.; Schmidt, S.D.; Gu, Y.; Yang, J.; Mazzella, M.J.; Coomaraswamy, J.; Horne, P.; Strome, B.; et al. Dissociated phenotypes in presenilin transgenic mice define functionally distinct gamma-secretases. Proceedings of the National Academy of Sciences of the United States of America 2005, 102, 8972–8977. [Google Scholar] [CrossRef] [PubMed]
- Sannerud, R.; Esselens, C.; Ejsmont, P.; Mattera, R.; Rochin, L.; Tharkeshwar, A.K.; De Baets, G.; De Wever, V.; Habets, R.; Baert, V.; et al. Restricted Location of PSEN2/gamma-Secretase Determines Substrate Specificity and Generates an Intracellular Abeta Pool. Cell 2016, 166, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Filadi, R.; Greotti, E.; Turacchio, G.; Luini, A.; Pozzan, T.; Pizzo, P. Presenilin 2 Modulates Endoplasmic Reticulum-Mitochondria Coupling by Tuning the Antagonistic Effect of Mitofusin 2. Cell reports 2016, 15, 2226–2238. [Google Scholar] [CrossRef]
- Yambire, K.F.; Rostosky, C.; Watanabe, T.; Pacheu-Grau, D.; Torres-Odio, S.; Sanchez-Guerrero, A.; Senderovich, O.; Meyron-Holtz, E.G.; Milosevic, I.; Frahm, J.; et al. Impaired lysosomal acidification triggers iron deficiency and inflammation in vivo. Elife 2019, 8. [Google Scholar] [CrossRef]
- Smith, M.A.; Harris, P.L.; Sayre, L.M.; Perry, G. Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proceedings of the National Academy of Sciences of the United States of America 1997, 94, 9866–9868. [Google Scholar] [CrossRef]
- Patel, A.; Malinovska, L.; Saha, S.; Wang, J.; Alberti, S.; Krishnan, Y.; Hyman, A.A. ATP as a biological hydrotrope. Science 2017, 356, 753–756. [Google Scholar] [CrossRef]
- David, D.C.; Ollikainen, N.; Trinidad, J.C.; Cary, M.P.; Burlingame, A.L.; Kenyon, C. Widespread protein aggregation as an inherent part of aging in C. elegans. PLoS Biol 2010, 8, e1000450. [Google Scholar] [CrossRef]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int J Mol Med 2019, 44, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.J.; Merlot, A.M.; Huang, M.L.; Bae, D.H.; Jansson, P.J.; Sahni, S.; Kalinowski, D.S.; Richardson, D.R. Cellular iron uptake, trafficking and metabolism: Key molecules and mechanisms and their roles in disease. Biochimica et biophysica acta 2015, 1853, 1130–1144. [Google Scholar] [CrossRef]
- Correia, S.C.; Moreira, P.I. Oxygen Sensing and Signaling in Alzheimer's Disease: A Breathtaking Story! Cell Mol Neurobiol 2022, 42, 3–21. [Google Scholar] [CrossRef] [PubMed]
- Oresic, M.; Hyotylainen, T.; Herukka, S.K.; Sysi-Aho, M.; Mattila, I.; Seppanan-Laakso, T.; Julkunen, V.; Gopalacharyulu, P.V.; Hallikainen, M.; Koikkalainen, J.; et al. Metabolome in progression to Alzheimer's disease. Translational psychiatry 2011, 1, e57. [Google Scholar] [CrossRef] [PubMed]
- Guglielmotto, M.; Tamagno, E.; Danni, O. Oxidative stress and hypoxia contribute to Alzheimer's disease pathogenesis: two sides of the same coin. ScientificWorldJournal 2009, 9, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Guglielmotto, M.; Aragno, M.; Autelli, R.; Giliberto, L.; Novo, E.; Colombatto, S.; Danni, O.; Parola, M.; Smith, M.A.; Perry, G.; et al. The up-regulation of BACE1 mediated by hypoxia and ischemic injury: role of oxidative stress and HIF1alpha. Journal of neurochemistry 2009, 108, 1045–1056. [Google Scholar] [CrossRef] [PubMed]
- Tamagno, E.; Guglielmotto, M.; Aragno, M.; Borghi, R.; Autelli, R.; Giliberto, L.; Muraca, G.; Danni, O.; Zhu, X.; Smith, M.A.; et al. Oxidative stress activates a positive feedback between the gamma- and beta-secretase cleavages of the beta-amyloid precursor protein. Journal of neurochemistry 2008, 104, 683–695. [Google Scholar] [CrossRef]
- Kontush, A.; Donarski, N.; Beisiegel, U. Resistance of human cerebrospinal fluid to in vitro oxidation is directly related to its amyloid-beta content. Free Radic Res 2001, 35, 507–517. [Google Scholar] [CrossRef]
- Kontush, A. Amyloid-beta: an antioxidant that becomes a pro-oxidant and critically contributes to Alzheimer's disease. Free Radic Biol Med 2001, 31, 1120–1131. [Google Scholar] [CrossRef]
- Smith, M.A.; Casadesus, G.; Joseph, J.A.; Perry, G. Amyloid-beta and tau serve antioxidant functions in the aging and Alzheimer brain. Free Radic Biol Med 2002, 33, 1194–1199. [Google Scholar] [CrossRef]
- Nadal, R.C.; Rigby, S.E.; Viles, J.H. Amyloid beta-Cu2+ complexes in both monomeric and fibrillar forms do not generate H2O2 catalytically but quench hydroxyl radicals. Biochemistry 2008, 47, 11653–11664. [Google Scholar] [CrossRef] [PubMed]
- Baruch-Suchodolsky, R.; Fischer, B. Abeta40, either soluble or aggregated, is a remarkably potent antioxidant in cell-free oxidative systems. Biochemistry 2009, 48, 4354–4370. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Pan, Y.; Kao, S.Y.; Li, C.; Kohane, I.; Chan, J.; Yankner, B.A. Gene regulation and DNA damage in the ageing human brain. Nature 2004, 429, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Iturria-Medina, Y.; Sotero, R.C.; Toussaint, P.J.; Mateos-Perez, J.M.; Evans, A.C.; Alzheimer's Disease Neuroimaging, I. Early role of vascular dysregulation on late-onset Alzheimer's disease based on multifactorial data-driven analysis. Nature communications 2016, 7, 11934. [Google Scholar] [CrossRef] [PubMed]
