
Submitted:
11 February 2023
Posted:
13 February 2023
You are already at the latest version
Abstract
Two related tumor suppressor genes, Brca1 and Brca2, attract a lot of attention from both fundamental and clinical points of view. Oncogenic hereditary mutations in these genes are firmly linked to the early onset of breast and ovarian cancers. However, the molecular mechanisms that drive extensive mutagenesis in these genes are not known. In this review we hypothesize that one of the potential mechanisms behind this phenomenon can be mediated by Alu mobile genomic elements. Linking mutations in the BRCA1 and BRCA2 genes to the general mechanism(s) of genome stability and DNA repair is critical to ensure the rationalized choice of anti-cancer therapy. Accordingly, we review the literature available on mechanisms of DNA damage repair where these proteins are involved in and how the inactivating mutations in these genes (BRCAness) can be exploited in anti-cancer therapy. We also propose a hypothesis that explains why breast and ovarian epithelial tissues are preferentially susceptible to mutations in BRCA genes. Finally, we discuss perspectives of novel therapeutic approaches for treating BRCAness cancers.
Keywords:
breast cancer
; ovarian cancer
; PARP inhibitors
; Alu repeats
; protein-protein interactions
1. Introduction
The first case of hereditary cancer was described back in 1866 by Pierre Paul Broca when he documented the development of breast and ovary cancers within his wife’s family. It took almost 130 years to decipher the genetic mechanism behind this hereditary cancer syndrome. This was done by Mary Claire-King and colleagues who published a linkage analysis of families with early onset of breast cancer and identified the gene locus of BRCA1 (BReast CAncer 1) at 17q21 [1]. The gene responsible for this phenotype was cloned in 1994. Shortly thereafter, the BRCA2 gene was identified and cloned, linking it to chromosome 13 [2]. The products of these genes are functionally classified as tumor suppressors meaning that inactivation of both copies of either gene is strongly associated with carcinogenesis. BRCA1 and BRCA2 proteins lack any structural homology, whereas a segment of BRCA1 is homologous to its partner, BARD1 protein. In contrast to the canonical tumor suppressor inactivation mechanism whereby one allele of a tumor suppressor gene is mutated and the other is either deleted or epigenetically inactivated (“loss of heterozygosity” principle, LOH), the BRCA mutated cancerous cells frequently bear the remaining alleles as wild-type [3]. In this case, mutations in the BRCA1/2 genes are often preceded by mutations in other critical tumor suppressor genes, PTEN and/or p53 [4]. Reversion of germline BRCA mutations in growing cancers is also common [5]. This indicates that haploinsufficiency may be the major basis for early development of breast cancer in BRCA1/2 pathogenic mutation carriers. Importantly, since the products of these genes are involved in the DNA damage response, the BRCA mutation status has the profound significance for selection of an appropriate therapeutic interventions.
2. Epidemiology of Cancer and BRCA
2.1. Pan-Cancer Overview
Despite their close functional connection, BRCA1 and BRCA2 have somewhat different effects on cancer development and progression. For example, BRCA1 and BRCA2 each correlate with different subtypes of breast cancer. BRCA1 mutations are linked preferentially to the triple negative form of breast cancer (estrogen receptor negative, progesterone receptor negative, and HER2 negative), whereas BRCA2-associated breast cancers are generally estrogen receptor positive. Furthermore, mutations in BRCA2 are more often associated with other types of epithelial cancer, including male breast cancer, pancreatic cancer, and prostate cancer than BRCA1 mutations.
Mutations in the BRCA1 gene are mostly associated with hereditary cancers and are rarely found in sporadic cancers. More than 300 germline mutations have been identified so far in patients with familial breast and/or ovarian cancer, and only a few somatic BRCA1 mutations have been identified in sporadic breast cancer [6]. However, there are reported cases of non-familial breast cancers with functional inactivation of BRCA1 which occurred due to attenuated gene expression of BRCA1 or its incorrect subcellular localization.
The expectancy of ovarian cancer to occur in either of these genes in various tumors is also different. For example, for BRCA1, the risk of ovarian cancer is 40 to 45% compared to 10 - 20% for BRCA2. In addition, the onset of ovarian cancer seems to occur earlier for BRCA1 cases than for the BRCA2 ones.
Since BRCA1 is a tumor suppressor and is directly involved in the double strand break (DSB) repair process, it is not surprising that the mutation status of this gene serves as a prediction marker of high risk of carcinogenesis. Carriers of germline mutations in the BRCA1 gene are prone to developing mostly breast and/or ovarian cancers [7]. Although BRCA mutations are also found in many other types of tumors they apparently do not have any detectable effect on cancer incidence in brain, colon, bladder, kidney, cervix, lungs, or melanoma [8,9]. Yet, the BRCA mutation status often correlates with the severity of the disease and shorter time of overall survival (whether the patient has the mutation or not can be associated with the severity of the disease and shorter times of overall survival// can influence the severity of the disease and the time of overall survival).
2.2. Ovarian Cancer
90% of ovarian cancers are epithelial ovarian cancer (EOC), which is further subdivided according to histological characteristics into low-grade serous, clear cell, endometrioid and mucinous [10], and the most common high-grade serous (HGSOC). The latter accounts for approximately 70% of all cases of EOC [11]. Importantly, approximately 15-20% of patients with HGSOC have germline mutations in BRCA1 or BRCA2 [12,13]. The presence of such BRCA mutations has been reported in other histological subtypes of EOC [13,14].
Hereditary ovarian cancers are characteristic to three autosomal dominant familial syndromes: breast/ovarian cancer, site-specific ovarian cancer, and Lynch (hereditary non-polyposis colorectal cancer) syndrome [15]. Meanwhile, a familial history of ovarian cancer, especially the ones associated with BRCA1 mutations, poses a significant lifetime risk of developing the disease.
Thus, 39–44% of women who inherit a BRCA1 oncogenic-driving mutation develop ovarian cancer by age 70–80 [16], and diagnosis at a later stage significantly worsens prognosis [17]. However, there is evidence that mutations in the BRCA1 gene are associated with an increase in the progression-free survival (PFS) [12,18,19,20]. This may be due to increased sensitivity of such patients to the treatment with platinum-containing drugs [7] or poly(ADP-ribose) polymerase (PARP) inhibitors.
Oncogenic mutations in BRCA1 can be not only germline, but also somatic. According to the results of several independent studies, they make up a significant part of all observed mutations in this gene among patients with ovarian cancer [21,22,23,24]. Overall, somatic BRCA mutations occur in approximately 5–7% of ovarian cancers [25]. The existence of somatic mutations fits into the concept of "BRCAness", in which germline mutations of BRCA1 or BRCA2 are not detected, but the DNA repair defect occurs due to the problems in the process of homologous recombination [26].
Studies do not reveal a significant difference in the course and aggressiveness of ovarian cancer in patients with somatic or germline BRCA1 mutations. Similar to patients with congenital BRCA1 mutations, patients with somatic BRCA1 mutations show increased sensitivity to platinum-containing drugs and olaparib, a PARP inhibitor [23,27].
2.3. Breast Cancer
Breast cancer is one of the most common types of cancer diagnosed in women. This disease can also occur in men, although much less frequently. Molecular subtypes of breast cancer include luminal A, luminal B, Her2 - positive, triple negative, claudin-low and normal-like with other molecular markers important for classification being ERα+, PR, EGFR, CK5/6, VEGF, KI67, TNBC, MES, IM, LAR [28]. Tumors associated with a BRCA1 mutation are more likely to be triple-negative breast cancer - more aggressive and difficult to treat [29].
Breast cancer caused by a mutation in the BRCA1 gene has a higher rate of mitosis and greater lymphatic permeability than sporadic breast cancer, as well as a higher frequency of somatic mutations in the p53 gene [28]. Women who inherit pathogenic BRCA1 mutations face a very high lifetime risk of developing breast cancer, 60% to 80% by the age of 80 years [28,30]. Two thirds of the BRCA1 mutations found in breast cancer are germline and the remaining third relates to somatic mutations [31,32,33]. Germline and somatic BRCA1 mutations are currently assumed as biologically equivalent, but this remains unproven despite significant effort [34]. There is evidence that tumors carrying BRCA1 germline mutations have similar biological signatures to tumors with somatic BRCA1 mutations [35,36]. However, there is also data showing that somatic mutations of the BRCA1 gene have not been identified in breast cancer without concurrent germline mutations [37], which may explain the small difference between tumors with somatic and germline BRCA1 mutations.
2.4. Pancreatic Cancer
Pancreatic cancer is reported to be the third most common cancer associated with BRCA mutations [38]. A family history of pancreatic cancer is found in 5–10% of patients with pancreatic cancer. Pancreatic ductal adenocarcinoma (PDAC) occurs especially frequently in families with ovarian or breast cancer [39]. Pathogenic mutations in BRCA2 occur in 2% of patients with pancreatic cancer, and mutations in BRCA1 in 1% patients. Approximately 7% of patients with pancreatic cancer carry germline mutations in BRCA1/2. In patients with hereditary pancreatic cancer, the prevalence of BRCA1/2 mutation carriers is estimated at 4.9–26%. Mutations in BRCA2 appear to be more common in pancreatic cancer. Furthermore, they are considered to be more dangerous and increase the risk of developing pancreatic cancer several fold [40].
2.5. Prostate Cancer
Mutations in the BRCA1 and BRCA2 genes increase the risk of developing prostate cancer. Some results indicate significantly lower survival rates and a more aggressive course of the disease [41,42]. Similar to pancreatic cancer, a higher risk of prostate cancer is more often associated with BRCA2 than with BRCA1. Male carriers of a BRCA2 gene mutation have a significantly increased risk of developing prostate cancer, while men with a BRCA1 mutation have a significantly lower risk of developing prostate cancer than patients with wild-type BRCA1/2 genes [43].
2.6. Mutations and the “Founder Effect”
Pathogenic mutations in BRCA1/2 have been found throughout the coding region of this gene and at splicing sites (Figure 1). Most mutations in both genes are small insertions or deletions resulting in frameshifts, nonsense mutations, or splice site changes that cause the stop codon to occur prematurely [15]. Therefore, it is quite difficult to isolate the regions that are most susceptible to deleterious mutations common among various types of cancer.
In respect to breast cancer, there are studies highlighting exon 10 (usually termed exon 11 for historical reasons), of BRCA1 as the most mutated in breast cancer patients [28,44]. According to The Breast Cancer Information Core (BIC), a catalog of BRCA1 and BRCA2 mutations identified worldwide, the most common BRCA1 mutations identified are 185delAG (16.5%), 5382insC (8.8%), and the C61G missense mutation (1,8%). However, exon 10 is longer than all other exons combined thus physically providing more opportunities for mutations to occur.
Of note, there is some variation in the distribution of BRCA1 mutations around the world. Some BRCA1 variants are limited to geographically isolated regions or specific populations, while other mutations are common in several countries at the same time, suggesting a spread by migration and perhaps a significantly older origin. This phenomenon is described as the “founder effect”. It facilitates diagnosis within the same population, but significantly complicates the study of BRCA1-associated cancers [45].
In some countries and ethnic communities, the spectrum of BRCA1 mutations is strictly limited to a few founder mutations. For example, the founder effect in the population of Ashkenazi Jews is well described: 3 mutations in the BRCA1 gene (BRCA1 c.68_69delAG, c.5266dupC and BRCA2 c.5946delT) account for 98–99% of the identified mutations and are found in approximately 2.6% of the Ashkenazi Jewish population [46].
In Russia, the most common BRCA1 mutation is c.5266dupC, accounting for about 90% of all BRCA1 mutations. Other less common mutations found in Western Russia are c.4035delA, c.181T>G and c.68_69delAG [47,48,49].
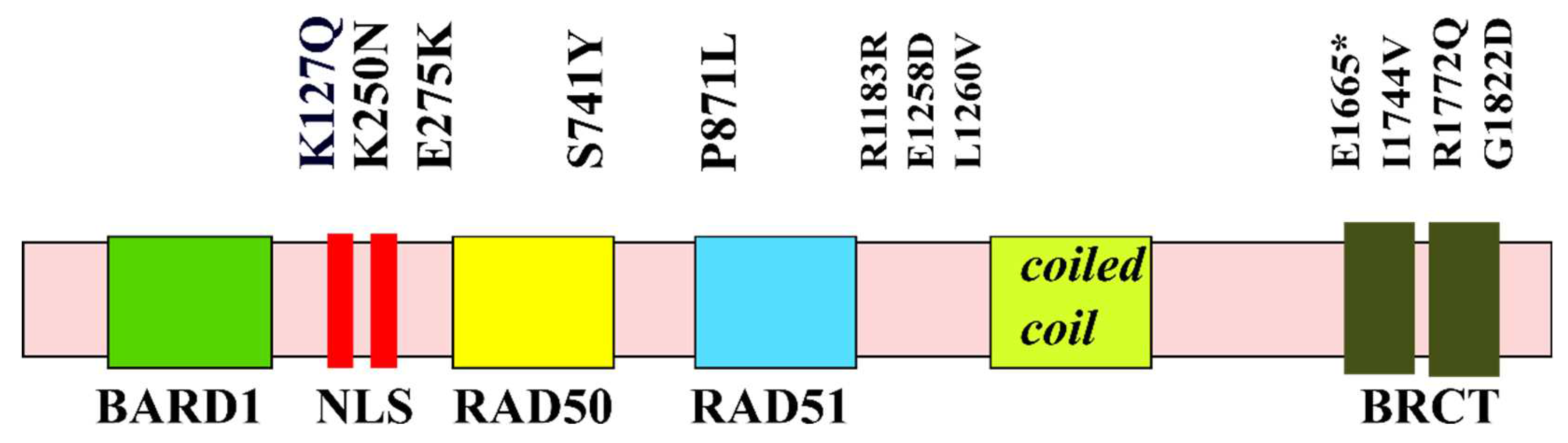
Figure 1.
Distribution of major pathogenic mutations along the domains of BRCA1.
3. Molecular Evolution of BRCA and Links to Human Cancers
Both BRCA1 and BRCA2 are ancient genes, which appeared early in evolution, and are indispensable for high-fidelity DSB DNA repair in most Eukaryota. However, it should be mentioned that BRCA1 seems to be absent from all fungi and BRCA2 was not found in yeast. Since harmful effects of mutations in the BRCA genes are developed only later in life, these mutations are likely to be passed on to future generations. Because these mutations do not affect reproductive fitness, the purging force of natural selection will be weak and insufficient to consistently eliminate these mutations [50]. Therefore, mutations in BRCA1 and BRCA2 may be considered as a good illustration of the mutation accumulation theory, especially because they are inherited in a dominant manner. In this situation, the dominant nature of BRCA1/2 mutations may decrease fertility of female carriers through an accelerated depletion of ovarian reserve as described in several independent reports (for example, [51,52]). Although the menopause onset is largely unaffected [52], and hence the magnitude of this effect may be overestimated [53], it is worth mentioning that even a small decrease in age-associated fertility may have drastic consequences on the evolutionary scale.
