Submitted:
03 March 2023
Posted:
09 March 2023
You are already at the latest version
Abstract
It is important to be familiar with the presentation of Turner syndrome and to understand the common causes of death associated with the disease. Using the autopsy case as an example, this case report will outline the classic presentation of Turner syndrome, go over its physiology, epidemiology, pathogenesis and explain the cause of death in this particular case while also highlighting other common causes of death/risk factors within Turner syndrome. Differential diagnosis and mimicking disease processes will also be discussed. This report will also highlight the molecular tests used for the diagnosis of Turner syndrome, discuss developments using in situ hybridization, and discuss why this method is best for the determination of Turner syndrome over conventional karyotyping.
Keywords:
Turner Syndrome
; Autopsy Case Report
History
A 46-year-old female was found unresponsive lying supine in her bed by her husband. Four days prior, the deceased started complaining of symptoms of vomiting and diarrhea which was attributed to food poisoning. Two days later she saw her doctor who prescribed her ondansetron for her vomiting symptoms. That same day she communicated to her husband that she was experiencing “pressure in her ears.” The night of her death, she took some Pepto Bismol® and went to bed at 0200 hours. When the husband went to bed next to her at 0600 hours, she was cold and unresponsive. He then promptly called 911.
Emergency responders found no vital signs and pronounced her dead at the scene (Figure 1). The medicolegal death investigator was consulted and responded to the scene to conduct an examination. No signs of trauma, abuse, neglect, or suspicion of foul-play were noted. There were no reported recent injuries, falls, or hospitalizations. It was revealed to the investigator that the patient had Turner syndrome.
Post-mortem external examination revealed a white, well-developed, female measuring 63 inches in length with a weight of 150 lb. At the time of examination, the body was well-preserved with rigor mortis and purple livor mortis covering the back which blanched upon manual pressure. The head and neck were unremarkable with the exception that the neck was wide and webbed in appearance (Figure 2). The torso had a horizontally-oriented 7.5 inch linear scar at the left mid axillary line. Inferior to this scar was a smaller 1 inch linear scar. Other then those findings the torso was unremarkable. The arms had multiple trivial scars with no injuries. The arms and hands were otherwise unremarkable. Superiorly, the lateral aspect of the right thigh had a 1.5 x 1.5 inch abraded contusion. The lateral aspect of the right knee had a 0.25 inch blue contusion.
CT scan of the body revealed bilateral opacification of the lungs with fluid in the chest cavities, blood in the pericardial sac, hyperdense material in the gastrointestinal tract, and air in the blood vessels of the liver (Figure 3).
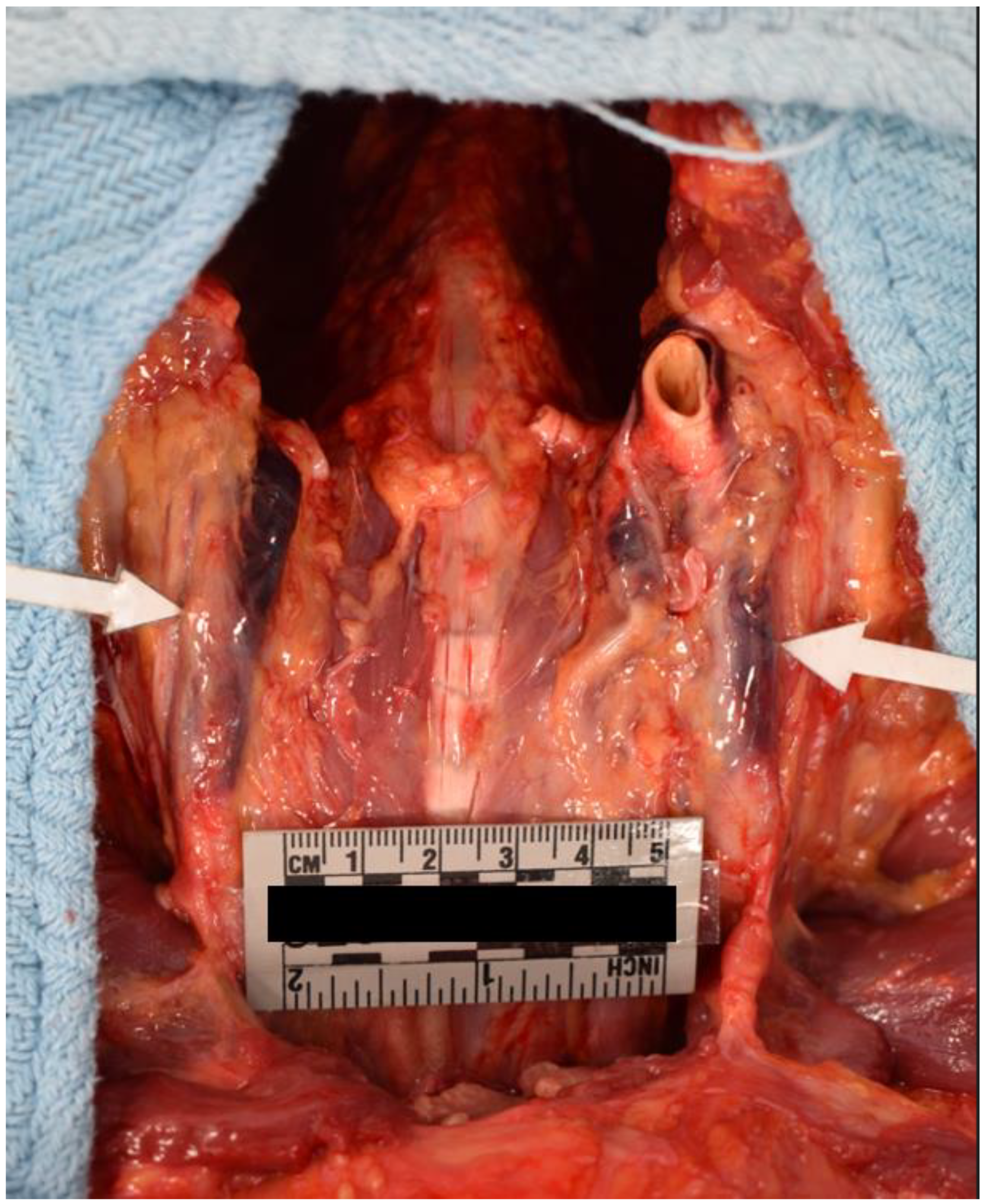
Internal examination of the body cavities revealed that the right pleural cavity contained 300 mL of serosanguineous fluid, and the left pleural cavity contained 200 mL of serosanguineous fluid. The left pleural cavity also had mild fibrous adhesions. The pericardial cavity contained 450 mL of blood with clots. The peritoneal cavity contained 250 mL of serous fluid. The lungs were both found to exhibit mild emphysematous changes with blebs of the right lower lung lobe. Internal examination of the cardiovascular system revealed left dominant circulation with coronary arteries following their normal distribution. The heart was 375 grams, and serial sections revealed no abnormalities, infarcts, cardiomegaly, thrombi, or dilations. The heart contained an aortic valve which was found to be bicuspid with thin pliant leaflets (Figure 4). The remaining heart valves were also thin and pliant but normal in form. The aorta had a 1.2 cm intimal defect within the ascending portion which was located 1.6 cm above the aortic valve (Figure 5). The ascending aorta had a circumference of 10 cm. Additionally