Submitted:
07 July 2021
Posted:
09 July 2021
You are already at the latest version
Abstract
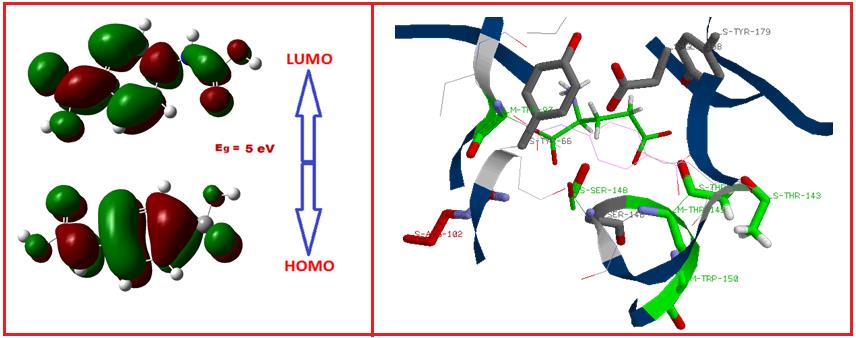
Structure based biological and chemical properties of 4-(carboxyamino)-benzoic acid has been studied by quantum chemical methods. The revamped geometric structure and its quantum chemical parameters were obtained by DFT-B3LYP/6-311G method. Normal mode analysis is performed to assign the fundamental vibrational frequencies as per the potential energy distribution (PED) by using the VEDA program. Simulation of IR and Raman spectral patterns are observed by refinement of scale factors. TD-DFT approach is used to explore the excited states of molecule and prediction of electronic absorption spectra. NMR chemical shifts of the molecule are determined by the gauge independent atomic orbital method. The molecular docking is performed to recognize the binding energy of the ligand with the dynamic site of protein. In our docking analysis, the protein 5DT6 shows the best results than other three proteins which could be used for further analysis. Further inter and intra molecular interactions, electrophilic, nucleophilic and chemical reactivity sites are found by molecular electrostatic potential, HOMO-LUMO and Global chemical reactivity descriptors. Thermodynamic property of the title compound is also reported. The determined quantum chemical parameters show high reactivity and the dipole moment was sufficiently high enough to induce nonlinear characteristics which are required for applications in optoelectronic devices.
Keywords:
DFT
; HOMO
; LUMO
; MEP
; FMO
; RDG
; ADMET
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