The reason for BRCA1 or BRCA2 mutations promoting carcinogenesis predominantly in breast and ovarian epithelia is assumed to be that since menstrual cycles periodically create a hormone-dependent enrichment in ROS in female hormone-responsive tissues, there would be a demand for an augmented expression of genes responsible for antioxidant defense and the DNA repair machinery against genotoxic metabolites including, for example, endogenous quinones derived from 2- and 4-hydroxyestradiols [54]. This may be a plausible explanation to the fact why mostly female hormone-responsive tissues are exquisitely sensitive to germline mutations in the BRCA1 and BRCA2 genes [55]. This, however, should be refined mechanistically, since the problem of tissue-specificity of oncogenic effects exerted by ubiquitously expressed genes is rather multifactorial [56].
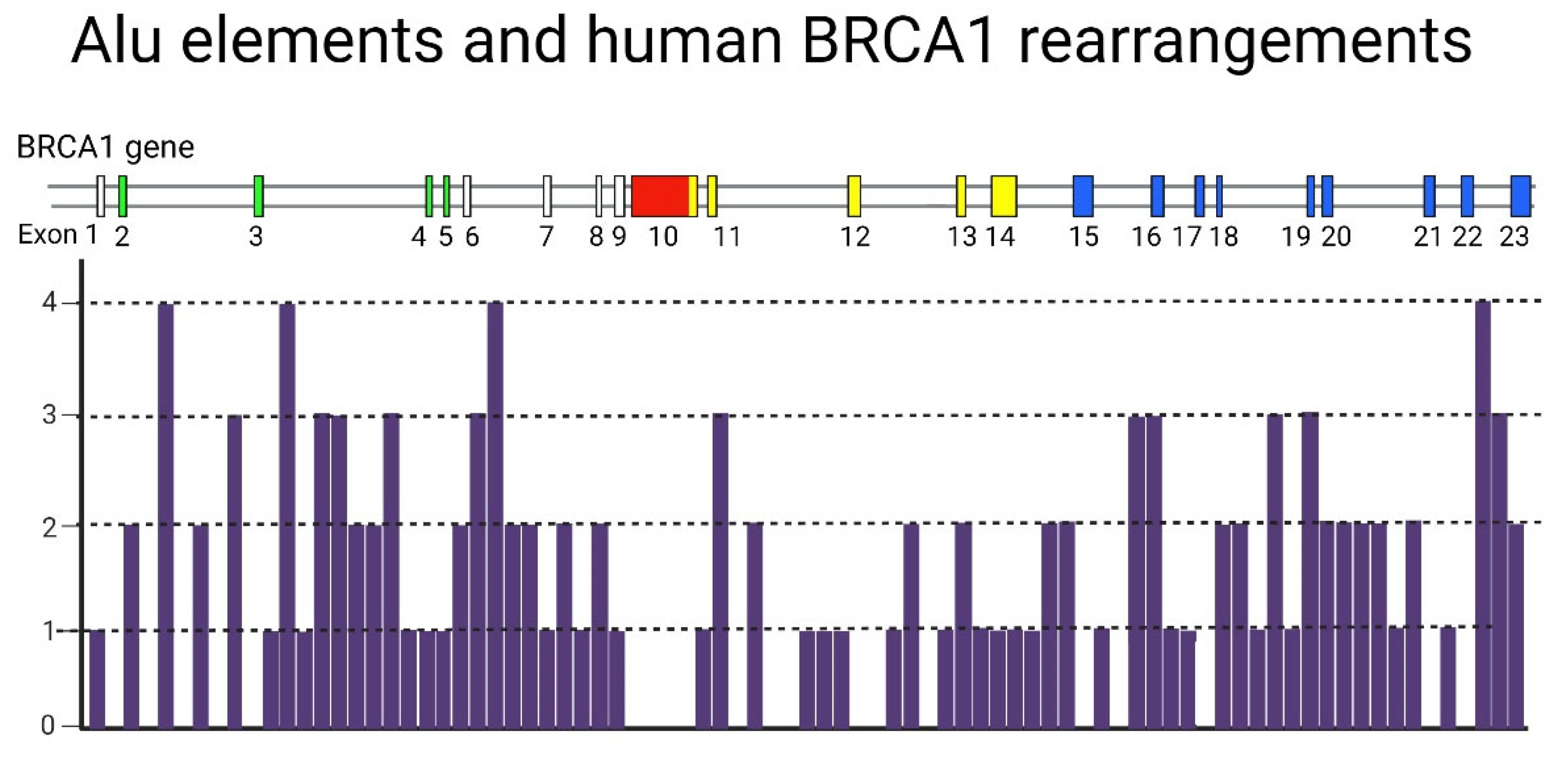
Figure 2.
Distribution of Alu repeats in the human BRCA1 gene.
4. A Potential Mechanism for Enrichment of Mutations in the BRCA Genes
Here we attempt to highlight the importance of intrinsic genetic mechanisms that control genomic instability in humans, specifically, by Alu repeat elements. Alu elements are primate-specific repeats and comprise 11% of the human genome. They have wide-ranging influences on gene expression. Alu elements are ~300 bp retrotransposon sequences that are normally silenced by DNA methylation and heterochromatin formation. However, in the germline, Alu elements are more active and may significantly contribute to genetic diseases and population diversity. In particular, we argue that Alu-repeats may significantly contribute to mutagenesis of BRCA1/2 genes by several mechanisms: through direct insertional mutagenesis and/or as an abundant source of repetitive sequences that, in turn, contribute to non-allelic homologous recombination which would result in genetic deletions and duplications [57].
Over the last 20 years, research has expanded to improve the understanding of BRCA-related breast and ovarian cancers, specifically how they respond to treatment as well as the expected clinical course. Better characterization of alterations in these genes may enable the development of new, targeted therapies, or broadening the clinical application of current therapies [7].
4.1. A Hypothetical Role of Transposable Elements (TEs) in BRCA-Associated Carcinogenesis
BRCA1/BRCA2 genes harbor a very high density of repetitive DNA elements that contribute to genetic instability [58]: BRCA1 gene contains 138 individual Alu elements [59], which occupy about 42% of intronic sequences (Figure 2). In addition, this gene includes 5% of another repeats [60,61]. BRCA2 contains approximately 47% repetitive DNA, but only 17-20% of them are Alu repeats. These genes show a high probability of mutations that are associated primarily with Alu-mediated genomic rearrangements [62,63]. These rearrangements are more frequent in BRCA1 than in BRCA2, probably due to the large number of Alu repeats in the gene sequence [64,65,66].
Although most genomic rearrangements are definitively pathogenic, causing frameshifts and premature termination codons, some rearrangements have more ambiguous effects, especially in-frame deletions of redundant exons [67] or some duplications, where additional copies of exons might be tolerated by the organism without deleterious effects [68].
About 10% breast cancer cases are related to defects in the BRCA1 or BRCA2 genes [69]. Women whose relatives have confirmed BRCA1 or BRCA2 defects have been shown to have a high lifetime risk of developing breast cancer (80% and 60%, respectively) [70,71]. It is shown than only BRCA1 large rearrangements (but not BRCA2) play a notable role in predisposition to breast and ovarian cancer in high-risk families of German origin [72]. Researchers analyzed 226 patients with high-risk hereditary breast and ovarian cancer and described 6 large genomic alterations in BRCA1 locus. BRCA1 mutations include a deletion of exon 5, a deletion comprising exons 5–7, deletion of exons 1A, 1B, and 2, two duplications of exon 13, and a deletion of exon 17. However, in BRCA2 gene nothing similar was found. In another study, in two families with breast and ovarian cancers there was found a 7.1 kb germline deletion, which includes exons 8 and 9. This deletion leads to a frameshift at the mRNA level [73].
Only a few other studies have investigated BRCA2 rearrangements [74]. To date, about 16 cases of BRCA2 germline rearrangements have been reported. It is shown that large genomic BRCA2 rearrangements are observed in male breast cancer families, predominantly [75]. Genomic rearrangements of the BRCA2 gene are present in 25 families with at least one man who has breast cancer. However, no BRCA2 gene rearrangements were found in 114 families among women with breast cancer [76]. These results raise the question of possible existence of sex-related mechanisms of gene rearrangements in the BRCA2 gene.
The Alu-indirect insertion in exon 3 of BRCA2 - c.156_157insAlu - is quite common in families with inherited predisposition to breast/ovarian cancer. Researchers found this mutation in 14 families (out of 208 tested) and it accounts for about a quarter of all mutations in the BRCA1/2 loci [77].
Thus, Alu-mediated rearrangements in the BRCA1 and BRCA2 genes, including deletions and insertions that lead to global genomic rearrangements of these genes, are closely associated with the predisposition and development of breast and ovarian cancers.
5. Structure-Function Analysis of Human BRCA1
BRCA1 is involved in vital processes in the nucleus, namely, transcription, DNA repair (including the repair of transcription-related DNA damage), and cell cycle control. Accordingly, BRCA1 is localized to discrete sub-nuclear structures associated with DNA replication or repair. DNA damage induces BRCA1 phosphorylation and recruitment to specific foci containing DNA repair proteins, where BRCA1 is deemed to act as a scaffold for the assembly of various multiprotein complexes. Despite the large molecular weight of BRCA1 (1863 amino acid residues [78]), only two conserved domains can be distinguished in its structure: the N-terminal RING domain (exons 2-6) [79] that encompasses 100 amino acid residues and two tandem C-terminal BRCT domains, 90 amino acid residues each [80], encoded by the end of exon 16, and exons 21-24, respectively. The region of the protein located between these two terminal domains is structurally variable between mammalian BRCA1 homologues. It is believed to be intrinsically disordered, yet, it is critical for the proper functioning of BRCA1, along with the other two conserved domains (Figure 3).
5.1. The RING Domain
The DNA-binding RING (Really Interesting New Gene) domain has an E3 ubiquitin ligase activity, being a scaffold for the interaction with the corresponding E2 ubiquitin ligases such as UbcH5, UbcH6, UbcH7, Ube2e2, UbcM2, Ube2w, and Ubc13 (Figure 4) [81]). The ubiquitin ligase activity of BRCA1 is stimulated by the formation of a heterodimer with the BARD1 protein [82]. The latter also contains a RING domain and tandem BRCT domains and shares some structural similarity to BRCA1 [83]. Like BRCA1, BARD1 tends to form specific foci in the nucleus in S-phase of the cell cycle that overlap with the ones formed by BRCA1, suggesting that the formation of the BRCA1-BARD1 complex is cell cycle-dependent [84].
The formation of a complex with BARD1 is necessary for stabilization of BRCA1 at the protein level. Furthermore, this interaction is apparently important for the nuclear localization of BRCA1. The BRCA1-BARD1 heterodimers are involved in the DNA repair of double-strand breaks and hence the preservation of DNA integrity, including the process of resolving impaired replication forks (for more details, see [81]). Mechanistically, the BRCA1/BARD1 complex is recruited by the RAP80 protein to sites of DNA damage [85] where the BRCA1/BARD1 ubiquitin ligase activity is utilized to modulate the activity and abundance of histones and cellular DNA damage response factors (Figure 5).
Importantly, the BRCA1-BARD1 heterodimers also interact with the RNA polymerase II holoenzyme. However, BRCA1 does not show increased affinity for specific DNA sequences except for some abnormal structures (some branched DNA formations) [86]. This does not allow BRCA1 to be considered a bona fide transcription factor. Taking into account the fact that in the central unstructured and C-terminal regions of BRCA1 there are many binding sites for various transcription factors, chromatin remodeling factors, and DNA-damage response factors, it would be fair to say that BRCA1 in complex with BARD1 forms a scaffold for the surveillance of genome integrity control during transcription [87]. However, there are also cases when BRCA1 acts as a corepressor: for example, the transcription factor ZBRK1 suppresses transcription of its target genes in a BRCA1/CtIP-dependent manner [88]. ZBRK1 acts as a metastatic suppressor by directly regulating MMP9 in cervical cancer.
5.2. The BRCT Domain
The C-terminal region of BRCA1 (1650-1863) is occupied by two BRCT (BRCA1-C-Terminal) tandem repeats domain connected by a 22 amino acid linker [89]. The BRCT domains are protein-binding modules that recognize the phosphorylated motif pSer-x-x-Phe [90]. Due to this, BRCA1 is included in the signaling cascades triggered by DNA damage as a scaffolding platform for the interactions of various kinases and other proteins involved in the regulation of the cell cycle [91]. In addition, BRCA1 itself undergoes reversible phosphorylation upon DNA damage [92] by key regulators of DNA damage response: PIKK kinases (ATM, ATR, DNA-PK) [93] and checkpoint effector kinases (Chk1, Chk2 and MK2) [94]. Phosphorylation of BRCA1 also creates new sites for complex protein-protein interactions (Figure 5).
BRCA1-BARD1 complex senses the ubiquitination status of histone H2A and works as a ubiquitin ligase of this histone. These activities play important roles in the choice between HR or NHEJ during DNA damage repair: BRCA1 acts as mediator for HR, antagonizing the 53BP1-mediated NHEJ pathway [95,96,97] (Figure 6A). BRCA1 interacts with BRCA2 that is complexed with SEM1/DSS1, ssDNA [98] (Figure 6B), recombinase RAD51, PALB2 and ssDNA-specific endonuclease XPG/ERCC5 [99].
5.3. BRCA1 and p53
P53 is arguably one of the major tumor suppressors in humans. The Tp53 gene is frequently mutated and several point mutations in its DNA binding domain convert the p53 protein into an oncogene. That p53 mutations occur in tumors bearing BRCA1 mutations suggests that the two genes may function in different signaling pathways to suppress tumorigenesis [100]. However, results from the experiments in mice have shown that tumorigenesis occurs much more efficiently when both BRCA1 and P53 are deleted compared to BRCA1 deletion alone [101], indicating that p53 is located downstream of Brca1 in the same signaling pathway. Accordingly, mutations in BRCA1 preceding mutations in the p53 gene, as seen in cases of familial breast cancer, are not sufficient for tumor progression. Since BRCA1-null cells display genomic instability, it is likely that persistent intrinsic DNA damage in the presence of wild-type p53 leads to extermination of such cells via p53-dependent cell cycle arrest and apoptosis.
Another fact that functionally links p53 and BRCA1 is that both p53 and Brca1 in response to various types of DNA damage become phosphorylated by DDR-dependent kinases, ATM and Chek1. Upon DNA damage, BRCA1 interacts with another kinase, c-Abl [102]. The C-terminus of BRCA1 is phosphorylated by c-Abl in vitro. In vivo, BRCA1 is phosphorylated at tyrosine residues depending on ATM and irradiation. However, tyrosine phosphorylation of BRCA1 does not disrupt the interaction between BRCA1 and c-Abl. Notably, cells with BRCA1 mutations exhibit constitutively high c-Abl kinase activity, which does not increase when cells are exposed to gamma radiation. Probably, BRCA1 mutations, due to defects in DNA repair, induce the kinase activity of c-Abl towards p53, which culminates in p53-dependent cell cycle arrest and cell death. In addition to phosphorylation and subsequent activation of p53 transcriptional activity, c-Abl also stabilizes p53 on the protein level by inactivating its major inhibitor, E3 ligase Mdm2 [103]. Curiously, c-Abl also phosphorylates another tyrosine kinase, BTK [104]. In this respect, we have recently shown that BTK can phosphorylate p53 leading to its stabilization and transcriptional activation [105] suggesting a novel role for BTK as a potential tumor suppressor [106].