a 2.1 cm adventitial defect was noted approximately 4 cm above the aortic valve (Figure 5). A suture line was noted of the descending aorta distal to the left subclavian artery; the anastomosis was intact. The aorta was free of atheroma (Figure 6). Blood was found to be within the aortic wall, tracking superiorly along both carotid arteries (Figure 7). The reflected scalp had a 2.5 x 2 cm area of transmural hemorrhage with underlying 3 x 2 cm subgaleal hemorrhage of the right posterior parietal aspect. The left occipital aspect had a 2 x 2 cm area of subgaleal hemorrhage. The ovaries were difficult to identify and were approximately 2 cm long and 0.5 cm in width. The ovaries displayed a fibrous cut surface. Lastly, the thyroid was found to consist of atrophic white tissue free of cyst and mass. Microscopic examination confirmed dissection with aortic sections revealing blood within the vessel wall. The myocardium sections were free of inflammatory infiltrate and necrosis. The rest of the internal findings were unremarkable and within the range of normal. Toxicology screen was negative. Based on the exam findings and medical history, the cause of death was determined to be hemopericardium due to rupture of the ascending aortic dissection, which was due to Turner syndrome. The manner of death was deemed natural.
Turner Syndrome
Turner syndrome (aka congenital ovarian hypoplasia syndrome) is a sex chromosome abnormality affecting females. It is associated with the full or partial loss of an X chromosome leading to karyotypes of 45,XO or 45,XO with mosaicism of cells that are 46,XX or 46,XY 1. The disease was first classified in 1938; it is the most common sex chromosome abnormality affecting approximately 3% of all females conceived 2. Amongst liveborn females, the estimated rate of Turner syndrome ranges from 1 in 1500 to 1 in 2500 2. The true proportion of patients with Turner syndrome remains unknown due to the range of presentation of the disease, which is due to the extent of mosaicism in patients; causing those with minor symptoms to go undiagnosed 3.
The genetic alteration behind Turner syndrome is not familial. In early embryonic development a sporadic chromosomal nondisjunction occurs. Depending on the point in embryonic development that this even occurs, it may affect all progeny cells leading to a monosomy (45,XO) or a subset of cells whose progeny is 45,XO mixed with a cohort of normal cells (46,XX or 46, XY) which generates a mosaicism 4.
The possibility of monosomy or mosaicism with consideration then of the percentage of mosaicism leads to a wide range of symptoms. Common presentations include short stature, broad “shield” shaped chest, widely spaced nipples, a webbed neck, infertility, atrophic “streaked” ovaries (containing mostly connective tissue), cubitus valgus, delayed puberty, reduced estrogen levels, hypertension, elevation of liver enzymes, cirrhosis of the liver, micrognathia, and renal malformations (horseshoe kidneys) or positional abnormalities 4–6. Importantly, there are serious congenital cardiovascular malformations associated with Turner syndrome such as coarctation of the aorta, bicuspid aortic valves, aortic dissection, elongation of the aortic arch, and mitral valve prolapse 6.
Furthermore, family history may reveal patients with possible learning disabilities, primary amenorrhea, premature ovarian failure, hearing loss due to frequent bouts of otitis media, or defects in the cochlea leading to sensorineural hearing loss. Patients may also have a history of ocular problems including nearsightedness or farsightedness, colorblindness, ptosis and/or strabismus. Patients may also have a history of fractures and low bone density. Patients are also at risk for autoimmune disease such as celiac disease, autoimmune thyroiditis and inflammatory bowel disease. The patient may also have a history of taking growth hormone for short stature and estrogen to simulate normal pubertal progression 5,6. Additionally it had been found that patients with Turner syndrome who have streaked ovaries with 45,XO and 46,XY mosaicism demonstrated increased risk of developing gonadoblastoma 7.