- Leenders, K.L.; Perani, D.; Lammertsma, A.A.; Heather, J.D.; Buckingham, P.; Healy, M.J.; Gibbs, J.M.; Wise, R.J.; Hatazawa, J.; Herold, S.; et al. Cerebral blood flow, blood volume and oxygen utilization. Normal values and effect of age. Brain : a journal of neurology. [CrossRef]
- Braz, I.D.; Fisher, J.P. The impact of age on cerebral perfusion, oxygenation and metabolism during exercise in humans. J Physiol 2016, 594, 4471–4483. [Google Scholar] [CrossRef]
- Solleiro-Villavicencio, H.; Rivas-Arancibia, S. Effect of Chronic Oxidative Stress on Neuroinflammatory Response Mediated by CD4(+)T Cells in Neurodegenerative Diseases. Front Cell Neurosci 2018, 12, 114. [Google Scholar] [CrossRef]
- Guay-Gagnon, M.; Vat, S.; Forget, M.F.; Tremblay-Gravel, M.; Ducharme, S.; Nguyen, Q.D.; Desmarais, P. Sleep apnea and the risk of dementia: A systematic review and meta-analysis. J Sleep Res 2022, 31, e13589. [Google Scholar] [CrossRef]
- Swinford, C.G.; Risacher, S.L.; Wu, Y.C.; Apostolova, L.G.; Gao, S.; Bice, P.J.; Saykin, A.J. Altered cerebral blood flow in older adults with Alzheimer's disease: a systematic review. Brain Imaging Behav, 1007. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.X. Decreased glucose transporters correlate to abnormal hyperphosphorylation of tau in Alzheimer disease. FEBS Lett 2008, 582, 359–364. [Google Scholar] [CrossRef]
- Qian, L.; Rawashdeh, O.; Kasas, L.; Milne, M.R.; Garner, N.; Sankorrakul, K.; Marks, N.; Dean, M.W.; Kim, P.R.; Sharma, A.; et al. Cholinergic basal forebrain degeneration due to sleep-disordered breathing exacerbates pathology in a mouse model of Alzheimer’s disease. Nature communications 2022, 13, 6543. [Google Scholar] [CrossRef]
- Yeo, T.T.; Chua-Couzens, J.; Butcher, L.L.; Bredesen, D.E.; Cooper, J.D.; Valletta, J.S.; Mobley, W.C.; Longo, F.M. Absence of p75NTR causes increased basal forebrain cholinergic neuron size, choline acetyltransferase activity, and target innervation. The Journal of neuroscience : the official journal of the Society for Neuroscience 1997, 17, 7594–7605. [Google Scholar] [CrossRef]
- Whitehouse, P.J.; Price, D.L.; Struble, R.G.; Clark, A.W.; Coyle, J.T.; Delon, M.R. Alzheimer's disease and senile dementia: loss of neurons in the basal forebrain. Science 1982, 215, 1237–1239. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Lee, H.G.; Perry, G.; Smith, M.A. Alzheimer disease, the two-hit hypothesis: an update. Biochimica et biophysica acta 2007, 1772, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Burns, K.A.; Ayoub, A.E.; Breunig, J.J.; Adhami, F.; Weng, W.L.; Colbert, M.C.; Rakic, P.; Kuan, C.Y. Nestin-CreER mice reveal DNA synthesis by nonapoptotic neurons following cerebral ischemia hypoxia. Cerebral cortex 2007, 17, 2585–2592. [Google Scholar] [CrossRef] [PubMed]
- Goda, N.; Ryan, H.E.; Khadivi, B.; McNulty, W.; Rickert, R.C.; Johnson, R.S. Hypoxia-inducible factor 1alpha is essential for cell cycle arrest during hypoxia. Molecular and cellular biology 2003, 23, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.D.; Tan, E.K. Iron regulatory protein (IRP)-iron responsive element (IRE) signaling pathway in human neurodegenerative diseases. Molecular neurodegeneration 2017, 12, 75. [Google Scholar] [CrossRef] [PubMed]
- Hin, N.; Newman, M.; Pederson, S.; Lardelli, M. Iron Responsive Element-Mediated Responses to Iron Dyshomeostasis in Alzheimer’s Disease. Journal of Alzheimer's Disease 2021, 84, 1597–1630. [Google Scholar] [CrossRef]
- Tsatsanis, A.; Wong, B.X.; Gunn, A.P.; Ayton, S.; Bush, A.I.; Devos, D.; Duce, J.A. Amyloidogenic processing of Alzheimer's disease beta-amyloid precursor protein induces cellular iron retention. Molecular psychiatry 2020, 25, 1958–1966. [Google Scholar] [CrossRef]
- Pigoni, M.; Wanngren, J.; Kuhn, P.H.; Munro, K.M.; Gunnersen, J.M.; Takeshima, H.; Feederle, R.; Voytyuk, I.; De Strooper, B.; Levasseur, M.D.; et al. Seizure protein 6 and its homolog seizure 6-like protein are physiological substrates of BACE1 in neurons. Molecular neurodegeneration 2016, 11, 67. [Google Scholar] [CrossRef]
- Smith, M.A.; Perry, G. Free radical damage, iron, and Alzheimer's disease. Journal of the neurological sciences 1995, 134 Suppl, 92–94. [Google Scholar] [CrossRef]
- Schubert, D.; Chevion, M. The role of iron in beta amyloid toxicity. Biochemical and biophysical research communications 1995, 216, 702–707. [Google Scholar] [CrossRef]
- Mullner, E.W.; Kuhn, L.C. A stem-loop in the 3' untranslated region mediates iron-dependent regulation of transferrin receptor mRNA stability in the cytoplasm. Cell 1988, 53, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Casey, J.L.; Hentze, M.W.; Koeller, D.M.; Caughman, S.W.; Rouault, T.A.; Klausner, R.D.; Harford, J.B. Iron-responsive elements: regulatory RNA sequences that control mRNA levels and translation. Science 1988, 240, 924–928. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Cleavage of APP by α- or β- then γ-secretases. β-secretase cleavage creates secreted APPβ (sAPPβ) and βCTF. βCTF is subsequently cleaved by γ-secretase to form Aβ and the APP intracellular domain (AICD) that can be imported into the nucleus to regulate transcription.