It is also known that BRCA1 and p53 are able to interact physically. Deletion analysis in the Brca1 gene allowed the identification of p53-interacting domains in the coiled-coiled region and in the second BRCT domain. On the other end, p53 interacts with BRCA1 at the C-terminus. BRCA1-mediated stabilization of the wild type p53 protein occurs through upregulation of the p14ARF gene product, which in turn upregulates mouse p53 phosphorylation at serine 18 (equivalent to human serine 15). Exon 10 (historical exon 11) of BRCA1 appears to be responsible for this, since cells with deletions of exon 10 in BRCA1 are defective in p53 stabilization after DNA damage [107].
Functionally, this interaction converts BRCA1 into a p53 coactivator [108]. Perhaps not surprisingly, both proteins, p53 and BRCA1, transcriptionally regulate the expression of the GADD45 gene, which induces growth arrest and DNA damage repair. Both BRCA1-deficient and GADD45-deficient cells have displayed a G2/M cell cycle checkpoint defect and increased genome instability [109].
Collectively, these results suggest that the phenotypic manifestation of BRCA1 tumorigenic mutations heavily relies on the spectrum of inactivation in other critical tumor suppressors, e. g. p53.
6. Survival of BRCA-Mutated Cancer Cells: Role of Tissue Microenvironment
One may wonder why the tumorigenic role of BRCA1/2 mutations is exemplified preferentially in breast cancer cells and not so much in other epithelial tissues. In this respect, it should be noted that mutated breast cancer cells are largely derived from luminal progenitor cells. Although germline BRCA1/2 mutations occur stochastically in many tissues [110], breast tissues of patients with oncogenic germline BRCA1/2 mutations have distinct histological features [111]. Premalignant lesions in this tissue also have certain molecular hallmarks such as upregulated expression of progesterone receptor A [112].
6.1. Hypothesis: Role of Breast Adipocytes in Early Progression of BRCA1/2 Mutated Microtumors
Survival of early malignant cells in the surrounding normal tissue is dependent on many factors, including escape from immune surveillance by natural killers etc. Indeed, it is physiological for breast ductal epithelium to invade into adipose tissue and partially displace it during lactation [113]. Thus, breast adipocytes may sense the invasion of micro-metastatic or circulating breast tumor cells as a normal process, which would prevent the inflammatory signaling in these niches.
In general, the role of adipocytes in cancer progression was highlighted in several excellent reviews [114,115,116,117,118]. It was suggested that adipocytes enhance cancer growth through secretion of exosomes that contain tumor promoting factors, e.g. TSP5 [119]. In this respect, breast cancer adipocytes may stimulate the onset of epithelial-mesenchymal transition (EMT) in breast cancer cells by expressing exosomal TSP5 [114,120]. Mechanistically, breast adipocytes protect early breast tumor cells from ferroptosis and other ROS-mediated forms of cell death by secretion of fatty acids [121], and the cross-talk between adipocytes and malignant cells may occur via secretion of leukemia inhibitory factor (LIF) and C-X-C subfamily chemokines in a positive feedback mode [122]. Also, these cancer-associated adipocytes undergo “browning” – the process of increasing the number of mitochondria [123]. This occurs concomitantly with inflammation-like signaling [124], and stimulation of vascularization [125]. Collectively, breast adipocytes may create a unique natural tumor niche for breast cancer cells with germline mutations in BRCA1/2 genes. Furthermore, breast cancer cells readily invade multiple tissues such as lungs, liver, bones etc. Again, adipocytes may play an important role in allowing invading cells to colonize and proliferate [126].
7. Vulnerabilities of BRCA-Mutated Cancer Cells
However aggressive may the BRCA mutant cancers be, these mutations also give fast growing cells certain features that may result in paradoxically better sensitivity to certain cytostatic and targeted therapies. Indeed, nowadays patients with triple-negative breast cancer have better prognosis for survival if they bear the BRCA mutations [127].
7.1. Platinum Complexes
Both platinum-containing drugs and PARP inhibitors are used to treat homologous recombination-deficient (HRD) cancers that have mutations in genes involved in double-strand DNA repair [128]. Platinum salts create DNA interstrand crosslinks that are extremely difficult to cleave in the absence of homologous recombination (HR), which leads to the death of HR-deficient (HRD) cancer cells [129]. Augmented sensitivity of BRCA1/2 mutated cancers to platinum salts has been well documented in numerous studies, for instance in those on ovarian cancer [130], pancreatic cancer [131], breast cancer [132]. If the normal copy of BRCA1 or BRCA2 is retained, the efficacy of platinum based therapies is decreased [133]. Also, platinum resistance may develop upon reverse mutations in BRCA1 [134].
7.2. PARP Inhibitors
The exact mechanism of action for PARP inhibitors (Figure 7) has not yet been fully understood. Initially, they were developed as dissipaters of DNA repair and potent sensitizers of cancer cells to chemotherapy, but they also showed a significant independent effect on patients with mutations in the HR genes, primarily BRCA1. The effect of synthetic lethality for PARP inhibitors was shown in cells with the loss-of-function mutations in BRCA1 [135]. There are several hypotheses about the mechanism of their combined action [128]. The main model posits that inhibitors bind to the PARP catalytic site, preventing its autoPARylation and further dissociation from DNA. The latter ultimately leads to the collapse of the replication fork and DNA double-strand breaks that cannot be repaired by HR in cancer cells [136]. The increased sensitivity to these drugs in tumor cells with either somatic or germline BRCA1 mutations suggests that the mechanism of HRD does not depend on whether the BRCA1 mutation was inherited or arose during the life of the patient.
Significant disturbances in the mechanism of DSB DNA repair in the absence of active BRCA1 or BRCA2 make cancer cells particularly sensitive to PARP inhibitors, especially in the case of LOH. In this case, the same molecular features that make these cancers more aggressive give them vulnerabilities that may be therapeutically exploited. There have been reports of some very encouraging examples of success of PARP inhibitors against BRCA mutated cancers, especially when they are used against relapsed cancer [137].
However, in treatment of certain types of tumours, for example, BRCA1/2 mutated and HER2 positive breast cancers, efficacy of talazoparib, a potent PARP1/2 inhibitor, did not surpass conventional chemotherapy [138]. This indicates that further personalization of anti-cancer therapy may improve effectiveness of PARP inhibitors as well as reduce its unwarranted use.
8. Future Perspectives and Conclusions
In the past few decades, the clinical significance of BRCA mutations for the rational choice of anti-cancer therapy has been firmly established. In this respect, synthetic lethal interactions between PARPi and BRCA mutation patients is a convincing example of how the fundamental discovery in molecular medicine can be translated into clinical cancer therapy. However, the next step problem is the multifariousness of PARPi resistance mechanisms (recently reviewed in depth by Jackson and Moldavan [139]) that eventually arise in BRCA mutant patients in response to the therapy. In particular, Alu mobile elements regulate the expression of many genes, including the ones that mediate DNA repair [140]. This observation poses an interesting question of whether Alu-repeats can be involved in the DNA damage repair process and serve as a potential mechanism for PARPi resistance in BRCA mutant cells [141].
Furthermore, the recently published data of the clinical trial RITA suggests that patients treated with a PARPi, niraparib, displayed significantly longer PFS compared to the placebo cohort, regardless of the presence or absence of intact HR repair [142]. This result indicates that PARPi might kill cancer cells in ways other than DNA repair.
Theoretically, it can be hypothesized that loss of BRCA by cancer cells should increase their susceptibility to various novel regiments of anti-cancer therapies due to the attenuated DNA repair. For example, viral therapeutical intervention seems as a plausible therapeutic approach to treat BRCAness cancers, especially in combination with PAPRi drugs [143]. However, it should be noted that PARP inhibition may activate genes linked to the normal interferon response in BRCA lacking cells [144], which may explain the molecular basis of interference between the treatment with oncolytic viruses and PARPi. Therefore, one should pay attention to the BRCA mutational status when implementing new oncolytic viruses against breast and/or ovarian cancers.
Managing BRCA1 and BRCA2 pathogenic mutations may include many options other than extensive testing and preventive surgery for such patients. The idea of long-term therapeutical interventions like hormone replacement has long been discussed but poses serious risks of adverse effects [145]. This concept is now re-emerging (discussed in [146]), due to the implementation of drug repurposing (Denosumab, Metformin, Letrozole, see Suppl. Table) as well as principally new approaches like adiponectin receptor targeting molecules [147].
Currently, there is a number of ongoing clinical trials with patients recruited based on their BRCA1/2 status (Table 1 excerpted from Supplementary Table to give a snapshot of modern approaches to employ co-targeting beyond standard cytostatic regimes). However, future perspectives for new specific therapies are much wider. For example, the ubiquitination activity of BRCA1 may become a perspective target for new synthetic lethality drugs [148]. PARP inhibition may be synergistically accompanied by blocking the RAD52 pathway of HR [149]. PARP inhibitors may be converted to more complex molecules with a double specificity mechanism of action [150]. Complex combinations, as expected, should be more effective, although more difficult and time-consuming to develop and adjust to practical regiments. For example, a combination of cisplatin, mitomycin C, and doxorubicin was reported to be more efficient than the respective double combinations [151]. The action of olaparib and other PARP inhibitors may be enhanced by many various supplements, even, for example, by some antioxidants [152]. Combining inhibition of PARP with blocking of ATR by ceralesertib may potentially boost the anticancer effect of already existing PARPi [153]. Further, DNA G-quadruplex binders such as pidnarulex may act in a similar manner, thus increasing the arsenal of drugs for BRCA mutated cancers [154]. Finally, there are multiple ways to boost standard neoadjuvant regiments, for example, by addition of bevacizumab to anthracycline and taxane for patients with BRCA mutations [155].
The p53 tumor suppressor plays an important role in inhibiting cancer progression, especially in response to chemotherapy or targeted therapy. Genomic inactivation of p53 by missense or nonsense mutations often leads to drug resistance in cancer cells. It was previously thought that since wild-type p53 transcriptionally induces the expression of genes involved in DNA repair [156], then p53-mutant cells with attenuated DNA repair would be more sensitive to PARP inhibitors which block homologous DNA repair. Accordingly, deficiency of or mutations in the p53 gene has been shown to enhance the cytotoxicity of PARP inhibition in various tumors with mutations in BRCA1/2 [157]. However, recent studies in colorectal cancer have shown that, contrary to previous findings, wild-type p53 activity appears to be important for a full cytotoxic response to PARP inhibition [158], as PARP inhibitors have been found to activate the p53 pathway [159]. One of the explanations for this phenomenon may be the fact that it is wild-type and not mutant p53 that promotes the export of BRCA1 from the nucleus, increasing cellular deficiency of homologous repair [160]. Another explanation could be that p53 encodes a large number of microRNAs that target genes responsible for the repair of double- and single-stranded DNA breaks [161,162] and thereby increasing the sensitivity of cancer cells to PARP inhibitors.
In this regard, the question arises of whether the combination of PARP and activators of p53 may have synergistic effect. Since Mdm2 is the principal p53-specific E3 ligase that degrades p53 [163], it will be interesting to see whether inhibitors of the p53-Mdm2 interaction can be combined with PARP inhibitors. A number of new Mdm2 inhibitors is currently undergoing clinical trials [164]. Notably, we and our colleagues have also discovered several new inhibitors of p53 interaction with Mdm2, and these molecules exhibited strong apoptotic effect [165,166,167]. Future experiments will show whether the combination of p53 activators and PARP inhibitors is a viable therapeutic approach to treat BRCAness cancers.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, Supplementary Table.
Author Contributions
Conceptualization, N.A.B., D.Y.G. and N.B.P.; writing—original draft preparation, A.P.L., L.S.A., S.D.Z.; writing—review and editing, D.Y.G., A.V.T., O.K.K., S.E.F. N.B.P.; visualization, T.V.K.; funding acquisition, N.A.B. All authors have read and agreed to the published version of the manuscript.
Funding
This work was financed by the Ministry of Science and Higher Education of the Russian Federation within the framework of state support for the creation and development of World-Class Research Centers ‘Digital Biodesign and Personalized Healthcare’ (No 75-15-2020-913).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Authors are grateful to Dr. Y.M. Rozenberg for critical reading of the manuscript.