Finally, the patient may have an extensive cardiac history including prolonged QT intervals, hypertension, MRI/ECHO assessment for cardiac abnormalities, coarctation of the aorta surgical management, aortic dissection management, aortic dilation management, blood pressure medication prescriptions including beta blockers, angiotensin-converting enzyme (ACE) inhibitors, calcium channel blockers, diuretics and/or angiotensin II receptor blockers 4,8.
In a forensic autopsy setting, it is important to note common causes of death due to Turner syndrome. A large cohort, 68 year-long retrospective review of Turner syndrome patients followed at the Mayo Clinic by Fuchs et al. demonstrated that the mean age of death in these patients were 53 +/- 17 years. Twenty-two percent (22%) of the causes of death were cardiac-related, 11% were due to malignancy, 9% were due to gastrointestinal bleeding, 4% were due to cirrhosis, 2% were due to end-stage renal failure, and 52% were unknown 9. Of the cardiac deaths, these were attributed to postoperative death after cardiovascular surgery [valve replacement, coronary artery bypass graft surgery (CABG), aortic arch and subclavian artery repair, or ascending aortic dissection], end stage heart failure, cardiac tamponade, aortic dissection, and sudden cardiac death 9. These findings echo previous studies reporting elevated rates of aortic dissection in patents with Turner syndrome 10.
Aortic dissection incidence in the above study by Fuchs et al. were calculated to be 1.9% with an average age of 40 years old 10. In comparison the average age of aortic dissection in the general population is 77 years old 10,11. As such for young patients who have suspected aortic dissection as a cause of death, Turner syndrome should be on the differential diagnosis list. In determining the etiology behind aortic dissection in Turner syndrome, it was found that in an assessment of 88 Turner syndrome patients who had an aortic dissection, there was an association between congenital heart disease (69% of the patients had coarctation of the aorta, bicuspid aortic valve or both), hypertension (54%), or both (75% of patients with hypertension also had a congenital heart disease) 12. Another factor which may correlate with the increased incidence of aortic dissection is the increased intima-media thickness and arterial stiffness observed amongst Turner syndrome patients. Interestingly, estrogen deficiency has been associated with both intimal thickening and is a well-known symptom of Turner syndrome highlighting a potential correlation 12,13.
Molecular Diagnosis
Blood lymphocyte karyotype of a minimum of 30 cells is the gold-standard method for the diagnosis of Turner syndrome 14. Importantly, karyotyping 30 cells has the possibility of missing cells with possible mosaicism creating an incorrect diagnosis of a 45,XO monosomy. Conversely, this assay may also only assess the mosaicism population 46,XX missing the diagnosis of Turner syndrome entirely. As such, cytogenetic studies of a second source of tissue (commonly taken from skin or a buccal smear) should be considered for patients who are suspected of Turner syndrome based on their presentation despite attaining a 46,XX karyotype 14. Additional cytogenetic studies should also be performed on a patient whose karyotype showed up as monosomy 45,XO and especially if the patient has virilizing features or clitoromegaly to assess for the presence of mosaicism of 46,XY 14.
Florescence in Situ hybridization (FISH) is the cytogenetic assay of choice, and probes for the X and Y centromeres should be performed on a minimum of 200 cells to detect low-level Y chromosome mosaicism 14. A recent study compared the rate of 45,XO cells comparatively using standard blood lymphocyte karyotyping and FISH analysis of buccal cells in 142 Turner syndrome patients 15. It was found that among 40 patients who were shown to have 45,XO monosomy via the blood lymphocyte test, FISH assessment of the buccal cells demonstrated that 20% of the patients were in fact 45,XO with 46,XX mosaicism highlighting the importance of preforming both assays to ensure correct diagnosis 15.
Differential Diagnosis
We have discussed the various phenotypes and presentations of Turner syndrome. There are many disease/genetic abnormalities such as Noonan syndrome and complete gonadal dysgenesis which can all resemble Turner syndrome and must be evaluated carefully. Noonan syndrome in particular can be easy to mistake as Turner syndrome due to shared presentations including: webbed neck, short stature, renal abnormalities, congenital heart disease, intellectual/developmental delay, strabismus, astigmatism, hearing issues, delayed puberty, and wide set nipples 16,17. Importantly, distinguishing features are that both males and females are affected by Noonan syndrome (as opposed to females only in Turner syndrome). Noonan syndrome has no chromosome abnormality, resulting in a normal karyotype assessment. The genetics behind the generation of Noonan syndrome is due to mutations in the RAS/MAPK signal pathway (frequently affecting PTPN11, SOS1, RAF1 or RIT1 genes) 16,18.
Complete gonadal dysgenesis is another mimicker of Turner syndrome. The defining feature of complete gonadal dysgenesis is bilateral streaked gonads in females 19. The patient would have a history of primary amenorrhea with an absence of secondary sexual characteristics or may be undergoing estrogen/progesterone therapy 19. This process is associated with sensorineural deafness 19. Importantly, these patients would have normal 46,XX karyotypes; the exact genetic abnormality is still unclear but there has been evidence to support that a potential cause is due to localized genetic alterations affecting the FSH receptor 20.
Case Conclusion
This case report discussed a fatal outcome of a patient with Turner syndrome. The presentation and pathologic findings of the decedent were consistent with classical findings expected in a patient with Turner syndrome. The cause of death, hemopericardium due to rupture of the dissected ascending aortic artery dissection, is a known cause of mortality amongst patients with Turner syndrome.