Figure 1.
Cleavage of APP by α- or β- then γ-secretases. β-secretase cleavage creates secreted APPβ (sAPPβ) and βCTF. βCTF is subsequently cleaved by γ-secretase to form Aβ and the APP intracellular domain (AICD) that can be imported into the nucleus to regulate transcription.
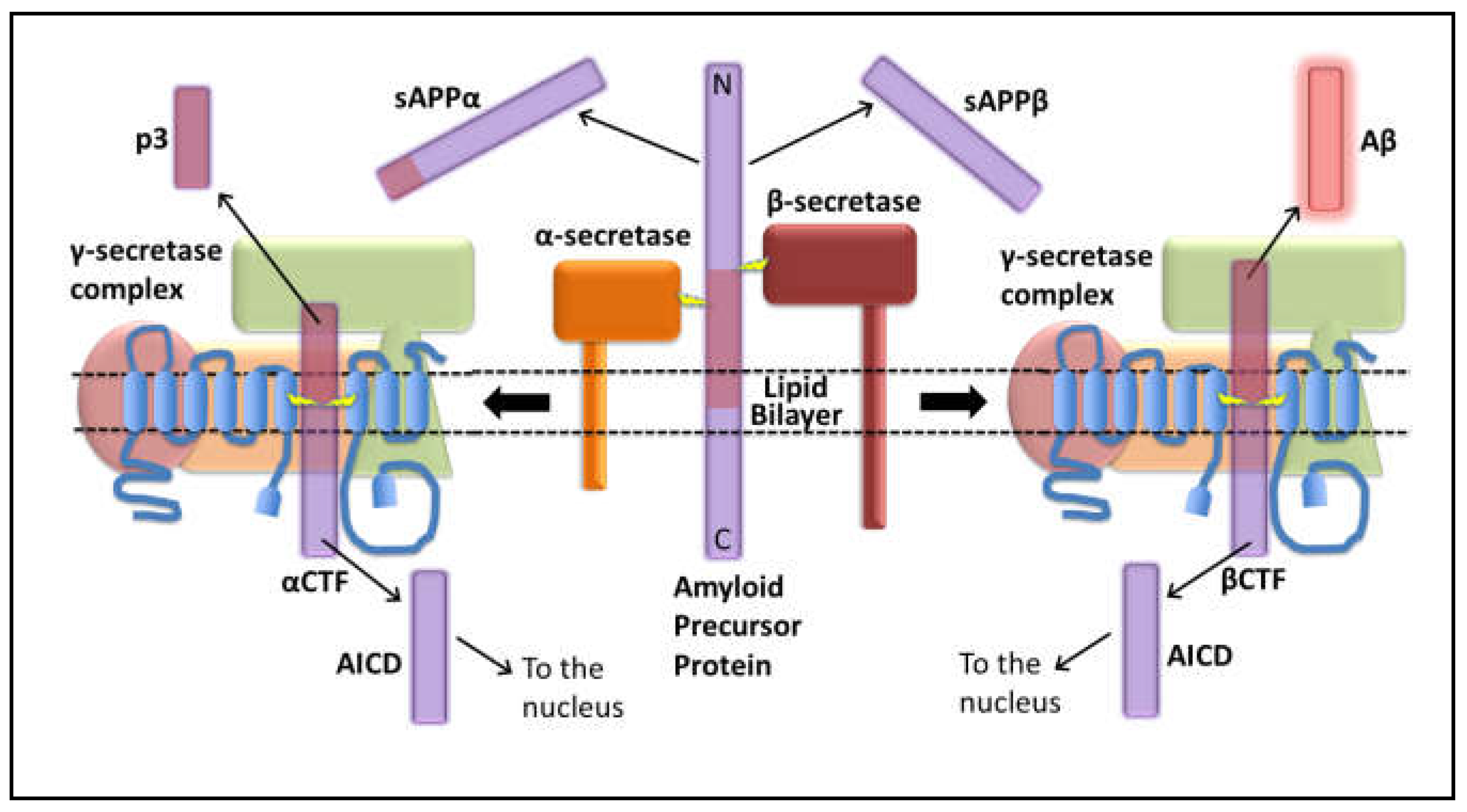
Figure 2.
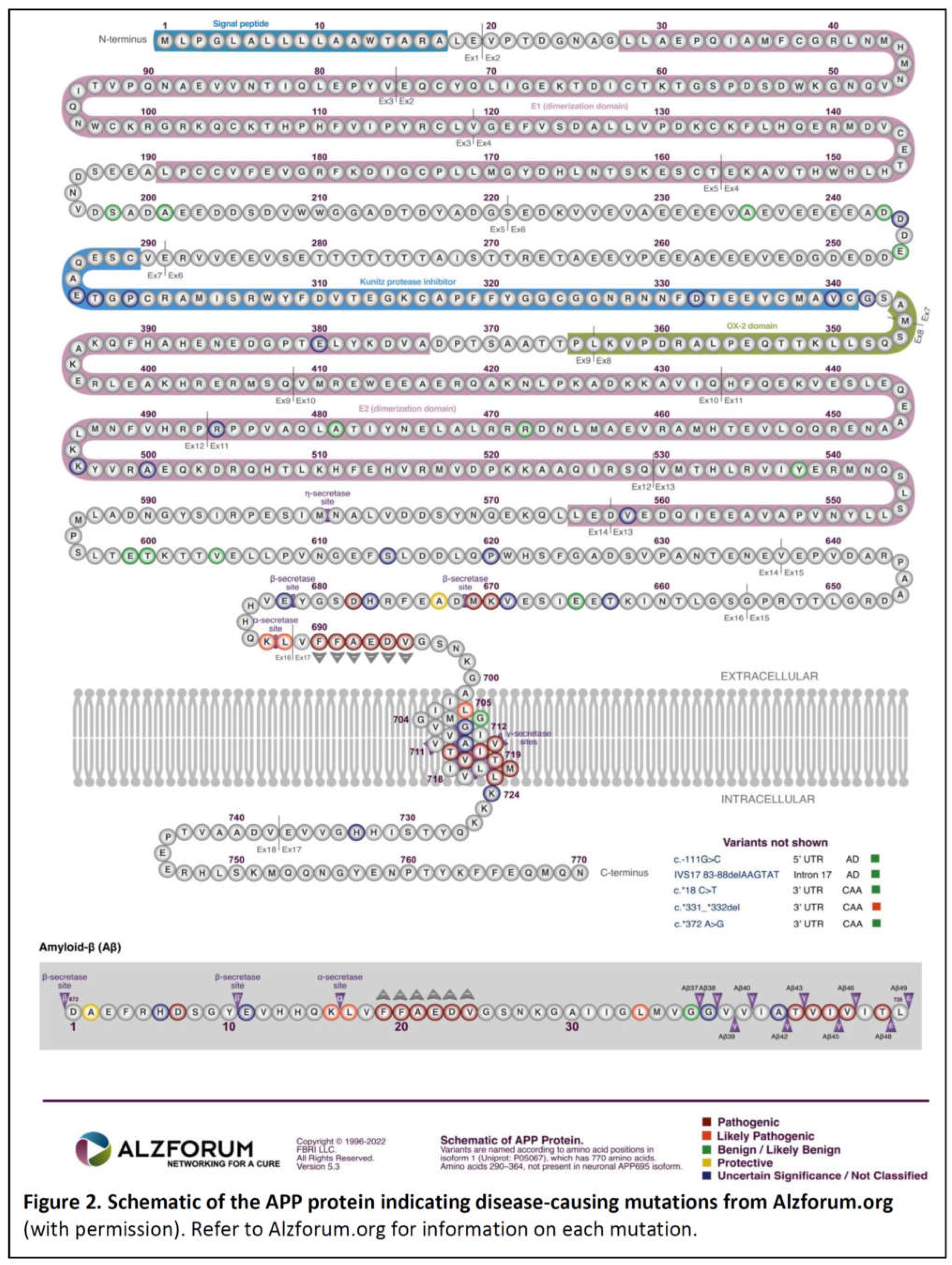
Figure 3.