Conflicts of Interest
The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Hall, J.M.; Lee, M.K.; Newman, B.; Morrow, J.E.; Anderson, L.A.; Huey, B.; King, M.C. Linkage of Early-Onset Familial Breast Cancer to Chromosome 17q21. Science 1990, 250, 1684–1689. [Google Scholar] [CrossRef]
- Wooster, R.; Neuhausen, S.L.; Mangion, J.; Quirk, Y.; Ford, D.; Collins, N.; Nguyen, K.; Seal, S.; Tran, T.; Averill, D. Localization of a Breast Cancer Susceptibility Gene, BRCA2, to Chromosome 13q12-13. Science 1994, 265, 2088–2090. [Google Scholar] [CrossRef] [PubMed]
- King, T.A.; Li, W.; Brogi, E.; Yee, C.J.; Gemignani, M.L.; Olvera, N.; Levine, D.A.; Norton, L.; Robson, M.E.; Offit, K.; et al. Heterogenic Loss of the Wild-Type BRCA Allele in Human Breast Tumorigenesis. Ann Surg Oncol 2007, 14, 2510–2518. [Google Scholar] [CrossRef]
- Martins, F.C.; De, S.; Almendro, V.; Gönen, M.; Park, S.Y.; Blum, J.L.; Herlihy, W.; Ethington, G.; Schnitt, S.J.; Tung, N.; et al. Evolutionary Pathways in BRCA1-Associated Breast Tumors. Cancer Discov 2012, 2, 503–511. [Google Scholar] [CrossRef]
- Kotoula, V.; Fostira, F.; Papadopoulou, K.; Apostolou, P.; Tsolaki, E.; Lazaridis, G.; Manoussou, K.; Zagouri, F.; Pectasides, D.; Vlachos, I.; et al. The Fate of BRCA1-Related Germline Mutations in Triple-Negative Breast Tumors. Am J Cancer Res 2017, 7, 98–114. [Google Scholar]
- Yang, Q.; Yoshimura, G.; Nakamura, M.; Nakamura, Y.; Suzuma, T.; Umemura, T.; Mori, I.; Sakurai, T.; Kakudo, K. BRCA1 in Non-Inherited Breast Carcinomas (Review). Oncol. Rep. 2002, 9, 1329–1333. [Google Scholar] [CrossRef] [PubMed]
- Neff, R.T.; Senter, L.; Salani, R. BRCA Mutation in Ovarian Cancer: Testing, Implications and Treatment Considerations. Ther Adv Med Oncol 2017, 9, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-C.; Lee, Y.-L.; Li, C.-Y. BRCA Genes and Related Cancers: A Meta-Analysis from Epidemiological Cohort Studies. Medicina (Kaunas) 2021, 57, 905. [Google Scholar] [CrossRef]
- Lee, Y.-C.; Lee, Y.-C.; Li, C.-Y.; Lee, Y.-L.; Chen, B.-L. BRCA1 and BRCA2 Gene Mutations and Lung Cancer Sisk: A Meta-Analysis. Medicina (Kaunas) 2020, 56, 212. [Google Scholar] [CrossRef]
- Gilks, C.B.; Prat, J. Ovarian Carcinoma Pathology and Genetics: Recent Advances. Hum Pathol 2009, 40, 1213–1223. [Google Scholar] [CrossRef]
- McCluggage, W.G. Morphological Subtypes of Ovarian Carcinoma: A Review with Emphasis on New Developments and Pathogenesis. Pathology 2011, 43, 420–432. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.I.; Lee, M.; Kim, H.S.; Chung, H.H.; Kim, J.-W.; Park, N.H.; Song, Y.-S. Effect of BRCA Mutational Status on Survival Outcome in Advanced-Stage High-Grade Serous Ovarian Cancer. J Ovarian Res 2019, 12, 40. [Google Scholar] [CrossRef] [PubMed]
- Risch, H.A.; McLaughlin, J.R.; Cole, D.E.; Rosen, B.; Bradley, L.; Kwan, E.; Jack, E.; Vesprini, D.J.; Kuperstein, G.; Abrahamson, J.L.; et al. Prevalence and Penetrance of Germline BRCA1 and BRCA2 Mutations in a Population Series of 649 Women with Ovarian Cancer. Am J Hum Genet 2001, 68, 700–710. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Royer, R.; Li, S.; McLaughlin, J.R.; Rosen, B.; Risch, H.A.; Fan, I.; Bradley, L.; Shaw, P.A.; Narod, S.A. Frequencies of BRCA1 and BRCA2 Mutations among 1,342 Unselected Patients with Invasive Ovarian Cancer. Gynecol Oncol 2011, 121, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Ramus, S.J.; Gayther, S.A. The Contribution of BRCA1 and BRCA2 to Ovarian Cancer. Mol Oncol 2009, 3, 138–150. [Google Scholar] [CrossRef]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.-A.; Mooij, T.M.; Roos-Blom, M.-J.; Jervis, S.; van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef] [PubMed]
- Soegaard, M.; Kjaer, S.K.; Cox, M.; Wozniak, E.; Høgdall, E.; Høgdall, C.; Blaakaer, J.; Jacobs, I.J.; Gayther, S.A.; Ramus, S.J. BRCA1 and BRCA2 Mutation Prevalence and Clinical Characteristics of a Population-Based Series of Ovarian Cancer Cases from Denmark. Clin Cancer Res 2008, 14, 3761–3767. [Google Scholar] [CrossRef] [PubMed]
- Alsop, K.; Fereday, S.; Meldrum, C.; deFazio, A.; Emmanuel, C.; George, J.; Dobrovic, A.; Birrer, M.J.; Webb, P.M.; Stewart, C.; et al. BRCA Mutation Frequency and Patterns of Treatment Response in BRCA Mutation-Positive Women with Ovarian Cancer: A Report from the Australian Ovarian Cancer Study Group. J Clin Oncol 2012, 30, 2654–2663. [Google Scholar] [CrossRef]
- Norquist, B.M.; Brady, M.F.; Harrell, M.I.; Walsh, T.; Lee, M.K.; Gulsuner, S.; Bernards, S.S.; Casadei, S.; Burger, R.A.; Tewari, K.S.; et al. Mutations in Homologous Recombination Genes and Outcomes in Ovarian Carcinoma Patients in GOG 218: An NRG Oncology/Gynecologic Oncology Group Study. Clin Cancer Res 2018, 24, 777–783. [Google Scholar] [CrossRef]
- Tan, D.S.P.; Rothermundt, C.; Thomas, K.; Bancroft, E.; Eeles, R.; Shanley, S.; Ardern-Jones, A.; Norman, A.; Kaye, S.B.; Gore, M.E. “BRCAness” Syndrome in Ovarian Cancer: A Case-Control Study Describing the Clinical Features and Outcome of Patients with Epithelial Ovarian Cancer Associated with BRCA1 and BRCA2 Mutations. J Clin Oncol 2008, 26, 5530–5536. [Google Scholar] [CrossRef]
- Eoh, K.J.; Kim, H.M.; Lee, J.-Y.; Kim, S.; Kim, S.W.; Kim, Y.T.; Nam, E.J. Mutation Landscape of Germline and Somatic BRCA1/2 in Patients with High-Grade Serous Ovarian Cancer. BMC Cancer 2020, 20, 204. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, B.T.J.; Timms, K.M.; Carey, M.S.; Gutin, A.; Meyer, L.A.; Flake, D.D.; Abkevich, V.; Potter, J.; Pruss, D.; Glenn, P.; et al. Somatic Mutations in BRCA1 and BRCA2 Could Expand the Number of Patients That Benefit from Poly (ADP Ribose) Polymerase Inhibitors in Ovarian Cancer. J Clin Oncol 2010, 28, 3570–3576. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib Maintenance Therapy in Platinum-Sensitive Relapsed Ovarian Cancer. N Engl J Med 2012, 366, 1382–1392. [Google Scholar] [CrossRef] [PubMed]
- Pennington, K.P.; Walsh, T.; Harrell, M.I.; Lee, M.K.; Pennil, C.C.; Rendi, M.H.; Thornton, A.; Norquist, B.M.; Casadei, S.; Nord, A.S.; et al. Germline and Somatic Mutations in Homologous Recombination Genes Predict Platinum Response and Survival in Ovarian, Fallopian Tube, and Peritoneal Carcinomas. Clin Cancer Res 2014, 20, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Moschetta, M.; George, A.; Kaye, S.B.; Banerjee, S. BRCA Somatic Mutations and Epigenetic BRCA Modifications in Serous Ovarian Cancer. Ann Oncol 2016, 27, 1449–1455. [Google Scholar] [CrossRef] [PubMed]
- Integrated Genomic Analyses of Ovarian Carcinoma. Nature 2011, 474, 609–615. [CrossRef] [PubMed]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.L.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib Maintenance Therapy in Patients with Platinum-Sensitive Relapsed Serous Ovarian Cancer: A Preplanned Retrospective Analysis of Outcomes by BRCA Status in a Randomised Phase 2 Trial. Lancet Oncol 2014, 15, 852–861. [Google Scholar] [CrossRef] [PubMed]
- Mehrgou, A.; Akouchekian, M. The Importance of BRCA1 and BRCA2 Genes Mutations in Breast Cancer Development. Med J Islam Repub Iran 2016, 30, 369. [Google Scholar]
- Aysola, K.; Desai, A.; Welch, C.; Xu, J.; Qin, Y.; Reddy, V.; Matthews, R.; Owens, C.; Okoli, J.; Beech, D.J.; et al. Triple Negative Breast Cancer—An Overview. Hered. Genet 2013, 2013, 001. [Google Scholar] [CrossRef]
- Kotsopoulos, J. BRCA Mutations and Breast Cancer Prevention. Cancers (Basel) 2018, 10, 524. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Brusco, L.; Daniels, M.; Wathoo, C.; Bailey, A.M.; Strong, L.; Shaw, K.; Lu, K.; Qi, Y.; Zhao, H.; et al. Incidental Germline Variants in 1000 Advanced Cancers on a Prospective Somatic Genomic Profiling Protocol. Ann Oncol 2016, 27, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Tutt, A.; Tovey, H.; Cheang, M.C.U.; Kernaghan, S.; Kilburn, L.; Gazinska, P.; Owen, J.; Abraham, J.; Barrett, S.; Barrett-Lee, P.; et al. Carboplatin in BRCA1/2-Mutated and Triple-Negative Breast Cancer BRCAness Subgroups: The TNT Trial. Nat Med 2018, 24, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Winter, C.; Nilsson, M.P.; Olsson, E.; George, A.M.; Chen, Y.; Kvist, A.; Törngren, T.; Vallon-Christersson, J.; Hegardt, C.; Häkkinen, J.; et al. Targeted Sequencing of BRCA1 and BRCA2 across a Large Unselected Breast Cancer Cohort Suggests That One-Third of Mutations Are Somatic. Ann Oncol 2016, 27, 1532–1538. [Google Scholar] [CrossRef] [PubMed]
- den Brok, W.D.; Schrader, K.A.; Sun, S.; Tinker, A.V.; Zhao, E.Y.; Aparicio, S.; Gelmon, K.A. Homologous Recombination Deficiency in Breast Cancer: A Clinical Review. JCO Precis Oncol 2017, 1, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Bodily, W.R.; Shirts, B.H.; Walsh, T.; Gulsuner, S.; King, M.-C.; Parker, A.; Roosan, M.; Piccolo, S.R. Effects of Germline and Somatic Events in Candidate BRCA-like Genes on Breast-Tumor Signatures. PLoS ONE 2020, 15, e0239197. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Tutt, A.; Ashworth, A. Hallmarks of “BRCAness” in Sporadic Cancers. Nat Rev Cancer 2004, 4, 814–819. [Google Scholar] [CrossRef] [PubMed]
- Rechsteiner, M.; Dedes, K.; Fink, D.; Pestalozzi, B.; Sobottka, B.; Moch, H.; Wild, P.; Varga, Z. Somatic BRCA1 Mutations in Clinically Sporadic Breast Cancer with Medullary Histological Features. J Cancer Res Clin Oncol 2018, 144, 865–874. [Google Scholar] [CrossRef]
- Greer, J.B.; Whitcomb, D.C. Role of BRCA1 and BRCA2 Mutations in Pancreatic Cancer. Gut 2007, 56, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Kowalewski, A.; Szylberg, Ł.; Saganek, M.; Napiontek, W.; Antosik, P.; Grzanka, D. Emerging Strategies in BRCA-Positive Pancreatic Cancer. J Cancer Res Clin Oncol 2018, 144, 1503–1507. [Google Scholar] [CrossRef]
- Luo, G.; Lu, Y.; Jin, K.; Cheng, H.; Guo, M.; Liu, Z.; Long, J.; Liu, C.; Ni, Q.; Yu, X. Pancreatic Cancer: BRCA Mutation and Personalized Treatment. Expert Rev Anticancer Ther 2015, 15, 1223–1231. [Google Scholar] [CrossRef]
- Castro, E.; Eeles, R. The Role of BRCA1 and BRCA2 in Prostate Cancer. Asian J Androl 2012, 14, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Messina, C.; Cattrini, C.; Soldato, D.; Vallome, G.; Caffo, O.; Castro, E.; Olmos, D.; Boccardo, F.; Zanardi, E. BRCA Mutations in Prostate Cancer: Prognostic and Predictive Implications. J Oncol 2020, 2020, 4986365. [Google Scholar] [CrossRef] [PubMed]
- Narod, S.A.; Neuhausen, S.; Vichodez, G.; Armel, S.; Lynch, H.T.; Ghadirian, P.; Cummings, S.; Olopade, O.; Stoppa-Lyonnet, D.; Couch, F.; et al. Rapid Progression of Prostate Cancer in Men with a BRCA2 Mutation. Br J Cancer 2008, 99, 371–374. [Google Scholar] [CrossRef] [PubMed]