Summary
In a forensic autopsy setting it is important to be familiar with the presentation of Turner syndrome and to understand the common causes of death associated with the disease. Information about the decedent’s medical history may reveal Turner syndrome which will allow for common presentations, risk factors and potential causes of death to be considered before an autopsy even begins. However, it is important to keep differentials open and for the forensic pathologist to keep a critical eye during the autopsy as the presentation of Turner syndrome can vary widely, and there are known mimicking syndromes. Additionally, patients may be undiagnosed; knowing the common presentations and diagnostic methodology can allow Turner syndrome to be considered in challenging cases where the presentation matches but the medical history does not.
Author Contributions
This publication was written by Andrew Sulaiman the autopsy was performed by Dr. Diane Peterson who also wrote up the autopsy report and the manuscript was reviewed and edited by Dr. Christine James, Dr. Diane Peterson, Tyla Gelman and Andrew Sulaiman.
Funding
This research received no specific funding.
Ethical approval
The Johnson County Medical Examiner’s Office has direct jurisdiction to investigate deaths under K.S.A. 22a-231 and K.S.A. 22a-242a.
Informed Consent Statement
No explicit informed consent is required for autopsy case reports as all data has been anonymized.
Data Availability Statement
Data are available upon request.
Conflicts of Interest
The authors declare no conflict of interest
References
- Kesler SRJC, America APCoN. Turner syndrome. 2007;16(3):709-722. [CrossRef]
- Saenger PJNEJoM. Turner’s syndrome. 1996;335(23):1749-1754. [CrossRef]
- Gunther DF, Eugster E, Zagar AJ, Bryant CG, Davenport ML, Quigley CA. Ascertainment bias in Turner syndrome: new insights from girls who were diagnosed incidentally in prenatal life. Pediatrics. 2004;114(3):640-644. [CrossRef] [PubMed]
- Kikkeri NS, Nagalli S. Turner Syndrome. In: StatPearls [Internet]. StatPearls Publishing; 2021.
- Kesler SR. Turner syndrome. Child and adolescent psychiatric clinics of North America. 2007;16(3):709-722. [CrossRef] [PubMed]
- Ackermann A, Bamba V. Current controversies in turner syndrome: Genetic testing, assisted reproduction, and cardiovascular risks. Journal of clinical & translational endocrinology. 2014;1(3):61-65. [CrossRef] [PubMed]
- Brant WO, Rajimwale A, Lovell MA, et al. Gonadoblastoma and Turner syndrome. The Journal of urology. 2006;175(5):1858-1860. [CrossRef] [PubMed]
- Sandahl K, Wen J, Erlandsen M, Andersen NH, Gravholt CH. Natural History of Hypertension in Turner Syndrome During a 12-Year Pragmatic Interventional Study. 2020;76(5):1608-1615. [CrossRef]
- Fuchs MM, Attenhofer Jost C, Babovic-Vuksanovic D, Connolly HM, Egbe AJJotAHA. Long-term outcomes in patients with Turner syndrome: a 68-year follow-up. 2019;8(11):e011501. [CrossRef]
- Price W, Clayton J, Collyer S, De Mey R, Wilson JJJoE, Health C. Mortality ratios, life expectancy, and causes of death in patients with Turner’s syndrome. 1986;40(2):97-102. [CrossRef]
- Gravholt CH, Landin-Wilhelmsen K, Stochholm K, et al. Clinical and epidemiological description of aortic dissection in Turner’s syndrome. 2006;16(5):430-436. [CrossRef] [PubMed]
- Carlson M, Silberbach M. Dissection of the aorta in Turner syndrome: two cases and review of 85 cases in the literature. 2007;44(12):745-749. [CrossRef] [PubMed]
- Ostberg JE, Donald AE, Halcox JPJ, Storry C, McCarthy C, Conway GS. Vasculopathy in Turner Syndrome: Arterial Dilatation and Intimal Thickening without Endothelial Dysfunction. The Journal of Clinical Endocrinology & Metabolism. 2005;90(9):5161-5166. [CrossRef]
- Wolff DJ, Van Dyke DL, Powell CM, Committee AWGotALQA. Laboratory guideline for Turner syndrome. Genetics in Medicine. 2010;12(1):52-55. [CrossRef] [PubMed]
- Graff A, Donadille B, Morel H, et al. Added value of buccal cell FISH analysis in the diagnosis and management of Turner syndrome. Human Reproduction. 2020;35(10):2391-2398. [CrossRef]
- Allen MJ, Sharma S. Noonan Syndrome. 2018.
- Romano AA, Allanson JE, Dahlgren J, et al. Noonan syndrome: clinical features, diagnosis, and management guidelines. 2010;126(4):746-759. [CrossRef] [PubMed]
- Jorge AAL, Malaquias AC, Arnhold IJP, Mendonca BB. Noonan Syndrome and Related Disorders: A Review of Clinical Features and Mutations in Genes of the RAS/MAPK Pathway. Hormone Research in Paediatrics. 2009;71(4):185-193. [CrossRef] [PubMed]
- Breehl L, Caban O. Genetics, gonadal dysgenesis. In: StatPearls [Internet]. StatPearls Publishing; 2021.
- Aittomäki K, Lucena JL, Pakarinen P, et al. Mutation in the follicle-stimulating hormone receptor gene causes hereditary hypergonadotropic ovarian failure. Cell. 1995;82(6):959-968. [CrossRef] [PubMed]
Figure 1.
Decedent as found at the scene.