γ-secretase cleavage of APP’s βCTF to generate Aβ. The entirety of βCTF is not shown. The figure was generated using information from [2]. The two alternative “product lines” for Aβ are shown. Less intense colours indicate lower relative prevalence. Grey and unfilled circles indicate transmembrane domain amino acid residues.
Figure 3.
γ-secretase cleavage of APP’s βCTF to generate Aβ. The entirety of βCTF is not shown. The figure was generated using information from [2]. The two alternative “product lines” for Aβ are shown. Less intense colours indicate lower relative prevalence. Grey and unfilled circles indicate transmembrane domain amino acid residues.
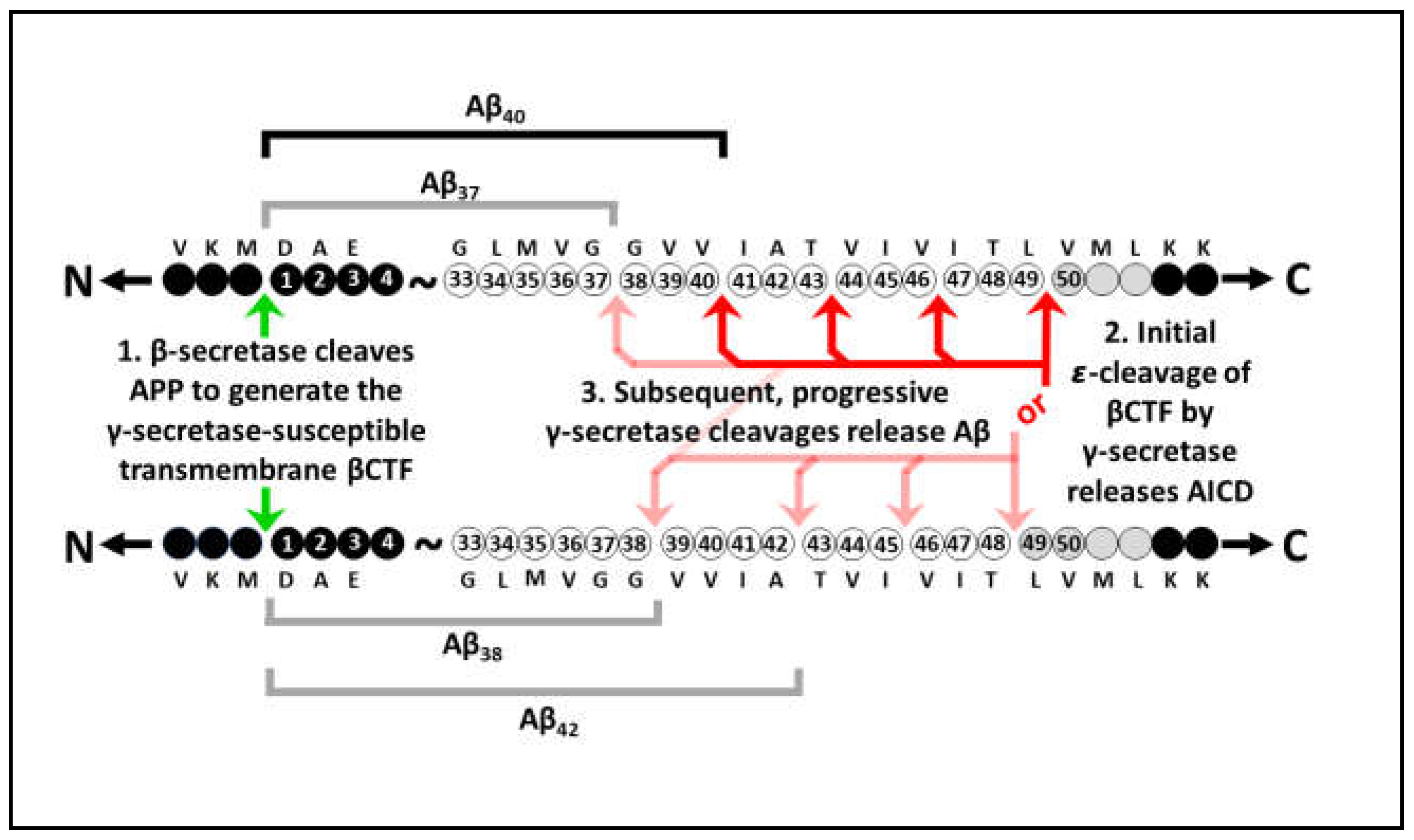
Figure 4.