- Pal, T.; Vadaparampil, S.; Betts, J.; Miree, C.; Li, S.; Narod, S.A. BRCA1/2 in High-Risk African American Women with Breast Cancer: Providing Genetic Testing through Various Recruitment Strategies. Genet Test 2008, 12, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Ferla, R.; Calò, V.; Cascio, S.; Rinaldi, G.; Badalamenti, G.; Carreca, I.; Surmacz, E.; Colucci, G.; Bazan, V.; Russo, A. Founder Mutations in BRCA1 and BRCA2 Genes. Ann Oncol 2007, 18 (Suppl. 6), vi93–vi98. [Google Scholar] [CrossRef]
- Roa, B.B.; Boyd, A.A.; Volcik, K.; Richards, C.S. Ashkenazi Jewish Population Frequencies for Common Mutations in BRCA1 and BRCA2. Nat Genet 1996, 14, 185–187. [Google Scholar] [CrossRef] [PubMed]
- Janavičius, R. Founder BRCA1/2 Mutations in the Europe: Implications for Hereditary Breast-Ovarian Cancer Prevention and Control. EPMA J 2010, 1, 397–412. [Google Scholar] [CrossRef]
- Sokolenko, A.P.; Sokolova, T.N.; Ni, V.I.; Preobrazhenskaya, E.V.; Iyevleva, A.G.; Aleksakhina, S.N.; Romanko, A.A.; Bessonov, A.A.; Gorodnova, T.V.; Anisimova, E.I.; et al. Frequency and Spectrum of Founder and Non-Founder BRCA1 and BRCA2 Mutations in a Large Series of Russian Breast Cancer and Ovarian Cancer Patients. Breast Cancer Res Treat 2020, 184, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Suspitsin, E.N.; Sherina, N.Y.; Ponomariova, D.N.; Sokolenko, A.P.; Iyevleva, A.G.; Gorodnova, T.V.; Zaitseva, O.A.; Yatsuk, O.S.; Togo, A.V.; Tkachenko, N.N.; et al. High Frequency of BRCA1, but Not CHEK2 or NBS1 (NBN), Founder Mutations in Russian Ovarian Cancer Patients. Hered Cancer Clin Pract. 2009, 7, 5. [Google Scholar] [CrossRef]
- Fabian, D.; Flatt, T. The Evolution of Aging; Nature Education Knowledge, 2011; Volume 3. [Google Scholar]
- Ben-Aharon, I.; Levi, M.; Margel, D.; Yerushalmi, R.; Rizel, S.; Perry, S.; Sharon, E.; Hasky, N.; Abir, R.; Fisch, B.; et al. Premature Ovarian Aging in BRCA Carriers: A Prototype of Systemic Precocious Aging? Oncotarget 2018, 9, 15931–15941. [Google Scholar] [CrossRef] [PubMed]
- Kępczyński, Ł.; Połatyńska, K.; Nykel, A.; Sałamunia, J.; Kałużewski, T.; Kużawczyk, A.; Gach, A. Age of Natural Menopause Onset in BRCA1/2 Carriers—Systematic Review and Meta-Analysis. Prz Menopauzalny 2020, 19, 171–173. [Google Scholar] [CrossRef] [PubMed]
- Drechsel Katja, C.E.; van Tilborg Theodora, C.; Eijkemans Marinus, J.C.; Lentjes Eef, G.W.M.; Irene, H.; Mariette, G.; van Golde Ron, J.T.; Willem, V.; Lichtenbelt Klaske, D.; Broekmans Frank, J.M.; et al. The Impact of BRCA1- and BRCA2 Mutations on Ovarian Reserve Status. Reprod Sci 2022. [Google Scholar] [CrossRef]
- Semmler, L.; Reiter-Brennan, C.; Klein, A. BRCA1 and Breast Cancer: A Review of the Underlying Mechanisms Resulting in the Tissue-Specific Tumorigenesis in Mutation Carriers. J Breast Cancer 2019, 22, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different Roles in a Common Pathway of Genome Protection. Nat Rev Cancer 2011, 12, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, M.H.; Serrano, L. Cell Type-Specific Properties and Environment Shape Tissue Specificity of Cancer Genes. Sci Rep 2016, 6, 20707. [Google Scholar] [CrossRef] [PubMed]
- Ade, C.; Roy-Engel, A.M.; Deininger, P.L. Alu Elements: An Intrinsic Source of Human Genome Instability. Curr Opin Virol 2013, 3, 639–645. [Google Scholar] [CrossRef]
- Welcsh, P.L.; King, M.C. BRCA1 and BRCA2 and the Genetics of Breast and Ovarian Cancer. Hum Mol Genet 2001, 10, 705–713. [Google Scholar] [CrossRef]
- Smith, T.M.; Lee, M.K.; Szabo, C.I.; Jerome, N.; McEuen, M.; Taylor, M.; Hood, L.; King, M.C. Complete Genomic Sequence and Analysis of 117 Kb of Human DNA Containing the Gene BRCA1. Genome Res 1996, 6, 1029–1049. [Google Scholar] [CrossRef]
- Montagna, M.; Santacatterina, M.; Torri, A.; Menin, C.; Zullato, D.; Chieco-Bianchi, L.; D’Andrea, E. Identification of a 3 Kb Alu-Mediated BRCA1 Gene Rearrangement in Two Breast/Ovarian Cancer Families. Oncogene 1999, 18, 4160–4165. [Google Scholar] [CrossRef]
- Girolimetti, G.; Perrone, A.M.; Santini, D.; Barbieri, E.; Guerra, F.; Ferrari, S.; Zamagni, C.; De Iaco, P.; Gasparre, G.; Turchetti, D. BRCA-Associated Ovarian Cancer: From Molecular Genetics to Risk Management. Biomed Res Int 2014, 2014, 787143. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.K.; Han, K.; Wang, J.; Lee, J.; Wang, H.; Callinan, P.A.; Dyer, M.; Cordaux, R.; Liang, P.; Batzer, M.A. Human Genomic Deletions Mediated by Recombination between Alu Elements. Am J Hum Genet 2006, 79, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Bozsik, A.; Pócza, T.; Papp, J.; Vaszkó, T.; Butz, H.; Patócs, A.; Oláh, E. Complex Characterization of Germline Large Genomic Rearrangements of the BRCA1 and BRCA2 Genes in High-Risk Breast Cancer Patients-Novel Variants from a Large National Center. Int J Mol Sci 2020, 21, 4650. [Google Scholar] [CrossRef] [PubMed]
- Unger, M.A.; Nathanson, K.L.; Calzone, K.; Antin-Ozerkis, D.; Shih, H.A.; Martin, A.M.; Lenoir, G.M.; Mazoyer, S.; Weber, B.L. Screening for Genomic Rearrangements in Families with Breast and Ovarian Cancer Identifies BRCA1 Mutations Previously Missed by Conformation-Sensitive Gel Electrophoresis or Sequencing. Am J Hum Genet 2000, 67, 841–850. [Google Scholar] [CrossRef]
- Nordling, M.; Karlsson, P.; Wahlström, J.; Engwall, Y.; Wallgren, A.; Martinsson, T. A Large Deletion Disrupts the Exon 3 Transcription Activation Domain of the BRCA2 Gene in a Breast/Ovarian Cancer Family. Cancer Res 1998, 58, 1372–1375. [Google Scholar] [PubMed]
- Ewald, I.P.; Ribeiro, P.L.I.; Palmero, E.I.; Cossio, S.L.; Giugliani, R.; Ashton-Prolla, P. Genomic Rearrangements in BRCA1 and BRCA2: A Literature Review. Genet Mol Biol 2009, 32, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-F.; Hu, Z.; Rao, N.-Y.; Song, C.-G.; Zhang, B.; Cao, M.-Z.; Su, F.-X.; Wang, Y.-S.; He, P.-Q.; Di, G.-H.; et al. The Prevalence of BRCA1 and BRCA2 Germline Mutations in High-Risk Breast Cancer Patients of Chinese Han Nationality: Two Recurrent Mutations Were Identified. Breast Cancer Res Treat 2008, 110, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Fachal, L.; Blanco, A.; Santamariña, M.; Carracedo, A.; Vega, A. Large Genomic Rearrangements of BRCA1 and BRCA2 among Patients Referred for Genetic Analysis in Galicia (NW Spain): Delimitation and Mechanism of Three Novel BRCA1 Rearrangements. PLoS ONE 2014, 9, e93306. [Google Scholar] [CrossRef]
- Lou, D.I.; McBee, R.M.; Le, U.Q.; Stone, A.C.; Wilkerson, G.K.; Demogines, A.M.; Sawyer, S.L. Rapid Evolution of BRCA1 and BRCA2 in Humans and Other Primates. BMC Evol Biol 2014, 14, 155. [Google Scholar] [CrossRef]
- O’Donovan, P.J.; Livingston, D.M. BRCA1 and BRCA2: Breast/Ovarian Cancer Susceptibility Gene Products and Participants in DNA Double-Strand Break Repair. Carcinogenesis 2010, 31, 961–967. [Google Scholar] [CrossRef]
- Hemel, D.; Domchek, S.M. Breast Cancer Predisposition Syndromes. Hematol Oncol Clin North Am 2010, 24, 799–814. [Google Scholar] [CrossRef] [PubMed]
- Preisler-Adams, S.; Schönbuchner, I.; Fiebig, B.; Welling, B.; Dworniczak, B.; Weber, B.H.F. Gross Rearrangements in BRCA1 but Not BRCA2 Play a Notable Role in Predisposition to Breast and Ovarian Cancer in High-Risk Families of German Origin. Cancer Genet Cytogenet 2006, 168, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Rohlfs, E.M.; Puget, N.; Graham, M.L.; Weber, B.L.; Garber, J.E.; Skrzynia, C.; Halperin, J.L.; Lenoir, G.M.; Silverman, L.M.; Mazoyer, S. An Alu-Mediated 7.1 Kb Deletion of BRCA1 Exons 8 and 9 in Breast and Ovarian Cancer Families That Results in Alternative Splicing of Exon 10. Genes Chromosomes Cancer 2000, 28, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Agata, S.; Dalla Palma, M.; Callegaro, M.; Scaini, M.C.; Menin, C.; Ghiotto, C.; Nicoletto, O.; Zavagno, G.; Chieco-Bianchi, L.; D’Andrea, E.; et al. Large Genomic Deletions Inactivate the BRCA2 Gene in Breast Cancer Families. J Med Genet 2005, 42, e64. [Google Scholar] [CrossRef] [PubMed]
- Karhu, R.; Laurila, E.; Kallioniemi, A.; Syrjäkoski, K. Large Genomic BRCA2 Rearrangements and Male Breast Cancer. Cancer Detect Prev 2006, 30, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Woodward, A.M.; Davis, T.A.; Silva, A.G.S.; Kirk, J.A.; Leary, J.A.; kConFab Investigators. Large Genomic Rearrangements of Both BRCA2 and BRCA1 Are a Feature of the Inherited Breast/Ovarian Cancer Phenotype in Selected Families. J Med Genet 2005, 42, e31. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, A.; Santos, C.; Rocha, P.; Pinto, P.; Bizarro, S.; Teixeira, M.R. Molecular Diagnosis of the Portuguese Founder Mutation BRCA2 c.156_157insAlu. Breast Cancer Res Treat 2009, 117, 215–217. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.A.; Reeves, J.R.; Cooke, T.G. BRCA1 and BRCA2 Proteins: Roles in Health and Disease. Mol Pathol 1998, 51, 237–247. [Google Scholar] [CrossRef]
- Brzovic, P.S.; Rajagopal, P.; Hoyt, D.W.; King, M.-C.; Klevit, R.E. Structure of a BRCA1–BARD1 Heterodimeric RING–RING Complex. Nat Struct Mol Biol 2001, 8, 833–837. [Google Scholar] [CrossRef]
- Wang, Y.; Bernhardy, A.J.; Johnson, N. Abstract A23: BRCA1 Mutations in the BRCT Domain Can Be Removed through Alternative Splicing and Induce PARP Inhibitor Resistance. Mol. Cancer Res. 2017, 15, A23. [Google Scholar] [CrossRef]
- Wu, W.; Koike, A.; Takeshita, T.; Ohta, T. The Ubiquitin E3 Ligase Activity of BRCA1 and Its Biological Functions. Cell Div 2008, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, R.; Fukuda, M.; Maeda, I.; Nishikawa, H.; Oyake, D.; Yabuki, Y.; Ogata, H.; Ohta, T. The RING Heterodimer BRCA1-BARD1 Is a Ubiquitin Ligase Inactivated by a Breast Cancer-Derived Mutation. J Biol Chem 2001, 276, 14537–14540. [Google Scholar] [CrossRef] [PubMed]
- Birrane, G.; Varma, A.K.; Soni, A.; Ladias, J.A.A. Crystal Structure of the BARD1 BRCT Domains. Biochemistry 2007, 46, 7706–7712. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Xu, X.L.; Yang, M.C.; Wei, F.; Ayi, T.C.; Bowcock, A.M.; Baer, R. Cell Cycle-Dependent Colocalization of BARD1 and BRCA1 Proteins in Discrete Nuclear Domains. Proc Natl Acad Sci U S A 1997, 94, 12075–12080. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Matsuoka, S.; Ballif, B.A.; Zhang, D.; Smogorzewska, A.; Gygi, S.P.; Elledge, S.J. Abraxas and RAP80 Form a BRCA1 Protein Complex Required for the DNA Damage Response. Science 2007, 316, 1194–1198. [Google Scholar] [CrossRef] [PubMed]
- Paull, T.T.; Cortez, D.; Bowers, B.; Elledge, S.J.; Gellert, M. Direct DNA Binding by Brca1. Proc Natl Acad Sci U S A 2001, 98, 6086–6091. [Google Scholar] [CrossRef]
- Starita, L.M.; Parvin, J.D. The Multiple Nuclear Functions of BRCA1: Transcription, Ubiquitination and DNA Repair. Curr Opin Cell Biol 2003, 15, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Pan, H.; Li, S.; Flesken-Nikitin, A.; Chen, P.L.; Boyer, T.G.; Lee, W.H. Sequence-Specific Transcriptional Corepressor Function for BRCA1 through a Novel Zinc Finger Protein, ZBRK1. Mol Cell 2000, 6, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.L.; Rodriguez, A.M.; Snyder, R.R.; Hankins, G.D.V.; Boehning, D. Structure-Function of the Tumor Suppressor BRCA1. Comput Struct Biotechnol J 2012, 1, e201204005. [Google Scholar] [CrossRef]
