
Submitted:
09 November 2020
Posted:
10 November 2020
You are already at the latest version
Abstract
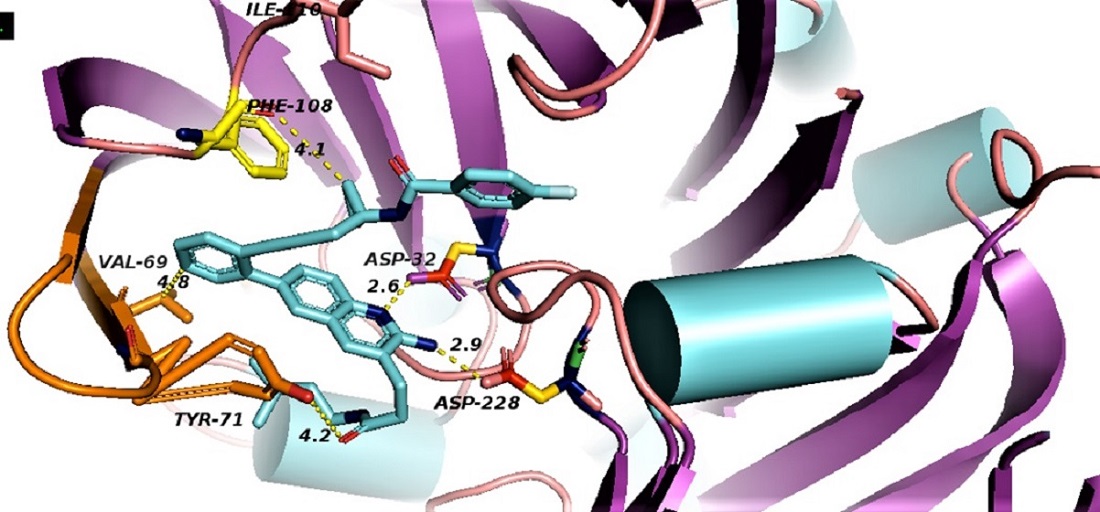
Alzheimer's disease (AD) is a progressive neurodegenerative brain disorder. One of the important therapeutic approaches of AD is the inhibition of β‐site APP cleaving enzyme‐1 (BACE1). This enzyme plays a central role in the synthesis of the pathogenic β-amyloid peptides (Aβ) in Alzheimer's disease. A group of potent BACE1 inhibitors with known x-ray structures (PDB ID 5i3X, 5i3Y, 5iE1, 5i3V, 5i3W, 4LC7, 3TPP) were studied by molecular dynamics simulation and binding energy calculation employing MM_GB(PB)SA. The calculated binding energies gave Kd values 0.139 µM, 1.39 nM, 4.39 mM, 24.3 nM, 1.39 mM, 29.13 mM and 193.07 nM, respectively. These inhibitors showed potent inhibitory activities in enzymatic and cell assays. The Kd values were compared with experimental values, the structures were discussed in view of the energy contributions to binding. Drug likeness of these inhibitors is also discussed. Accommodation of ligands in the catalytic site of BACE1 is discussed depending on the type of fragment involved in each structure. Molecular dynamics (MD) simulations and energy studies were used to explore the recognition of the selected BACE1 inhibitors by Asp 32, Asp228 and the hydrophobic flap. The results show that selective BACE1 inhibition may be due to the formation of strong electrostatic interactions with Asp32 and Asp228 and a large number of hydrogen bonds, π-π and Van der Waals interactions with the amino acid residues located inside the catalytic cavity. Interactions with the ligands show a similar binding mode with BACE1. These results help to rationalize the design of selective BACE1 inhibitors.
Keywords:
Alzheimer's disease
; BACE1
; Molecular dynamics
; MM/GBSA
; Inhibitors
; Drug likeness
; Ligand efficiency
; Kd.
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