Age of disease onset for fAD mutations in APP (modified from Lumsden et al. [1]). Secretase cleavage sites are indicted in yellow. “X” indicates reported fAD onset ages of specific individuals. Onset ages for duplications of the entire APP gene are shown on the left.
Figure 4.
Age of disease onset for fAD mutations in APP (modified from Lumsden et al. [1]). Secretase cleavage sites are indicted in yellow. “X” indicates reported fAD onset ages of specific individuals. Onset ages for duplications of the entire APP gene are shown on the left.
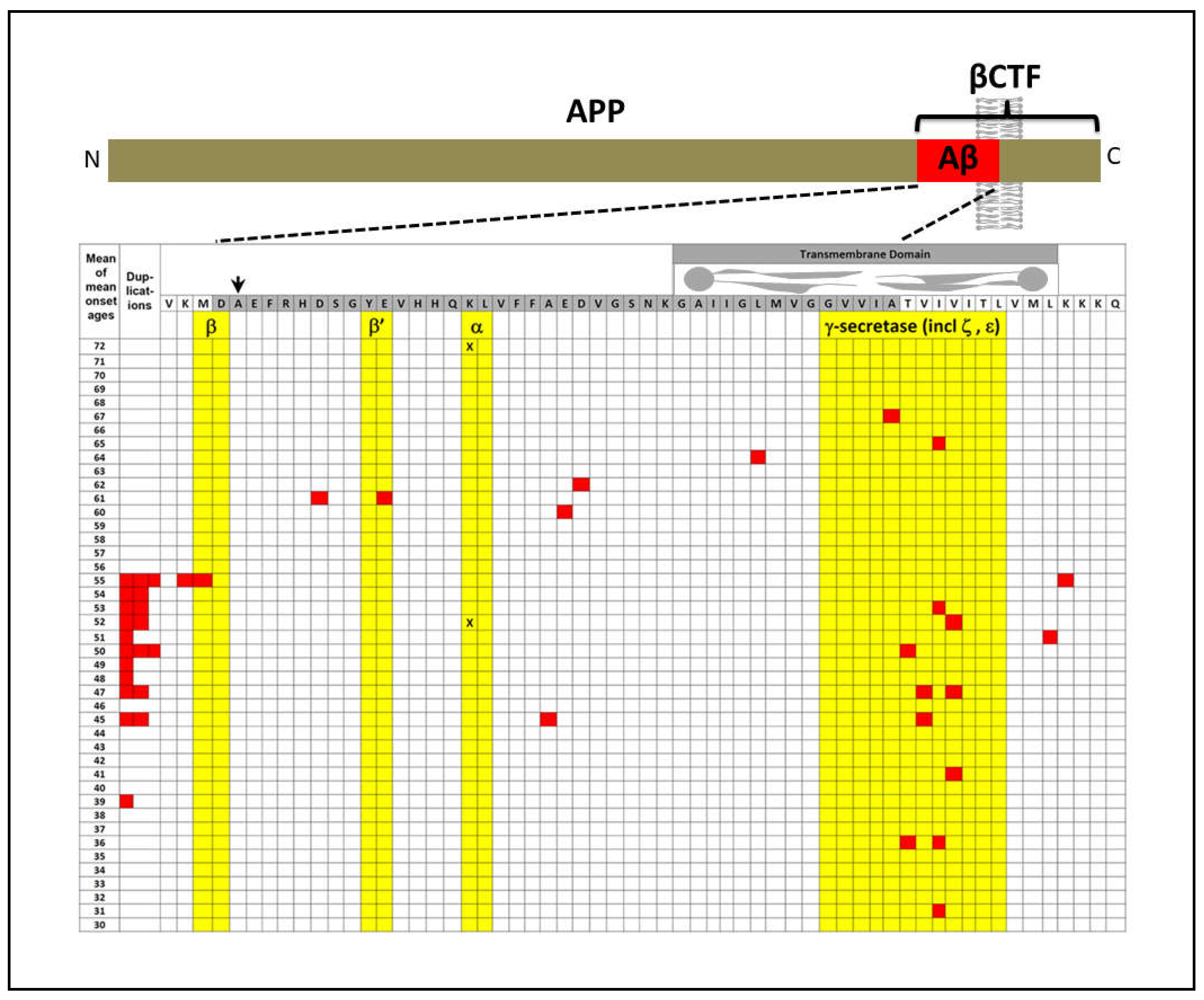
Figure 5.