- Lee, M.S.; Green, R.; Marsillac, S.M.; Coquelle, N.; Williams, R.S.; Yeung, T.; Foo, D.; Hau, D.D.; Hui, B.; Monteiro, A.N.A.; et al. Comprehensive Analysis of Missense Variations in the BRCT Domain of BRCA1 by Structural and Functional Assays. Cancer Res 2010, 70, 4880–4890. [Google Scholar] [CrossRef]
- Williams, R.S.; Bernstein, N.; Lee, M.S.; Rakovszky, M.L.; Cui, D.; Green, R.; Weinfeld, M.; Glover, J.N.M. Structural Basis for Phosphorylation-Dependent Signaling in the DNA-Damage Response. Biochem Cell Biol 2005, 83, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Willers, H.; Feng, Z.; Ghosh, J.C.; Kim, S.; Weaver, D.T.; Chung, J.H.; Powell, S.N.; Xia, F. Chk2 Phosphorylation of BRCA1 Regulates DNA Double-Strand Break Repair. Mol Cell Biol 2004, 24, 708–718. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR Substrate Analysis Reveals Extensive Protein Networks Responsive to DNA Damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, H.C.; Yaffe, M.B. Kinases That Control the Cell Cycle in Response to DNA Damage: Chk1, Chk2, and MK2. Curr Opin Cell Biol 2009, 21, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Botuyan, M.V.; Zhao, D.; Cui, G.; Mer, E.; Mer, G. Mechanisms of BRCA1-BARD1 Nucleosome Recognition and Ubiquitylation. Nature 2021, 596, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.R.; Clifford, G.; Bonnet, C.; Groth, A.; Wilson, M.D.; Chapman, J.R. BARD1 Reads H2A Lysine 15 Ubiquitination to Direct Homologous Recombination. Nature 2021, 596, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Witus, S.R.; Burrell, A.L.; Farrell, D.P.; Kang, J.; Wang, M.; Hansen, J.M.; Pravat, A.; Tuttle, L.M.; Stewart, M.D.; Brzovic, P.S.; et al. BRCA1/BARD1 Site-Specific Ubiquitylation of Nucleosomal H2A Is Directed by BARD1. Nat Struct Mol Biol 2021, 28, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Jeffrey, P.D.; Miller, J.; Kinnucan, E.; Sun, Y.; Thoma, N.H.; Zheng, N.; Chen, P.-L.; Lee, W.-H.; Pavletich, N.P. BRCA2 Function in DNA Binding and Recombination from a BRCA2-DSS1-SsDNA Structure. Science 2002, 297, 1837–1848. [Google Scholar] [CrossRef]
- Trego, K.S.; Groesser, T.; Davalos, A.R.; Parplys, A.C.; Zhao, W.; Nelson, M.R.; Hlaing, A.; Shih, B.; Rydberg, B.; Pluth, J.M.; et al. Non-Catalytic Roles for XPG with BRCA1 and BRCA2 in Homologous Recombination and Genome Stability. Mol Cell 2016, 61, 535–546. [Google Scholar] [CrossRef]
- Scully, R.; Livingston, D.M. In Search of the Tumour-Suppressor Functions of BRCA1 and BRCA2. Nature 2000, 408, 429–432. [Google Scholar] [CrossRef]
- Deng, C.X.; Scott, F. Role of the Tumor Suppressor Gene Brca1 in Genetic Stability and Mammary Gland Tumor Formation. Oncogene 2000, 19, 1059–1064. [Google Scholar] [CrossRef] [PubMed]
- Foray, N.; Marot, D.; Randrianarison, V.; Venezia, N.D.; Picard, D.; Perricaudet, M.; Favaudon, V.; Jeggo, P. Constitutive Association of BRCA1 and C-Abl and Its ATM-Dependent Disruption after Irradiation. Mol Cell Biol 2002, 22, 4020–4032. [Google Scholar] [CrossRef] [PubMed]
- Levav-Cohen, Y.; Goldberg, Z.; Zuckerman, V.; Grossman, T.; Haupt, S.; Haupt, Y. C-Abl as a Modulator of P53. Biochem Biophys Res Commun 2005, 331, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Hantschel, O.; Rix, U.; Schmidt, U.; Bürckstümmer, T.; Kneidinger, M.; Schütze, G.; Colinge, J.; Bennett, K.L.; Ellmeier, W.; Valent, P.; et al. The Btk Tyrosine Kinase Is a Major Target of the Bcr-Abl Inhibitor Dasatinib. Proc Natl Acad Sci U S A 2007, 104, 13283–13288. [Google Scholar] [CrossRef] [PubMed]
- Althubiti, M.; Rada, M.; Samuel, J.; Escorsa, J.M.; Najeeb, H.; Lee, K.-G.; Lam, K.-P.; Jones, G.D.D.; Barlev, N.A.; Macip, S. BTK Modulates P53 Activity to Enhance Apoptotic and Senescent Responses. Cancer Res 2016, 76, 5405–5414. [Google Scholar] [CrossRef] [PubMed]
- Rada, M.; Barlev, N.; Macip, S. BTK: A Two-Faced Effector in Cancer and Tumour Suppression. Cell Death Dis 2018, 9, 1064. [Google Scholar] [CrossRef]
- Xu, X.; Qiao, W.; Linke, S.P.; Cao, L.; Li, W.M.; Furth, P.A.; Harris, C.C.; Deng, C.X. Genetic Interactions between Tumor Suppressors Brca1 and P53 in Apoptosis, Cell Cycle and Tumorigenesis. Nat Genet 2001, 28, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Somasundaram, K.; Peng, Y.; Tian, H.; Zhang, H.; Bi, D.; Weber, B.L.; El-Deiry, W.S. BRCA1 Physically Associates with P53 and Stimulates Its Transcriptional Activity. Oncogene 1998, 16, 1713–1721. [Google Scholar] [CrossRef]
- Pietrasik, S.; Zajac, G.; Morawiec, J.; Soszynski, M.; Fila, M.; Blasiak, J. Interplay between BRCA1 and GADD45A and Its Potential for Nucleotide Excision Repair in Breast Cancer Pathogenesis. Int J Mol Sci 2020, 21, 870. [Google Scholar] [CrossRef]
- Clarke, C.L.; Sandle, J.; Jones, A.A.; Sofronis, A.; Patani, N.R.; Lakhani, S.R. Mapping Loss of Heterozygosity in Normal Human Breast Cells from BRCA1/2 Carriers. Br J Cancer 2006, 95, 515–519. [Google Scholar] [CrossRef]
- Armes, J.E.; Egan, A.J.M.; Southey, M.C.; Dite, G.S.; McCredie, M.R.E.; Giles, G.G.; Hopper, J.L.; Venter, D.J. The Histologic Phenotypes of Breast Carcinoma Occurring before Age 40 Years in Women with and without BRCA1 or BRCA2 Germline Mutations. Cancer 1998, 83, 2335–2345. [Google Scholar] [CrossRef]
- Mote, P.A.; Leary, J.A.; Avery, K.A.; Sandelin, K.; Chenevix-Trench, G.; Kirk, J.A.; Clarke, C.L. Germ-Line Mutations in BRCA1 or BRCA2 in the Normal Breast Are Associated with Altered Expression of Estrogen-Responsive Proteins and the Predominance of Progesterone Receptor A. Genes Chromosomes Cancer 2004, 39, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Ingthorsson, S.; Traustadottir, G.A.; Gudjonsson, T. Cellular Plasticity and Heterotypic Interactions during Breast Morphogenesis and Cancer Initiation. Cancers (Basel) 2022, 14, 5209. [Google Scholar] [CrossRef] [PubMed]
- Avşar Abdik, E. Differentiated Pre-Adipocytes Promote Proliferation, Migration and Epithelial-Mesenchymal Transition in Breast Cancer Cells of Different P53 Status. Mol Biol Rep 2021, 48, 5187–5198. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Cha, Y.J.; Koo, J.S. Adipocyte Biology in Breast Cancer: From Silent Bystander to Active Facilitator. Prog Lipid Res 2018, 69, 11–20. [Google Scholar] [CrossRef]
- Takehara, M.; Sato, Y.; Kimura, T.; Noda, K.; Miyamoto, H.; Fujino, Y.; Miyoshi, J.; Nakamura, F.; Wada, H.; Bando, Y.; et al. Cancer-Associated Adipocytes Promote Pancreatic Cancer Progression through SAA1 Expression. Cancer Sci 2020, 111, 2883–2894. [Google Scholar] [CrossRef]
- Kothari, C.; Diorio, C.; Durocher, F. The Importance of Breast Adipose Tissue in Breast Cancer. Int J Mol Sci 2020, 21, 5760. [Google Scholar] [CrossRef]
- Yao, H.; He, S. Multi-faceted Role of Cancer-associated Adipocytes in the Tumor Microenvironment (Review). Mol Med Rep 2021, 24, 866. [Google Scholar] [CrossRef] [PubMed]
- Jafari, N.; Kolla, M.; Meshulam, T.; Shafran, J.S.; Qiu, Y.; Casey, A.N.; Pompa, I.R.; Ennis, C.S.; Mazzeo, C.S.; Rabhi, N.; et al. Adipocyte-Derived Exosomes May Promote Breast Cancer Progression in Type 2 Diabetes. Sci Signal 2021, 14, eabj2807. [Google Scholar] [CrossRef]
- Lee, Y.; Jung, W.H.; Koo, J.S. Adipocytes Can Induce Epithelial-Mesenchymal Transition in Breast Cancer Cells. Breast Cancer Res Treat 2015, 153, 323–335. [Google Scholar] [CrossRef]
- Xie, Y.; Wang, B.; Zhao, Y.; Tao, Z.; Wang, Y.; Chen, G.; Hu, X. Mammary Adipocytes Protect Triple-Negative Breast Cancer Cells from Ferroptosis. J Hematol Oncol 2022, 15, 72. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; He, X.; Tong, C.; Li, H.; Xie, C.; Wu, Y.; Wang, L.; Yan, X.; Luo, D.; Tang, Y.; et al. Cancer-Associated Adipocytes Promote the Invasion and Metastasis in Breast Cancer through LIF/CXCLs Positive Feedback Loop. Int J Biol Sci 2022, 18, 1363–1380. [Google Scholar] [CrossRef] [PubMed]
- Jones, L.P.; Buelto, D.; Tago, E.; Owusu-Boaitey, K.E. Abnormal Mammary Adipose Tissue Environment of Brca1 Mutant Mice Show a Persistent Deposition of Highly Vascularized Multilocular Adipocytes. J Cancer Sci Ther 2011, 004. [Google Scholar] [CrossRef] [PubMed]
- Miran, I.; Scherer, D.; Ostyn, P.; Mazouni, C.; Drusch, F.; Bernard, M.; Louvet, E.; Adam, J.; Mathieu, M.-C.; Haffa, M.; et al. Adipose Tissue Properties in Tumor-Bearing Breasts. Front Oncol 2020, 10, 1506. [Google Scholar] [CrossRef]
- Koellensperger, E.; Bonnert, L.-C.; Zoernig, I.; Marmé, F.; Sandmann, S.; Germann, G.; Gramley, F.; Leimer, U. The Impact of Human Adipose Tissue-Derived Stem Cells on Breast Cancer Cells: Implications for Cell-Assisted Lipotransfers in Breast Reconstruction. Stem Cell Res Ther 2017, 8, 121. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; He, Y.; Yu, X. Bone Marrow Adipocyte: An Intimate Partner With Tumor Cells in Bone Metastasis. Front Endocrinol (Lausanne) 2018, 9, 339. [Google Scholar] [CrossRef] [PubMed]
- De Talhouet, S.; Peron, J.; Vuilleumier, A.; Friedlaender, A.; Viassolo, V.; Ayme, A.; Bodmer, A.; Treilleux, I.; Lang, N.; Tille, J.-C.; et al. Clinical Outcome of Breast Cancer in Carriers of BRCA1 and BRCA2 Mutations According to Molecular Subtypes. Sci Rep 2020, 10, 7073. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Ceccaldi, R.; Shapiro, G.I.; D’Andrea, A.D. Homologous Recombination Deficiency: Exploiting the Fundamental Vulnerability of Ovarian Cancer. Cancer Discov 2015, 5, 1137–1154. [Google Scholar] [CrossRef] [PubMed]
- Torrisi, R.; Zuradelli, M.; Agostinetto, E.; Masci, G.; Losurdo, A.; De Sanctis, R.; Santoro, A. Platinum Salts in the Treatment of BRCA-Associated Breast Cancer: A True Targeted Chemotherapy? Crit Rev Oncol Hematol 2019, 135, 66–75. [Google Scholar] [CrossRef]
- Giannone, G.; Scotto, G.; Katsaros, D.; De Giorgi, U.; Farolfi, A.; Borella, F.; Cosma, S.; Ferrero, A.; Mangiacotti, S.; Villa, M.; et al. Hypersensitivity to Platinum Salts According to BRCA Status in Ovarian Cancer: A Retrospective Analysis of Clinical Outcomes and Systematic Review of Literature. Gynecol Oncol 2021, 162, 80–87. [Google Scholar] [CrossRef]
- Wattenberg, M.M.; Asch, D.; Yu, S.; O’Dwyer, P.J.; Domchek, S.M.; Nathanson, K.L.; Rosen, M.A.; Beatty, G.L.; Siegelman, E.S.; Reiss, K.A. Platinum Response Characteristics of Patients with Pancreatic Ductal Adenocarcinoma and a Germline BRCA1, BRCA2 or PALB2 Mutation. Br J Cancer 2020, 122, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Byrski, T.; Huzarski, T.; Dent, R.; Gronwald, J.; Zuziak, D.; Cybulski, C.; Kladny, J.; Gorski, B.; Lubinski, J.; Narod, S.A. Response to Neoadjuvant Therapy with Cisplatin in BRCA1-Positive Breast Cancer Patients. Breast Cancer Res Treat 2009, 115, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, K.N.; Wubbenhorst, B.; Wenz, B.M.; De Sloover, D.; Pluta, J.; Emery, L.; Barrett, A.; Kraya, A.A.; Anastopoulos, I.N.; Yu, S.; et al. BRCA Locus-Specific Loss of Heterozygosity in Germline BRCA1 and BRCA2 Carriers. Nat Commun 2017, 8, 319. [Google Scholar] [CrossRef] [PubMed]