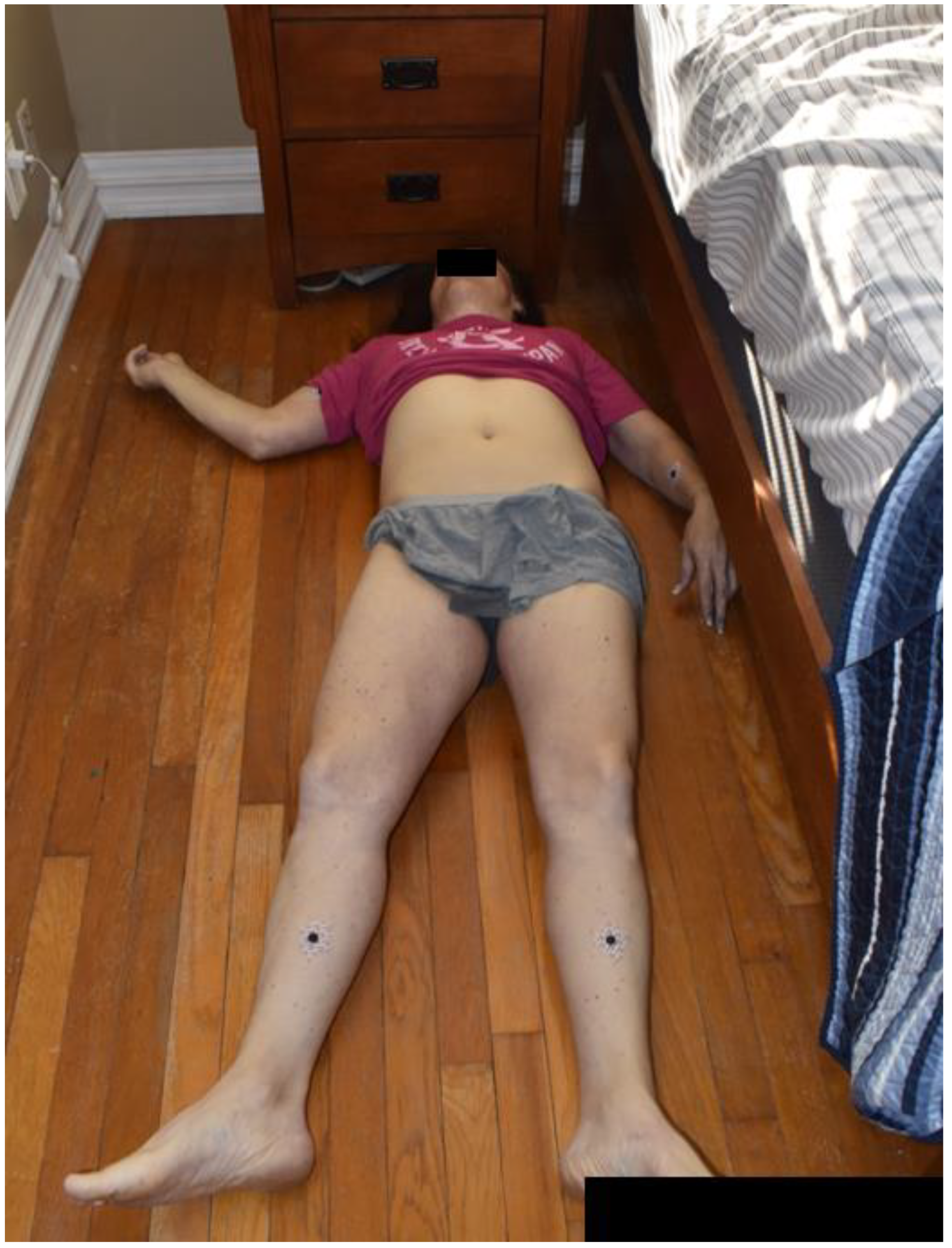
Figure 2.
Depiction of the Decedent’s Wide and Webbed Neck.
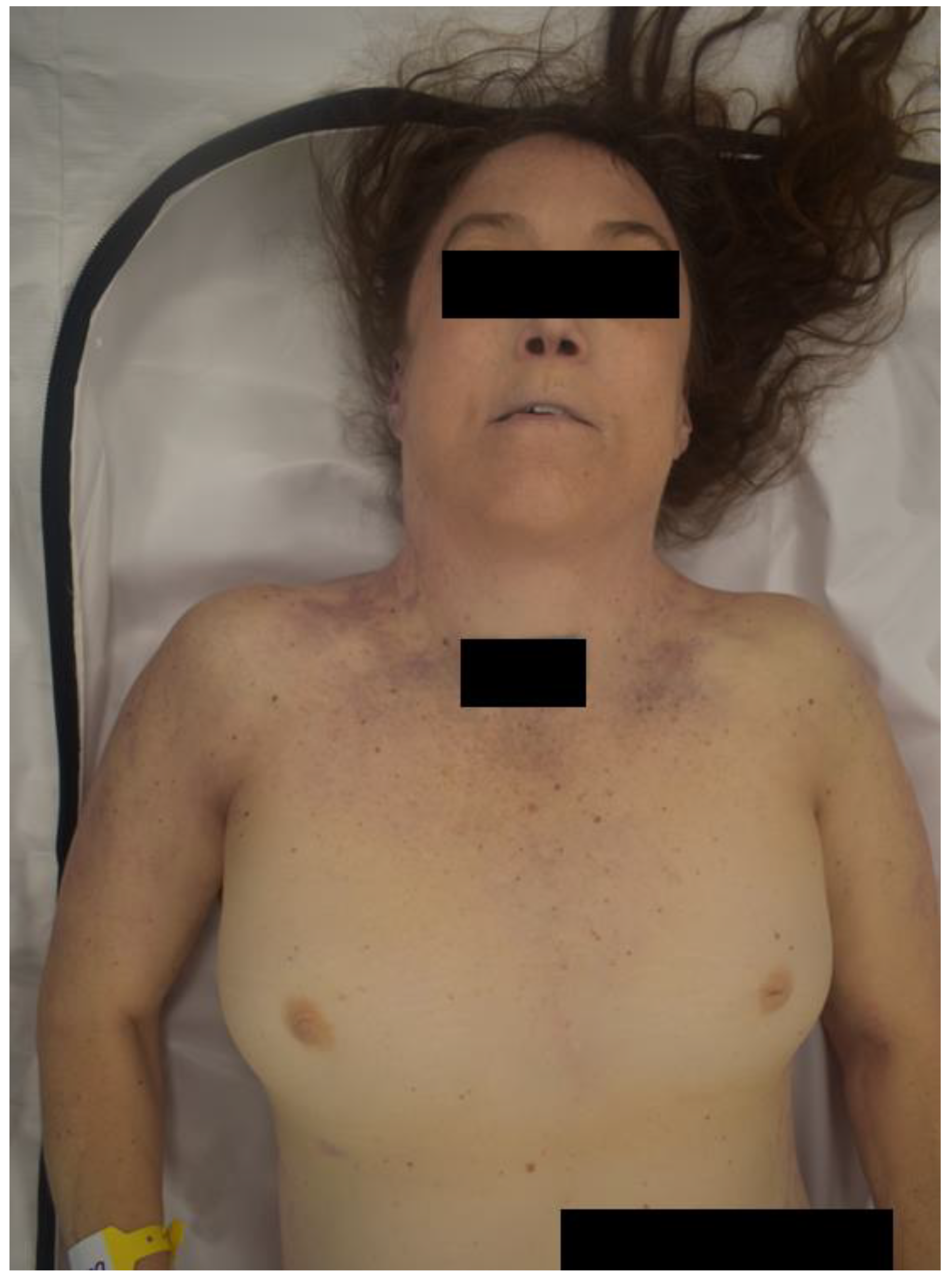