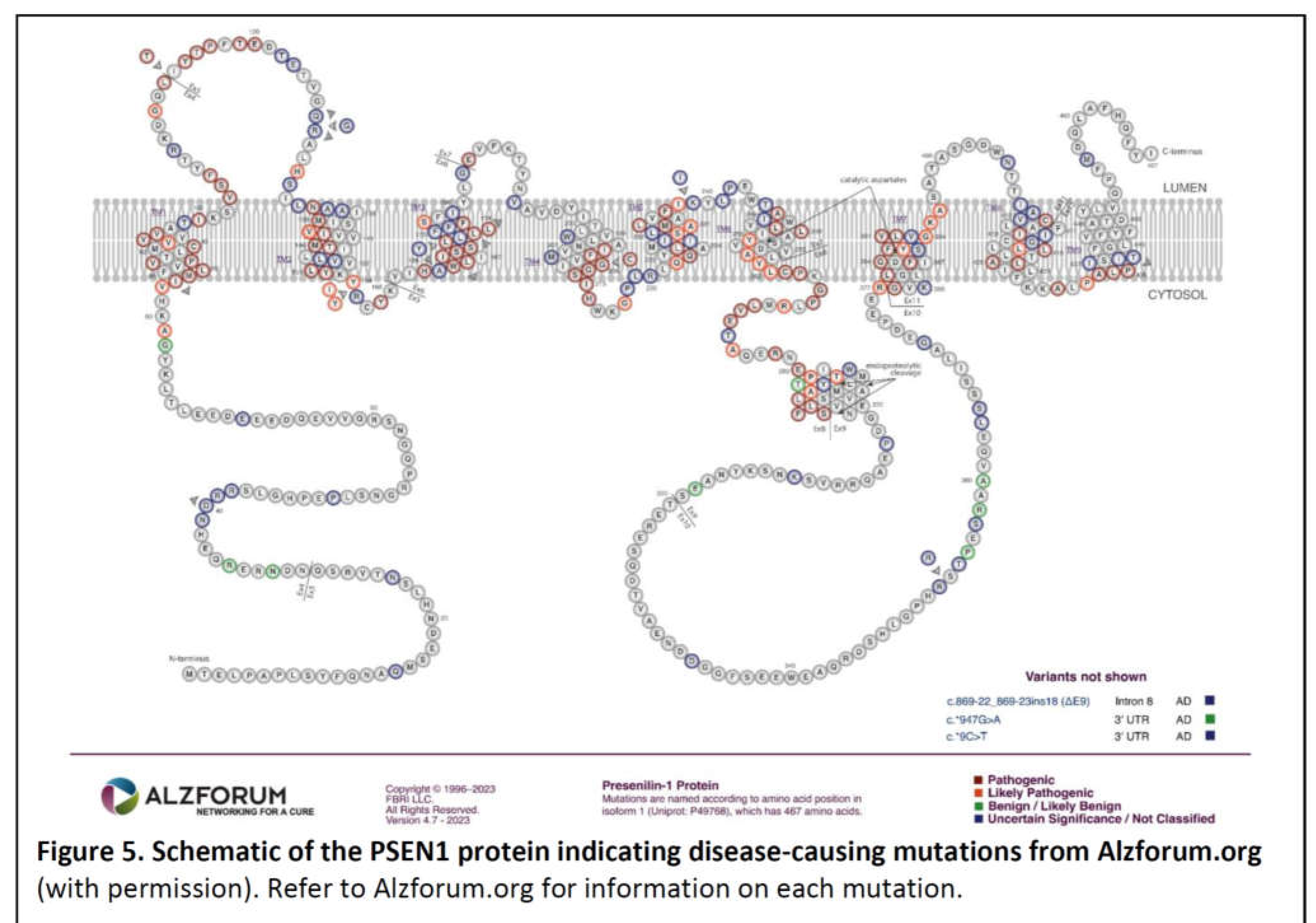
Figure 6.
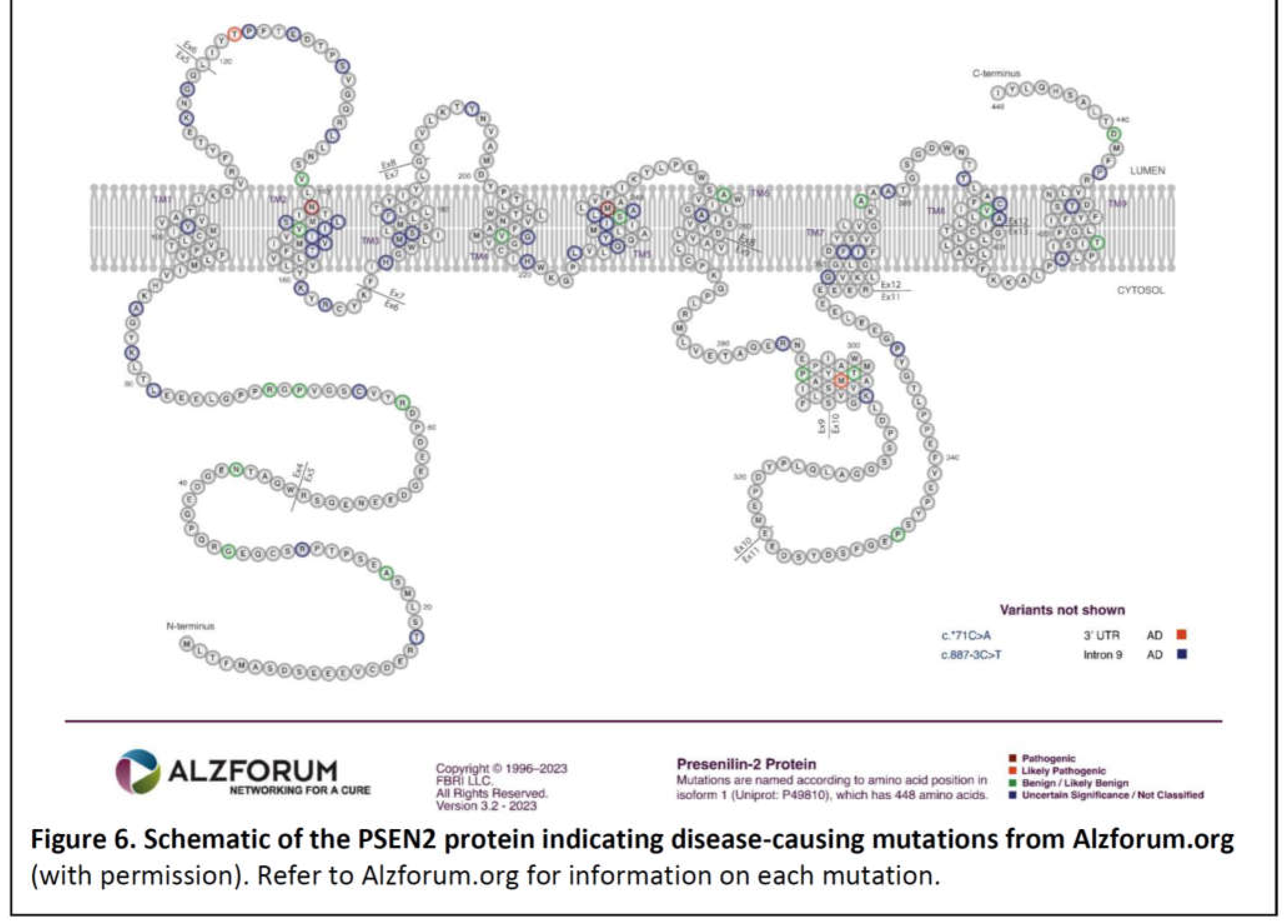
Figure 7.
γ-secretase complex assembly, activation, and action. NCSTN, PSENEN (PEN-2), and APH1A/B assemble with the presenilin holoprotein. Auto-endoproteolysis occurs which activates the ability to endoproteolyse other γ-secretase substrates.
Figure 7.
γ-secretase complex assembly, activation, and action. NCSTN, PSENEN (PEN-2), and APH1A/B assemble with the presenilin holoprotein. Auto-endoproteolysis occurs which activates the ability to endoproteolyse other γ-secretase substrates.
