
Submitted:
24 October 2019
Posted:
27 October 2019
You are already at the latest version
Abstract
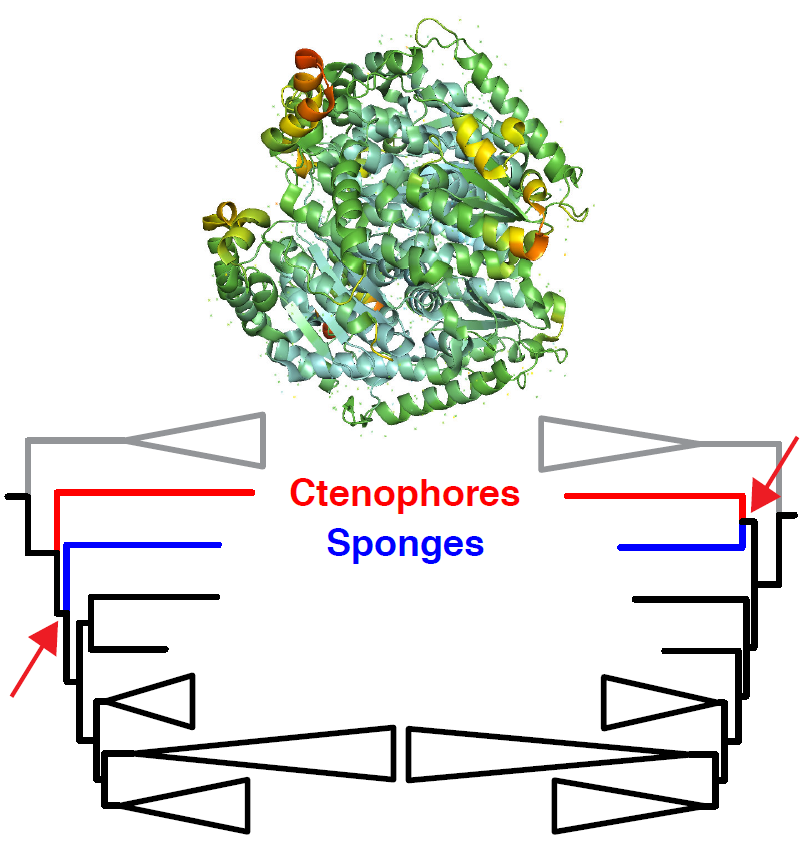
Phylogenomics, the use of large datasets to examine phylogeny, has revolutionized the study of evolutionary relationships. However, genome-scale data have not been able to resolve all relationships in the tree of life; this could reflect, at least in part, the poor-fit of the models used to analyze heterogeneous datasets. Some of the heterogeneity may reflect the different patterns of selection on proteins based on their structures. To test that hypothesis, we developed a pipeline to divide phylogenomic protein datasets into subsets based on secondary structure and relative solvent accessibility. We then tested whether amino acids in different structural environments had distinct signals for the topology of the deepest branches in the metazoan tree. The most striking difference in phylogenetic signal reflected relative solvent accessibility; analyses of exposed sites (on the surface of proteins) yielded a tree that placed ctenophores sister to all other animals whereas sites buried inside proteins yielded a tree with a sponge-ctenophore clade. These differences in phylogenetic signal were not ameliorated when we repeated our analyses using the CAT model, a mixture model that is often used for analyses of protein datasets. In fact, the heterogeneous CAT model resulted in several rearrangements that are unlikely to represent evolutionary history. However, analyses conducted after recoding amino acids to limit the impact of deviations from compositional stationarity increased the congruence in the estimates of phylogeny for exposed and buried sites; after recoding amino acids both trees supported placement of ctenophores sister to all other animals. These results provide striking evidence that it is necessary to achieve a better understanding of the constraints due to protein structure to improve phylogenetic estimation.
Keywords:
phylogenomics
; protein structure
; secondary structure
; relative solvent accessibility CAT model
; non-stationary models
; RY coding
; metazoan phylogeny
; ctenophora
; porifera
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