Figure 3.
CT scan revealing fluid in the chest cavity as well as blood in the pericardial sac.
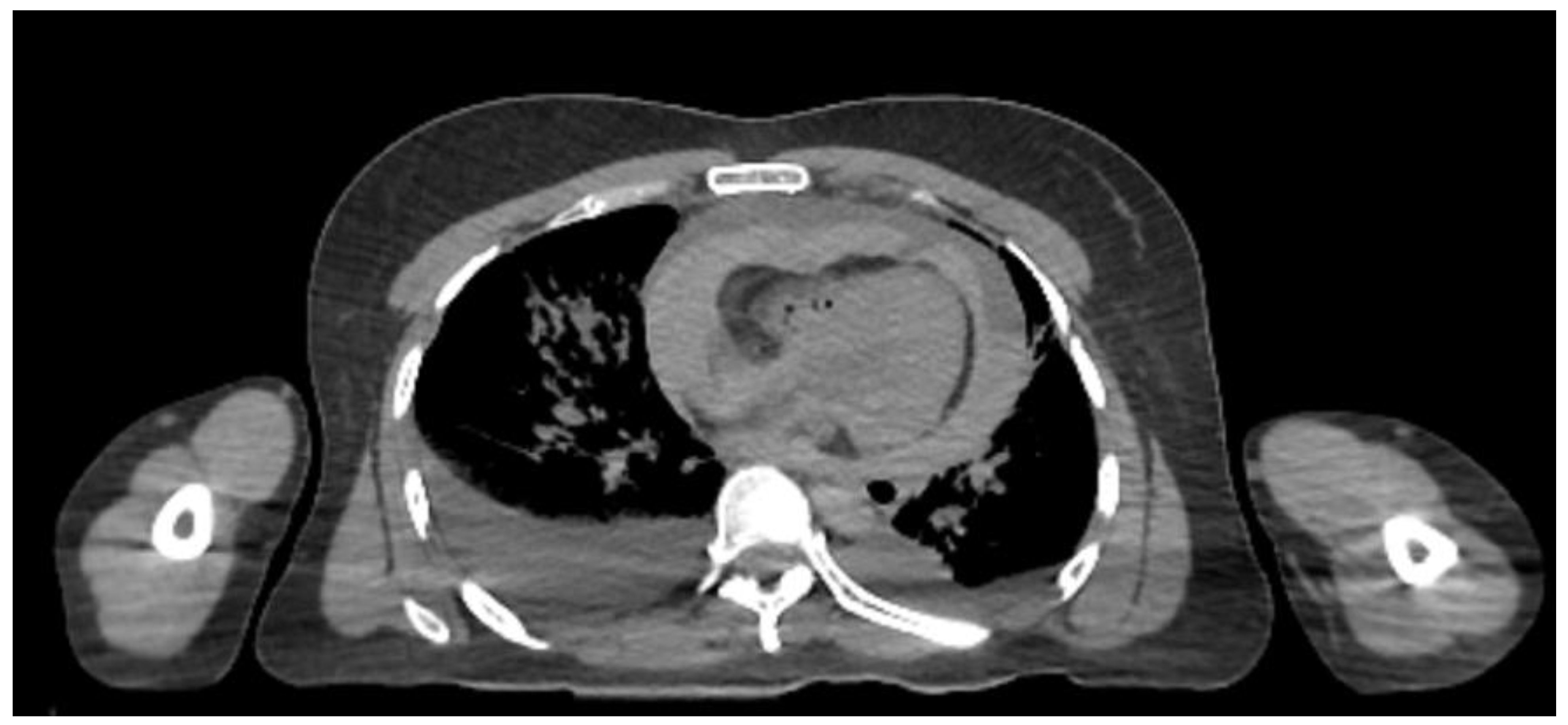
Figure 4.