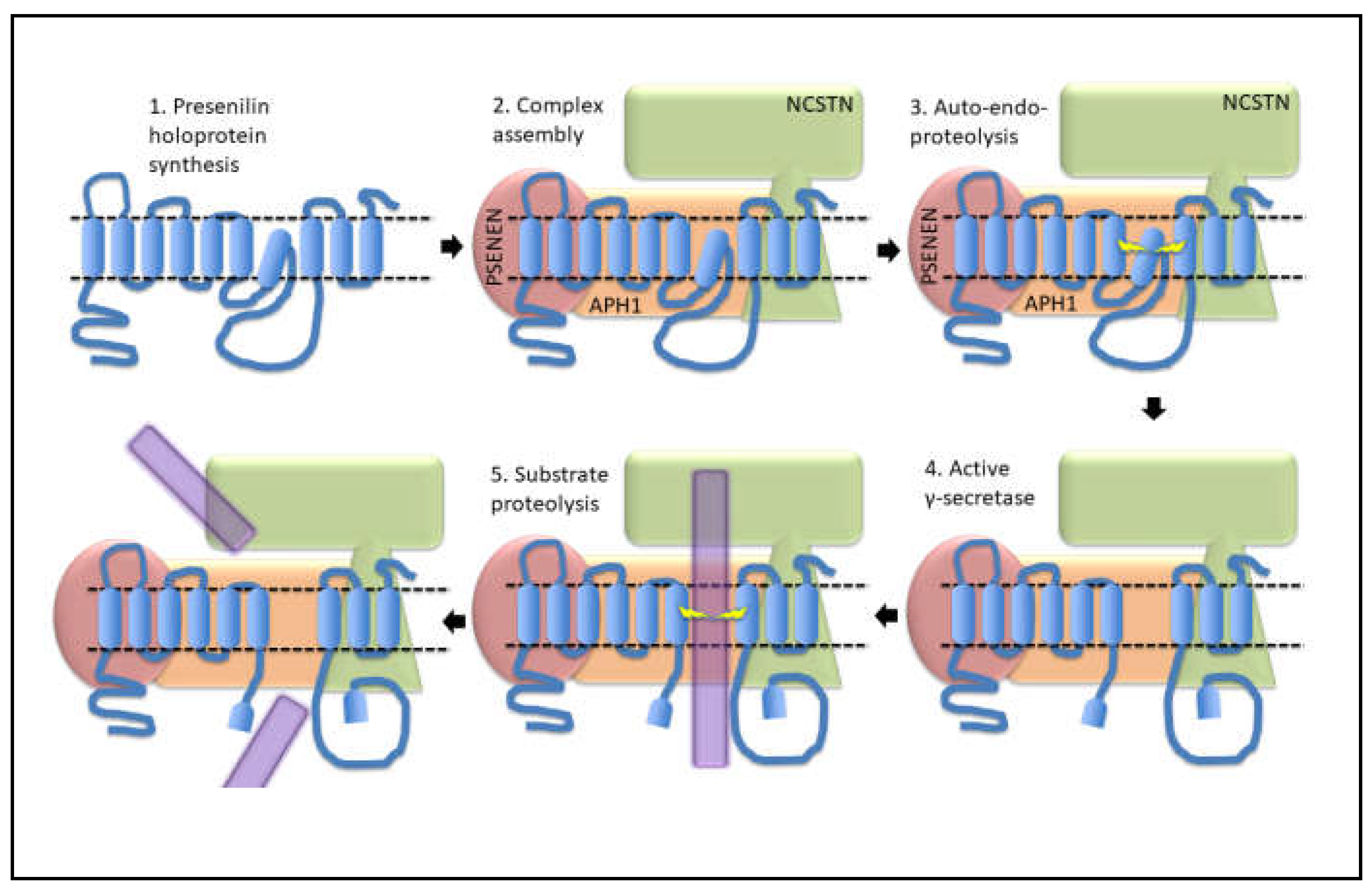
Figure 8.
fAD mutations in the presenilins and in APP both affect acidification of the lysosome with consequences for iron importation, the availability of Fe2+ ions, and recycling (for Fe2+ recovery through autophagy) of mitochondria and of Fe3+ stored in ferritin.
Figure 8.
fAD mutations in the presenilins and in APP both affect acidification of the lysosome with consequences for iron importation, the availability of Fe2+ ions, and recycling (for Fe2+ recovery through autophagy) of mitochondria and of Fe3+ stored in ferritin.