- Afghahi, A.; Timms, K.M.; Vinayak, S.; Jensen, K.C.; Kurian, A.W.; Carlson, R.W.; Chang, P.-J.; Schackmann, E.; Hartman, A.-R.; Ford, J.M.; et al. Tumor BRCA1 Reversion Mutation Arising during Neoadjuvant Platinum-Based Chemotherapy in Triple-Negative Breast Cancer Is Associated with Therapy Resistance. Clin Cancer Res 2017, 23, 3365–3370. [Google Scholar] [CrossRef] [PubMed]
- Pilié, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-Art Strategies for Targeting the DNA Damage Response in Cancer. Nat Rev Clin Oncol 2019, 16, 81–104. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Lord, C.J.; Serra, V.; Tutt, A.; Balmaña, J.; Castroviejo-Bermejo, M.; Cruz, C.; Oaknin, A.; Kaye, S.B.; de Bono, J.S. A Decade of Clinical Development of PARP Inhibitors in Perspective. Ann Oncol 2019, 30, 1437–1447. [Google Scholar] [CrossRef] [PubMed]
- Bredow, K.; Blümcke, B.; Schneider, S.; Püsken, M.; Schmutzler, R.; Rhiem, K. Long-Term Survival of a BRCA2 Mutation Carrier Following Second Ovarian Cancer Relapse Using PARPi Therapy: A Case Report. Mol Clin Oncol 2022, 17, 137. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Yu, X. Comparison between Talazoparib and Conventional Chemotherapy in the Treatment of HER2-Positive Breast Cancer Patients: A Retrospective Study. Front Immunol 2022, 13, 901636. [Google Scholar] [CrossRef] [PubMed]
- Jackson, L.M.; Moldovan, G.-L. Mechanisms of PARP1 Inhibitor Resistance and Their Implications for Cancer Treatment. NAR Cancer 2022, 4, zcac042. [Google Scholar] [CrossRef]
- Mustafina, O.E. The Possible Roles of Human Alu Elements in Aging. Front Genet 2013, 4, 96. [Google Scholar] [CrossRef]
- Morales, M.E.; White, T.B.; Streva, V.A.; DeFreece, C.B.; Hedges, D.J.; Deininger, P.L. The Contribution of Alu Elements to Mutagenic DNA Double-Strand Break Repair. PLoS Genet 2015, 11, e1005016. [Google Scholar] [CrossRef] [PubMed]
- González-Martín, A.; Pothuri, B.; Vergote, I.; DePont Christensen, R.; Graybill, W.; Mirza, M.R.; McCormick, C.; Lorusso, D.; Hoskins, P.; Freyer, G.; et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N Engl J Med 2019, 381, 2391–2402. [Google Scholar] [CrossRef] [PubMed]
- Tookman, L.A.; Browne, A.K.; Connell, C.M.; Bridge, G.; Ingemarsdotter, C.K.; Dowson, S.; Shibata, A.; Lockley, M.; Martin, S.A.; McNeish, I.A. RAD51 and BRCA2 Enhance Oncolytic Adenovirus Type 5 Activity in Ovarian Cancer. Mol Cancer Res 2016, 14, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Reisländer, T.; Lombardi, E.P.; Groelly, F.J.; Miar, A.; Porru, M.; Di Vito, S.; Wright, B.; Lockstone, H.; Biroccio, A.; Harris, A.; et al. BRCA2 Abrogation Triggers Innate Immune Responses Potentiated by Treatment with PARP Inhibitors. Nat Commun 2019, 10, 3143. [Google Scholar] [CrossRef] [PubMed]
- Loizzi, V.; Dellino, M.; Cerbone, M.; Arezzo, F.; Cazzato, G.; Damiani, G.R.; Pinto, V.; Silvestris, E.; Kardhashi, A.; Cicinelli, E.; et al. The Role of Hormonal Replacement Therapy in BRCA Mutated Patients: Lights and Shadows. Int J Mol Sci 2023, 24, 764. [Google Scholar] [CrossRef] [PubMed]
- Singer, C.F. Nonsurgical Prevention Strategies in BRCA1 and BRCA2 Mutation Carriers. Breast Care (Basel) 2021, 16, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Nehme, R.; Diab-Assaf, M.; Decombat, C.; Delort, L.; Caldefie-Chezet, F. Targeting Adiponectin in Breast Cancer. Biomedicines 2022, 10, 2958. [Google Scholar] [CrossRef]
- Zong, D.; Adam, S.; Wang, Y.; Sasanuma, H.; Callén, E.; Murga, M.; Day, A.; Kruhlak, M.J.; Wong, N.; Munro, M.; et al. BRCA1 Haploinsufficiency Is Masked by RNF168-Mediated Chromatin Ubiquitylation. Mol Cell 2019, 73, 1267–1281.e7. [Google Scholar] [CrossRef]
- Sullivan-Reed, K.; Bolton-Gillespie, E.; Dasgupta, Y.; Langer, S.; Siciliano, M.; Nieborowska-Skorska, M.; Hanamshet, K.; Belyaeva, E.A.; Bernhardy, A.J.; Lee, J.; et al. Simultaneous Targeting of PARP1 and RAD52 Triggers Dual Synthetic Lethality in BRCA-Deficient Tumor Cells. Cell Rep 2018, 23, 3127–3136. [Google Scholar] [CrossRef]
- Kayumov, M.; Jia, L.; Pardaev, A.; Song, S.-S.; Mirzaakhmedov, S.; Ding, C.; Cheng, Y.-J.; Zhang, R.I.; Bao, X.; Miao, Z.-H.; et al. Design, Synthesis and Pharmacological Evaluation of New PARP1 Inhibitors by Merging Pharmacophores of Olaparib and the Natural Product Alantolactone. Eur J Med Chem 2022, 240, 114574. [Google Scholar] [CrossRef]
- Gorodnova, T.V.; Sokolenko, A.P.; Kotiv, K.B.; Sokolova, T.N.; Ivantsov, A.O.; Guseynov, K.D.; Nekrasova, E.A.; Smirnova, O.A.; Berlev, I.V.; Imyanitov, E.N. Neoadjuvant Therapy of BRCA1-Driven Ovarian Cancer by Combination of Cisplatin, Mitomycin C and Doxorubicin. Hered Cancer Clin Pr. 2021, 19, 14. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Chatterjee, S.; Paul, S.; Das, B.; Dash, S.R.; Das, C.; Kundu, C.N. Olaparib Enhances the Resveratrol-Mediated Apoptosis in Breast Cancer Cells by Inhibiting the Homologous Recombination Repair Pathway. Exp Cell Res 2022, 420, 113338. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Xu, H.; George, E.; Hallberg, D.; Kumar, S.; Jagannathan, V.; Medvedev, S.; Kinose, Y.; Devins, K.; Verma, P.; et al. Combining PARP with ATR Inhibition Overcomes PARP Inhibitor and Platinum Resistance in Ovarian Cancer Models. Nat Commun 2020, 11, 3726. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Hurley, L.H. A First-in-Class Clinical G-Quadruplex-Targeting Drug. The Bench-to-Bedside Translation of the Fluoroquinolone QQ58 to CX-5461 (Pidnarulex). Bioorg Med Chem Lett 2022, 77, 129016. [Google Scholar] [CrossRef] [PubMed]
- Fasching, P.A.; Loibl, S.; Hu, C.; Hart, S.N.; Shimelis, H.; Moore, R.; Schem, C.; Tesch, H.; Untch, M.; Hilfrich, J.; et al. BRCA1/2 Mutations and Bevacizumab in the Neoadjuvant Treatment of Breast Cancer: Response and Prognosis Results in Patients With Triple-Negative Breast Cancer From the GeparQuinto Study. J Clin Oncol 2018, 36, 2281–2287. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of P53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Williamson, C.T.; Kubota, E.; Hamill, J.D.; Klimowicz, A.; Ye, R.; Muzik, H.; Dean, M.; Tu, L.; Gilley, D.; Magliocco, A.M.; et al. Enhanced Cytotoxicity of PARP Inhibition in Mantle Cell Lymphoma Harbouring Mutations in Both ATM and P53. EMBO Mol Med 2012, 4, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Smeby, J.; Kryeziu, K.; Berg, K.C.G.; Eilertsen, I.A.; Eide, P.W.; Johannessen, B.; Guren, M.G.; Nesbakken, A.; Bruun, J.; Lothe, R.A.; et al. Molecular Correlates of Sensitivity to PARP Inhibition beyond Homologous Recombination Deficiency in Pre-Clinical Models of Colorectal Cancer Point to Wild-Type TP53 Activity. EBioMedicine 2020, 59, 102923. [Google Scholar] [CrossRef]
- Hong, T.; Lei, G.; Chen, X.; Li, H.; Zhang, X.; Wu, N.; Zhao, Y.; Zhang, Y.; Wang, J. PARP Inhibition Promotes Ferroptosis via Repressing SLC7A11 and Synergizes with Ferroptosis Inducers in BRCA-Proficient Ovarian Cancer. Redox Biol 2021, 42, 101928. [Google Scholar] [CrossRef]
- Feng, Z.; Kachnic, L.; Zhang, J.; Powell, S.N.; Xia, F. DNA Damage Induces P53-Dependent BRCA1 Nuclear Export. J Biol Chem 2004, 279, 28574–28584. [Google Scholar] [CrossRef]
- Parfenyev, S.; Singh, A.; Fedorova, O.; Daks, A.; Kulshreshtha, R.; Barlev, N.A. Interplay between P53 and Non-Coding RNAs in the Regulation of EMT in Breast Cancer. Cell Death Dis 2021, 12, 17. [Google Scholar] [CrossRef] [PubMed]
- Barlev, N.A.; Sayan, B.S.; Candi, E.; Okorokov, A.L. The MicroRNA and P53 Families Join Forces against Cancer. Cell Death Differ 2010, 17, 373–375. [Google Scholar] [CrossRef] [PubMed]
- Lezina, L.; Aksenova, V.; Fedorova, O.; Malikova, D.; Shuvalov, O.; Antonov, A.V.; Tentler, D.; Garabadgiu, A.V.; Melino, G.; Barlev, N.A. KMT Set7/9 Affects Genotoxic Stress Response via the Mdm2 Axis. Oncotarget 2015, 6, 25843–25855. [Google Scholar] [CrossRef] [PubMed]
- Portman, N.; Milioli, H.H.; Alexandrou, S.; Coulson, R.; Yong, A.; Fernandez, K.J.; Chia, K.M.; Halilovic, E.; Segara, D.; Parker, A.; et al. MDM2 Inhibition in Combination with Endocrine Therapy and CDK4/6 Inhibition for the Treatment of ER-Positive Breast Cancer. Breast Cancer Res 2020, 22, 87. [Google Scholar] [CrossRef] [PubMed]
- Bulatov, E.; Sayarova, R.; Mingaleeva, R.; Miftakhova, R.; Gomzikova, M.; Ignatyev, Y.; Petukhov, A.; Davidovich, P.; Rizvanov, A.; Barlev, N.A. Isatin-Schiff Base-Copper (II) Complex Induces Cell Death in P53-Positive Tumors. Cell Death Discov 2018, 4, 103. [Google Scholar] [CrossRef] [PubMed]
- Davidovich, P.; Aksenova, V.; Petrova, V.; Tentler, D.; Orlova, D.; Smirnov, S.; Gurzhiy, V.; Okorokov, A.L.; Garabadzhiu, A.; Melino, G.; et al. Discovery of Novel Isatin-Based P53 Inducers. ACS Med Chem Lett 2015, 6, 856–860. [Google Scholar] [CrossRef]
- Fedorova, O.; Daks, A.; Petrova, V.; Petukhov, A.; Lezina, L.; Shuvalov, O.; Davidovich, P.; Kriger, D.; Lomert, E.; Tentler, D.; et al. Novel Isatin-Derived Molecules Activate P53 via Interference with Mdm2 to Promote Apoptosis. Cell Cycle 2018, 17, 1917–1930. [Google Scholar] [CrossRef]
Figure 3.
Alignment of the exon-intron structure of BRCA1 and the domain composition of the corresponding protein. Note that exon 10 is usually referred to as exon 11 for historical reasons.
Figure 3.
Alignment of the exon-intron structure of BRCA1 and the domain composition of the corresponding protein. Note that exon 10 is usually referred to as exon 11 for historical reasons.
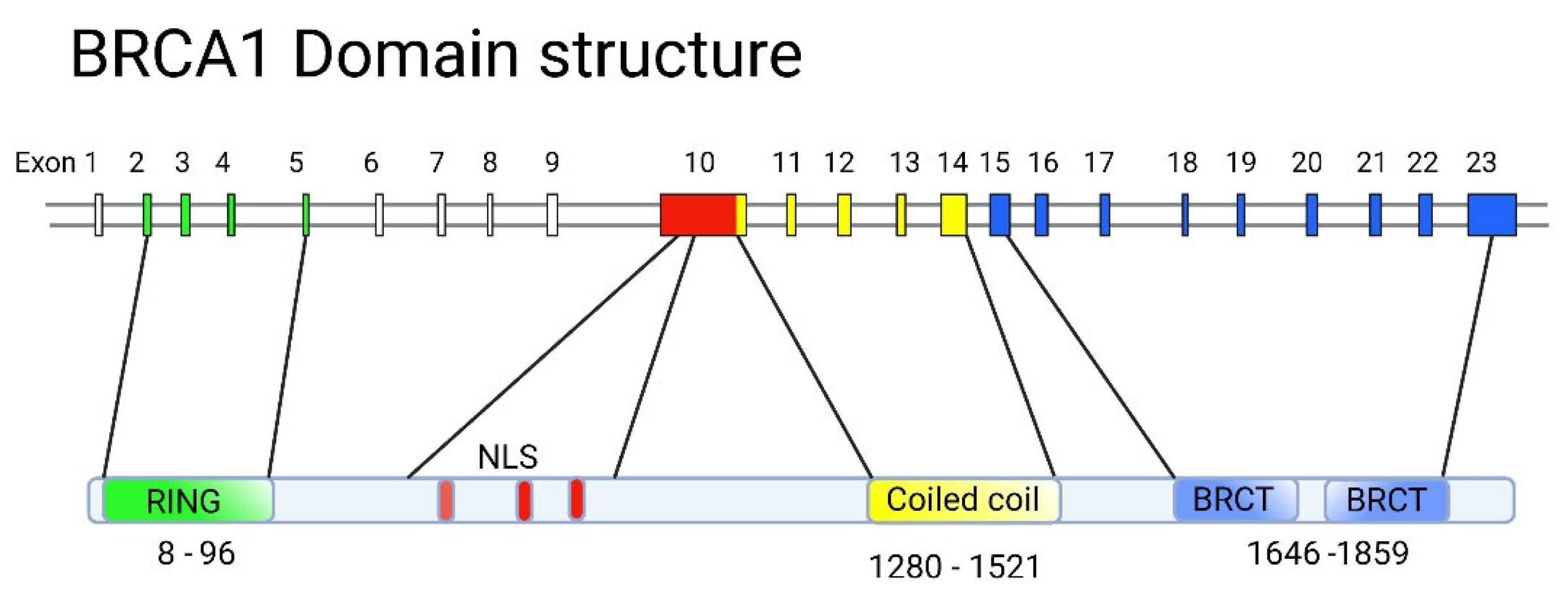
Figure 4.
Major protein interactions aligned with regions of BRCA1.
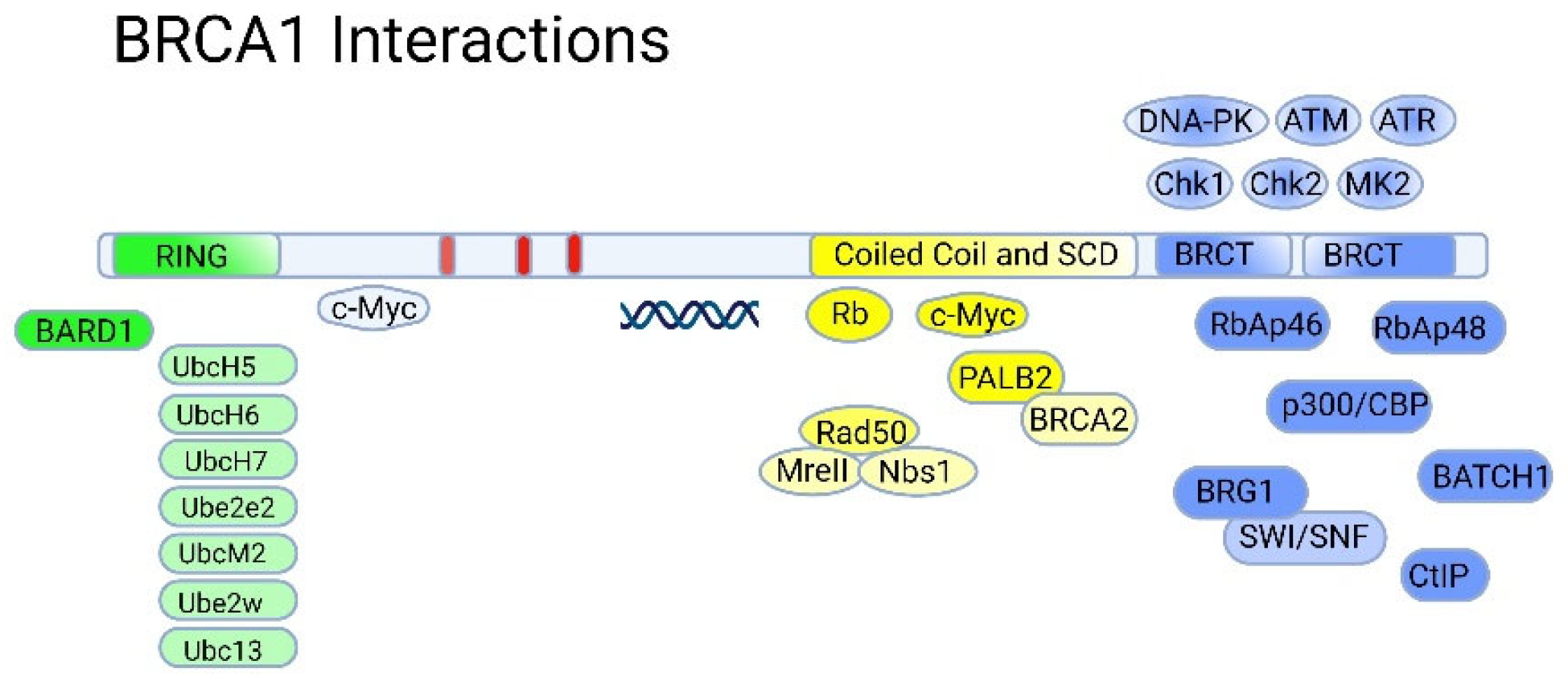
Figure 5.
Major functional protein-protein interactions of BRCA1 and BRCA2 and functional roles of these complexes. A – DNA homologous recombination; B - G2/M transition after recovery from DNA damage; C - DNA damage resistance, and G2-M checkpoint control; D – regulation of immediate cellular response to single-strand DNA damage; E – histone ubiquitination concurrently with transcription regulation and DNA repair; G – response to double strand DNA damage.
Figure 5.
Major functional protein-protein interactions of BRCA1 and BRCA2 and functional roles of these complexes. A – DNA homologous recombination; B - G2/M transition after recovery from DNA damage; C - DNA damage resistance, and G2-M checkpoint control; D – regulation of immediate cellular response to single-strand DNA damage; E – histone ubiquitination concurrently with transcription regulation and DNA repair; G – response to double strand DNA damage.