Heart containing a Bicuspid Aortic Valve with intimal defect of the aorta.
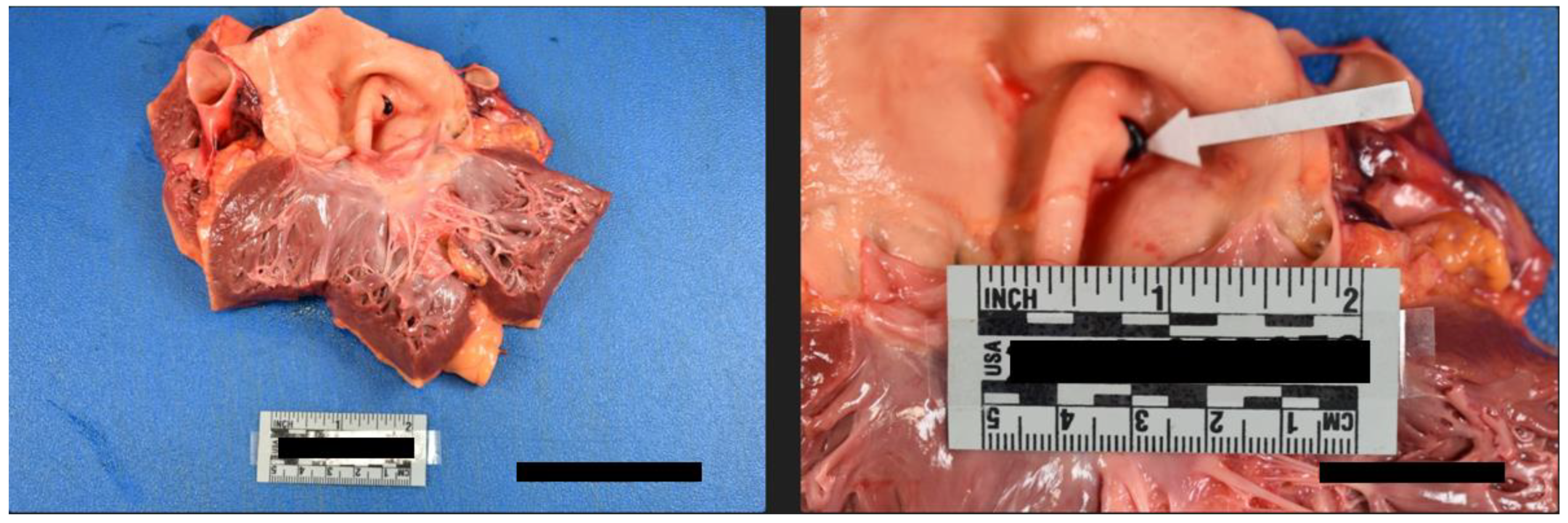
Figure 5.
Adventitia defect of the Aorta.
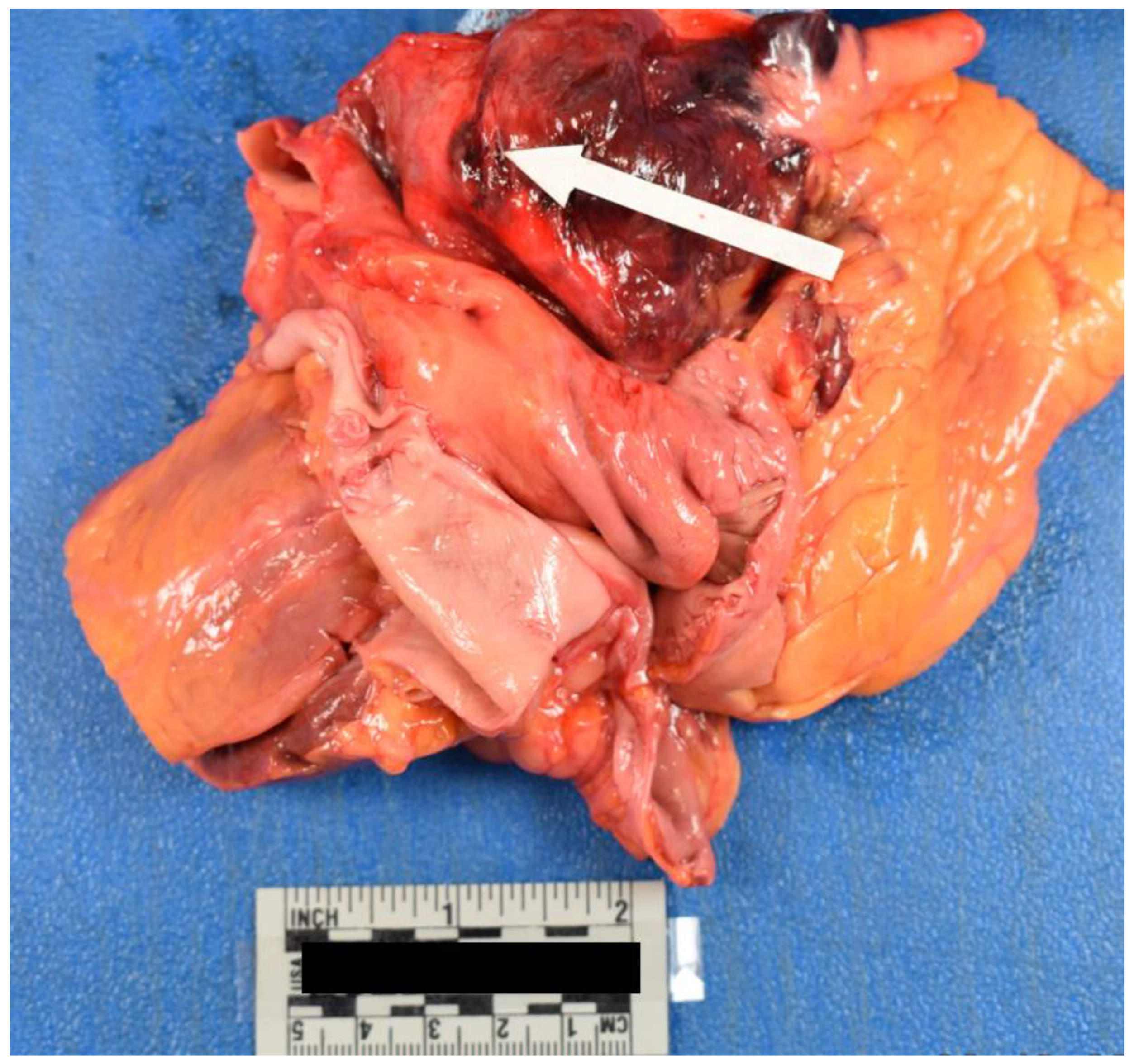
Figure 6.