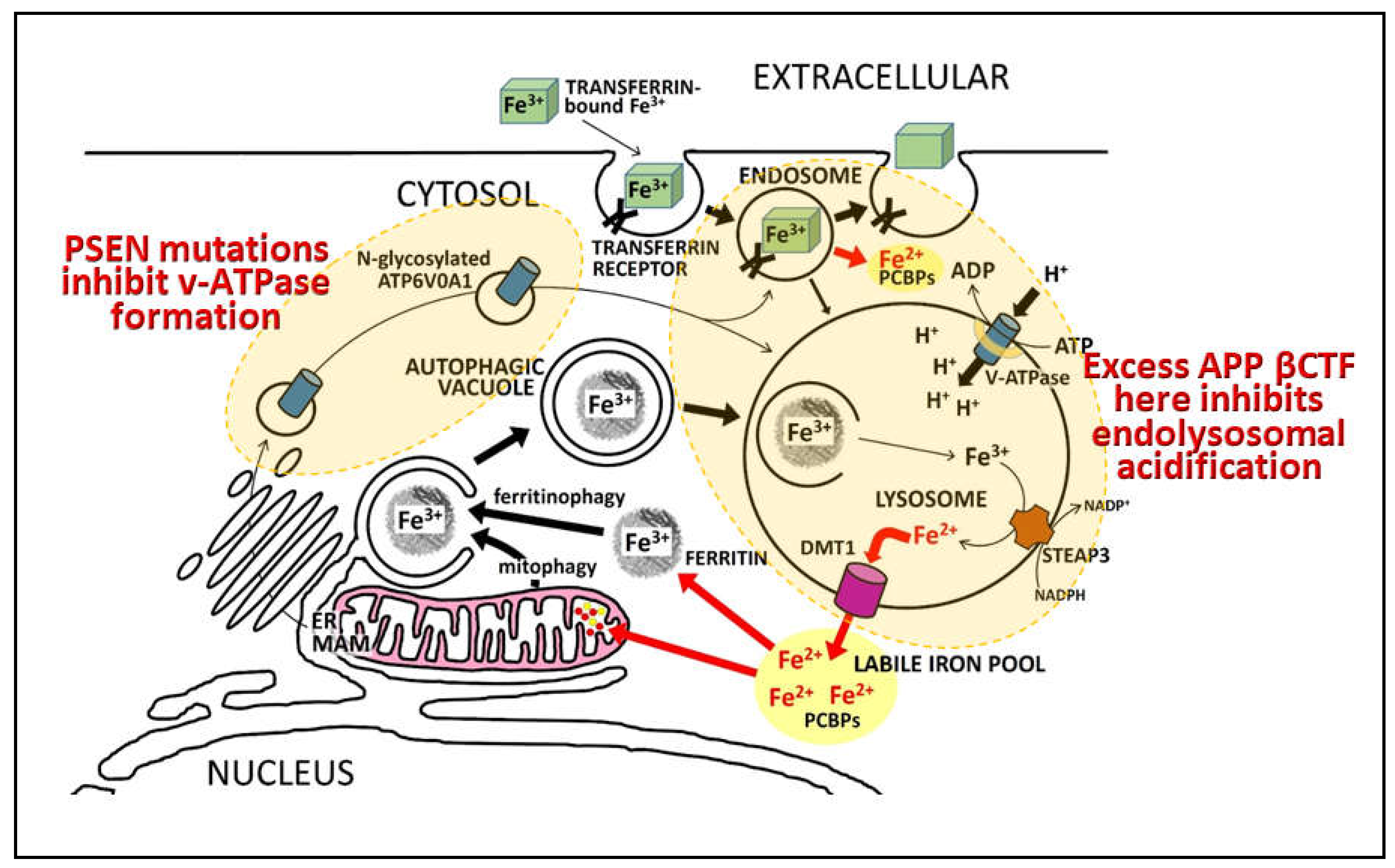
Figure 9.
A multimerisation model for how heterozygosity for fAD mutations in the presenilins may increase γ-secretase activity by destabilising multimers of holoproteins or even not-yet-active γ-secretase complexes to give excessive γ-secretase activation.
Figure 9.
A multimerisation model for how heterozygosity for fAD mutations in the presenilins may increase γ-secretase activity by destabilising multimers of holoproteins or even not-yet-active γ-secretase complexes to give excessive γ-secretase activation.
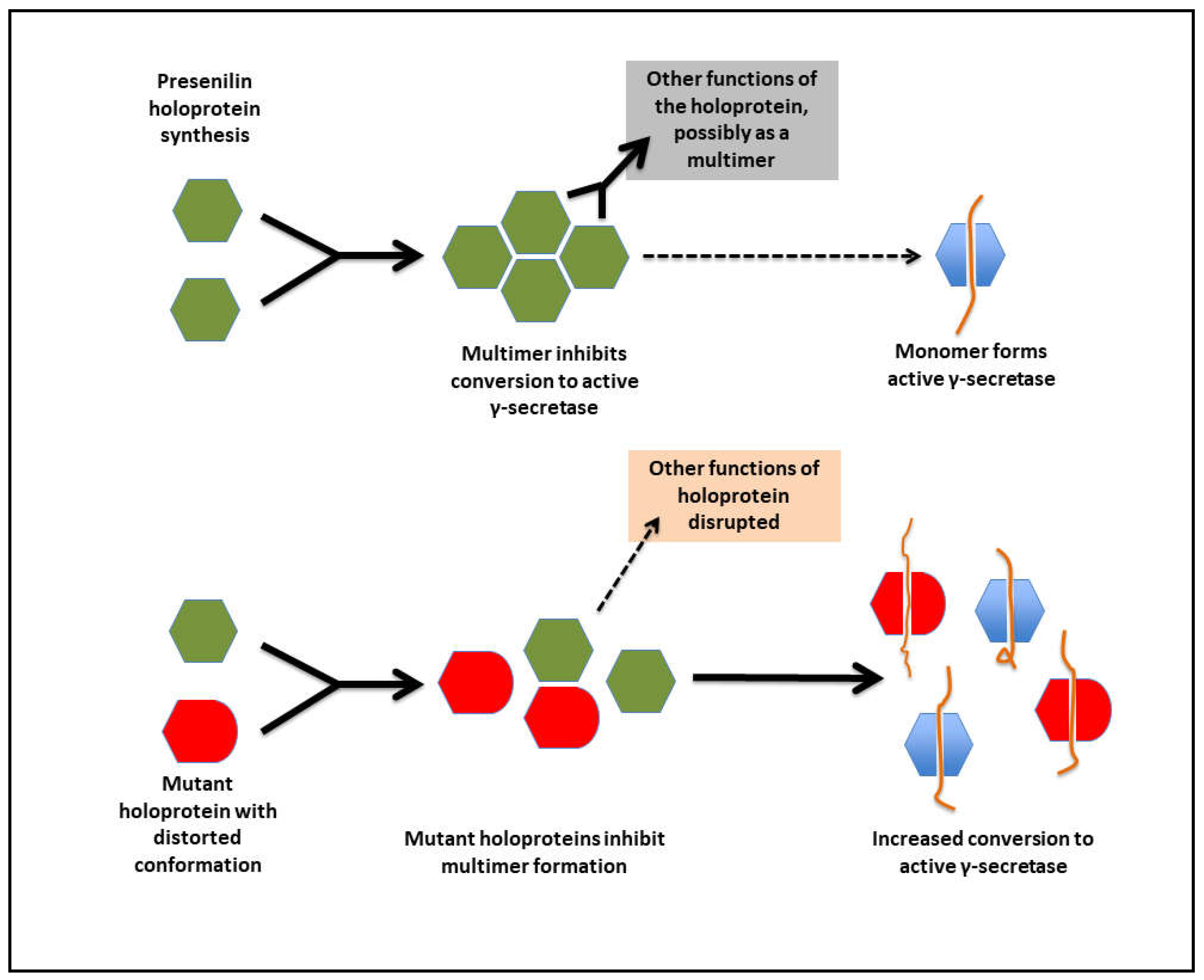
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



