
Submitted:
17 December 2019
Posted:
19 December 2019
You are already at the latest version
Abstract
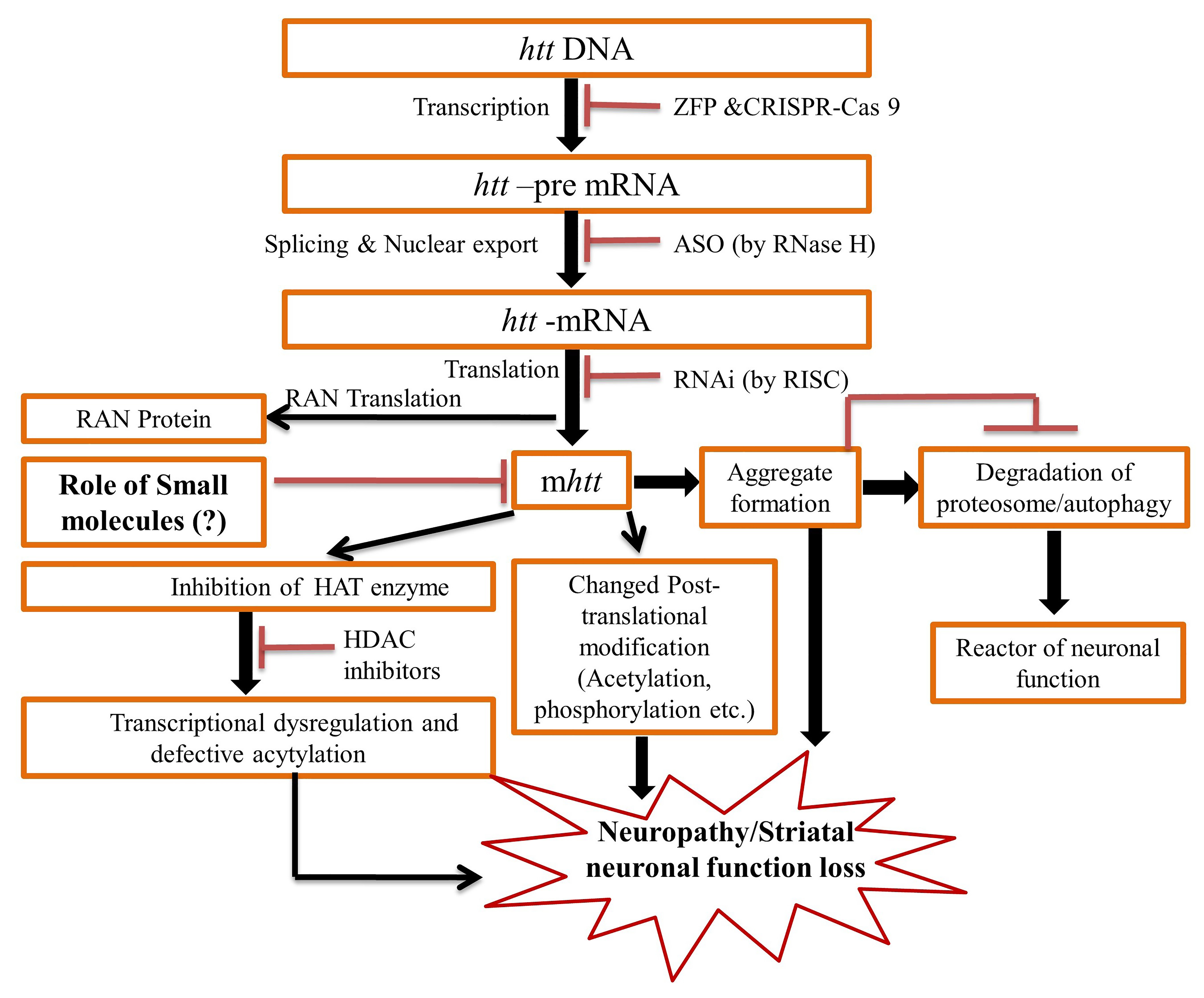
Huntington disease (HD) is an autosomal dominantly inherited fatal neurodegenerative disease. It affects motor, cognitive and psychiatric functions, and ultimately leads to death. The pathology of the disease is due to an expansion of CAG repeats in exon 1 of the huntingtin gene on chromosome 4, which produces a mutant huntingtin protein (mhtt). HD patients manifest a typical phenotype of sporadic, rapid, involuntary control of limb movement, stiffness of limbs, impaired cognition and severe psychiatric disturbances. A variety of symptomatic treatments (which target excitotoxicity, the dopamine pathway, caspases, aggregation, mitochondrial dysfunction, transcriptional dysregulation, mHtt, nucleic acid, neurodegeneration, fetal neural transplants, etc.) are currently available, and new symptomatic and potentially disease-modifying therapies are being actively developed. Recent advances in novel therapeutic strategies, including targeting mutant huntingtin (mhtt) and the htt gene, promise another wave of disease-modifying trials in the near future. A better appreciation of heterogeneous clinical phenomenology and immediate tractable treatment goals coupled with advances in new therapeutics heralds a golden age of HD treatment that will positively impact the quality of life and longevity of HD patients and inform advances in other inherited and neurodegenerative neurological disorders. In the present review literature, our aims to address the latest research on promising therapeutics based on influencing the hypothesized pathological mechanisms associated with HD.
Keywords:
huntington disease
; cag repeat
; mutant huntingtin (mhtt)
; therapeutics
; neurodegeneration
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