Aorta with Suture Line.
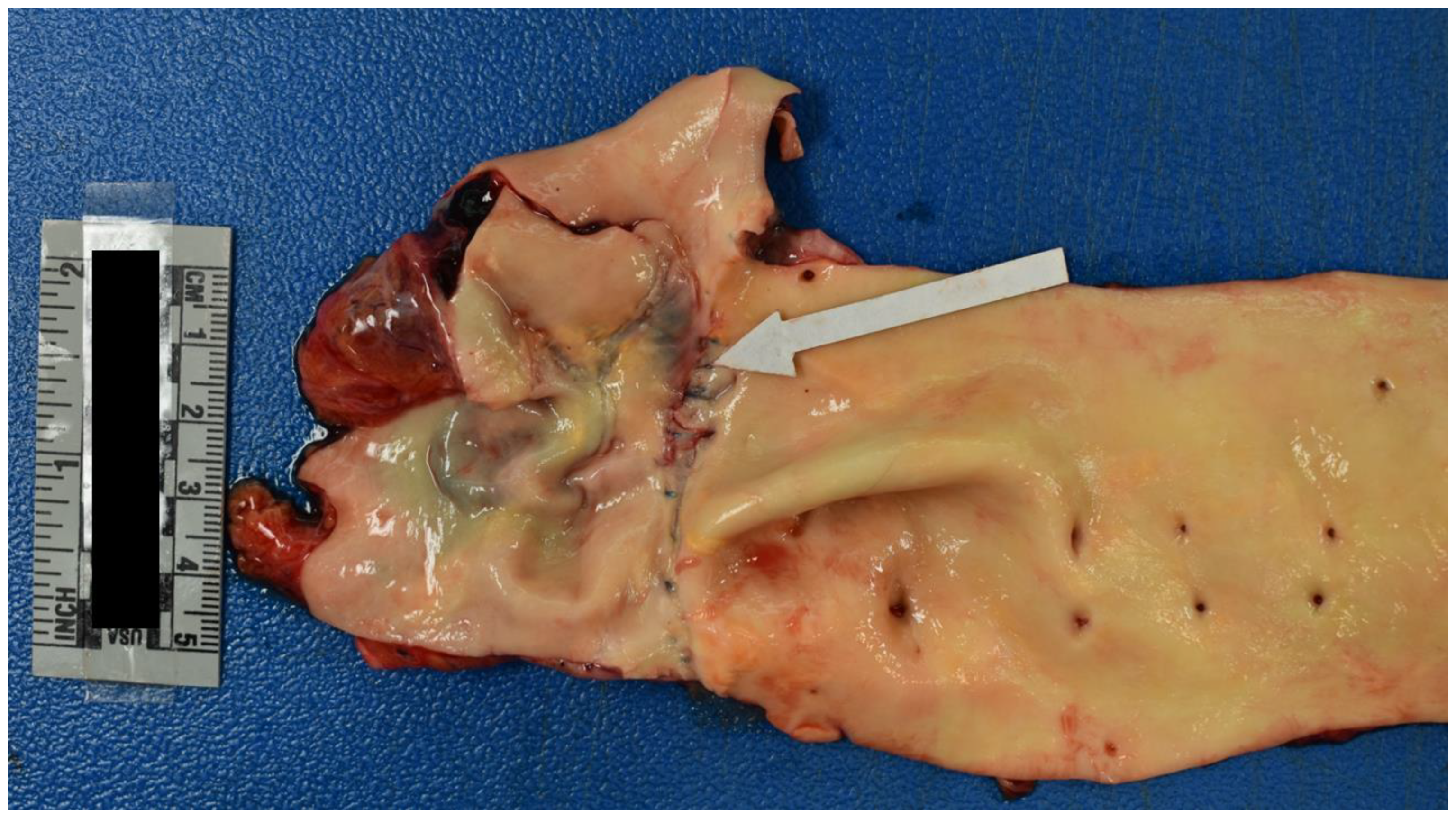
Figure 7.
Blood Tracking Along both Carotid Arteries.
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