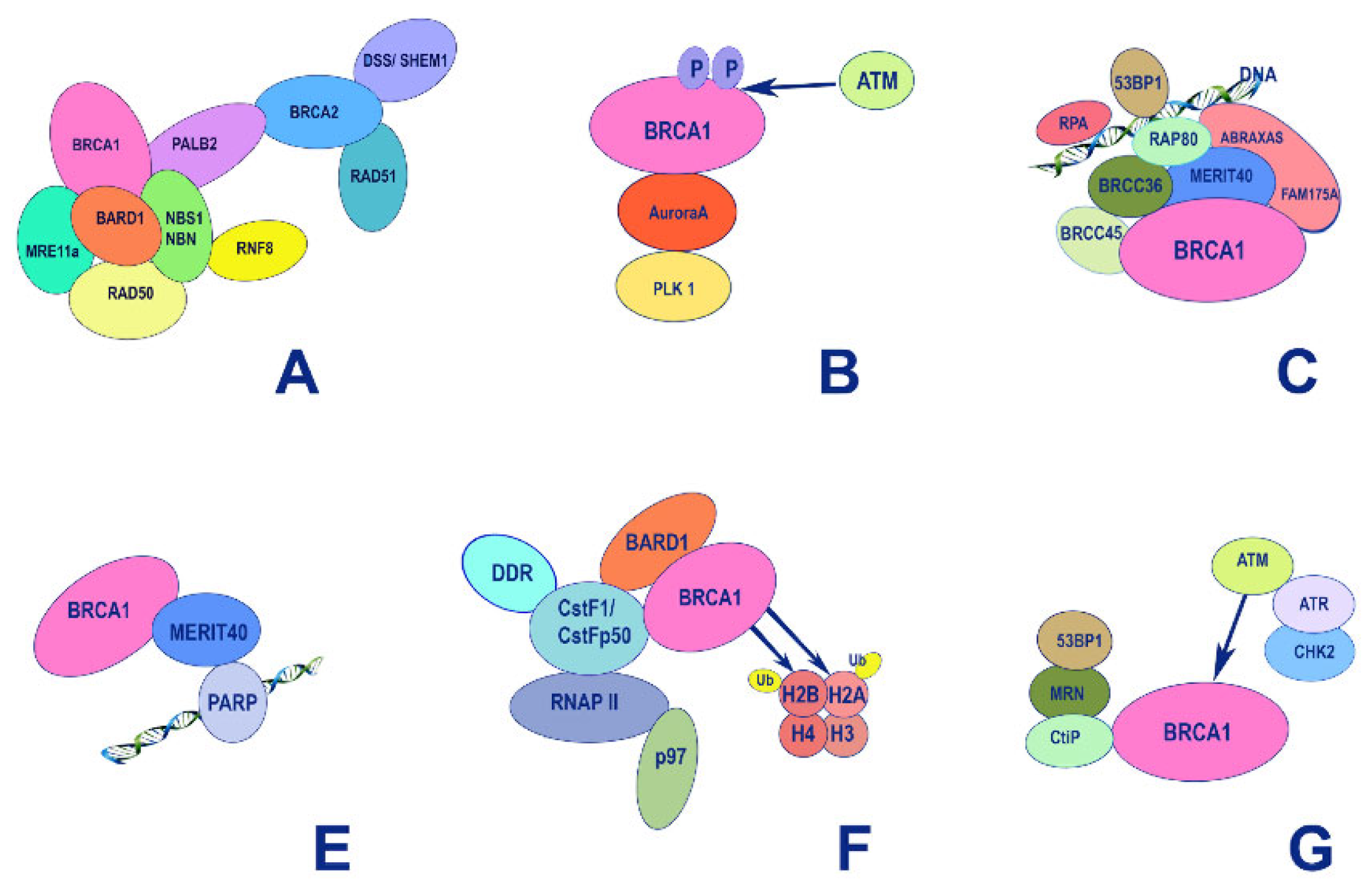
Figure 6.
A—Three-dimensional structure of the nucleosome in complex with BRCA1 and BARD1 fragments (BRCA1 – red, BARD1 – blue, histone 2A – green); B—structure of BRCA2 with DSS and ssDNA (DSS1—red, ssDNA—black) (adopted from XXX).
Figure 6.
A—Three-dimensional structure of the nucleosome in complex with BRCA1 and BARD1 fragments (BRCA1 – red, BARD1 – blue, histone 2A – green); B—structure of BRCA2 with DSS and ssDNA (DSS1—red, ssDNA—black) (adopted from XXX).
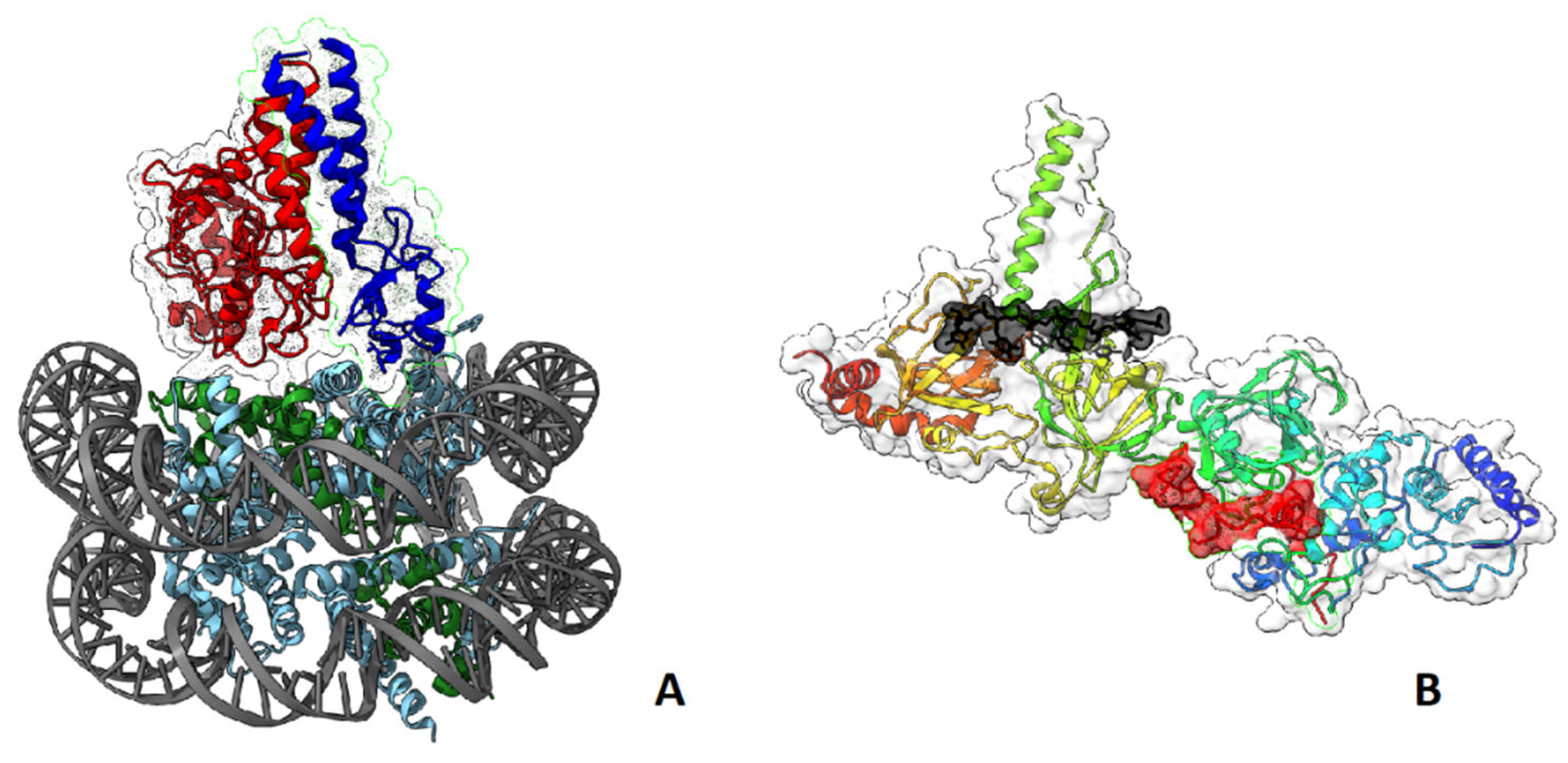
Figure 7.
Structural formulae of PARP inhibitors. Upper row – PARP inhibitors already approved for clinical use. Lower row – some new perspective compounds with sponsored trials. Arrow indicates prodrug conversion. Box indicates unsatisfactory clinical data.
Figure 7.
Structural formulae of PARP inhibitors. Upper row – PARP inhibitors already approved for clinical use. Lower row – some new perspective compounds with sponsored trials. Arrow indicates prodrug conversion. Box indicates unsatisfactory clinical data.
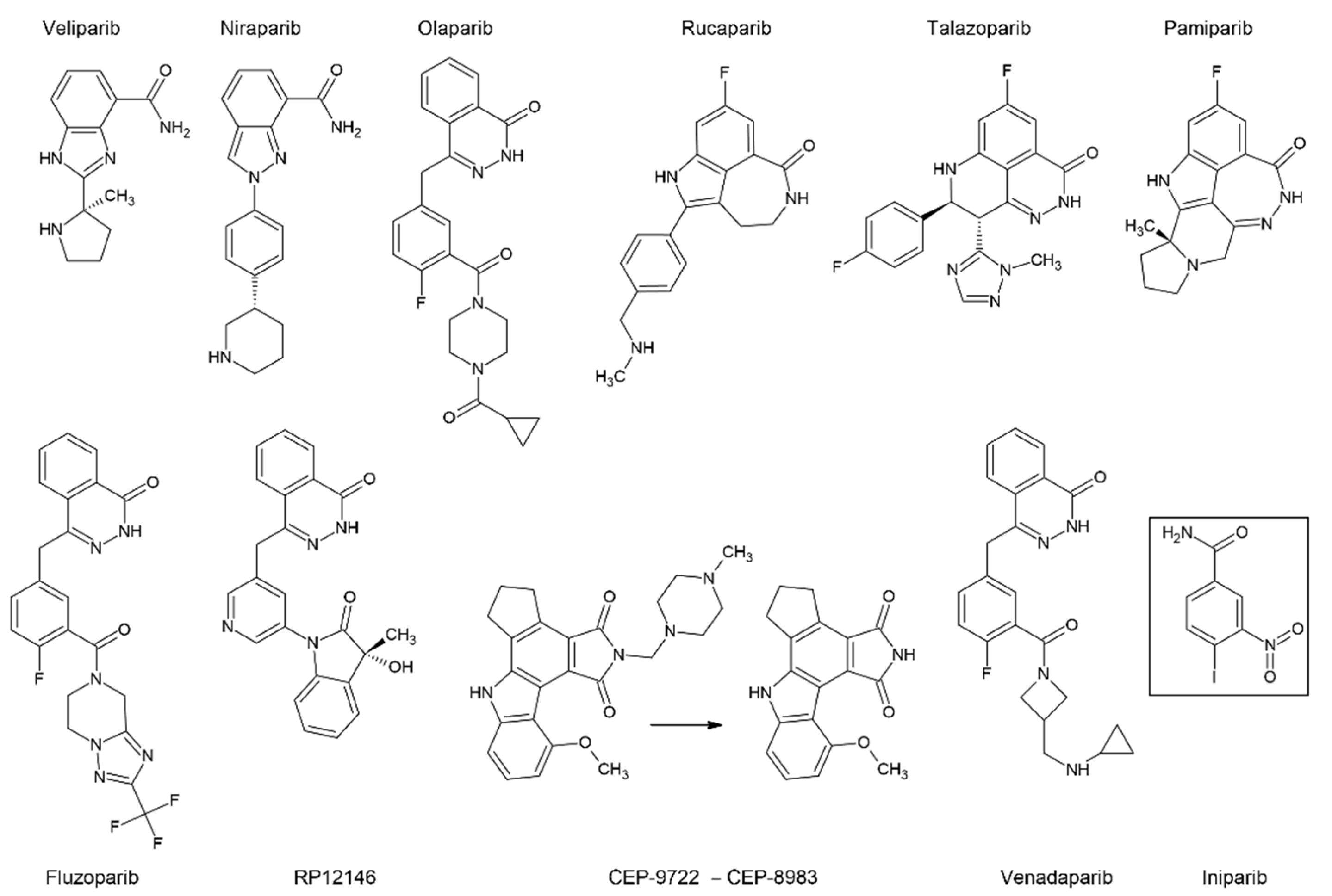

Table 1.
Representative ongoing clinical trials of drug combinations (PARPi and targeted therapies) against cancers with BRCA1,2 inactivating mutations.
Table 1.
Representative ongoing clinical trials of drug combinations (PARPi and targeted therapies) against cancers with BRCA1,2 inactivating mutations.
| PARP Inhibitor |
Cancer Type | Co-Target | Co-Treatment | Phase | Register Number |
|---|---|---|---|---|---|
| Olaparib | BC,OC,FTC,EndA, UCC | mTORC1/2 or AKT |
Vistusertib or Capivasertib | 1b | NCT02208375 |
| Olaparib | OC, FTC, PPC | CTLA-4 | Tremelimumab | 2 | NCT02571725 |
| Talazoparib | TNBC | mTOR/PI3K | Gedatolisib | 2 | NCT03911973 |
| Olaparib | BC | CDK4,6 and HR |
Palbociclib, Fulvestrant | 1 | NCT03685331 |
| Niraparib | FTC,OC,EndA,PPC | PI3K | Copanlisib | 1 | NCT03586661 |
| Olaparib | TNBC | PD-L1 | Durvalumab | 2 | NCT05209529 |
| Niraparib | PanC | PD-1 | Dostarlimab | 2 | NCT04493060 |
| Talazoparib | melanoma | PD-1 | Nivolumab | 2 | NCT04187833 |
| Niraparib | rare tumors | PD-1 | Sintilimab | 2 | NCT04423185 |
| Olaparib | BC | VEGFR or ATR |
Cediranib or Ceralasertib | 2 | NCT04090567 |
| Fluzoparib | HER2- BC | VEGFR | Apatinib | 3 | NCT04296370 |
| Olaparib | OC, FTC, genital neoplasms |
multiple receptor tyrosine kinases | Anlotinib | 1 | NCT04566952 |
| Olaparib | serous OC | ATR | Ceralasertib | 2 | NCT03462342 |
| Rucaparib | mesothelioma | - | - | 2 | NCT03654833 |
| Olaparib | Pt-resistant OC | CDK4,6 | Abemaciclib | 1/1b | NCT04633239 |
| Olaparib | prostate cancer | LRHL | Leuprolide | 2 | NCT05498272 |
| Talazoparib | OC and other | BRD2,3,4 | ZEN-3694 | 2 | NCT05327010 |
| Veliparib | BC | BRCA1,2 | Temozolomide | 2 | NCT01009788 |
BC – breast cancer, EndA – endometrial adenocarcinoma, FTC – fallopian tube cancer, OC – ovarian cancer, PanC – pancreatic cancer, PPC – primary peritoneal cancer, Pt – platinum-based chemotherapy, TNBC – triple negative breast cancer, UCC – uterine corpus carcinoma.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



